
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 04 September 2019
Sec. Primary Immunodeficiencies
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02108
This article is part of the Research TopicApplication of Cytometry in Primary ImmunodeficienciesView all 21 articles
 Cindy S. Ma1,2,3*
Cindy S. Ma1,2,3* Stuart G. Tangye1,2,3*
Stuart G. Tangye1,2,3*The advent of flow cytometry has revolutionized the way we approach our research and answer specific scientific questions. The flow cytometer has also become a mainstream diagnostic tool in most hospital and pathology laboratories around the world. In particular the application of flow cytometry has been instrumental to the diagnosis of primary immunodeficiencies (PIDs) that result from monogenic mutations in key genes of the hematopoietic, and occasionally non-hematopoietic, systems. The far-reaching applicability of flow cytometry is in part due to the remarkable sensitivity, down to the single-cell level, of flow-based assays and the extremely user-friendly platforms that enable comprehensive analysis, data interpretation, and importantly, robust and rapid methods for diagnosing PIDs. A prime example is the absence of peripheral blood B cells in patients with agammaglobulinemia due to mutations in BTK or related genes in the BCR signaling pathway. Similarly, the development of intracellular staining protocols to detect expression of SAP, XIAP, or DOCK8 expedites the rapid diagnosis of the X-linked lymphoproliferative diseases or an autosomal recessive form of hyper-IgE syndrome (HIES), respectively. It has also become evident that distinct cohorts of PID patients exhibit unique “lymphocyte phenotypic signatures” that are often diagnostic even prior to identifying the genetic lesion. Flow cytometry-based sorting provides a technique for separating specific subsets of immune cells such that they can be studied in isolation. Thus, flow-based assays can be utilized to measure immune cell function in patients with PIDs, such as degranulation by cytotoxic cells, cytokine expression by many immune cells (i.e., CD4+ and CD8+ T cells, macrophages etc.), B-cell differentiation, and phagocyte respiratory burst in vitro. These assays can also be performed using unfractionated PBMCs, provided the caveat that the composition of lymphocytes between healthy donors and the PID patients under investigation is recognized. These functional deficits can assist not only in the clinical diagnosis of PIDs, but also reveal mechanisms of disease pathogenesis. As we move into the next generation of multiparameter flow cytometers, here we review some of our experiences in the use of flow cytometry in the study, diagnosis, and unraveling the pathophysiology of PIDs.
The application of new technologies to basic research and clinical investigations has greatly improved biochemical and molecular analyses of cellular physiology and identified defects in these processes that underpin human disease. Thus, technological advances have enabled not only fundamental discoveries of basic immunology, but also a greater understanding of disease pathogenesis, rapid diagnoses of these conditions as well as providing opportunities for the development and/or implementation of improved therapies. Great examples of this can be found in the fields of clinical immunology and immunodeficiency.
Using electrophoretic analysis of serum from a young boy with severe and recurrent bacterial infections led to the discovery by Col Ogden Bruton of the first case of agammaglobulinemia (1). Importantly, treating this patient with monthly subcutaneous infusions of concentrated human immune serum globulin prevented further infections (1). These observations established that agammaglobulinemia caused this patient's recurrent infections, and that the gammaglobulin fraction of serum contained antibodies capable of prophylactically preventing infection. Critically, this finding in a single patient—which pre-dated the discovery of B cells by more than a decade (2)—led to the identification 40 years later that mutations in BTK, encoding Bruton's tyrosine kinase, cause X-linked agammaglobulinemia (XLA) (3).
Extending this work by Bruton, the advent of serological reagents capable of reacting with specific lymphocyte subsets—such as polyclonal antisera raised against surface Ig molecules—enabled the identification of B cells in the peripheral blood of healthy individuals, and the absence of Ig-expressing cells in XLA (4–7). Importantly, XLA patients were found to have near-normal frequencies of precursor B cells in their bone marrow (BM), the site of B-cell development (8), establishing that the very early—but not later—stages of B-cell development were intact in XLA patients. Similarly, advances in techniques to fractionate human peripheral blood leukocyte subsets by density gradient centrifugation allowed the isolation of populations enriched for B cells, T cells and monocytes. This elegant approach also revealed a stark paucity of B cells in XLA (9). These analytical approaches demonstrating an absence of peripheral blood Ig-expressing B cells in XLA patients provided a clear explanation for their agammaglobulinemia.
These serological studies using anti-Ig not only defined B cells as the cellular deficiency in XLA, but also gave greater clarity to other immune deficient conditions. For instance, investigation of X-linked or autosomal recessive (AR) severe combined immunodeficiency (SCID) revealed that, despite persistent lymphopenia and hypogammaglobulinemia, the majority (>90%) of lymphocytes in these individuals were actually B cells (7, 10). This established that these conditions were likely due to a deficiency of T cells, thus defining T−B+ SCID. Likewise, studies of males who were hypo- or agammaglobulinemic but had normal frequencies of B cells, along with T cells, delineated an X-linked PID distinct from XLA that probably represented X-linked hyper-IgM syndrome (6, 7).
The ability to generate immortalized cells lines (hybridomas) producing monoclonal Abs (mAbs) with defined and distinct specificities (11) led to quantum leaps in basic and clinical immunology. Thus, it quickly became possible to identify immune cell populations and subsets not only according to the differential expression of surface Ig, but also by the presence, or absence of other cell surface molecules. By using mAbs against surface markers that are coordinately expressed during B-cell development, it was found that the very few B cells present in the circulation of XLA patients resembled immature B cells in BM and were distinct from mature B cells in the peripheral blood of healthy donors (12). This finding was critical in identifying the stage at which B cell development is blocked by inactivating mutations in BTK (3).
This approach of studying immune cell subsets by immunofluorescent microscopy was also critical in understanding pathophysiology of HIV infection and subsequent progression to AIDS. Here, a reduction in the number of peripheral blood CD4+ T cells, and a corresponding inversion of the CD4:CD8 T cell ratio, became a defining clinical characteristic of individuals infected with HIV (13–16). Furthermore, the steady decline in numbers of CD4+ T cells in HIV infection became predictive of progression to full blown AIDS, revealing the need to longitudinally track CD4+ T cells as a biomarker of disease progression following HIV infection (16, 17).
While methodologies such as density gradient centrifugation and immunofluorescent microscopy advanced our understanding of basic immunology and disease, they were laborious and often lacked the level of sensitivity and quantitation required to make definitive interpretations of the data. By simultaneously enabling the rapid analysis of large numbers of immune cells, flow cytometry has had a profound impact on immunology (18), including its application to the study of primary and acquired immunodeficiencies.
It became possible to quickly assess the status of CD4+ T cells in HIV infection (17), accurately define the stages and phenotypes of B cell development in human BM and how mutations in genes such as BTK, BLNK, IGHM, IGLL1, CD79A, CD79B, PIK3R1, and TCF3 differentially affect this process (3, 19, 20), and delineate distinct types of SCID due to different gene mutations according to the presence and absence of specific lymphocyte populations, such as B+T−NK– SCID (IL2RG, JAK3), B−T−NK+ SCID (RAG1, RAG2), B+T−NK+ SCID (IL7RA), or B−T−NK− SCID (ADA) (20, 21).
The discoveries of surface molecules that are induced on activated lymphocytes, or distinguish discrete subsets of T and B cells, also led to major advances in the discovery, diagnosis, management, and classification of PIDs (20). Thus, males with X-linked hyper IgM syndrome could be identified by the inability of their CD4+ T cells to upregulate expression of functional CD40L following anti-CD3/CD28 or PMA/ionomycin-mediated activation (22, 23). Common variable immunodeficiency (CVID) due to ICOS mutations was discovered by the identification of a small number of patients whose T cells lacked ICOS expression following in vitro stimulation (24). The finding that CD27 is expressed on human memory, but not naïve, B cells (25, 26) enabled an entirely new stratification system of CVID that could reliably classify patients with various pathologies (27, 28).
