
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 24 September 2019
Sec. B Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02136
This article is part of the Research TopicNext Generation B cell Targeting Therapies in Autoimmune DiseasesView all 5 articles
 Sarah Y. Weißenberg1,2
Sarah Y. Weißenberg1,2 Franziska Szelinski1,2
Franziska Szelinski1,2 Eva Schrezenmeier1Ana-Luisa Stefanski1
Eva Schrezenmeier1Ana-Luisa Stefanski1 Annika Wiedemann1
Annika Wiedemann1 Hector Rincon-Arevalo1,2,3Anna Welle4Annemarie Jungmann4
Hector Rincon-Arevalo1,2,3Anna Welle4Annemarie Jungmann4 Karl Nordström4Jörn Walter4
Karl Nordström4Jörn Walter4 Juliana Imgenberg-Kreuz5
Juliana Imgenberg-Kreuz5 Gunnel Nordmark5
Gunnel Nordmark5 Lars Rönnblom5Prathyusha Bachali6Michelle D. Catalina6
Lars Rönnblom5Prathyusha Bachali6Michelle D. Catalina6 Amrie C. Grammer6
Amrie C. Grammer6 Peter E. Lipsky6
Peter E. Lipsky6 Andreia C. Lino1,2†
Andreia C. Lino1,2† Thomas Dörner1,2*†
Thomas Dörner1,2*†Autoimmune diseases (AID) such as systemic lupus erythematosus (SLE), primary Sjögren's syndrome (pSS), and rheumatoid arthritis (RA) are chronic inflammatory diseases in which abnormalities of B cell function play a central role. Although it is widely accepted that autoimmune B cells are hyperactive in vivo, a full understanding of their functional status in AID has not been delineated. Here, we present a detailed analysis of the functional capabilities of AID B cells and dissect the mechanisms underlying altered B cell function. Upon BCR activation, decreased spleen tyrosine kinase (Syk) and Bruton's tyrosine kinase (Btk) phosphorylation was noted in AID memory B cells combined with constitutive co-localization of CD22 and protein tyrosine phosphatase (PTP) non-receptor type 6 (SHP-1) along with hyporesponsiveness to TLR9 signaling, a Syk-dependent response. Similar BCR hyporesponsiveness was also noted specifically in SLE CD27− B cells together with increased PTP activities and increased transcripts for PTPN2, PTPN11, PTPN22, PTPRC, and PTPRO in SLE B cells. Additional studies revealed that repetitive BCR stimulation of normal B cells can induce BCR hyporesponsiveness and that tissue-resident memory B cells from AID patients also exhibited decreased responsiveness immediately ex vivo, suggesting that the hyporesponsive status can be acquired by repeated exposure to autoantigen(s) in vivo. Functional studies to overcome B cell hyporesponsiveness revealed that CD40 co-stimulation increased BCR signaling, induced proliferation, and downregulated PTP expression (PTPN2, PTPN22, and receptor-type PTPs). The data support the conclusion that hyporesponsiveness of AID and especially SLE B cells results from chronic in vivo stimulation through the BCR without T cell help mediated by CD40–CD154 interaction and is manifested by decreased phosphorylation of BCR-related proximal signaling molecules and increased PTPs. The hyporesponsiveness of AID B cells is similar to a form of functional anergy.
Breach of self-tolerance, maintenance of autoimmune memory, and continuous autoantibody production are important pathologic features of B cells in autoimmune diseases (AID) (1) such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and primary Sjögren's syndrome (pSS). A number of phenotypic abnormalities of peripheral B cell subsets have been reported in AID (2–10), but their relationship to the functional abnormalities of the B cell axis has not been fully delineated. As antigen-induced B cell receptor (BCR) signaling is crucial for B cell fate (11–13), the BCR plays a pivotal role in the development and maintenance of autoimmunity. Therefore, BCR signaling has been extensively studied and there is general consensus that pathologically increased BCR signaling contributes to B cell overactivity and autoimmunity. This was the rationale for testing the impact of the anti-CD22 monoclonal antibody, epratuzumab, in SLE, since it was thought that CD22 engagement would impose negative regulation of BCR signaling (14). Since this trial failed to meet its primary endpoint (15), it is possible that hyperreactive BCR signaling is not central to SLE and perhaps other AID.
The BCR acts as a signal transducer, integrating receptor occupancy to downstream events that regulate cellular survival and activation. The strength of the signal determines cell fate and is tightly balanced by the activities of stimulatory and inhibitory molecules, including various phosphotyrosine kinases (PTKs) and phosphotyrosine phosphatases (PTPs) (11–13). Antigen binding induces phosphorylation of the BCR-associated Igα (CD79a) and Igβ (CD79b) chains leading to downstream Lyn and spleen tyrosine kinase (Syk) phosphorylation (16). This activates 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 (PLCγ2), Bruton's tyrosine kinase (Btk), and protein kinase B (Akt), which results in Ca2+- and Akt-dependent transcription (17–19). As a negative feedback loop, e.g., CD22 becomes phosphorylated and recruits PTP non-receptor type 6 (SHP-1) to the BCR, which dephosphorylates BCR downstream targets (14, 20, 21).
Genome-wide association studies (GWAS) identified polymorphisms of BCR downstream scaffold proteins, PTKs and PTPs, as associated with AID, supporting the idea of intrinsically defective BCR signaling in AID (22–29). Despite these suggestions, the major understanding of BCR signaling in autoimmunity is mainly based on studies in mice, in which BCR hyperreactivity can be a main driver of autoimmunity (30). However, in humans, the data are contradictory. There are studies reporting increased BCR signaling measured by Ca2+ release and downstream tyrosine phosphorylation related to a lack of negative regulation, such as by low-affinity immunoglobulin gamma Fc receptor II-b (FcγRIIb), phosphatase and tensin homolog (PTEN), or Lck/Yes novel tyrosine kinase (Lyn), in B cells from SLE patients (31–37). In contrast, a growing body of literature provides evidence that the BCR signal in autoimmunity is impaired, at least in some B cell subsets, with reduced tyrosine phosphorylation, Ca2+ release, and recruitment of signaling kinases to lipid rafts upon BCR stimulation (38–41).
In addition to abnormalities in the BCR pathway of stimulation, T cell-independent responses, such as TLR9 responses, have been reported to enhance B cell activation, in particular when autoantigen/ribonucleoprotein-immune complexes simultaneously engage BCR and TLR9 (42). However, recent reports indicate that SLE B cells also display low reactivity following TLR9 signaling (43–45). In addition to intrinsic B cell abnormalities, alteration of the functional status of other cells involved in regulating antibody production may also contribute to the development of AID. In this regard, abnormal germinal center (GC) reactions in autoimmune tissues (46), disturbances of regulatory T cells (1), increased TFH (47), abnormalities of CD4+ (48), and CD8+ T cells (49, 50) with diminished T cell responses have also been reported. Therefore, the precise set of abnormalities underlying B cell dysfunction in AID remains to be fully delineated.
Here, we carried out a comparative analysis of peripheral as well as tissue-resident B cells from different AID patients and demonstrate that AID B cells share a phenotype of hyporesponsiveness toward BCR and TLR9 stimulation. This suggests a common signaling dysfunction between these diseases. However, SLE B cells appear to display a more prominent phenotype as not only antigen experienced conventional CD27+ memory but also CD27− B cells exhibit BCR dysfunction. Of note, within this study, we compare characteristics of CD27− and CD27+ B cells that can also include CD27− isotype switched B cells or switched and non-switched CD27+ memory B cells (7, 8, 51, 52). The composition of CD27− and CD27+ B cell subsets may differ among AID patients and healthy donors (HD) (53). Therefore, analyzing CD27− and CD27+ subsets as a whole prevented us from determining whether there were subtle changes in different subsets. This decreased responsiveness, likely induced by chronic BCR engagement in vivo, can be partially overcome by CD40 engagement, which reduced the expression of PTPs, such as PTPN22. Therefore, the functional B cell anergy detected in vitro in AID B cells appears to reflect intensive BCR engagement in vivo.
EDTA anticoagulated peripheral blood samples were obtained from 85 SLE, 42 RA, and 51 pSS patients and 118 HD. Donor details are listed in Table S1. Patients with RA fulfilling the ACR/EULAR criteria (54), SLE meeting the SLICC criteria (55), and pSS fulfilling the AECG criteria (56) were included in this study. Tissue samples were obtained from surgeries: four spleen samples from patients with immune thrombocytopenia (ITP), seven spleen samples and four tonsil samples from patients without autoimmune background, and one parotid sample from a pSS patient. All patients and donors gave their consent according to the approval of the local ethics' committee at the Charité University Hospital Berlin. Written consensus was obtained from all patients and controls.
Staining antibodies, stimulation reagents, media, and other reagents were purchased from BD Bioscience (Franklin Lakes, NJ, USA), BioLegend (San Diego, CA, USA), eBioscience/Thermo Fisher (Carlsbad, CA, USA), Illumina (San Diego, CA, USA), Invitrogen/Thermo Fisher (Carlsbad, CA, USA), Life Technologies/Thermo Fisher (Carlsbad, CA, USA), Miltenyi Biotec (Bergisch Gladbach, Germany), Jackson ImmunoResearch (West Grove, FL, USA), PeproTech (Rocky Hill, NJ, USA), Promega Corporation (Madison, WI, USA), Selleck Chemicals (Houston, TX, USA), Sigma-Aldrich (St. Louis, MO, USA), and UCB Pharma (Slough, UK) and listed in Tables S2, S3. Quality control of flow cytometry stainings was performed using SPHERO Rainbow Calibration Particles (BD Bioscience, Franklin Lakes, NJ, USA) and Cytometer Setup and Tracking beads (BD Biosciences, Franklin Lakes, NJ, USA) for stable MFIs over time (57).
Fresh peripheral whole blood (100 μl) was lysed and fixed in 1 ml pre-warmed Lyse/Fix buffer (BD Bioscience) for 10 min at 37°C. Permeabilization and staining were performed as previously described (40). Cells were stained with anti-CD3, -CD14, -CD19, -CD20, and -CD27 or combinations of anti-protein and anti-phospho-protein antibodies: Syk/pSyk(Y352), Akt1/pAkt(S473), Btk/pBtk(Y223), and PLCγ2/pPLCγ2(Y759), respectively. Flow cytometry analysis was performed using a FACSCanto II flow cytometer (BD Bioscience Franklin Lakes, NJ, USA). The gating strategy used is shown in Figure S1. As an internal negative control, CD3+ T cells were included for all protein and phospho-protein antibodies assays except Akt1, where an isotype control was conducted, as Akt1 is widely expressed among lymphocytes.
