 John A. Hamilton1,2*
John A. Hamilton1,2*- 1The University of Melbourne, Department of Medicine, Royal Melbourne Hospital, Parkville, VIC, Australia
- 2Australian Institute for Musculoskeletal Science (AIMSS), The University of Melbourne and Western Health, St. Albans, VIC, Australia
Pre-clinical models and clinical trials demonstrate that targeting the action of the cytokine, granulocyte macrophage-colony stimulating factor (GM-CSF), can be efficacious in inflammation/autoimmunity reinforcing the importance of understanding how GM-CSF functions; a significant GM-CSF-responding cell in this context is likely to be the monocyte. This article summarizes critically the literature on the downstream cellular pathways regulating GM-CSF interaction with monocytes (and macrophages), highlighting some contentious issues, and conclusions surrounding this biology. It also suggests future directions which could be undertaken so as to more fully understand this aspect of GM-CSF biology. Given the focus of this collection of articles on monocytes, the following discussion in general will be limited to this population or to its more mature progeny, the macrophage, even though GM-CSF biology is broader than this.
Introduction
The glycoprotein, granulocyte macrophage-colony stimulating factor (GM-CSF) or CSF2, was originally defined as a hemopoietic growth factor based upon its ability to form colonies in vivo of granulocytes, and macrophages from bone marrow precursor cells (1). However, subsequently, it has been viewed more as a cytokine acting via a specific receptor, expressed mainly on myeloid cell populations, such as monocytes/macrophages, neutrophils and eosinophils, to enhance their survival and/or to activate/differentiate them (2–5). While not having a significant effect on steady state myelopoiesis, in the lung GM-CSF signaling normally maintains surfactant homeostasis and its disruption causes pulmonary alveolar proteinosis (PAP) most likely due to compromised alveolar macrophage development (6, 7). This GM-CSF-driven development of lung alveolar macrophages is of fetal monocyte origin (8). Recently it has been proposed that GM-CSF is required for cholesterol clearance in alveolar macrophages with reduced cholesterol clearance being the primary macrophage defect driving PAP pathogenesis (9). There is evidence that GM-CSF also controls non-lymphoid tissue dendritic cell (DC) homeostasis (10).
Seeing that this Review resides within a collection of articles on monocytes its content will generally be focussed on this population and its tissue counterpart, the macrophage, even though GM-CSF biology is broader involving other responding cell types such as neutrophils and eosinophils.
GM-CSF and Autoimmune/Inflammatory Disease
Based mainly in data using GM-CSF gene deficient mice or neutralizing monoclonal antibody (mAb) in models of autoimmunity and chronic inflammation, it is apparent that GM-CSF can be a key driver of tissue inflammation and its associated pain. Examples include arthritis, EAE, cardiovascular disease, and lung disease. The data summarizing these findings have been reviewed recently (11–14) although some of this data more pertinent to the main topic of this Review will be mentioned. It should also be noted that systemically administered GM-CSF can have beneficial effects in inflammatory disease (for example, colitis) and host defense (for example, as an adjuvant) although caution should be exercised in assessing the significance of such administration for the role of endogenous GM-CSF in inflamed tissue (5, 14).
Given the potentially wide range of GM-CSF biology involving multiple cellular sources and responding myeloid cell types (5), human conditions that involve both acquired and/or innate immunity could fall within the realm of GM-CSF influence. As a result of some of the basic biology from pre-clinical models and GM-CSF expression in the corresponding human condition, a number of clinical trials using neutralizing mAbs to target GM-CSF or its receptor in autoimmune/inflammatory diseases have been performed and are continuing. There have been beneficial effects on disease severity in rheumatoid arthritis and asthma trials but, for reasons yet to be elucidated, not in plaque psoriasis—the data from these trials have been reviewed recently (11, 13–15).
GM-CSF Receptor and Signaling
The GM-CSF receptor (GM-CSFR) is a type I cytokine receptor comprising in a multimeric complex a binding (α) subunit and a signaling (β) subunit, the latter shared with interleukin 3 (IL-3) and interleukin-5 (IL-5) receptors. These pathways have been linked to key residues in the intracellular regions of GM-CSFR using mainly receptor mutants expressed in cell lines (16–18). Key downstream signaling pathways from GM-CSFR are often those involving JAK2/STAT5 and ERK (16, 17, 19–21) with ERK activity linked to GM-CSF enhancement of human monocyte survival in vivo (21). The GM-CSF-driven development of lung alveolar macrophages is dependent on the transcription factors, PU.1 (22) and PPARγ (23). The debated contribution of other transcription factors, namely interferon regulatory factor (IRF) 4 and IRF5, to GM-CSF-driven monocyte/macrophage polarization (24–26), is discussed below.
