 Bettina Hohberger1*
Bettina Hohberger1* Rudolf Kunze2Gerd Wallukat3
Rudolf Kunze2Gerd Wallukat3 Katja Kara1Christian Y. Mardin1Robert Lämmer1Ursula Schlötzer-Schrehardt1Sami Hosari1Folkert Horn1Luis Munoz4
Katja Kara1Christian Y. Mardin1Robert Lämmer1Ursula Schlötzer-Schrehardt1Sami Hosari1Folkert Horn1Luis Munoz4 Martin Herrmann4
Martin Herrmann4- 1Department of Ophthalmology, Friedrich-Alexander-University of Erlangen-Nürnberg, Erlangen, Germany
- 2Science Office, Berlin-Buch, Campus Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 3Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 4Department of Internal Medicine III, Institute of Clinical Immunology and Rheumatology, University of Erlangen-Nürnberg, Erlangen, Germany
Recently, agonistic autoantibodies (agAAb) activating the β2-adrenergic receptor were detected in primary open-angle glaucoma (POAG) or ocular hypertension (OHT) patients and were linked to intraocular pressure (IOP) (1). The aim of the present study was to quantify β2-agAAb in the sera of glaucoma suspects and patients with primary and secondary glaucoma. Patients with OHT (n = 33), pre-perimetric POAG (pre-POAG; n = 11), POAG (n = 28), and 11 secondary OAG (SOAG) underwent ophthalmological examinations including examinations with Octopus G1 perimetry and morphometry. Twenty-five healthy individuals served as controls. Serum-derived IgG samples were analyzed for β2-agAAb using a functional bioassay. The beat-rate-increase of spontaneously beating cultured neonatal rat cardiomyocytes was monitored with 1.6 beats/15 s as cut-off. None of the sera of normal subjects showed β2-agAAb. In POAG or OHT patients increased beating rates of 4.1 ± 2.2 beats/15 s, and 3.7 ± 2.8 beats/15 s were detected (p > 0.05). Glaucoma patients with (POAG) and without perimetric (pre-POAG) defects did not differ (pre-POAG 4.4 ± 2.6 beats/15 s, POAG 4.1 ± 2.0 beats/15 s, p > 0.05). Patients with SOAG yielded mean beating rates of 4.7 ± 1.7 beats/15 s (p > 0.05). β2-agAAb were seen in 73% of OHT, 82% of pre-POAG, 82% of POAG, and 91% SOAG patients (p < 0.001). Clinical data did not correlate with beating rate (p > 0.05). The robust β2-agAAb seropositivity in patients with OHT, pre-POAG, POAG, and SOAG suggest a primary common role for β2-agAAb starting early in glaucoma pathophysiology and turned out to be a novel marker identifying all patients with increased IOP independent of glaucoma stage and entity.
Introduction
The pathophysiology of glaucoma is known to be multifactorial. In addition to oxidative stress (2), vascular dysregulation (3), or morphological alterations of the trabecular meshwork (4), several further factors may be involved. Intraocular pressure (IOP) is the main risk factor for glaucoma. However, patients with glaucoma developed progressive disease despite optimal pharmaceutical and surgical IOP lowering therapies (5). The β2-adrenergic receptors (β2AR) is likely be involved in glaucoma pathogenesis, as topical application of the β-blocker timolol is an important module of anti-glaucomatous therapies. β2AR are to be found on several ocular tissues. β2AR is expressed by cells of the trabecular meshwork and the ciliary body, responsible for outflow and secretion of aqueous humor (AH), respectively (6, 7). Furthermore, microvessels and optic nerve tissue (8–10) reportedly express β2AR and thus influence microcirculation and neurons, respectively.
Recently, we described specific agonistic autoantibodies (agAAb) activating the β2AR (β2-agAAb) in patients with primary open-angle glaucoma (POAG) and ocular hypertension (OHT). We hypothesize that autoimmunity contributes to the etiopathogenesis of glaucoma. Adsorption of β2-agAAb may influence production and outflow of aqueous humor and, consequently, IOP (11).
The exact role of agonistic agAAb activating adrenergic receptors in disease is still elusive. Chagas' disease (12), allergic asthma (13), idiopathic dilated cardiomyopathy (13) in patients with heart failure (14) are associated with agonistic agAAb. Furthermore, in sera of patients, who suffer from Alzheimer's or vascular dementia β2-agAAb have been reported (15). The β2-agAAb, observed in patients with POAG or OHT, were directed against the second extracellular loop of β2AR (peptides 181–187 and 186–192); the major subclass was IgG3. In a principal-of-proof study IOP decreased transiently after unspecific removal by immunoadsorption of IgG, including the potentially pathogenic β2-agAAb (1). These observations lead us to postulate the hypothesis of an involvement of agonistic β2-agAAb in the pathogenesis of glaucoma. The aim of the present study was to investigate the distribution of agonistic β2-agAAb in glaucoma suspects and in patients with primary and secondary open-angle glaucoma.
