 Kata P. Szilveszter
Kata P. Szilveszter Tamás Németh
Tamás Németh Attila Mócsai
Attila Mócsai- Department of Physiology, Semmelweis University School of Medicine, Budapest, Hungary
Tyrosine kinases relay signals from diverse leukocyte antigen receptors, innate immune receptors, and cytokine receptors, and therefore mediate the recruitment and activation of various leukocyte populations. Non-receptor tyrosine kinases of the Jak, Src, Syk, and Btk families play major roles in various immune-mediated disorders, and small-molecule tyrosine kinase inhibitors are emerging novel therapeutics in a number of those diseases. Autoimmune and inflammatory skin diseases represent a broad spectrum of immune-mediated diseases. Genetic and pharmacological studies in humans and mice support the role of tyrosine kinases in several inflammatory skin diseases. Atopic dermatitis and psoriasis are characterized by an inflammatory microenvironment which activates cytokine receptors coupled to the Jak-Stat signaling pathway. Jak kinases are also implicated in alopecia areata and vitiligo, skin disorders mediated by cytotoxic T lymphocytes. Genetic studies indicate a critical role for Src-family kinases and Syk in animal models of autoantibody-mediated blistering skin diseases. Here, we review the various tyrosine kinase signaling pathways and their role in various autoimmune and inflammatory skin diseases. Special emphasis will be placed on identification of potential therapeutic targets, as well as on ongoing preclinical and clinical studies for the treatment of inflammatory skin diseases by small-molecule tyrosine kinase inhibitors.
Introduction
Tyrosine kinases are intracellular enzymes mediating tyrosine phosphorylation of downstream molecules. They play a critical role in signal transduction by various cell surface receptors including, among others, growth factor receptors, adhesion receptors, immunoreceptors, and cytokine receptors. Given their role in multiple signaling processes and disease pathogenesis, tyrosine kinases have emerged as excellent therapeutic targets for the targeted therapy of various diseases. Indeed, small molecule tyrosine kinase inhibitors became important contributors to the pharmacological control of a diverse array of diseases including various malignant processes and immune-mediated diseases such as autoimmune and inflammatory conditions (1).
Inflammatory joint diseases such as rheumatoid arthritis have been in the focus of the development of tyrosine kinase inhibitors for therapeutic purposes within the area of immune mediated diseases (2). This has culminated in the regulatory approval of Jak inhibitors for the treatment of rheumatoid arthritis and certain related disease states (3). Besides inflammatory arthritis, inflammatory skin diseases are another major group of diseases with a major pathogenetic component of various immune cells and immunological pathways. Several lines of evidence indicate the contribution of various tyrosine kinases to the development and progression of diverse inflammatory skin diseases. Those issues suggest that tyrosine kinase inhibitors may provide therapeutic benefit in inflammatory skin diseases.
In this review article, we summarize our current knowledge and understanding of the role of tyrosine kinases in autoimmune and inflammatory skin diseases. We first provide an overview of the various receptor and non-receptor tyrosine kinases and their role in immunological and inflammatory processes. We then summarize the role of the various tyrosine kinases in specific autoimmune or inflammatory skin diseases. Special emphasis is placed on genetic studies in mice and humans indicating a role for tyrosine kinase pathways in inflammatory skin diseases, as well as the preclinical and clinical development of tyrosine kinases inhibitors for the targeted pharmacological therapy of those diseases.
Signal Transduction by Tyrosine Kinases
Non-receptor Tyrosine Kinases
Non-receptor tyrosine kinases are intracellular tyrosine kinases without a direct role in sensing extracellular cues. Nevertheless, these tyrosine kinases are often coupled to various cell surface receptors and are intimately involved in the transmission of extracellular signals to downstream intracellular signaling pathways and cellular effector functions.
There are a total of 10 different non-receptor tyrosine kinase families. Of those, we will discuss Janus kinases (also known as Jak-family kinases), Src-family kinases, and the Syk tyrosine kinase, as well as members of the Btk kinase family.
Janus Kinase Family
The Janus kinase family consists of four members: Jak1, Jak2, Jak3, and Tyk2. While Jak1, Jak2, and Tyk2 are ubiquitously expressed, the expression of Jak3 is limited to the hematopoietic compartment.
These kinases are primarily involved in the signal transduction of various cytokine receptors which are grouped into type I (extracellular WSXWS sequence present; e.g., IL-2, IL-6, GH, EPO, G-CSF, and GM-CSF receptors) and type II (no extracellular WSXWS sequence; e.g., IFN-α, IFN-β, and IL-10 receptors) cytokine receptors. Since Jak-coupled cytokine receptors act as dimers, Jak family kinase activity is also mediated by involving two Jak-family kinases. In most cases, the two cooperating kinases are different (“heterodimers,” although they do not form a firm dimer), although Jak2 can also cooperate with another Jak2 molecule (“homodimer”). Upon ligand binding, the conformational changes of the receptor and/or ligand-induced dimerization promotes Jak activation, which leads to autophosphorylation of tyrosine residues of the kinase itself, further augmenting its kinase activity. Jaks then phosphorylate the receptor chains, allowing the recruitment of various signal transducer and activator of transcription (Stat) transcription factors. Stat molecules are then also phosphorylated by Jak kinases, leading to dimerization and translocation to the nucleus where they activate or repress gene expression and influence epigenetic alterations (Figure 1). These basic signaling principles are conserved across the diverse array of different biological functions of the Jak-Stat signaling pathway.
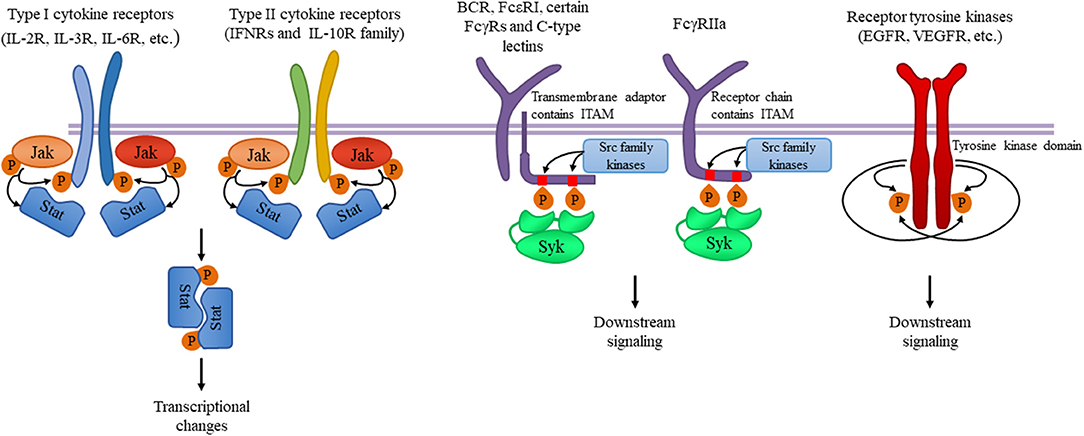
Figure 1. Tyrosine kinases and their signaling pathways. Type I and type II cytokine receptors utilize Janus kinases for the initiation of downstream signaling. Type I cytokine receptor superfamily shares a common amino acid motif WSXWS. Within this family receptors containing the common γ chain (γc) recognize IL-2, IL-4, and IL-13 among others utilizing Jak1 and Jak3. Cytokines including IL-3 and IL-6 are also recognized by type I cytokine receptors acting through Jak1/Jak2 heterodimers or Jak2 homodimers, respectively. The type II cytokine receptor family includes receptors activated by interferons (IFNs) and the IL-10 family utilizing heterodimers of Jak1 along with Jak2 or Tyk2. Ligand binding leads to Jak activation resulting in phosphorylation of the receptor and downstream signal transducers and activators known as Stats mediating transcriptional changes (more information in the text). Src-family kinases and Syk are involved in several immune cell signaling pathways like immunoreceptor, integrin and C-type lectin signaling. Upon ligand binding, activation of Src-family kinases leads to the phosphorylation of tyrosine residues in immunoreceptor tyrosine-based activation motifs (ITAMs), that can be part of a transmembrane adaptor molecule like in case of B cell receptor (BCR), FcεRI, and certain FcγRs and C-type lectins, or of the receptor chain itself like in FcγRIIa in humans. Syk is recruited to the dually phosphorylated ITAMs and becomes activated resulting in the recruitment and activation of various further adapter proteins promoting downstream signaling. Receptor tyrosine kinases, for example EGFR and VEGFR have intrinsic tyrosine kinase activity leading to auto-and transphosphorylation of the receptor chains upon ligand binding. Recruitment of several adaptors and effector molecules through SH2 and phosphotyrosine binding domains mediate downstream signaling.
Despite the complex and promiscuous nature of receptor association of the different Jak family kinases, human inherited traits and mouse genetic studies have revealed several critical functions of the different Jak kinases. Jak1 is essential for the signaling through type II cytokine receptors (such as IFN receptors), as well as through receptors that utilize the common γ-chain (γc) or the shared gp130 subunit. Jak1 deficiency in mice leads to defective lymphoid development and neurological defects resulting in perinatal lethality without disturbing other hemopoietic lineages (4). Signaling downstream of type II IFNs and receptors with shared gp130 subunit also require Jak2 (besides Jak1), whereas, Jak2 mediated signaling is not required for lymphoid development. IL-3 receptor and several hormone-like receptors (EPO, TPO, GH, PRL) signal through Jak2 alone. Deletion of Jak2 leads to embryonic lethality due to failure of definitive erythropoiesis in mice, likely due to the role of Jak2 in signaling by cytokine receptors involved in the regulation of hematopoiesis and, especially, erythropoiesis (5, 6). Jak3 expression is essentially limited to hematopoietic cells and it is known that it constitutively and exclusively binds to γc-containing receptors including IL-2 and IL-4 receptors. Jak3 mutation in humans leads to severe combined immunodeficiency (7, 8) and studies using Jak3−/− mice further confirmed the critical role of Jak3 in lymphoid development (9, 10). Tyk2 is important in IL-12 and IL-23-mediated T cell responses and IFN signaling (11, 12). Tyk2−/− mice are viable but susceptible to infections, and macrophages fail to respond to LPS both in vitro and in vivo (13, 14).
Given their central role in cytokine signaling it is not surprising that Jaks have a role in several immune mediated diseases involving autoimmunity, transplant rejection, and malignancies. Therefore, pharmacological targeting of Jaks was plausible and Jak inhibitors have been extensively studied in several clinical studies. A critical aspect of Jak inhibitors is their selectivity profile for the different Jak family kinases which determines the spectrum of their biological effects. Table 1 provides a list and the selectivity profile of currently available Jak inhibitors based on cell-free assays. The mechanism of action of those drugs is competitive binding to the ATP binding site of the kinase domain therefore inhibiting phosphorylation and activation of Jaks, except for the case of PF6615600 and BMS986165 (15). PF6615600 mediates a covalent, irreversible Jak3 inhibition through a non-conserved Cys residue in the ATP binding pocket, whereas BMS986165 binds to the pseudokinase domain of Tyk2 (15). First generation Jak inhibitors (tofacitinib, ruxolitinib, baricitinib, and oclacitinib) tend to be less selective among the Jak family kinases due to structural similarities in the ATP binding site of different Jaks, whereas more selective inhibitors were developed during later stages of drug development. Discrepancies between biochemical and cellular potencies of Jak inhibitors have been reported, potentially due to the dominant role of one Jak over another in certain cytokine signaling pathways (16).
