 Federico Perdomo-Celis
Federico Perdomo-Celis Natalia A. Taborda
Natalia A. Taborda Maria T. Rugeles
Maria T. Rugeles- 1Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia, Medellin, Colombia
- 2Grupo de Investigaciones Biomédicas Uniremington, Programa de Medicina, Facultad de Ciencias de la Salud, Corporación Universitaria Remington, Medellin, Colombia
Although the combined antiretroviral therapy (cART) has decreased the deaths associated with the immune deficiency acquired syndrome (AIDS), non-AIDS conditions have emerged as an important cause of morbidity and mortality in HIV-infected patients under suppressive cART. Since these conditions are associated with a persistent inflammatory and immune activation state, major efforts are currently made to improve the immune reconstitution. CD8+ T-cells are critical in the natural and cART-induced control of viral replication; however, CD8+ T-cells are highly affected by the persistent immune activation and exhaustion state driven by the increased antigenic and inflammatory burden during HIV infection, inducing phenotypic and functional alterations, and hampering their antiviral response. Several CD8+ T-cell subsets, such as interleukin-17-producing and follicular CXCR5+ CD8+ T-cells, could play a particular role during HIV infection by promoting the gut barrier integrity, and exerting viral control in lymphoid follicles, respectively. Here, we discuss the role of CD8+ T-cells and some of their subpopulations during HIV infection in the context of cART-induced viral suppression, focusing on current challenges and alternatives for reaching complete reconstitution of CD8+ T-cells antiviral function. We also address the potential usefulness of CD8+ T-cell features to identify patients who will reach immune reconstitution or have a higher risk for developing non-AIDS conditions. Finally, we examine the therapeutic potential of CD8+ T-cells for HIV cure strategies.
Epidemiology of HIV Infection and Impact of Antiretroviral Therapy
The human immunodeficiency virus type-1 (HIV) infection remains an important public health problem, particularly in developing countries. As a result of the long-term subclinical presentation of this infection, a high rate of shortfall at diagnosis is evidenced worldwide, and almost half of the HIV-infected patients are not aware of their HIV status (1). Therefore, most of the available data on HIV epidemiology is based on estimates, particularly in resource-limited settings (2). This issue dramatically affects public health policies; representing a major barrier for HIV global control. Thus, according to UNAIDS 2017 data, at the end of 2016, 36.7 million people lived with HIV, and of these, 17.8 million were women, and 2.1 million were children. The problem is increasing, as the global annual number of new HIV infections is 1.8 million, and deaths associated with the acquired immune deficiency syndrome (AIDS) reach 1 million.
The most important advance in the clinical management of HIV infection was the development of antiretroviral drugs and their therapeutic combination to suppress systemic viral load. Indeed, the combined antiretroviral therapy (cART), i.e., combination of three or more antiretroviral drugs, generally including at least two drugs of different mechanisms of action (1), has been effective in decreasing the deaths associated with AIDS. According to UNAIDS 2017 estimates, global AIDS-related deaths declined in 48% between 2005 and 2016, with near to eight million deaths prevented since the introduction of the therapy in 1995. Moreover, the decrease in the viral load by cART and consequent lower transmission risk, has led to the reduction of 16% of new HIV infections between 2010 and 2016. In addition, pre-exposure and post-exposure prophylaxis are recommended in some countries for prevention of HIV infection (3). Thus, cART constitutes a critical strategy for the control of HIV-associated morbidity and mortality, as well as a transmission prevention approach.
The World Health Organization (WHO) and Centers for Disease Control and Prevention (CDC) recommends the initiation of cART as soon as possible after diagnosis, resulting in improved viral control and prevention of AIDS conditions (available at https://www.who.int/hiv/topics/treatment/en/ and https://aidsinfo.nih.gov/guidelines). However, guidelines for management HIV-infected patients in some Latin American countries, such as Colombia, have clearly defined indications for initiation of cART, based on CD4+ T-cell counts and viral load (4). Importantly, from the total diagnosed patients in Colombia, 89.9% received at least one dose of antiretroviral drugs, although the number of patients receiving continuous therapy and with viral suppression (plasma or serum viral load <20 copies RNA/mL) only reached 66.6% of treated patients. This reflects the poor health system that is not able to sustain antiretroviral supply, in addition to problems with therapy adherence; these problems might be common in other Latin American countries. Overall, the strategies to increase the rate of diagnosis and the rapid initiation and maintenance of cART, will help to achieve the 90-90-90 target for HIV-infected patients (2).
Current Challenges of the Antiretroviral Therapy
In addition to important issues in prevention, screening, and diagnosis of HIV-infection, the treatment of diagnosed patients has faced several challenges and pitfalls. These issues can be divided as following: (i) operative, related to coverage, adherence, and adequate monitoring of therapy; (ii) virological, related to generation of viral resistance to antiretroviral drugs, and (iii) immunological, related to immune reconstitution failure. Operative challenges include those associated with the lack of the required financial resources for implementing integral HIV programs; inadequate stocks of antiretroviral drugs; limited health system infrastructures; low acceptance of cART initiation and long-term adherence with absence of proven methods to ensure treatment adherence and an adequate follow-up. The complexity of cART regimens and the side effects of antiretroviral drugs are also a major complaint in HIV-infected patients. Moreover, the persistent stigma associated with HIV infection limits the timely detection of cases and early initiation of cART (5). The second type of cART challenge is the antiretroviral drugs resistance, which is the most frequent type of therapy failure, inducing a change in the first-line cART scheme (6). Drug resistance results from the high rate of viral mutations, enhanced by a poor cART adherence, pharmacokinetics limitations, inadequate dosing or drug interactions (7). In addition, a weak monitoring and limited indicators for antiretroviral drugs resistance reduce the rate of successful cART (8). Finally, partial immune reconstitution by cART is a major concern in the setting of viral suppression. It has recently attracted more attention, as it has been associated with increased non-AIDS conditions and related mortality, despite the reduction in AIDS-related deaths (9). Indeed, non-AIDS conditions, such as cardiovascular disease or stroke, are responsible for around 42% of deaths among HIV-infected patients with viral suppression induced by cART. Moreover, the mortality rate in these individuals is higher than in the general population, even excluding the AIDS-related conditions (10, 11).
Importantly, the success of cART in reducing AIDS-related deaths and increasing the life expectancy of HIV-infected patients, has resulted in a high number of patients over 50 years living with HIV (available at https://www.cdc.gov/hiv/group/age/olderamericans/index.html). Thus, the presence of concomitant diseases, such as metabolic or cardiovascular pathologies, and their respective medications, influence the choice of antiretroviral drugs for cART regimens, and increase the risk of side effects due to drug interactions (12).
CD8+ T-Cells, Effector Functions, and Subpopulations
Activation and Differentiation of CD8+ T-Cells
CD8+ T-cells are part of the adaptive immune system, playing a critical role for protection against foreign organisms and malignancies (13). The activation of naïve CD8+ T-cells requires three signals, provided by antigen-presenting cells (APC). Initially, there is the recognition of the antigen by the T-cell receptor (TCR); in the case of CD8+ T-cells, the peptides are 8–10 amino acids in length and are presented by major histocompatibility complex (MHC) class I molecules. Second, costimulatory signals are required, such as the binding of CD80 or CD86 from the APC to the CD28 molecule expressed by the CD8+ T-cell, and finally an alarm signal produced in response to pathogens, such as IL-12 and type I interferon (IFN), among others (14, 15).
After activation, CD8+ T-cells undergo clonal expansion, generating a large pool of effector cells. These cells exhibit high effector functions, such as the expression of cytokines, cytotoxic molecules and a high capacity for degranulation (16–19). Additionally, at this point, effector CD8+ T-cells acquire the ability to migrate to peripheral tissues (20). After clonal expansion, effector cells suffer massive apoptosis, in a period known as contraction phase. Finally, the remaining antigen-specific CD8+ T-cells constitute the pool of memory cells, which decrease their effector profile, remaining in a quiescent state expecting a new antigenic challenge. This period, which can last the whole life of the individual, is known as the memory phase (21). Several CD8+ T-cells differentiation models propose that memory cells can differentiate into effector cells once they are exposed to activation signals, acquiring a high capacity for cytokine production and cytotoxic potential (22).
CD8+ T-Cells Effector Mechanisms
CD8+ T-cell effector mechanisms can be classified in lytic (cytotoxicity) and non-lytic (cytokine production) (Figure 1). These effector functions are primarily regulated by the balance between the T-bet, Eomes, and Runx families of transcription factors (23–26). Indeed, these transcription factors not only bind to DNA sequences of effector molecules such as granzyme B and IFN-γ, but also cooperate with chromatin remodeling proteins to regulate chromatin accessibility of key genes in activated CD8+ T-cells (27). The cytotoxic capacity of CD8+ T-cells depends on the content of their lytic granules and their degranulation capacity (28, 29). Lytic granules are secretory lysosomes containing effector molecules, such as granzymes and perforin (30). The core of the granule is surrounded by a lipid bilayer containing lysosome-associated membrane glycoproteins (LAMP), including LAMP-1 (CD107a), LAMP-2 (CD107b), and LAMP-3 (CD63) (31). During the degranulation process, the granule membrane is fused with the plasma membrane of the activated CD8+ T-cell, and the content of the granule is released into the immunological synapse between the CD8+ T-cell and the target cell (29). Of note, LAMP molecules are not found on the surface of resting CD8+ T-cells. Therefore, the evaluation of the surface expression of CD107a/b allows to identify the degranulation of CD8+ T-cells (28, 29). In addition, lytic granules also contain the membrane pore-forming protein granulysin (32), the proteoglycan matrix protein serglycin (33), the perforin inhibitor calreticulin (34), and the lysosomal enzymes cathepsins (35). Moreover, apoptosis-inducing Fas ligand (CD95L) is stored in specialized secretory lysosomes of CD8+ T-cells and the degranulation process controls its expression at the cell surface (36).
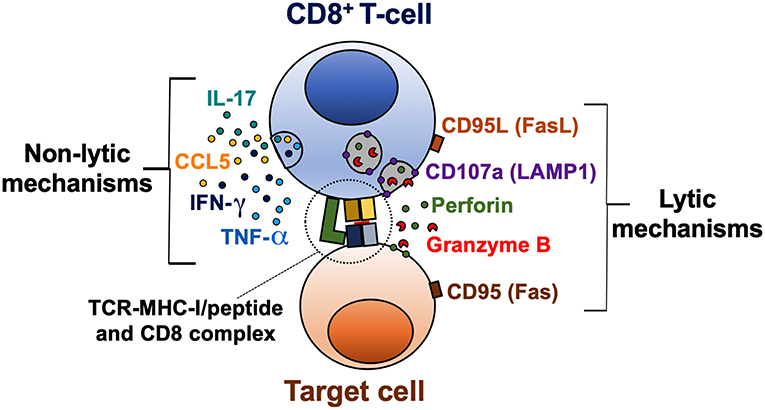
Figure 1. Lytic and non-lytic effector mechanisms of CD8+ T-cells. CD8+ T-cells are activated after recognition of an MHC-I/peptide complex, which binds to the TCR and CD8 molecules. The cytotoxic potential of CD8+ T-cells is determined by the expression of cytotoxic molecules granzyme B and perforin, and a coordinated degranulation process, which can be evaluated by the cell membrane expression of CD107a (LAMP1). Receptor-mediated cell death via CD95L/CD95 interaction is also a lytic mechanism of CD8+ T-cells. Activated CD8+ T-cells can also produce cytokines such as IFN-γ, TNF-α, and IL-17, and the β-chemokine CCL5, which exert a variety of antiviral, inflammatory, and regulatory functions.
Perforin and granzymes are the most abundant proteins within the lytic granules of CD8+ T-cells and cooperate for inducing death of target cells (37). Apparently, granzymes work in a perforin-dependent manner, since initial studies in mast cell lines, the presence of perforin was required for granzyme B-induced death of target cells (38). Thus, the pore formation by perforin facilitates the entry of granzymes into target cells (39). However, granzymes could be internalized by endocytosis, after binding to the mannose-6-phosphate receptor in target cells (40). Molecular effector mechanisms can vary among granzyme subtypes, inducing apoptosis through caspase-dependent and independent mechanisms (41). Once in the cytosol of target cells, granzymes cleave the BH3-interacting domain death agonist (BID) and pro-caspase 3 (42, 43). Truncated BID alters the mitochondrial membrane, inducing the release of pro-apoptotic factors such as cytochrome C, involved in the formation of the apoptosome (44), and endonuclease G, which causes DNA fragmentation (45). In addition, active caspase 3 also induces endonuclease and protease activation, causing degradation of cellular DNA and cytoskeletal proteins (46).
Cytokines secreted by CD8+ T-cells include IFN-γ, TNF-α, and IL-2, important for promoting antiviral, inflammatory responses, and T-cell survival and proliferation, respectively (47). Additionally, CD8+ T-cells secrete the β-chemokines macrophage inflammatory protein (MIP)-1α (CCL3), MIP-1β (CCL4), and regulated upon activation, normal T-cell expressed and secreted (RANTES; CCL5). These chemokines bind to the receptors CCR1, CCR3, CCR4, and CCR5, some of them, previously characterized as HIV co-receptors (48); therefore they play an important role in this infection by blocking viral binding and entry into target cells (49, 50).
CD8+ T-Cell Subpopulations
Several CD8+ T-cell subsets have been described, based on the expression of the differentiation markers CD45RA, CD45RO, CCR7, CD62L, CD27, and CD28, among others. The most extended classification divides them into naïve, central memory, effector memory, and terminal effector cells; each of them has different effector capacities, particularly evaluated by the expression of granzymes, perforin, IFN-γ, the degranulation ability, and in vitro cytotoxicity [Table 1; (18, 51)]. The lowest functional capacity is observed for naïve CD8+ T-cells, which have low expression of effector molecules, reduced degranulation capacity and low in vitro cytotoxicity. Central memory cells have a low/intermediate cytotoxic potential, given their low basal expression of granzymes and perforin, which confers them a limited immediate in vitro cytotoxicity. However, these cells can degranulate and express de novo effector molecules after polyclonal or antigen-specific stimulation (22, 52). Finally, effector memory and terminal effector cells are characterized by a high cytotoxic capacity (Table 1).