Materials and Methods
Patients
One hundred-eight individuals were recruited from the Department of Ophthalmology and Eye Hospital, Friedrich-Alexander-University Erlangen-Nürnberg [Erlangen Glaucoma Registry, ISSN 2191-5008, CS-2011; NTC00494923 (5)]. We recruited 33 patients with OHT (54.8 ± 12.8 years, 17 female, 16 male), 11 pre-perimetric POAG (58.8 ± 10.9 years, 5 female, 6 male), 28 POAG (62.2 ± 13.7 years, 11 female, 17 male), and 11 SOAG (50.6 ± 12.5 years, 3 female, 8 male; 6 pigmentary glaucoma, PG; 5 Pseudoexfoliation glaucoma, PEXG). Twenty-five healthy subjects served as controls.
The probands underwent a complete ophthalmological examination including slit-lamp biomicroscopy, funduscopy, and white-on-white perimetry (Octopus 500, Interzeag, Schlieren, Switzerland; program G1). IOP was measured by Goldmann applanation tonometry at five times of the day (10.00 a.m., 12.00 a.m., 4.00 p.m., 9.00 p.m., and 12.00 p.m.). The retinal nerve fiber layer thickness, RNFL, was quantified by Spectralis Optical Coherence Tomography (Spectralis® OCT Version 1.9.10.0, Heidelberg Engineering, Heidelberg, Germany). The study protocol was done in accordance with the tenets of the Declaration of Helsinki and was approved by the Local Ethic Committee.
Control Subjects
Controls were screened with slit-lamp biomicroscopy, tonometry, fundoscopy, and papillometry for normal values.
Ocular Hypertension (OHT) Patients
Patients with OHT had an increased IOP (>21 mmHg, confirmed at least once), yet no sign of glaucomatous optic disc alterations, and normal perimetric white-on-white findings (G1 perimetry).
Primary Open-Angle Glaucoma (POAG) Patients
Diagnosis of POAG was based on an open anterior chamber angle, IOP > 21 mmHg (confirmed at least once), and a glaucomatous optic disc, classified after Jonas (16). Additionally, functional perimetric field loss had to be confirmed at least once according to the following criteria:
(I) at least three adjacent test points having a deviation ≥5 dB and with one test point with a deviation >10 dB lower than normal, or (II) at least two adjacent test points with a deviation ≥10 dB, or (III) at least three adjacent test points with a deviation ≥5 dB abutting the nasal horizontal meridian or (IV) a mean visual field defect of >2.6 dB.
Secondary Open-Angle Glaucoma (SOAG) Patients
Secondary open-angle glaucoma (SOAG) were classified according to POAG, yet with additional PEX material (PEXG) or pigmentary dispersion (PG). Additionally, SOAG patients displayed loss of the visual field, according to the before mentioned criteria (see POAG).
Primary Pre-perimetric Open-Angle Glaucoma (pre-POAG) Patients
Patients with pre-POAG showed the same morphological findings like those with POAG, however, their visual field was (still) normal.
Affinity Purification of agAAb
Affinity purification of agAAb was done by a biotinylated peptide biotin-AINCYANETCCD corresponding to the second extracellular loop of the β2AR. After incubation of 1 ml IgG with 300 μl of the peptide (100 μg/ml; for 1 h), this procedure was followed by an incubation with streptavidin-coated magnetic particles (Roche, Germany; for 30 min). The separation was performed with a magnetic separator (Dynal, Germany). After washing with PBS, eluation with 3 M KSCN in two 0.5 ml steps was done. Afterwards the agAAb were dialyzed with PBS (48 h, 4°C).
Cardiomyocyte Bioassay
Cardiac myocytes were prepared from heart ventricle of 1–2 day-old Sprague-Dawley rats (13). By digestion with a 0.25% solution of crude porcine trypsin (Serva, Germany) the myocardial cells were dispersed and suspended in SM20-I medium (Biochrom, Germany), which contained penicillin (Heyl, Germany), 10% heat-inactivated neonatal calf serum (Gibco, Germany), glutamine (Serva, Germany), streptomycin (HEFA Pharma; Germany), hydrocortisone (Merck, Germany), and fluorodeoxyuridine (Serva, Germany). After seeding the cardiomyocytes with a field density of 160.000 cells/cm2, the culture medium was refaced after 24 h. The cells were cultured for 3–4 days at 37°C previous to stimulation. Two hours before onset of the experiments, the medium was renewed with fresh culture solution. Target point was the beating rate for 15 s of 7–10 selected cardiomyocte cells or synchronously contracting cell clusters per flask, placed on a heated stage inverted microscope at 37°C (basal contraction rate: 162 ± 4 beats/min). The cardiomyocytes were incubated with the IgG fractions of probands' sera (duration: 60 min; dilution of 1:40). The target was loop II (HWYRATHQEAINCYANETCCDFFTNQ). The two epitopes were AINCYAN (AS181–187) and ANETCCD (AS186–192).