PBMCs were isolated using density gradient centrifugation method as described previously (40). Freshly isolated cells were directly suspended in ice-cold MACS rinsing buffer (with BSA; Miltenyi) for B and T cell purification, in pre-warmed RPMI 1640 (with GlutaMAX, Life Technologies) for short-term stimulation assays, or in pre-warmed phosphate buffered saline (PBS) for carboxyfluorescein succinimidyl ester (CFSE, Invitrogen/Thermo Fisher, Carlsbad, CA, USA) staining and long-term in vitro culture. Cells from at least one HD and one patient were analyzed simultaneously to enhance reliability.
MNCs from tissues were isolated from spleens, tonsils, and parotid as described previously (58). Cells were released from minced tissue samples by shaking with ice-cold MACS buffer. Samples were filtered (70 μm cell strainer, Corning, NY, USA) and MNCs were isolated using density gradient centrifugation. Residual erythrocytes were removed using EL Buffer (Quiagen, Venlo, Netherlands). Cells were stored at −20°C within FBS/DMSO buffer.
B and T cell enrichment from PBMCs was carried out using human B cell Kit II or human Pan T cell kit (Miltenyi Biotec, Bergisch-Gladbach, Germany) for magnetic cell sorting according to the manufacturer's protocols. B and T cell purities were checked by flow cytometry after staining with anti-biotin and anti-CD19 or anti-CD3 antibodies. Cell suspensions with 82% purity were used for further experiments.
Purified B or T cells were lysed for 30 min on ice with Halt Protease Inhibitor Cocktail (1% in Pierce IP Lysis Buffer; Thermo Fisher). Then, the assay was processed according to the manufacturer's protocol and as described previously (59) using a commercial PTP and protein serine/threonine phosphatase (PSP) activity kit (Promega Corporation); 25,000 cells/well (PTP) and 80,000 cells/well (PSP) were used. In order to ensure the specificity of the PTPs and PSP activity, the same experiments were performed using the inhibitors monovanadate (10 mM) and sodium fluoride (10 mM) (Sigma-Aldrich), respectively. Cell lysates were analyzed at 600 nm using a Spectramax Plus 384 micro plate reader (Molecular Devices, San Jose, CA, USA). Phosphatase activity was quantified by the release of free phosphate. Concentrations were assessed from standard dilution series.
For functional phosphorylation kinetics, PBMCs or thawed MNCs (106 cells) were rested for 1 h at 37°C in RPMI and stimulated with anti-IgG/IgM F(ab′)2 (13 μg/ml) for 2, 5, 8, 15, and 30 min, respectively. An additional RPMI control served as control at baseline. BCR stimulation was stopped by adding 1 ml of pre-warmed Lys/Fix buffer (BD Bioscience). Lysis, fixation, permeabilization, and staining were performed as described previously (40). Cells were stained with anti-CD3, -CD14, -CD19, -CD20, -CD27, and combinations of Syk/pSyk(Y352), Syk/pAkt(S473), or Btk/pBtk(Y223), respectively. Flow cytometry analysis was performed using a FACSCanto II flow cytometer. MFIs were used to assess phosphorylation intensity of phospho-proteins within different B cell subsets (gating strategy see Figure S1). Previously reported CD27−Syk++ cells (60) were excluded in pSyk(Y352) and pAkt(S473) kinetics, because they have been shown to represent a population of memory-like B cells.
For chronic BCR stimulation experiments, cells were pre-incubated with anti-IgG/IgM (2 μg/ml), CpG (0.5 μg/ml) or RPMI for 24, 48, or 72 h (37°C, 5% CO2) and subsequently stimulated with anti-IgG/IgM or RPMI as a control for 5 min. For co-stimulation experiments, cells were pre-incubated with recombinant human IL-4 (20 ng/ml) or IL-21 (20 ng/ml) or CD40L (500 ng/ml, human CD40L Multimer kit, Miltenyi Biotec) or combinations thereof for 48 h (37°C, 5% CO2) and subsequently stimulated with anti-IgG/IgM or RPMI as a control for 5 min. Cells were lysed, fixed, permeabilized, stained for Syk/pSyk(Y352), and analyzed as described above. Flow cytometry analysis was performed using a FACSCanto II or LSRFortessa flow cytometer.
Purified B cells were suspended in 100 μl of RPMI (0.2 × 106 cells) equilibrated for 1 h at 37°C, stimulated with anti-CD22 F(ab′)2 epratuzumab-A488 (10 μg/ml) (UCB) or left unstimulated, fixed, and permeabilized as described above. Cells were stained with mouse-anti-human-SHP-1 (BD Bioscience) and, for unstimulated control, with anti-CD22 F(ab′)2 epratuzumab-A488. Washed cells were stained with the secondary antibodies donkey-anti-mouse-RRX. Finally, cells were centrifuged onto slides and covered with Vectashield HardSet mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, USA) to stain the nucleus. Co-localization/fluorescence overlap of SHP-1/CD22 was evaluated (0 = no co-localization, 1 = all pixels co-localized) using a Nikon A1-Rsi confocal microscope and NIS Elements C imaging software (Nikon, Tokyo, Japan).
For cell proliferation analysis upon B cell stimulation, PBMCs (2 × 106 cells/ml in PBS) were stained with CFSE (5 μM; Invitrogen) for 4 min at 37°C prior stimulation. After staining, cells were washed and incubated with 10 ml of RPMI for 30 min at 37°C. Before proceeding to the stimulation protocol, cells were washed with RPMI (10% FBS, 1% P/S).
PBMCs (106 cells per well) were rested for 1 h at 37°C and subsequently stimulated with CpG (0.5 μg/ml), anti-IgG/IgM (2 μg/ml), or CD40L (500 ng/ml) and the combinations CpG/CD40L, CpG/anti-IgG/IgM, or CpG/anti-IgG/IgM/CD40L for 5 days (37°C, 5% CO2) in RPMI (10% FBS, 1% P/S). For B cell proliferation, recombinant human IL-2 (20 ng/ml) and IL-10 (20 ng/ml) (Miltenyi Biotec) were added to the culture. After stimulation, cells were washed with MACS buffer and stained for 15 min at 4°C. Cells were stained with CD3, CD14, CD19, CD20, CD27, and CD38. Before flow cytometry analysis, DAPI was added to exclude dead cells. Cells were analyzed using a FACSCanto II flow cytometer (BD Bioscience; see gating strategy on Figure S5).
Idat files of the Illumina Infinium HumanMethylation450 BeadChip data (61–63) were processed with RnBeads (v1.6.1). Data were aligned to hg 19 reference genome, and SNPs and sex chromosomes were excluded from further processing. A bead count cutoff of ≥10 and a greedy cut p-value cutoff of ≤ 0.01 was set to filter the data for high quality. The data were normalized by using the swan method. DMCs were defined as CpGs with ≥5% DNA methylation difference and p ≤ 0.01.
Isolated PBMCs were cultured overnight with CD40L (500 ng/ml) and IL-4 (20 ng/ml) or medium as a control. FcR blocking reagent was added to cell suspension before cells were stained with anti-CD14, -CD27, -CD20, -CD19, -CD3, -CD16, and DAPI, and CD19+ B cells were sorted directly into Arcturus PicoPure Extraction Buffer (Thermo Fisher) using a FACSAria™ II Sorter (BD Bioscience). Stranded sequencing library preparation of total mRNA was done using TruSeq RNA library prep kit (Illumina). Around 60 million passed filter reads per library were collected in a paired end read mode with 50-bp sequences each by the HiSeq2500 Illumina system.
RNA-Seq from stimulated and control CD19+ B cells was performed on two SLE and one HD and deposited under PRJNA564980 at NCBI sequence read archive. Three technical replicates were included for each cohort and time point; files were obtained from FASTQC. FASTQC, Trimmomatic, STAR, Sambamba, and featureCounts were done separately. After careful examination of the PCA plots, three technical replicates of each cohort and condition were averaged into one and then used to perform relative gene expression. After FASTQC quality control analysis, Trimmomatic was used to cut adapter sequences, low-quality reads, and the first 14 reads of each sequence due to non-random primer bias. Reads were aligned to the human reference genome hg38 in STAR, and the.sam files were converted to sorted.bam files using Sambamba. Relative DE counts were generated in featureCounts. FastQC, Trimmomatic, STAR, Sambamba, and the featureCounts programs are all free, open source programs available at the following web addresses:
FastQC—https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Trimmomatic—http://www.usadellab.org/cms/?page$=$~trimmomatic
STAR—https://github.com/alexdobin/STAR
http://labshare.cshl.edu/shares/gingeraslab/www-data/dobin/STAR/STAR.posix/doc/STARmanual.pdf
Sambamba—http://lomereiter.github.io/sambamba/
FeatureCounts—http://subread.sourceforge.net/
Data were derived from publicly available data sets: GSE4588 CD20+ B cells from SLE and HD (6 SLE, 7 HD), GSE117935 CD19+ B cells from multiple sclerosis (MS) patients and HD (10 MS, 10 HD), E-MTAB-2713 CD4+ T cells from SLE and HD (53 SLE, 41 HD), and E-MTAB-2713 CD8+ T cells from SLE and HD (22 SLE, 31 HD). DE was done for each data set of SLE patients and HD. GCRMA normalized expression values were variance corrected using local empirical Bayesian shrinkage before calculation of DE using the ebayes function in the open source package BioConductor LIMMA package (https://www.bioconductor.org/packages/release/bioc/html/limma.html). Resulting p-values were adjusted for multiple hypothesis testing and filtered to retain DE probes with an FDR < 0.2 (64).
All flow cytometry data were analyzed with FlowJo (version 10.3, TreeStar, Ashland, OR. USA). Statistical analysis was performed with GraphPad Prism (version 5.04, GraphPad Software, La Jolla, CA. USA). For all data sets, Gaussian distribution was assumed. For the comparison of two groups, unpaired t-test was applied, and for paired analysis, paired t-test was applied. When multiple groups were compared, one-way ANOVA with Dunnett's test for multiple comparisons (DMCT) was applied. For the comparison of time-dependent kinetics and multiple groups, two-way ANOVA with Bonferroni test for multiple comparisons (BMCT) was used.
BCR signaling is regulated by a finely tuned balance of PTKs and PTPs (16, 65). In a previous study, we found that B cells from SLE patients have enhanced PTP activities and reduced Syk phosphorylation compared to HDs (40). Most SLE patients exhibited this abnormality, independent of disease activity. To determine whether these abnormalities are unique to SLE, we assessed PTP and PSP activities in SLE, RA, pSS, and HD CD19+ B cells (Figure 1A). SLE B cells exhibited significantly increased PTP activities for both PTP-specific substrates (TyrPP1 and TyrPP2), whereas no significantly increased PTP activities were found in RA and pSS B cells (Figure 1A). Similarly, we found that PSP activity was uniquely increased in SLE, but not in RA or pSS patients compared to HD. We employed specific PTP and PSP inhibitors to ensure specificity of the PTPs/PSPs analyzed. PTP activity could be blocked and PSP inhibition was dose dependent (Figures 1A,B). PTP and PSP activities in CD3+ T cells from AID patients were similar to HD (Figure S1), suggesting that enhanced PTP/PSP activity is a unique characteristic of SLE B cells.