We have also exploited this finding, together with the availability of patients with PIDs, to identify non-redundant molecular and cellular requirements for the generation and/or maintenance of memory B cells in humans. Thus, mutations that disrupt (i) CD4+ T cell/B cell interactions and thus delivery of CD4+ T cell-mediated B cell help (e.g., loss of function [LOF] CD40L, ICOS, CD40, NEMO, SH2D1A, RLTPR [CARMIL2], CD27/CD70), (ii) cytokine signaling particularly through IL-21 and STAT3 (i.e., IL21, IL21R, IL2RG, ZNF341, IL10R LOF; STAT3 dominant negative [DN]; STAT1 gain of function [GOF]), or (iii) other intracellular signaling pathways (STK4, DOCK8, SP110 LOF; PIK3CD GOF) all reduce memory B cells (defined as CD19+CD20+CD10−CD27+ cells) in affected individuals (29–41) (Figure 1). Similar studies performed by other investigators have established that signaling via CARD11/BCL10/MALT1 (45), CD19/CD81 (46), and NIK/NFKB2 (47, 48) are also key regulators of the generation and/or maintenance of human memory B cells. Importantly, this approach also established that, for instance, IL12Rβ1/2, IL-23R, TYK2, and STAT1 signaling (32, 42), nor SPPL2A (43) or GINS1 (44), are required for generating and/or maintaining the memory B cell pool in humans (Figure 1).
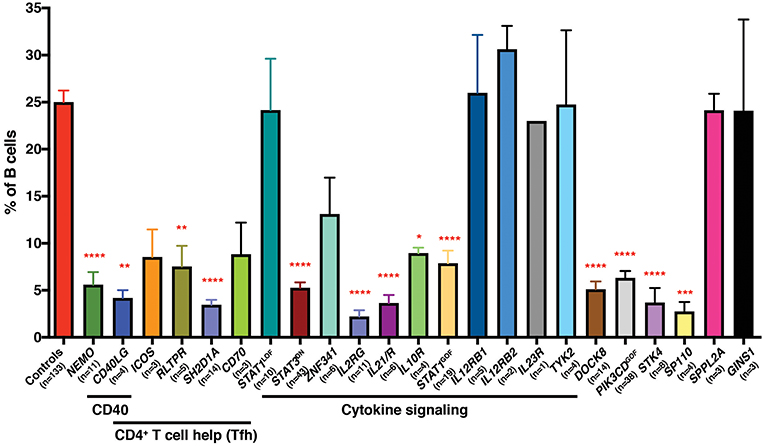
Figure 1. Impact of inborn errors of immunity on the generation of memory B cells. PBMCs from the indicated numbers of healthy donor controls or from individuals with pathogenic mutations in the indicated genes were stained with mAbs against CD19, CD20, CD10, and CD27. The proportions of B cells exhibiting a CD19+CD10−CD27+ memory phenotype was determined by flow cytometric gating and analysis. Significant differences were determined by ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). The age range of the healthy donors is from birth (cord blood) to ~65 years old. These data are compiled from findings previously reported in the following publications: (29–37, 39–44).
Further advances in flow cytometric methodologies enabled detection of intracellular proteins (49), as well as post-translational modifications to proteins involved in cell signaling (50). These developments were also embraced by the clinical immunology field to facilitate rapid diagnosis of PIDs and discover patients with novel immune defects. The ability to quantify expression of SAP, XIAP, BTK, FOXP3, and DOCK8 proteins by intracellular staining and flow cytometric analysis expedited detection and diagnosis of patients with X-linked lymphoproliferative disease (XLP) type 1 (LOF mutations in SH2D1A), XLP-2 (LOF mutations in XIAP/BIRC4), XLA (BTK), IPEX (FOXP3), and an AR form of HIES (DOCK8), respectively (51–56) (Figure 2). Furthermore, this facilitated the identification of female carriers of some X-linked traits, such as XLP-1, XLP-2, and XLA (51, 55–57, 59) (Figure 2), as well as the discovery of somatic reversion in XLP-1 (60), DOCK8-deficiency (54), and leukocyte adhesion deficiency-1 due to mutations in ITGB2 encoding CD18 (61, 62). Similarly, the detection of intracellular phosphorylated STAT proteins in response to cytokine-mediated stimulation of lymphocyte populations has been developed as a functional screen to identify individuals with LOF mutations in IL10R, IL12RB1, or IFNGR1 (63–65), or GOF mutations in STAT1 (66). Importantly, this technique has been applied to discover patients with novel inborn errors of immunity, including LOF mutations in IL21R (67), IL6ST (68), and IL6R (69). Clinical flow cytometry has also played a valuable role in studying PIDs affecting innate immunity. Assessing respiratory burst in phagocytes using oxidation of fluorescent probes such as dihydrorhodamine 123 (DHR) following leukocyte activation in vitro is the gold standard for diagnosing individuals with either X-linked or AR forms of chronic granulomatous disease (70), as well as females carriers of the X-linked form of this condition (59). Lastly, the use of cell permeable fluorescent dyes such as carboxyfluorescein succinimidyl ester (CFSE), Cell trace violet and Cell trace yellow, to label intracellular molecules and then track the dilution and concomitant reduction in fluorescence intensity of these dyes with each cell division (71, 72) has enabled detailed analysis of the role of cell division in lymphocyte differentiation and how these events can be uncoupled or compromised in various PIDs (29–31, 33, 35–37, 40, 58, 60, 73–76).
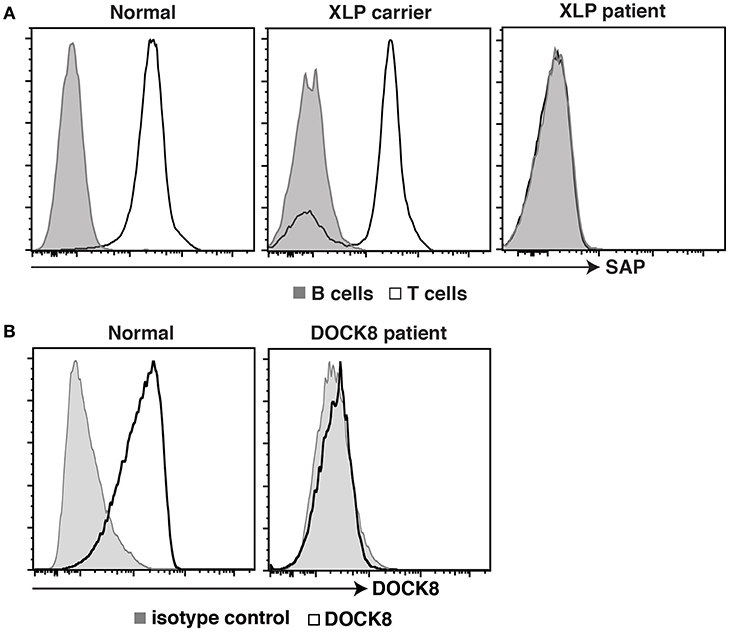
Figure 2. Intracellular staining for SAP and DOCK8 in controls, patients, and female carriers. (A) PBMCs from healthy donors, female carriers of the XLP trait, or individuals with pathogenic mutations in SH2D1A, or were stained with mAbs against CD3, CD20, and SAP. Expression of SAP in B cells (tinted histogram) and T cells (open histogram) was then determined. (B) PBMCs from healthy donors, or from an individual suspected of having DOCK8-deficiency were stained with an isotype control Ab (tinted histogram) or a mAb against DOCK8 (open histogram). Expression of DOCK8 in lymphocytes was then determined. These data are compiled from findings previously reported in the following publications: (37, 57, 58).