Statistical Analysis
Statistical analysis was done using SPSS (version 21.0). Data are displayed as mean ± standard deviation and percentages. Mann-Whitney-U-tests were calculated. Correlation analyses were performed.
Results
The data of this study is displayed in Figure 1 and Table 1. The beating rate of the cardiomyocytes was 0.15 ± 0.5 beats/15 s in normal subjects. However, patients with glaucoma yielded significant alterations of the cardiomyocytes in the bioassay: patients with OHT and POAG showed increased beating rates of 3.7 ± 2.8 and 4.1 ± 2.2 beats/15 s, respectively. Subgroup analysis for patients with glaucoma with (POAG) and without visual field defect (pre-perimetric POAG) showed mean beating rates of 4.4 ± 2.6 beats/15 s (pre-perimetric POAG) and 4.1 ± 2.0 beats/15 s (POAG). Patients with SOAG yielded mean beating rates of 4.7 ± 1.7 beats/15 s. Subdividing into PEXG and PD showed mean beating rates of 5.2 ± 0.8 beats/15 s and 4.2 ± 2.2 beats/15 s, respectively. No difference in beating rates was seen for group and subgroup analysis (p > 0.05, Mann-Whitney-U-test).
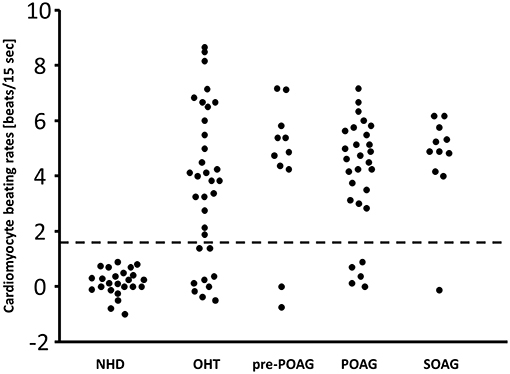
Figure 1. β2-agAAb in patients with OHT, primary, and secondary glaucoma. Cardiomyocyte beating rates [beats/15 s] modulated by IgG from sera of control subjects (normal healthy donor, NHD), patients with ocular hypertension (OHT), pre-perimetric POAG (primary open-angle glaucoma), POAG, and SOAG (secondary open-angle glaucoma) (cut-off 1.6 beats/15 s); cardiomyocyte beating rate is counted as difference of the baseline value (i.e., spontaneously beating rate of cultured cardiomyocytes) and the activated beating rate after addition of the test samples; therefore, some negative values were recorded.
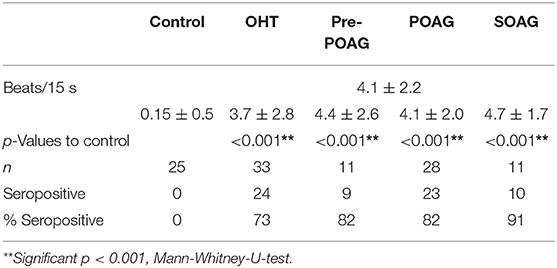
Table 1. Mean beating rates [beats/15 s], absolute number and percentage of β2-agAAb seropositivity of ocular hypertension (OHT), pre-perimetric POAG (pre-perimetric primary open-angle glaucoma), POAG, and SOAG patients in comparison to control subjects. Level of significance was reached for all observed patients' group compared to control.
With the cut-off value of 1.6 beats/15 s, β2-agAAb were to be observed in 24/33 (73%) of patients with OHT, 9/11 (82%) with pre-perimetric POAG, and 23/28 (82%) with POAG. SOAG patients yielded agAAb in 10/11 (91%). Subdivision into PD and PEXG showed 5/6 (83%) and 5/5 (100%), respectively (Figure 1).
Clinical data of all patients' groups can be seen in Table 2. Beating rates of β2-agAAb seropositive patients showed no correlation with morphometric measurements (RNFL; p > 0.05) and functional parameters (mean defect MD; loss variance LV p > 0.05). Additionally, no correlation was seen for beating rate of β2-agAAb seropositive patients and maximum IOP of the day of examination (p > 0.05). Additionally, not a single significant correlation was to be observed for the stages of glaucoma disease and beating rates (p > 0.05). Subgroup analysis yielded no significant correlation of beating rates with MD in POAG, SOAG (p > 0.05), respectively. Additionally, beating rates showed no correlation with maximum IOP of the day of examination in β2-agAAb seropositive OHT, pre-perimetric POAG, POAG, and SOAG (p > 0.05). RNFL was not correlated with beating rates in β2-agAAb seropositive pre-perimetric POAG, POAG, and SOAG (p > 0.05). Thus, the beat rate discriminated between all patients with increased IOP independent of glaucoma stage and entity.