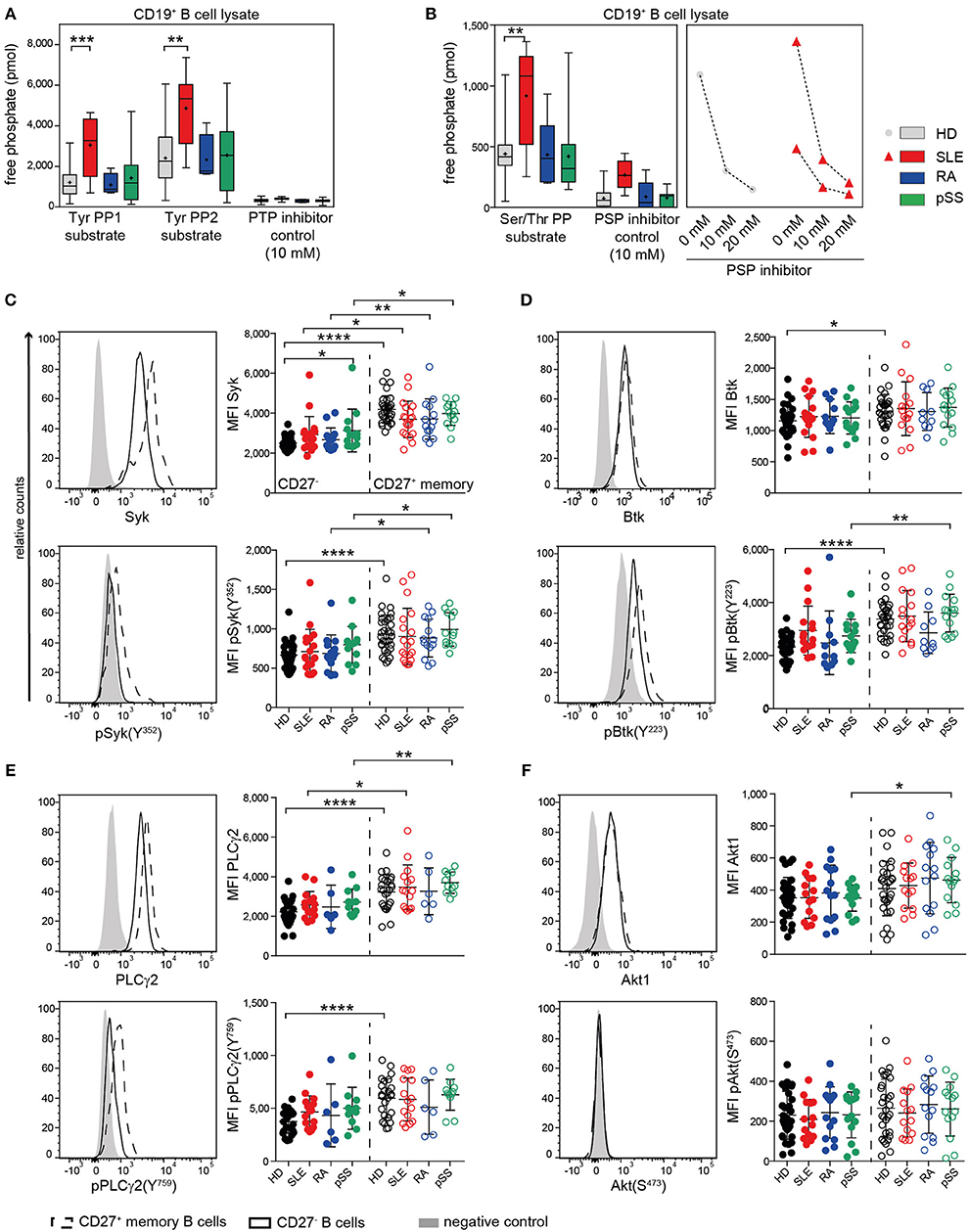
Figure 1. Enhanced PTP activity in SLE B cells and similar kinase expression and phosphorylation in AID among CD27− and CD27+ memory B cells. (A) PTP [n(HD/SLE/RA/pSS) = 22/8/4/13] and (B) PSP [n(HD/SLE/RA/pSS) = 19/8/5/10] activities of CD19+ B cells in HD (gray), SLE (red), RA (blue), and pSS (green). CD27− (dots) and CD27+ memory (open circles) B cells from HD (black), SLE (red), RA (blue), and pSS (green) patients were analyzed for the expression of (C) Syk/pSyk(Y352) [n(HD/SLE/RA/pSS) = 32/19/14/13]; (D) Btk/pBtk(Y223) [n(HD/SLE/RA/pSS) = 30/16/11/15]; (E) PLCγ2/pPLCγ2(Y759) [n(HD/SLE/RA/pSS) = 25/15/6/11] and (F) Akt1/pAkt(S473) [n(HD/SLE/RA/pSS) 30/14/14/13]. Representative histograms of CD27− (solid line); CD27+ (dashed line), and negative control (gray) are shown. Box whisker plots represent median (line), mean (plus), and the range from minimum to maximum; lines in scatter dot plots represent means ± SD (ANOVA with DMCT; t-test; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
The location of a PTP, such as SHP-1, is important for BCR signaling regulation. Here, we analyzed the co-localization of SHP-1 (PTPN6) with CD22 with and without anti-CD22 engagement, which recruits and activates SHP-1. Cap formation was not confined to one or the other AID, with increased baseline co-localization of SHP-1 and CD22 in SLE, pSS, and RA patients. Notably, the degree of co-localization constitutively present in SLE B cells appeared at a maximum and could not be further increased upon CD22 engagement. In contrast, CD22 engagement increased co-localization of SHP-1 in RA, pSS, and HD B cells (Figure S1). The data suggest that a functionally active PTP complex of SHP-1 was substantially increased in SLE B cells at baseline.
Next, we studied the status of BCR downstream kinases and phospholipase PLCγ2 in CD27− and CD27+ memory B cells of AID patients. We analyzed the baseline phosphorylation of Syk, Btk, and PLCγ2, which are regulated by PTPs. Furthermore, we analyzed Akt1 regulated by PSPs. We found similar basal Syk expression and phosphorylation at the activation site Syk(Y352) within CD27− and CD27+ memory B cells in patients and HD (Figure 1C). Syk expression and baseline phosphorylation were generally higher in CD27+ memory than in CD27− B cells across all groups. For the downstream signaling molecules Btk and PLCγ2 and their phosphorylation sites Btk(Y223) and PLCγ2(Y759), we found similar expression and phosphorylation levels among all patients and HD (Figures 1D,E). Akt1 expression and phosphorylation at Akt(S473) were comparable among CD27− and CD27+ B cells from all donor groups (Figure 1F). Constitutive expression and phosphorylation levels of BCR-associated kinases (Figure 1) were overall similar to HD in all AID tested in both B cell subsets, despite increased constitutive phosphatase activity in SLE B cells.
Subsequently, we assessed phosphorylation of Syk(Y352), Btk(Y223), and Akt(S473) following BCR engagement in AID and HD CD27− and CD27+ memory B cells (Figure 2, Figure S2). Even though the composition of switched and non-switched cells among CD27− and CD27+ B cells may differ among patients and HD, we did not observe substantial heterogeneity on the functional level, except within the recently reported CD27−Syk++ cells, which were excluded from this analysis.
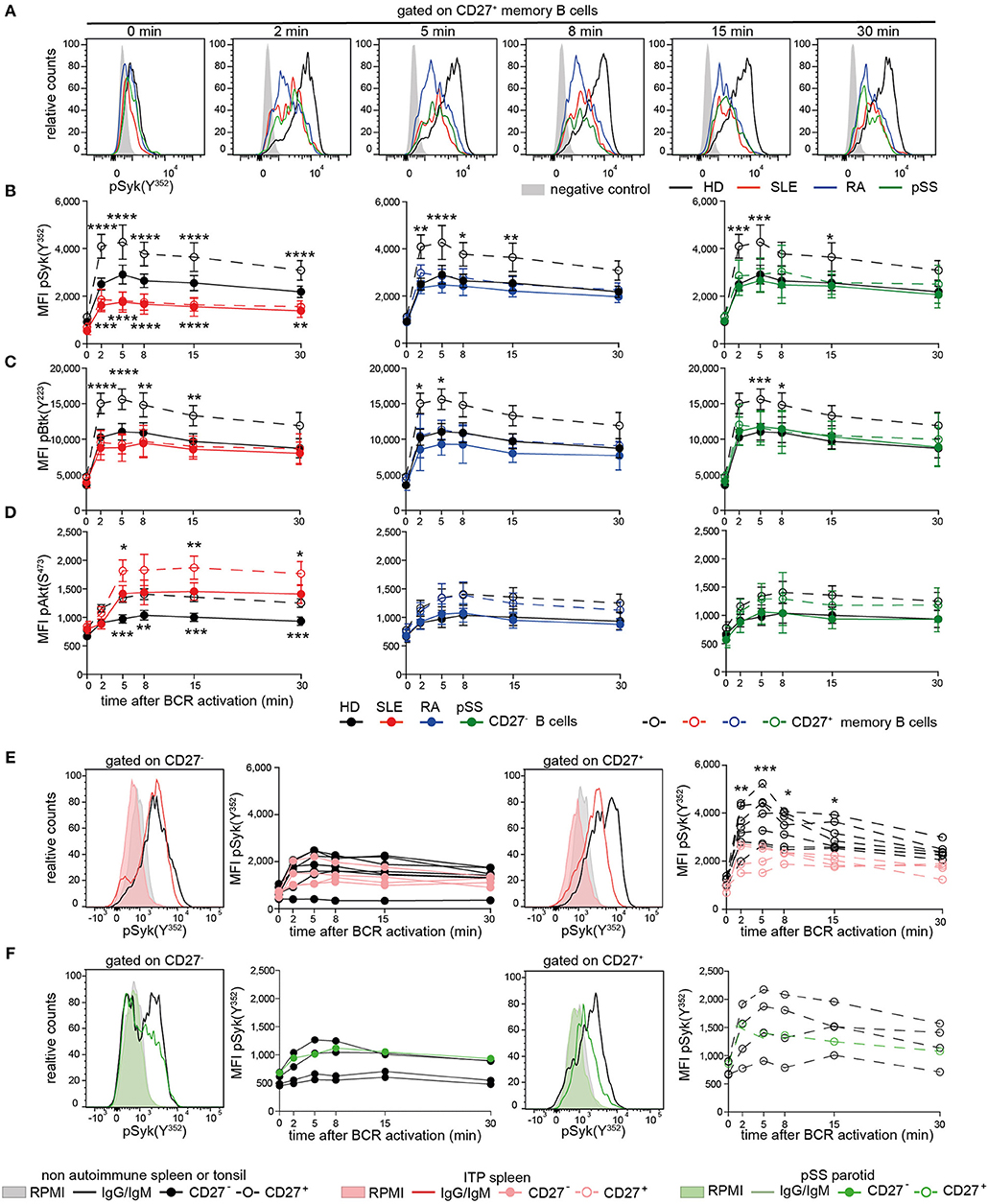
Figure 2. Reduced PTK phosphorylation upon BCR signaling in peripheral and tissue-resident CD27+ B cells from patients with AID. PBMCs or MNCs from HD (black), SLE (red), RA (blue), pSS (green), and ITP (pink) vs. non-ITP (black) spleens and pSS parotid gland (green) vs. tonsils (black) were stimulated with anti-IgG/IgM F(ab)2. (A) Representative pSyk(Y352) histograms from CD27+ memory B cells are shown. CD3+ T cells served as control (gray). Phospho-kinetics of (B) Syk(Y352) [n(HD/SLE/RA/pSS) = 19/11/11/10], (C) Btk(Y223) [n(HD/SLE/RA/pSS) = 21/10/6/14], and (D) Akt(S473) [n(HD/SLE/RA/pSS) = 29/15/11/10] in CD27+ memory (dashed lines) and CD27− (solid lines) B cells are shown. Phospho-Syk(Y352) kinetics in B cells from (E) spleen and (F) tonsil/parotid with representative histograms [nspleens(non-ITP/ITP) = 7/4; n(tonsils/parotid) = 4/1]. Histograms show unstimulated (filled areas) vs. stimulated (solid lines) cells. Data show mean ±95% CI (two-way ANOVA with BMCT for the comparison of AID vs. HD CD27− or CD27+ memory B cells, respectively; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
BCR-induced phospho-(p)Syk(Y352) in AID CD27+ memory B cells and SLE CD27− B cells was significantly lower compared to HDs (Figures 2A,B). Btk(Y223) phosphokinetics were qualitatively similar to that of pSyk(Y352), with a maximum phosphorylation after 5 min (Figure 2C). AID CD27+ memory B cells revealed significantly reduced pBtk(Y223) in comparison to HDs, whereas Btk(Y223) phosphorylation was reduced in SLE and RA CD27− B cells. SLE B cells do not differ in surface IgG and IgM expression compared to HD (40), indicating that this phenotype is not related to reduced Ig expression. Furthermore, we observed reduced phosphorylation in pSS patients who had not received any treatment, suggesting that these effects were not treatment dependent.