However, there are important caveats to consider when using flow cytometry to determine potential molecular causes of PIDs. While the vast majority of gene variants found to cause PIDs abolish protein expression, and thus a lack of the encoded protein can be used as a surrogate to confirm the pathogenicity of a mutation, several variants do not affect protein expression and are pathogenic due to them being LOF. An example of this can be found in one of the original descriptions of mutations in CD40LG, which reported detectable expression of CD40L on activated CD4+ T cells from one patient with X-linked hyper-IgM syndrome despite the presence of a predicted inactivating mutation (23). Similarly, pathogenic mutations have been identified in SH2D1A (77), DOCK8 (78), BTK (55, 56), and IL10RA (65) in patients with XLP, AR HIES, XLA and very early onset inflammatory bowel disease, respectively, yet expression of the encoded proteins is unaffected. Despite this, flow cytometry was still valuable in some of these cases to establish the LOF nature of variants that did not affect protein expression. Here, quantifying binding of soluble CD40 to the surface of activated CD4+ T cells established that a specific mutation in CD40LG that preserved protein expression, as determined flow cytometrically using an anti-CD40L mAb, was unable to bind CD40L (23). Likewise, LOF of expressed but mutant IL-10RA was confirmed by demonstrating an inability of IL-10 to induce phosphorylation of STAT3 in PBMCs from this patient (65). Thus, like all aspects of research and clinical immunology, awareness of the limitations of specific assays needs to be borne in mind, and multiple approaches adopted to ensure the most accurate and clinically-actionable results are obtained. But the contribution of flow cytometry to clinical medicine remains central and unquestioned.
To study the impact of gene mutations on the human immune system of individuals with PIDs, it is important to first be able to identify distinct populations of immune cells in peripheral blood (PB), as this is the predominant source of cells that is readily obtainable from patients. In line with this, mAbs directed against a myriad of cell surface markers can be used to identify, isolate, and characterize distinct in vivo-generated immune cell subsets (Table 1). Of the main conventional lymphocyte populations CD3, CD4, CD8, CD20, and CD56 have been used to identify total CD3+ T, CD4+ helper T (Th), CD8+ cytotoxic T, CD20+ B, and CD3−CD56+ natural killer (NK) cells (Table 1). In addition to this, unconventional T cell subsets, which contribute to both innate and adaptive immune responses, have been identified within the T cell compartment as CD3+Vα24+Vβ11+ NKT, CD3+γδ+αβ− γδ T and CD3+Vα7.2+CD161+ mucosal associated invariant T (MAIT) cells (79) (Table 1).

Table 1. Immune cell populations identified in human peripheral blood according to specific phenotypes.
Importantly, differential expression of specific cell surface markers can be used to determine the maturation status of distinct subsets of CD4+ and CD8+ T and B cells. Specifically CCR7 and CD45RA delineate naive (CD45RA+CCR7+), central memory (TCM; CD45RA−CCR7+), and effector memory (TEM; CD45RA−CCR7−) CD4+ T cells, and naive (CD45RA+CCR7+), TCM (CD45RA−CCR7+), TEM (CD45RA− CCR7−), and revertant memory (TEMRA; CD45RA+CCR7−) CD8+ T cell populations (80, 81) (Table 1). The CD4+ T cell compartment can also be resolved into different “helper” subsets based on expression of CD25, CD127, CD45RA, CXCR5, CXCR3, and CCR6. As such, CD4+ T cell populations corresponding to regulatory T cells (Tregs; CD25hiCD127lo), T follicular helper (Tfh; CD45RA−CXCR5+), Th1 (CD45RA−CXCR5−CXCR3+CCR6−), Th2 (CD45RA−CXCR5−CXCR3−CCR6−), and Th17 (CD45RA−CXCR5−CXCR3−CCR6+) subsets can be identified in PB of healthy individuals (32, 81, 82).
CD20+ human B cells can be divided into transitional (CD10+CD27−), naïve (CD10−CD27−), and memory (CD10−CD27+) subsets by their differential expression of CD10 and CD27 (25, 26, 83, 84). Early/immature and late/mature subsets of transitional B cells can also be identified according to differential expression of CD21 or CD44 (35, 83, 84). Furthermore, memory B cells can remain IgMhi or undergo Ig isotype switching to become IgG- or IgA-expressing cells (25, 26) (Table 1). Plasmablasts (CD19+CD38hiCD27hiCD20lo) can also be detected, though these cells persist at very low frequencies in the PB at the basal state (85). A population of CD19hiCD21lo B cells can also be detected within the B cell compartment. These CD19hiCD21lo have been referred to as “atypical” and/or “aged memory” B cells, and have been associated with both health and disease (86–88) (Table 1). Thus, on one hand they have been proposed as being plasmablast precursors that are rapidly re-activated and differentiate into plasmablasts during anamnestic immune responses to specific Ag (86). On the other hand they have been considered to be pathogenic in the setting of chronic infection (e.g., HIV, malaria, Hepatitis B) as they are “exhausted” and unable to clear these pathogens, or self-reactive in Ab-mediated autoimmune disease (SLE, RA, Sjogren's syndrome) (27, 87, 88).
Over the past two decades, our flow cytometric-based studies of various PIDs have provided substantial insight and understanding into the non-redundant roles of specific genes, molecules, and signaling pathways in the development, differentiation and effector function of human B cells, CD4+ T cells, CD8+ T cells and innate-like lymphocytes. These findings have not only identified critical requirements for establishing robust primary and long-lived immunity against various pathogens, but have elucidated mechanisms underlying infectious susceptibility in the setting of these PIDs, and defined specialized functions of discrete subsets of immune cells host defense. Some of our key findings from these studies are summarized below:
AD HIES is characterized by recurrent opportunistic bacterial (Staphylococcal) and fungal (Candida sps.) infections, recurrent cyst-forming pneumonia and impaired generation of Ag-specific Abs following vaccination or infection, despite dramatically increased levels of serum IgE (89). Affected individuals also present with non-immune connective tissue defects such as broad facial features, high palate, retention of primary teeth, hyperextensibility, scoliosis, osteoporosis, and recurrent fractures (89). AD-HIES was found to result from heterozygous germline DN mutations in STAT3 (STAT3DN) (90, 91). Examination of PBMCs from these patients has revealed a “lymphocyte phenotype signature” that is distinct from healthy donors.
Specifically, while there were normal frequencies of total CD4+ T cells in STAT3DN, we found increases in proportions of naïve and decreases in proportions of TCM cells (Figures 3A,B, 4A,B) (32). Further investigations into CD4+ T cell subsets revealed a significant decrease in CXCR5+ Tfh and CCR6+ Th17 cells and to a lesser extent an increase in the CXCR5−CXCR3−CCR6− memory population (32, 92, 94, 95), which contains Th2 cells, in patients with STAT3DN mutations (Figures 3C, 4C,D). Frequencies of total CD8+ T cells are comparable in healthy donors and STAT3DN patients (Figure 3D), however STAT3DN patients have an increase in naïve and a corresponding decrease in TCM, TEM, and TEMRA CD8+ T cells (Figures 3E, 4E,F) (73). Examination of the B cell compartment of STAT3DN patients also revealed stark differences to healthy donors. Despite normal frequencies of total B cells (Figure 3F), they tended to be more immature as revealed by an increase in transitional and naïve B cells and a concurrent decrease in memory B cells (Figures 1, 3G) (31, 32, 74). Interestingly, despite the severe reduction in memory B cells, those that do develop in STAT3DN patients have undergone Ig isotype switching, albeit with a trend toward IgG and away from IgA (Figure 3H) (31, 74).