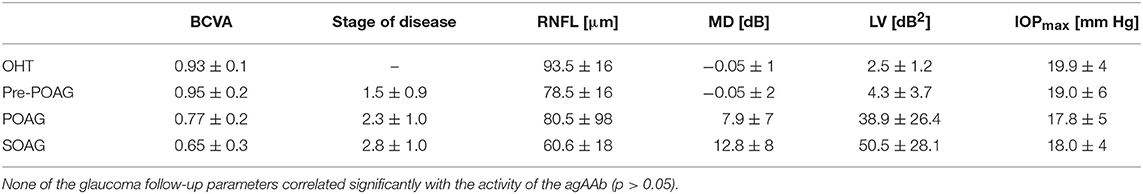
Table 2. Clinical data of OHT (ocular hypertension), pre-perimetric POAG (primary open-angle glaucoma), POAG, and SOAG (secondary open-angle glaucoma) patients: best corrected visual acuity (BCVA), disease stage (classified after Jonas), retinal nerve fiber layer thickness (RNFL), mean defect (MD), loss variance (LV), and maximum IOP (IOPmax).
Discussion
As shown in a previous study, pathophysiology of glaucoma is multifactorial and includes immunological components (1). Agonistic β2-agAAb were to be observed in sera of patients with OHT and POAG. Transient removal of circulating IgG including the agAAb transiently reduced the IOP, suggesting a link between β2-agAAb and IOP. As a cardiomyocyte bioassay detects the presence and function of autoantibodies, this method was used with the focus of showing functional active β2-agAAb. In the present study, we screened sera of glaucoma suspects and patients with primary and secondary glaucoma for the presence of functional active β2-agAAb. A percentage of 82 of POAG patients, and 91 of SOAG patients showed these β2-agAAb, whereas all healthy donors were seronegative. As β2-agAAb were also to be detected in sera of pre-perimetric POAG, and OHT patients, β2-agAAb, we assume that β2-agAAb are involved in the early etiology of glaucoma by a shared mechanism.
Immunological mechanisms were hypothesized to operate in glaucoma disease by several studies (17–19). However, the exact pathomechanisms of this neurodegenerative disease are still elusive. An elevated, not regulated IOP is known to be the major risk factor for onset and progression of glaucoma. Yet, most of patients with glaucoma show a disease progression despite optimal conservative and surgical IOP lowering therapies (5). Thus, several other factors contribute to its multifactorial pathogenesis (e.g., oxidative stress, vascular dysregulation) (20, 21).
It is supposed that agonistic β2-agAAb influence IOP (1), as therapy with β-blockers is common in clinical praxis. Increased IOP is caused by an imbalance between production and outflow of AH, both influenced by β2-agAAb. Experimental data showed that catecholamines can increase AH flow (22). Epinephrine induced AH flow, especially at nighttime (day: 15%: night 47%) (23). The molecular target for these clinical observations can be seen in the activity, expression and regulation of the Na-K-Cl co-transporter. As isoproterenol, a non-selective βAR agonist, is able to stimulate chloride transport via the ciliary epithelial layer (24) and Na-K-Cl co-transporter activity in pigmented ciliary body (25), it is justified to assume an influence of adrenergic agonists on the Na-K-Cl cotransporter in the ciliary body.
Considering the adrenergic subtype, application of a specific β2-blocker (ICI 118.551) diminished the stimulation of the Na-K-Cl co-transporter. We hypothesize that β2-agAAb hyperactivate the Na-K-Cl co-transporter in the ciliary body, as a similar effect of epinephrine on Na-K-Cl co-transporter activity was reported and this stimulation was inhibited by blocking the β2AR (25). The Na-K-Cl co-transporter activity then causes an increased AH flow especially at nighttime. This hypothesis is supported by clinical data of increased AH flow in OAG patients compared to normal subjects during the night hours (26). The Na-K-Cl co-transporter of the trabecular meshwork (TM), the main outflow pathway of AH, is a further potential target of the β2-agAAb which expose βAR predominantly of β2-subtype (7, 27, 28). Changes in the pore sizes of the TM, and thus of the AH outflow, were influenced by swelling and shrinkage of TM cells. The Na-K-Cl co-transporter is supposed to be a major regulator of the cellular volume and monolayer permeability regulation of TM cells, via ion transport (29). Inhibition and stimulation of the Na-K-Cl co-transporter in human TM cells leads to a reduction and increase of the volumes of TM cells, respectively (29). In cultured glaucomatous TM cells the cellular volume was reportedly enlarged when compared with healthy tissues and is reduced after incubation with bumetanide, a blocker of the Na-K-Cl co-transporter (30). Consistent with this finding the inhibition of the Na-K-Cl-co-transporter in human TM cells also led to an increased AH outflow facility (31). We hypothesize that β2-agAAb stimulate the Na-K-Cl-co-transporter in the TM cells, increase the TM cell volume with a consecutive decrease of extracellular space and outflow. This elevates the IOP.
