
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 September 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02122
 Esmaeil Mortaz1,2
Esmaeil Mortaz1,2 Seyed Sajjad Zadian1
Seyed Sajjad Zadian1 Mehri Shahir1
Mehri Shahir1 Gert Folkerts3
Gert Folkerts3 Johan Garssen3,4
Johan Garssen3,4 Sharon Mumby5
Sharon Mumby5 Ian M. Adcock5,6*
Ian M. Adcock5,6*According to the World Health Organization (WHO), trauma is responsible for 10% of deaths and 16% of disabilities worldwide. This is considerably higher than those for malaria, tuberculosis, and HIV/AIDS combined. While the human suffering and death caused by injury is well-recognized, injury has a significant medical care cost. Better prediction of the state of trauma patients in the days immediately after trauma may reduce costs. Traumatic injuries to multiple organs can cause dysfunction in all systems of the body especially the immune system placing patients at high risk of infections and inflammatory complications which are often fatal. Neutrophils are the most abundant leukocyte in the human circulation and are crucial for the prevention of microbial disease. Significant changes in neutrophil functions such as enhanced chemotaxis, Neutrophil extracellular trap (NET)-induced cell death (NETosis), and phagocytosis occur early after injury followed by prolonged functional defects such as phagocytosis, killing mechanisms, and receptor expression. Analysis of these changes may improve the prediction of the patient's condition over time. We provide a comprehensive and up-to-date review of the literature investigating the effect of trauma on neutrophil phenotype with an underlying goal of using this knowledge to examine the predictive potential of neutrophil alterations on secondary complications in patients with traumatic injuries. We conclude that alterations in neutrophil surface markers and functions may be potential biomarkers that predict the outcome of trauma patients.
Neutrophils are the most abundant leukocyte in humans (60–70% of circulating leukocytes) and have a major role in the innate immune response against invading pathogens and are important mediators of inflammation-induced injury (1). In healthy adults, circulating neutrophils are considered as dormant cells but they are activated when they encounter damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) and thereby maintain homeostasis within the immune system (2).
Neutrophils are equipped with a range of anti-microbial mechanisms including phagocytosis, degranulation, and release of reactive oxygen species (ROS), neutrophil extracellular traps (NETs), and cytokine production that deliver lethal hits against microorganisms (3, 4). Neutrophils rapidly adapt to changes in microenvironmental signals and show different functional phenotypes (neutrophil heterogeneity) during inflammation (5, 6). Trauma affects the phenotype and function of neutrophils (7) although similar changes in neutrophil phenotype have been observed with acute coronary syndromes and in patients with the acute phase of autoimmune disease [Figure 1; (8, 9)]. The World Health Organization (WHO) estimated that trauma causes 5.8 million deaths annually (10, 11) although mortality rates related to trauma have significantly reduced in recent years due to improvements in treatment particularly in those treating coagulopathy and blood loss. However, secondary complications, such as sepsis, multiple organ failure (MOF), and nosocomial infections can impact upon the condition of trauma patients and result in death (12). The induction of altered immune system responses particularly changes in neutrophil phenotypes is increasingly recognized as an important factor in the response to trauma (7).
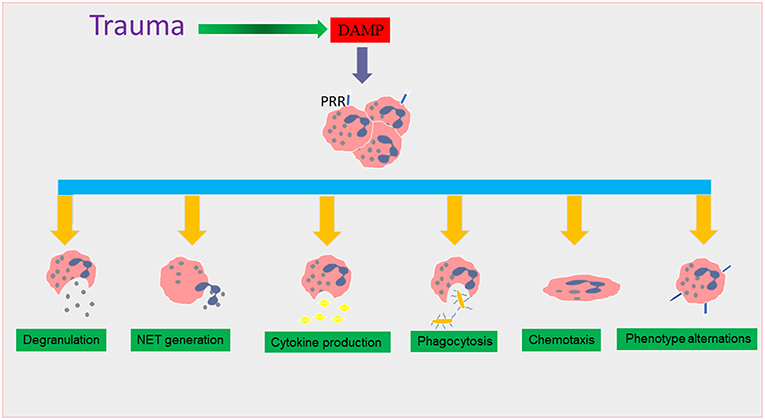
Figure 1. The effect of trauma on neutrophil functions. During traumatic injury, damage associated molecular patterns (DAMPs) are released into the systemic circulation as a result of tissue damage. DAMPs interact with surface pattern recognition receptors (PRR), causing neutrophil recruitment which triggers many functional responses that are thought to lead to the induction of the systemic inflammatory response (SIRS) phase in trauma. These neutrophil phenotype changes include alterations in degranulation, neutrophil extracellular trap (NET) formation, cytokine production, and chemotaxis and phagocytosis.
Marked alterations in a range of neutrophil functions and in phenotypic markers occur following trauma, which causes a massive release of neutrophils, banded cells and sometimes even immature cells (i.e., metamyelocytes) from the bone marrow into the circulation (13, 14). Experimental studies strongly suggest a direct relationship is present between trauma severity and subsequent tissue damage and neutrophils dysfunction (15). In addition, impaired neutrophil chemotaxis, due in part to release of immature neutrophils into the circulation, was seen in children following blunt force trauma (16).
In this review we provide a comprehensive overview of the most recent studies examining the role of neutrophils in severe trauma with the goal of identifying a link between trauma severity and neutrophil phenotype and function. This knowledge of the correlation between neutrophil phenotype and the prediction of patient survival may provide better early biomarkers for the clinical outcome and treatment of trauma patients.
Trauma activates innate immune responses to produce pro- and anti-inflammatory cytokines primarily by cells of the innate immune response such as neutrophils. The systemic inflammatory response syndrome (SIRS) and the Compensatory Anti-inflammatory Response syndrome (CARS) may be induced with severe trauma without being accompanied by sepsis and multiple organ failure (17). CARS is considered an imprecise term that does not truly reflect the key role that the neutrophil plays in immune tolerance during trauma. The multifunctional aspects of neutrophil biology in this process is critical to induction of immune tolerance by acting as danger- or damage-sensing cells in multiple organs which reflects their Janus-like effects in trauma (17).
While SIRS is a pro-inflammatory syndrome that is associated with killing infectious organisms through activation of the immune system, immune tolerance represents is a complex pattern of immunologic responses characterized by deactivation of the systemic immune system. As such, immune tolerance is not simply the reversal of SIRS, but it can exist separately from SIRS. Immune tolerance can be dangerous when its effects are unchecked, leaving the host vulnerable to a secondary exposure to pathogens because of increased immune effector cell apoptosis leading to auto-immunity (18, 19). Studies have also shown that immune tolerance reduces the severity of the SIRS pro-inflammatory response, but after trauma, tolerance can lead to increased immunosuppression (17). The uptake of apoptotic cells by dendritic cells and macrophages promotes tolerance by suppressing the release of pro-inflammatory cytokines, and increasing the release of anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β (TGF-β) (20, 21).
One of the important factors that increases morbidity in post-traumatic cases is an imbalance between the systemic inflammatory response SIRS and tolerance (22). In the first hours of trauma, the severity of the SIRS is associated with early MOF and infections however, in the following days, immune tolerance plays an important role in the increased incidence of nosocomial infections and late organ failure and late sepsis [(23); Figure 2].
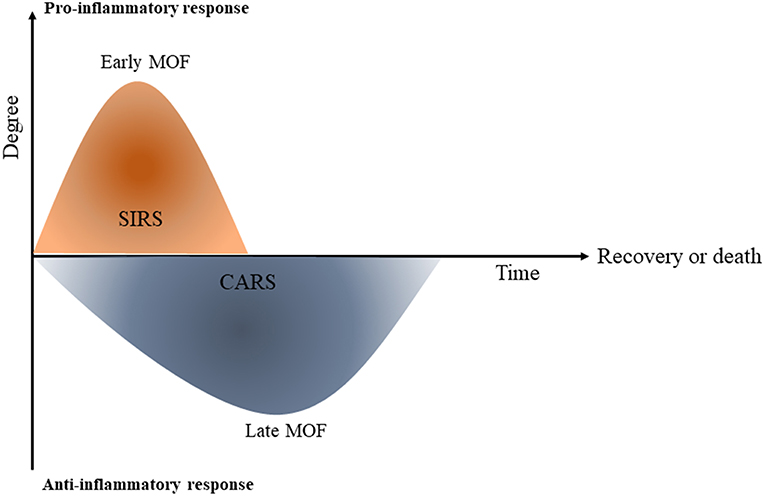
Figure 2. Systemic inflammatory response (SIRS) and compensatory immune tolerance in trauma (CARS). Schematic diagram shows that once trauma has occurred, a primary systemic pro-inflammatory response (SIRS) is initiated which can contribute to early multi-orgn failure (MOF). The compensatory immune tolerance or anti-inflammatory response syndrome (CARS) can begin while the pro-inflammatory SIRS is still present. At the later phase CARS can lead to immune paralysis and following that, late multi organ failure [This figure is adapted from Hietbrink et al. (23)].
A number of studies propose that dysregulated apoptotic immune cell death may have an important role in the severity of multiple organ dysfunction and sepsis following a variety of traumatic events (24, 25). Understanding the role of altered apoptotic cell death in contributing to immune and organ dysfunction as seen in sepsis and shock is essential. Apoptosis is increased following various traumas such as ischemia, burn, sepsis, and traumatic brain injury (26–28). The incidence of programmed cell death following traumatic brain and spinal injury has been studied and re-enforced the importance of apoptosis in the pathogenesis of post-traumatic outcomes although no data yet relates this to neutrophil function (26–28).