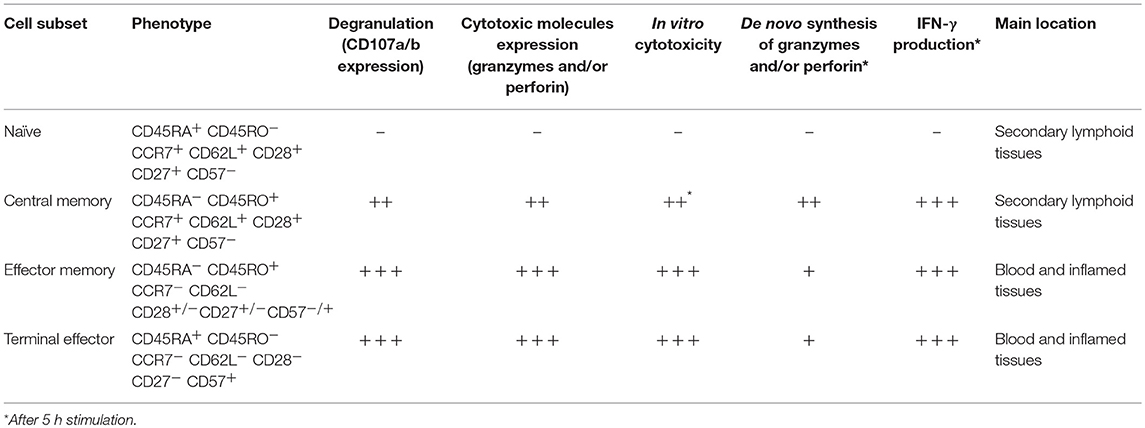
Table 1. Effector capacity of CD8+ T-cells according to their differentiation stage.
Of note, the distribution of CD8+ T-cell subsets and their effector machinery also vary according to the body tissues and compartments, with effector CD8+ T-cells mainly located in blood and inflamed tissues, and naïve and central memory cells primarily found in secondary lymphoid organs (Table 1). Particularly, their transcriptional program determines this cell distribution and effector function. Thus, CD8+ T-cells in lymphoid tissues, as well as in gastrointestinal mucosa, from healthy individuals, have a lower expression of perforin and granzyme B compared with blood cells, which is associated with a low expression of T-bet in these tissues (53–55). Interestingly, CD8+ T-cells in lymphoid tissues are poised to upregulate cytotoxic molecules and rapidly exert effector functions, similar to long-lived memory CD8+ T-cells (22). However, they also upregulate trafficking markers such as CXCR3 to egress from lymphoid tissues (53). Moreover, compared with blood cells, memory CD8+ T-cells from tonsil, lymph nodes, and spleen exhibit higher tissue residency markers such as CD69, CD103, and CD49a, but lower expression of T-bet, eomes, perforin, and granzyme B. Therefore, cells from blood and tissue compartment form distinct phenotypic and functional clusters (56).
According to the cytokines they produce, several subpopulations of CD8+ T-cells have been described. Similar to CD4+ T-cells, IFN-γ-producing, and IL-5/IL-13-producing CD8+ T-cells are designated as Tc1 and Tc2 cells, respectively (57). Regulatory CD8+ T-cell subsets have also been described (58). Moreover, IL-17-producing CD8+ T-cells (Tc17) can be induced by polarizing cytokines such as Transforming Growth Factor (TGF)-β1 and IL-6 (59). This population is characterized by the expression of the C-type lectin receptor CD161 (60), the transcription factor retinoic acid receptor-related orphan nuclear receptor (ROR)-γt (61), and its main localization is the intestinal tract (62–64). Importantly, the IL-17 cytokine family is constituted by six proteins that share homology (IL-17A through IL-17F) (65). The most widely studied is IL-17A (here referred as IL-17), and pro-inflammatory and immunomodulatory effects have been described (66). In the context of HIV infection, this cytokine has attracted attention due to its beneficial effects in the gut mucosa, such as the promotion of the conformation of tight junctions in epithelial cells (67), secretion of antimicrobial peptides (68), as well as recruitment of immune cells to sites of mucosal injury (69). In this sense, Tc17 cells could promote gut homeostasis, exerting a protective mechanism during HIV infection. Finally, similar to CD4+ T-cells, follicular CXCR5-expressing CD8+ T-cells have been characterized (70), and their role during HIV infection is examined below.
Progressive Dysfunction of CD8+ T-Cells During HIV Infection and Poor Reconstitution Despite cART
CD8+ T-cells are important in the control of HIV replication (71), as evidenced by: (i) emergence of HIV-specific CD8+ T-cells coinciding with the decrease of viremia in acutely infected patients (72); (ii) a potent HIV-specific CD8+ T cell response contribute to the reduction of the pool of HIV-infected cells and the HIV reservoir (73); (iii) increase in viral load in SIV-infected macaques after CD8+ T-cell depletion (74, 75); (iii) associations between the frequency and/or functional capacity of HIV-specific CD8+ T-cells and limited viral replication and/or disease non-progression in HIV-infected patients (76, 77); (iv) appearance of viral escape mutations to evade the immune pressure of HIV-specific CD8+ T-cells (78); (v) requirement of CD8+ T-cells for maintaining therapy-induced viral suppression in SIV-infected macaques (79). In fact, some CD8+ T-cells features have been proposed as correlate of protection in HIV-infected patients (80).
While HIV-specific CD8+ T-cells control the virus during acute HIV/SIV infection, their cytotoxic potential dramatically decreases along with disease progression and are no longer capable of exerting an appropriate antiviral response (81, 82). Certainly, CD8+ T-cells suffer important alterations in their frequency, differentiation, and activation profile, undergoing immune exhaustion, and progressive dysfunction (24, 83–87). Compared with seronegative individuals, the total pool of circulating CD8+ T-cells is persistently increased in untreated HIV-infected patients (88), with higher frequency of memory subsets and reduction of naïve cells (89, 90). In addition, patients exhibit higher expression of the activation markers HLA-DR, CD38, and Ki-67 (91), and immune exhaustion markers such as programmed death (PD)-1 and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) (92). Remarkably, the HLA-DR+ CD38+ Ki-67+ PD-1+ phenotype in CD8+ T-cells characterizes the effector phase after acute viral infections or vaccination, which is associated with disease control (16, 93–96). Nonetheless, the expression of these molecules during chronic HIV infection is accompanied by the impairment of CD8+ T-cell lytic and non-lytic mechanisms (24, 83, 97), as well as the proliferative ability and survival (98–100). Specific subpopulations, such as Tc17 cells, are decreased in gut mucosa and blood during HIV/SIV infections (101–103). Importantly, these alterations are observed in HIV-specific CD8+ T-cells (76, 104), as well as cells specific for other pathogens, such as cytomegalovirus (CMV), Epstein-Barr virus, influenza virus, and adenovirus (105, 106). Taking into account that HIV-specific cells constitute <20% of the total CD8+ T-cell population (107, 108), these data reflect the massive bystander activation that CD8+ T-cells undergo during HIV infection.
Considering that a large size of viral burden is a product of infected cells in lymph nodes and gastrointestinal mucosa (109), and there is a low distribution of antiretroviral drugs to these compartments (110), an effective CD8+ T-cell response in these tissues is required to control viral replication and the reservoir size. Nonetheless, the cytolytic activity of lymph node CD8+ T-cells is reduced in chronically HIV-infected patients (both untreated and on cART) compared with seronegative controls (53). Furthermore, lymph node and rectal HIV-specific CD8+ T-cells have lower expression of granzyme B and perforin, as well as T-bet, compared with blood cells (53, 55). Thus, while there is a regulated cytolytic function in CD8+ T-cells in lymphoid tissues and gut mucosa, there is apparently a poorer cytotoxic response in HIV-infected patients. Intriguingly, HIV controllers maintain low viral load in the absence of therapy and despite this low cytotoxic potential of CD8+ T-cells in lymphoid tissues (53, 111). Thus, it is possible that non-lytic mechanisms and a polyfunctional response, including IFN-γ, TNF-α, and MIP-1β production, as well as degranulation, are critical in the control of HIV infection in lymphoid and gastrointestinal tissues (111, 112).
Along with the suppression of viral load and the increase in the CD4+ T-cell counts, cART induces improvement of some of the CD8+ T-cell alterations found in HIV-infected patients (113–115), whereas treatment discontinuation causes the increase of CD8+ T-cell activation and dysfunction (116, 117). However, compared with seronegative individuals, cART does not fully reconstitute the CD8+ T-cell counts (88, 118), the proportions of memory subsets, the levels of activation and exhaustion markers (91, 118–121), and their functional capacity (24, 122). In addition, the loss of Tc17 cells and CD161-expressing CD8+ T-cells is not restored in HIV-infected patients under suppressive cART (101, 102, 120). Interestingly, in seronegative individuals, HLA-DR+ CD38+ cells constitute the main IL-17-producing subset among CD8+ T-cells (120), consistent with an effector memory profile of HLA-DR-expressing CD8+ T-cells (123). However, this subset is decreased in HIV-infected patients on cART (120). Remarkably, early initiation of cART is associated with improved reconstitution of CD8+ T-cell counts and activation levels (124, 125), whereas a long treatment is required for improvement of some CD8+ T-cell phenotypic and functional disturbances (120, 123).
It is important to mention that the decrease in antigen load with ART induces a decline in the frequency of circulating HIV-specific CD8+ T-cells (108, 115, 126, 127), with subsequent increases after treatment interruption or failure (108, 127, 128). Changes in the frequencies of circulating CMV-specific CD8+ T-cells are also observed in patients with treatment interruption (108, 115, 127). HIV-specific CD8+ T-cells are maintained in lymph nodes (128) and exert potent antiviral responses in ex vivo assays (129), but are not able to suppress viral replication in the absence of therapy. This issue represents a major challenge in the setting of supervised treatment interruptions strategies in the search of HIV cure strategies (130). Certainly, memory virus-specific CD8+ T-cells respond to the changes in antigen load and the inflammatory milieu during cART, actively migrating within body compartments and possibly modulating their effector response.
Causes of CD8+ T-Cell Dysfunction During HIV Infection
Three factors are major determinants of CD8+ T-cell dysfunction during chronic infections (131): persistent antigen, negative costimulation, and chronic inflammation. In addition, the loss of CD4+ T-cell help enhances CD8+ T-cell dysfunction during HIV infection (132), whereas cell-intrinsic defects may also contribute to this pathogenic process (133). Remarkably, even in the context of cART-induced viral suppression, the three major determinants of CD8+ T-cells dysfunction are present, since there is a residual HIV replication and microbial burden that maintain a persistent antigen load (110, 134, 135), induce continuous expression of inhibitory receptors (131), and the secretion of inflammatory mediators (136–138).
Persistent Antigen and Chronic Inflammation
Chronic immune activation is a hallmark of HIV infection (139), and it is associated with phenotypic and functional changes of immune cell populations (140), impairment of antiviral mechanisms (141), increase in the number of target cells (142), CD4+ T-cell regenerative failure (143, 144), and risk of organ damage (145). HIV-associated immune activation is explained by the persistent viral replication and reactivation of HIV reservoirs, recurrence of co-infections, loss of the integrity of the gut mucosa, and increased systemic levels of pro-inflammatory cytokines [such as IL-6, IL-1β, and tumor necrosis factor (TNF)-α], among other factors (139). Of note, the maintenance of low immune activation levels characterizes non-pathogenic simian immunodeficiency virus (SIV) infection in natural hosts despite sustained viral replication (146). Moreover, the levels of immune activation predict the magnitude of CD4+ T-cell depletion better than viral loads in HIV-infected patients (91, 147), and are associated with disease progression, the development of AIDS-defining and non-AIDS conditions, and mortality (148–150).
Tissue reservoirs (i.e., tissues containing cells with integrated HIV) promote a persistent antigenic burden during HIV infection, even during cART. In a macaque model of SIV infection, the primary reservoir sites of infection were lymphoid tissues (~98% of total RNA+ cells), including lymph nodes, spleen, and gut-associated lymphoid tissues (GALT) (109). Other tissues, such as brain, kidney, heart or lung, individually contributed to <1% of RNA+ cells (109). Similarly HIV tissue reservoirs are predominant in GALT (151), and lymph nodes (152). In lymphoid follicles, CD4+ T-cells, particularly of the CXCR5+ follicular subset, exhibit high levels of infection (153), and free virions are captured by follicular dendritic cells (154). Remarkably, although cART decreases the HIV reservoir size in lymph nodes, HIV RNA and DNA can still be detected after years of therapy (152, 155), whereas there is minimal cART-induced change in HIV DNA in gut tissues (109). Thus, viral reservoirs constitute an important source of persistent antigen, even during cART.
The loss of the integrity of the gut mucosa is also responsible for a high antigenic burden that consequently drives immune dysfunction (156). Mechanistically, this process can be viewed as follows (Figure 2A):
1. Alteration in the gut mucosa: during HIV/SIV infection, it is induced by the decrease of mucosal IL-17/IL-22-producing cells (157, 158), loss of gut junctional complexes (159, 160), changes in the microbiota composition (161), and persistence of HIV reservoirs in GALT (151).
2. Microbial translocation: Gut barrier disruption allows the passage of microbial products from the intestinal lumen to the lamina propria and systemic circulation (162).
3. Activation of immune cells by microbial-associated molecular patterns (MAPS) via pathogen-recognition receptors (PRRs): innate immune cells are activated by microbial components via PRRs, such as Toll-like receptors (TLR) (163, 164). Importantly, in chronic inflammatory settings (165, 166), as well as during HIV infection (167), CD4+, and CD8+ T-cells may upregulate TLR-2, 3, 4, 7, and 9 expression (167, 168), and human T-cells can respond in vitro to several MAPS (168, 169). In the case of CD4+ T-cells, this TLR-mediated activation renders them more susceptible to HIV replication (170) and apoptosis (169), whereas in the case of CD8+ T cells, TLR engagement lowers the activation threshold (171), which can be deleterious in a chronic setting. Thus, T-cell exposure to TLR agonists may directly contribute to increased T-cell activation during chronic HIV infection.