The various cellular responses (survival, proliferation, activation and/or differentiation) appear to be explained by dose-dependent and sequential activation by GM-CSF of specific signaling pathways downstream of the activated receptor (16, 27). For example, physiological picomolar concentrations of GM-CSF are able to promote Ser585 phosphorylation in the cytoplasmic domain of the GM-CSFR β subunit to regulate cell survival via phosphoinositide 3-kinase activity and in the absence of other biological responses which occur at higher GM-CSF concentrations (18, 28). A time- and dose-dependent licensing process in mouse and human monocytes by GM-CSF in vivo has been described that disables their inflammatory functions and promotes their conversion into suppressor cells (29): this two-step licensing requires activation of the AKT/mTOR/mTORC1 signaling cascade by GM-CSF followed by signaling through the IFN-γR/IRF-1 pathway. Consistent with these dose-dependent signaling responses, dose dependent effects of a neutralizing anti-GM-CSF mAb on monocyte-derived activation/polarization vs. cell number levels were found in an inflammation model—indications were that higher local GM-CSF concentrations were needed for the activation/polarization response (30). Monocytes/macrophages generated in vivo from mouse bone marrow precursors with different concentrations of GM-CSF differed in function with possible implications for GM-CSF-dependent pathology (31)—cells generated with a high concentration of GM-CSF were more potent in generating cytokines and chemokines. The links between the various signaling pathways listed and their dependence on GM-CSF concentration in monocytes/macrophages requires further analysis to assess their contribution to the various cellular responses mentioned above. Additional signal transduction findings, particularly linked with the role of GM-CSF in inflammation, are described below.
Cellular Sources of GM-CSF and “Networks”
Both hemopoietic [e.g., T and B lymphocytes (12, 32–35) and innate lymphoid cells such as ILC3] (36–38) and non-hemopoietic cell types (e.g., fibroblast, endothelial, and epithelial populations) can produce GM-CSF although usually requiring an activating stimulus (5, 12, 14, 32, 37, 39–43). In accord with this requirement, in the steady state GM-CSF circulates at low levels and tends to be expressed basally in non-sterile tissues such as skin, lung and gut (44, 45). Even though in inflammation GM-CSF can serve as a communication conduit between tissue-invading lymphocytes and myeloid cells, there is some controversy as to which factors can induce GM-CSF production in T helper (Th) cells (12).
To help understand the chronicity of certain inflammatory/autoimmune responses, a “CSF network” hypothesis was originally proposed in which there is an interdependent co-regulation of proinflammatory cytokines, such as IL-1 and TNF, with GM-CSF as part of a positive feedback “loop” involving communication between monocytes/macrophages and neighboring cell populations, such as fibroblasts, endothelial cells etc. (3–5, 46); this concept has been expanded to include cytokines, such as IL-23 and IL-6, as components of an autocrine/paracrine “network” involving macrophages, DCs and Th cells (45, 47, 48). Recently, positive feedback “loops” have also been put forward involving GM-CSF in inflammatory-dilated cardiomyopathy and breast cancer metastasis (49, 50).
GM-CSF and Monocyte/Macrophage Function
Macrophage Polarization
Based only on increased expression of pro-inflammatory cytokines, GM-CSF-treated monocytes/macrophages have been termed “M1-like” (51). However, such cells have also been considered to have characteristics of both M1 and M2 cells, for example, as regards their cytokine expression (39, 52), and GM-CSF-activated mouse monocytes have been reported to alleviate experimental colitis (52). Partly on account of the modest overlap with classical M1 polarization and their dual M1/M2 characteristics, it has been recommended that the M1/M2 polarization terminology not be applied to GM-CSF-treated monocytes/macrophages (14, 25, 26, 53). Even though increased mRNA expression for TNF, IL-1β, and IL-6 is readily observed in GM-CSF-treated (primed) monocytes/macrophages in vivo, significant cytokine secretion usually requires another stimulus, such as lipopolysaccharide (26, 54, 55).
Endogenous mediators can contribute to the phenotypes of GM-CSF-treated monocytes/macrophages (25). As an example, GM-CSF-mediated macrophage polarization of human monocytes in vivo has been reported to be modulated by endogenous activin A (25, 56); it also has been proposed that the GM-CSF-induced PPARγ expression in human macrophages is primarily regulated in this way (57). Endogenous TGF-β has also been invoked to have a similar role in the development and homeostasis of mouse alveolar macrophages (58). Since most, if not all, mediators involved in the host inflammatory response to injury and/or infection are endeavoring to be beneficial by restoring homeostasis, it is important to explore such a role for GM-CSF in its action on monocytes/macrophages.