Hypoxia can induce apoptosis in various organs, but studies have shown no increase in apoptotic liver cells in trauma animals, despite the presence of systemic hypoxia. Therefore, it is likely that local mechanisms are responsible for the induction of apoptotic cell death (29, 30). Oxidative stress, ischemia, and some mediators such as steroids, tumor necrosis factor (TNF), nitric oxide, complement C5a, and Fas ligand (FasL) have been identified as important factors that induce apoptotic cell death (31–33). Caspase 8 is activated mainly by extracellular signaling through the Fas/FasL system (CD95/CD95L) or via TNF-α receptors suggesting that the reported enhanced expression of caspase 8 indicates an extracellular stimulation of apoptosis (34–36). Immune cells that undergo modified apoptotic changes include neutrophils, macrophages, dendritic cells, and lymphocytes although epithelial and endothelial cell apoptotic changes have also been reported (37). Although neutrophils are conventionally thought of as short-lived cells which die following apoptosis, they may persist for several days (38, 39), and whilst most studies focus on programmed cell death following different traumatic insults they do not determine the mechanisms of neutrophil accumulation at the region of interest (38, 39).
The initial role of neutrophils is innate defense against infection by eliminating pathogens. They kill pathogens using reactive oxygen species (ROS) and lytic enzymes, so they can potentially contribute to bystander organ injury. Therefore, neutrophil apoptosis reduces neutrophil-mediated tissue damage (40). Activation of neutrophils, which induced by trauma, is directly linked to the immune response. Trauma is usually associated with over-activation of innate immune responses followed by a further immune-suppression, which leads to elevated susceptibility to infection, sepsis, and multiple organ dysfunction syndrome (MODS) and act as one of the leading causes of human death (23, 41–43).
Inflammatory mediators may prolong the circulation half-life of neutrophils from 6 h up to several days based on the decreased level of pro-apoptotic proteins, including apoptosis regulator Bax and upregulation of anti-apoptotic proteins, such as myeloid cell leukemia 1 (Mcl-1) (44–46). Delayed apoptosis of neutrophils induces tissue damage by the release of ROS and neutrophil elastase (NE) (47, 48). Inhibition of neutrophil apoptosis has been reported in SIRS (49), sepsis (46, 50), and burn injuries (51).
In shock or injury, exposure to pro-inflammatory factors are thought to prime circulating neutrophils and induce tissue injury (52, 53). Studies have shown that following hemorrhage or endo-toxemia, a remarkable decrease in lung neutrophil apoptosis has been seen for up to 24 h after the insult (54). Jimenez et al. showed a significant decrease in apoptosis in SIRS patients and that this resulted in amplified neutrophil-mediated killing which, in a non-inflammatory environment, may result in SIRS and subsequent organ failure. Therefore, suppression of neutrophil apoptosis increases the potential for tissue injury (55). It is unclear whether the death receptor or mitochondrial-mediated, apoptotic pathway predominates in controlling neutrophil apoptosis in animals exposed to septic shock (56). Studies of neutrophil apoptosis in sepsis cases demonstrated that delayed apoptosis seems to occur as a result of the activation of anti-apoptotic factors and NF-κB and then suppression of caspases 9 and 3 (57–62).
The development of sepsis after major trauma is associated with changes in the expression of apoptosis-related factors (63). After trauma, in the early phase, neutrophil apoptosis is mainly regulated by anti-apoptotic B-cell lymphoma 2 (Bcl-2) members that inhibit the intrinsic mitochondrial-dependent pathway (63). However, neutrophil apoptosis is not always associated with the expression of the anti-apoptotic factor Mcl-1. Neutrophils in patients with sepsis 10-days after the initial trauma displayed reduced neutrophil apoptosis despite decreased levels of Mcl-1 and the Bcl-2-associated A1 protein. The association between reduced neutrophil apoptosis and the severity of illness supports the importance of neutrophils activity in the pathophysiology of sepsis (63).
The soluble form of Fas (sFas) is derived by alternative splicing from the membrane form or by proteolytic cleavage of membrane-bound receptors. Serum sFas has been shown to inhibit neutrophils apoptosis in vitro (64) and plays an important role in the inhibition of neutrophil extrinsic apoptosis associated with increased levels of polymorphonuclear leukocyte elastase (PMNE), a marker for systemic inflammation. The results show a high relationship between sFas and patients' Sequential Organ Failure Assessment (SODA) and Multiple Organ Dysfunction (MOD) stage in sepsis and provide evidence for the clinical significance of the risk for the development of sepsis and MOF. Trauma patients with and without sepsis development demonstrated a significant reduction in the apoptosis of circulating neutrophils at least until 10-days after trauma. So, sFas may be a feasible target for new therapeutic strategies to limit neutrophil life span and hyperactivity (65).
NETs may have an important role in the regulation of inflammatory responses to injury. Accumulation of activated neutrophils occurs in the damaged tissue following injury and these may form NETs (66). Recent studies have shown the potential role of NETs in the pathogenesis of an extensive range of non-infectious inflammations including post-injury sterile inflammation (67). Margraf and co-workers in 2008 showed that NETs levels in plasma may predict sepsis and MOF on the intensive care unit in patients after multiple trauma (68, 69).
The severity of tissue damage in cases of transfusion-related acute lung injury (TRALI) is associated with the degree of NETs formation with NETs detectable in the plasma and lung of TRALI patients (70). Mitochondrial DNA can trigger NETosis via activation of Toll Like Receptor (TLR)9 after severe trauma, independent of the NADPH oxidase system (71) and mitochondrial (mt)DNA is found in NETs formed after trauma (66). The detailed molecular mechanism of mtDNA-NETs release is unknown (67).
As NETs are rapidly degraded by DNase in the circulation, it is possible that NETs are actively produced throughout the 5-days after trauma and surgery (72).
However, surgery alone can stimulate NETs formation independent of prior trauma as evidenced by NETs formation after elective total hip replacement. This suggests that sepsis may not have been an initiating factor for the NETs formation. NETs formation in these patients can be viewed as part of the sterile inflammatory response of the innate immune system (72).
The importance of neutrophil-neutrophil cross-talk and connection with other cells related to NETs formation has been shown. Platelets are the most well-defined players in NETosis. Many platelet-derived soluble factors and ligand/receptor pairs maintain neutrophil activation (73). Among these soluble factors, alarmins such as platelet-derived high-mobility group protein box 1 (HMGB1) and chemokines including platelet factor 4 (PF4)/CXCL4) produced by platelets activate neutrophil NETs formation in vitro and in animal models (74).
In human neutrophils, P-selectin may drive sterile NETs formation (74). Other platelet-localized cell adhesion molecules such as β2 integrin (CD18) may also play a critical role in this process (74, 75). Indeed, platelet biology impacts upon many aspects of inflammation which makes the identification of their direct or indirect contribution to NETosis not readily predictable (67).
Improved methodologies are needed for the better understanding of detailed mechanisms of NETs. The current techniques combine fluorescent microscopy or fluorescent intensity measurements and generally use DNA-intercalating dyes, while taking the risk of visualizing necrotic cells with dye permeable cell membrane. Antibody-based techniques are required to detect activated, non-necrotic cells with intact cell membrane, such as flow cytometry-cell-sorting, supported by microscopic imaging. Additionally, a consensus on the structural and behavioral definition of NETs formation is essential for future NETs research, due to their fragility, their highly dynamic nature and their morphological heterogeneity (67).
Within hours and days after trauma, the expression of neutrophil markers become noticeably distinct compared to those from healthy individuals. The various markers show distinct dynamics over time. In this regard, the severity of changes in function and phenotype of neutrophils in trauma depends upon the severity of injury as measured by indices such as the injury severity score (ISS) and the new injury severity score (NISS) (76, 77).
Some studies report a significantly higher percentage of neutrophils positive for CD11a in trauma patients than in controls at 3 and 96 h after injury (78). However, another study showed that there is a significant decrease in CD11a expression 24 h after trauma (79). Integrins (e.g., Mac-1 also known as alpha M beta 2 integrin which is composed of CD11b and CD18) are involved in leukocyte adhesion to the endothelium. Functional integrins are only expressed upon neutrophils activation (80) and neutrophil Mac-1 expression has been proposed as a marker of injury severity in several studies (81–83). Circulating neutrophils show upregulation of CD11b, after injury with a second peak of CD11b expression at day 5 (79). Contrarily, Scannell and colleagues have reported attenuation in expression of ICAM-1, CD11b, and CD18 on circulating leukocytes 2 h after injury (84). The level of metabolic acidosis after trauma correlates directly with CD11b expression on circulating neutrophils which may provide a mechanism whereby post-traumatic shock results in neutrophil-mediated organ failure (82).
Increased Mac-1 expression is found on neutrophils from patients who were admitted with an ISS >16 as compared to trauma patients with an ISS <16 which could be a useful marker for prediction of survival of trauma patients but need more investigation (78). In addition, neutrophils sampled at various time points pre- and post-operative days had increased expression of CD11b when treated with Platelet-activating factor (PAF) and/or fMLP (N-formyl-methionyl-leucyl-phenylalanine (fMLP) although the expression of CD11b in unstimulated conditions did not change with surgery, suggesting minimal activation in vivo and a failure of PAF to act as an agonist on human neutrophils (85).
CD18 expression follows the same expression pattern following trauma as CD11b although this did not reach significance (78). However, similar to the in vitro findings, the numbers of CD11a surface receptors do not increase synchronously with CD11b/CD18 receptors although their affinity may increase (78).
The expression level of CD62L (L-selectin), a receptor that mediates the initial step of the adhesion cascade, the capture and rolling of leukocytes on endothelial cells, was decreased up to 24 h after injury (85, 86). No correlation was demonstrated between immune cell CD62L expression and trauma severity scores although a meta-analysis by Stengel and colleagues indicated that soluble L-selectin levels were correlated with ISS (87). Some studies have reported an association between decreased L-selectin expression on leukocytes and the occurrence of SIRS or early MOF. These studies also show a correlation between the degree of neutrophils activation and the severity of complications occurring during the pro-inflammatory phase (88, 89). These molecules can be found as soluble factors in serum (sL-selectin). The level of sL-selectin in the blood is correlated with the activation level of the neutrophil population (23).