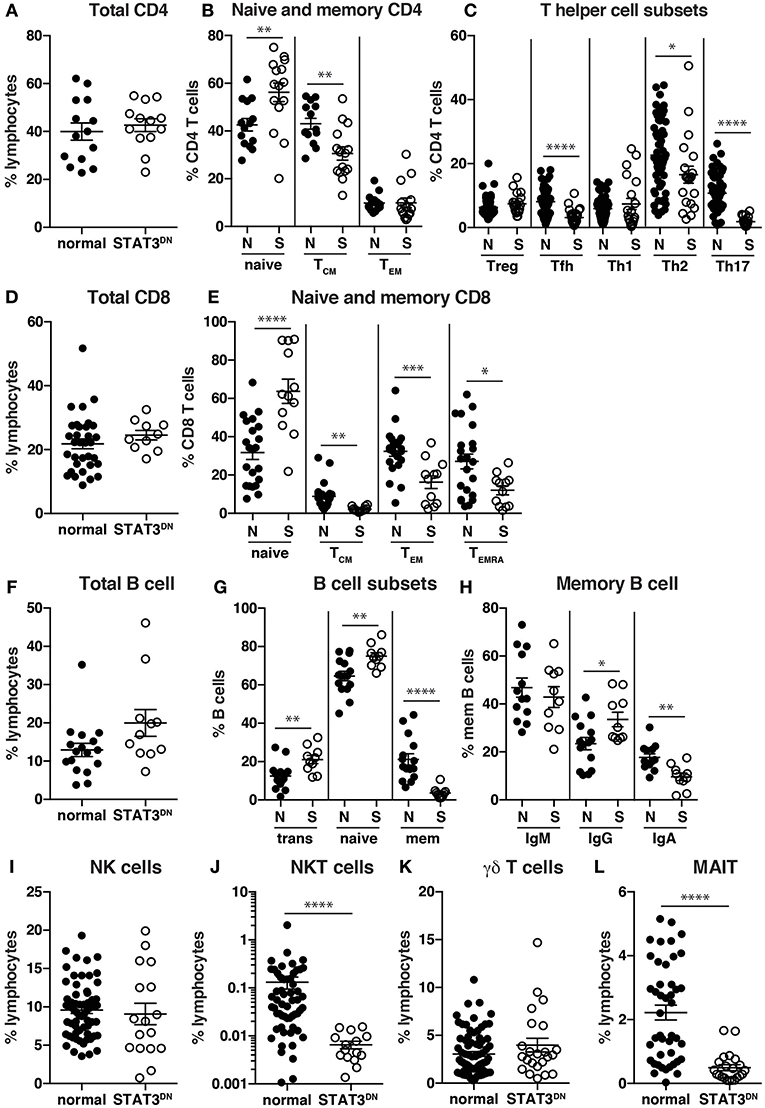
Figure 3. Identification of immune cell populations in STAT3DN patients. (A) Total CD4 T cells, (B) naïve, central memory (TCM), effector memory (TEM) CD4+ T cells, (C) regulatory T cells (Treg) T follicular helper (Tfh) cells, Th1, Th2, Th17 CD4+ T cells, (D) total CD8+ T cells, (E) naïve, TCM, TEM, and revertant memory (TEMRA) CD8+ T cells, (F) total B cells, (G) transitional, naive and memory B cells, (H) IgM, IgG and IgA memory B cells, (I) NK cells, (J) NKT cells, (K) γδ T cells and (L) mucosal associated invariant T (MAIT) cells were identified in normal donors (N) and STAT3DN patients (S). Each point represents a different sample and horizontal line represents the average; statistics were performed in Prism using t-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). These data are compiled from findings previously reported in the following publications: (31, 32, 73–75, 92, 93).
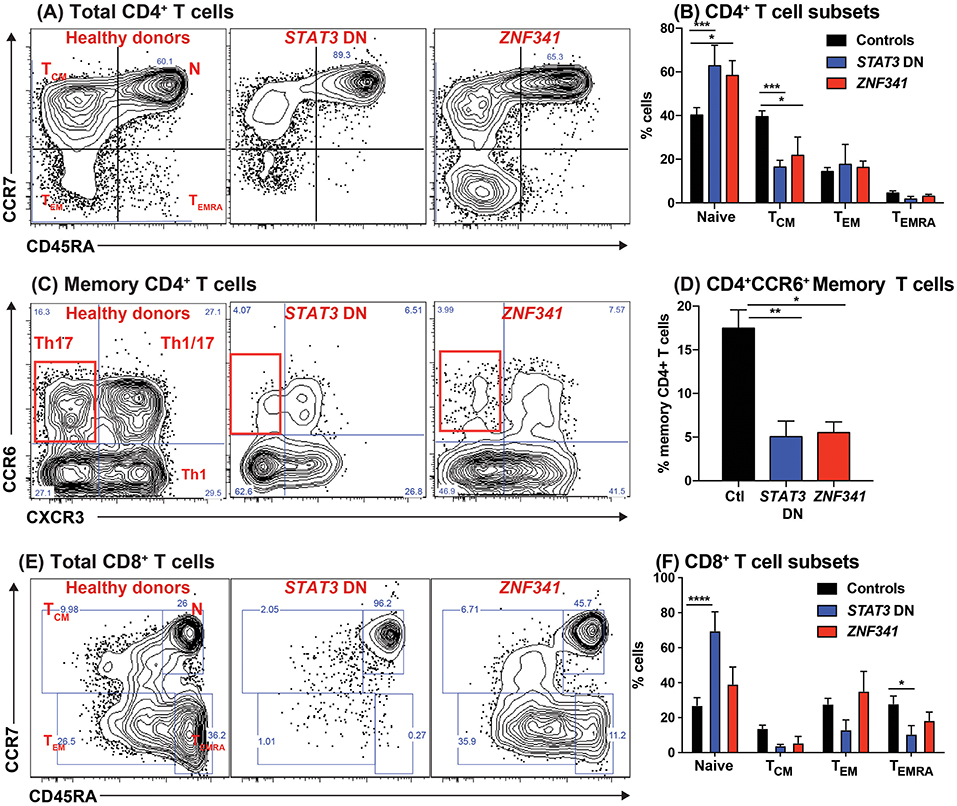
Figure 4. Recessive mutations in ZNF341 phenocopy clinical and cellular defects due to autosomal dominant STAT3 mutations to cause AR HIES. PBMCs from healthy donors or from individuals with pathogenic STAT3 DN or ZNF341 LOF mutations were stained with mAbs against CD4, CD8, CD45RA, CCR7, CXCR3, CCR6, CXCR5, CD127, and CD25. (A,B) Total CD4+ T cells were identified as CD4+CD8− cells, and then proportions of naïve, TCM, TEM, and TEMRA cells were determined. (C,D) the memory compartment (CD4+CD45RA−) of CD4+ T cells was analyzed to quantify the proportions of cells with a CXCR3−CCR6+ Th17-, CXCR3+CCR6− Th1-, CXCR3+CCR6+ Th1/17-, and CXCR3−CCR6− Th2-type phenotype. (E,F) Total CD8+ T cells were identified as CD4−CD8+ cells, and then proportions of naïve, TCM, TEM, and TEMRA cells were determined. The contour plots in (A,C,E) are representative of 1 healthy donor, and 1 patient each with mutations in STAT3 or ZNF341. The graphs represent the mean ± sem of CD4+ and CD8+ T cell subsets detected in the PB of 17 healthy donors, 8 STAT3DN patients or 4 ZNF341-deficient patients. Significant differences were determined by ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). These data are compiled from our findings previously reported in Beziat et al. (40).
In regards to innate-like lymphocytes, the frequencies of NK and γδ T cells in the PB of STAT3DN patients is normal (Figures 3I,K). However, we found a severe reduction in NKT and MAIT cells in the absence of intact STAT3 signaling (Figures 3J,L) (93, 95). Taken together this analysis revealed a distinct phenotype for STAT3DN lymphocytes compared to healthy controls. This lymphocyte signature has not only aided in the identification of potential STAT3DN patients, but also provided valuable insights into disease susceptibility and a cellular and molecular explanation for the clinical features of STAT3 deficiency. For instance, CCR6+ Th17 cells are implicated in protective immunity against fungal infections (81, 95). Thus, the severe reduction in Th17 cells in STAT3DN patients explains their extreme susceptibility to Candida albicans and subsequent chronic mucocutaneous candidiasis. Furthermore, the significant reductions in memory B cells and Tfh cells due to STAT3 deficiency are likely to account for defects in long-lived humoral immunity in STAT3DN patients (95).