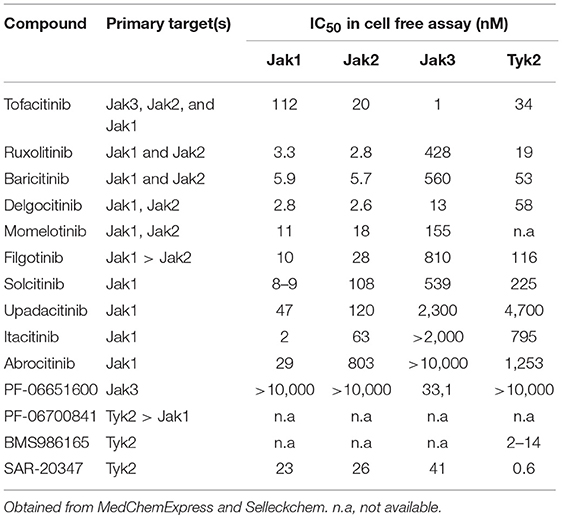
Table 1. Jak inhibitors and their selectivity profile.
The most studied Jak inhibitor is tofacitinib, which ameliorated autoimmune arthritides in various animal models (17–20) and proved to be effective in several phase II and III studies in the treatment of rheumatoid arthritis, leading to regulatory approval by both the FDA and EMA (2, 21). Jak inhibitors are also currently under investigation in other immune mediated diseases like inflammatory bowel disease, transplant rejection, and multiple dermatological disorders. Excellent reviews about the current state of Jak inhibitors and ongoing clinical trials have been published recently (3, 15, 22).
Src-Family Kinases and the Syk Tyrosine Kinase
The Src kinase family includes nine members (including Hck, Fgr, Lyn, and Lck) which are involved in many signaling pathways in immune cells including immunoreceptor as well as integrin signaling. Src-family kinase activity is regulated by tyrosine phosphorylation and Src homology 2 and 3 (SH2 and SH3) mediated protein-protein interactions with partner proteins containing phosphotyrosine or proline-rich motifs, respectively. Src-family kinases are ubiquitously expressed, although different cells express different family members. Within the immune system, T cells express Lck and Fyn, B cells express Fyn, Lyn, and Blk, and myeloid cells express Hck, Fgr, and Lyn (23). Spleen tyrosine kinase (Syk) is a tandem SH2 domain-containing enzyme acting mostly downstream of Src-family kinases. Syk is expressed in most hematopoietic lineage cells except for T-cells (and, partially, NK-cells) where a closely related kinase, ZAP-70 is expressed and performs a similar function (24).
Immunoreceptors such as B cell receptors (BCR), T cell receptors (TCR), and various activating Fc receptors of innate immune cells are physically associated with transmembrane adapter proteins carrying immunoreceptor tyrosine-based activation motifs (ITAMs). Ligand-receptor interaction results in the enzymatic activation of Src-family kinases phosphorylating the ITAM motifs in the receptor subunits (24). Dually phosphorylated ITAMs are recognized by the tandem SH2 domains of Syk (or ZAP-70 in T cells and NK cells), leading to the recruitment and activation of various further adapter proteins and the activation of several downstream signaling pathways leading to cellular responses (Figure 1). Leukocyte integrin “outside-in” signaling also requires Src family kinases and Syk, resulting in adhesion-induced activation of immune cells (24–28). Some inhibitory receptors containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs) also act through Src-family kinases (mostly Lyn), resulting in phosphatase activation and downmodulating of activating signals.
T cell development requires TCR-based signaling and Src-family kinases, particularly Lck. In case of B lymphocytes, Lyn kinase has primary role in BCR signaling. Paradoxically, B cell-specific deletion of Lyn not only results in the expected defects in B cell development, but also leads to autoimmunity (29). Myeloid cells primarily express Hck, Fgr, and Lyn which have a critical but overlapping role in the activation of neutrophils and macrophages through Fcγ receptors, as well as through β1 and β2 integrins. A prominent feature of Src-family kinases is a significant functional overlap between individual family members. Therefore, in contrast to Jak kinases, individual Src-family kinases are not essential for a given response and complete inhibition of a signaling pathway often requires combinational deletion of multiple kinases in myeloid cells. FcγR mediated phagocytosis is slowed in macrophages lacking Hck, Fgr, and Lyn (30). Adhesion-induced activation is also abrogated in neutrophils lacking Hck, Fgr, and Lyn including oxidative burst, degranulation, and cell spreading (28). Their role has also been shown in chemokine and cytokine responses (31). Moreover, Hck−/−Lyn−/−Fgr−/− triple knockout but not single or double knockout animals were completely protected from autoantibody induced arthritis due to the defective generation of inflammatory environment without affecting the intrinsic migratory capacity of myeloid cells (32).
Currently available Src-family inhibitors have limited selectivity, also inhibiting various other tyrosine kinases such as c-Kit, EGFR, or Abl (summarized in Table 2). Those inhibitors are often used in cancer therapy based on their effects on kinases other than Src-family kinases. As an example, dasatinib and bosutinib are potent multi-target inhibitors of Abl, Kit, and several members of the Src kinase family. Beside their therapeutic use in hematological malignancies, they have been found to be relevant in inflammatory conditions as well both in vitro and in vivo in immune-mediated experimental models (48–51).
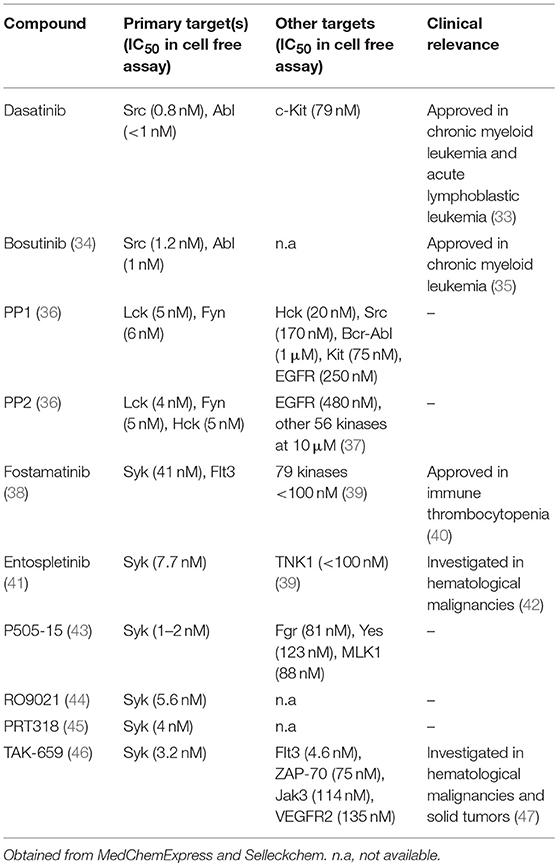
Table 2. Inhibitors of the Src-family and Syk.
Syk and ZAP-70 are also essential for the development of mature B and T cells, respectively (24, 52, 53). Syk deficiency leads to perinatal lethality due to defective separation of lymphoid and blood vessels (54). In the myeloid compartment, Syk is a key protein mediating Fc receptor and integrin mediated signaling and also mediates downstream signaling of C-type lectins like Dectin-1 recognizing fungal antigens (55, 56). Deficiency of the Syk kinase produces profound defects in neutrophil/macrophage integrin signaling and responses to immune complexes, resulting in significantly reduced stimulation of respiratory burst, degranulation and cell spreading (57–59). Syk-deficient bone marrow chimeras proved to be completely protected from autoantibody-induced arthritis that is due to enzymes specifically expressed in neutrophils (60–62). The partially selective Syk inhibitor fostamatinib showed clinical benefit in rheumatoid arthritis patients (63) and has been also investigated in other autoimmune and allergic diseases but considerable adverse events possibly due to its poor selectivity profile led to the suspension of further investigations in RA. New and more specific Syk inhibitors have been developed in the past few years (summarized in Table 2) that show promising results in this regard according to in vitro results, animal models of autoimmune arthritis and phase I clinical trials (41, 43, 44, 64).
Cerdulatinib and gusacitinib represent dual inhibitors of Syk and Jak kinases and cerdulatinib demonstrated efficacy in experimental arthritis (65). The concept that dual inhibition may result in a stronger therapeutic response is favorable, however it can also represent a limitation by the increased risk of toxicity.
Bruton's Tyrosine Kinase
Bruton's tyrosine kinase (Btk) is involved in the development and activation of B cells through BCR and Toll-like receptor (TLR) signaling (66). Patients with loss-of-function mutations in the Btk gene suffer from immunodeficiency due to the absence of mature B cells and immunoglobulins (67, 68). Similarly, deficiency of Btk in mice results in an impaired differentiation of B cells (69). In addition, transgenic mice that overexpress human Btk display systemic autoimmune response with spontaneous germinal center formation, increased cytokine production (IFNγ and IL-6) and anti-nuclear autoantibodies (ANAs) (70). Btk and other members of the Btk family like Tec kinase are also expressed in myeloid cells regulating maturation and effector function (71). Btk inhibitors interacting with the ATP binding site have been developed and proved to be effective in several systemic autoimmune mouse models like arthritis and lupus models (72, 73).
Receptor Tyrosine Kinases
Receptor tyrosine kinases represent a large family of receptors recognizing various hormones, cytokines, and growth factors (74). They form dimeric combinations upon ligand binding resulting in auto- and transphosphorylation and the recruitment and activation of effectors containing SH2 and phosphotyrosine binding domains, leading to multiple downstream signaling (Figure 1).
EGFR and its related receptors, PDGFRs, VEGF receptors and their intact signaling are essential for normal embryonic development and adult tissue homeostasis including cell survival, proliferation, adhesion and migration. Their deregulation has been associated with many human diseases, including immune-mediated disorders and cancer. Targeted therapy by receptor tyrosine kinase inhibitors revolutionized cancer therapy (75). VEGF receptors mediate angiogenesis and lymphangiogenesis during the inflammation process regulating immune cell recruitment and resolution of inflammation.
Tyrosine Kinases in Inflammatory Skin Diseases
Atopic Dermatitis
Atopic dermatitis (AD) is the most common inflammatory skin disease. A TH2 dominated immune response is essential in the pathogenesis causing eczematous dermatitis with intense pruritus accompanied by elevated serum concentrations of IgE. AD is commonly associated with other TH2 mediated allergic diseases called the “atopic-allergic march” (76).