Monocytes, Macrophages, and DCs
It is debated as to whether GM-CSF can give rise to monocyte-derived DCs (MoDCs) in vivo or not (10, 14, 30, 59–61) even though GM-CSF, often in combination with IL-4, is widely used in vivo to generate mouse and human DC populations from bone marrow precursors and blood monocytes, respectively (20, 62–64). Two major types of GM-CSF-dependent phagocytes, termed macrophages and inflammatory DCs, have been claimed to have arisen in vivo from mouse CD209− and CD209+ monocyte subsets (65)—their relationships to the in vivo generated populations (see below) also need further analysis. Mouse CD103+ DCs (also called cDC1) from different lymphoid and non-lymphoid tissues have distinct functional activities and there has been disagreement about the contribution of GM-CSF to their development in vivo (10, 14, 66, 67) with perhaps varying levels of GM-CSF helping to explain the discrepancies between different studies (68). Obviously, more work needs to be done to understand the role of GM-CSF in cDC development in the steady state and during inflammation. It has been proposed that the effector functions of GM-CSF-expanded myeloid cells in vivo are guided by their tissue microenvironment (69).
Mouse populations generated by GM-CSF from bone marrow precursors are heterogeneous with cells having both DC and macrophage features being observed—such features include surface markers, morphology, motility, antigen presentation, T cell activation, cytokine production, and gene expression profiles (51, 70–73); in fact their nomenclature is debated as to whether they should be termed DCs or macrophages (25, 73–75). As an advance on the use and interpretation of the data from such cultures, cell sorting strategies have isolated populations from them with macrophage and DC properties (73). Again the GM-CSF concentrations employed likely contribute to the phenotypes of the resulting populations (31). The in vivo relationships of the in vivo generated populations from mouse bone marrow and human monocyte cultures are not fully defined.
Inflammation/Autoimmunity
In chronic inflammation and autoimmunity myeloid populations, for example, monocyte/macrophages and neutrophils, the cell populations which are potentially responsive to GM-CSF, are likely candidates to be regulating tissue damage and inflammation, being capable of releasing mediators, such as cytokines, chemokines, proteases and reactive oxygen species, as part of this response (5, 12, 26, 76, 77) (Figure 1). Of likely relevance to its function in inflammation/autoimmunity, GM-CSF upregulates class II MHC (21, 78, 79) and CD1 expression (80, 81) in human monocytes. However, it is worth noting that it cannot be assumed that monocytes/macrophages are the only myeloid cell types via which GM-CSF functions to regulate autoimmunity/inflammation (5). Amongst members of the macrophage lineage, GM-CSF initiates cardiac disease in resident mouse tissue macrophages (40) while in contrast only CCR2+ Ly6C+ monocytes require GM-CSF to lead to a pathogenic signature for EAE progression characterized by the induction of genes linked to inflammasome function, phagocytosis and chemotaxis, i.e., they become pathogenic DCs (76). Interestingly, it was reported that intrinsic GM-CSFR signaling by mouse monocytes and their precursors is not a prerequisite for the differentiation of monocytes into inflammatory monocyte-derived DCs in vivo during acute injuries (10). Nevertheless, moDCs do become more abundant in mice in which levels of GM-CSF are increased indicating again that GM-CSF can still be a critical factor influencing moDC differentiation, particularly under conditions where GM-CSF levels are elevated (61). GM-CSF-responsive CCR2+ moDCs and not Csf2rb−/− moDCs are critical for Th17 induction and EAE progression (60). In addition to being able to preferentially control putative moDC numbers in antigen-induced mouse peritonitis, GM-CSF could also regulate macrophage numbers in the inflamed peritoneal cavity (30, 82, 83). Whether this regulation of monocyte-derived populations was due to effects of GM-CSF on cell trafficking in or out of a lesion and/or cell survival is unknown (30, 83) although effects on the latter parameter in other inflammatory/autoimmune models have been discounted (40, 60). Interestingly, in this context it has been suggested that GM-CSF controls mouse DC survival in non-lymphoid tissues as the mechanism for their homeostasis (10). There is also evidence that during an inflammatory response GM-CSF may act systemically to promote hemopoietic cell mobilization and development (40, 84–87).