β2AR are also present on pericytes and thus β2-agAAb may also influence the vascular microcirculation. Activation of β2AR on retinal blood vessels reportedly induce vasodilatation in vivo (32). A “direct” autonomic innervation is not present in retinal blood vessels (33). Yet, sympathetic activation can activate β2AR “indirectly” via its transmitter adrenaline (34). Especially, β2- and β3AR agonists have been shown to mediate retinal vasodilatation (35). According to the current data available in literature, β2AR mediate vasodilation and regulate the retinal microcirculation. We hypothesize that β2-agAAb mediated changes in retinal microcirculation further contributes to onset and progression of glaucoma, as blood flow in the optic nerve head and choroid were reduced in patients with POAG and OHT (36).
β2AR of human astrocytes and neurons of the optic nerve (9) are a third candidate target of β2-agAAb. After optic nerve transection in rabbits and rats, β2AR expression increased and peaked 90 days after intervention (9). A regulative function of astrocyte-borne β2AR has been assumed, as immature astrocytes suppressed glial scar formation (9, 37). Further support for this hypothesis was provided by an in vivo study on neuronal damage showing a reduced astrocyte hypertrophy and consecutive glial scar formation after application of βAR antagonists (38). Recent data showed that just an elevated IOP induced oxidative stress (via reduction in glutathione), axon degeneration of the optic nerve head and autophagy in the retinas. Thus, hypoxic glial cells can be detected in animals with OHT, even in the absence of a glaucomatous pathology (39). β2AR of astrocytes were also involved in the regulation of the glucose metabolism (40, 41) and are discussed to contribute to neuronal degeneration (42, 43).
In summary, β2-agAAb influence three factors of onset and progression of glaucoma: IOP, retinochoridal microcirculation as well as astrocyte and neuronal degeneration. All these targets are common findings in patients with OHT and POAG. Thus, β2-agAAb seemed to be a very early factor in the etiopathogenesis of glaucoma disease. Autoantibodies are hallmarks of a plethora of chronic diseases, like systemic lupus erythematosus (SLE) (44), refractory hypertension (45), type 2 diabetes (46), and dementia (15). Agonistic autoantibodies usually belong to the IgG subclasses 1–3. They are characterized by their binding to the extracellular loop 1 or 2 of neighboring G protein-coupled receptor molecules (GPCRs). This homodimerization non-canonically activates them in a manner similar to their natural ligands (47).
In contrast to the short-term activating canonical agonists, the binding of the agAAb delivers a rather stable signal, as observed in cell culture with neonatal rat cardiomyocytes. The uncontrolled, prolonged activation of the GPCR and its pathophysiological consequences was intensively investigated, especially for the AR. In a rat model, the cardiotoxic effects of agAAb against ß1AR have been demonstrated (48). Accordingly, fragments from the loop 2 of the β1AR induce dilated cardiomyopathy (DCM), also frequently associated with these agAAb in humans. In most autoimmune diseases, the pathogenic contribution of the agAAb is still elusive. Relatively clear clinical findings are present in DCM. Therapeutic apheresis (immunoadsorption) not only removed agAAb against the ß1AR from the bloodstream but also improves cardiac output for months (49). The main therapeutic target in glaucoma is the elevated IOP. Reduction of each single mmHg delays glaucoma onset and/or progression (50). Previously we reported that unspecific adsorption of IgG was accompanied by a transient decrease of IOP in patients, refractory to maximal conservative therapies (1). We argue that a specific elimination of β2-agAAb is a candidate option for the therapy of medical refractory glaucoma.
Glaucoma can be detected in up to 13% of patients with SLE. This may be caused by (I) a general predisposition to autoimmunity, (II) the interaction of the various AAb and agAAb, and (III) the therapeutic administration of glucocorticoids (51, 52). It is of interest to analyze the consequences of the interaction of different agAAb being simultaneously present in a patient.
Functional active agonistic autoantibodies directed against the GPCR are measured by using a cardiomyocyte bioassay, yet alternatives failed until now. However, western blot, immune precipitation and ELISA were unable to discriminate between agonistic autoantibodies and those that merely bind the autoantigene. None of the assays, including ours (unpublished) were able to reliably identify the agonistic antibodies. This might be due to the potential different conformation of human recombinant receptors (β2AR) compared to the highly steric specific epitope, binding agAAb. Additionally, the molecular weight of the antibody binding loop peptide (instead of the whole ß2AR) is too low for precipitation.
Conclusion
Immunologic factors are increasingly being discussed to be involved in the etiopathogenesis of glaucoma (1, 17). Here we present data on β2-agAAb seropositivity in patients with POAG, SOAG, pre-perimetric POAG, and OHT and argue for a critical role of β2-agAAb already in an early asymptomatic phase of glaucoma disease. Further studies are justified to clarify the molecular basis of the β2-agAAb in glaucoma disease and therapy.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the recommendations of Declaration of Helsinki and ethic committee of the university of Erlangen with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the ethic committee of the University of Erlangen.