Pathogenesis—Pivotal Role of Barrier Disruption and Subsequent TSLP Production
The fundamental lesion is currently thought to be an impaired barrier function that can be due to disrupted expression of essential barrier proteins like filaggrin (77, 78). Subsequent increased penetration of cutaneous and environmental antigens leads to the production of keratinocyte-derived cytokines including thymic stromal lymphopoietin (TSLP). TSLP is thought to be a critical factor driving the pathogenesis of atopic diseases. TSLP receptor is widely expressed in cells that contribute to AD (dendritic cells, T cells, B cells, mast cells, eosinophils, epithelial cells, and sensory neurons) utilizing the Jak-Stat pathway in humans. However, interestingly, it seems that murine TSLP receptor activates Stats by the Btk-family kinase Tec without the involvement of Janus kinases (79). Tamoxifen-induced keratinocyte-specific TSLP-deficient mice displayed drastically reduced allergic skin inflammation in a tape-stripping- and ovalbumin-induced AD model accompanied by the impairment of TH2 response and allergen-induced sensitization (80). In contrast, overexpression of TSLP in keratinocytes triggered massive itching behavior together with the development of AD-like dermatitis (81). Therefore, intradermal TSLP injection is often used to induce AD-like dermatitis in mice. Wilson et al. showed that it triggers itch sensation within minutes independently from the presence of adaptive immunity or mast cells (82). They also showed that TSLP receptors are present in sensory neurons innervating the skin and TSLP-evoked neuronal activation was responsible for itch sensation.
TH2 Mediated Allergic Responses—Key Role of TH2 Cytokines in AD
The importance of the TH2 pathway is further supported by the observation that transgenic mice overexpressing the TH2 cytokines IL-4 or IL-13 spontaneously develop skin inflammation that is frequently used as an animal model for AD (83). IL-4 and IL-13 are recognized by the type I cytokine receptor (IL-4 receptor) containing the γc subunit that signals through Jak1 and Jak3. Besides regulating IgE production in B cells and promoting the differentiation of TH2 lymphocytes, the IL4 receptor is also constitutively expressed by keratinocytes. Stimulation of IL-4 receptors leads to cytokine and chemokine production and downregulation of genes involved in keratinocyte differentiation (such as filaggrin) in vitro. This suggests that IL-4 receptor signaling in keratinocytes can further contribute to barrier impairment and inflammation in AD (84–87). Moreover, type 2 cytokines are also capable of activating sensory neurons directly depending on IL-4 receptor and Jak signaling, thus contributing to the development of chronic itch in AD (88). Dupilumab, a monoclonal antibody against the α subunit of the IL-4 receptors that blocks signaling from both IL-4 and IL-13 was effective in phase III studies (89, 90) and it was the first biologic agent approved by the FDA and EMA in the treatment of adults with moderate-to-severe atopic dermatitis (91).
Contribution of IgE-Mediated Signaling to the Allergic Response in AD
Elevated IgE levels and IgE autoreactivity were also suggested to contribute to the development and severity of AD. Beside mast cells and basophils, significant FcεRI expression has been shown in professional antigen presenting cells in atopic skin lesions. FcεRIs crosslinked with IgE are expected to use ITAM-dependent pathways including Src-family kinases and Syk to facilitate degranulation, internalization of allergens and antigen presentation promoting TH2 immunity (92).
Chronic Phase—Transition Into TH1-Type Inflammation
TH1 and IFNγ-mediated responses are thought to dominate the chronic phase of the disease (83). Animal models of hapten-, or allergen-induced contact dermatitis resemble pathogenetic features of both acute and chronic AD involving the disruption of the barrier as a primary event followed by sensitization, inflammation, increased epidermal proliferation, and changes in keratinocyte-differentiation (83).
The Jak-Stat Pathway in Preclinical Studies
The above mentioned pathogenetic features strongly suggest the role of the Jak-Stat pathway in AD (Figure 2). Accordingly, Yasuda et al. showed that gain of function mutation in Jak1 resulted in a spontaneous dermatitis phenotype (93). Generation of bone marrow chimeras revealed that Jak1 expression in non-hematopoietic cells was responsible for the development of dermatitis, but Jak1 acting in immune cells exacerbated the dermatitis symptoms and disease severity. They claimed that a possible molecular mechanism behind these findings was that hyperactivation of Jak1 pathway in epidermal keratinocytes resulted in a skin barrier defect due to the overexpression of serine proteases (93). The Jak3 inhibitor tofacitinib reduced ear-swelling and scratching behavior in an allergen-induced dermatitis model, especially upon topical application (94). Inhibition of Jak1 and Jak2 by topically applied ruxolitinib or momelotinib successfully decreased inflammation in a hapten-induced hypersensitivity model as well as in TSLP-induced dermatitis in mice together with the down-regulation of mRNA expression of IL-4, IL-5, IFNγ, and TSLP in the skin (95, 96). Oral administration of delgocitinib (JTE-052) efficiently reduced inflammation in a murine model of hapten-induced hypersensitivity. Moreover, delgocitinib inhibited proliferation and activation of T cells, but did not affect the number of DCs migrated to the draining lymph node during the sensitization phase (97). Delgocitinib was even found to be superior to conventional therapeutic agents like cyclosporine or tacrolimus ointment in hapten- and in TSLP-induced murine dermatitis models with respect to ear thickness, microscopic phenotype, inflammatory cytokine production, and serum IgE level (98). Another study suggested that delgocitinib might directly enhance keratinocyte differentiation in vitro (87). Delgocitinib treatment increased the expression of filaggrin and loricrin, genes that are known to be involved in keratinocyte differentiation, in primary human keratinocytes. In addition, IL-4 receptor-mediated downregulation of these genes was reversed upon delgocitinib treatment. Transepidermal water loss was also reduced upon delgocitinib treatment in vivo in a murine dry skin model which does not induce immune cell infiltration, indicating that Jak inhibition can improve skin barrier function independently of affecting immune cell activation (87). Taken together, the Jak-Stat pathway seems to be a central component of AD development, mediating multiple aspects of the pathogenesis such as type II cytokine signaling and TSLP-mediated inflammation, itching, keratinocyte disruption, and barrier impairment together with IFNγ-driven responses in the chronic phase. These findings strongly suggest a therapeutic utility of Jak inhibitors in human AD.
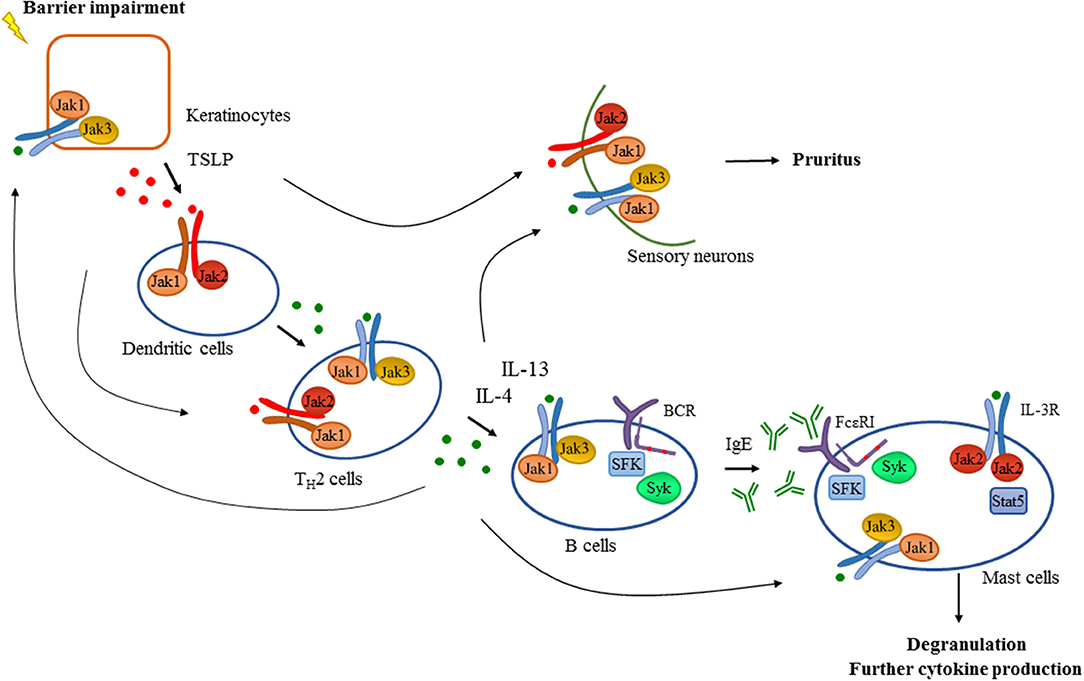
Figure 2. Tyrosine kinases in the development of atopic dermatitis. Barrier impairment is one of the initial steps in the pathogenesis of atopic dermatitis which leads to the production of TSLP by keratinocytes that activates dendritic cells in a Jak1/Jak2 dependent manner in the skin. Proinflammatory mediators produced by dendritic cells and keratinocytes drive the activation and differentiation of CD4+ T cells toward the TH2 phenotype. Activated TH2 cells produce several cytokines including IL-4 and IL-13 which promote isotype switch and IgE production in B cells acting through receptors with the common γ chain (γc) utilizing the Jak1/Jak3 heterodimer. Additionally, intact B cell receptor (BCR) signaling involving Src-family kinases (SFK) and Syk is essential for normal B cell development. Downstream effector cells like mast cells recognize IgE through FcεRI in a Src-family and Syk dependent manner leading to degranulation and the production of proinflammatory mediators contributing to the development of atopic dermatitis. There are several feedback loops and shortcuts in this pathway facilitating inflammation utilizing tyrosine kinases. TH2 cytokines also promote the activation of keratinocytes and effector immune cells like mast cells requiring intact Jak signaling. Activation of mast cells through IL-3R/Jak2/Stat5 pathway was also found to be important in allergen-induced dermatitis in mice. Furthermore, TSLP and TH2 cytokines acting through a Jak dependent manner in sensory neurons were found to mediate pruritus in atopic dermatitis in addition to mast cell-derived compounds such as histamine.
Jak Inhibitors in Clinical Trials
Given the substantial interest toward Jak inhibitors, a large amount of data is available from case reports, retrospective studies and open-label studies in the field of treatment of inflammatory skin diseases. In this review we are focusing on randomized, placebo-controlled phase II-III clinical trials that meet the standards of accepted evidence-based medicine (Table 3).