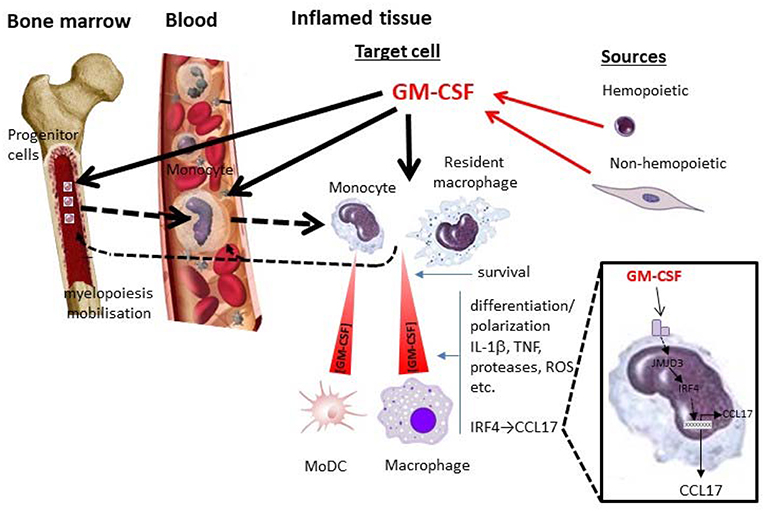
Figure 1. GM-CSF and monocytes/macrophages in inflammation. Depicted are some potential local and systemic actions of GM-CSF on monocyte/macrophage populations during an inflammatory reaction. Whether particular actions operate are currently debated and are likely to depend on the nature of the inflammatory reaction and the levels of GM-CSF attained from hemopoietic (e.g., lymphocyte) and non-hemopoietic (e.g., fibroblast) cell populations. Locally GM-CSF can act in a concentration—dependent manner on target cells (resident macrophages and/or blood-derived monocytes) to promote their survival and/or polarization/differentiation; the latter cell target can give rise to MoDCs. Their polarization/differentiation can be characterized by the production of proinflammatory mediators such as cytokines (e.g., IL-1β, TNF), proteases, reactive oxygen species (ROS), etc. One interesting pathway (zoomed), which seems to be important for GM-CSF-dependent inflammation and associated pain, leads to CCL17 production via JMJD3 and IRF4. GM-CSF can also act systemically in the blood and/or bone marrow, either directly or indirectly ( ) via its cellular targets in the tissue, leading to migration/mobilization of monocytes or their precursors and/or monocyte development from these precursors (myelopoiesis) (
) via its cellular targets in the tissue, leading to migration/mobilization of monocytes or their precursors and/or monocyte development from these precursors (myelopoiesis) ( ). MoDC, monocyte-derived DC.
). MoDC, monocyte-derived DC.
GM-CSF vs. M-CSF (CSF-1)
The gene expression profiles of human monocytes differentiated for 7 days in GM-CSF or M-CSF (CSF-1) differ substantially (25) and display distinct bioenergetic profiles (88). Since monocytes/macrophages are in general likely to be exposed to CSF-1 in the steady state, it has been proposed that pro-inflammatory stimuli, such as GM-CSF and interferon γ, lead to a cellular state of “CSF-1 resistance” or compromised CSF-1 signaling (5). CSF-1 could also be another endogenous mediator contributing to the phenotype of GM-CSF-treated human monocytes (89). Human monocytes differentiated in CSF-1 are widely used as a model for steady state tissue macrophages. In contrast to this widely used practice of employing CSF-1 as the differentiation stimulus, human monocytes treated in vivo with GM-CSF for 3 days have been used as a starting population of “macrophages” to analyse the transcriptional regulator networks upon cellular activation by a diverse range of stimuli (75, 90), stressing the need for researchers in the macrophage field to be conscious of the terminology used in any particular article.
GM-CSF and Interferon Regulatory Factors (IRFs)
Based on a number of reports (91–94), the hemopoietic-specific transcription factor, IRF4 (95), appears to be a key signaling molecule regulating the adoption of DC-like properties in GM-CSF-treated precursors such as monocytes. Ly6Chi Trem4neg mouse monocytes can differentiate into Zbtb46+ MoDCs in response to GM-CSF and IL-4 in an IRF4 dependent manner (96). Also, GM-CSF-IRF4 signaling upregulates MHC Class II expression in mouse macropahges (97). However, IRF5 rather than IRF4, has been reported to be important for GM-CSF-mediated macrophage polarization (24) although there is disagreement with this conclusion in that IRF4 is considered to be more important based on the divergent data for the relative enhanced expression of the two IRFs by GM-CSF in human monocytes (25, 26). There is no obvious reason for this divergence although subtle differences in culture conditions could perhaps play a role. In support of the importance of IRF4, there is recent evidence that IRF4, most likely acting in monocytes/macrophages, is important in controlling how GM-CSF promotes arthritis and associated pain, as well as inflammatory pain per se (26, 98). There is evidence in turn that enhanced JMJD3 histone demethylase activity is required for GM-CSF-induced IRF4 transcription to occur in monocytes/macrophages as well as for GM-CSF-induced inflammatory pain (26) (see below).
GM-CSF/CCL17 Axis
We recently found that the chemokine, CCL17, is the most highly up-regulated gene in GM-CSF-treated human monocytes and, unlike TNF and IL-1β, is secreted at high levels by GM-CSF-treated monocytes and mouse macrophages (26). It was also found surprisingly that CCL17 mediated GM-CSF-driven inflammatory pain as well as GM-CSF-driven and GM-CSF-dependent arthritic pain and disease. These pro-inflammatory actions of GM-CSF via CCL17 in turn required IRF4 and JMJD3 activity (26) (Figure 1). This proposed pro-inflammatory effect of IRF4 in macrophages was also surprising as IRF4 is usually considered to have an anti-inflammatory role in such cells since it down-regulates their production of pro-inflammatory cytokines such as TNF and IL-1β (99–101). Thus, GM-CSF joins the list of cytokines, such as IL-4 and TSLP, which can up-regulate CCL17 expression in monocytes/macrophages. This new GM-CSF → CCL17 pathway appears to be active in rheumatoid arthritis patients since circulating CCL17 levels are dramatically reduced upon anti-GM-CSF receptor monoclonal antibody therapy (102).