Author Contributions
BH, RK, MH, and GW had the idea and planned the study. US-S planned the study. GW performed the laboratory work. RL, CM, LM, SH, and FH performed and acquired the clinical trial. BH performed data acquisition and statistical analysis. BH and KK were responsible for the draft of the manuscript.
Funding
The Erlangen Glaucoma Registry was funded by the German Research Society (DFG) from 1991 to 2009, NCT00494923.
Conflict of Interest Statement
GW, MH, and RK do have a patent (EP1832600A1).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Junemann A, Hohberger B, Rech J, Sheriff A, Fu Q, Schlotzer-Schrehardt U, et al. Agonistic autoantibodies to the β2-adrenergic receptor involved in the pathogenesis of open-angle glaucoma. Front Immunol. (2018) 9:145. doi: 10.3389/fimmu.2018.00145
2. Hohberger B, Chaudhri MA, Michalke B, Lucio M, Nowomiejska K, Schlotzer-Schrehardt U, et al. Levels of aqueous humor trace elements in patients with open-angle glaucoma. J Trace Elem Med Biol. (2018) 45:150–5. doi: 10.1016/j.jtemb.2017.10.003
3. Flammer J. The vascular concept of glaucoma. Surv Ophthalmol. (1994) 38(Suppl):S3–6. doi: 10.1016/0039-6257(94)90041-8
4. Tektas OY, Lutjen-Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. (2009) 88:769–75. doi: 10.1016/j.exer.2008.11.025
5. Hohberger B, Monczak E, Mardin CY. 26 years of the erlangen glaucoma registry: demographic and perimetric characteristics of patients through the ages. Klin Monbl Augenheilkd. (2017) 236:691–8. doi: 10.1055/s-0043-112856
6. Wax MB, Molinoff PB. Distribution and properties of beta-adrenergic receptors in human iris-ciliary body. Invest Ophthalmol Vis Sci. (1987) 28:420–30.
7. Crider JY, Sharif NA. Adenylyl cyclase activity mediated by beta-adrenoceptors in immortalized human trabecular meshwork and non-pigmented ciliary epithelial cells. J Ocul Pharmacol Ther. (2002) 18:221–30. doi: 10.1089/108076802760116142
8. Ferrari-Dileo G. Beta 1 and beta 2 adrenergic binding sites in bovine retina and retinal blood vessels. Invest Ophthalmol Vis Sci. (1988) 29:695–9.
9. Mantyh PW, Rogers SD, Allen CJ, Catton MD, Ghilardi JR, Levin LA, et al. Beta 2-adrenergic receptors are expressed by glia in vivo in the normal and injured central nervous system in the rat, rabbit, and human. J Neurosci. (1995) 15:152–64. doi: 10.1523/JNEUROSCI.15-01-00152.1995
10. Feher LZ, Kalman J, Puskas LG, Gyulveszi G, Kitajka K, Penke B, et al. Impact of haloperidol and risperidone on gene expression profile in the rat cortex. Neurochem Int. (2005) 47:271–80. doi: 10.1016/j.neuint.2005.04.020
11. Chan KKW, Tang F, Tham CCY, Young AL, Cheung CY. Retinal vasculature in glaucoma: a review. BMJ Open Ophthalmol. (2017) 1:e000032. doi: 10.1136/bmjophth-2016-000032
12. Borda E, Pascual J, Cossio P, Delavega M, Arana R, Sterinborda L. A circulating Igg in chagas-disease which binds to beta-adrenoceptors of myocardium and modulates their activity. Clin Exp Immunol. (1984) 57:679–86.
13. Wallukat G, Wollenberger A. Effects of the serum gamma globulin fraction of patients with allergic asthma and dilated cardiomyopathy on chronotropic beta adrenoceptor function in cultured neonatal rat heart myocytes. Biomed Biochim Acta. (1987) 46:S634–9.
14. Zhang L, Hu D, Shi X, Li J, Zeng W, Xu L, et al. Autoantibodies against the myocardium beta 1-adrenergic and M2-muscarinic receptors in patients with heart failure. Zhonghua Nei Ke Za Zhi. (2001) 40:445–7.
15. Karczewski P, Hempel P, Kunze R, Bimmler M. Agonistic autoantibodies to the alpha(1) -adrenergic receptor and the beta(2) -adrenergic receptor in Alzheimer's and vascular dementia. Scand J Immunol. (2012) 75:524–30. doi: 10.1111/j.1365-3083.2012.02684.x
16. Jonas JB, Gusek GC, Naumann GO. Optic disc morphometry in chronic primary open-angle glaucoma. I. Morphometric intrapapillary characteristics. Graefes Arch Clin Exp Ophthalmol. (1988) 226:522–30. doi: 10.1007/BF02169199
17. Grus FH, Joachim SC, Hoffmann EM, Pfeiffer N. Complex autoantibody repertoires in patients with glaucoma. Mol Vis. (2004) 10:132–7.