Compared to control individuals, traumatic patients were characterized by a statistically significant decreased responsiveness of active FcγRII (a marker of neutrophil priming) on neutrophils toward the fMLP (90, 91). High levels of active- FcγRII expression was indicative of increased responsiveness to bacterial products (92). The decreased responsiveness of active FcγRII toward the fMLP on the circulating neutrophils after trauma may impact on downstream inflammatory events. Indeed, the degree of this reduced responsiveness correlated with trauma severity as measured by ISS (91). Furthermore, the degree of decrease in fMLP-induced active FcγRII on neutrophils is related to severity of the clinical response and to SIRS (91).
In contrast, the expression of FcγRIII (CD16) on neutrophils was suppressed during severe trauma. Furthermore, soluble CD16 increased significantly at day 1 in multi-trauma patients who later developed infection (93). Similar results were also seen during the first 24 h after chest trauma (92). FcγRIII is normally expressed on banded neutrophils at lower levels compared with mature neutrophils. Therefore, this decline in FcγRIII may reflect an influx of young neutrophils (90).
CD43 (Leukosialin) is expressed on early hematopoietic progenitors and is one of the most abundant transmembrane sialoglycoproteins on neutrophils (86). CD43 prevents interactions of surface molecules and acts as a negative regulator of cell function (94). CD43 membrane expression was decreased by up to 80% upon exposure to phorbol-12-myristate-13-acetate (PMA) or fMLP in the presence of cytochalasin B (95). PMA activation significantly reduced neutrophils CD43 expression. Downregulation of CD43 has been seen in hemodialysis, neutrophil activation, and during neutrophil migration whereas, fMLP normally causes CD43 shedding from neutrophils (96).
The key regulatory step in neutrophils tissue migration occurs at the level of bone marrow release and association with the endothelial layer (23). There is a large storage pool of mature neutrophils in the bone marrow which is rapidly mobilized during inflammation causing a dramatic rise in circulating neutrophil numbers (97). CXCR4 and its ligand, CXCL12 (or stromal cell derived factor-1), trigger neutrophils release from the bone marrow under normal conditions (98). In addition, CXCL12 acts as a chemokine by attracting neutrophils to the site of inflammation (99) and not surprisingly, therefore, activated CXCR4 has been described as having multiple biological functions including chemotaxis, differentiation and survival (100).
During trauma, the release of DAMPs enhances neutrophils migration through the 600 μm sinusoids and they then elongate and squeeze themselves through the tissue to reach the site of injury (101, 102). High mobility group box-1 (HMGB-1), a recently identified inflammatory cytokine, is implicated in the pathogenesis of several inflammatory diseases where it acts as an important DAMP (103). The interaction of HMGB1 with specific receptors on numerous cell types results in increased production and release of proinflammatory cytokines and chemokines (104). In some studies, HMGB-1 expression correlates with ISS (105) but this is not replicated elsewhere (106). In light of these contradictory data, it is important to examine the potential of HMGB-1 as a biomarker of trauma in much larger homogenous cohorts and after therapeutic intervention.
Generally, the leukocyte recruitment cascade contains the following steps: (a) selectin-mediated rolling, (b) chemokine-triggered activation, and (c) integrin-dependent arrest (107). Rolling is mediated by L-selectin (CD62L), P-selectin (CD62P), and E-selectin (CD62E), which interact with glycosylated ligands such as P-selectin glycoprotein ligand 1 (PSGL1), ESL1 (E-selectin ligand 1), and CD44 (108). The interaction of selectins with their ligands enables leukocytes to adhere to the inflamed endothelium (109). Consequently, down regulation of L-selectin expression on leukocytes and induction of SIRS or MOF, may suggest cross talk between the development of complications occurring during SIRS and the degree of neutrophils activation (91, 92). Importantly, the serum levels of sL-selectin are higher 6 h after trauma during which time neutrophils migrate to the tissue (78).
High concentrations of the neutrophil chemotactic factor IL-8 have been reported in trauma patients (110, 111). IL-8 activates and recruits neutrophils to the site of inflammation by interacting with its receptors CXC receptor 1 (CXCR1) and CXCR2 on the neutrophils cell membrane [Figure 3; (112)]. CXCR1 responses are lower in trauma patients than in control subjects but the activity of CXCR2 is higher and may be implicated in the later clinical complications seen with neutrophil activation. Indeed, CXCR2 activity correlates with neutrophils hyperactivity and with outcomes such as acute respiratory distress syndrome (ARDS) whereas reduced CXCR2 function seen in inflammatory environments may impair neutrophil functions (46, 113, 114). Activation of circulating endogenous factor VII-activating protease (FSAP) in multiple trauma patients led to increased complement (C)5a anaphylatoxin generation and modulation of the posttraumatic SIRS in vivo (115). C5a is a potent chemoattractant involved in the activation and recruitment of neutrophils at the site of trauma (116). Robust C5a generation during trauma may cause defects in neutrophil defense systems and C5a might be considered as a potential target for therapeutic intervention to prevent immune dysfunctions that occur in the days following trauma (117).
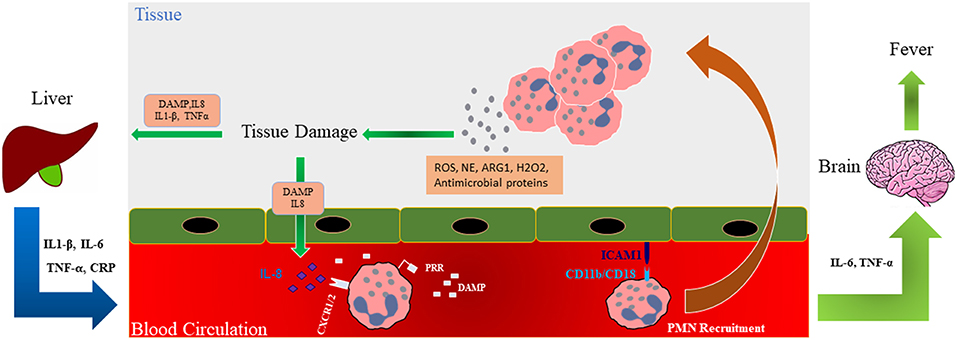
Figure 3. Role of neutrophils in tissue damage. Schematic illustration shows that during trauma, damage associated molecular patterns (DAMPs) are generated and these along with released mediators (ROS; NE; ARG1; hydrogen peroxide, H2O2, and antimicrobial proteins) recruit neutrophils to the site of injury where they release factors such as additional ROS and NE involved in the development of organ failure in a feedforward manner. Interleukin (IL)-6 and TNF-α can affect the brain causing fever in traumatic patients. Trauma-generated DAMPs also affect the liver and cause the release of inflammatory cytokines into the circulation which, in turn, further modulate neutrophil numbers and activation. DAMP, Damage-associated molecular pattern; ROS, Reactive oxygen species; PRR, pattern recognition receptor; ICAM-1, Intercellular Adhesion Molecule 1; ARG1, Arginase 1; NE, Neutrophil elastase; TNF-α, Tumor necrosis factor alpha.
Integrins are important components of the transmigration process and are only expressed on activated neutrophils (80). Thus, the expression of Mac-1 is increased on neutrophils from multi trauma patients as compared to mild trauma patients suggesting neutrophil activation (78). In contrast, during late organ failure the expression of Mac-1 is down-regulated (118) and is associated with the development of MOF (119).
The formation of free radicals and ROS is an important component of activated neutrophils following trauma and is involved in phagocytosis.
Neutrophils are the major cellular producers of ROS and alterations in the levels of ROS production reflect neutrophils activation status. The production and role of the oxidative burst in neutrophils during brain trauma has been extensively reviewed (120–122). Increased oxidative burst correlates with the incidence of SIRS and MOF (123). On the other hand, ROS have been identified as a necessary component of the NLRP3 inflammasome activator in various diseases including hepatic ischemia/reperfusion injury (124). The NLRP3 inflammasome is essential for the onset of acute sterile inflammation such as that seen in trauma injury (125).
Increased ROS production by neutrophils may lead to an uncontrolled inflammatory response which results in tissue damage. The highest levels of ROS production were found between 3 and 24 h after trauma after in vitro stimulation of the cells (126). Furthermore, the expression of inducible nitric oxide (NO) synthase (iNOS or NOS2) and of NADPH oxidase (gp91phox) in leukocytes is reduced following traumatic brain injury (TBI), whereas, after 24 h the expression of iNOS, cyclooxygenase (COX)-2 and of gp91phox is significantly increased in monocytes and neutrophils (127). This leads to increased prostaglandin (PG) induction either directly or indirectly. Spinal cord injury (SCI) causes a systemic inflammatory response resulting in increased oxidative burst within neutrophils and monocytes from 12 to 48 h and 1 week post-injury and the maximal increase was at 24 h in neutrophils. In addition, the expression of gp91phox, COX-2 and iNOS were significantly increased 24 h after trauma in neutrophils and monocytes of these patients (128). Furthermore, the newly formed ROS in injured tissue leads to the migration and subsequent activation of neutrophils resulting in the accumulation of activated neutrophils in the spine (129). Table 1 indicates the markers that reflect changes in neutrophil functions/phenotype of neutrophil biology that occur during trauma.