More recent studies have indicated that the GM-CSF → CCL17 pathway can be linked with TNF activity (103) as well as regulating experimental osteoarthritic pain and optimal disease (98)—the latter model data have led to a clinical trial being initiated in osteoarthritis using a CCL17 antagonist (NCT03485365 ClinicalTrials.gov). Interestingly, CCL17 may not necessarily be functioning as a chemokine in its regulation of inflammatory pain and arthritic pain/disease (98, 103).
Conclusions
It would appear from the above that GM-CSF-dependent inflammatory pathways in monocytes (and macrophages) are likely to be critical for the purported role of GM-CSF in inflammation, autoimmunity and host defense. In addition to attempting to summarize the relevant literature on this topic I have tried to highlight some of the contentious issues which are currently being debated. Such issues, which I have endeavored to represent diagrammatically (Figure 1), are: (i) when, how and at what concentrations GM-CSF controls cell number and/or activation/differentiation (polarization) in vivo, (ii) whether GM-CSF controls MoDC development in vivo, (iii) the nature of GM-CSF-induced cell polarization, (iv) whether IRF4- or IRF5- dependent pathways are more important for GM-CSF-dependent biology, (v) when and how endogenous GM-CSF can act systemically in addition to locally in tissues, and (vi) how relevant are the effects of systemically administered GM-CSF to the actions of endogenous GM-CSF.
In order to understand better the role of GM-CSF-dependent pathways, future studies in some of the following areas are likely to be informative: (i) additional clinical trials targeting GM-CSF action and that of other putative downstream mediators, such as CCL17, (ii) human monocyte/macrophage studies, (iii) cellular metabolic responses to GM-CSF, and (iv) the significance of GM-CSF → IRF4 signaling.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
R. Sallay and A. Cook are thanked for their assistance in the preparation of the manuscript.
References
1. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. (1980) 56:947–58.
2. Hamilton JA, Stanley ER, Burgess AW, Shadduck RK. Stimulation of macrophage plasminogen activator activity by colony-stimulating factors. J Cell Physiol. (1980) 103:435–45. doi: 10.1002/jcp.1041030309
3. Hamilton JA. A colony-stimulating factor network involving mononuclear phagocytes and other cells. In: Furth VR, editor. Haematopoietic Growth Factors and Mononuclear Phagocytes. Basel: Karger (1993). p. 29–35. doi: 10.1159/000422228
4. Hamilton JA. Rheumatoid arthritis: opposing actions of haemopoietic growth factors and slow-acting anti-rheumatic drugs. Lancet. (1993) 342:536–9. doi: 10.1016/0140-6736(93)91653-4
5. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. (2008) 8:533–44. doi: 10.1038/nri2356
6. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
7. Kitamura T, Tanaka N, Watanabe J, Uchida KS, Yamada Y, Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. (1999) 190:875–80. doi: 10.1084/jem.190.6.875
8. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
9. Sallese A, Suzuki T, Mccarthy C, Bridges J, Filuta A, Arumugam P, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep. (2017) 7:10211. doi: 10.1038/s41598-017-10879-w
10. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls non-lymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. (2012) 36:1031–46. doi: 10.1016/j.immuni.2012.03.027
11. Hamilton JA. GM-CSF as a target in inflammatory/autoimmune disease: current evidence and future therapeutic potential. Expert Rev Clin Immunol. (2015) 11:457–65. doi: 10.1586/1744666X.2015.1024110
12. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
13. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
14. Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. (2017) 16:53–70. doi: 10.1038/nrd.2016.231
15. Cook AD, Hamilton JA. Investigational therapies targeting the granulocyte macrophage colony-stimulating factor receptor-alpha in rheumatoid arthritis: focus on mavrilimumab. Ther Adv Musculoskelet Dis. (2018) 10:29–38. doi: 10.1177/1759720X17752036
16. Hansen G, Hercus TR, Mcclure BJ, Stomski FC, Dottore M, Powell J, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. (2008) 134:496–507. doi: 10.1016/j.cell.2008.05.053
17. Perugini M, Brown AL, Salerno DG, Booker GW, Stojkoski C, Hercus TR, et al. Alternative modes of GM-CSF receptor activation revealed using activated mutants of the common beta-subunit. Blood. (2010) 115:3346–53. doi: 10.1182/blood-2009-08-235846
18. Thomas D, Powell JA, Green BD, Barry EF, Ma Y, Woodcock J, et al. Protein kinase activity of phosphoinositide 3-kinase regulates cytokine-dependent cell survival. PLoS Biol. (2013) 11:e1001515. doi: 10.1371/journal.pbio.1001515
19. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. (2002) 71:511–9. doi: 10.1189/jlb.71.3.511
20. Van De Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. (2012) 119:3383–93. doi: 10.1182/blood-2011-11-370130