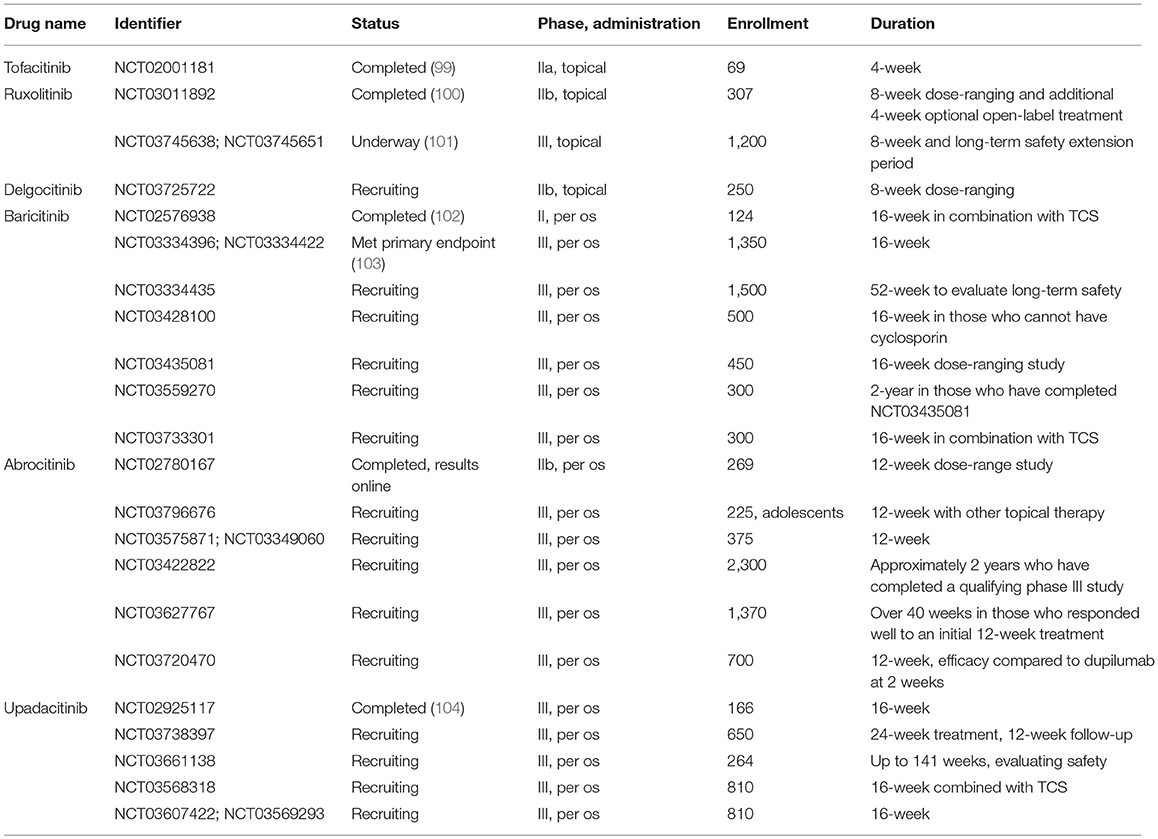
Table 3. Phase II and III clinical trials studying Jak inhibitors in atopic dermatitis.
Both topical tofacitinib and ruxolitinib treatment significantly improved skin inflammation and pruritus in AD patients in phase II studies (99, 100). In addition, the TRuE-AD phase III clinical trial has just started with the aim of assessing the efficacy of topical ruxolitinib treatment with long-term safety extension period in a larger cohort (101). Studies evaluating efficacy and safety of novel, more selective Jak inhibitors have also shown promising results. Patients treated with baricitinib in combination with topical corticosteroids achieved greater reduction in disease severity than corticosteroids alone (102). In another phase III trial, baricitinib as a monotherapy met its primary endpoint in AD patients (103). Per os abrocitinib and upadacitinib also significantly improved the Eczema Area and Severity Index and reduced pruritus compared to placebo [NCT02780167, (104), respectively]. Emerging number of studies are currently recruiting patients to evaluate efficacy and long-term safety up to 2 years in AD patients (Table 3).
Psoriasis
IL-23 Is Crucial to Activate IL-17-Mediated Effector Responses
Psoriasis is a common chronic inflammatory skin disease characterized by epidermal hyperplasia and parakeratosis together with the accumulation of inflammatory cells in the dermis, clinically causing red scaly papules and plaques. Activation of the IL-23/IL-17 axis plays a pivotal role in the pathogenesis of the disease (Figure 3). Resident immune cells such as dendritic cells and macrophages are considered to be initially activated through Toll like receptors that lead to the production of cytokines including IL-23 and TNFα that are known to be critical for IL-17 production in human psoriatic skin (105, 106). In fact, intradermal injection of IL-23 alone results in a skin pathology that strongly mimics human disease and is therefore commonly used as an animal model of psoriasis (107). IL-23 is known to induce the differentiation of TH17 cells that are considered as a major source of IL-17 in humans (106). However, activated dermal γδ T cells were also implicated as major IL-17-producing cells upon IL-23 stimulation in mouse models of psoriasis (108). IL-17 and IL-22 (another TH17 cytokine) then act as effectors inducing keratinocyte proliferation and hyperkeratosis. They also enhance the production of inflammatory cytokines and chemokines (including IL-1β, CCL20, and IL-8) by keratinocytes, leading to recruitment of other effector cells like neutrophils, further contributing to tissue damage and establishing the inflammatory milieu (109).
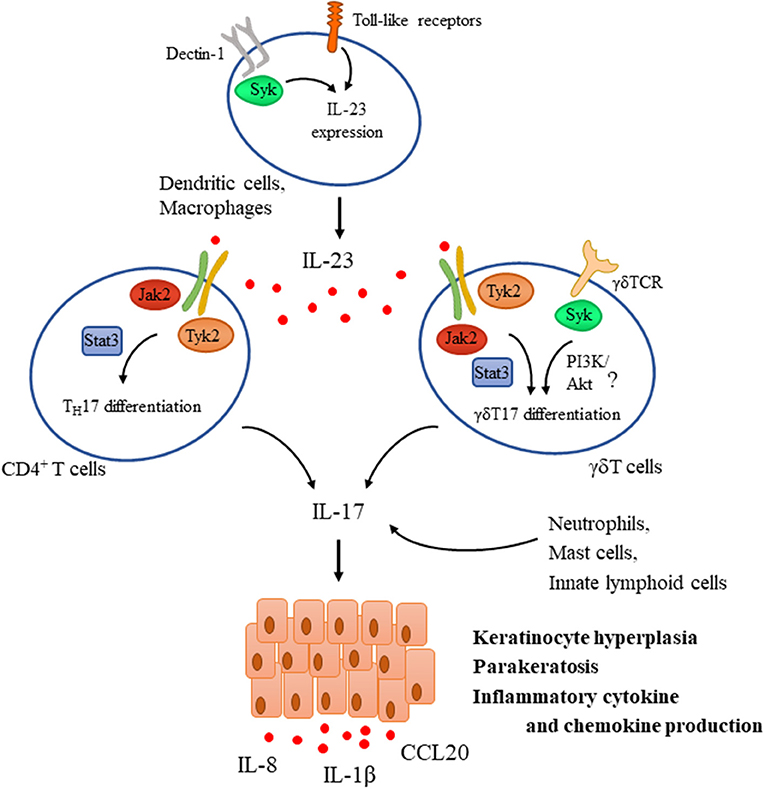
Figure 3. Tyrosine kinases in the IL-23/IL-17 axis during psoriasis pathogenesis. IL-23 is a key mediator driving psoriasis pathogenesis. It is expressed by dendritic cells upon their activation through Toll-like receptors (TLR) and Dectin-1, the latter one utilizing Syk. IL-23 promotes the differentiation of CD4+ T cells and γδT cells as well toward TH17 and γδT17, respectively via a Jak2/Tyk2-Stat3-mediated pathway and also induces the expression of IL-17. TCR activation in γδ T cells was also proposed to be important for mediating IL-17 production and psoriatic skin lesions, utilizing Syk and probably the PI3K/Akt pathway. In addition to TH17 and γδT17 cells, other sources of IL-17 include neutrophils, mast cells, and innate lymphoid cells. IL-17 then acts as an effector acting mainly on keratinocytes mediating hyperproliferation, parakeratosis, and production of several inflammatory chemokines and cytokines like IL-1β, IL-8, and CCL20.
Jak-Stat Signaling in Psoriasis
A number of novel biological therapies (including monoclonal antibodies against IL-23, IL-17, and IL-17R) have been approved for the treatment of psoriasis in the last few years (110). IL-23 receptors rely on Jak2/Tyk2 heterodimer-mediated signaling, implicating their role in the pathogenesis of the disease. Several genes of Jak-Stat signaling pathway have also shown to be associated with psoriasis (111). This was further supported by a genetic approach showing that Tyk2-deficient mice had significantly reduced ear swelling and epidermal hyperplasia upon injection with IL-23. In addition, infiltration of various leukocytes (including different T cell subsets, neutrophils, and macrophages) and the production of the pro-inflammatory cytokines IL-17 and IL-22 were also impaired in the absence of Tyk2 (112). Downstream signaling components like Stat3, a key factor in TH17 differentiation, was also found to be upregulated in human psoriatic lesions (113). Sano et al. showed that constitutive expression of Stat3 in keratinocytes resulted in a dermatitis phenotype closely resembling psoriasis (113). Epidermal hyperplasia, parakeratosis and dermal infiltration of immune cells occurred upon tape stripping or wounding of the skin, and in some mice, it occurred even spontaneously. The development of psoriatic lesions in these mice required both hyperactive Stat3 in keratinocytes and activated T cells in the dermis. Furthermore, inhibition of Stat3 with decoy oligonucleotides successfully inhibited disease development and even reversed disease severity showing that Stat3 may be an important regulator of genes in keratinocytes in the development of psoriasis cooperating with T cells (113).
Jak Inhibitors in the Treatment of Psoriasis
Pharmacological inhibition of the Jak-Stat pathway showed promising results in murine models of psoriasis reducing disease pathology, keratinocyte-activation, and proinflammatory cytokine levels (95, 98, 114). In addition, a number of clinical trials investigate the effect of different Jak inhibitors in psoriasis (Table 4). Short-term oral tofacitinib therapy resulted in significant clinical improvement in patients with moderate-to-severe plaque psoriasis (116) along with reducing epidermal thickness, DC and T cell numbers and the expression of psoriasis-related genes in the lesional skin (115). Several additional phase III studies were completed evaluating the long-term safety and efficacy of the treatment. Oral administration of tofacitinib was non-inferior to parenteral etanercept indicating that tofacitinib may provide a more convenient therapeutic option (117). Tofacitinib also demonstrated sustained efficacy in patients with psoriasis through up to 52 months and was well-tolerated with an acceptable safety profile detailed later (118, 121, 122, 130). In a 56-week withdrawal and retreatment study, patients who received continuous treatment maintained a response more effectively, however, 60% of patients who relapsed upon tofacitinib withdrawal recaptured a response with tofacitinib (119).
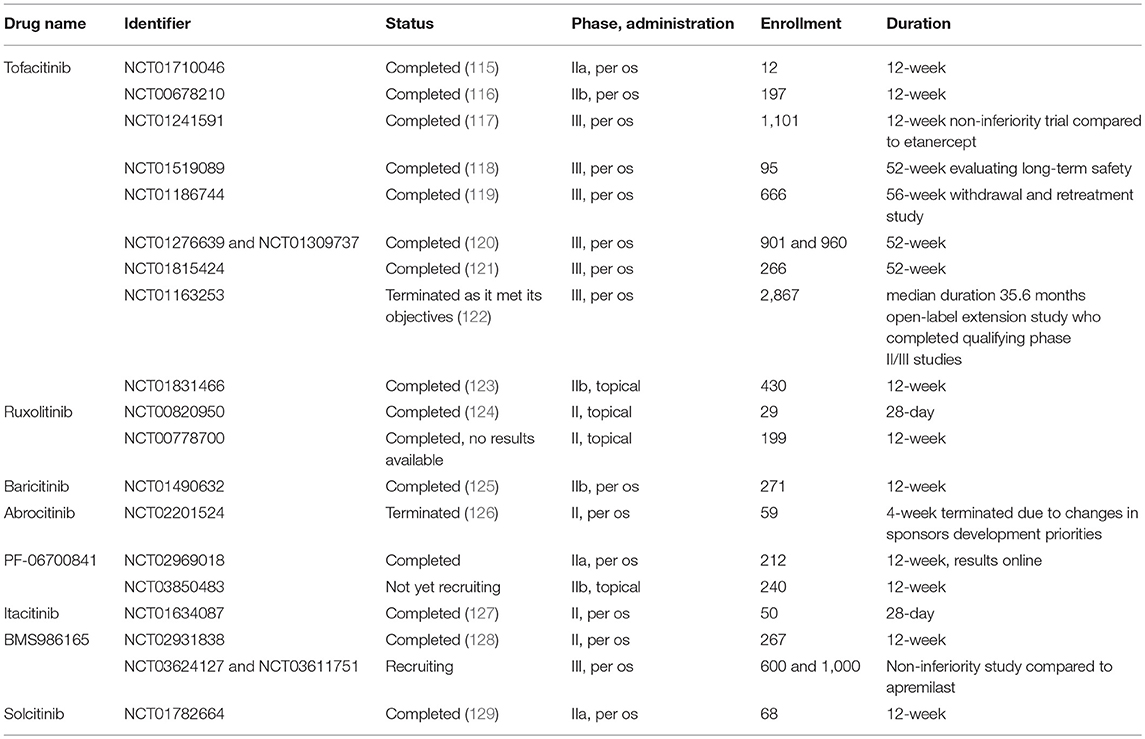
Table 4. Phase II and III clinical trials studying Jak inhibitors in psoriasis.