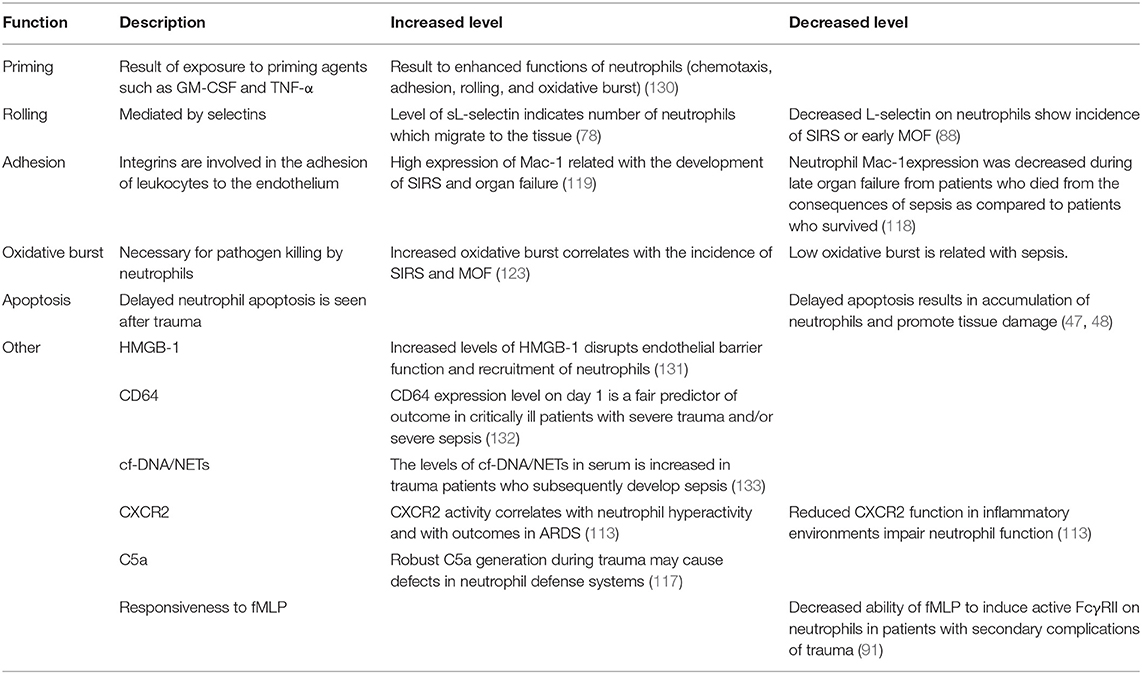
Table 1. Functions and markers that reflect changes in neutrophil functions/phenotype in trauma.
A fundamental step in the host defense response against infection is phagocytosis (134). Given that perturbation in immune responses followed by multi trauma may lead to sepsis, investigation of neutrophils phagocytosis seems necessary. Changes in neutrophils phagocytic activity following trauma vary depending on the microbial species which explain the conflicting results observed with phagocytic kinetics (135–137).
Some reports have demonstrated decreased neutrophil phagocytosis following trauma and injury, for example, the lowest rates of neutrophil phagocytosis were observed at 48 and 72 h after TBI (127). In contrast, the neutrophil phagocytosis index (FI) was higher 1–3 days after severe tissue injury (138). Generally, after trauma, ingestion of E coli was enhanced, whereas phagocytosis of K pneumoniae was depressed. Ingestion of S aureus, however, was unaffected (134). This effect may also involve platelets which also express TLR2 ns TLR4 enabling the generation of NETs and the killing of gram positive and negative bacteria (139).
The lifespan of circulating neutrophils is classically considered to be short (<1 day) however, recent observations reported that the median peripheral blood (PB) human neutrophils lifespan is up to 10 times longer (5.4 days) (140–142). Neutrophils lifespan is further increased at sites of inflammation due to inhibition of cell apoptosis by inflammatory factors such as cytokines. Lifespan extension of neutrophils during inflammatory conditions may also alter neutrophils function and phenotype (143).
Trauma and subsequent complications affect the phenotype and function of circulating neutrophils, and, particularly, in case of severe trauma, the development of dysfunctional neutrophils might play a detrimental role. Indeed, severe posttraumatic inflammation induces a boost in the release of banded and immature neutrophils into the circulation, leading to bone marrow exhaustion, and a compromised immune response, both associated with a poor outcome. Additionally, morphological changes were observed after trauma, including increased cell size and membrane plasticity and a modified shape, wherein neutrophils become more elongated (144). In trauma, there are immunosuppressive low-density neutrophils (LDNs), a subtype of neutrophil named after their discovery in the PB mononuclear cell (PBMC) (145, 146). These granulocytes are not only activated but express a high level of arginase activity, which in turn might be linked to T-cell function, providing a possible explanation for the impairment of the adaptive immunity mediated by neutrophils during trauma (146).
In sepsis, it has been demonstrated that this granulocyte subset inhibits T-cells, possibly via arginase release and/or ROS production (145, 147, 148). In contrast, there might be subsets of neutrophils, which are beneficial to repair the initial trauma impact. For example, a population of CD11b+/Gr-1+/CXCR4hi neutrophils likely recruited by vascular endothelial growth factor A (VEGF-A) induce revascularization via MMP-9 (149). While neutrophil heterogeneity is often described in the context of chronic inflammation, this is also seen in other scenarios such as cancer (145, 150).
A quick and reliable prediction of prognosis is important particularly in the emergency room. Posttraumatic organ failure, is thought to be triggered by the initial inflammatory response (151). The time between the life span of peripheral neutrophils (5 days) and the time to produce new neutrophils from myelocytes (7 days) is critical in determining the risk of infectious complications such as septic shock (152). Early identification and prediction of septic shock may be greatly helpful in the identification of patients for the adaptation of the treatment regime (153, 154). Patients with subsequent MOF showed significantly higher mean circulating concentrations of C3a and thromboxane B2 at the first day post injury compared to the patients without MOF. Neopterin/creatinine ratios were also significantly higher in patients with multiple organ failure when MOF had already become established (155). So, seems to these mediators are useful for prediction of occurrence of secondary complications in traumatic patients but for realization more studies are needed.
Interestingly, in patients with major trauma, there was no significant difference in systemic C-reactive peptide and IL-6 levels between survivor and non-survivor groups. Furthermore, no differences between these groups were found for terminal complement complex, thromboxane B2, and neopterin/creatinine ratios (156). In contrast, some studies have shown plasma concentrations of neutrophil elastase, lactate, antithrombin III, IL-6, and IL-8 were significantly higher in non-survivors compared with survivors 24 h after trauma (151). Authors suggested that early alterations in serum levels of IL-6 constitute a useful predictive marker for identifying traumatic patients (156). Nevertheless, the role of IL-6 in critically ill patients has been discussed controversially in recent studies (157, 158). These data suggest that the overall level of the initial inflammatory response correlates with the development of post-traumatic organ failure.
Elevated serum NO and of its oxidation products (NOx) and of blood lactate in polytrauma patients are markers of a serious clinical course. However, a normal NOx combined with a very high lactate level may indicate a fatal prognosis in these patients (159). This suggests that elevated lactate levels may be an important prognostic factor rather than NOx itself. Decreased neutrophil responsiveness appears to be a prerequisite for septic shock after trauma. Indeed, the initial decreased responsiveness of circulating neutrophils to fMLP-induced FcγRII activation was related to the development of late onset septic complications after >5 days (14).
CD11b and active FcγRII/CD32 levels on unstimulated neutrophils, however, did not correlate with the severity of injury. Rather, there was a significant inverse correlation between the neutrophils ability to activate FcγRII in response to fMLP and the severity of injury. In addition, a decreased ability of fMLP to induce active FcγRII on neutrophils was found in patients who developed secondary complications of trauma such as SIRS or acute lung injury (ALI) and it seems to be a useful marker which could show occurrence of secondary complications (91).
For 9 of 10 septic shock patients, initial shock symptoms became evident between days 8 and 10 after admission. Measuring the kinetics of the neutrophil response demonstrated that the lowest neutrophil responsiveness to fMLP was found within the first 7 days after injury. Therefore, the impaired responsiveness to fMLP and other changes in receptor expression clearly preceded clinical symptoms of sepsis (160). On the other hand, CD64 expression level on day 1 is a fairly good predictor of outcome in critically ill patients with severe trauma and/or severe sepsis (132). Also, one study showed that CD64 expression level for neutrophils on postoperative day 1 is the best early predictor of intra-abdominal infection after colorectal cancer surgery (161).
Finally, the pre-operative surface expression of adhesion molecules involved in the migration of neutrophils such as CD99 and CD47 correlates with post-operative creatinine levels, a measurement of renal injury (162).
Recent clinical studies have shown that the levels of circulating free-DNA (cf-DNA)/NETs can be potentially used for predicting injury severity following trauma with sepsis. It is important to note, however, that cf-DNA is not synonymous with NETs. The levels of cf-DNA/NETs in serum is increased in trauma patients who subsequently develop sepsis (133) whilst the levels of cf-DNA/NETs in synovial fluid are also increased in patients with septic arthritis (163). It is more recently used in the prediction of mortality in patients with severe burn injury (69).
DNase is naturally present in human blood and produced as a defense mechanism associated with NETs. The expression of DNase is increased in the early stages of sepsis after major trauma (69). DNase degrades NETs in a concentration dependent manner and DNase levels may be a potential biomarker of NETs formation (164).