21. Achuthan A, Aslam ASM, Nguyen Q, Lam PY, Fleetwood AJ, Frye AT, et al. Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity. Cell Death Dis. (2018) 9:267. doi: 10.1038/s41419-018-0332-4
22. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
23. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
24. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12:231–8. doi: 10.1038/ni.1990
25. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vivo models. J Immunol. (2012) 188:5752–65. doi: 10.4049/jimmunol.1103426
26. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. (2016) 126:3453–66. doi: 10.1172/JCI87828
27. Broughton SE, Hercus TR, Nero TL, Dottore M, Mcclure BJ, Dhagat U, et al. Conformational changes in the GM-CSF receptor suggest a molecular mechanism for affinity conversion and receptor signaling. Structure. (2016) 24:1271–81. doi: 10.1016/j.str.2016.05.017
28. Guthridge MA, Powell JA, Barry EF, Stomski FC, Mcclure BJ, Ramshaw H, et al. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. Embo J. (2006) 25:479–89. doi: 10.1038/sj.emboj.7600948
29. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. (2017) 1:947–60. doi: 10.1182/bloodadvances.2017006858
30. Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, et al. Specific contributions of CSF-1 and GM-CSF to the dynamics of the mononuclear phagocyte system. J Immunol. (2015) 195:134–44. doi: 10.4049/jimmunol.1500369
31. Sun L, Rautela J, Delconte RB, Souza-Fonseca-Guimaraes F, Carrington EM, Schenk RL, et al. GM-CSF quantity has a selective effect on granulocytic vs. monocytic myeloid development and function. Front Immunol. (2018) 9:1922. doi: 10.3389/fimmu.2018.01922
32. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. (2012) 335:597–601. doi: 10.1126/science.1215173
33. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. (2014) 6:241ra280. doi: 10.1126/scitranslmed.3008706
34. Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J Exp Med. (2014) 211:1243–56. doi: 10.1084/jem.20131471
35. Yamada H, Haraguchi A, Sakuraba K, Okazaki K, Fukushi JI, Mizu-Uchi H, et al. Th1 is the predominant helper T cell subset that produces GM-CSF in the joint of rheumatoid arthritis. RMD Open. (2017) 3:e000487. doi: 10.1136/rmdopen-2017-000487
36. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol. (2014) 15:354–64. doi: 10.1038/ni.2830
37. Pearson C, Thornton EE, Mckenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. (2016) 5:e10066. doi: 10.7554/eLife.10066
38. Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. (2018) 48:1220–32 e1225. doi: 10.1016/j.immuni.2018.04.009
39. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. (2012) 209:1505–17. doi: 10.1084/jem.20112691
40. Stock AT, Hansen JA, Sleeman MA, Mckenzie BS, Wicks IP. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. (2016) 213:1983–98. doi: 10.1084/jem.20151853
41. Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med. (2017) 214:3293–310. doi: 10.1084/jem.20170689
42. Sheih A, Parks WC, Ziegler SF. GM-CSF produced by the airway epithelium is required for sensitization to cockroach allergen. Mucosal Immunol. (2017) 10:705–15. doi: 10.1038/mi.2016.90
43. Chen G, Bracamonte-Baran W, Diny NL, Hou X, Talor MV, Fu K, et al. Sca-1(+) cardiac fibroblasts promote development of heart failure. Eur J Immunol. (2018) 48:1522–38. doi: 10.1002/eji.201847583
44. Metcalf D, Nicola NA. The Hemopoietic Colony-Stimulating Factors: From Biology to Clinical Applications. Cambridge: University Press (1995). doi: 10.1017/CBO9780511663376
45. Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. (2013) 34:81–9. doi: 10.1016/j.it.2012.08.006
46. Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol. (2002) 23:403–8. doi: 10.1016/S1471-4906(02)02260-3
47. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. (2008) 205:2281–94. doi: 10.1084/jem.20071119
48. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
49. Su S, Wu W, He C, Liu Q, Song E. Breaking the vicious cycle between breast cancer cells and tumor-associated macrophages. Oncoimmunology. (2014) 3:e953418. doi: 10.4161/21624011.2014.953418
50. Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. (2014) 211:1449–64. doi: 10.1084/jem.20132126
51. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. GM-CSF and M-CSF (CSF-1)-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities—implications for CSF blockade in inflammation. J Immunol. (2007) 178:5245–52. doi: 10.4049/jimmunol.178.8.5245
52. Dabritz J, Weinhage T, Varga G, Wirth T, Walscheid K, Brockhausen A, et al. Reprogramming of monocytes by GM-CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol. (2015) 194:2424–38. doi: 10.4049/jimmunol.1401482
53. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
54. Hart PH, Whitty GA, Piccoli DS, Hamilton JA. Synergistic activation of human monocytes by granulocyte-macrophage colony-stimulating factor and IFN-gamma. Increased TNF-alpha but not IL-1 activity. J Immunol. (1988) 141:1516–21.