While these findings established critical roles for STAT3 signaling in the generation and/or maintenance of various populations of effector lymphocytes, they did not directly identify the upstream STAT3-activating cytokine(s) required for these processes. However, this has now been achieved by the identification and analysis of PID patients with inactivating mutations in specific cytokines or their receptors that signal through STAT3. Thus, IL-21/IL-21R/STAT3 signaling is required for establishing the pool of memory B cells (31, 32, 74, 95) and NKT cells (93, 95), IL-23R/IL-12Rβ1/STAT3 (but not IL-12Rβ2) signaling is necessary for MAIT cells (42, 93), and IL-23R/IL-12Rβ1/STAT3 and IL-21/IL-21R/STAT3 signaling likely co-operate to generate Th17 cells (32, 42, 95).
Recently, 2 studies identified 19 patients with an AR form of HIES who essentially clinically phenocopied individuals with STAT3DN mutations (40, 96). The molecular lesion underlying this form of recessive HIES was found to be bi-allelic mutations in the novel transcription factor ZNF341. The link between ZNF341 and STAT3 function was provided by the finding that ZNF341 binds to the STAT3 promoter and regulates STAT3 expression. Consequently, ZNF341-deficient patients have low levels of STAT3 mRNA and protein and poor responses following stimulation with STAT3-activating cytokines (40, 96).
When PBMCs from ZNF341-deficient patients were examined, we identified a lymphocyte signature very similar to that of STAT3DN patients. The CD4+ T cell compartment was comprised of increased frequencies of naïve and decreased frequencies of TCM cells (Figures 4A,B) (32, 40). This could be further broken down to reveal decreases in CCR6+ Th17 (Figures 4C,D) and CXCR5+ Tfh cells and increases in Th2 cells (40, 96). The paucity of Th17 phenotype cells in ZNF341- and STAT3-deficient patients (i.e., CD4+CCR6+CXCR3− memory T cells) was confirmed functionally by demonstrating by flow cytometry reductions in proportions of their memory CD4+ T cells that expressed intracellular IL17A, IL17F, and IL22 (Figures 5A,B) (32, 40, 92), canonical cytokines produced by Th17 cells (81, 95). Similarly, ZNF341-deficient and STAT3DN patients had increased proportions of memory CD4+ T cells expressing the characteristic Th2 cytokines IL-4 and IL-13 (Figures 5C,D) (40), consistent with the finding of increased CD4+CD45RA−CXCR5−CCR6−CXCR3− memory cells (Figure 3C), as well as the hyper-IgE phenotype and Th2-associated pathologies, in these individuals (40, 95, 96). However, in contrast to STAT3DN patients, the CD8+ T cell compartment in ZNF341-deficient patients was relatively comparable to that of normal donors (Figures 4E,F) (40, 73). ZNF341-deficient patients have similar decreases in memory B cells as STAT3DN patients (Figure 1), and their few memory B cells are predominantly IgG+ rather than IgA+ (31, 40, 74). When populations of innate-like T cells were examined, ZNF341-deficient patients had comparable frequencies of γδ T and NKT cells, but fewer MAIT cells than healthy donors (40). In contrast to STAT3, ZNF341 is likely to be important for NK cell development, as these cells are significantly decreased in ZNF341-deficient patients compared to healthy controls (40).
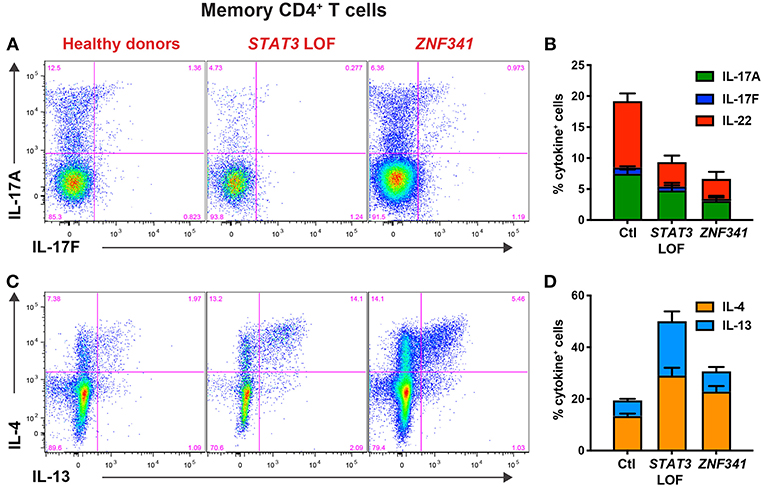
Figure 5. CD4+ T cell defects detectable by immunophenotyping of STAT3DN and ZNF341-deficient patients correlate with compromised function determined flow cytometrically in vitro. Sort-purified CD4+ memory T cells isolated from healthy donors or individuals with pathogenic STAT3 DN or ZNF341 LOF mutations were cultured in vitro with anti-CD2/CD3/CD28 mAb conjugated to beads. Following 5 days, the cells were harvested, washed, and then restimulated with PMA/ionomycin for 6 h, with Brefeldin A being added for the final 4 h of culture. Cells were then harvested, fixed, permeabilised, and stained with fluorescently-labeled mAbs specific for (A,B) IL-17A and IL-17F, or (C,D) IL-4 and IL-13. The proportions of cytokine-expressing cells were then determined by flow cytometric analysis. Contour plots (A,C) are representative of 1 healthy donor, and 1 patient with mutations in STAT3 or ZNF341. The graphs (B,D) represent the mean ± sem of CD4+ memory T cells expressing the indicated cytokine from 23–28 healthy donors, 6–7 STAT3DN patients or 4–6 ZNF341-deficient patients. These data are compiled from our findings previously reported in Beziat et al. (40).
Dedicator of cytokinesis 8 (DOCK8) deficiency is a combined immunodeficiency caused by AR LOF mutations in DOCK8 (97). This disorder is characterized by recurrent cutaneous viral, bacterial and fungal infections, increased serum IgE levels, and severe atopic disease, including food-induced anaphylaxis (97). Similar to SAP expression in XLP, the use of mAbs to detect DOCK8 expression has been crucial for the diagnosis of DOCK8-deficient patients (37, 53, 97) (Figure 2). Intracellular flow cytometry for DOCK8 expression has also detected somatic reversion in these patients (54).
DOCK8-deficient patients also have a unique lymphocyte phenotype, with decreased frequencies of CD4+ T cells, but normal frequencies of CD8+ T cells, resulting in an inverted CD4:CD8 ratio compared to healthy donors (37, 58, 76). When DOCK8-deficient CD4+ and CD8+ T cells were further investigated, we found a decrease in naïve CD4+ and CD8+ T cells and a corresponding increase in CD4+ TEM and CD8+ TEM and TEMRA populations compared to healthy donors (37, 58, 76). Furthermore, memory T cells in DOCK8-deficiency contained reduced frequencies of CD27+, CD28+, and CD127+ and higher frequencies of CD57+, CD95+, and PD1+ cells, indicating these cells had undergone premature exhaustion or senescence compared to their counterparts in healthy donors (37, 58, 76). There were also reductions in frequencies of CD4+CCR6+ memory T cells in DOCK8-deficient patients, suggesting a role for DOCK8 in Th17 cells (37, 58, 78). This loss of CCR6+ Th17 cells also provides a cellular explanation for increased susceptibility of DOCK8-deficient patients to infections with Candida sp. (81, 95). The B cell compartment in DOCK8-deficient patients comprises normal frequencies of total and transitional B cells, however there are increases in naïve and decreases in memory B cells compared to healthy donors (37, 98), thus highlighting a requirement for DOCK8 for B cell differentiation. DOCK8-deficient patients were also found to have decreased αβ T, NKT, and MAIT cells, normal frequencies of NK cells, but increased γδ T cells (37). Importantly, this cellular phenotype has been able to predict individuals with AR HIES who may have pathogenic mutations in DOCK8, and provide explanations for clinical features of this condition (37, 58, 76).