Akt(S473) phosphokinetics displayed a maximum after 8 min (Figure 2D). Phospho-Akt(S473) in both B cell compartments from RA and pSS patients was comparable to those from HDs, whereas SLE patients showed increased pAkt(S473) compared to HD, at all-time points. Of particular note, memory B cells from HD expressed higher levels of tested phospho-PTKs and pAkt(S473) than CD27− B cells upon BCR engagement as a general characteristic.
Taken together, diminished pSyk(Y352) and pBtk(Y223), but not pAkt(S473), upon BCR stimulation is a common characteristic of AID memory B cells, whereas it is also present in SLE CD27− B cells, suggesting that the SLE B cell compartment is more globally abnormal.
To determine whether these findings are restricted to circulating B cells, we analyzed tissue-resident CD27− and CD27+ B cells from autoimmune and non-autoimmune patients (Figures 2E,F). ITP patients' spleens were compared to control spleens from non-autoimmune patients. Control tonsils were obtained from patients undergoing tonsillectomy. Parotid tissue was received from one pSS patient undergoing parotidectomy to exclude lymphoma.
We found reduced pSyk(Y352) kinetics in ITP CD27+ memory B cells compared to control spleens, whereas pSyk(Y352) kinetics in CD27− B cells were comparable to controls (Figure 2E). Phospho-Syk(Y352) was also diminished in tonsillar and parotid B cells (Figure 2F). These data indicate that B cells with diminished BCR-induced PTK phosphorylation particularly in the CD27+ memory B cell compartment are present in tissues of AID patients and in the tonsil.
Subsequently, we tested whether reduced BCR signaling observed in SLE B cells and RA and pSS memory B cells can be induced in HD B cells by continuous stimulation with anti-BCR or TLR9 antagonists (Figure 3). Previously, we showed that ex vivo IgG and IgM expression does not differ between SLE and HD B cells (40). Here, continuous stimulation of HD B cells with anti-BCR, but not with CpG, resulted in reduced Syk(Y352) phosphorylation upon subsequent anti-BCR stimulation. This was observed for CD27− and CD27+ memory HD B cells (Figures 3A–C).
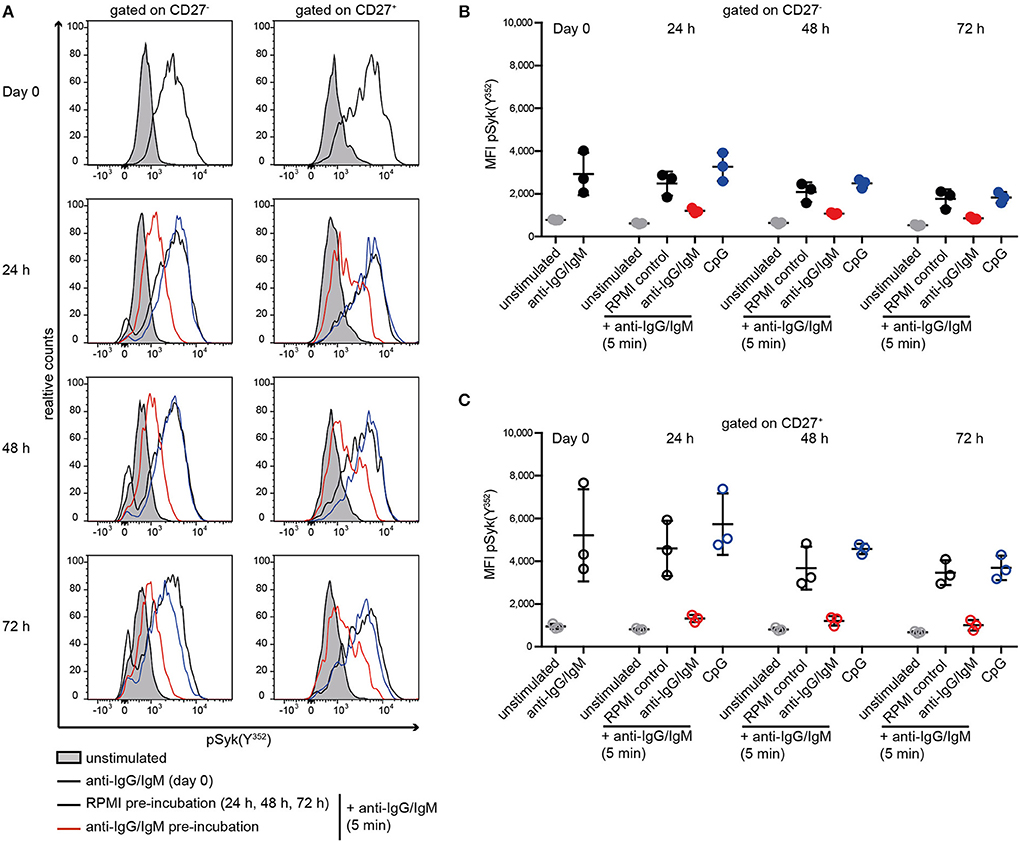
Figure 3. Reduced Syk(Y352) phosphorylation upon re-stimulation of the BCR. PBMCs from HD were incubated with anti-IgG/IgM, CpG, or RPMI as a control for 24, 48, or 72 h and re-stimulated with anti-IgG/IgM for 5 min. (A) Representative histograms of Syk(Y352) phosphorylation at day 0 and without (black line) or with anti-IgG/IgM (red) or CpG (blue) pre-incubation for 24, 48, or 72 h. The unstimulated control is shown as a gray peak. Anti-IgG/IgM induced pSyk(Y352) MFIs in (B) CD27− B cells (solid dots) and (C) CD27+ B cells (open circles) at day 0 or after pre-incubation with RPMI as a control (black), anti-IgG/IgM (red), or CpG (blue) for 24, 48, and 72 h. Unstimulated controls are displayed as gray dots. Horizontal lines represent means ± SD [n(HD) = 3].
Next, we tested the impact of CD40 co-stimulation on BCR-mediated signaling of AID B cells (66). We measured pSyk(Y352) upon CD40 co-stimulation followed by BCR engagement in CD27− and CD27+ memory B cells from patients and HD (Figure 4).
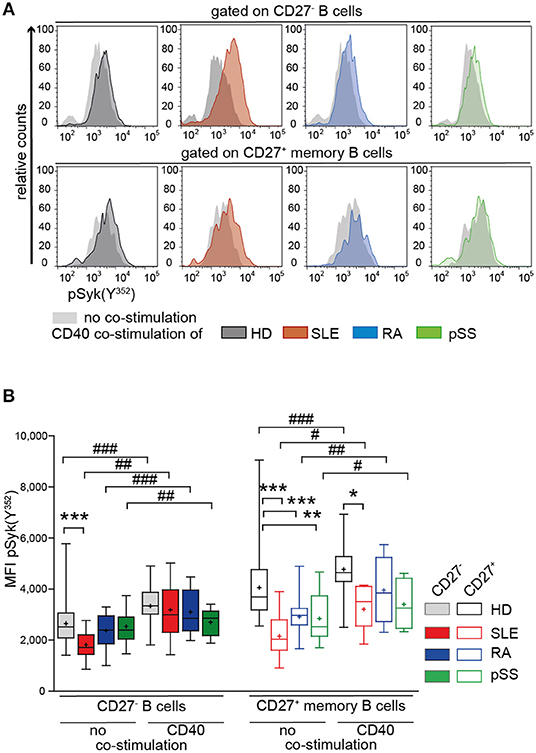
Figure 4. Increased Syk(Y352) phosphorylation reflecting BCR responsiveness of CD27+ memory B cells from HD, SLE, RA, and pSS upon co-stimulation with CD40L. PBMCs from HD (black), SLE (red), RA (blue), and pSS (green) patients were treated with CD40L and subsequently stimulated with anti-IgG/IgM F(ab)2. (A) Representative histograms of pSyk(Y352) in CD27− (top row) and CD27+ memory (bottom row) B cells in the presence (colored areas) or absence (gray areas) of prior CD40L co-stimulation. (B) pSyk(Y352) in CD27− (filled boxes) and CD27+ memory (clear boxes) B cells with [n(HD/SLE/RA/pSS) = 11/7/5/5] and without [n(HD/SLE/RA/pSS) = 30/18/16/15] CD40 co-stimulation. Box whisker plots represent median (line), mean (plus), and the range from minimum to maximum (ANOVA with DMCT, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; paired t-test, #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001).