4. Cytokine secretion and immune cells activation and dysfunction: IL-1β, IL-18, IL-6, TNF-α, and type-I IFN induced by PRRs ligation promote the activation of innate and adaptive immune cells (172, 173). Moreover, cytokines such as IL-15 and IL-12 are an important signal for bystander activation of CD8+ T-cells, particularly memory subsets (105).
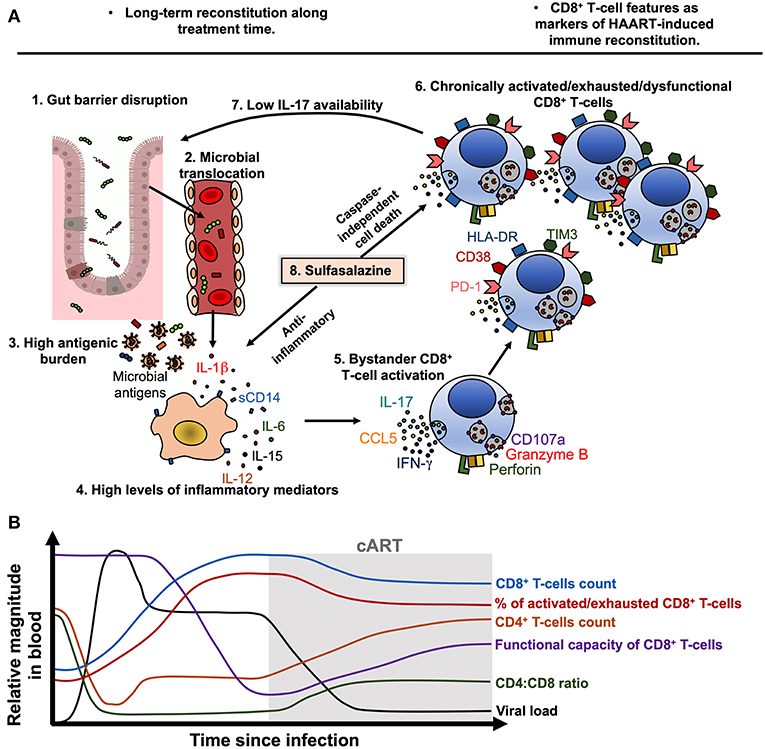
Figure 2. Model of CD8+ T-cell activation, exhaustion and dysfunction during treated HIV infection. (A) During chronic HIV, and despite cART-induced viral suppression, gut barrier disruption (1) causes the passage of microbial products to systemic circulation (microbial translocation, 2), contributing to a high antigenic burden together with residual HIV replication and ongoing co-infections (3). Microbial products and other antigens activate innate immune cells such as monocytes/macrophages, which activate and release soluble CD14 (sCD14) and inflammatory cytokines such as IL-1β, IL-6, IL-15, and IL-12 (4). Particularly, IL-15 and IL-12 induce bystander activation of CD8+ T-cells (5), which in early stages of disease may exhibit potent cytotoxic capacity and cytokine production. Nonetheless, chronic bystander stimulation induces exhaustion and dysfunction of CD8+ T-cells (6), which have an increased expression of HLA-DR, CD38, PD-1 and TIM3, and lower degranulation capacity and IL-17 secretion, among other alterations. The low secretion of IL-17 causes a low availability of this cytokine and its beneficial effects on gut mucosa, worsening the gut barrier disruption (7). In this setting, sulfasalazine could be a therapeutic approach to target the inflammatory environment and activated/dysfunctional CD8+ T-cells, through the inhibition of inflammatory cytokine secretion and induction of caspase-independent cell death (8). (B) Model of the dynamic of viral load, CD4+ T-cells count, CD8+ T-cells count, CD4:CD8 ratio, the frequency of activated/exhausted CD8+ T-cells and their functional capacity in HIV-infected patients before and after ART beginning. Of note, the viral load is efficiently suppressed and the CD4+ T-cells count are recovered after treatment beginning, but the CD8+ T-cells features remain disturbed despite therapy.
Negative Costimulation
As previously mentioned, CD8+ T-cells from HIV-infected patients exhibit increased expression of inhibitory receptors such as PD-1, TIM-3, lymphocyte Activation Gene-3 (LAG-3), CD160, and 2B4 (86, 120, 174). Overall, these receptors interfere with TCR signaling by competing with costimulatory ligands, modulating intracellular pathways, or inducing inhibitory molecules in T-cells; these mechanisms decrease the response of antigen-specific cells to cognate stimulation (175). Expression of inhibitory receptors, particularly PD-1, is also associated with a reduced functional capacity of total CD8+ T-cells after polyclonal stimulation, such as lower degranulation capacity, with consequent decreased release of the cytotoxic molecules granzyme B and perforin (123), and a lower proportion of IL-17-producing cells [(120); Figure 2A]. Moreover, activated/exhausted CD8+ T-cells present higher susceptibility to apoptosis (104, 176, 177), as in the case of Tc17 cells, which are highly activated and exhausted during HIV infection and are susceptible to activation-induced cell death (102, 178). Finally, cell-intrinsic defects, such as an impaired signaling machinery (133), and an altered transcriptional, epigenetic, and metabolic profile may also account for the observed dysfunction in activated/exhausted CD8+ T-cells during HIV infection (24, 179, 180).
Potential Immunomodulatory Strategies for Improving CD8+ T-Cells Immune Reconstitution During Antiretroviral Therapy
cART Intensification
Some studies indicate that the intensification of the cART with HIV integrase inhibitors, such as raltegrevir promotes the normalization of the level of activation of CD8+ T-cells (181, 182). Indeed, according with their mechanism of action, integrase inhibitors decrease the levels of proviral DNA (182–185), impacting in the reservoir size and the consequent antigenic burden and immune activation. The effect of raltegravir intensification of cART is also evidenced by the decrease in the levels of the coagulation marker D-dimer (184).
Anti-inflammatory Agents
Since chronic immune activation and systemic inflammation are important determinants of HIV-associated morbidity and mortality, some anti-inflammatory drugs have been explored in the setting of HIV infection, including acetylsalicylic acid, statins, and hydroxychloroquine [reviewed in (186)]. Among them, the treatment with the statins atorvastatin or rosuvastatin in the presence of cART has shown a reduction in the levels of HLA-DR/CD38 and/or PD-1-expressing CD8+ T-cells compared with placebo controls (187–189). We also explored the effect of sulfasalazine (SSZ) on the reconstitution of CD8+ T-cells functional capacity, focused on IL-17 production (120). Sulfasalazine molecule combines the antibiotic sulphapyridine with the anti-inflammatory drug 5-aminosalicylic acid, and has been used in the treatment of chronic inflammatory diseases (190). Previous reports indicated that SSZ not only inhibits macrophage activation and secretion of TNF-α (191, 192), but also induces apoptosis of activated—and possibly dysfunctional—T-cells (193). Certainly, in vitro analyses with cells derived from HIV-infected patients on cART, showed that SSZ induces caspase-independent cell death of CD8+ T-cells co-expressing HLA-DR and CD38 (120), possibly through the mitochondrio-nuclear translocation of the apoptosis- inducing factor (AIF), that induces DNA fragmentation (193). Moreover, SSZ decreased the levels of LPS-induced IL-1β. These mechanisms were associated with an increase in the proportion of Tc17 cells in HIV-infected patients [(120); Figure 2A]. Interestingly, clinical observations from Colombian health care programs for HIV indicate that SSZ improves the symptoms of HIV-infected patients suffering wasting syndrome. This effect could be related with the abnormal T-cell activation in this group of individuals (194), that is targeted by SSZ. Thus, the use of SSZ and/or other anti-inflammatory drugs could be considered for managing patients with poor response to cART or with advance/progressive disease; large clinical studies that address its usefulness are needed.
Cytokines
Due to the high relevance of Tc17 cells in the context of HIV infection, previous studies have evaluated different strategies to reconstitute this population, as well as mucosa-associated invariant T (MAIT) cells, which are an important IL-17-producing CD161hi CD8+ subset. The administration of IL-7, a member of the γ-common chain family of cytokines, to HIV-infected patients on cART increases the frequency and number of circulating MAIT cells (195). In vitro, IL-7 also promotes the cytotoxic capacity and cytokine production of MAIT cells (196). This approach could be relevant in the clinical setting to limit the deterioration of the gut barrier, most likely preventing microbial co-infections. Another member of the γ-common chain, IL-21, promotes the survival, expansion, and cytotoxic responses of antigen-specific CD8+ T-cells (197–199). Accordingly, the administration of IL-21 to SIV-infected rhesus macaques increased the expression of cytotoxic molecules and polyfunctional CD8+ T-cells, compared with untreated controls (200). A functionally-related cytokine, IL-15, also increased the levels of granzyme B+ and perforin+ CD8+ T-cells after administration (in the form of complex between IL-15 and IL-15 receptor α) in uninfected and SHIV-infected rhesus macaques, both in cells from peripheral blood, lymph nodes, and mucosa (201). Interestingly, IL-15 treatment also promoted the migration of cytotoxic CD8+ T-cells to lymphoid follicles (201), whereas administration of an IL-15 superagonist to SIV-infected macaques induced this effect through the induction of the expression of the follicle-homing chemokine receptor CXCR5 in CD8+ T-cells (202). Thus, γ-common chain cytokines are potentially useful as immunotherapies for promoting CD8+ T-cells function HIV-infected patients, and clinical studies are required to evaluate their effectiveness.
Checkpoint Therapy
Checkpoint therapy (i.e., blockade of the aforementioned inhibitory receptors or their ligands) may constitute an important strategy to improve CD8+ T-cell function in HIV-infected patients on cART. Certainly, in vitro blockade of PD-1 or its ligands PD-L1/L2 promotes HIV or SIV-specific CD8+ T-cells function and proliferative capacity (83, 203). Similarly, evidence in macaque models of SIV infection indicates that anti-PD-1 antibody therapy increases the frequencies of SIV-specific CD8+ T-cells in blood and gut, with improved polyfunctional capacity and proliferative potential; this effect is associated with significant reductions in viral load and increased survival of infected macaques (204). Interestingly, PD-1 blockade also promotes the response of CD8+ T-cells against gut-resident pathogens, as well as contribute to reduce gut epithelial damage, microbial translocation, and immune activation (205). Moreover, the administration of anti-PD-1 antibody to rhesus macaques prior initiation of cART enhanced the antiviral function of CD8+ T-cells, whereas promoted the expansion of CXCR5+ perforin+ granzyme B+ CD8+ T-cells after cART interruption, resulting in a better control of viremia (206). Interestingly, the CXCR5+ subset is the main CD8+ T-cell population responding to PD-1 blockade (207), which would be important in the context of HIV infection due to the role of follicles as tissue reservoirs (discussed below). Another effect of PD-1 axis blockade would be the reversal of HIV latency (208), that could contribute to recognition and elimination of infected cells by functionally improved HIV-specific CD8+ T-cells. In line with this evidence, a phase I clinical trial of an anti-PD-L1 antibody in 8 HIV-infected patients on cART showed that the frequency of Gag-specific CD8+ T-cells expressing IFN-γ or CD107a increased from baseline to day 28 post-treatment, although there was no statistical significance, most likely due to high inter-individual variability. There were no changes in the CD4+ T-cell counts or CD4:CD8 ratio (209). Importantly, an overall safety has been shown for the anti-PD-L1 or anti-PD1 antibody therapy, even in patients with concomitant malignancies (209, 210). Together, these studies indicate that PD-1 axis blockade could improve the antiviral function of CD8+ T-cells during HIV infection, along with other beneficial effects on the levels of microbial translocation, immune activation and viral reservoirs.
CD8+ T-Cells as Correlates of Immune Reconstitution During Antiretroviral Therapy
A large body of evidence indicate that monitoring CD4+ T-cell counts, and viral load is informative of the effectiveness of cART-induced viral suppression in HIV-infected patients. Typically, CD4+ T-cells increase rapidly in the first weeks of treatment, followed by a more gradual increase, depending on the level of nadir CD4+ T-cells or the naïve subpopulation at the time of treatment initiation (211–215). Moreover, HIV-infected patients who maintain viral suppression and had CD4+ T-cells ≥300 cells/μL are unlikely to experience counts <200 cells/μL (threshold for opportunistic infection risk), suggesting that routine CD4+ T-cells monitoring may be unnecessary in some scenarios (216). Certainly, undetectable viral load or CD4+ T-cell counts do not reflect the immunological alterations that are present in treated HIV-infected patients. In contrast, the persistently high CD8+ T-cell counts and low CD4:CD8 ratio are indicative of partial immune reconstitution in treated HIV-infected patients, and, due to their accessibility in low-income settings and predictive power of adverse clinical outcomes (88, 217, 218), could be useful tools for immune monitoring of these individuals. Moreover, in HIV-infected patients under long-term suppressive cART (more than 25 months) there is a recovery in the proportion of CD107a+ and IL-17+ CD8+ T-cells, reaching similar levels to those seen in seronegative individuals (120, 123). These findings suggest a potential usefulness of these CD8+ T-cell functional markers to predict immune reconstitution in HIV-infected patients on cART, in addition to others currently used such as the CD8+ T-cell counts and the CD4:CD8 ratio (88, 217, 218).
In summary, cART-induced immune reconstitution is a long-term and incomplete process. After treatment initiation, viral load is rapidly controlled and CD4+ T-cell counts are progressively recovered. However, CD8+ T-cell counts remain increased despite cART, along with a low CD4:CD8 ratio (Figure 2B). Moreover, the frequency of activated/exhausted CD8+ T-cells do not completely return to basal levels, and some grade of dysfunction remains in several CD8+ T-cell subsets (Figure 2B). The improvement of cART effectiveness may require the combination of strategies such as an early beginning of therapy, rigorous clinical monitoring, the use of reliable biomarkers and possibly the use of immunomodulatory therapies.