Topical treatment provides an excellent opportunity to overcome possible systemic adverse effects. Tofacitinib ointment also showed greater efficacy compared to vehicle at week 8, but failed to be superior to placebo at week 12 in a phase IIb study (123). Topical ruxolitinib was found to be well-tolerated, safe, and efficacious in short-term treatment in a smaller cohort of patients (124).
Oral treatment of novel, more selective inhibitors also improved symptoms and were well-tolerated in patients with psoriasis in phase II trials (125–127, 129). Since IL-12/IL-23-mediated signaling relies on Jak2/Tyk2 heterodimers, specific inhibition of these kinases may provide further improvement in psoriasis patients. Due to the critical role of Jak2 in hemopoietic development, inhibition of Tyk2 seem to be plausible in the treatment of psoriatic patients. Indeed, BMS986165 showed promising results in a 12-week phase II trial enrolling 267 patients (128) and a phase III study is currently recruiting patients to compare oral BMS986165 treatment to placebo and currently available treatment apremilast (NCT03624127 and NCT03611751).
Syk Tyrosine Kinase in IL-17-Mediated Inflammation
Little is known about the role of Syk in psoriasis. The pattern recognition receptor Dectin-1 was implicated in the disease process, suggesting that recognition of fungal antigens in a Syk and CARD9 dependent manner promotes the maturation of DCs and their ability to induce IL-17 production by TH17 cells (55). In addition, another source of IL-17 is γδ T cells, which utilize Syk as a dominant proximal kinase of the γδ TCR signaling pathway. Furthermore, skin inflammation was ameliorated in mice lacking the adaptor molecule RhoH that recruits Syk to the TCR in imiquimod-induced psoriasis model (131). These findings suggest that Syk may contribute to IL-17 production, but its actual relevance in case of psoriasis needs further investigation.
Role of Growth Factor Receptors in AD and Psoriasis
EGF receptor family members in keratinocytes facilitate epidermal differentiation and plays a crucial role in wound healing as well as in carcinogenesis. Though epidermal hyperplasia is a hallmark of both AD and psoriasis, the contribution of EGF receptor signaling to inflammatory skin disorders is poorly understood. Psoriatic lesions are known to overexpress EGFR and ligands like amphiregulin. Transgenic overexpression of amphiregulin in either basal or suprabasal epidermis causes severe psoriasis-like hyperplasia and skin inflammation in mice (132, 133). In line with that, neutralizing antibodies against amphiregulin reduce epidermal thickness of human psoriatic lesions transplanted onto mice with severe combined immunodeficiency (134). However, mice lacking another EGFR ligand epiregulin develop severe chronic dermatitis showing complicated modulating role of EGFR signaling pathways in the epidermis (135). Mice lacking epidermal EGFR spontaneously develop skin inflammation, decreased host defense and deficient skin barrier function (136). Proinflammatory cytokines such as IFNγ was also suggested to transactivate EGFR leading to the downregulation of the expression of chemokines like CCL2, CCL5, and CXCL10. In line with that, EGFR inhibition resulted in the aggravation of allergic contact dermatitis in mice by enhanced chemokine production of keratinocytes, promoting subsequent leukocyte recruitment (137). In contrast, IL-8 gene expression is actively induced by the EGFR ligands in keratinocytes (135, 137). In turn, IL-8 could contribute to activating the metalloprotease-dependent release of EGFR ligands by acting on its specific receptor in cancer cells (138), indicating the possibility of a positive feedback loop both for epidermal hyperplasia and neutrophil accumulation.
It is also well-known that EGFR inhibitor therapy in malignancies often causes inflammatory or toxic effects on the skin, and such side effects even act as strong predictors of good response to treatment (139).
Psoriasis and atopic dermatitis are both characterized by altered angiogenesis and lymphangiogenesis (140). Hyperplastic hyperpermeable dermal blood vessels can be detected in psoriatic skin lesions and transgenic delivery of VEGF to the skin results in a profound inflammatory skin condition resembling psoriasis (141) while topical application of VEGFR inhibitor successfully prevented disease development in the mouse model of psoriasis (142). Interestingly, stimulation of lymphangiogenesis by VEGFR-3 or via administration of its ligand VEGF-C inhibited inflammatory cell infiltration by oxazolone-induced skin inflammation (143, 144). This indicates that blood vessels contribute to the development of inflammatory environment by helping inflammatory cell infiltration while lymphatic vessels may limit skin inflammation by helping their elimination. Thus, selectivity can be especially important upon targeting VEGFRs in malignancies.
Alopecia Areata and Vitiligo
Both alopecia areata and vitiligo are characterized by IFNγ producing autoreactive cytotoxic T lymphocytes that attack hair follicles and melanocytes, respectively.
Alopecia areata (AA) is the main cause of non-scarring hair loss most commonly occurring in the scalp. The upregulation of several Jak-Stat pathway components downstream of γ-chain containing cytokines (which are known to promote the activity and survival of IFNγ-producing cytotoxic T cells) was detected in AA skin both from humans and mice. In a mouse model of AA, systemic administration of Jak inhibitors successfully prevented the development of disease. Moreover, both systemic and topical administration was able to reverse established disease and even promoted hair regrowth (145). Gene expression and immunofluorescent studies from mouse skin showed that expression and activity of the Jak-Stat pathway is dynamically changing during the hair follicle cycle. Moreover, Jak inhibition promoted the entry into the hair cycle and subsequent hair regrowth by activating hair follicle stem cells in healthy mice and also in lymphocyte-deficient mouse strains (146). This dual effect further justifies the investigation of Jak inhibitors in the treatment of AA. Some open label phase II clinical studies showed that Jak inhibitor treatment resulted in significant hair regrowth and improvement of AA in patients with better response if administered orally instead of topical formulations. Hair loss typically reoccurred after discontinuation of therapy within months (147–149). There are several active randomized, double-blind, placebo-controlled phase II clinical trials on the efficacy and safety of topical and oral Jak inhibitors in AA (Table 5), promising further insight into these issues in the near future.
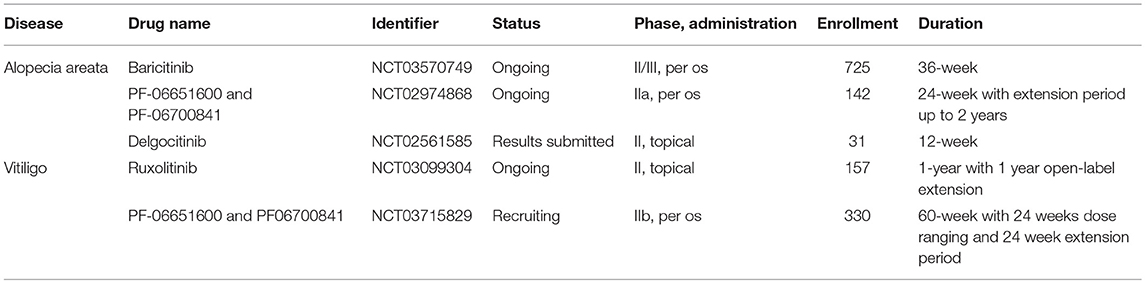
Table 5. Phase II and III clinical trials studying Jak inhibitors in alopecia areata and vitiligo.
In case of vitiligo cytotoxic T cell-mediated melanocyte destruction causes depigmentation leading to the occurrence of white spots throughout the body surface of patients. IFNγ produced by activated melanocyte-specific cytotoxic T lymphocytes is strongly implicated in the disease pathogenesis by promoting further T cell accumulation in the skin through IFNγ-dependent chemokines like CXCL10 (150). In addition, IFNγ was found to directly induce melanocyte senescence and apoptosis of primary human melanocytes which could be attenuated by siRNA against Jak2 and Stat1, but not Jak1. IFNγ treatment also resulted in the accumulation of reactive oxygen species and the production of proinflammatory cytokines like IL-6 which are considered as important contributing factors facilitating a vitiligo-prone environment in the skin (151).
Given the multiple role of IFNγ in vitiligo, inhibition of the Jak-Stat pathway may represent a promising therapeutic strategy. Beside some case reports and retrospective studies showing benefits of Jak inhibitors treating vitiligo patients (152, 153), so far only one open label phase II proof of concept pilot trial has been completed. This showed that topical ruxolitinib treatment provided significant repigmentation in facial vitiligo in a small cohort of patients (154). A randomized double-blind, dose-ranging, placebo-controlled phase II trial is now ongoing for evaluating the efficacy of ruxolitinib cream in vitiligo (NCT03099304). Another randomized controlled phase IIb trial is currently recruiting patients for evaluating per os treatment with novel selective Jak inhibitors (NCT03715829, also see in Table 5).
An additional important aspect of AA and vitiligo is that these diseases mostly cause cosmetic concern with emotional problems. Therefore, analyzing the safety and benefit profile of potential treatments is extremely important and Jak inhibitors should be very carefully tested in this regard.
Pemphigus
Pemphigus vulgaris and foliaceus are severe autoimmune blistering skin diseases with autoantibodies that are directed against the desmosomal cadherins, mainly desmoglein 3 (Dsg3) and Dsg1. These are required for the proper intercellular adhesion of keratinocytes and autoantibody deposition results in flaccid blister formation within the epidermis affecting mucous membranes and skin. Pemphigus vulgaris (PV) can be life threatening causing significant loss of the physical barrier. Application of artificial barriers like ointments can improve barrier function but there is an unmet need for definitive treatment.
Given the central role of Btk and Syk in B cell development and activation, these kinases may promote pathogenic antibody production in pemphigus. An inhibitor of Btk is under current evaluation in an open-label phase II clinical trial in pemphigus (NCT02704429) moreover, a randomized, double-blind placebo-controlled phase III study is already recruiting patients (NCT03762265).