Increased pSyk(Y352) was observed in CD27− and CD27+ memory B cells from AID patients and HDs after treatment with CD40L compared to BCR stimulation alone (Figure 4A). After CD40L co-stimulation, Syk(Y352) phosphorylation in RA and pSS CD27+ memory B cells and in SLE CD27− B cells was comparable to HD (Figure 4B), whereas BCR engagement alone gave rise to diminished pSyk(Y352) (Figures 2B, 4B). pSyk(Y352) expression by SLE CD27+ memory B cells increased upon CD40L co-stimulation compared to BCR stimulation alone, even though the phosphorylation amplitude remained lower than those in HD (Figure 4B). These data suggest improved BCR responsiveness by CD40L co-stimulation in memory B cells and in CD27− SLE cells.
It has been reported that Th2 signals restore BCR signaling in a small population of anergic IgM−IgD+CD27− B cells present in blood of HD and SLE patients (67). Therefore, we tested whether additional stimulation with IL-4 could further modulate pSyk(Y352) responses. IL-21, a key cytokine driving GC reactions (68), served as control. The presence of IL-4 or IL-21 alone led to modest effects on the pSyk(Y352) response to BCR engagement. IL-4 in combination with CD40L, however, led to higher responses than with CD40L alone in all groups, whereas the addition of IL-21 in combination with CD40L increased pSyk(Y352) only in CD27− B cells in all donors (Figure S3).
We also tested whether co-stimulation affects basal expression and phosphorylation of Syk. We found that CD40L co-stimulation (with and without IL-4 or IL-21) caused a minor upregulation of Syk, whereas pSyk(Y352) was increased. SLE CD27− B cells showed somewhat increased basal Syk and pSyk(Y352) upon CD40L co-stimulation with and without IL-4 or IL-21 (Figure S3). However, the amplitude of pSyk(Y352) changes upon CD40L stimulation was very modest.
TLR9-mediated activation has been described to be impaired in SLE, including reduced production of cytokines upon TLR9 stimulation (43, 44). Thus, we tested whether reduced TLR9 responsiveness is a shared abnormality among AID B cells (Figure 5).
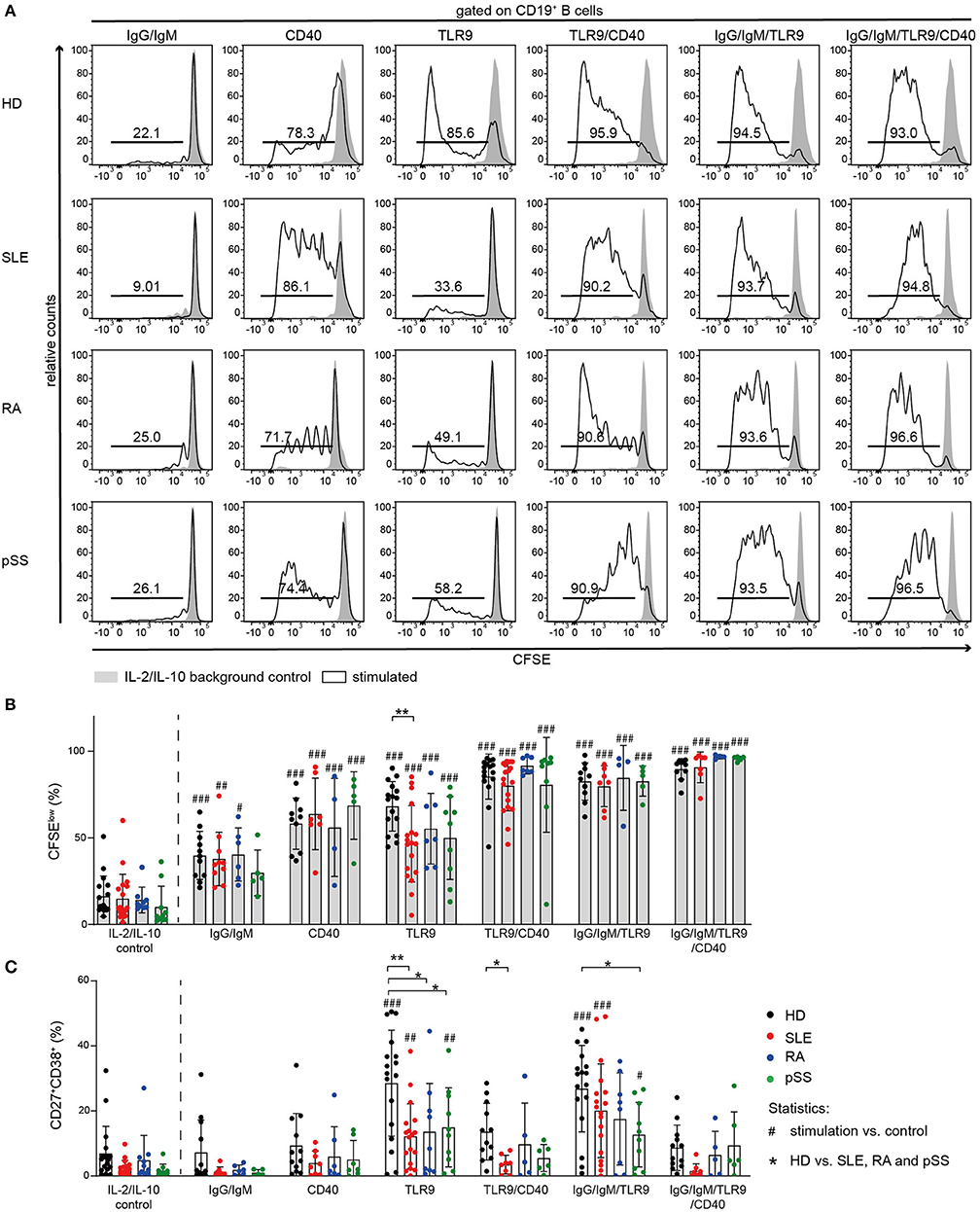
Figure 5. Proliferation of AID B cells upon CD40 stimulation or combined stimulation of BCR and TLR9 is comparable to HD. B cells from HD (black), SLE (red), RA (blue), and pSS (green) patients were stimulated with anti-IgG/IgM, CD40L, or CpG and the combinations CD40L/CpG, CpG/anti-IgG/IgM, or CD40L/CpG/anti-IgG/IgM. (A) Representative CFSE histograms of HD, SLE, RA, and pSS B cells after 5 days of culture for the indicated stimulus (lines) compared to background control (gray). (B) Percentages of CFSElow B cells [n(HD/SLE/RA/pSS) = 17/19/9/10 (IL-2/IL-10 control); 11/10/6/5 (IgG/IgM); 10/7/4/5 (CD40); 16/18/7/9 (TLR9); 10/7/4/5 (TLR9/CD40); 16/19/7/9 (IgG/IgM/TLR9); 10/7/4/5 (IgG/IgM/TLR9/CD40)]. (C) Resulting frequency of CD27+CD38+ B cells upon activation in culture [n(HD/SLE/RA/pSS) = 18/18/11/10 (IL-2/IL-10 control); 12/8/6/5 (IgG/IgM); 12/8/7/7 (CD40); 17/18/9/10 (TLR9); 12/8/5/6 (TLR9/CD40); 17/18/8/10 (IgG/IgM/TLR9); 12/8/5/6 (IgG/IgM/TLR9/CD40)]. Bars shown represent mean ± SD (ANOVA with DMCT, *p ≤ 0.05, **p ≤ 0.01; #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001).
TLR9-induced B cell proliferation was decreased in AID B cells and diminished responsiveness was most pronounced in SLE B cells (Figures 5A,B). Moreover, TLR9-induced differentiation into ASCs was impaired in all AID B cells compared to HD (Figure 5C, Figure S4). Further, AID B cells gave rise to a twofold lower frequency of CD27+CD38+ cells compared to HDs when stimulated with CpG (Figure 5C, Figure S4).
It has been reported that Syk is necessary for TLR9 signaling (69, 70). To test whether Syk has a critical role in B cell activation following TLR9 stimulation, we stimulated HD B cells with CpG in the presence or absence of the Syk inhibitor entospletenib. Syk inhibition resulted in reduced frequencies of HD CD27+CD38+ B cells upon TLR9 stimulation, mimicking the B cell hyporesponsiveness of AID memory B cells, and SLE B cells (Figure S4).
Next, we investigated the effect of combined TLR9 and BCR stimulation because of the crucial role of TLR9 engagement in breaking tolerance against nuclear antigens and driving B cell activation (71–73). As both BCR and TLR9 appear to depend on Syk activity, combined stimulation increased differentiation of CD27+CD38+ ASC only modestly, but the response remained generally lower than HD B cell responses (Figure 5C). However, co-stimulation increased proliferation of all AID B cells to the level observed with HD B cells (Figure 5B).
Notably, CD40L activation induced B cell proliferation in all groups (Figure 5B). In line with previous reports (74), differentiation into CD27+CD38+ B cells was not observed upon stimulation with CD40L alone (Figure 5C, Figure S4).
In order to further investigate the effect of CD40 engagement on AID B cells and whether their hyporesponsiveness to TLR9 agonists can be overcome through CD40/CD40L interaction, we cultured AID and HD B cells in the presence or absence of CD40L together with CpG or the combination of CpG with anti-IgG/IgM (Figure 5).
Co-stimulation of CD40 increased TLR9-induced proliferation of AID B cells. Stimulation of AID B cells with CD40L and CpG compared to CpG alone resulted in increased frequencies of CFSElow cells, similar to that induced in HD B cells (Figures 5A,B). However, the frequencies of CD27+CD38+ ASCs in CD40L co-stimulation cultures were lower compared to TLR9 or TLR9/BCR stimulation (Figure 5C) consistent with previous reports that CD40L stimulation blocks CpG induced in vitro B cell differentiation (74). Therefore, CD40 engagement provided the co-stimulation signal that allowed AID B cells, especially SLE B cells, to proliferate when co-stimulated through TLR9, but did not promote differentiation into CD27+CD38+ ASC.
Subsequently, we addressed whether reduced responsiveness of AID B cells is epigenetically controlled. Individual epigenome-wide association studies (EWAS) identified differentially methylated regions (DMR) in B cells from SLE (61), RA (63), and pSS patients (62) based on a meta-analysis of Infinium HumanMethylation450K BeadChip data of CD19+ cells.
The global methylation values of AID patients correlated strikingly with methylation values of HD samples (r = 0.99; Figure S5). We were interested in the methylation state of certain genes that are related to CD40 and BCR signaling, B cell activation, and, in addition, the methylation status of kinases and phosphatases (Figure S5). Except for the hypomethylation of multiple CpGs at the IFITM1 locus, we could not detect substantial differences among selected CpGs. However, we found kinase EIF2AK2 (cg14126601) and kinase modulator TRIP6 (cg19279257) to be hypomethylated in AID.