55. Borriello F, Iannone R, Di Somma S, Loffredo S, Scamardella E, Galdiero MR, et al. GM-CSF and IL-3 modulate human monocyte TNF-alpha production and renewal in in vivo models of trained immunity. Front Immunol. (2016) 7:680. doi: 10.3389/fimmu.2016.00680
56. Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, et al. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood. (2011) 117:5092–101. doi: 10.1182/blood-2010-09-306993
57. Nieto C, Bragado R, Municio C, Sierra-Filardi E, Alonso B, Escribese MM, et al. The activin A-peroxisome proliferator-activated receptor gamma axis contributes to the transcriptome of GM-CSF-conditioned human macrophages. Front Immunol. (2018) 9:31. doi: 10.3389/fimmu.2018.00031
58. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine TGF-beta promotes the development and homeostasis of alveolar macrophages. Immunity. (2017) 47:903–12 e904. doi: 10.1016/j.immuni.2017.10.007
59. Zhan Y, Vega-Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur J Immunol. (2012) 42:2889–900. doi: 10.1002/eji.201242477
60. Ko HJ, Brady JL, Ryg-Cornejo V, Hansen DS, Vremec D, Shortman K, et al. GM-CSF-responsive monocyte-derived dendritic cells are pivotal in Th17 pathogenesis. J Immunol. (2014) 192:2202–9. doi: 10.4049/jimmunol.1302040
61. Chow KV, Delconte RB, Huntington ND, Tarlinton DM, Sutherland RM, Zhan Y, et al. Innate allorecognition results in rapid accumulation of monocyte-derived dendritic cells. J Immunol. (2016) 197:2000–8. doi: 10.4049/jimmunol.1600181
62. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
63. Suzuki H, Katayama N, Ikuta Y, Mukai K, Fujieda A, Mitani H, et al. Activities of granulocyte-macrophage colony-stimulating factor and interleukin-3 on monocytes. Am J Hematol. (2004) 75:179–89. doi: 10.1002/ajh.20010
64. Conti L, Gessani S. GM-CSF in the generation of dendritic cells from human blood monocyte precursors: recent advances. Immunobiology. (2008) 213:859–70. doi: 10.1016/j.imbio.2008.07.017
65. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6C(hi) monocytes controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity. (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
66. King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. (2010) 207:953–61. doi: 10.1084/jem.20091844
67. Edelson BT, Bradstreet TR, Kc W, Hildner K, Herzog JW, Sim J, et al. Batf3-dependent CD11b(low/-) peripheral dendritic cells are GM-CSF-independent and are not required for Th cell priming after subcutaneous immunization. PLoS ONE. (2011) 6:e25660. doi: 10.1371/journal.pone.0025660
68. Jiao Z, Bedoui S, Brady JL, Walter A, Chopin M, Carrington EM, et al. The closely related CD103+ dendritic cells. (DCs) and lymphoid-resident CD8+ DCs differ in their inflammatory functions. PLoS ONE. (2014) 9:e91126. doi: 10.1371/journal.pone.0091126
69. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity. (2017) 46:245–60. doi: 10.1016/j.immuni.2017.01.007
70. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
71. Na YR, Jung D, Gu GJ, Seok SH. GM-CSF grown bone marrow derived cells are composed of phenotypically different dendritic cells and macrophages. Mol Cells. (2016) 39:734–41. doi: 10.14348/molcells.2016.0160
72. Rogers PB, Driessnack MG, Hiltbold Schwartz E. Analysis of the developmental stages, kinetics, and phenotypes exhibited by myeloid cells driven by GM-CSF in vivo. PLoS ONE. (2017) 12:e0181985. doi: 10.1371/journal.pone.0181985
73. Erlich Z, Shlomovitz I, Edry-Botzer L, Cohen H, Frank D, Wang H, et al. Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat Immunol. (2019) 20:397–406. doi: 10.1038/s41590-019-0313-5
74. Hume DA, Mabbott N, Raza S, Freeman TC. Can DCs be distinguished from macrophages by molecular signatures? Nat Immunol. (2013) 14:187–9. doi: 10.1038/ni.2516
75. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
76. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
77. Croxford AL, Spath S, Becher B. GM-CSF in neuroinflammation: licensing myeloid cells for tissue damage. Trends Immunol. (2015) 36:651–62. doi: 10.1016/j.it.2015.08.004
78. Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Cytokines in chronic inflammatory arthritis. IV Granulocyte/macrophage colony-stimulating factor-mediated induction of class II MHC antigen on human monocytes: a possible role in rheumatoid arthritis. J Exp Med. (1989) 170:865–75. doi: 10.1084/jem.170.3.865
79. Hornell TM, Beresford GW, Bushey A, Boss JM, Mellins ED. Regulation of the class II MHC pathway in primary human monocytes by granulocyte-macrophage colony-stimulating factor. J Immunol. (2003) 171:2374–83. doi: 10.4049/jimmunol.171.5.2374
80. Kasinrerk W, Baumruker T, Majdic O, Knapp W, Stockinger H. CD1 molecule expression on human monocytes induced by granulocyte-macrophage colony-stimulating factor. J Immunol. (1993) 150:579–84.
81. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:899–907. doi: 10.1136/annrheumdis-2014-206578
82. Cook AD, Braine EL, Hamilton JA. Stimulus-dependent requirement for granulocyte-macrophage colony-stimulating factor in inflammation. J Immunol. (2004) 173:4643–51. doi: 10.4049/jimmunol.173.7.4643
83. Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony-stimulating factor receptor alpha expression and its targeting in antigen-induced arthritis and inflammation. Arthritis Res Ther. (2016) 18:287. doi: 10.1186/s13075-016-1185-9
84. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. (2009) 113:3190–7. doi: 10.1182/blood-2008-07-168575
85. Cook AD, Turner AL, Braine EL, Pobjoy J, Lenzo JC, Hamilton JA. Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. (2011) 63:2340–51. doi: 10.1002/art.30354
86. Griseri T, Mckenzie BS, Schiering C, Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. (2012) 37:1116–29. doi: 10.1016/j.immuni.2012.08.025
87. Wang M, Subramanian M, Abramowicz S, Murphy AJ, Gonen A, Witztum J, et al. Interleukin-3/granulocyte macrophage colony-stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler Thromb Vasc Biol. (2014) 34:976–84. doi: 10.1161/ATVBAHA.113.303097
88. Izquierdo E, Cuevas VD, Fernandez-Arroyo S, Riera-Borrull M, Orta-Zavalza E, Joven J, et al. Reshaping of human macrophage polarization through modulation of glucose catabolic pathways. J Immunol. (2015) 195:2442–51. doi: 10.4049/jimmunol.1403045
89. Hamilton JA. Coordinate and non-coordinate colony stimulating factor formation by human monocytes. J Leukoc Biol. (1994) 55:355–61. doi: 10.1002/jlb.55.3.355
90. Schmidt SV, Krebs W, Ulas T, Xue J, Bassler K, Gunther P, et al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. (2016) 26:151–70. doi: 10.1038/cr.2016.1
91. Lehtonen A, Veckman V, Nikula T, Lahesmaa R, Kinnunen L, Matikainen S, et al. Differential expression of IFN regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J Immunol. (2005) 175:6570–9. doi: 10.4049/jimmunol.175.10.6570
92. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity. (2013) 39:722–32. doi: 10.1016/j.immuni.2013.08.028
93. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. (2013) 4:2990. doi: 10.1038/ncomms3990
94. Yashiro T, Yamaguchi M, Watanuki Y, Kasakura K, Nishiyama C. The transcription factors PU.1 and IRF4 determine dendritic cell-specific expression of RALDH2. J Immunol. (2018) 201:3677–82. doi: 10.4049/jimmunol.1800492
95. Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, et al. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS ONE. (2011) 6:e25812. doi: 10.1371/journal.pone.0025812
96. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep. (2016) 15:2462–74. doi: 10.1016/j.celrep.2016.05.025
97. Lee MC, Lacey DC, Fleetwood AJ, Achuthan A, Hamilton JA, Cook AD. GM-CSF- and IRF4-dependent signaling can regulate myeloid cell numbers and the macrophage phenotype during inflammation. J Immunol. (2019) 202:3033–40. doi: 10.4049/jimmunol.1801549
98. Lee MC, Saleh R, Achuthan A, Fleetwood AJ, Forster I, Hamilton JA, et al. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res Ther. (2018) 20:62. doi: 10.1186/s13075-018-1560-9
99. Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci USA. (2005) 102:16001–6. doi: 10.1073/pnas.0504226102
100. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
101. Eguchi J, Kong X, Tenta M, Wang X, Kang S, Rosen ED. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. (2013) 62:3394–403. doi: 10.2337/db12-1327
102. Guo X, Wang S, Godwood A, Close D, Ryan PC, Roskos LK, et al. Pharmacodynamic biomarkers and differential effects of TNF- and GM-CSF-targeting biologics in rheumatoid arthritis. Int J Rheum Dis. (2019) 22:646–53. doi: 10.1111/1756-185X.13395
Keywords: cell survival, polarization, inflammation, pain, IRF4, CCL17
Citation: Hamilton JA (2019) GM-CSF-Dependent Inflammatory Pathways. Front. Immunol. 10:2055. doi: 10.3389/fimmu.2019.02055
Received: 01 March 2019; Accepted: 14 August 2019;
Published: 04 September 2019.
Edited by:
Calum C. Bain, University of Edinburgh, United KingdomReviewed by:
Alexander Mildner, Helmholtz Association of German Research Centers (HZ), GermanyOlof Rådmark, Karolinska Institute, Sweden
Martin Guilliams, VIB-UGent Center for Inflammation Research (IRC), Belgium
Copyright © 2019 Hamilton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John A. Hamilton, amFoYW1pQHVuaW1lbGIuZWR1LmF1