IgG4 is the major subclass of autoantibodies in pemphigus and it is known to have limited ability to activate complement. Accordingly, acantholysis in pemphigus has been demonstrated to be independent from complement and Fc receptors (155, 156). Anti-Dsg3 IgG was able to directly induce the destruction of desmosomes by steric hindrance, promoting the internalization of Dsg3 (157, 158) and interfering with desmosome turnover (159). Recently it has been shown that Dsg3 transcription is negatively regulated by Stat3 in keratinocytes and corticosteroid treatment upregulates Dsg3 expression by inhibiting Stat3 through a yet unknown mechanism possibly involving Jaks (160). Those results suggest that specific inhibition of the Jak-Stat3 pathway may also be beneficial without the known adverse effects of steroids. TH2 cytokines like IL-4, IL-9, and IL-21, that utilize Jak-family kinases are known to contribute to the induction and regulation of autoantibody production in pemphigus (161, 162) therefore Jak inhibitors also serve as a potential treatment, but preclinical or clinical studies are yet to be done (163).
Binding of pathogenic IgG can also trigger outside-in signaling in keratinocytes eliciting acantholysis and apoptosis. Moreover, autoantibody binding also promoted secretion of inflammatory cytokines from keratinocytes, which may augment the pathogenic autoimmune response (164). However, the precise molecular mechanism is at present unclear. Several downstream mediators were implicated involving activation of EGFR-mediated signaling and focal adhesion kinase in keratinocytes (165–169). In a recent report, Src family kinases were implicated in autoantibody-mediated desmosome disassembly (170). Src phosphorylation was induced in keratinocytes upon in vitro antibody treatment obtained from PV patients. Loss of cell cohesion caused by anti-Dsg3 antibody was abolished upon Src inhibition by PP2 both in vitro and in vivo in a neonatal mouse model. However, inhibition of Src was not protective in some cases against PV-Ig-induced loss-of-keratinocyte-cohesion in keratinocyte monolayer, nor in intact human skin (170). These results underlie the need for further investigations of keratinocyte signaling in pemphigus pathogenesis.
Pemphigoid Diseases
Pemphigoid diseases are characterized by autoantibody production against distinct components of the dermal-epidermal junction leading to dermal-epidermal separation and tense blister formation (171).
Bullous pemphigoid (BP) is the most prevalent autoimmune bullous disease. Antibody formation directed against key hemidesmosomal components BP180 (also called type XVII collagen), and/or BP230 results in subepidermal blistering phenotype. Urticarial plaques and pruritus are also present in most of the cases which is a unique symptom among the pemphigoid group. Epidermolysis bullosa acquisita (EBA) is a very rare disease characterized by autoantibodies directed against type VII collagen (C7), the key anchoring fibril in the upper dermis, also causing subepidermal blister formation (172).
Essential Role of Antibody-Deposition in Pemphigoid Diseases
Antibody deposition to the basement membrane is the key feature in the pathogenesis, demonstrated by the fact that passive transfer of either anti-BP180 or anti-C7 antibodies isolated either from human patients or generated against human or murine antigens result in a severe blistering phenotype in mice (173–176). These approaches became useful experimental models to investigate the effector phase of autoantibody-mediated pemphigoid diseases. Several studies showed that deposition of antibodies are followed by the activation of the complement system and the recruitment of neutrophils mediating tissue damage and blister formation (177–181). Eosinophils and mast cells are also considered as contributors of blister formation by producing mediators facilitating further neutrophil recruitment and tissue damage (182–184). However, there are controversies in the literature especially regarding the role of mast cells (185). It has been proposed that autoantibodies can also directly affect epidermal cell—extracellular matrix integrity (186–188) and can trigger morphological and functional changes in keratinocytes (189) including IL-6 and IL-8 production (190) which can promote the recruitment of neutrophils (Figure 4).
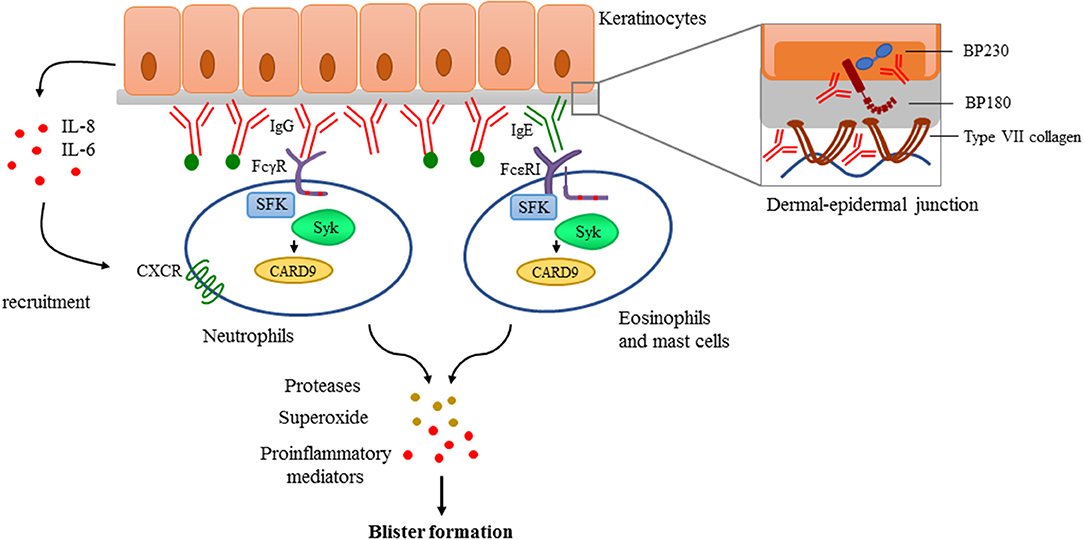
Figure 4. Antibody-mediated signaling in pemphigoid diseases. Pemphigoid diseases are characterized by autoantibody production against components of the dermal-epidermal junction (DEJ) like BP180 and BP230 in case of bullous pemphigoid, or against type VII collagen in patients with epidermolysis bullosa acquisita (Structure of the DEJ with special attention to the above-mentioned autoantigens is shown on the upper right panel). Autoantibody deposition along the DEJ results in the activation of the complement system (indicated with green dots) and initiation of keratinocyte-responses involving IL-6 and IL-8 production. This leads to recruitment of innate effector cells like neutrophils, eosinophils, and mast cells. Recognition of deposited immune complexes through Fc receptors utilizing Src-family kinases (SFK), Syk, and CARD9 is critical for the development of pemphigoid diseases, as shown by genetic studies using transgenic animals (see more detail in the text). Immune complex-mediated activation of effectors leads to the release of reactive oxygen species, proteases, and the production of proinflammatory mediators culminating into blister formation.
Pathways Mediating Immune-Complex Recognition and Leukocyte Migration
Signaling through complement receptors and recognition of deposited immune complexes by neutrophils through activating Fcγ receptors was essential for blister formation both in vitro using human cryosections and in in vivo mouse models (191–193). FcεRI signaling in mast cells, eosinophils and basophils have also been implicated in immune complex recognition because IgE isotypes of autoantibodies can also be detected in many patients in addition to IgG (194, 195). Actual pathogenetic relevance of IgE and FcεRI was further supported by the fact that omalizumab treatment was able to reduce the number of blisters and itching in BP patients (196). However, the role of other potential participants, namely β2 integrins, which are known to be involved in the migration of neutrophils to the inflamed area, is not entirely clear. Mac-1-deficient neonatal mice developed impaired neutrophil infiltration and were resistant to blister formation after 24 h of a single anti-BP180 treatment (197). In contrast, another study in the experimental model of EBA induced by repeated anti-C7 injections found that the absence of Mac-1 led to an even exacerbated disease phenotype (198). Therefore, further experiments are needed to reveal the role of β2 integrins in pemphigoid diseases.
Src-Family Kinases and Syk in Pemphigoid Diseases
Fc receptor-mediated signaling is strongly dependent of intracellular tyrosine kinases like Src-family kinases and Syk (24, 25). Given the central role of immune complex recognition by Fc receptors of resident and recruited innate immune cells, Src-family kinases and Syk may have an important role in the development of pemphigoid diseases. Indeed, triple knockout mice lacking Hck, Fgr, and Lyn, three Src-family kinases expressed in the myeloid compartment, were completely protected in an autoantibody induced model of EBA (32). Neutrophils lacking Hck, Fgr, and Lyn also failed to produce superoxide in response to C7-containing immune complexes (199), which has been shown to be important in mediating tissue damage in the skin (200). The Syk tyrosine kinase, which is recruited to ITAM sequences phosphorylated by Src-family kinases, was also found indispensable for the effector phase of the disease (199, 201). Analysis of Syk-deficient bone marrow chimeras revealed that Syk deficiency completely protected mice from anti-C7 antibody-induced skin disease and abrogated the accumulation of key cytokines and chemokines, as well as the infiltration of leukocytes, at the site of inflammation (199). Moreover, Syk deficient neutrophils failed to release CXCL2 or leukotriene B4 upon activation by immobilized C7-anti-C7 immune complexes in vitro. Integrin signaling also acts through Src kinases and Syk, however, in vivo migratory capacity either of Syk deficient or Src-family triple knockout neutrophils remained unaffected (199). Furthermore, neutrophil-specific expression of the CARD9 adaptor protein was also found to contribute to the development of the disease, as complete or neutrophil-specific CARD9 deletion partially protected mice from anti-C7 antibody-induced skin inflammation, likely due to CARD9-dependent regulation of neutrophil gene expression changes (202).
Taken together, Fc receptors and β2 integrins signal through Src-family kinases and Syk which are indispensable for immune-complex and adhesion-induced activation of effector cells (neutrophils, macrophages and, possibly, eosinophils and mast cells) without affecting their intrinsic migratory capacity. Src and Syk kinases are also responsible for the amplification of the inflammation process through the release of mediators that recruit neutrophils and/or directly damage dermal-epidermal junction in experimental pemphigoid models like proteases and superoxide. Therefore, these non-receptor tyrosine kinases may be good candidates for therapeutic intervention in the future, even though the development of specific inhibitors has yet to be solved.
Presence of Jaks in Pemphigoid Diseases
Several proinflammatory cytokine levels are elevated in blister fluid of BP patients such as IL-1β, IL-4, IL-6, IL-8, and TSLP (172, 203–205), and many of them act through the Jak-Stat pathway. In line with that, the expression of Jak-Stat proteins was found to be also elevated in skin lesions of BP patients (206). A meeting abstract discussed that pharmacological inhibition of Jak2 impaired the induction of EBA by antibody transfer and had therapeutic effects too in immunization-induced EBA model (207). However, there are no clinical studies using Jak inhibitors in pemphigoid diseases. A case report has been published about successfully treating a BP patient with anti-IL-4 antibody (208). The low number of clinical studies in general with novel therapeutic options is possibly due to the fact that BP usually affects elderly patients, therefore one should very carefully balance risk-benefit ratio in case of novel systemic treatments with special attention to inflammation and carcinogenesis.