XLP-1 is a rare X-linked PID whereby affected males present with extreme sensitivity to disease resulting from Epstein-Barr virus (EBV) infection. Following exposure to EBV, XLP patients develop severe infectious mononucleosis, leading to often-fatal hemophagocytic lymphohistiocytosis (99). XLP-1 patients also exhibit hypogammaglobulinemia and a heightened risk of developing B-cell lymphoma, both of which occur independently of exposure to EBV (99). XLP-1 results from LOF mutations in SH2D1A, encoding signaling lymphocytic activation molecule (SLAM)-associated protein (SAP) (20). SAP is a small intracellular adaptor protein that binds to tyrosine-based motifs in the intracellular domain of SLAM family members and regulates signaling downstream of these receptors (99). The availability of mAbs specific for SAP has been instrumental in expanding our knowledge of this condition. Thus, flow cytometry has (i) accurately defined the cell types that express SAP (predominantly T, NK, and NKT cells, but rarely B cells), (ii) offered a rapid and sensitive diagnostic tool to detect not only XLP patients whom lack SAP expression, but also female carriers of the XLP trait who have bimodal SAP expression in their T and NK cells (Figure 2) (57), and (3) revealed lymphocyte defects in XLP-1, thereby providing insight into disease pathogenesis. XLP patients have a paucity for CD27+ memory B cells (Figure 1), and the few memory B cells detected express IgM, thereby indicating an inability to undergo class switching to express IgG or IgA (29, 30). Within the CD4+ T cell compartment, XLP patients have a comparable frequency of naïve, TCM and TEM populations to healthy donors (29). Although XLP patients have a normal frequency of Tfh cells (100, 101), the inability of these cells to support B cell differentiation in an in vitro helper assay indicates defective B-helper function (30). Together, these observations identify defective CD4+ T cell help as the cellular basis for hypogammaglobulinemia in XLP-1 patients, rather than a B-cell intrinsic defect. This is indeed consistent with the fact that B cells do not express SAP (57, 60) and XLP B cells function normally when provided with the correct T-dependent stimuli in vitro (30).
CD8+ T cells from XLP patients are skewed toward TEM and TEMRA cells, usually at the expense of naïve T cells. Interestingly, by utilizing intracellular flow cytometry to detect SAP expression, we were able to show that some XLP patients undergo somatic reversion and a population of SAP+ CD45RA−CCR7− TEM cells is detectable within their CD8+ T cells (60). These reverted SAP+ CD8+ T cells in XLP patients could respond to EBV and kill-EBV infected B cells, suggesting they were able to provide protective immunity against ongoing EBV infection (60). These functional features were detected by concomitantly tracking proliferation and degranulation of EBV-specific CD8+ T cells (60), further illustrating the utility of flow cytometry in establishing functionality of immune cells in PIDs.
XLP patients also lack NKT cells, revealing an essential role for SAP in the development of this cell type and potentially implicating NKT cells in some of the clinical manifestations of XLP such as impaired anti-viral and anti-tumor immune responses (102). Consistent with this essential role of SAP in NKT cell development, female XLP carriers undergo X chromosome inactivation within NKT cells, but not T or NK cells, resulting in all NKT cells in these carriers expressing only the WT allele (57, 102).
Flow cytometry has been central to enhancing our understanding of PIDs. It has enabled diagnoses and provided mechanistic insights into disease pathogenesis in many PIDs. For example, the severe reduction in Th17 cells and memory B cells in STAT3DN, ZNF341-, and DOCK8-deficient patients explains susceptibility to recurrent opportunistic bacterial and fungal infections, and impaired long-lived protective humoral immunity, respectively. While our focus has been on lymphocyte populations, flow cytometry has been used to identify other populations such as monocytes (CD14+), dendritic cell (DC) subsets [plasmacytoid DCs (pDCs; lineage−HLA-DR+CD123+), CD1c+ myeloid DCs (CD1c+ mDCs; lineage−HLA-DR+CD11c+CD1c+), and CD141+ mDC (CD141+ mDC; lineage−HLA-DR+CD11c+CD141+)], and innate lymphoid cells [(ILCs), ILC1, ILC2, and ILC precursors (ILCPs)]. Beyond deep immunophenotypic analysis by examining expression of specific cell surface molecules, flow cytometry has also been used to diagnose PIDs by assessing expression of specific intracellular proteins (SAP, DOCK8, XIAP, BTK) as well as quantifying cytokine-induced STAT phosphorylation and Ag-receptor induced calcium flux. As we move toward the next generation of flow cytometers, which are capable of simultaneously detecting upward of 28 fluorochromes, the future is looking brighter, fluorescent even, in terms of the applicability of flow cytometry in the study and diagnosis of PIDs.
CM and ST designed, conceptualized, and wrote this review.
Research performed in the Tangye/Ma lab has been supported by the National Health and Medical Council of Australia, the Office of Health and Medical Research of the New South Wales Government, the Jeffrey Modell Foundation, the Job Research Foundation, and the Cancer Council NSW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the authors.
We thank our many clinical colleagues and collaborators, as well as the many patients and families, who have made it possible for our laboratories to pursue many of the questions discussed in this review. We are particularly indebted to Jean-Laurent Casanova, Anne Puel, Stephanie Boisson-Dupuis, Emmanuelle Jouanguy, Vivien Beziat, Satoshi Okada, Peter Arkwright, Kaan Boztug, Isabelle Meyts, Qiang Pan-Hammarstrom, Melanie Wong, Paul Gray, Steve Holland, Gulbu Uzel, and Elissa Deenick for ongoing support and input into these projects, and members of our lab who have contributed to these studies over the years!
2. Cooper MD, Peterson RD, Good RA. Delineation of the thymic and bursal lymphoid systems in the chicken. Nature. (1965) 205:143–6. doi: 10.1038/205143a0
3. Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. (2009) 27:199–227. doi: 10.1146/annurev.immunol.021908.132649
4. Cooper MD, Lawton AR, Bockman DE. Agammaglobulinaemia with B lymphocytes. Specific defect of plasma-cell differentiation. Lancet. (1971) 2:791–4. doi: 10.1016/S0140-6736(71)92742-5
5. Grey HM, Rabellino E, Pirofsky B. Immunoglobulins on the surface of lymphocytes. IV. Distribution in hypogammaglobulinemia, cellular immune deficiency, and chronic lymphatic leukemia. J Clin Invest. (1971) 50:2368–75. doi: 10.1172/JCI106735
6. Siegal FP, Pernis B, Kunkel HG. Lymphocytes in human immunodeficiency states: a study of membrane-associated immunoglobulins. Eur J Immunol. (1971) 1:482–6. doi: 10.1002/eji.1830010615
7. Cooper MD, Lawton AR. Circulating B-cells in patients with immunodeficiency. Am J Pathol. (1972) 69:513–28.
8. Pearl ER, Vogler LB, Okos AJ, Crist WM, Lawton AR III, Cooper MD. B lymphocyte precursors in human bone marrow: an analysis of normal individuals and patients with antibody-deficiency states. J Immunol. (1978) 120:1169–75.