CXCR5+ CD8+ T-Cells and Their Role During HIV Infection
Another subpopulation of CD8+ T-cells that is relevant in the setting of HIV infection is that expressing CXCR5, since they could be a potential therapeutic cell-based strategy to eradicate HIV in follicles (219), an important body viral reservoir (153, 220). CXCR5-expressing CD8+ T-cells follow a particular dynamic during HIV/SIV infection. A common observation across studies is the increase in the proportion of CXCR5+ CD8+ T-cells in lymphoid follicles in HIV-infected patients, compared with seronegative controls (70, 221–223). A similar accumulation of follicular CXCR5+ CD8+ T-cells has been observed in SIV-infected macaques (224, 225). This accumulation is associated with the inflammatory/immune activation environment within follicles and increased levels of the CXCR5-ligand CXCL13 in HIV-infected patients, but not with the levels of viral replication or antigens, both during HIV (222) and SIV infection (225). Moreover, CXCR5+ CD8+ T-cells could also migrate within lymph nodes in response to CXCL10, which is ligand of CXCR3, as has been observed in SIV-infected macaques (224). Interestingly, CXCR5+ CD4+ T-cells also accumulate during chronic HIV infection (153, 223, 226), whereas this process is also driven by the local inflammation in SIV-infected macaques (227). Accordingly, increased immune activation and inflammatory cytokines secretion is present in lymph nodes from chronically HIV-infected patients (228). Interestingly, lymphotoxin α and β, and TNF are required for the expression of CXCL13 by murine follicular stromal cells (229), CXCL13 is expressed in chronic inflammation (230), and is a marker of chronic inflammation in HIV-infected patients, even during cART (231). Thus, the antigen burden in lymphoid tissues, both by infected CD4+ T-cells or virion-bearing follicular dendritic cells, may induce an inflammatory environment that in turn promote the production of CXCL13, contributing to the accumulation of CXCR5+ CD8+ T-cells.
Importantly, ART does not decrease the frequency of CXCR5+ CD8+ T-cells in human lymph nodes (222), which is in agreement with the low drug distribution to these tissues and consequent unchanged inflammatory environment, in comparison with blood (110). In addition, the compromise in follicles architecture during HIV infection may also be responsible for the increased passage of CD8+ T-cells and other cell types to these compartments (232).
On the other hand, circulating CXCR5-expressing CD8+ T-cells, which exhibit a transitional memory phenotype, are decreased in untreated HIV-infected patients compared with seronegative controls, but are maintained in patients under suppressive cART (233). Interestingly, the decrease in CXCR5+ CCR7− CD8+ T-cells is associated with the increase in CXCR5− CCR7− cells, which are most likely an effector memory population. Moreover, the frequency of CXCR5hi CCR7−/lo CD8+ T-cells is inversely correlated with the level of systemic HIV replication in untreated HIV-infected patients, particularly in elite controllers (233). Strikingly, while similar correlations between the number of circulating human CXCR5+ CD8+ T-cells and systemic viral load have been reported (221), comparable associations were obtained between the frequencies of lymph node-confined memory CXCR5+ CD8+ T-cells and systemic viral load in HIV controllers patients (53). Thus, immune activation and local inflammation, but not viral replication, are apparently major drivers of CXCR5+ CD8+ T-cells (and CXCR5+ CD4+ T-cells) accumulation in lymphoid follicles during chronic HIV/SIV infection (Figure 3). Once in the lymphoid follicles, antigen-specific CXCR5+ CD8+ T-cells could partially eliminate infected cells, being reflected in lower local and systemic viral load, as has been observed in SIV-infected macaques (234). In fact, a role of this subpopulation in the control of HIV (70, 221) and SIV (225, 235) replication has been proposed, their antiviral function in follicles could be an important mechanism of disease protection, and its frequency could be as useful correlate of limited viral replication. If the systemic viral load, immune activation and/or tissue inflammation decrease, these cells could egress from the follicles, enter systemic circulation and search for other lymphoid follicles or tissues (Figure 3). However, it is yet unclear which other factors may influence the induction of CXCR5 and follicle migration of CD8+ T-cells.
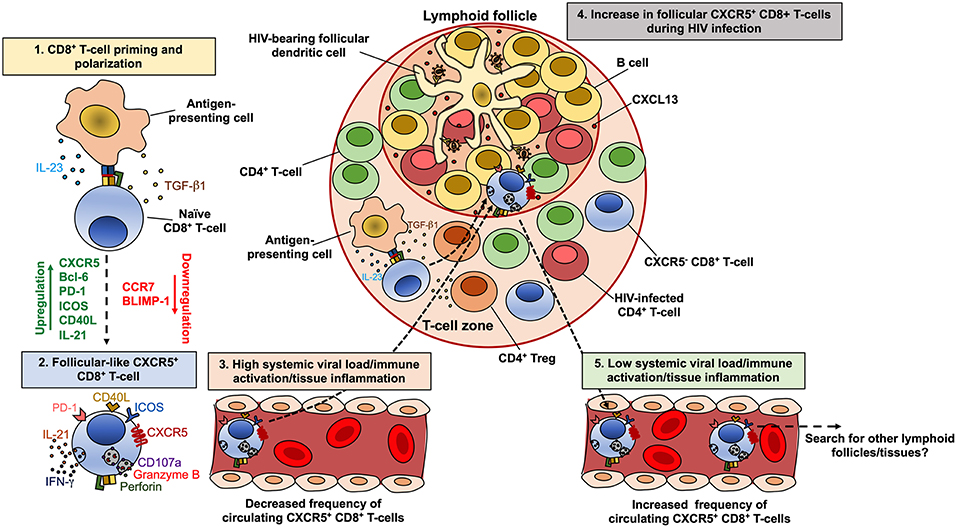
Figure 3. Model of the dynamics of CXCR5+ CD8+ T-cells during HIV infection. CD8+ T-cells are polarized to a follicular-like CXCR5+ profile after priming by an antigen-presenting cell that secrete TGF-β1 and IL-23 (1). These cytokines induce the up-regulation of CXCR5, Bcl-6, PD-1, ICOS, CD40L and IL-21, with down-regulation of CCR7 and BLIMP-1 (2). In circulation or peripheral tissues, follicular-like CXCR5+ CD8+ T-cells also exhibit a cytotoxic potential and production of IFN-γ. During chronic HIV infection and in the setting of high viral load and consequent increased immune activation and tissue inflammation, there is an accumulation of CXCR5+ CD8+ T-cells in lymphoid follicles, in response to CXCL13 (4). The high levels of TGF-β1 produced by antigen-presenting cells or regulatory T cells (Treg), may also induce the differentiation and accumulation of CXCR5+ CD8+ T-cells. Once in the follicles, CXCR5+ CD8+ T-cells exert an antiviral effect, which can be reflected in lower viral replication and the subsequent decrease in the immune activation/tissue inflammation and the egress of this subset from follicles. In circulation, CXCR5+ CD8+ T-cells possibly search for other lymphoid follicles or peripheral tissues (5).
In order to explore the differentiation conditions of human CXCR5+ CD8+ T-cells, we evaluated the effect of TGF-β1, IL-23, and IL-12, which promote the differentiation of human follicular CD4+ T-cells (236) and induce the expression of CXCR5 in non-human primate CD8+ T-cells (234). In vitro, TGF-β1 plus IL-23 (in the presence of TCR stimulation) induced the expression of CXCR5 in purified CD8+ T-cells from healthy individuals, as well as a follicular-like phenotypic and transcriptional profile, such as the up-regulation of PD-1, inducible T-cell costimulatory (ICOS), CD40L and BCL6, and the down-regulation of CCR7 and PRDM1 (Perdomo-Celis F, In press). These data suggest that, in vivo, TGF-β1 and IL-23 may be polarizing factors for follicular-like CXCR5+ CD8+ T-cells, which could then migrate to lymphoid follicles for exerting immune surveillance (Figure 2). Considering the increased levels of regulatory T cells (Treg)-derived TGF-β1 in lymphoid tissues during HIV infection (237), there could be an appropriate environment for the differentiation and accumulation of follicular-like CXCR5+ CD8+ T-cells (Figure 3). Importantly, the induction of the expression of CXCR5 in CD8+ T-cells through cytokine stimulation or genetic engineering may be a useful strategy for improving the migration of these cells to lymphoid follicles and boost follicular antiviral responses that help to eradicate viral reservoirs in these structures (202, 238, 239).
CD8+ T-Cell-Based Strategies To Treat Or Cure HIV Infection
Current strategies for CD8+ T-cell-based strategies to treat or cure HIV infection are based on stimulating pre-existing and/or inducing de novo HIV-specific immune responses through vaccine therapies or redirecting HIV-specific CD8+ T-cells and improving their function and persistence after adoptive transfer. The latter adoptive transfer strategies can be divided in (i) Expansion of HIV-specific CD8+ T-cells; (ii) Artificial CD8+ TCR; (iii) Chimeric antigen receptors (CAR); (iv) Selection of CD8+ T-cells with long-term persistence; (v) Induction of HIV-specific CD8+ T-cells from virus-naïve donor cells. Of note, these strategies may also be accompanied by the aforementioned immunomodulatory approaches, in order to reach a global reconstitution of the CD8+ T-cell response, as well as with latency-reversing agents, to induce the reactivation of viral reservoir (240), particularly in the case of patients receiving cART.
CD8+ T-Cell-Based Vaccines
Several HIV vaccine candidates have been focused on the induction of CD8+ T-cell responses in order to control viremia, i.e., as a therapeutic approach. They range from whole attenuated virus, vector viruses containing HIV proteins, DNA plasmids, and HIV peptides [reviewed in (241)]. Importantly, immunization should be performed with multiple selected peptides based on conserved regions of HIV proteins, which induce subdominant but effective CD8+ T-cell responses, and are restricted by the most common HLA alleles in a specific population (242, 243). Nonetheless, in general, a low and transient reduction of viral load has been observed with most therapeutic vaccines (241), indicating that a combination of this and other approaches is required for a relevant impact on disease progression.
HIV-Specific CD8+ T-Cells Adoptive Transfer
First reports including small cohorts of HIV-infected patients (some of them receiving cART) demonstrated that the infusion of autologous CD8+ T-cells enriched for HIV-specific cells targeting several viral proteins (expanded ex vivo through polyclonal stimulation), transiently decreased plasma viremia or productively infected cells, and increased CD4+ T-cell counts (244, 245). Nonetheless, in other studies, a rapid apoptosis of adoptively transferred HIV-specific CD8+ T-cells was associated with failure in the reduction of viral load (246). Moreover, transfer of HIV-specific CD8+ T-cells induced selection of escape mutant HIV variants, which was associated with rise in viral load and decrease of CD4+ T-cells (247). More recently, other approaches based on infusion of CD8+ T-cells specific for multiple HIV antigens (248) demonstrated efficacy in the control of autologous reservoir virus (249). Similarly, CD8+ T-cells transduced with vectors expressing a TCR targeting the relevant HIV epitope SL9 in Gag protein, showed lysis capacity and reduced infectious cells in vivo in SCID mice (250), as well as functional capacity in cells derived from cART-receiving patients (251). However, engineered TCR may have recognition of self-epitopes, resulting in severe reactions such as cardiac toxicity (252), which limits the use of this approach. The use of CAR CD8+ T-cells may overcome the problems of viral escape, HIV-induced HLA downregulation, and immune exhaustion (253). First-generation CAR T-cells containing the extracellular domain of human CD4 linked to the CD3ζ chain (CD4ζ) showed prolonged survival and trafficking to gut mucosa, although did not change viral load in HIV-infected patients (254). More recently, the CD4ζ CAR was re-engineered to express the 4-1BB costimulatory domain, inducing a more potent HIV suppressing capacity in vitro and in vivo in a humanized mouse model (255). CAR T-cells have also been designed with expression of single-chain variable fragments of broadly neutralizing antibodies (256), or carbohydrate-recognition domain of C-type lectin, both targeting gp120 (257). Another approach to improve the survival of adoptively transferred HIV-specific CD8+ T-cells is to select those with a long-term memory profile. Accordingly, ex vivo expanded HIV-specific CD28+ CD8+ T-cells survived for at least 84 days after infusion in cART-receiving patients and maintained central memory characteristics (258). Finally, functional HIV-specific CD8+ T-cells can be generated from HIV-negative donors through priming with peptide-pulsed dendritic cells, followed by polyclonal expansion. This approach could be useful in the setting of hematopoietic stem cell transplantation in HIV-infected patients to recover or improve the virus-specific CD8+ T-cell response (259).
In summary, these studies demonstrate the potential of CD8+ T-cell-based therapeutic strategies for HIV treatment or cure strategies. More importantly, combination of different strategies, such as vaccine induction of virus-specific CD8+ T-cells and subsequent adoptive transfer, could limit viral replication and/or rebound after cART interruption, as recently evidenced in a macaque model (260).
Conclusions and Future Perspectives
Despite intense research on HIV, there is no preventing vaccine and cure available for this infection. In fact, a high incidence is still reported worldwide, underlying the importance of providing an optimal cART and health care management to HIV-infected patients, not only for improving their quality of life and preventing several co-morbidities, but also for breaking the chain of transmission. Thus, the characterization of HIV-infected patients under suppressive cART is required to understand the benefits, pitfalls, and challenges of cART, to identify novel immunomodulatory targets, and to elucidate potential strategies for reaching a functional or sterilizing cure for this infection. Despite complete suppression of viral load and reconstitution of CD4+ T-cells, HIV-infected patients exhibit increased systemic inflammation levels and a compromised CD8+ T-cells response, characterized by high activation/exhaustion and impaired lytic and non-lytic mechanisms. Some of the phenotypic and/or functional CD8+ T-cell features could be used in the clinical setting to identify those patients reaching immune reconstitution during continuous suppressive cART. Some of the CD8+ T-cell alterations, such as low IL-17 production, could be targeted by immunomodulatory drugs. Finally, follicular CXCR5+ CD8+ T-cells could be a therapeutic weapon for targeting viral reservoirs in lymphoid follicles, along with other CD8+ T-cell-based strategies.