Pentraxin 3 (PTX3) is a member of pentraxin family and acts as a soluble pattern recognition receptor (PRR) in the innate immune response (165). PTX3 is an extrahepatic acute-phase protein that has been implicated in the pathophysiology of trauma due to its extrahepatic formation and induction by different trauma-associated cytokines such as IL-1, IL-6 and TNF-α however this needs to be confirmed. Studies have shown that circulating PTX3 levels are associated with the injury severity and may reflect the immunological changes arising during soft tissue injury. Further studies are needed to prove PTX3 is a surrogate factor for soft tissue damage. PTX3 concentrations were higher in poly-traumatized compared to mono-traumatized and healthy individuals (166). PTX3 and some of the other components of NETs form a complex to enhance the actions of other NETs component proteins (164). Thus, new roles of PTX3 in the innate immune response, together with a pattern of binding to the NETs component proteins suggest an important role in NETosis (167).
Neutrophils are main players in the context of inflammatory complications during and after traumatic injuries. Marked alterations in a range of neutrophils functions and in phenotypic markers occur following trauma, which causes a massive release of neutrophils, banded cells, and sometimes even immature cells from the bone marrow into the circulation. In trauma injury, neutrophils are able to modify the phenotype and function based on the body's requirements. In particular, CD11b is considered as important marker of poor prognosis (82) whilst the increased expression and activity of CXCR2 on neutrophils also correlates with neutrophil function and poor outcomes in ARDS (113). In this respect, the specific neutrophil phenotype and function could be considered as a biomarker of patient survival. Altered neutrophils phenotypes include increased expression of key cell surface molecules or enhanced life span and the expression of NETs and NETS-associated factors.
We conclude that neutrophils not only play a pivotal role in the regulation and modulation of trauma but that delineation of their particular phenotype, the expression of specific cell surface markers and the release of NETS-related factors could be used as a predictive tool for the management of trauma patients. Future studies should aim to identify key proteomic or transcriptomic markers that define each phenotype so that more rapid assessment of these can be made. Finally, randomized controlled studies using drugs directed against specific neutrophil subtypes will be essential to confirm this tenet.
EM, SZ, and MS wrote first draft. EM and IA revised the manuscript. SM, GF, JG, and IA has revised final version and added extra information.
SM and IA were supported by the British Heart Foundation(PG/14/27/30679) and the Dunhill Medical Trust (R368/0714). IA was also supported by the Wellcome Trust (093080/Z/10/Z) and by EPSRC (EP/T003189/1) and SM by EU project 853850.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LL declared a shared affiliation, with no collaboration, with several of the authors, JG and GF, to the handling editor at time of review.
ALI, Acute Lung Injury; ARDS, Acute respiratory distress syndrome; CARS, Compensatory anti-inflammatory response; Cf-DNA, Circulating Free DNA; COX-2, Cyclooxygenase 2; DAMP, Damage-associated molecular pattern; ESL1, E-selectin ligand 1; FasL, Fas ligand; FI, phagocytosis index; fMLP, N-formyl-methionyl-leucyl-phenylalanine; FSAP, factor VII-activating protease; HMGB-1, High mobility group box-1; ISS, Injury Severity Score; LDN, Low-Density Neutrophils; Mac-1, alpha M beta 2 integrin; Mcl-1, Myeloid cell leukemia 1; MMP, Matric MetalloProteinase; MODS, Multiple organ dysfunction syndrome; MOF, Multiple organ failure; MPO, Myeloperoxidase; mtDNA, mitochondrial DNA; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; NE, Neutrophil elastase; NETs, Neutrophil Extracellular Traps; NETosis, cell death characterized by release of decondensed chromatin and granular contents into the extracellular space; NF-κB, Nuclear Factor κB; NISS, New Injury Severity Score; NLRP3, NLR (NOD-like receptor) Family Pyrin Domain Containing 3; NO, Nitric Oxide; NOx, Oxidized NO products; NOS2, Inducible nitric oxide synthase or iNOS; PAF, Platelet-activating factor; PAMP, Pathogen-associated molecular pattern; PB, Peripheral Blood; PBMC, Peripheral Blood Mononuclear Cells; PECAM-1, Platelet endothelial cell adhesion molecule 1; PF4, Platelet Factor 4/CXC chemokine ligand 4; PG, Prostaglandin; PMA, phorbol-12-myristate-13-acetate; PMNE, Polymorphonuclear leukocyte elastase; PRR, Pattern Recognition Receptor; PSGL1, P-selectin glycoprotein ligand 1; PTX3, Pentraxin 3; ROS, Reactive oxygen species; SCI, Spinal cord injury; SIRS, Systemic inflammatory response syndrome; sL-selectin, soluble L-selectin; SOFA, Sequential Organ Failure Assessment; TBI, Traumatic brain injury; TGF-β, Transforming Growth Factor beta; TLR, Toll Like Receptor; TRALI, Transfusion-related acute lung injury; WHO, World Health Organization; VEGF-A, Vascular endothelial growth factor A.
1. Kobayashi SD, DeLeo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. (2009) 1:309–33. doi: 10.1002/wsbm.32
2. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
3. Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. (2013) 210:1283–99. doi: 10.1084/jem.20122220
4. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
5. Hellebrekers P, Hietbrink F, Vrisekoop N, Leenen LPH, Koenderman L. Neutrophil functional heterogeneity: identification of competitive phagocytosis. Front Immunol. (2017) 8:1498. doi: 10.3389/fimmu.2017.01498
6. Tak T, Wijten P, Heeres M, Pickkers P, Scholten A, Heck AJR, et al. Human CD62Ldimneutrophils identified as a separate subset by proteome profiling and in vivo pulse-chase labeling. Blood. (2017) 129:3476–85. doi: 10.1182/blood-2016-07-727669
7. Hazeldine J, Hampson P, Lord JM. The impact of trauma on neutrophil function. Injury. (2014) 45:1824–33. doi: 10.1016/j.injury.2014.06.021
8. Maugeri N, Rovere-Querini P, Evangelista V, Godino C, Demetrio M, Baldini M, et al. An intense and short-lasting burst of neutrophil activation differentiates early acute myocardial infraction from systemic inflammatory syndromes. PLoS ONE. (2012) 7:e39484. doi: 10.1371/journal.pone.0039484
9. Allen E, Bakke AC, Purtzer MZ, Deodhar A. Neutrophil CD64 expression: distinguishing acute inflammatory autoimmune disease from systemic infections. Ann Rheum Dis. (2002) 61:522–5. doi: 10.1136/ard.61.6.522
10. Lord JM, Midwinter MJ, Chen YF, Belli A, Brohi K, Kovacs EJ, et al. The systemic immune response to trauma: An overview of pathophysiology and treatment. Lancet. (2014) 384:1455–65. doi: 10.1016/S0140-6736(14)60687-5
11. Rossaint R, Bouillon B, Cerny V, Coats TJ, Duranteau J, Fernández-Mondéjar E, et al. The European guideline on management of major bleeding and coagulopathy following trauma: fourth edition. Crit Care. (2016) 20:100. doi: 10.1186/s13054-016-1265-x
12. Glance LG, Stone PW, Mukamel DB, Dick AW. Increases in mortality, length of stay, and cost associated with hospital-acquired infections in trauma patients. Arch Surg. (2011) 146:794–801. doi: 10.1001/archsurg.2011.41
13. Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Shiu MY, Baker AJ, et al. Resuscitation of traumatic hemorrhagic shock patients with hypertonic saline - without dextran - inhibits neutrophil and endothelial cell activation. Shock. (2012) 38:341–50. doi: 10.1097/SHK.0b013e3182635aca
14. Groeneveld KM, Koenderman L, Warren BL, Jol S, Leenen LPH, Hietbrink F. Early decreased neutrophil responsiveness is related to late onset sepsis in multitrauma patients: an international cohort study. PLoS ONE. (2017) 12:e0180145. doi: 10.1371/journal.pone.0180145
15. Wang X, Li ZY, Zeng L, Zhang AQ, Pan W, Gu W, et al. Neutrophil CD64 expression as a diagnostic marker for sepsis in adult patients: a meta-analysis. Crit Care. (2015) 19:245. doi: 10.1186/s13054-015-0972-z
16. Krause PJ, Woronick CL, Burke G, Slover N, Kosciol C, Kelly T, et al. Depressed neutrophil chemotaxis in children suffering blunt trauma. Pediatrics. (1994) 93:807–9.
17. Binkowska AM, Michalak G, Slotwinski R. Current views on the mechanisms of immune responses to trauma and infection. Cent Eur J Immunol. (2015) 40:206–16. doi: 10.5114/ceji.2015.52835
18. Van Der Poll T, Meijers JCM. Systemic inflammatory response syndrome and compensatory anti-inflammatory response syndrome in sepsis. J Innate Immun. (2010) 2:379–80. doi: 10.1159/000318190
19. Toliver-Kinsky T, Kobayashi M, Suzuki F, Sherwood ER. The systemic inflammatory response syndrome. In: Herndon DN, editor. Total Burn Care. 5th ed. Philadelphia, PA: Elsevier (2018). p. 205–20.
20. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR. Immunosuppressive effects of apoptotic cells. Nature. (1997) 390:350–1. doi: 10.1038/37022
21. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. (1998) 101:890–8. doi: 10.1172/JCI1112
22. Stoecklein VM, Osuka A, Lederer JA. Trauma equals danger–damage control by the immune system. J Leukoc Biol. (2012) 92:539–51. doi: 10.1189/jlb.0212072
23. Hietbrink F, Koenderman L, Rijkers GT, Leenen LPH. Trauma: the role of the innate immune system. World J Emerg Surg. (2006) 1:15. doi: 10.1186/1749-7922-1-15
24. Chung CS, Yang S, Song GY, Lomas J, Wang P, Simms HH, et al. Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow during sepsis. Surgery. (2001) 130:339–45. doi: 10.1067/msy.2001.116540
25. Ayala A, Evans TA, Chaudry IH. Does hepatocellular injury in sepsis involve apoptosis? J Surg Res. (1998) 76:165–73. doi: 10.1006/jsre.1998.5314
26. Hotchkiss RS, Schmieg RE Jr, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. (2000) 28:3207–17. doi: 10.1097/00003246-200009000-00016
27. Ikeda H, Suzuki Y, Suzuki M, Koike M, Tamura J, Tong J, Nomura M, Itoh G. Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut. (1998) 42:530–7. doi: 10.1136/gut.42.4.530
28. Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. (2000) 17:927–38. doi: 10.1089/neu.2000.17.927
29. Tamura DY, Moore EE, Partrick DA, Johnson JL, Offner PJ, Silliman CC. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock. (2002) 17:269–73. doi: 10.1097/00024382-200204000-00005
30. Jung F, Weiland U, Johns RA, Ihling C, Dimmeler S. Chronic hypoxia induces apoptosis in cardiac myocytes: a possible role for Bcl-2-like proteins. Biochem Biophys Res Commun. (2001) 286:419–25. doi: 10.1006/bbrc.2001.5406
31. Janeway CA. How the immune system works to protect the host from infection: a personal view. Proc Natl Acad Sci USA. (2001) 98:7461–8. doi: 10.1073/pnas.131202998
32. Mica L, Härter L, Trentz O, Keel M. Endotoxin reduces CD95-induced neutrophil apoptosis by cIAP-2-mediated caspase-3 degradation. J Am Coll Surg. (2004) 199:595–602. doi: 10.1016/j.jamcollsurg.2004.05.272
33. Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. (2003) 10:26–35. doi: 10.1038/sj.cdd.4401186
34. Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. (1998) 281:1305–8. doi: 10.1126/science.281.5381.1305
35. Jaeschke H, Farhood A, Cai SX, Tseng BY, Bajt ML. Protection against TNF-induced liver parenchymal cell apoptosis during endotoxemia by a novel caspase inhibitor in mice. Toxicol Appl Pharmacol. (2000) 169:77–83. doi: 10.1006/taap.2000.9035
36. Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem. (1999) 274:5053–60. doi: 10.1074/jbc.274.8.5053
37. Wesche DE. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. (2005) 78:325–37. doi: 10.1189/jlb.0105017
38. Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. (1998) 228:518–27. doi: 10.1097/00000658-199810000-00008
39. Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and GUT by an endotoxin and TNF- independent pathway. Shock. (1997) 7:247–53. doi: 10.1097/00024382-199704000-00002
40. Wickel DJ, Cheadle WG, Mercer-Jones MA, Neal Garrison R. Poor outcome from peritonitis is caused by disease acuity and organ failure, not recurrent peritoneal infection. Ann Surg. (1997) 225:744–56. doi: 10.1097/00000658-199706000-00012
41. Zhou JY, Krovvidi RK, Gao Y, Gao H, Petritis BO, De AK, et al. Trauma-associated human neutrophil alterations revealed by comparative proteomics profiling. Proteomics Clin Appl. (2013) 7:571–83. doi: 10.1002/prca.201200109
42. Matsuda N, Hattori Y. Systemic inflammatory response syndrome (SIRS): molecular pathophysiology and gene therapy. J Pharmacol Sci. (2006) 101:189–98. doi: 10.1254/jphs.CRJ06010X
43. Stahel PF, Smith WR, Moore EE. Role of biological modifiers regulating the immune response after trauma. Injury. (2007) 38:1409–22. doi: 10.1016/j.injury.2007.09.023
44. Wang J, Arase H. Regulation of immune responses by neutrophils. Ann N Y Acad Sci. (2014) 19:66–81. doi: 10.1111/nyas.12445
45. Härter L, Mica L, Stocker R, Trentz O, Keel M. Mcl-1 correlates with reduced apoptosis in neutrophils from patients with sepsis. J Am Coll Surg. (2003) 197:964–73. doi: 10.1016/j.jamcollsurg.2003.07.008
46. Kovtun A, Messerer DAC, Scharffetter-Kochanek K, Huber-Lang M, Ignatius A. Neutrophils in tissue trauma of the skin, bone, and lung: two sides of the same coin. J Immunol Res. (2018) 2018:8173983. doi: 10.1155/2018/8173983
47. Donnelly SC, Macgregor I, Zamani A, Gordon MWG, Robertson CE, Steedman DJ, et al. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am J Respir Crit Care Med. (1995) 151:1428–33. doi: 10.1164/ajrccm.151.5.7735596
48. Bhatia R, Dent C, Topley N, Pallister I. Neutrophil priming for elastase release in adult blunt trauma patients. J Trauma. (2006) 60:590–6. doi: 10.1097/01.ta.0000205614.51885.ff
49. Papathanassoglou ED, Moynihan JA, McDermott MP, Ackerman MH. Expression of Fas (CD95) and Fas ligand on peripheral blood mononuclear cells in critical illness and association with multiorgan dysfunction severity and survival. Crit Care Med. (2001) 29:709–18. doi: 10.1097/00003246-200104000-00002
50. Taneja R, Parodo J, Song HJ, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. (2004) 32:1460–9. doi: 10.1097/01.CCM.0000129975.26905.77
51. Chitnis D, Dickerson C, Munster AM, Winchurch RA. Inhibition of apoptosis in polymorphonuclear neutrophils from burn patients. J Leukoc Biol. (1996) 59:835–9. doi: 10.1002/jlb.59.6.835
52. Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, et al. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. (2003) 19:358–65. doi: 10.1097/00024382-200304000-00011
53. Ayala A, Chung C-S, Lomas JL, Song GY, Doughty LA, Gregory SH, et al. Shock-induced neutrophil mediated priming for acute lung injury in mice. Am J Pathol. (2002) 161:2283–94. doi: 10.1016/S0002-9440(10)64504-X
54. Parsey MV, Kaneko D, Shenkar R, Abraham E. Neutrophil apoptosis in the lung after hemorrhage or endotoxemia: apoptosis and migration are independent of IL-1β. Clin Immunol. (1999) 91:219–25. doi: 10.1006/clim.1999.4693
55. Jimenez MF, Watson RWG, Parodo J, Evans D, Foster D, Steinberg M, et al. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg. (1997) 132:1263–70. doi: 10.1001/archsurg.1997.01430360009002
56. Ayala A, Karr SM, Evans TA, Chaudry IH. Factors responsible for peritoneal granulocyte apoptosis during sepsis. J Surg Res. (1997) 69:67–75. doi: 10.1006/jsre.1997.5027
57. Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis. J Leukoc Biol. (2000) 68:158–66.
58. Dibbert B, Weber M, Nikolaizik WH, Vogt P, Schoni MH, Blaser K, et al. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci USA. (1999) 96:13330–5. doi: 10.1073/pnas.96.23.13330
59. Villunger A, O'Reilly L, Holler N, Adams J, Strasser A. Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase: regulators of distinct cell death and survival pathways in granulocytes. J Exp Med. (2000) 192:647–58. doi: 10.1084/jem.192.5.647
60. Weinmann P, Gaehtgens P, Walzog B. Bcl-XL– and bax-alpha–mediated regulation of apoptosis of human neutrophils via caspase-3. Blood. (1999) 93:3106–15.
61. Moulding D, Quayle J, Hart C, Edwards S. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. (1998) 90:2495–502.
62. Chuang PI, Yee E, Karsan A, Winn RK, Harlan JM. A1 is a constitutive and inducible Bcl-2 homologue in mature human neutrophils. Biochem Biophys Res Commun. (1998) 249:361–5. doi: 10.1006/bbrc.1998.9155
63. Paunel-Görgülü A, Kirichevska T, Lögters T, Windolf J, Flohé S. Molecular mechanisms underlying delayed apoptosis in neutrophils from multiple trauma patients with and without sepsis. Mol Med. (2012) 18:325–35. doi: 10.2119/molmed.2011.00380
64. Cascino I, Papoff G, Eramo A, Ruberti G. Soluble FAS/APO-1 splicing variants and apoptosis. Front Biosci. (1996) 1:D12-18. doi: 10.2741/A112
65. Paunel-Görgülü A, Flohé S, Scholz M, Windolf J, Lögters T. Increased serum soluble Fas after major trauma is associated with delayed neutrophil apoptosis and development of sepsis. Crit Care. (2011) 15:R20. doi: 10.1186/cc9965
66. Liu FC, Chuang YH, Tsai YF, Yu HP. Role of neutrophil extracellular traps following injury. Shock. (2014) 41:491–8. doi: 10.1097/SHK.0000000000000146
67. Tuboly E, Briggs GD, Balogh ZJ. The role of neutrophil extracellular traps in post-injury inflammation. In: Khajah MA, editor. Role of Neutrophils in Disease Pathogenesis. IntechOpen (2017). doi: 10.5772/intechopen.68906
68. Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. (2009) 40:912–8. doi: 10.1016/j.injury.2009.05.024
69. Margraf S, Lögters T, Reipen J, Altrichter J, Scholz M, Windolf J. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock. (2008) 30:352–8. doi: 10.1097/SHK.0b013e31816a6bb1
70. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. (2012) 119:6335–43. doi: 10.1182/blood-2012-01-405183
71. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
72. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care. (2014) 29:1133.e1–e5. doi: 10.1016/j.jcrc.2014.07.013
73. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
74. Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. (2016) 7:271. doi: 10.3389/fimmu.2016.00271
75. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusionrelated acute lung injury. J Clin Invest. (2012) 122:2661–71. doi: 10.1172/JCI61303
76. Samin OA, Civil ID. The new injury severity score versus the injury severity score in predicting patient outcome: a comparative evaluation on trauma service patients of the auckland hospital. Annu Proc Assoc Adv Automot Med. (1999) 43:1–15.
77. Palmer C. Major trauma and the injury severity score–where should we set the bar? Annu Proc Assoc Adv Automot Med. (2007) 51:13–29.