18. Joachim SC, Pfeiffer N, Grus FH. Autoantibodies in patients with glaucoma: a comparison of IgG serum antibodies against retinal, optic nerve, and optic nerve head antigens. Graefes Arch Clin Exp Ophthalmol. (2005) 243:817–23. doi: 10.1007/s00417-004-1094-5
19. Reichelt J, Joachim SC, Pfeiffer N, Grus FH. Analysis of autoantibodies against human retinal antigens in sera of patients with glaucoma and ocular hypertension. Curr Eye Res. (2008) 33:253–61. doi: 10.1080/02713680701871157
20. Grieshaber MC, Mozaffarieh M, Flammer J. What is the link between vascular dysregulation and glaucoma? Surv Ophthalmol. (2007) 52(Suppl 2):S144–54. doi: 10.1016/j.survophthal.2007.08.010
21. Fick A, Junemann A, Michalke B, Lucio M, Hohberger B. Levels of serum trace elements in patients with primary open-angle glaucoma. J Trace Elem Med Biol. (2019) 53:129–34. doi: 10.1016/j.jtemb.2019.02.006
22. Brubaker RF. Flow of aqueous humor in humans [The Friedenwald Lecture]. Invest Ophthalmol Vis Sci. (1991) 32:3145–66.
23. Topper JE, Brubaker RF. Effects of timolol, epinephrine, and acetazolamide on aqueous flow during sleep. Invest Ophthalmol Vis Sci. (1985) 26:1315–9.
24. Crook RB, Takahashi K, Mead A, Dunn JJ, Sears ML. The role of NaKCl cotransport in blood-to-aqueous chloride fluxes across rabbit ciliary epithelium. Invest Ophthalmol Vis Sci. (2000) 41:2574–83.
25. Hochgesand DH, Dunn JJ, Crook RB. Catecholaminergic regulation of Na-K-Cl cotransport in pigmented ciliary epithelium: differences between PE and NPE. Exp Eye Res. (2001) 72:1–12. doi: 10.1006/exer.2000.0927
26. Larsson LI, Rettig ES, Brubaker RF. Aqueous flow in open-angle glaucoma. Arch Ophthalmol. (1995) 113:283–6. doi: 10.1001/archopht.1995.01100030037018
27. Jampel HD, Lynch MG, Brown RH, Kuhar MJ, De Souza EB. Beta-adrenergic receptors in human trabecular meshwork. Identification and autoradiographic localization. Invest Ophthalmol Vis Sci. (1987) 28:772–9.
28. Wax MB, Molinoff PB, Alvarado J, Polansky J. Characterization of beta-adrenergic receptors in cultured human trabecular cells and in human trabecular meshwork. Invest Ophthalmol Vis Sci. (1989) 30:51–7.
29. O'donnell ME, Brandt JD, Curry FR. Na-K-Cl cotransport regulates intracellular volume and monolayer permeability of trabecular meshwork cells. Am J Physiol. (1995) 268:C1067–74. doi: 10.1152/ajpcell.1995.268.4.C1067
30. Putney LK, Vibat CR, O'donnell ME. Intracellular Cl regulates Na-K-Cl cotransport activity in human trabecular meshwork cells. Am J Physiol. (1999) 277:C373–83. doi: 10.1152/ajpcell.1999.277.3.C373
31. Al-Aswad LA, Gong H, Lee D, O'donnell ME, Brandt JD, Ryan WJ, et al. Effects of Na-K-2Cl cotransport regulators on outflow facility in calf and human eyes in vitro. Invest Ophthalmol Vis Sci. (1999) 40:1695–701.