Author Contributions
FP-C, NT, and MR reviewed the literature and wrote the manuscript.
Funding
This study was supported by Universidad de Antioquia, UdeA, COLCIENCIAS (Codes: 111571249724 and 111577757051, to MR) (http://www.colciencias.gov.co) and Corporación Universitaria Remington—Uniremington (Codes: 4000000063-16 and 4000000121-17 to NT) (http://www.uniremington.edu.co). FP-C was supported by Universidad de Antioquia, Corporacion Universitaria Remington, and Fundacion Sapiencia in Medellin.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Juan C. Zapata for critically reviewing this manuscript. Thanks to Angela Betancur for her administrative support.
References
1. WHO Guidelines Approved by the Guidelines Review Committee. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection: Recommendations for a Public Health Approach. Geneva: World Health OrganizationCopyright (c) World Health Organization (2013).
2. Levi J, Raymond A, Pozniak A, Vernazza P, Kohler P, Hill A. Can the UNAIDS 90-90-90 target be achieved? A systematic analysis of national HIV treatment cascades. BMJ Glob Heal. (2016) 1:e000010. doi: 10.1136/bmjgh-2015-000010
3. Saag MS, Benson CA, Gandhi RT, Hoy JF, Landovitz RJ, Mugavero MJ, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the International Antiviral Society-USA Panel. JAMA. (2018) 320:379–96. doi: 10.1001/jama.2018.8431
4. Ministerio de Salud y Protección Social Fondo de Población de las Naciones Unidas - UNFPA. Guía de Práctica Clínica Basada en la Evidencia Científica Para la Atención de la Infección por VIH/Sida en Adolescentes (con 13 años o más de edad) y Adultos. (2014)
5. Bigna JJR, Plottel CS, Koulla-Shiro S. Challenges in initiating antiretroviral therapy for all HIV-infected people regardless of CD4 cell count. Infect Dis Poverty. (2016) 5:85. doi: 10.1186/s40249-016-0179-9
6. DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents With HIV. Washington, DC: Department of Health and Human Services (2018).
7. Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. (2012) 72:e1–25. doi: 10.2165/11633630-000000000-00000
8. Aldous JL, Haubrich RH. Defining treatment failure in resource-rich settings. Curr Opin HIV AIDS. (2009) 4:459–66. doi: 10.1097/COH.0b013e328331dea5
9. Wilson EMP, Sereti I. Immune restoration after antiretroviral therapy: the pitfalls of hasty or incomplete repairs. Immunol Rev. (2013) 254:343–54. doi: 10.1111/imr.12064
10. Croxford S, Kitching A, Desai S, Kall M, Edelstein M, Skingsley A, et al. Mortality and causes of death in people diagnosed with HIV in the era of highly active antiretroviral therapy compared with the general population: an analysis of a national observational cohort. Lancet Public Heal. (2017) 2:e35–46. doi: 10.1016/S2468-2667(16)30020-2
11. Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996-2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis. (2010) 50:1387–1396. doi: 10.1086/652283
12. Hughes CA, Tseng A, Cooper R. Managing drug interactions in HIV-infected adults with comorbid illness. CMAJ. (2015) 187:36–43. doi: 10.1503/cmaj.131626
13. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. (2011) 35:161–8. doi: 10.1016/j.immuni.2011.07.010
14. Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. (2003) 171:5165–71. doi: 10.4049/jimmunol.171.10.5165
15. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Cutting edge: type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. (2005) 174:4465–9. doi: 10.4049/jimmunol.174.8.4465
16. Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. (2008) 28:710–22. doi: 10.1016/j.immuni.2008.02.020
17. Hamann D, Baars PA, Rep MHG, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. (1997) 186:1407–18. doi: 10.1016/S0165-2478(97)85793-8
18. Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med. (2004) 199:925–36. doi: 10.1084/jem.20031799
19. Bachmann MF, Barner M, Viola A, Kopf M. Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur J Immunol. (1999) 29:291–9. doi: 10.1002/(SICI)1521-4141(199901)29:01<291::AID-IMMU291>;3.0.CO;2-K
20. Weninger W, Manjunath N, Von Andrian UH. Migration and differentiation of CD8+ T cells. Immunol Rev. (2002) 186:221–3. doi: 10.1034/j.1600-065X.2002.18618.x
21. Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. (2004) 78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
22. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature. (2017) 552:362–367. doi: 10.1038/nature24633
23. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. (2012) 338:1220–5. doi: 10.1126/science.1229620
24. Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-bet and eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog. (2014) 10:e1004251. doi: 10.1371/journal.ppat.1004251
25. McLane LM, Banerjee PP, Cosma GL, Makedonas G, Wherry EJ, Orange JS, et al. Differential localization of T-bet and eomes in CD8 T cell memory populations. J Immunol. (2013) 190:3207–15. doi: 10.4049/jimmunol.1201556
26. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. (2009) 206:51 LP−59. doi: 10.1084/jem.20081242
27. van der Veeken J, Zhong Y, Sharma R, Mazutis L, Dao P, Pe'er D, et al. Natural genetic variation reveals key features of epigenetic and transcriptional memory in virus-specific CD8 T cells. Immunity. (2019) 50:1202–17.e7. doi: 10.1016/j.immuni.2019.03.031
28. Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. (2003) 281:65–78. doi: 10.1016/S0022-1759(03)00265-5
29. Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol. (2004) 75:497–512. doi: 10.1016/S0091-679X(04)75020-7
30. Peters PJ. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. (1991) 173:1099–109. doi: 10.1084/jem.173.5.1099
31. Fukuda M. Lysosomal membrane glycoproteins: structure, biosynthesis, and intracellular trafficking. J Biol Chem. (1991) 266:21327–30.
32. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. (1998) 282:121–5. doi: 10.1126/science.282.5386.121
33. Metkar SS, Wang B, Aguilar-Santelises M, Raja SM, Uhlin-Hansen L, Podack E, et al. Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme b-serglycin complexes into target cells without plasma membrane pore formation. Immunity. (2002) doi: 10.1016/S1074-7613(02)00286-8
34. Dupuis M, Schaerer E, Krause KH, Tschopp J. The calcium-binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J Exp Med. (1993) 16:417–28. doi: 10.1084/jem.177.1.1
35. Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med. (2002) 196:493–503. doi: 10.1084/jem.20011836
36. Bossi G, Griffiths GM. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat Med. (1999) 5:90–6. doi: 10.1038/4779
37. Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. (2003) 3:361–70. doi: 10.1038/nri1083
38. Shiver JW, Henkart PA. A noncytotoxic mast cell tumor line exhibits potent IgE-dependent cytotoxicity after transfection with the cytolysin/perforin gene. Cell. (1991) 64:1175–81. doi: 10.1016/0092-8674(91)90272-Z
39. Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RHP, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood. (2013) doi: 10.1182/blood-2012-07-446146
40. Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, et al. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. (2000) 103:491–500. doi: 10.1016/S0092-8674(00)00140-9
41. Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC. Granzyme B: a natural born killer. Immunol Rev. (2003) 193:31–8. doi: 10.1034/j.1600-065X.2003.00044.x
42. Barry M, Heibein JA, Pinkoski MJ, Lee SF, Moyer RW, Green DR, et al. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol Cell Biol. (2000) 20:3781–94. doi: 10.1128/MCB.20.11.3781-3794.2000
43. Atkinson EA, Barry M, Darmon AJ, Shostak I, Turner PC, Moyer RW, et al. Cytotoxic T lymphocyte-assisted suicide: caspase 3 activation is primarily the result of the direct action of granzyme B. J Biol Chem. (1998) 273:21261–6. doi: 10.1074/jbc.273.33.21261
44. Zou H, Li Y, Liu X, Wang X. An APAf-1. cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. (1999) 274:11549–56. doi: 10.1074/jbc.274.17.11549
45. Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. (2001) 412:95–9. doi: 10.1038/35083620
46. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
47. Gulzar N, Copeland K. CD8+ T-cells: function and response to HIV infection. Curr HIV Res. (2004) 2:23–37. doi: 10.2174/1570162043485077
48. Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, et al. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. (1996) 85:1135–48. doi: 10.1016/S0092-8674(00)81313-6
49. Saunders KO, Ward-Caviness C, Schutte RJ, Freel SA, Overman RG, Thielman NM, et al. Secretion of MIP-1β and MIP-1α by CD8+ T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell Immunol. (2011) 266:154–64. doi: 10.1016/j.cellimm.2010.09.011
50. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science. (1995) 27:1811–5. doi: 10.1126/science.270.5243.1811
51. Romero P, Zippelius A, Kurth I, Pittet MJ, Touvrey C, Iancu EM, et al. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J Immunol. (2007) 178:4112–9. doi: 10.4049/jimmunol.178.7.4112
52. Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. (2017) 214:1593–606 doi: 10.1084/jem.20161760
53. Reuter MA, Del Rio Estrada PM, Buggert M, Petrovas C, Ferrando-Martinez S, Nguyen S, et al. HIV-specific CD8+ T cells exhibit reduced and differentially regulated cytolytic activity in lymphoid tissue. Cell Rep. (2017) 21:3458–70. doi: 10.1016/j.celrep.2017.11.075
54. Kiniry BE, Ganesh A, Critchfield JW, Hunt PW, Hecht FM, Somsouk M, et al. Predominance of weakly cytotoxic, T-bet(Low)Eomes(Neg) CD8(+) T-cells in human gastrointestinal mucosa: implications for HIV infection. Mucosal Immunol. (2017) 10:1008–20. doi: 10.1038/mi.2016.100
55. Kiniry BE, Hunt PW, Hecht FM, Somsouk M, Deeks SG, Shacklett BL. Differential expression of CD8(+) T cell cytotoxic effector molecules in blood and gastrointestinal mucosa in HIV-1 infection. J Immunol. (2018) 200:1876–88. doi: 10.4049/jimmunol.1701532
56. Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, et al. Identification and characterization of HIV-specific resident memory CD8(+) T cells in human lymphoid tissue. Sci Immunol. (2018) 3:eaar4526. doi: 10.1126/sciimmunol.aar4526
57. Mittrücker HW, Visekruna A, Huber M. Heterogeneity in the differentiation and function of CD8+ T cells. Arch Immunol Ther Exp. (2014) 62:449–58. doi: 10.1007/s00005-014-0293-y
58. Yu Y, Ma X, Gong R, Zhu J, Wei L, Yao J. Recent advances in CD8(+) regulatory T cell research. Oncol Lett. (2018) 15:8187–94. doi: 10.3892/ol.2018.8378
59. Srenathan U, Steel K, Taams LS. IL-17+ CD8+ T cells: differentiation, phenotype and role in inflammatory disease. Immunol Lett. (2016) 178:20–6. doi: 10.1016/j.imlet.2016.05.001
60. Billerbeck E, Kang Y-H, Walker L, Lockstone H, Grafmueller S, Fleming V, et al. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci USA. (2010) 107:3006–11. doi: 10.1073/pnas.0914839107
61. Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, et al. Th17-like developmental process leads to CD8+Tc17 cells with reduced cytotoxic activity. Eur J Immunol. (2009) 39:1716–25. doi: 10.1002/eji.200939412
62. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol. (2010) 40:2174–81. doi: 10.1002/eji.200940257
63. Kondo T, Takata H, Matsuki F, Takiguchi M. Cutting edge: phenotypic characterization and differentiation of human CD8+ T cells producing IL-17. J Immunol. (2009) 182:1794–8. doi: 10.4049/jimmunol.0801347
64. Fergusson JR, Hühn MH, Swadling L, Walker LJ, Kurioka A, Llibre A, et al. CD161int CD8+ T cells: a novel population of highly functional, memory CD8+ T cells enriched within the gut. Mucosal Immunol. (2016) 9:401–13. doi: 10.1038/mi.2015.69
65. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. (2011) 34:149–62. doi: 10.1016/j.immuni.2011.02.012
66. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
67. Lee JS, Tato CM, Joyce-Shaikh B, Gulan F, Cayatte C, Chen Y, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
68. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
69. Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol. (2003) 170:4665–72. doi: 10.4049/jimmunol.170.9.4665
70. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. (2016) 17:1187–96. doi: 10.1038/ni.3543
71. McBrien JB, Kumar NA, Silvestri G. Mechanisms of CD8+ T cell-mediated suppression of HIV/SIV replication. Eur J Immunol. (2018) 48:898–914. doi: 10.1002/eji.201747172
72. Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. (1994) 68:6103–10.
73. Takata H, Buranapraditkun S, Kessing C, Fletcher JLK, Muir R, Tardif V, et al. Delayed differentiation of potent effector CD8+ T cells reducing viremia and reservoir seeding in acute HIV infection. Sci Transl Med. (2017) 9:eaag1809. doi: 10.1126/scitranslmed.aag1809
74. Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. (1999) 189:991–8. doi: 10.1084/jem.189.6.991
75. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. (1999) 283:857–60.
76. Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci USA. (2007) 104:6776–81. doi: 10.1073/pnas.0611244104
77. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. (2006) 107:4781–89. doi: 10.1182/blood-2005-12-4818
78. Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. (1997) 3:205–11. doi: 10.1038/nm0297-205
79. Cartwright EK, Spicer L, Smith SA, Lee D, Fast R, Paganini S, et al. CD8+ lymphocytes are required for maintaining viral suppression in SIV-infected macaques treated with short-term antiretroviral therapy. Immunity. (2016) 45:656–68. doi: 10.1016/j.immuni.2016.08.018
80. Makedonas G, Betts MR. Living in a house of cards: re-evaluating CD8+ T-cell immune correlates against HIV. Immunol Rev. (2011) 239:109–24. doi: 10.1111/j.1600-065X.2010.00968.x
81. Roberts ER, Carnathan DG, Li H, Shaw GM, Silvestri G, Betts MR. Collapse of cytolytic potential in SIV-specific CD8+ T cells following acute siv infection in rhesus macaques. PLoS Pathog. (2016) 12:e1006135. doi: 10.1371/journal.ppat.1006135
82. Demers KR, Makedonas G, Buggert M, Eller MA, Ratcliffe SJ, Goonetilleke N, et al. Temporal dynamics of CD8+ T cell effector responses during primary HIV infection. PLoS Pathog. (2016) 12:e1005805. doi: 10.1371/journal.ppat.1005805
83. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
84. Appay V, Papagno L, Spina CA, Hansasuta P, King A, Jones L, et al. Dynamics of T cell responses in HIV infection. J Immunol. (2002) 168:3660–6. doi: 10.4049/jimmunol.168.7.3660
85. Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. (2001) 410:106–11. doi: 10.1038/35065118
86. Hoffmann M, Pantazis N, Martin GE, Hickling S, Hurst J, Meyerowitz J, et al. Exhaustion of activated CD8 T cells predicts disease progression in primary HIV-1 infection. PLoS Pathog. (2016) 12:1–19. doi: 10.1371/journal.ppat.1005661
87. Papagno L, Spina CA, Marchant A, Salio M, Rufer N, Little S, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. (2004) 2:e20. doi: 10.1371/journal.pbio.0020020
88. Helleberg M, Kronborg G, Ullum H, Ryder LP, Obel N, Gerstoft J. Course and clinical significance of CD8+ T-cell counts in a large cohort of HIV-infected individuals. J Infect Dis. (2015) 211:1726–34. doi: 10.1093/infdis/jiu669
89. Roederer M, Dubs JG, Anderson MT, Raju PA, Herzenberg LA, Herzenberg LA. CD8 naive T cell counts decrease progressively in HIV-infected adults. J Clin Invest. (1995) 95:2061–6. doi: 10.1172/JCI117892
90. Chen G, Shankar P, Lange C, Valdez H, Skolnik PR, Wu L, et al. CD8 T cells specific for human immunodeficiency virus, Epstein-Barr virus, and cytomegalovirus lack molecules for homing to lymphoid sites of infection. Blood. (2001) 98:156–64. doi: 10.1182/blood.V98.1.156
91. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. (2003) 187:1534–43. doi: 10.1086/374786
92. Kuchroo VK, Anderson AC, Petrovas C. Coinhibitory receptors and CD8 T cell exhaustion in chronic infections. Curr Opin HIV AIDS. (2014) 9:439–45. doi: 10.1097/COH.0000000000000088
93. Lindgren T, Ahlm C, Mohamed N, Evander M, Ljunggren H-G, Bjorkstrom NK. Longitudinal analysis of the human T cell response during acute hantavirus infection. J Virol. (2011) 85:10252–60. doi: 10.1128/JVI.05548-11
94. McElroy AK, Akondy RS, Harmon JR, Ellebedy AH, Cannon D, Klena JD, et al. A case of human lassa virus infection with robust acute T-cell activation and long-term virus-specific T-cell responses. J Infect Dis. (2017) 215:1862–72. doi: 10.1093/infdis/jix201
95. Chandele A, Sewatanon J, Gunisetty S, Singla M, Onlamoon N, Akondy RS, et al. Characterization of human CD8 T cell responses in dengue virus-infected patients from India. J Virol. (2016) 90:11259–78. doi: 10.1128/JVI.01424-16
96. Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. (2009) 183:7919–30. doi: 10.4049/jimmunol.0803903
97. Peretz Y, He Z, Shi Y, Yassine-Diab B, Goulet JP, Bordi R, et al. CD160 and PD-1 co-expression on HIV-specific CD8 T cells defines a subset with advanced dysfunction. PLoS Pathog. (2012) 8:e1002840. doi: 10.1371/journal.ppat.1002840
98. Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, et al. Loss of HIV-1–specific CD8 + T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1–specific CD4 + T cells. J Exp Med. (2004) 200:701–12. doi: 10.1084/jem.20041270
99. Day CL, Kiepiela P, Leslie AJ, van der Stok M, Nair K, Ismail N, et al. Proliferative capacity of epitope-specific CD8 T-cell responses is inversely related to viral load in chronic human immunodeficiency virus type 1 infection. J Virol. (2007) 81:434–8. doi: 10.1128/JVI.01754-06
100. Yan J, Sabbaj S, Bansal A, Amatya N, Shacka JJ, Goepfert PA, et al. HIV-specific CD8+ T cells from elite controllers are primed for survival. J Virol. (2013) 87:5170–81. doi: 10.1128/JVI.02379-12
101. Gaardbo JC, Hartling HJ, Thorsteinsson K, Ullum H, Nielsen SD. CD3+CD8+CD161high Tc17 cells are depleted in HIV-infection. AIDS. (2013) 27:659–62. doi: 10.1097/QAD.0b013e32835b8cb3
102. Cosgrove C, Ussher JE, Rauch A, Gärtner K, Kurioka A, Hühn MH, et al. Early and nonreversible decrease of CD161++/MAIT cells in HIV infection. Blood. (2013) 121:951–61. doi: 10.1182/blood-2012-06-436436
103. Nigam P, Kwa S, Velu V, Amara RR. Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection. J Immunol. (2011) 186:745–53. doi: 10.4049/jimmunol.1002807
104. Hua S, Lécuroux C, Sáez-Cirión A, Pancino G, Girault I, Versmisse P, et al. Potential role for HIV-specific CD38-/HLA-DR+ CD8+ T cells in viral suppression and cytotoxicity in HIV controllers. PLoS ONE. (2014) 9:2–11. doi: 10.1186/1471-2334-14-S2-P64
105. Bastidas S, Graw F, Smith MZ, Kuster H, Gunthard HF, Oxenius A. CD8+ T cells are activated in an antigen-independent manner in HIV-infected individuals. J Immunol. (2014) 192:1732–44. doi: 10.4049/jimmunol.1302027
106. Doisne J-M, Urrutia A, Lacabaratz-Porret C, Goujard C, Meyer L, Chaix M-L, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. (2004) 173:2410–8. doi: 10.4049/jimmunol.173.4.2410
107. Betts MR, Ambrozak DR, Douek DC, Bonhoeffer S, Brenchley JM, Casazza JP, et al. Analysis of total human immunodeficiency virus (HIV)-specific CD4+ and CD8+ T-cell responses: relationship to viral load in untreated HIV infection. J Virol. (2001) 75:11983–91. doi: 10.1128/JVI.75.24.11983-11991.2001
108. Casazza JP, Betts MR, Picker LJ, Koup RA. Decay kinetics of human immunodeficiency virus-specific CD8? T cells in peripheral blood after initiation of highly active antiretroviral therapy. J Virol. (2001) 75:6508–16. doi: 10.1128/JVI.75.14.6508-6516.2001
109. Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. (2017) 23:1271–6. doi: 10.1038/nm.4411
110. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci USA. (2014) 111:2307–12. doi: 10.1073/pnas.1318249111
111. Ferre AL, Hunt PW, Critchfield JW, Young DH, Morris MM, Garcia JC, et al. Mucosal immune responses to HIV-1 in elite controllers: a potential correlate of immune control. Blood. (2009) 113:3978–89. doi: 10.1182/blood-2008-10-182709
112. Critchfield JW, Young DH, Hayes TL, Braun JV, Garcia JC, Pollard RB, et al. Magnitude and complexity of rectal mucosa HIV-1-specific CD8+ T-cell responses during chronic infection reflect clinical status. PLoS ONE. (2008) 3:e3577. doi: 10.1371/journal.pone.0003577
113. Rehr M, Cahenzli J, Haas A, Price DA, Gostick E, Huber M, et al. Emergence of polyfunctional CD8+ T cells after prolonged suppression of human immunodeficiency virus replication by antiretroviral therapy. J Virol. (2008) 82:3391–404. doi: 10.1128/JVI.02383-07
114. Streeck H, Brumme ZL, Anastario M, Cohen KW, Jolin JS, Meier A, et al. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. (2008) 5:e100. doi: 10.1371/journal.pmed.0050100
115. Rinaldo CRJ, Huang XL, Fan Z, Margolick JB, Borowski L, Hoji A, et al. Anti-human immunodeficiency virus type 1 (HIV-1) CD8(+) T-lymphocyte reactivity during combination antiretroviral therapy in HIV-1-infected patients with advanced immunodeficiency. J Virol. (2000) 74:4127–38. doi: 10.1128/JVI.74.9.4127-4138.2000
116. Garcia F, Plana M, Vidal C, Cruceta A, O'Brien WA, Pantaleo G, et al. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS. (1999) 13:F79–86. doi: 10.1097/00002030-199907300-00002
117. Serwanga J, Mugaba S, Betty A, Pimego E, Walker S, Munderi P, et al. CD8 T-cell responses before and after structured treatment interruption in ugandan adults who initiated ART with CD4 T cells <200 cell/muL: the DART trial STI substudy. AIDS Res Treat. (2011) 2011:875028. doi: 10.1155/2011/875028
118. Tanko RF, Soares AP, Masson L, Garrett NJ, Samsunder N, Abdool Karim Q, et al. Residual T cell activation and skewed CD8+ T cell memory differentiation despite antiretroviral therapy-induced HIV suppression. Clin Immunol. (2018) 195:127–38. doi: 10.1016/j.clim.2018.06.001
119. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. (2006) 12:1198–202. doi: 10.1038/nm1106-1329b
120. Perdomo-Celis F, Feria MG, Taborda NA, Rugeles MT. A low frequency of IL-17-producing CD8+ T-cells is associated with persistent immune activation in people living with HIV despite HAART-induced viral suppression. Front Immunol. (2018) 9:2502. doi: 10.3389/fimmu.2018.02502
121. Migueles SA, Weeks KA, Nou E, Berkley AM, Rood JE, Osborne CM, et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J Virol. (2009) 83:11876–89. doi: 10.1128/JVI.01153-09
122. Kelleher AD, Sewell WA, Cooper DA. Effect of protease therapy on cytokine secretion by peripheral blood mononuclear cells (PBMC) from HIV-infected subjects. Clin Exp Immunol. (1999) 115:147–52. doi: 10.1046/j.1365-2249.1999.00761.x
123. Perdomo-Celis F, Velilla PA, Taborda NA, Rugeles MT. An altered cytotoxic program of CD8+ T-cells in HIV-infected patients despite HAART-induced viral suppression. PLoS ONE. (2019) 14:e0210540. doi: 10.1371/journal.pone.0210540
124. Cao W, Mehraj V, Trottier B, Baril JG, Leblanc R, Lebouche B, et al. Early initiation rather than prolonged duration of antiretroviral therapy in hiv infection contributes to the normalization of CD8 T-cell counts. Clin Infect Dis. (2016) 62:250–7. doi: 10.1093/cid/civ809
125. Jenabian MA, El-Far M, Vyboh K, Kema I, Costiniuk CT, Thomas R, et al. Immunosuppressive tryptophan catabolism and gut mucosal dysfunction following early HIV infection. J Infect Dis. 212:355–66. doi: 10.1093/infdis/jiv037
126. Ogg GS, Jin X, Bonhoeffer S, Moss P, Nowak MA, Monard S, et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J Virol. (1999) 73:797–800.