Systemic Lupus Erythematosus
Almost all patients with systemic lupus erythematosus (SLE) develop lupus-specific cutaneous symptoms at some point in the disease course. There are also patients with cutaneous lupus that do not meet other diagnostic criteria for SLE. SLE is characterized by autoreactive B cells and autoantibody formation which proposes the role for Syk and Btk, non-receptor tyrosine kinases mediating BCR signaling, in disease development. The partially selective Syk inhibitor fostamatinib prevented the development of skin disease and significantly reduced established skin disease in lupus-prone mice (209). In addition, Btk inhibiton significantly attenuated the lupus-associated cutaneous disease phenotypes in mice (210). Systemic inflammatory conditions mediated by secretion of proinflammatory cytokines that act through Jaks is also important in SLE cutaneous manifestations (211–215). Indeed, Jak inhibition by ruxolitinib prevented the development of cutaneous lupus lesions in lupus-prone mice (216). However, baricitinib failed to improve skin manifestations in systemic lupus patients in a phase II trial, despite the fact that the overall systemic symptoms were effectively reduced by the drug (217).
Neutrophilic Dermatoses
Neutrophilic dermatoses represent a group of disorders characterized by massive neutrophil infiltration in the skin without evidence of infection. Signs of systemic inflammation often accompanies skin pathology and they are often associated with malignancies and autoimmune diseases like RA and inflammatory bowel diseases. Interestingly, autoinflammatory diseases share various common features with neutrophilic dermatoses suggesting some similarities in the pathogenesis (218). Since an excellent review has been currently published about mechanisms of inflammation in neutrophil dermatoses (219), here we are focusing on evidence about the importance of tyrosine kinases.
Abnormalities in neutrophil function is obviously implicated in the pathogenesis. The SH2 domain-containing tyrosine phosphatase SHP-1 is essential for inhibiting proinflammatory signal transduction and loss-of-function mutation of SHP-1 in mice causes severe cutaneous inflammation resembling human neutrophilic dermatoses. Kanneganti and her group showed that IL-1α signaling through IL-1R and adaptor protein Myd88 drives inflammation in this model where tyrosine phosphorylation of Myd88 is counterregulated by SHP-1 and Syk (220). Dysfunction of SHP-1 leads to the release of Syk from inhibition resulting in excessive expression of proinflammatory mediators and other effector molecules. Downstream of Syk, the CARD9 adaptor protein was also found to be a key mediator in cutaneous inflammation in the aforementioned model (221).
Cytokine dysregulation are considered as contributing factors to the development of the disease. Elevation of serum G-CSF was detected in patients with active disease of unknown origin (222), moreover, G-CSF is a common cause of drug-induced neutrophilic dermatosis (223). G-CSFR signals through Jak2 suggesting a possible role of Jak2 creating cytokine dysregulation. In addition, increased expression of IFNγ was also described in patients and rare genetic autoinflammatory diseases characterized by high IFNγ production also represent neutrophilic dermatosis where Jak inhibitor therapy significantly improved symptoms in some cases (224).
Adverse Events During JAK Inhibitor Treatment in Inflammatory Diseases
Safety information from long-term studies of Jak inhibitor treatment are limited due to the novelty of these drugs. Most data are available from RA patients (225), but there is slowly emerging information from patients with skin inflammation and several long-term safety-assessing trials are currently recruiting patients (Tables 3–5).
Safety profile is considered generally acceptable, infections and laboratory abnormalities can be observed as major adverse events (226). The increased risk of infections was similar to that observed by the use of biologics, with the exception of higher risk for varicella zoster infection which should be taken into consideration. Cytopenias, mostly anemia and neutropenia relatively often occur likely due to Jak2 inhibition but rarely severe. Elevated serum cholesterol level has been also mentioned in patients treated with Jak inhibitors (227). It was usually sustained after the first few months and was controllable by statin therapy. Among less frequent adverse events thromboembolism was documented, however most patients already suffer from increased risk due to chronic systemic inflammation which further complicates the picture (228). One of the most serious concerns is the possibility of the development of malignancies upon long-term Jak inhibitor treatment. Current studies have not shown higher risk (225), but longer follow-up is needed to properly address this matter.
Taken together, Jak inhibitor treatment has not higher risk than biologics overall. However, skin diseases often present as less severe but chronic symptoms where risk/benefit ratio should be carefully considered upon choosing appropriate treatment. Nevertheless, the possibility of topical application represents an excellent opportunity which deserve further elaboration.
Conclusion and Future Directions
The highly effective treatment of various malignancies by tyrosine kinase inhibitors and the regulatory approval of Jak inhibitors for the targeted therapy of rheumatoid arthritis has generated major interest in the therapeutic targeting of other diseases, including inflammatory skin diseases, by tyrosine kinase inhibitors. Besides the most extensively studied Jak inhibitors, compounds targeting other kinases such as Syk, Src-family kinases, or Btk are also expected to emerge as new therapeutics for inflammatory dermatitis. The more and more detailed understanding of individual kinase family members and the development of novel inhibitors with more specifically tailored specificities toward individual kinases are expected to lead to more refined therapies driving the field toward personalized targeted therapeutic approaches. Skin diseases also provide unique opportunities for the development of novel small molecule therapeutics, mostly through the opportunity of topical application without systemic side effects. On the other hand, the cosmetic aspects of skin diseases and the role of skin and mucous membranes as critical barriers between the internal and external environment also present substantial challenges during the development of novel therapeutics. Taken together, the scientific community may expect exciting major advances in the field of understanding and targeting tyrosine kinases in inflammatory skin diseases in the coming years.
Author Contributions
KS and AM wrote the manuscript. TN provided intellectual advice.
Funding
This work was supported by the Hungarian National Research, Development and Innovation Office (NVKP_16-2016-1-0039 to AM), the Hungarian Ministry of National Economy (VEKOP-2.3.2-16-2016-00002 to AM), and the Hungarian Higher Education Institutional Excellence Program (to AM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of our research group for inspiring discussions. We dedicate this work to the memory of Tibor Vántus who played a fundamental role in promoting the development of tyrosine kinase inhibitors to target inflammatory processes and inflammatory disease pathogenesis in our group.
References
1. Gadina M. Advances in kinase inhibition: treating rheumatic diseases and beyond. Curr Opin Rheumatol. (2014) 26:237–43. doi: 10.1097/BOR.0000000000000023
2. Mocsai A, Kovacs L, Gergely P. What is the future of targeted therapy in rheumatology: biologics or small molecules? BMC Med. (2014) 12:43. doi: 10.1186/1741-7015-12-43
3. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. (2017) 16:843–62. doi: 10.1038/nrd.2017.201
4. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. (1998) 93:373–83. doi: 10.1016/S0092-8674(00)81166-6
5. Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. (1998) 93:385–95. doi: 10.1016/S0092-8674(00)81167-8
6. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. (1998) 93:397–409. doi: 10.1016/S0092-8674(00)81168-X
7. Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. (1995) 377:65–8. doi: 10.1038/377065a0
8. Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. (1995) 270:797–800. doi: 10.1126/science.270.5237.797
9. Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science. (1995) 270:800–2. doi: 10.1126/science.270.5237.800
10. Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. (1995) 3:771–82. doi: 10.1016/1074-7613(95)90066-7
11. Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. (2000) 13:561–71. doi: 10.1016/S1074-7613(00)00055-8
12. Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. (2000) 13:549–60. doi: 10.1016/S1074-7613(00)00054-6
13. Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. (2003) 4:471–7. doi: 10.1038/ni910
14. Shaw MH, Boyartchuk V, Wong S, Karaghiosoff M, Ragimbeau J, Pellegrini S, et al. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc Natl Acad Sci USA. (2003) 100:11594–9. doi: 10.1073/pnas.1930781100
15. Virtanen A, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. (2019) 33:15–32. doi: 10.1007/s40259-019-00333-w
16. Yu V, Pistillo J, Archibeque I, Han Lee J, Sun B-C, Schenkel LB, et al. Differential selectivity of JAK2 inhibitors in enzymatic and cellular settings. Exp Hematol. (2013) 41:491–500. doi: 10.1016/j.exphem.2013.01.005
17. Yarilina A, Xu K, Chan C, Ivashkiv LB. Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum. (2012) 64:3856–66. doi: 10.1002/art.37691
18. LaBranche TP, Jesson MI, Radi ZA, Storer CE, Guzova JA, Bonar SL, et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. (2012) 64:3531–42. doi: 10.1002/art.34649
19. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. (2011) 186:4234–43. doi: 10.4049/jimmunol.1003668
20. Milici AJ, Kudlacz EM, Audoly L, Zwillich S, Changelian P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther. (2008) 10:R14. doi: 10.1186/ar2365
21. Gomez-Puerta JA, Mocsai A. Tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Curr Top Med Chem. (2013) 13:760–73. doi: 10.2174/15680266113139990094
22. Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: the present and future of jakinibs. J Leukoc Biol. (2018) 104:499–514. doi: 10.1002/JLB.5RI0218-084R
23. Lowell CA. Src-family kinases: rheostats of immune cell signaling. Mol Immunol. (2004) 41:631–43. doi: 10.1016/j.molimm.2004.04.010
24. Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
25. Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. (2005) 26:208–14. doi: 10.1016/j.it.2005.02.002
26. Futosi K, Mocsai A. Tyrosine kinase signaling pathways in neutrophils. Immunol Rev. (2016) 273:121–39. doi: 10.1111/imr.12455
27. Jakus Z, Fodor S, Abram CL, Lowell CA, Mocsai A. Immunoreceptor-like signaling by β2 and β3 integrins. Trends Cell Biol. (2007) 17:493–501. doi: 10.1016/j.tcb.2007.09.001
28. Mocsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. (1999) 162:1120–6.
29. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol. (2014) 192:919–28. doi: 10.4049/jimmunol.1301979
30. Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, et al. Fcγ receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J Exp Med. (2000) 191:669–82. doi: 10.1084/jem.191.4.669
31. Mocsai A, Jakus Z, Vantus T, Berton G, Lowell CA, Ligeti E. Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol. (2000) 164:4321–31. doi: 10.4049/jimmunol.164.8.4321
32. Kovacs M, Nemeth T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med. (2014) 211:1993–2011. doi: 10.1084/jem.20132496
33. Lindauer M, Hochhaus A. Dasatinib. Recent Results Cancer Res. (2018) 212:29–68. doi: 10.1007/978-3-319-91439-8_2
34. Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. (2006) 66:11314–22. doi: 10.1158/0008-5472.CAN-06-1199
35. Amsberg GK, Schafhausen P. Bosutinib in the management of chronic myelogenous leukemia. Biologics. (2013) 7:115–22. doi: 10.2147/BTT.S30182
36. Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. (1996) 271:695–701. doi: 10.1074/jbc.271.2.695
37. Brandvold KR, Steffey ME, Fox CC, Soellner MB. Development of a highly selective c-Src kinase inhibitor. ACS Chem Biol. (2012) 7:1393–8. doi: 10.1021/cb300172e
38. Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. (2006) 319:998–1008. doi: 10.1124/jpet.106.109058
39. Sharman J, Hawkins M, Kolibaba K, Boxer M, Klein L, Wu M, et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. (2015) 125:2336–43. doi: 10.1182/blood-2014-08-595934
40. Connell NT, Berliner N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood. (2019) 133:2027–30. doi: 10.1182/blood-2018-11-852491
41. Currie KS, Kropf JE, Lee T, Blomgren P, Xu J, Zhao Z, et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J Med Chem. (2014) 57:3856–73. doi: 10.1021/jm500228a
42. Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol. (2017) 10:145. doi: 10.1186/s13045-017-0512-1
43. Coffey G, DeGuzman F, Inagaki M, Pak Y, Delaney SM, Ives D, et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J Pharmacol Exp Ther. (2012) 340:350–9. doi: 10.1124/jpet.111.188441
44. Liao C, Hsu J, Kim Y, Hu DQ, Xu D, Zhang J, et al. Selective inhibition of spleen tyrosine kinase (SYK) with a novel orally bioavailable small molecule inhibitor, RO9021, impinges on various innate and adaptive immune responses: implications for SYK inhibitors in autoimmune disease therapy. Arthritis Res Ther. (2013) 15:R146. doi: 10.1186/ar4329
45. Reilly MP, Sinha U, Andre P, Taylor SM, Pak Y, Deguzman FR, et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. (2011) 117:2241–6. doi: 10.1182/blood-2010-03-274969
46. Lam B, Arikawa Y, Cramlett J, Dong Q, de Jong R, Feher V, et al. Discovery of TAK-659 an orally available investigational inhibitor of Spleen Tyrosine Kinase (SYK). Bioorg Med Chem Lett. (2016) 26:5947–50. doi: 10.1016/j.bmcl.2016.10.087
47. Kaplan JB, Gordon LI, Infante JR, Popat R, Rambaldi A, Madan S, et al. Updated results from a phase 1 study of TAK-659, an investigational and reversible SYK inhibitor, in Patients (Pts) with advanced solid tumor or lymphoma malignancies. Blood. (2016) 128:624.