On the global level, genes encoding interferon-induced proteins, such as IFITM1, IFI44L, and MX1, were found to be the most differentially methylated CpGs, with hypomethylation in all AID groups (Table S4). These genes are hypomethylated mainly in SLE and pSS and to a lesser extent in RA compared to HD (Figure S5).
Increased PTP/PSP activity was uniquely found in SLE CD19+ B cells (Figures 1A,B). Furthermore, we found that CD40/IL-4R co-signaling substantially improved BCR signaling (Figure S3). Based on this, we suggested that CD40/IL-4R signaling may be involved in the modulation of PTP and PSP expression. To test this hypothesis, we analyzed the expression of non-receptor (NR)- and receptor (R)-type PTPs and PSPs in SLE and the impact of CD40/IL-4R co-stimulation on their expression.
First, we analyzed the expression of selected PTPs within publicly available gene expression data sets from SLE and HD CD20+ B cells, CD4+, and CD8+ T cells, and MS and HD CD19+ B cells as controls (Figure 6). Expression of PTPN2, PTPN11, PTPN22, PTPRC, and PTPRO was found to be specifically increased in SLE CD20+ B cells, as we found no differentially expressed PTP in MS B cells compared to HD. Further, the data point to a B cell-specific abnormality, as we found no differentially expressed RPTP and only marginal differential expressed NRPTP among CD4+ and CD8+ T cells from SLE compared to HD (Figure 6). These data correspond to the comparable PTP activity measured in SLE and HD CD3+ T cells (Figure S1).
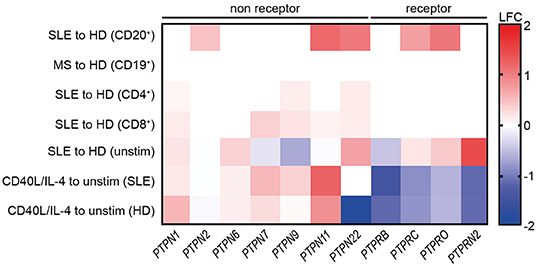
Figure 6. Common reduction of receptor-type PTP expression upon co-stimulation with CD40L/IL-4. Differential expression of selected genes related to receptor-type and non receptor-type PTPs in SLE vs. HD CD20+ B cells (6 SLE, 7 HD), MS vs. HD CD19+ B cells (10 MS, 10 HD), SLE vs. HD CD4+ (53 SLE, 41 HD), and CD8+ T cells (22 SLE, 31 HD) (lines 1–4 are from publicly available data). Differential gene expression from un-stimulated SLE vs. HD and CD40L/IL-4 stimulated for SLE vs. un-stimulated SLE or HD CD19+ B cells, respectively [n(HD/SLE) = 1/2], (lines 4–7).
Next, increased expression of PTPN6 and PTPRN2 in unstimulated SLE CD19+ B cells was detected. However, expression of PTPRB was decreased. Of note, PTPN2 and PTPN22 and all RPTP (except PTPRB) were downregulated upon CD40L/IL-4 stimulation compared to their overexpression before stimulation.
We tested the possibility that PSPs may be involved in abnormal SLE B cell signaling as we found increased PSP activity in SLE (Figure 1B). We analyzed genes related to the PP2 and PP2C family. Unstimulated B cells displayed small differences in PSP expression between SLE and HD. CD40L/IL-4 co-stimulation led to heterogeneous changes (increased expression of PP2 family genes except PPP2R3B (unaffected) and decreased PPP2R5D, whereas PP2C family members were unaffected (Figure S6).
These data show that T cell help by CD40/IL-4R engagement alters the expression of NRPTPs such as PTPN22 and different RPTPs and further underlines the crucial role of T cell co-stimulation in defining B cell dysfunction in SLE.
Despite information in the literature suggesting increased BCR signaling in AID (31, 37, 75), here we provide extensive evidence that BCR signaling and responses to TLR9 stimulation are reduced in AID memory B cells but more broadly in SLE B cells. Furthermore, it was demonstrated that this BCR signaling abnormality is not only characteristic of peripheral B cells, but is also present in tissue-resident B cells from ITP spleens and pSS parotid gland. In contrast to RA and pSS, in which BCR signaling defects are present only in antigen-experienced memory B cells, in SLE, this abnormality is also manifested in CD27− B cells. Moreover, the SLE B cell abnormality extended to increased expression of PTPs, globally enhanced PTP and PSP activities, and maximal constitutive recruitment of the PTP SHP-1 to CD22. As a proof of concept, we compared PTP expression under stimulated and unstimulated conditions in two SLE and one healthy control subject. Although considered preliminary, results of this analysis were consistent with that obtained from larger publicly available data sets. Notably, CD40 co-stimulation resulted in normalization of BCR signaling, increased B cell proliferation, together with reduced expression of PTPs such as PTPN22 in SLE. The B cell abnormalities are likely to reflect continuous stimulation through the BCR in vivo without appropriate co-stimulation. These results indicate that there is a spectrum of abnormalities in BCR signaling in AID, with SLE manifesting the most extensive B cell dysfunction and that the B cell dysfunction detected ex vivo and in vitro reflects varying degrees of BCR stimulation in vivo, without appropriate T cell-derived co-stimulation.
Reduced BCR signaling together with increased co-localization of the inhibitory co-receptor CD22 and SHP-1 in RA, pSS, and most pronounced in SLE B cells and increased PTP activities in SLE B cells may reflect a phenotype related to anergy (76–80). This conclusion is favored by the observation that repeated BCR engagement in vitro induces BCR hyporesponsiveness together with studies demonstrating that continuous signaling via SHP-1 is required to maintain anergy (80, 81). Moreover, the anergy phenotype extends to TLR9 signaling but does not involve the CD40 pathway. Finally, the anergy phenotype appears to be overcome by CD40 engagement, suggesting that it derives from incomplete signaling through the BCR in vivo without appropriate T cell-derived co-stimulation. Notably, the data suggest that when the proper series of signals are provided, AID B cells function in a largely normal manner, indicating that intrinsic B cell defects in these conditions may be minimal.
It has been reported that reduced signaling of phosphoinositide 3-kinase (PI3K) suppresses phosphatidylinositol (3,4,5)-trisphosphate (PIP3) generation by PI3K and is key for B cell anergy (82). In our studies, Akt signaling, which is downstream of PI3K, was not abnormal in RA and pSS, but was found to be increased in SLE. Another study demonstrated that proliferating GC B cells lack active BCR signaling which is induced and maintained by increased phosphatase activity and persistent co-localization of SHP-1 with the BCR after ligation (59). Therefore, reduced BCR signaling in peripheral blood and tissue-resident AID CD27+ B cells is a characteristic of what might be considered a post-activated functionally anergic phenotype. This status could be induced in HD B cells by chronic BCR stimulation, indicating that repetitive stimulation by self-antigens or immune complexes in the absence of appropriate co-stimulation can induce this functional status. Importantly, induction of this state of anergy appears to be specific for signaling through the BCR, as stimulation with CpG did not cause B cells to become anergic, even though anergic B cells exhibited decreased activation through TLR9.
Interestingly, reduced BCR-induced Syk phosphorylation in both CD27− B cells and CD27+ memory B cells was uniquely found in SLE. Iwata et al. have previously reported increased phosphorylation of Syk in total SLE CD19+ compared to healthy controls (83). One difference to our data could be that Iwata et al. included in their analysis CD27−Syk++ memory-like B cells that are increased in SLE patients compared to HD (60). This population has been excluded in the current analysis. However, the more extensive abnormality appeared to be related to increased PTP activity and expression. This may reflect the more persistent stimulation of B cells in SLE, perhaps reflecting the enhanced expression of T follicular helper cells in SLE (47, 84) and recent findings that IL-21 promotes CD11chiT-bet+ B cell development enriched of autoreactive cells in SLE (85). Even though increased TFH and increased expression of CD40L have been reported in SLE, there may be compartmentalization of these cells away from sites of B cell activation, permitting persistent engagement of the BCR with appropriation T cell-derived T cell help.
Lymphocyte hyporesponsiveness in AID is not restricted to B cells. Impaired cytotoxic function and exhaustion of CD8+ T cells, a characteristic of viral infections, has been reported in SLE (49, 50, 86). SLE T cells display abnormal T cell signaling (48, 87) and reduced Th1/Th2 Ca2+ responses were reported in pSS (88). Although hyporesponsiveness is not restricted to B cells, in the current study, T cells from AID patients did not show enhanced PTP/PSP activities, indicating a dysfunctional regulatory process restricted to B cells.
TLR9 activation facilitates the development of autoantibodies against dsDNA and ribonucleoproteins, indicating a critical role of this receptor in the activation of B cells in autoimmunity (89, 90). It activates memory B cells and drive in vitro proliferation and differentiation of B cells into ASCs (71, 72) and is considered to be involved in type I interferon production in autoimmunity (91). However, we found that AID B cells responded weakly to TLR9 activation in vitro. This is consistent with other studies describing reduced responses of SLE B cells against pokeweed mitogen (92); reduced IL-6, IL-10, vascular endothelial growth factor (VEGF), and IL-1ra production and reduced Ki-67 expression (43, 44); and reduced frequencies of CD69+CD86+ and TACI+CD25+ B cells after TLR9 in vitro stimulation (44). We and others (69, 70) found a functional connection between TLR9 and BCR signaling since Syk inhibition had an impact on TLR9 responses of normal B cells. This finding suggests that the abnormality in TLR9 signaling may be part of the anergic phenotype in SLE and reflect the reduced Syk signaling induced by repetitive BCR engagement in vivo in this disease.
Some reports indicate a role of reduced BCR signaling in the development and progression of autoimmunity (93, 94). Interestingly, inhibition of PTPN22 could reset central B cell tolerance in NOD scid gamma chain knock out (NSG) mice, which were engrafted with human hematopoietic stem cells carrying the gain of function mutation of PTPN22 (94). Whereas, this study indicates that a normalized BCR signal could restore immune tolerance, the strong role of CD40 activation in PTPN22 risk gene carriers is also consistent with the idea that this pathway is critical for censoring the overly active immune system in autoimmunity (95). Whether PTPN22 variant increases or decreases BCR signaling is a matter of debate. Mice expressing this mutation displayed enhanced BCR and CD40 responses (96). CD40 seems to be a critical context-dependent co-stimulatory molecule regulating both the full activation of BCR-stimulated B cells as well as their subsequent censoring. In this context, the current result that CD40 co-stimulation can render AID dysfunctional B cells susceptible to BCR stimulation and that CD40 treatment of SLE B cells diminished the expression of, e.g., PTPN22 further support that modulation of the CD40 pathway is of critical importance in regulating B cell function at many levels.
The effectiveness of B cell-directed therapies, such as rituximab (anti-CD20+) in RA, or belimumab (anti-BAFF/BLyS) in SLE, underscores the role of B cells in these diseases (97, 98). Our study suggests that blocking CD40/CD40L interaction by fostering B cell anergy holds promise to interfere with the cycle of B cell activation, tissue damage, and inflammation in AID. In fact, a study using an anti-CD154 antibody could prevent ASC generation in SLE patients (99). A second-generation PEGylated monoclonal antibody is currently in clinical development for SLE (100). Our data support that this treatment would be beneficial in RA and pSS as well.