127. Mollet L, Li TS, Samri A, Tournay C, Tubiana R, Calvez V, et al. Dynamics of HIV-specific CD8+ T lymphocytes with changes in viral load.The RESTIM and COMET Study Groups. J Immunol. (2000) 165:1692–704. doi: 10.4049/jimmunol.165.3.1692
128. Altfeld M, Van Lunzen J, Frahm N, Yu XG, Schneider C, Eldridge RL, et al. Expansion of pre-existing, lymph node-localized CD8+ T cells during supervised treatment interruptions in chronic HIV-1 infection. J Clin Invest. (2002) 109:837–43. doi: 10.1172/JCI14789
129. Killian MS, Roop J, Ng S, Hecht FM, Levy JA. CD8+ cell anti-HIV activity rapidly increases upon discontinuation of early antiretroviral therapy. J Clin Immunol. (2009) 29:311–8. doi: 10.1007/s10875-009-9275-y
130. Kaufmann DE, Lichterfeld M, Altfeld M, Addo MM, Johnston MN, Lee PK, et al. Limited durability of viral control following treated acute HIV infection. PLoS Med. (2004) 1:e36. doi: 10.1371/journal.pmed.0010036
131. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
132. Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol. (2011) 85:733–41. doi: 10.1128/JVI.02030-10
133. Schweneker M, Favre D, Martin JN, Deeks SG, Joseph M, McCune JM. HIV-induced changes in T cell signaling pathways. J Immunol. (2008) 180:6490–500. doi: 10.4049/jimmunol.180.10.6490
134. Dornadula G, Zhang H, VanUitert B, Stern J, Livornese LJ, Ingerman MJ, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. (1999) 282:1627–32. doi: 10.1001/jama.282.17.1627
135. Ouedraogo DE, Makinson A, Vendrell JP, Casanova ML, Nagot N, Cezar R, et al. Pivotal role of HIV and EBV replication in the long-term persistence of monoclonal gammopathy in patients on antiretroviral therapy. Blood. (2013) 122:3030–3. doi: 10.1182/blood-2012-12-470393
136. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. (2008) 5:e203. doi: 10.1371/journal.pmed.0050203
137. Baker JV, Neuhaus J, Duprez D, Kuller LH, Tracy R, Belloso WH, et al. Changes in inflammatory and coagulation biomarkers: a randomized comparison of immediate versus deferred antiretroviral therapy in patients with HIV infection. J Acquir Immune Defic Syndr. (2011) 56:36–43. doi: 10.1097/QAI.0b013e3181f7f61a
138. Neuhaus J, Jacobs DR Jr, Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. (2010) 201:1788–95. doi: 10.1086/652749
139. Paiardini M, Müller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev. (2013) 254:78–101. doi: 10.1111/imr.12079
140. Kestens L, Vanham G, Gigase P, Young G, Hannet I, Vanlangendonck F, et al. Expression of activation antigens, HLA-DR and CD38, on CD8 lymphocytes during HIV-1 infection. AIDS. (1992) 6:793–7. doi: 10.1097/00002030-199208000-00004
141. Boasso A, Shearer GM, Chougnet C. Immune dysregulation in human immunodeficiency virus infection: know it, fix it, prevent it? J Intern Med. (2009) 265:78–96. doi: 10.1111/j.1365-2796.2008.02043.x
142. Biancotto A, Iglehart SJ, Vanpouille C, Condack CE, Lisco A, Ruecker E, et al. HIV-1-induced activation of CD4+T cells creates new targets for HIV-1 infection in human lymphoid tissue ex vivo. Blood. (2008) 111:699–704. doi: 10.1182/blood-2007-05-088435
143. Schacker TW, Brenchley JM, Beilman GJ, Reilly C, Pambuccian SE, Taylor J, et al. Lymphatic tissue fibrosis is associated with reduced numbers of naïve CD4+ T cells in human immunodeficiency virus type 1 infection. Clin Vaccine Immunol. (2006) 13:556–60. doi: 10.1128/CVI.13.5.556-560.2006
144. Douek DC, Betts MR, Hill BJ, Little SJ, Lempicki R, Metcalf JA, et al. Evidence for increased T cell turnover and decreased thymic output in HIV infection. J Immunol. (2001) 167:6663–8. doi: 10.4049/jimmunol.167.11.6663
145. Moroni M, Antinori S. HIV and direct damage of organs: disease spectrum before and during the highly active antiretroviral therapy era. AIDS. (2003) 17 (Suppl 1):S51–64. doi: 10.1097/00002030-200304001-00008
146. Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. (2012) 335:1188–93. doi: 10.1126/science.1217550
147. Goicoechea M, Smith D, Liu L. Determinants of CD4+ T cell recovery during suppressive antiretroviral therapy: association of immune activation, T cell maturation markers, and cellular HIV-1 DNA. J Infect. (2006) 194:29–37. doi: 10.1086/504718
148. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. (2011) 203:780–90. doi: 10.1093/infdis/jiq118
149. Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J Infect Dis. (2014) 210:1228–38. doi: 10.1093/infdis/jiu238
150. Cheru LT, Park EA, Saylor CF, Burdo TH, Fitch KV, Looby S, et al. I-FABP is higher in people with chronic hiv than elite controllers, related to sugar and fatty acid intake and inversely related to body fat in people with HIV. Open forum Infect Dis. (2018) 5:ofy288. doi: 10.1093/ofid/ofy288
151. Chun T-W, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. (2008) 197:714–20. doi: 10.1086/527324
152. Wong JK, Gunthard HF, Havlir DV, Zhang ZQ, Haase AT, Ignacio CC, et al. Reduction of HIV-1 in blood and lymph nodes following potent antiretroviral therapy and the virologic correlates of treatment failure. Proc Natl Acad Sci USA. (1997) 94:12574–9. doi: 10.1073/pnas.94.23.12574
153. Perreau M, Savoye A-L, De Crignis E, Corpataux J-M, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. (2013) 216:143–56. doi: 10.1084/jem.20121932
154. Spiegel H, Herbst H, Niedobitek G, Foss HD, Stein H. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am J Pathol. (1992) 140:15–22.
155. Gunthard HF, Havlir DV, Fiscus S, Zhang ZQ, Eron J, Mellors J, et al. Residual human immunodeficiency virus (HIV) Type 1 RNA and DNA in lymph nodes and HIV RNA in genital secretions and in cerebrospinal fluid after suppression of viremia for 2 years. J Infect Dis. (2001) 183:1318–27. doi: 10.1086/319864
156. Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. (2008) 1:23–30. doi: 10.1038/sj.mi.2007.1750001
157. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of mucosal CD103 DCs and IL-17 and IL-22 lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. (2012) 5:646–57. doi: 10.1038/mi.2012.38
158. Ryan ES, Micci L, Fromentin R, Paganini S, McGary CS, Easley K, et al. Loss of function of intestinal IL-17 and IL-22 producing cells contributes to inflammation and viral persistence in SIV-infected rhesus macaques. PLoS Pathog. (2016) 12:e1005412. doi: 10.1371/journal.ppat.1005412
159. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. (2010) 6:e1001052. doi: 10.1371/journal.ppat.1001052
160. Chung CY, Alden SL, Funderburg NT, Fu P, Levine AD. Progressive proximal-to-distal reduction in expression of the tight junction complex in colonic epithelium of virally-suppressed HIV+ individuals. PLoS Pathog. (2014) 10:e1004198. doi: 10.1371/journal.ppat.1004198
161. Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. Intestinal Microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. (2015) 211:19–27. doi: 10.1093/infdis/jiu409
162. Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. (2012) 10:655–66. doi: 10.1038/nrmicro2848
163. Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, et al. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J Immunol. (2003) 171:4320–8. doi: 10.4049/jimmunol.171.8.4320
164. Svard J, Paquin-Proulx D, Buggert M, Noyan K, Barqasho B, Sonnerborg A, et al. Role of translocated bacterial flagellin in monocyte activation among individuals with chronic HIV-1 infection. Clin Immunol. (2015) 161:180–9. doi: 10.1016/j.clim.2015.08.018
165. Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Fernandez S, et al. Toll-like receptor (TLR) expression on CD4+ and CD8+ T-cells in patients chronically infected with hepatitis C virus. Cell Immunol. (2010) 264:150–5. doi: 10.1016/j.cellimm.2010.06.001
166. Tripathy A, Khanna S, Padhan P, Smita S, Raghav S, Gupta B. Direct recognition of LPS drive TLR4 expressing CD8(+) T cell activation in patients with rheumatoid arthritis. Sci Rep. (2017) 7:933. doi: 10.1038/s41598-017-01033-7
167. Miller Sanders C, Cruse JM, Lewis RE. Toll-like receptor and chemokine receptor expression in HIV-infected T lymphocyte subsets. Exp Mol Pathol. (2010) 88:26–31. doi: 10.1016/j.yexmp.2009.09.006
168. Song Y, Zhuang Y, Zhai S, Huang D, Zhang Y, Kang W, et al. Increased expression of TLR7 in CD8(+) T cells leads to TLR7-mediated activation and accessory cell-dependent IFN-gamma production in HIV type 1 infection. AIDS Res Hum Retroviruses. (2009) 25:1287–95. doi: 10.1089/aid.2008.0303
169. Funderburg N, Luciano AA, Jiang W, Rodriguez B, Sieg SF, Lederman MM. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS ONE. (2008) 3:e1915. doi: 10.1371/journal.pone.0001915
170. Thibault S, Tardif MR, Barat C, Tremblay MJ. TLR2 signaling renders quiescent naive and memory CD4+ T cells more susceptible to productive infection with X4 and R5 HIV-type 1. J Immunol. (2007) 179:4357–66. doi: 10.4049/jimmunol.179.7.4357
171. Mercier BC, Cottalorda A, Coupet C-A, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol. (2009) 182:1860–7. doi: 10.4049/jimmunol.0801167
172. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
173. Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. (2009) 83:3719–33. doi: 10.1128/JVI.01844-08
174. Yamamoto T, Price DA, Casazza JP, Ferrari G, Nason M, Chattopadhyay PK, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. (2011) 117:4805–15. doi: 10.1182/blood-2010-11-317297
175. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
176. Chun T-W, Justement JS, Sanford C, Hallahan CW, Planta MA, Loutfy M, et al. Relationship between the frequency of HIV-specific CD8+ T cells and the level of CD38+CD8+ T cells in untreated HIV-infected individuals. Proc Natl Acad Sci USA. (2004) 101:2464–9. doi: 10.1073/pnas.0307328101
177. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. (2006) 203:2281–92. doi: 10.1084/jem.20061496
178. Leeansyah E, Ganesh A, Quigley MF, Sönnerborg A, Andersson J, Hunt PW, et al. Activation, exhaustion, and persistent decline of the antimicrobial MR1-restricted MAIT-cell population in chronic HIV-1 infection. Blood. (2013) 121:1124–35. doi: 10.1182/blood-2012-07-445429
179. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. (2016) 354:1165–9. doi: 10.1126/science.aae0491
180. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity. (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
181. Llibre JM, Buzon MJ, Massanella M, Esteve A, Dahl V, Puertas MC, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study. Antivir Ther. (2012) 17:355–64. doi: 10.3851/IMP1917
182. Buzón JM, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med. (2010) 16:460–5. doi: 10.1038/nm.2111
183. Nicastri E, Tommasi C, Abbate I, Bonora S, Tempestilli M, Bellagamba R, et al. Effect of raltegravir on the total and unintegrated proviral HIV DNA during raltegravir-based HAART. Antivir Ther. (2011) 16:797–803. doi: 10.3851/IMP1833
184. Hatano H, Strain MC, Scherzer R, Bacchetti P, Wentworth D, Hoh R, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV Infection: a randomized, placebo-controlled trial. J Infect Dis. (2013) 208:1436–42. doi: 10.1093/infdis/jit453
185. Arponen S, Benito J, Lozano S, Blanco F, Garrido C, De Mendoza C. More pronounced effect of integrase inhibitor Raltegravir on proviral DNA reduction that other antiretroviral drugs in patients achieving undetectable viremia. In: 15TH Conference on Retroviruses and Opportunistic Infections # 796. Boston, MA (2008).
186. Bandera A, Colella E, Rizzardini G, Gori A, Clerici M. Strategies to limit immune-activation in HIV patients. Expert Rev Anti Infect Ther. (2017) 15:43–54. doi: 10.1080/14787210.2017.1250624
187. Nakanjako D, Ssinabulya I, Nabatanzi R, Bayigga L, Kiragga A, Joloba M, et al. Atorvastatin reduces T-cell activation and exhaustion among HIV-infected cART-treated suboptimal immune responders in Uganda: a randomised crossover placebo-controlled trial. Trop Med Int Health. (2015) 20:380–90. doi: 10.1111/tmi.12442
188. Ganesan A, Crum-Cianflone N, Higgins J, Qin J, Rehm C, Metcalf J, et al. High dose atorvastatin decreases cellular markers of immune activation without affecting HIV-1 RNA levels: results of a double-blind randomized placebo controlled clinical trial. J Infect Dis. (2011) 203:756–64. doi: 10.1093/infdis/jiq115
189. Funderburg NT, Jiang Y, Debanne SM, Labbato D, Juchnowski S, Ferrari B, et al. Rosuvastatin reduces vascular inflammation and T-cell and monocyte activation in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. (2015) 68:396–404. doi: 10.1097/QAI.0000000000000478
190. Plosker GL, Croom KF. Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs. (2005) 65:1825–49. doi: 10.2165/00003495-200565130-00008
191. Szabo C, Haskó G, Szabó C, Németh ZH, Deitch EA. Sulphasalazine inhibits macrophage activation: inhibitory effects on inducible nitric oxide synthase expression, interleukin-12 production and major histocompatibility complex II expression. Immunology. (2001) 103:473–8. doi: 10.1046/j.1365-2567.2001.01272.x
192. Rodenburg RJT, Ganga A, Van Lent PLEM, Van De Putte LBA, Van Venrooij WJ. The antiinflammatory drug sulfasalazine inhibits tumor necrosis factor alpha expression in macrophages by inducing apoptosis. Arthritis Rheum. (2000) 43:1941–50. doi: 10.1002/1529-0131(200009)43:9<1941::AID-ANR4>3.0.CO;2-O
193. Liptay S, Bachem M, Häcker G, Adler G, Debatin KM, Schmid RM. Inhibition of nuclear factor kappa B and induction of apoptosis in T-lymphocytes by sulfasalazine. Br J Pharmacol. (1999) 128:1361–9. doi: 10.1038/sj.bjp.0702937
194. Schneider T, Ullrich R, Bergs C, Schmidt W, Riecken EO, Zeitz M. Abnormalities in subset distribution, activation, and differentiation of T cells isolated from large intestine biopsies in HIV infection. The Berlin Diarrhoea/Wasting Syndrome Study Group. Clin Exp Immunol. (1994) 95:430–5.