78. Maekawa K, Futami S, Nishida M, Terada T, Inagawa H, Suzuki S, et al. Effects of trauma and sepsis on soluble L-selectin and cell surface expression of L-selectin and CD11b. J Trauma. (1998) 44:460–8. doi: 10.1097/00005373-199803000-00007
79. Lo SK, Van Seventer GA, Levin SM, Wright SD. Two leukocyte receptors (CD11a/CD18 and CD11b/CD18) mediate transient adhesion to endothelium by binding to different ligands. J Immunol. (1989) 143:3325–9.
80. Parkos CA. Molecular events in neutrophil transepithelial migration. BioEssays. (1997) 19:865–73. doi: 10.1002/bies.950191006
81. Deitch EA, Feketeova E, Adams JM, Forsythe RM, Xu DZ, Itagaki K, et al. Lymph from a primate baboon trauma hemorrhagic shock model activates human neutrophils. Shock. (2006) 25:460–3. doi: 10.1097/01.shk.0000209551.88215.1e
82. Botha AJ, Moore FA, Moore EE, Peterson VM, Goode AW. Base deficit after major trauma directly relates to neutrophil CD11b expression: a proposed mechanism of shock-induced organ injury. Intens Care Med. (1997) 23:504–9. doi: 10.1007/s001340050365
83. Moore EE, Johnson JL, Cheng AM, Masuno T, Banerjee A. Insights from studies of blood substitutes in trauma. Shock. (2005) 24:197–205. doi: 10.1097/01.shk.0000180075.76766.fe
84. Scannell G, Waxman K, Vaziri ND, Zhang J, Kaupke CJ, Jalali M, et al. Effects of trauma on leukocyte intercellular adhesion molecule-1, CD11b, and CD18 expressions. J Trauma. (1995) 39:641–4. doi: 10.1097/00005373-199510000-00004
85. Edelman JJB, Fung YL, Pennings GJ, Reddel CJ, Bannon PG, Bayfield MS, et al. Off-pump coronary artery bypass surgery induces prolonged alterations to host neutrophil physiology. Shock. (2013) 39:149–54. doi: 10.1097/SHK.0b013e31827c2aba
86. Fung YL, Silliman CC, Minchinton RM, Wood P, Fraser JF. Cardiopulmonary bypass induces enduring alterations to host neutrophil physiology: a single-center longitudinal observational study. Shock. (2008) 30:642–8. doi: 10.1097/SHK.0b013e318173e717
87. Stengel D, Bauwens K, Keh D, Gerlach H, Ekkernkamp A, Tauber R, et al. Prognostic value of an early soluble L-selectin (sCD62L) assay for risk assessment in blunt multiple trauma: a metaanalysis. Clin Chem. (2005) 51:16–24. doi: 10.1373/clinchem.2004.040097
88. Barkhausen T, Krettek C, Van Griensven M. L-selectin: adhesion, signalling and its importance in pathologic posttraumatic endotoxemia and non-septic inflammation. Exp Toxicol Pathol. (2005) 57:39–52. doi: 10.1016/j.etp.2005.02.007
89. Seidelin JB, Nielsen OH, Strøm J. Soluble L-selectin levels predict survival in sepsis. Intensive Care Med. (2002) 28:1613–8. doi: 10.1007/s00134-002-1501-5
90. Visser T, Hietbrink F, Groeneveld KM, Koenderman L, Leenen LPH. Isolated blunt chest injury leads to transient activation of circulating neutrophils. Eur J Trauma Emerg Surg. (2011) 37:177–84. doi: 10.1007/s00068-010-0041-x
91. Hietbrink F, Koenderman L, Althuizen M, Leenen LPH. Modulation of the innate immune response after trauma visualised by a change in functional PMN phenotype. Injury. (2009) 40:851–5. doi: 10.1016/j.injury.2008.11.002
92. Groeneveld KM, Hietbrink F, Hardcastle TC, Warren BL, Koenderman L, Leenen LPH. Penetrating thorax injury leads to mild systemic activation of neutrophils without inflammatory complications. Injury. (2014) 45:522–7. doi: 10.1016/j.injury.2013.09.030
93. Simms HH, D'Amico R. Posttraumatic auto-oxidative polymorphonuclear neutrophil receptor injury predicts the development of nosocomial infection. Arch Surg. (1997) 132:171–7. doi: 10.1001/archsurg.1997.01430260069016
94. Remold-O'Donnell E, Parent D. Two proteolytic pathways for down-regulation of the barrier molecule CD43 of human neutrophils. J Immunol. (1994) 152:3595–605.
95. Rieu P, Porteu F, Bessou G, Lesavre P, Halbwachs-Mecarelli L. Human neutrophils release their major membrane sialoprotein, leukosialin (CD43), during cell activation. Eur J Immunol. (1992) 22:3021–6. doi: 10.1002/eji.1830221138
96. Fung YL, Fraser JF, Wood P, Minchinton RM, Silliman MD, Silliman CC. The systemic inflammatory response syndrome induces functional changes and relative hyporesponsiveness in neutrophils. J Crit Care. (2008) 23:542–9. doi: 10.1016/j.jcrc.2007.09.004
97. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. (2008) 125:281–8. doi: 10.1111/j.1365-2567.2008.02950.x
98. Petit I, Ponomaryov T, Zipori D, Tsvee L. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. (2002) 3:687–94. doi: 10.1038/ni813
99. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. (2008) 9:46–56. doi: 10.1038/nrn2297
100. Tilton B, Ho L, Oberlin E, Loetscher P, Baleux F, Clark-Lewis I, et al. Signal transduction by Cxc chemokine receptor 4. J Exp Med. (2000) 192:313–24. doi: 10.1084/jem.192.3.313
101. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17:1381–90. doi: 10.1038/nm.2514
102. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
103. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, James H, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. (2012) 2652–60. doi: 10.1182/blood-2002-05-1300
104. Andersson U, Wang H, Palmblad K, Aveberger A-C, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (Hmg-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. (2002) 192:565–70. doi: 10.1084/jem.192.4.565
105. Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock. (2009) 32:17–22. doi: 10.1097/SHK.0b013e3181997173
106. Giannoudis PV, Mallina R, Harwood P, Perry S, Sante ED, Pape HC. Pattern of release and relationship between HMGB-1 and IL-6 following blunt trauma. Injury. (2010) 41:1323–7. doi: 10.1016/j.injury.2010.09.012
107. Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. (1991) 67:1033–6. doi: 10.1016/0092-8674(91)90279-8
108. McEver RP. Rolling back neutrophil adhesion. Nat Immunol. (2010) 11:282–4. doi: 10.1038/ni0410-282
109. Alon R, Hammer DA, Springer TA. Lifetime of the p-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature. (1995) 374:539–42. doi: 10.1038/374539a0
110. Mimasaka S, Funayama M, Hashiyada M, Nata M, Tsunenari S. Significance of levels of IL-6 and IL-8 after trauma: a study of 11 cytokines post-mortem using multiplex immunoassay. Injury. (2007) 38:1047–51. doi: 10.1016/j.injury.2007.02.045
111. Pallister I, Dent C, Topley N. Increased neutrophil migratory activity after major trauma: a factor in the etiology of acute respiratory distress syndrome? Crit Care Med. (2002) 30:1717–21. doi: 10.1097/00003246-200208000-00007
112. Murphy PM. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin Hematol. (1997) 34:311–8.
113. Adams JM, Hauser CJ, Livingston DH, Lavery RF, Fekete Z, Deitch EA. Early trauma polymorphonuclear neutrophil responses to chemokines are associated with development of sepsis, pneumonia, and organ failure. J Trauma. (2001) 51:452–6; discussion 452–7. doi: 10.1097/00005373-200109000-00005
114. Bastian OW, Mrozek MH, Raaben M, Leenen LPH, Koenderman L, Blokhuis TJ. Serum from the human fracture hematoma contains a potent inducer of neutrophil chemotaxis. Inflammation. (2018) 41:1084–92. doi: 10.1007/s10753-018-0760-4
115. Etscheid M, Kanse SM, Lochnit G, Gallenmueller A, Stephan F, Denk S, et al. Factor VII-activating protease is activated in multiple trauma patients and generates anaphylatoxin C5a. J Immunol. (2012) 188:2858–65. doi: 10.4049/jimmunol.1103029
116. Guo R-F, Ward PA. Role of c5a in inflammatory responses. Annu Rev Immunol. (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
117. Flierl MA, Perl M, Rittirsch D, Bartl C, Schreiber H, Fleig V, et al. The role of C5A in the innate immune response after experimental blunt chest trauma. Shock. (2008) 29:25–31. doi: 10.1097/shk.0b013e3180556a0b
118. Muller Kobold AC, Tulleken JE, Zijlstra JG, Sluiter W, Hermans J, Kallenberg CGM, Cohen Tervaert JW. Leukocyte activation in sepsis: correlations with disease state and mortality. Intensive Care Med. (2000) 26:883–92. doi: 10.1007/s001340051277
119. Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma Acute Care Surg. (1996) 40:501–10; discussion 510–2. doi: 10.1097/00005373-199604000-00001
120. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. (2016) 2016:2183026. doi: 10.1155/2016/2183026
121. Zang Q, Maass DL, White J, Horton JW. Cardiac mitochondrial damage and loss of ROS defense after burn injury: the beneficial effects of antioxidant therapy. J Appl Physiol. (2006) 102:103–12. doi: 10.1152/japplphysiol.00359.2006
122. Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Shear-dependent attenuation of cellular ROS levels can suppress proinflammatory cytokine injury to human brain microvascular endothelial barrier properties. J Cereb Blood Flow Metab. (2015) 35:1648–56. doi: 10.1038/jcbfm.2015.102
123. Partrick DA, Moore FA, Moore EE, Barnett CC Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. N Horizons Sci Pract Acute Med. (1996) 4:194–210.