In conclusion, we found that SLE B cells and RA and pSS memory B cells exhibit diminished responsiveness to BCR and TLR9 signaling, which may reflect a status of post-activation functional anergy owing to in vivo engagement of the BCR without appropriate T cell-derived co-stimulation. CD40 activation of B cells is critical to overcome this state of diminished responsiveness and restore BCR responses. Our findings support the investigation of new therapeutic options that interfere with the interaction between CD40/CD154 and, thereby, foster functional B cell anergy and decreased disease activity.
The RNASeq data sets analyzed from the literature can be found in NCBI gene expression omnibus (GSE4588, CD20+ B cells; GSE117935, CD19+ B cells) and ArrayExpress platform (E-MTAB-2713, CD4+ and CD8+ T cells). The RNASeq data generated for this study are available in NCBI sequence read archive (PRJNA564980).
This study was carried out in accordance with the recommendations of the ethics' committee at the Charité University Hospital Berlin with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
The concept of the study was developed by AL, PL, JW, LR, and TD. Data were obtained and analyzed by SW, AL, FS, AWi, HR-A, A-LS, PB, AWe, AJ, KN, JW, ES, GN, JI-K, MC, and AG. All authors developed, read, and approved the current manuscript.
TD laboratory was supported by DFG grants (project Do491/10-1, TR130, SFB650, CRC Immunobone) and the DRFZ, a Leibniz Institute was supported by the Senate of Berlin. ES was supported by a Fellowship of the Berlin Institutes of Health. HR-A was supported by COLCIENCIAS scholarship No. 727, 2015. KN was supported by the German Federal Ministry of Research and Education grant for de.NBI (031L0101D). LR was supported by The Swedish Research Council and The Swedish Society of Medicine (the Ingegerd Johansson donation) grants. We acknowledge support from the DFG and the Open Access Publication Fund of the Charité—Universitätsmedizin Berlin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Karine Serre for critical reading of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02136/full#supplementary-material
1. Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. (2013) 382:819–31. doi: 10.1016/S0140-6736(13)60954-X
2. Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. (2000) 165:5970–9. doi: 10.4049/jimmunol.165.10.5970
3. Bohnhorst JO, Bjorgan MB, Thoen JE, Natvig JB, Thompson KM. Bm1–Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjogren's syndrome. J Immunol. (2001) 167:3610–8. doi: 10.4049/jimmunol.167.7.3610
4. Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, et al. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. (2002) 46:2160–71. doi: 10.1002/art.10445
5. Jacobi AM, Odendahl M, Reiter K, Bruns A, Burmester GR, Radbruch A, et al. Correlation between circulating CD27 high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. (2003) 48:1332–42. doi: 10.1002/art.10949
6. Odendahl M, Keitzer R, Wahn U, Hiepe F, Radbruch A, Dorner T, et al. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis. (2003) 62:851–8. doi: 10.1136/ard.62.9.851
7. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
8. Jacobi AM, Reiter K, Mackay M, Aranow C, Hiepe F, Radbruch A, et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. (2008) 58:1762–73. doi: 10.1002/art.23498
9. Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, et al. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. (2010) 69:305–8. doi: 10.1136/ard.2008.096495
10. Yeo L, Lom H, Juarez M, Snow M, Buckley CD, Filer A, et al. Expression of FcRL4 defines a pro-inflammatory, RANKL-producing B cell subset in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:928–35. doi: 10.1136/annrheumdis-2013-204116
11. Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. (2004) 5:317–27. doi: 10.1038/ni1036
12. Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med. (2006) 203:1081–91. doi: 10.1084/jem.20060087
13. Shinnakasu R, Kurosaki T. Regulation of memory B and plasma cell differentiation. Curr Opin Immunol. (2017) 45:126–31. doi: 10.1016/j.coi.2017.03.003
14. Sieger N, Fleischer SJ, Mei HE, Reiter K, Shock A, Burmester GR, et al. CD22 ligation inhibits downstream B cell receptor signaling and Ca(2+) flux upon activation. Arthritis Rheum. (2013) 65:770–9. doi: 10.1002/art.37818
15. Clowse ME, Wallace DJ, Furie RA, Petri MA, Pike MC, Leszczynski P, et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: results from two phase III randomized, double-blind, placebo-controlled trials. Arthritis Rheumatol. (2017) 69:362–75. doi: 10.1002/art.39856
16. Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, et al. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell. (2002) 10:1057–69. doi: 10.1016/S1097-2765(02)00739-6
17. Pogue SL, Kurosaki T, Bolen J, Herbst R. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J Immunol. (2000) 165:1300–6. doi: 10.4049/jimmunol.165.3.1300
18. Scharenberg AM, Humphries LA, Rawlings DJ. Calcium signalling and cell-fate choice in B cells. Nat Rev Immunol. (2007) 7:778–89. doi: 10.1038/nri2172
19. Toapanta FR, Bernal PJ, Sztein MB. Diverse phosphorylation patterns of B cell receptor-associated signaling in naive and memory human B cells revealed by phosphoflow, a powerful technique to study signaling at the single cell level. Front Cell Infect Microbiol. (2012) 2:128. doi: 10.3389/fcimb.2012.00128
20. Poe JC, Fujimoto Y, Hasegawa M, Haas KM, Miller AS, Sanford IG, et al. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat Immunol. (2004) 5:1078–87. doi: 10.1038/ni1121
21. Muller J, Obermeier I, Wohner M, Brandl C, Mrotzek S, Angermuller S, et al. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. Proc Natl Acad Sci USA. (2013) 110:12402–7. doi: 10.1073/pnas.1304888110
22. Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. (2008) 40:211–6. doi: 10.1038/ng.79
23. Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun. (2009) 10:397–403. doi: 10.1038/gene.2009.19
24. Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, et al. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. (2011) 70:463–8. doi: 10.1136/ard.2010.137174
25. Jarvinen TM, Hellquist A, Zucchelli M, Koskenmies S, Panelius J, Hasan T, et al. Replication of GWAS-identified systemic lupus erythematosus susceptibility genes affirms B-cell receptor pathway signalling and strengthens the role of IRF5 in disease susceptibility in a Northern European population. Rheumatology. (2012) 51:87–92. doi: 10.1093/rheumatology/ker263
26. Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet. (2012) 44:1227–30. doi: 10.1038/ng.2439
27. Vaughn SE, Kottyan LC, Munroe ME, Harley JB. Genetic susceptibility to lupus: the biological basis of genetic risk found in B cell signaling pathways. J Leukoc Biol. (2012) 92:577–91. doi: 10.1189/jlb.0212095
28. Sun F, Xu J, Wu Z, Li P, Chen H, Su J, et al. Polymorphisms in the FAM167A-BLK, but not BANK1, are associated with primary Sjogren's syndrome in a Han Chinese population. Clin Exp Rheumatol. (2013) 31:704–10.
29. Burbelo PD, Ambatipudi K, Alevizos I. Genome-wide association studies in Sjogren's syndrome: what do the genes tell us about disease pathogenesis? Autoimmun Rev. (2014) 13:756–61. doi: 10.1016/j.autrev.2014.02.002
30. Zikherman J, Weiss A. Antigen receptor signaling in the rheumatic diseases. Arthritis Res Ther. (2009) 11:202. doi: 10.1186/ar2528
31. Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest. (1996) 98:2549–57. doi: 10.1172/JCI119073
32. Enyedy EJ, Mitchell JP, Nambiar MP, Tsokos GC. Defective FcgammaRIIb1 signaling contributes to enhanced calcium response in B cells from patients with systemic lupus erythematosus. Clin Immunol. (2001) 101:130–5. doi: 10.1006/clim.2001.5104
33. Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Invest Med. (2001) 49:157–65. doi: 10.2310/6650.2001.34042
34. Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. (2003) 4:287–94. doi: 10.1038/ni892
35. Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med. (2006) 203:2157–64. doi: 10.1084/jem.20051503
36. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. (2007) 178:3272–80. doi: 10.4049/jimmunol.178.5.3272
37. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. (2014) 6:246ra299. doi: 10.1126/scitranslmed.3009131
38. Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. (2009) 182:3343–7. doi: 10.4049/jimmunol.0713370
39. Dam EM, Habib T, Chen J, Funk A, Glukhova V, Davis-Pickett M, et al. The BANK1 SLE-risk variants are associated with alterations in peripheral B cell signaling and development in humans. Clin Immunol. (2016) 173:171–80. doi: 10.1016/j.clim.2016.10.018
40. Fleischer SJ, Daridon C, Fleischer V, Lipsky PE, Dorner T. Enhanced tyrosine phosphatase activity underlies dysregulated B cell receptor signaling and promotes survival of human lupus B cells. Arthritis Rheumatol. (2016) 68:1210–21. doi: 10.1002/art.39559
41. Vasquez A, Baena A, Gonzalez LA, Restrepo M, Munoz CH, Vanegas-Garcia A, et al. Altered recruitment of Lyn, Syk and ZAP-70 into lipid rafts of activated B cells in systemic lupus erythematosus. Cell Signal. (2019) 58:9–19. doi: 10.1016/j.cellsig.2019.03.003
42. Bengtsson AA, Ronnblom L. Role of interferons in SLE. Best Pract Res Clin Rheumatol. (2017) 31:415–28. doi: 10.1016/j.berh.2017.10.003
43. Sieber J, Daridon C, Fleischer SJ, Fleischer V, Hiepe F, Alexander T, et al. Active systemic lupus erythematosus is associated with a reduced cytokine production by B cells in response to TLR9 stimulation. Arthritis Res Ther. (2014) 16:477. doi: 10.1186/s13075-014-0477-1
44. Gies V, Schickel JN, Jung S, Joublin A, Glauzy S, Knapp AM, et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight. (2018) 3:96795. doi: 10.1172/jci.insight.96795
45. Stefanski AL, Wiedemann A, Reiter K, Hiepe F, Lino AC, Dorner T. Enhanced PD-1 and diminished PD-L1 upregulation capacity mark post-activated lupus B cells. Arthritis Rheumatol. (2019). 71:1539–44. doi: 10.1002/art.40897.