32. Group CN-TGS. The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Am J Ophthalmol. (1998) 126:498–505. doi: 10.1016/S0002-9394(98)00272-4
33. Delaey C, Van De Voorde J. Regulatory mechanisms in the retinal and choroidal circulation. Ophthalmic Res. (2000) 32:249–56. doi: 10.1159/000055622
34. Nakazawa T, Sato A, Mori A, Saito M, Sakamoto K, Nakahara T, et al. Beta-adrenoceptor-mediated vasodilation of retinal blood vessels is reduced in streptozotocin-induced diabetic rats. Vascul Pharmacol. (2008) 49:77–83. doi: 10.1016/j.vph.2008.06.001
35. Mori A, Nakahara T, Sakamoto K, Ishii K. Role of beta3-adrenoceptors in regulation of retinal vascular tone in rats. Naunyn Schmiedebergs Arch Pharmacol. (2011) 384:603–8. doi: 10.1007/s00210-011-0682-2
36. Fuchsjager-Mayrl G, Wally B, Georgopoulos M, Rainer G, Kircher K, Buehl W, et al. Ocular blood flow and systemic blood pressure in patients with primary open-angle glaucoma and ocular hypertension. Invest Ophthalmol Vis Sci. (2004) 45:834–9. doi: 10.1167/iovs.03-0461
37. Smith GM, Miller RH. Immature type-1 astrocytes suppress glial scar formation, are motile and interact with blood vessels. Brain Res. (1991) 543:111–22. doi: 10.1016/0006-8993(91)91054-5
38. Sutin J, Griffith R. Beta-adrenergic receptor blockade suppresses glial scar formation. Exp Neurol. (1993) 120:214–22. doi: 10.1006/exnr.1993.1056
39. Jassim AH, Inman DM. Evidence of hypoxic glial cells in a model of ocular hypertension. Invest Ophthalmol Vis Sci. (2019) 60:1–15. doi: 10.1167/iovs.18-24977
40. Gibbs ME, Hutchinson DS, Summers RJ. Role of β-adrenoceptors in memory consolidation: β3-adrenoceptors act on glucose uptake and β2-adrenoceptors on glycogenolysis. Neuropsychopharmacology. (2008) 33:2384–97. doi: 10.1038/sj.npp.1301629
41. Catus SL, Gibbs ME, Sato M, Summers RJ, Hutchinson DS. Role of β-adrenoceptors in glucose uptake in astrocytes using β-adrenoceptor knockout mice. Br J Pharmacol. (2011) 162:1700–15. doi: 10.1111/j.1476-5381.2010.01153.x
42. Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. (2006) 2:679–89. doi: 10.1038/ncpneuro0355
43. Steele ML, Robinson SR. Reactive astrocytes give neurons less support: implications for Alzheimer's disease. Neurobiol Aging. (2012) 33:423.e1–13. doi: 10.1016/j.neurobiolaging.2010.09.018
44. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. (2015) 14:75–9. doi: 10.1016/j.autrev.2014.10.003
45. Wenzel K, Haase H, Wallukat G, Derer W, Bartel S, Homuth V, et al. Potential relevance of alpha(1)-adrenergic receptor autoantibodies in refractory hypertension. PLoS ONE. (2008) 3:e3742. doi: 10.1371/journal.pone.0003742
46. Hempel P, Karczewski P, Kohnert KD, Raabe J, Lemke B, Kunze R, et al. Sera from patients with type 2 diabetes contain agonistic autoantibodies against G protein-coupled receptors. Scand J Immunol. (2009) 70:159–60. doi: 10.1111/j.1365-3083.2009.02280.x
47. Wallukat GNE, Müller J, Brinckmann R, Schimke J, Kunze R. The Pathophysiological Role of Autoantibodies Directed to G-Protein Coupled Receptors and Therapeutic Strategies of Antibody Removal. Lengerich: Pabst Science Publishers (2002).
48. Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, et al. Direct evidence for a beta(1)-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest. (2004) 113:1419–29. doi: 10.1172/JCI200420149
49. Muller J, Wallukat G, Dandel M, Bieda H, Brandes K, Spiegelsberger S, et al. Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation. (2000) 101:385–91. doi: 10.1161/01.CIR.101.4.385
50. Leske MC, Heijl A, Hussein M, Bengtsson B, Hyman L, Komaroff E, et al. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch Ophthalmol. (2003) 121:48–56. doi: 10.1001/archopht.121.1.48
51. Fel AAE, Le Jeunne C. Indications et complications des corticoïdes en ophtalmologie. Presse Med. (2012) 41:414–421. doi: 10.1016/j.lpm.2012.02.001
52. Carli L, Tani C, Querci F, Della Rossa A, Vagnani S, Baldini C, et al. Analysis of the prevalence of cataracts and glaucoma in systemic lupus erythematosus and evaluation of the rheumatologists' practice for the monitoring of glucocorticoid eye toxicity. Clin Rheumatol. (2013) 32:1071–3. doi: 10.1007/s10067-013-2214-6
Keywords: autoantibodies, glaucoma, ocular hypertension, β2-adrenergic receptor, primary open-angle glaucoma, secondary open-angle glaucoma
Citation: Hohberger B, Kunze R, Wallukat G, Kara K, Mardin CY, Lämmer R, Schlötzer-Schrehardt U, Hosari S, Horn F, Munoz L and Herrmann M (2019) Autoantibodies Activating the β2-Adrenergic Receptor Characterize Patients With Primary and Secondary Glaucoma. Front. Immunol. 10:2112. doi: 10.3389/fimmu.2019.02112
Received: 24 April 2019; Accepted: 21 August 2019;
Published: 01 October 2019.
Edited by:
Marco Emilio Bianchi, Vita-Salute San Raffaele University, ItalyReviewed by:
Luciano Quaranta, University of Pavia, ItalyAndras Perl, Upstate Medical University, United States
Copyright © 2019 Hohberger, Kunze, Wallukat, Kara, Mardin, Lämmer, Schlötzer-Schrehardt, Hosari, Horn, Munoz and Herrmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bettina Hohberger, bettina.hohberger@uk-erlangen.de