9. Geha RS, Rosen FS, Merler E. Identification and characterization of subpopulations of lymphocytes in human peripheral blood after fractionation on discontinuous gradients of albumin. The cellular defect in X-linked agammaglobulinemia. J Clin Invest. (1973) 52:1726–34. doi: 10.1172/JCI107354
10. Griscelli C, Durandy A, Virelizier JL, Ballet JJ, Daguillard F. Selective defect of precursor T cells associated with apparently normal B lymphocytes in severe combined immunodeficiency disease. J Pediatr. (1978) 93:404–11. doi: 10.1016/S0022-3476(78)81146-9
11. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. (1975) 256:495–7. doi: 10.1038/256495a0
13. Stahl RE, Friedman-Kien A, Dubin R, Marmor M, Zolla-Pazner S. Immunologic abnormalities in homosexual men. Relationship to Kaposi's sarcoma. Am J Med. (1982) 73:171–8. doi: 10.1016/0002-9343(82)90174-7
14. Ammann AJ, Abrams D, Conant M, Chudwin D, Cowan M, Volberding P, Lewis B, Casavant C. Acquired immune dysfunction in homosexual men: immunologic profiles. Clin Immunol Immunopathol. (1983) 27:315–25. doi: 10.1016/0090-1229(83)90084-3
15. Modlin RL, Meyer PR, Hofman FM, Mehlmauer M, Levy NB, Lukes RJ, et al. T-lymphocyte subsets in lymph nodes from homosexual men. JAMA. (1983) 250:1302–5. doi: 10.1001/jama.250.10.1302
16. Detels R, Visscher BR, Fahey JL, Sever JL, Gravell M, Madden DL, et al. Predictors of clinical AIDS in young homosexual men in a high-risk area. Int J Epidemiol. (1987) 16:271–6. doi: 10.1093/ije/16.2.271
17. Fauci AS, Macher AM, Longo DL, Lane HC, Rook AH, Masur H, et al. NIH conference. Acquired immunodeficiency syndrome: epidemiologic, clinical, immunologic, and therapeutic considerations. Ann Intern Med. (1984) 100:92–106. doi: 10.7326/0003-4819-100-1-92
18. Herzenberg LA, Herzenberg LA. Genetics, FACS, immunology, and redox: a tale of two lives intertwined. Annu Rev Immunol. (2004) 22:1–31. doi: 10.1146/annurev.immunol.22.012703.104727
19. Boisson B, Wang YD, Bosompem A, Ma CS, Lim A, Kochetkov T, et al. A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(-) B cells. J Clin Invest. (2013) 123:4781–5. doi: 10.1172/JCI71927
20. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
21. Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. (2004) 22:625–55. doi: 10.1146/annurev.immunol.22.012703.104614
22. DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, de Saint Basile G. CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature. (1993) 361:541–3. doi: 10.1038/361541a0
23. Korthauer U, Graf D, Mages HW, Briere F, Padayachee M, Malcolm S, et al. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. (1993) 361:539–41. doi: 10.1038/361539a0
24. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
25. Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. (1998) 188:1679–89. doi: 10.1084/jem.188.9.1679
26. Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. (1998) 188:1691–703. doi: 10.1084/jem.188.9.1691
27. Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. (2002) 99:1544–51. doi: 10.1182/blood.V99.5.1544
28. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
29. Ma CS, Hare NJ, Nichols KE, Dupre L, Andolfi G, Roncarolo MG, et al. Impaired humoral immunity in X-linked lymphoproliferative disease is associated with defective IL-10 production by CD4+ T cells. J Clin Invest. (2005) 115:1049–59. doi: 10.1172/JCI23139
30. Ma CS, Pittaluga S, Avery DT, Hare NJ, Maric I, Klion AD, et al. Selective generation of functional somatically mutated IgM+CD27+, but not Ig isotype-switched, memory B cells in X-linked lymphoproliferative disease. J Clin Invest. (2006) 116:322–33. doi: 10.1172/JCI25720
31. Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med. (2013) 210:2739–53. doi: 10.1084/jem.20130323
32. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. (2015) 136:993–1006 e1. doi: 10.1016/j.jaci.2015.05.036
33. Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. (2016) 213:2413–35. doi: 10.1084/jem.20160576
34. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. (2017) 214:91–106. doi: 10.1084/jem.20160849
35. Avery DT, Kane A, Nguyen T, Lau A, Nguyen A, Lenthall H, et al. Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med. (2018) 215:2073–95. doi: 10.1084/jem.20180010
36. Moran I, Avery DT, Payne K, Lenthall H, Davies EG, Burns S, et al. B cell-intrinsic requirement for STK4 in humoral immunity in mice and human subjects. J Allergy Clin Immunol. (2019) 143:2302–5. doi: 10.1016/j.jaci.2019.02.010
37. Pillay BA, Avery DT, Smart JM, Cole T, Choo S, Chan D, et al. Hematopoietic stem cell transplant effectively rescues lymphocyte differentiation and function in DOCK8-deficient patients. JCI Insight. (2019) 5:127527. doi: 10.1172/jci.insight.127527
38. Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. (2006) 107:3045–52. doi: 10.1182/blood-2005-07-2955
39. Recher M, Berglund LJ, Avery DT, Cowan MJ, Gennery AR, Smart J, et al. IL-21 is the primary common gamma chain-binding cytokine required for human B-cell differentiation in vivo. Blood. (2011) 118:6824–35. doi: 10.1182/blood-2011-06-362533
40. Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. (2018) 3:eaat4956. doi: 10.1126/sciimmunol.aat4956
41. Cliffe ST, Bloch DB, Suryani S, Kamsteeg EJ, Avery DT, Palendira U, et al. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol. (2012) 130:735–42 e6. doi: 10.1016/j.jaci.2012.02.054
42. Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F, et al. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. (2018) 3:eaau6759. doi: 10.1126/sciimmunol.aau6759
43. Kong XF, Martinez-Barricarte R, Kennedy J, Mele F, Lazarov T, Deenick EK, et al. Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol. (2018) 19:973–85. doi: 10.1038/s41590-018-0178-z
44. Cottineau J, Kottemann MC, Lach FP, Kang YH, Vely F, Deenick EK, et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J Clin Invest. (2017) 127:1991–2006. doi: 10.1172/JCI90727
45. Lu HY, Bauman BM, Arjunaraja S, Dorjbal B, Milner JD, Snow AL, et al. The CBM-opathies-a rapidly expanding spectrum of human inborn errors of immunity caused by mutations in the CARD11-BCL10-MALT1 complex. Front Immunol. (2018) 9:2078. doi: 10.3389/fimmu.2018.02078
46. Wentink MWJ, van Zelm MC, van Dongen JJM, Warnatz K, van der Burg M. Deficiencies in the CD19 complex. Clin Immunol. (2018) 195:82–7. doi: 10.1016/j.clim.2018.07.017
47. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. (2019) 10:297. doi: 10.3389/fimmu.2019.00297
48. Willmann KL, Klaver S, Dogu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. (2014) 5:5360. doi: 10.1038/ncomms6360
49. Pala P, Hussell T, Openshaw PJ. Flow cytometric measurement of intracellular cytokines. J Immunol Methods. (2000) 243:107–24. doi: 10.1016/S0022-1759(00)00230-1
50. Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. (2003) 55:61–70. doi: 10.1002/cyto.a.10072
51. Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. (2010) 362:1–9. doi: 10.1016/j.jim.2010.08.010
52. Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. (2007) 120:744–50; quiz 51–2. doi: 10.1016/j.jaci.2007.08.044
53. Pai SY, de Boer H, Massaad MJ, Chatila TA, Keles S, Jabara HH, et al. Flow cytometry diagnosis of dedicator of cytokinesis 8 (DOCK8) deficiency. J Allergy Clin Immunol. (2014) 134:221–3. doi: 10.1016/j.jaci.2014.02.023
54. Jing H, Zhang Q, Zhang Y, Hill BJ, Dove CG, Gelfand EW, et al. Somatic reversion in dedicator of cytokinesis 8 immunodeficiency modulates disease phenotype. J Allergy Clin Immunol. (2014) 133:1667–75. doi: 10.1016/j.jaci.2014.03.025
55. Futatani T, Miyawaki T, Tsukada S, Hashimoto S, Kunikata T, Arai S, et al. Deficient expression of Bruton's tyrosine kinase in monocytes from X-linked agammaglobulinemia as evaluated by a flow cytometric analysis and its clinical application to carrier detection. Blood. (1998) 91:595–602.