195. Sortino O, Richards E, Dias J, Leeansyah E, Sandberg JK, Sereti I. IL-7 treatment supports CD8+ mucosa-associated invariant T-cell restoration in HIV-1-infected patients on antiretroviral therapy. AIDS. (2018) 32:825–8. doi: 10.1097/QAD.0000000000001760
196. Leeansyah E, Svärd J, Dias J, Buggert M, Nyström J, Quigley MF, et al. Arming of MAIT cell cytolytic antimicrobial activity is induced by IL-7 and defective in HIV-1 infection. PLoS Pathog. (2015) 11:e1005072. doi: 10.1371/journal.ppat.1005072
197. Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. (2005) 175:2261–9. doi: 10.4049/jimmunol.175.4.2261
198. Novy P, Huang X, Leonard WJ, Yang Y. Intrinsic IL-21 signaling is critical for CD8 T cell survival and memory formation in response to vaccinia viral infection. J Immunol. (2011) 186:2729–38. doi: 10.4049/jimmunol.1003009
199. White L, Krishnan S, Strbo N, Liu H, Kolber MA, Lichtenheld MG, et al. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV). Blood. (2007) 109:3873–80. doi: 10.1182/blood-2006-09-045278
200. Pallikkuth S, Rogers K, Villinger F, Dosterii M, Vaccari M, Franchini G, et al. Interleukin-21 administration to rhesus macaques chronically infected with simian immunodeficiency virus increases cytotoxic effector molecules in T cells and NK cells and enhances B cell function without increasing immune activation or viral replication. Vaccine. (2011) 29:9229–38. doi: 10.1016/j.vaccine.2011.09.118
201. Watson DC, Moysi E, Valentin A, Bergamaschi C, Devasundaram S, Fortis SP, et al. Treatment with native heterodimeric IL-15 increases cytotoxic lymphocytes and reduces SHIV RNA in lymph nodes. PLoS Pathog. (2018) 14:e1006902. doi: 10.1371/journal.ppat.1006902
202. Webb GM, Li S, Mwakalundwa G, Folkvord JM, Greene JM, Reed JS, et al. The human IL-15 superagonist ALT-803 directs SIV-specific CD8+ T cells into B-cell follicles. Blood Adv. (2018) 2:76–84. doi: 10.1182/bloodadvances.2017012971
203. Velu V, Kannanganat S, Ibegbu C, Chennareddi L, Villinger F, Freeman GJ, et al. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol. (2007) 81:5819–28. doi: 10.1128/JVI.00024-07
204. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. (2009) 458:206–10. doi: 10.1038/nature07662
205. Dyavar Shetty R, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, et al. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest. (2012) 122:1712–6. doi: 10.1172/JCI60612
206. Mylvaganam GH, Chea LS, Tharp GK, Hicks S, Velu V, Iyer SS, et al. Combination anti-PD-1 and antiretroviral therapy provides therapeutic benefit against SIV. JCI Insight. (2018) 3:122940. doi: 10.1172/jci.insight.122940
207. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+T cells that provide the proliferative burst after PD-1 therapy. Nature. (2016) 537:417–21. doi: 10.1038/nature19330
208. Fromentin R, DaFonseca S, Costiniuk CT, El-Far M, Procopio FA, Hecht FM, et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4 + T cells from ART-suppressed individuals. Nat Commun. (2019) 10:814. doi: 10.1038/s41467-019-08798-7
209. Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis. (2017) 215:1725–33. doi: 10.1093/infdis/jix191
210. Ostios-Garcia L, Faig J, Leonardi GC, Adeni AE, Subegdjo SJ, Lydon CA, et al. Safety and efficacy of PD-1 inhibitors among HIV-positive patients with non-small cell lung cancer. J Thorac Oncol. (2018) 13:1037–42. doi: 10.1016/j.jtho.2018.03.031
211. Pakker NG, Kroon EDMB, Roos MTL, Otto SA, Hall D, Wit FWNM, et al. Immune restoration does not invariably occur following long-term HIV-1 suppression during antiretroviral therapy. AIDS. (1999) 13:203–12. doi: 10.1097/00002030-199902040-00008
212. Notermans DW, Pakker NG, Hamann D, Foudraine NA, Kauffmann RH, Meenhorst PL, et al. Immune reconstitution after 2 years of successful potent antiretroviral therapy in previously untreated human immunodeficiency virus type 1-infected adults. J Infect Dis. (1999) 180:1050–6. doi: 10.1086/315013
213. Lok JJ, Bosch RJ, Benson CA, Collier AC, Robbins GK, Shafer RW, et al. Long-term increase in CD4+ T-cell counts during combination antiretroviral therapy for HIV-1 infection. AIDS. (2010) 24:1867–76. doi: 10.1097/QAD.0b013e32833adbcf
214. Negredo E, Massanella M, Puig J, Perez-Alvarez N, Gallego-Escuredo JM, Villarroya J, et al. Nadir CD4 T cell count as predictor and high CD4 T cell intrinsic apoptosis as final mechanism of poor CD4 T cell recovery in virologically suppressed HIV-infected patients: clinical implications. Clin Infect Dis. (2010) 50:1300–8. doi: 10.1086/651689
215. Robbins GK, Spritzler JG, Chan ES, Asmuth DM, Gandhi RT, Rodriguez BA, et al. Incomplete reconstitution of T cell subsets on combination antiretroviral therapy in the AIDS clinical trials group protocol 384. Clin Infect Dis. (2009) 48:350–61. doi: 10.1086/595888
216. Gale HB, Gitterman SR, Hoffman HJ, Gordin FM, Benator DA, Labriola AM, et al. Is frequent CD4+ T-lymphocyte count monitoring necessary for persons with counts > = 300 cells/muL and HIV-1 suppression? Clin Infect Dis. (2013) 56:1340–3. doi: 10.1093/cid/cit004
217. Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog. (2014) 10:e1004078. doi: 10.1371/journal.ppat.1004078
218. Serrano-Villar S, Pérez-Elías MJ, Dronda F, Casado JL, Moreno A, Royuela A, et al. Increased risk of serious non-AIDS-related events in HIV-infected subjects on antiretroviral therapy associated with a low CD4/CD8 ratio. PLoS ONE. (2014) 9:e85798. doi: 10.1371/journal.pone.0085798
219. Perdomo-Celis F, Taborda NA, Rugeles MT. Follicular CD8+ T cells: origin, function and importance during HIV infection. Front Immunol. (2017) 8:1241. doi: 10.3389/fimmu.2017.01241
220. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med. (2016) 22:754–61. doi: 10.1038/nm.4113
221. He R, Ye L. Follicular CXCR5-expressing CD8+ T-cells curtail chronic viral infection. Nature. (2016) 537:412–6. doi: 10.1038/nature19317
222. Petrovas C, Ferrando-Martinez S, Gerner MY, Casazza JP, Pegu A, Deleage C, et al. H I V Follicular CD8 T cells accumulate in HIV infection and can kill infected cells in vitro via bispecific antibodies. Sci Transl Med. (2017) 9:eaag2285. doi: 10.1126/scitranslmed.aag2285
223. Moysi E, Pallikkuth S, De Armas LR, Gonzalez LE, Ambrozak D, George V, et al. Altered immune cell follicular dynamics in HIV infection following influenza vaccination. J Clin Invest. (2018) 128:3171–85. doi: 10.1172/JCI99884
224. Ferrando-Martinez S, Moysi E, Pegu A, Andrews S, Makamdop KN, Ambrozak D, et al. Accumulation of follicular CD8+T cells in pathogenic SIV infection. J Clin Invest. (2018) 128:2089–103. doi: 10.1172/JCI96207
225. Rahman MA, McKinnon KM, Karpova TS, Ball DA, Venzon DJ, Fan W, et al. Associations of simian immunodeficiency virus (SIV)-specific follicular CD8 + T cells with other follicular T cells suggest complex contributions to SIV viremia control. J Immunol. (2018) 200:2714–26. doi: 10.4049/jimmunol.1701403
226. Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest. (2012) 122:3271–80. doi: 10.1172/JCI64314
227. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. (2012) 122:3281–94. doi: 10.1172/JCI63039
228. Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood. (2007) 109:4272–9. doi: 10.1182/blood-2006-11-055764
229. Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. (1999) 189:403–12. doi: 10.1084/jem.189.2.403
230. Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol. (2000) 156:1133–8. doi: 10.1016/S0002-9440(10)64981-4
231. Mehraj V, Ramendra R, Isnard S, Dupuy FP, Lebouché B, Costiniuk C, et al. CXCL13 as a biomarker of immune activation during early and chronic HIV infection. Front Immunol. (2019) 10:289. doi: 10.3389/fimmu.2019.00289
232. Wood GS. The immunohistology of lymph nodes in HIV infection: a review. Prog AIDS Pathol. (1990) 2:25–32.
233. Perdomo-Celis F, Taborda NA, Rugeles MT. Circulating CXCR5-expressing CD8+T-cells are major producers of IL-21 and associate with limited HIV replication. J Acquir Immune Defic Syndr. (2018) 78:473–82. doi: 10.1097/QAI.0000000000001700
234. Mylvaganam GH, Rios D, Abdelaal HM, Iyer S, Tharp G, Mavinger M, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc Natl Acad Sci USA. (2017) 114:1976–81. doi: 10.1073/pnas.1621418114
235. Li S, Folkvord JM, Rakasz EG, Abdelaal HM, Wagstaff RK, Kovacs KJ, et al. Simian immunodeficiency virus-producing cells in follicles are partially suppressed by CD8 + cells in vivo. J Virol. (2016) 90:11168–80. doi: 10.1128/JVI.01332-16
236. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol. (2014) 15:856–65. doi: 10.1038/ni.2947
237. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood. (2006) 108:3808–17. doi: 10.1182/blood-2006-05-021576
238. Ayala VI, Deleage C, Trivett MT, Jain S, Coren LV, Breed MW, et al. CXCR5-dependent entry of CD8 T cells into rhesus macaque B-cell follicles achieved through T-cell engineering. J Virol. (2017) 91:e02507–16. doi: 10.1128/JVI.02507-16
239. Haran KP, Hajduczki A, Pampusch MS, Mwakalundwa G, Vargas-Inchaustegui DA, Rakasz EG, et al. Simian immunodeficiency virus (SIV)-specific chimeric antigen receptor-T cells engineered to target B cell follicles and suppress SIV replication. Front Immunol. (2018) 9:492. doi: 10.3389/fimmu.2018.00492
240. Chomont N, Okoye AA, Favre D, Trautmann L. Wake me up before you go: a strategy to reduce the latent HIV reservoir. AIDS. (2018) 32:293–8. doi: 10.1097/QAD.0000000000001695
241. Pantaleo G, Levy Y. Therapeutic vaccines and immunological intervention in HIV infection: a paradigm change. Curr Opin HIV AIDS. (2016) 11:576–84. doi: 10.1097/COH.0000000000000324
242. Karlsson I, Kloverpris H, Jensen KJ, Stryhn A, Buus S, Karlsson A, et al. Identification of conserved subdominant HIV Type 1 CD8(+) T Cell epitopes restricted within common HLA Supertypes for therapeutic HIV Type 1 vaccines. AIDS Res Hum Retroviruses. (2012) 28:1434–43. doi: 10.1089/aid.2012.0081
243. Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther. (2014) 22:464–75. doi: 10.1038/mt.2013.248
244. Lieberman J, Skolnik PR, Parkerson GR III, Fabry JA, Landry B, Bethel J, et al. Safety of autologous, ex vivo-expanded human immunodeficiency virus (HIV)-specific cytotoxic T-lymphocyte infusion in HIV-infected patients. Blood. (1997) 90:2196–206.
245. Brodie SJ, Lewinsohn DA, Patterson BK, Jiyamapa D, Krieger J, Corey L, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. (1999) 5:34–41. doi: 10.1038/4716
246. Tan R, Xu X, Ogg GS, Hansasuta P, Dong T, Rostron T, et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood. (1999) 93:1506–10.
247. Koenig S, Conley AJ, Brewah YA, Jones GM, Leath S, Boots LJ, et al. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat Med. (1995) 1:330–6. doi: 10.1038/nm0495-330
248. Lam S, Sung J, Cruz C, Castillo-Caro P, Ngo M, Garrido C, et al. Broadly-specific cytotoxic T cells targeting multiple HIV antigens are expanded from HIV+ patients: implications for immunotherapy. Mol Ther. (2015) 23:387–95. doi: 10.1038/mt.2014.207
249. Sung JA, Lam S, Garrido C, Archin N, Rooney CM, Bollard CM, et al. Expanded cytotoxic T-cell lymphocytes target the latent HIV reservoir. J Infect Dis. (2015) 212:258–63. doi: 10.1093/infdis/jiv022
250. Joseph A, Zheng JH, Follenzi A, Dilorenzo T, Sango K, Hyman J, et al. Lentiviral vectors encoding human immunodeficiency virus type 1 (HIV-1)-specific T-cell receptor genes efficiently convert peripheral blood CD8 T lymphocytes into cytotoxic T lymphocytes with potent in vitro and in vivo HIV-1-specific inhibitory activity. J Virol. (2008) 82:3078–89. doi: 10.1128/JVI.01812-07
251. Mummert C, Hofmann C, Huckelhoven AG, Bergmann S, Mueller-Schmucker SM, Harrer EG, et al. T-cell receptor transfer for boosting HIV-1-specific T-cell immunity in HIV-1-infected patients. AIDS. (2016) 30:2149–58. doi: 10.1097/QAD.0000000000001176
252. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
253. Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. (2018) 18:605–16. doi: 10.1038/s41577-018-0042-2
254. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. (2000) 96:785–93.
255. Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. (2017) 13:e1006613. doi: 10.1371/journal.ppat.1006613
256. Liu B, Zou F, Lu L, Chen C, He D, Zhang X, et al. Chimeric antigen receptor T cells guided by the single-chain Fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4+ T lymphocytes isolated from HIV-1-infected individuals receiving suppressive C. J Virol. (2016) 90:9712–24. doi: 10.1128/JVI.00852-16
257. Ghanem MH, Bolivar-Wagers S, Dey B, Hajduczki A, Vargas-Inchaustegui DA, Danielson DT, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. (2018) 20:407–19. doi: 10.1016/j.jcyt.2017.11.001
258. Chapuis AG, Casper C, Kuntz S, Zhu J, Tjernlund A, Diem K, et al. HIV-specific CD8+ T cells from HIV+ individuals receiving HAART can be expanded ex vivo to augment systemic and mucosal immunity in vivo. Blood. (2011) 117:5391–402. doi: 10.1182/blood-2010-11-320226
259. Patel S, Lam S, Cruz CR, Wright K, Cochran C, Ambinder RF, et al. Functionally active HIV-specific T cells that target gag and Nef can be expanded from virus-naive donors and target a range of viral epitopes: implications for a cure strategy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2016) 22:536–41. doi: 10.1016/j.bbmt.2015.12.007
Keywords: CD8+ T-cell, HIV, antiretroviral therapy, IL-17, exhaustion, CXCR5
Citation: Perdomo-Celis F, Taborda NA and Rugeles MT (2019) CD8+ T-Cell Response to HIV Infection in the Era of Antiretroviral Therapy. Front. Immunol. 10:1896. doi: 10.3389/fimmu.2019.01896
Received: 11 April 2019; Accepted: 26 July 2019;
Published: 09 August 2019.
Edited by:
Rosana Pelayo, Mexican Social Security Institute (IMSS), MexicoReviewed by:
Perla Mariana Del Rio Estrada, National Institute of Respiratory Diseases (Mexico), MexicoAikaterini Alexaki, United States Food and Drug Administration, United States
Fernando Roger Esquivel-Guadarrama, Autonomous University of the State of Morelos, Mexico
Copyright © 2019 Perdomo-Celis, Taborda and Rugeles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria T. Rugeles, bWFyaWEucnVnZWxlc0B1ZGVhLmVkdS5jbw==