124. Kim HY, Kim SJ, Lee SM. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. (2015) 282:259–70. doi: 10.1111/febs.13123
125. Han S, Cai W, Yang X, Jia Y, Zheng Z, Wang H, et al. ROS-mediated NLRP3 inflammasome activity is essential for burn-induced acute lung injury. Mediators Inflamm. (2015) 2015:720457. doi: 10.1155/2015/720457
126. Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation: an early vulnerable window. Surgery. (1995) 118:358–65. doi: 10.1016/S0039-6060(05)80345-9
127. Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS ONE. (2013) 8:e68963. doi: 10.1371/journal.pone.0068963
128. Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Dekaban GA, et al. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol. (2009) 215:308–16. doi: 10.1016/j.expneurol.2008.10.022
129. Swain SD, Rohn TT, Quinn MT. Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid Redox Signal. (2002) 4:69–83. doi: 10.1089/152308602753625870
130. Tanaka H, Ishikawa K, Nishino M, Shimazu T, Yoshioka T. Changes in granulocyte colony-stimulating factor concentration in patients with trauma and sepsis. J Trauma. (1996) 40:718–25; discussion 718–6. doi: 10.1097/00005373-199605000-00006
131. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. (2000) 165:2950–4. doi: 10.4049/jimmunol.165.6.2950
132. Djordjevic D, Pejovic J, Surbatovic M, Jevdjic J, Radakovic S, Veljovic M, et al. Prognostic value and daily trend of interleukin-6, neutrophil CD64 expression, C-reactive protein and lipopolysaccharide-binding protein in critically Ill patients: reliable predictors of outcome or not? J Med Biochem. (2015) 34:431–9. doi: 10.1515/jomb-2015-0002
133. Meng W, Paunel-Görgülü A, Flohé S, Witte I, Schädel-Höpfner M, Windolf J, yet al. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediators Inflamm. (2012) 2012:149560. doi: 10.1155/2012/149560
134. Taylor JV, Gordon LE, Hall H, Heinzelmann M, Polk HC Jr. Differences between bacterial species shown by simultaneous assessment of neutrophil phagocytosis and generation of reactive oxygen intermediates in trauma patients. Arch Surg. (1999) 134:1222–8. doi: 10.1001/archsurg.134.11.1222
135. Grogan JB. Altered neutrophil phagocytic function in burn patients. J Trauma. (1976) 16:734–8. doi: 10.1097/00005373-197609000-00009
136. Bjerknes R, Vindenes H, Pitkänen J, Ninnemann J, Lærum OD, Åbyholm F. Altered polymorphonuclear neutrophilic granulocyte functions in patients with large burns. J Trauma. (1989) 29:847–55. doi: 10.1097/00005373-198906000-00024
137. Shijo H, Iwabuchi K, Hosoda S, Watanabe H, Nagaoka I, Sakakibara N. Evaluation of neutrophil functions after experimental abdominal surgical trauma. Inflamm Res. (1998) 47:67–74. doi: 10.1007/s000110050278
138. Pap G, Fûrész J, Fennt J, Kovács GC, Nagy L, Hamar J. Self-regulation of neutrophils during phagocytosis is modified after severe tissue injury. Int J Mol Med. (2006) 17:649–54. doi: 10.3892/ijmm.17.4.649
139. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
140. Tofts PS, Chevassut T, Cutajar M, Dowell NG, Peters AM. Doubts concerning the recently reported human neutrophil lifespan of 5.4 days. Blood. (2011) 117:6050–2. doi: 10.1182/blood-2010-10-310532
141. Pillay J, Den Braber I, Vrisekoop N, Kwast LM, De Boer RJ, Borghans JAM, et al. in vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
142. Simon SI, Kim MH. A day (or 5) in a neutrophil's life. Blood. (2010) 116:511–2. doi: 10.1182/blood-2010-05-283184
143. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
144. Lam SW, Leenen LPH, van Solinge WW, Hietbrink F, Huisman A. Comparison between the prognostic value of the white blood cell differential count and morphological parameters of neutrophils and lymphocytes in severely injured patients for 7-day in-hospital mortality. Biomarkers. (2012) 17:642–7. doi: 10.3109/1354750X.2012.712161
145. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
146. Bryk JA, Popovic PJ, Zenati MS, Munera V, Pribis JP, Ochoa JB. Nature of myeloid cells expressing arginase 1 in peripheral blood after trauma. J Trauma. (2010) 68:843–52. doi: 10.1097/TA.0b013e3181b026e4
147. Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care. (2014) 18:article R163. doi: 10.1186/cc14003
148. Janols H, Bergenfelz C, Allaoui R, Larsson AM, Rydén L, Björnsson S, et al. High frequency of myeloid-derived suppressor cells in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol. (2014) 69:685–93. doi: 10.1189/jlb.5HI0214-074R
149. Christoffersson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
150. Garley M, Jabłonska E. Heterogeneity among neutrophils. Arch Immunol Ther Exp. (2018) 66:21–30. doi: 10.1007/s00005-017-0476-4
151. Nast-Kolb D, Waydhas C, Gippner-Steppert C, Schneider I, Trupka A, Ruchholtz S, et al. Indicators of the posttraumatic inflammatory response correlate with organ failure in patients with multiple injuries. J Trauma. (1997) 42:446–54. doi: 10.1097/00005373-199703000-00012
152. Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest. (1976) 58:705–15. doi: 10.1172/JCI108517
153. Pillay J, Hietbrink F, Koenderman L, Leenen LP. The systemic inflammatory response induced by trauma is reflected by multiple phenotypes of blood neutrophils. Injury. (2007) 38:1365–72. doi: 10.1016/j.injury.2007.09.016
154. Maier B, Lefering R, Lehnert M, Laurer HL, Steudel WI, Neugebauer EA, et al. Early versus late onset of multiple organ failure is associated with differing patterns of plasma cytokine biomarker expression and outcome after severe trauma. Shock. (2007) 28:668–74. doi: 10.1097/shk.0b013e318123e64e
155. Roumen RMH, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, et al. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Crit Care Med. (1995) 23:474–80. doi: 10.1097/00003246-199503000-00010
156. Altrichter J, Zedler S, Kraft R, Faist E, Mitzner SR, Sauer M, et al. Neutrophil-derived circulating free DNA (cf-DNA/NETs), a potential prognostic marker for mortality in patients with severe burn injury. Eur J Trauma Emerg Surg. (2010) 36:551–7. doi: 10.1007/s00068-010-0013-1
157. Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. (2006) 2:619–26. doi: 10.1038/ncprheum0338
158. Gomez CR, Nomellini V, Baila H, Oshima K, Kovacs EJ. Comparison of the effects of aging and IL-6 on the hepatic inflammatory response in two models of systemic injury: scald injury versus I.P. LPS administration. Shock. (2009) 31:178–84. doi: 10.1097/SHK.0b013e318180feb8
159. Beitl E, Banasova A, Vlcek M, Mikova D, Hampl V. Nitric oxide as an indicator for severity of injury in polytrauma. Bratislava Med J. (2016) 117:217–20. doi: 10.4149/BLL_2016_041
160. Hietbrink F, Koenderman L, Althuizen M, Pillay J, Kamp V, Leenen LPH. Kinetics of the innate immune response after trauma: implications for the development of late onset sepsis. Shock. (2013) 40:21–7. doi: 10.1097/SHK.0b013e318295a40a
161. Povsic MK, Beovic B, Ihan A. Perioperative increase in neutrophil CD64 expression is an indicator for intra-abdominal infection after colorectal cancer surgery. Radiol Oncol. (2017) 51:211–20. doi: 10.1515/raon-2016-0016
162. Kennedy SA, McEllistrem B, Kinsella A, Fan Y, Boyce S, Murphy K, et al. EuroSCORE and neutrophil adhesion molecules predict outcome post-cardiac surgery. Eur J Clin Invest. (2012) 42:881–90. doi: 10.1111/j.1365-2362.2012.02666.x
163. Logters T, Paunel-Gorgulu A, Zilkens C, Altrichter J, Scholz M, Thelen S, et al. Diagnostic accuracy of neutrophil-derived circulating free DNA (cf-DNA/NETs) for septic arthritis. J Orthop Res. (2009) 27:1401–7. doi: 10.1002/jor.20911
164. Napirei M, Ricken A, Eulitz D, Knoop H, Mannherz HG. Expression pattern of the deoxyribonuclease 1 gene: lessons from the Dnase1 knockout mouse. Biochem J. (2004) 380:929–37. doi: 10.1042/bj20040046
165. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. (2010) 11:328–34. doi: 10.1038/ni.1854
166. Kleber C, Becker CA, Schmidt-Bleek K, Schaser KD, Haas NP. Are pentraxin 3 and transsignaling early markers for immunologic injury severity in polytrauma? Clin Orthop Relat Res. (2013) 471:352–8. doi: 10.1007/s11999-013-2922-x
Keywords: trauma, neutrophil subtype, injury, survival, neutrophils
Citation: Mortaz E, Zadian SS, Shahir M, Folkerts G, Garssen J, Mumby S and Adcock IM (2019) Does Neutrophil Phenotype Predict the Survival of Trauma Patients? Front. Immunol. 10:2122. doi: 10.3389/fimmu.2019.02122
Received: 24 February 2019; Accepted: 23 August 2019;
Published: 06 September 2019.
Edited by:
Anja Fuchs, Washington University in St. Louis, United StatesReviewed by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesCopyright © 2019 Mortaz, Zadian, Shahir, Folkerts, Garssen, Mumby and Adcock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian M. Adcock, aWFuLmFkY29ja0BpbXBlcmlhbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.