56. Kanegane H, Futatani T, Wang Y, Nomura K, Shinozaki K, Matsukura H, et al. Clinical and mutational characteristics of X-linked agammaglobulinemia and its carrier identified by flow cytometric assessment combined with genetic analysis. J Allergy Clin Immunol. (2001) 108:1012–20. doi: 10.1067/mai.2001.120133
57. Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. (2011) 9:e1001187. doi: 10.1371/journal.pbio.1001187
58. Tangye SG, Pillay B, Randall KL, Avery DT, Phan TG, Gray P, et al. Dedicator of cytokinesis 8-deficient CD4(+) T cells are biased to a TH2 effector fate at the expense of TH1 and TH17 cells. J Allergy Clin Immunol. (2017) 139:933–49. doi: 10.1016/j.jaci.2016.07.016
59. Marciano BE, Zerbe CS, Falcone EL, Ding L, DeRavin SS, Daub J, et al. X-linked carriers of chronic granulomatous disease: illness, lyonization, and stability. J Allergy Clin Immunol. (2018) 141:365–71. doi: 10.1016/j.jaci.2017.04.035
60. Palendira U, Low C, Bell AI, Ma CS, Abbott RJ, Phan TG, et al. Expansion of somatically reverted memory CD8+ T cells in patients with X-linked lymphoproliferative disease caused by selective pressure from Epstein-Barr virus. J Exp Med. (2012) 209:913–24. doi: 10.1084/jem.20112391
61. Tone Y, Wada T, Shibata F, Toma T, Hashida Y, Kasahara Y, et al. Somatic revertant mosaicism in a patient with leukocyte adhesion deficiency type 1. Blood. (2007) 109:1182–4. doi: 10.1182/blood-2007-08-039057
62. Uzel G, Tng E, Rosenzweig SD, Hsu AP, Shaw JM, Horwitz ME, et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1). Blood. (2008) 111:209–18. doi: 10.1182/blood-2007-04-082552
63. Fleisher TA, Dorman SE, Anderson JA, Vail M, Brown MR, Holland SM. Detection of intracellular phosphorylated STAT-1 by flow cytometry. Clin Immunol. (1999) 90:425–30. doi: 10.1006/clim.1998.4654
64. Uzel G, Frucht DM, Fleisher TA, Holland SM. Detection of intracellular phosphorylated STAT-4 by flow cytometry. Clin Immunol. (2001) 100:270–6. doi: 10.1006/clim.2001.5078
65. Lee CH, Hsu P, Nanan B, Nanan R, Wong M, Gaskin KJ, et al. Novel de novo mutations of the interleukin-10 receptor gene lead to infantile onset inflammatory bowel disease. J Crohns Colitis. (2014) 8:1551–6. doi: 10.1016/j.crohns.2014.04.004
66. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. (2013) 131:1611–23. doi: 10.1016/j.jaci.2012.11.054
67. Kotlarz D, Zietara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. (2013) 210:433–43. doi: 10.1084/jem.20111229
68. Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. (2017) 214:2547–62. doi: 10.1084/jem.20161810
69. Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. (2019). doi: 10.1084/jem.20190344. [Epub ahead of print].
70. Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods. (1995) 178:89–97. doi: 10.1016/0022-1759(94)00247-T
71. Hasbold J, Gett AV, Rush JS, Deenick E, Avery D, Jun J, et al. Quantitative analysis of lymphocyte differentiation and proliferation in vitro using carboxyfluorescein diacetate succinimidyl ester. Immunol Cell Biol. (1999) 77:516–22. doi: 10.1046/j.1440-1711.1999.00874.x
72. Tempany JC, Zhou JH, Hodgkin PD, Bryant VL. Superior properties of CellTrace Yellow as a division tracking dye for human and murine lymphocytes. Immunol Cell Biol. (2018) 96:149–59. doi: 10.1111/imcb.1020
73. Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8(+) T-cell memory formation and function. J Allergy Clin Immunol. (2013) 132:400–11 e9. doi: 10.1016/j.jaci.2013.05.029
74. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. (2010) 207:155–71. doi: 10.1084/jem.20091706
75. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. (2012) 119:3997–4008. doi: 10.1182/blood-2011-11-392985
76. Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. (2011) 208:2305–20. doi: 10.1084/jem.20110345
77. Morra M, Simarro-Grande M, Martin M, Chen AS, Lanyi A, Silander O, et al. Characterization of SH2D1A missense mutations identified in X-linked lymphoproliferative disease patients. J Biol Chem. (2001) 276:36809–16. doi: 10.1074/jbc.M101305200
78. Keles S, Charbonnier LM, Kabaleeswaran V, Reisli I, Genel F, Gulez N, et al. Dedicator of cytokinesis 8 regulates signal transducer and activator of transcription 3 activation and promotes TH17 cell differentiation. J Allergy Clin Immunol. (2016) 138:1384–94 e2. doi: 10.1016/j.jaci.2016.04.023
79. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol. (2015) 16:1114–23. doi: 10.1038/ni.3298
80. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
81. Sallusto F. Heterogeneity of human CD4(+) T cells against microbes. Annu Rev Immunol. (2016) 34:317–34. doi: 10.1146/annurev-immunol-032414-112056
82. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
83. Cuss AK, Avery DT, Cannons JL, Yu LJ, Nichols KE, Shaw PJ, et al. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. (2006) 176:1506–16. doi: 10.4049/jimmunol.176.3.1506
84. Suryani S, Fulcher DA, Santner-Nanan B, Nanan R, Wong M, Shaw PJ, et al. Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood. (2010) 115:519–29. doi: 10.1182/blood-2009-07-234799
85. Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. (2002) 99:2154–61. doi: 10.1182/blood.V99.6.2154
86. Lau D, Lan LY, Andrews SF, Henry C, Rojas KT, Neu KE, et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci Immunol. (2017) 2:eaai8153. doi: 10.1126/sciimmunol.aai8153
87. Karnell JL, Kumar V, Wang J, Wang S, Voynova E, Ettinger R. Role of CD11c(+) T-bet(+) B cells in human health and disease. Cell Immunol. (2017) 321:40–5. doi: 10.1016/j.cellimm.2017.05.008
88. Portugal S, Obeng-Adjei N, Moir S, Crompton PD, Pierce SK. Atypical memory B cells in human chronic infectious diseases: an interim report. Cell Immunol. (2017) 321:18–25. doi: 10.1016/j.cellimm.2017.07.003
89. Kane A, Deenick EK, Ma CS, Cook MC, Uzel G, Tangye SG. STAT3 is a central regulator of lymphocyte differentiation and function. Curr Opin Immunol. (2014) 28C:49–57. doi: 10.1016/j.coi.2014.01.015
90. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
91. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
92. Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. (2008) 205:1551–7. doi: 10.1084/jem.20080218
93. Wilson RP, Ives ML, Rao G, Lau A, Payne K, Kobayashi M, et al. STAT3 is a critical cell-intrinsic regulator of human unconventional T cell numbers and function. J Exp Med. (2015) 212:855–64. doi: 10.1084/jem.20141992
94. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. (2012) 209:1241–53. doi: 10.1084/jem.20120994
95. Tangye SG, Pelham SJ, Deenick EK, Ma CS. Cytokine-mediated regulation of human lymphocyte development and function: insights from primary immunodeficiencies. J Immunol. (2017) 199:1949–58. doi: 10.4049/jimmunol.1700842
96. Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. (2018) 3:eaat4941. doi: 10.1126/sciimmunol.aat4941
97. Su HC, Jing H, Angelus P, Freeman AF. Insights into immunity from clinical and basic science studies of DOCK8 immunodeficiency syndrome. Immunol Rev. (2019) 287:9–19. doi: 10.1111/imr.12723
98. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links Toll-like receptor–MyD88 signaling to B cell activation. Nat Immunol. (2012) 13:612–20. doi: 10.1038/ni.2305
99. Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol. (2014) 34:772–9. doi: 10.1007/s10875-014-0083-7
100. Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. (2010) 33:241–53. doi: 10.1016/j.immuni.2010.07.015
101. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
Keywords: flow cytometry, B cells, T cells, immunodeficencies, phenotpye
Citation: Ma CS and Tangye SG (2019) Flow Cytometric-Based Analysis of Defects in Lymphocyte Differentiation and Function Due to Inborn Errors of Immunity. Front. Immunol. 10:2108. doi: 10.3389/fimmu.2019.02108
Received: 12 June 2019; Accepted: 21 August 2019;
Published: 04 September 2019.
Edited by:
Roshini Sarah Abraham, Nationwide Children's Hospital, United StatesReviewed by:
Kohsuke Imai, Tokyo Medical and Dental University, JapanCopyright © 2019 Ma and Tangye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cindy S. Ma, Yy5tYUBnYXJ2YW4ub3JnLmF1; Stuart G. Tangye, cy50YW5neWVAZ2FydmFuLm9yZy5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.