46. Tipton CM, Hom JR, Fucile CF, Rosenberg AF, Sanz I. Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: A B-cell immunomics approach. Immunol Rev. (2018) 284:120–31. doi: 10.1111/imr.12660
47. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. (2015) 67:988–99. doi: 10.1002/art.39020
48. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
49. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. (2015) 523:612–6. doi: 10.1038/nature14468
50. Comte D, Karampetsou MP, Yoshida N, Kis-Toth K, Kyttaris VC, Tsokos GC. Signaling lymphocytic activation molecule family member 7 engagement restores defective effector CD8+ T cell function in systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:1035–44. doi: 10.1002/art.40038
51. Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M, Peter HH, et al. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. (2004) 113:161–71. doi: 10.1016/j.clim.2004.05.010
52. Rodriguez-Bayona B, Ramos-Amaya A, Perez-Venegas JJ, Rodriguez C, Brieva JA. Decreased frequency and activated phenotype of blood CD27 IgD IgM B lymphocytes is a permanent abnormality in systemic lupus erythematosus patients. Arthritis Res Ther. (2010) 12:R108. doi: 10.1186/ar3042
53. Wei C, Jenks S, Sanz I. Polychromatic flow cytometry in evaluating rheumatic disease patients. Arthritis Res Ther. (2015) 17:46. doi: 10.1186/s13075-015-0561-1
54. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. (2010) 62:2569–81. doi: 10.1002/art.27584
55. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2677–86. doi: 10.1002/art.34473
56. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. (2002) 61:554–8. doi: 10.1136/ard.61.6.554
57. Kalina T, Flores-Montero J, Van Der Velden VH, Martin-Ayuso M, Bottcher S, Ritgen M, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. (2012) 26:1986–2010. doi: 10.1038/leu.2012.122
58. Giesecke C, Frolich D, Reiter K, Mei HE, Wirries I, Kuhly R, et al. Tissue distribution and dependence of responsiveness of human antigen-specific memory B cells. J Immunol. (2014) 192:3091–100. doi: 10.4049/jimmunol.1302783
59. Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. (2012) 336:1178–81. doi: 10.1126/science.1213368
60. Fleischer SJ, Giesecke C, Mei HE, Lipsky PE, Daridon C, Dorner T. Increased frequency of a unique spleen tyrosine kinase bright memory B cell population in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:3424–35. doi: 10.1002/art.38854
61. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. (2013) 9:e1003678. doi: 10.1371/journal.pgen.1003678
62. Imgenberg-Kreuz J, Sandling JK, Almlof JC, Nordlund J, Signer L, Norheim KB, et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjogren's syndrome reveals regulatory effects at interferon-induced genes. Ann Rheum Dis. (2016) 75:2029–36. doi: 10.1136/annrheumdis-2015-208659
63. Julia A, Absher D, Lopez-Lasanta M, Palau N, Pluma A, Waite Jones L, et al. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum Mol Genet. (2017) 26:2803–11. doi: 10.1093/hmg/ddx177
64. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
65. Alsadeq A, Hobeika E, Medgyesi D, Klasener K, Reth M. The role of the Syk/Shp-1 kinase-phosphatase equilibrium in B cell development and signaling. J Immunol. (2014) 193:268–76. doi: 10.4049/jimmunol.1203040
66. Ying H, Li Z, Yang L, Zhang J. Syk mediates BCR- and CD40-signaling integration during B cell activation. Immunobiology. (2011) 216:566–70. doi: 10.1016/j.imbio.2010.09.016
67. Szodoray P, Stanford SM, Molberg O, Munthe LA, Bottini N, Nakken B. T-helper signals restore B-cell receptor signaling in autoreactive anergic B cells by upregulating CD45 phosphatase activity. J Allergy Clin Immunol. (2016) 138839-851:e838. doi: 10.1016/j.jaci.2016.01.035
68. Zotos D, Coquet JM, Zhang Y, Light A, D'costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. (2010) 207:365–78. doi: 10.1084/jem.20091777
69. Iwata S, Yamaoka K, Niiro H, Nakano K, Wang SP, Akashi K, et al. Amplification of Toll-like receptor-mediated signaling through spleen tyrosine kinase in human B-cell activation. J Allergy Clin Immunol. (2012) 1291594-1601:e1592. doi: 10.1016/j.jaci.2012.03.014
70. Kremlitzka M, Macsik-Valent B, Erdei A. Syk is indispensable for CpG-induced activation and differentiation of human B cells. Cell Mol Life Sci. (2015) 72:2223–36. doi: 10.1007/s00018-014-1806-x
71. Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. (2003) 101:4500–4. doi: 10.1182/blood-2002-11-3569
72. Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. (2003) 19:837–47. doi: 10.1016/S1074-7613(03)00323-6
73. Busconi L, Bauer JW, Tumang JR, Laws A, Perkins-Mesires K, Tabor AS, et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J Immunol. (2007) 179:7397–405. doi: 10.4049/jimmunol.179.11.7397
74. Marasco E, Farroni C, Cascioli S, Marcellini V, Scarsella M, Giorda E, et al. B-cell activation with CD40L or CpG measures the function of B-cell subsets and identifies specific defects in immunodeficient patients. Eur J Immunol. (2017) 47:131–43. doi: 10.1002/eji.201646574
75. Galligan CL, Siebert JC, Siminovitch KA, Keystone EC, Bykerk V, Perez OD, et al. Multiparameter phospho-flow analysis of lymphocytes in early rheumatoid arthritis: implications for diagnosis and monitoring drug therapy. PLoS ONE. (2009) 4:e6703. doi: 10.1371/journal.pone.0006703
76. Nossal GJ, Pike BL. Evidence for the clonal abortion theory of B-lymphocyte tolerance. J Exp Med. (1975) 141:904–17. doi: 10.1084/jem.141.4.904
77. Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. (1988) 334:676–82. doi: 10.1038/334676a0
78. Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. (2010) 237:249–63. doi: 10.1111/j.1600-065X.2010.00936.x
79. Khoder A, Alsuliman A, Basar R, Sobieski C, Kondo K, Alousi AM, et al. Evidence for B cell exhaustion in chronic graft-versus-host disease. Front Immunol. (2017) 8:1937. doi: 10.3389/fimmu.2017.01937
80. Franks SE, Cambier JC. Putting on the brakes: regulatory kinases and phosphatases maintaining B cell anergy. Front Immunol. (2018) 9:665. doi: 10.3389/fimmu.2018.00665
81. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med. (2016) 213:751–69. doi: 10.1084/jem.20150537
82. Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity. (2009) 31:749–60. doi: 10.1016/j.immuni.2009.08.026
83. Iwata S, Yamaoka K, Niiro H, Jabbarzadeh-Tabrizi S, Wang SP, Kondo M, et al. Increased Syk phosphorylation leads to overexpression of TRAF6 in peripheral B cells of patients with systemic lupus erythematosus. Lupus. (2015) 24:695–704. doi: 10.1177/0961203314560424
84. Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet ME, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Front Immunol. (2018) 9:1637. doi: 10.3389/fimmu.2018.01637
85. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat Commun. (2018) 9:1758. doi: 10.1038/s41467-018-03750-7
86. Kis-Toth K, Comte D, Karampetsou MP, Kyttaris VC, Kannan L, Terhorst C, et al. Selective loss of signaling lymphocytic activation molecule family member 4-positive CD8+ T cells contributes to the decreased cytotoxic cell activity in systemic lupus erythematosus. Arthritis Rheumatol. (2016) 68:164–73. doi: 10.1002/art.39410
87. Sharabi A, Kasper IR, Tsokos GC. The serine/threonine protein phosphatase 2A controls autoimmunity. Clin Immunol. (2018) 186:38–42. doi: 10.1016/j.clim.2017.07.012
88. Legany N, Toldi G, Orban C, Megyes N, Bajnok A, Balog A. Calcium influx kinetics, and the features of potassium channels of peripheral lymphocytes in primary Sjogren's syndrome. Immunobiology. (2016) 221:1266–72. doi: 10.1016/j.imbio.2016.06.004
89. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. (2005) 202:321–31. doi: 10.1084/jem.20050338
90. Lartigue A, Courville P, Auquit I, Francois A, Arnoult C, Tron F, et al. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. (2006) 177:1349–54. doi: 10.4049/jimmunol.177.2.1349
91. Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity. (2015) 42:1159–70. doi: 10.1016/j.immuni.2015.05.012
92. Ginsburg WW, Finkelman FD, Lipsky PE. Circulating and pokeweed mitogen-induced immunoglobulin-secreting cells in systemic lupus erythematosus. Clin Exp Immunol. (1979) 35:76–88.
93. Konigsberger S, Prodohl J, Stegner D, Weis V, Andreas M, Stehling M, et al. Altered BCR signalling quality predisposes to autoimmune disease and a pre-diabetic state. EMBO J. (2012) 31:3363–74. doi: 10.1038/emboj.2012.169
94. Schickel JN, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, et al. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol. (2016) 1:aaf7153. doi: 10.1126/sciimmunol.aaf7153
95. Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. (2011) 121:3635–44. doi: 10.1172/JCI45790
96. Metzler G, Dai X, Thouvenel CD, Khim S, Habib T, Buckner JH, et al. The autoimmune risk variant PTPN22 C1858T alters B cell tolerance at discrete checkpoints and differentially shapes the naive repertoire. J Immunol. (2017) 199:2249–60. doi: 10.4049/jimmunol.1700601
97. Burmester GR, Feist E, Dorner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:77–88. doi: 10.1038/nrrheum.2013.168
98. Dorner T, Lipsky PE. Beyond pan-B-cell-directed therapy—New avenues and insights into the pathogenesis of SLE. Nat Rev Rheumatol. (2016) 12:645–57. doi: 10.1038/nrrheum.2016.158
99. Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, et al. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154–CD40 interactions. J Clin Invest. (2003) 112:1506–20. doi: 10.1172/JCI200319301
100. Chamberlain C, Colman PJ, Ranger AM, Burkly LC, Johnston GI, Otoul C, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis. (2017) 76:1837–44. doi: 10.1136/annrheumdis-2017-211388
Keywords: systemic lupus erythematosus, rheumatoid arthritis, primary Sjögren's syndrome, B cell receptor signaling, toll-like receptor 9, CD40, post-activation, anergy
Citation: Weißenberg SY, Szelinski F, Schrezenmeier E, Stefanski A-L, Wiedemann A, Rincon-Arevalo H, Welle A, Jungmann A, Nordström K, Walter J, Imgenberg-Kreuz J, Nordmark G, Rönnblom L, Bachali P, Catalina MD, Grammer AC, Lipsky PE, Lino AC and Dörner T (2019) Identification and Characterization of Post-activated B Cells in Systemic Autoimmune Diseases. Front. Immunol. 10:2136. doi: 10.3389/fimmu.2019.02136
Received: 30 April 2019; Accepted: 27 August 2019;
Published: 24 September 2019.
Edited by:
Ignacio Sanz, Emory University, United StatesReviewed by:
Paolo Casali, University of Texas Health Science Center San Antonio, United StatesCopyright © 2019 Weißenberg, Szelinski, Schrezenmeier, Stefanski, Wiedemann, Rincon-Arevalo, Welle, Jungmann, Nordström, Walter, Imgenberg-Kreuz, Nordmark, Rönnblom, Bachali, Catalina, Grammer, Lipsky, Lino and Dörner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Dörner, dGhvbWFzLmRvZXJuZXJAY2hhcml0ZS5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.