48. Futosi K, Nemeth T, Pick R, Vantus T, Walzog B, Mocsai A. Dasatinib inhibits proinflammatory functions of mature human neutrophils. Blood. (2012) 119:4981–91. doi: 10.1182/blood-2011-07-369041
49. Azizi G, Goudarzvand M, Afraei S, Sedaghat R, Mirshafiey A. Therapeutic effects of dasatinib in mouse model of multiple sclerosis. Immunopharmacol Immunotoxicol. (2015) 37:287–94. doi: 10.3109/08923973.2015.1028074
50. da Silva AL, Magalhaes RF, Branco VC, Silva JD, Cruz FF, Marques PS, et al. The tyrosine kinase inhibitor dasatinib reduces lung inflammation and remodelling in experimental allergic asthma. Br J Pharmacol. (2016) 173:1236–47. doi: 10.1111/bph.13430
51. Guo K, Bu X, Yang C, Cao X, Bian H, Zhu Q, et al. Treatment effects of the second-generation tyrosine kinase inhibitor dasatinib on autoimmune arthritis. Front Immunol. (2018) 9:3133. doi: 10.3389/fimmu.2018.03133
52. Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. (1995) 378:298–302. doi: 10.1038/378298a0
53. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. (1995) 378:303–6. doi: 10.1038/378303a0
54. Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, Mocsai A, et al. Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science. (2003) 299:247–51. doi: 10.1126/science.1079477
55. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. (2007) 8:630–8. doi: 10.1038/ni1460
56. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. (2009) 459:433–6. doi: 10.1038/nature07965
57. Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. (2002) 16:547–58. doi: 10.1016/S1074-7613(02)00303-5
58. Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. (2006) 7:1326–33. doi: 10.1038/ni1407
59. Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. (1997) 186:1027–39. doi: 10.1084/jem.186.7.1027
60. Jakus Z, Simon E, Balazs B, Mocsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum. (2010) 62:1899–910. doi: 10.1002/art.27438
61. Nemeth T, Futosi K, Szilveszter K, Vilinovszki O, Kiss-Papai L, Mocsai A. Lineage-specific analysis of Syk function in autoantibody-induced arthritis. Front Immunol. (2018) 9:555. doi: 10.3389/fimmu.2018.00555
62. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. (2011) 187:4319–30. doi: 10.4049/jimmunol.1100341
63. Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, et al. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. (2008) 58:3309–18. doi: 10.1002/art.23992
64. Coffey G, Rani A, Betz A, Pak Y, Haberstock-Debic H, Pandey A, et al. PRT062607 achieves complete inhibition of the spleen tyrosine kinase at tolerated exposures following oral dosing in healthy volunteers. J Clin Pharmacol. (2017) 57:194–210. doi: 10.1002/jcph.794
65. Coffey G, Betz A, DeGuzman F, Pak Y, Inagaki M, Baker DC, et al. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J Pharmacol Exp Ther. (2014) 351:538–48. doi: 10.1124/jpet.114.218164
66. Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front Immunol. (2019) 10:95. doi: 10.3389/fimmu.2019.00095
67. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. (1993) 72:279–90. doi: 10.1016/0092-8674(93)90667-F
68. Vetrie D, Vorechovsky I, Sideras, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. (1993) 361:226–33. doi: 10.1038/364362a0
69. Middendorp S, Dingjan GM, Hendriks RW. Impaired precursor B cell differentiation in Bruton's tyrosine kinase-deficient mice. J Immunol. (2002) 168:2695–703. doi: 10.4049/jimmunol.168.6.2695
70. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. (2012) 119:3744–56. doi: 10.1182/blood-2011-12-397919
71. Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, et al. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. (2011) 117:1329–39. doi: 10.1182/blood-2010-04-281170
72. Corneth OBJ, Klein Wolterink RGJ, Hendriks RW. BTK Signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. (2016) 393:67–105. doi: 10.1007/82_2015_478
73. Horwood NJ, Urbaniak AM, Danks L. Tec family kinases in inflammation and disease. Int Rev Immunol. (2012) 31:87–103. doi: 10.3109/08830185.2012.670334
74. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
75. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. (2014) 79:34–74. doi: 10.1016/j.phrs.2013.11.002
76. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. (2018) 4:1. doi: 10.1038/s41572-018-0001-z
77. Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38:441–6. doi: 10.1038/ng1767
78. Sandilands A, O'Regan GM, Liao H, Zhao Y, Terron-Kwiatkowski A, Watson RM, et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J Invest Dermatol. (2006) 126:1770–5. doi: 10.1038/sj.jid.5700459
79. Wohlmann A, Sebastian K, Borowski A, Krause S, Friedrich K. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol Chem. (2010) 391:181–6. doi: 10.1515/bc.2010.029
80. Leyva-Castillo JM, Hener, Jiang H, Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol. (2013) 133:154–63. doi: 10.1038/jid.2012.239
81. Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. (2005) 202:541–9. doi: 10.1084/jem.20041503
82. Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. (2013) 155:285–95. doi: 10.1016/j.cell.2013.08.057
83. Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. (2009) 129:31–40. doi: 10.1038/jid.2008.106
84. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. (2007) 120:150–5. doi: 10.1016/j.jaci.2007.04.031
85. Bao L, Shi VY, Chan LS. IL-4 regulates chemokine CCL26 in keratinocytes through the Jak1, 2/Stat6 signal transduction pathway: implication for atopic dermatitis. Mol Immunol. (2012) 50:91–7. doi: 10.1016/j.molimm.2011.12.008
86. Bao L, Shi VY, Chan LS. IL-4 up-regulates epidermal chemotactic, angiogenic, and pro-inflammatory genes and down-regulates antimicrobial genes in vivo and in vitro: relevant in the pathogenesis of atopic dermatitis. Cytokine. (2013) 61:419–25. doi: 10.1016/j.cyto.2012.10.031
87. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. (2015) 136:667–77.e7. doi: 10.1016/j.jaci.2015.03.051
88. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. (2017) 171:217–28.e13. doi: 10.1016/j.cell.2017.08.006
89. Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. (2014) 371:130–9. doi: 10.1056/NEJMoa1314768
90. Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. (2017) 389:2287–303. doi: 10.1111/imj.67_13578
91. Shirley M. Dupilumab: first global approval. Drugs. (2017) 77:1115–21. doi: 10.1007/s40265-017-0768-3
92. Novak N, Tepel C, Koch S, Brix K, Bieber T, Kraft S. Evidence for a differential expression of the FcepsilonRIgamma chain in dendritic cells of atopic and nonatopic donors. J Clin Invest. (2003) 111:1047–56. doi: 10.1172/JCI200315932
93. Yasuda T, Fukada T, Nishida K, Nakayama M, Matsuda M, Miura I, et al. Hyperactivation of JAK1 tyrosine kinase induces stepwise, progressive pruritic dermatitis. J Clin Invest. (2016) 126:2064–76. doi: 10.1172/JCI82887
94. Fukuyama T, Ehling S, Cook E, Baumer W. Topically administered Janus-Kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. (2015) 354:394–405. doi: 10.1124/jpet.115.223784
95. Fridman JS, Scherle PA, Collins R, Burn T, Neilan CL, Hertel D, et al. Preclinical evaluation of local JAK1 and JAK2 inhibition in cutaneous inflammation. J Invest Dermatol. (2011) 131:1838–44. doi: 10.1038/jid.2011.140
96. Jin W, Huang W, Chen L, Jin M, Wang Q, Gao Z, et al. Topical application of JAK1/JAK2 inhibitor momelotinib exhibits significant anti-inflammatory responses in DNCB-induced atopic dermatitis model mice. Int J Mol Sci. (2018) 19:E3973. doi: 10.3390/ijms19123973
97. Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, et al. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. (2016) 84:258–65. doi: 10.1016/j.jdermsci.2016.09.007
98. Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, et al. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: comparison with conventional therapeutic agents. Exp Dermatol. (2018) 27:22–29. doi: 10.1111/exd.13370
99. Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. (2016) 175:902–11. doi: 10.1111/bjd.14871
100. Incyte Corporation. Incyte Announces Positive Data from Phase 2b Trial of Ruxolitinib Cream in Patients with Atopic Dermatitis (2018). Available online at: https://www.businesswire.com/news/home/20180913005058/en/Incyte-Announces-Positive-Data-Phase-2b-Trial
101. Incyte Corporation. Incyte Announces First Patient Treated in Phase 3 Clinical Trial Program for Ruxolitinib Cream in Atopic Dermatitis (2018). Available online at: https://www.businesswire.com/news/home/20181220005644/en/
102. Incyte Corporation. Baricitinib Meets Primary Endpoint in Phase 2 Study of Patients with Moderate-to-Severe Atopic Dermatitis (2017). Available onlilne at: https://www.prnewswire.com/news-releases/baricitinib-meets-primary-endpoint-in-phase-2-study-of-patients-with-moderate-to-severe-atopic-dermatitis-300518896.html
103. Lilly Announces Top-Line Phase 3 Results for Baricitinib in Patients with Moderate to Severe Atopic Dermatitis. Eli Lilly & Company (2019). Available online at: https://www.prnewswire.com/news-releases/lilly-announces-top-line-phase-3-results-for-baricitinib-in-patients-with-moderate-to-severe-atopic-dermatitis-300788783.html
104. AbbVie. AbbVie's Upadacitinib (ABT-494) Meets Primary Endpoint in Phase 2b Study in Atopic Dermatitis (2017). Available online at: https://news.abbvie.com/news/abbvies-upadacitinib-abt-494-meets-primary-endpoint-in-phase-2b-study-in-atopic-dermatitis.htm
105. Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, et al. Psoriasis. Nat Rev Dis Primers. (2016) 2:16082. doi: 10.1038/nrdp.2016.82
106. Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. (2009) 129:1339–50. doi: 10.1038/jid.2009.59
107. Singh TP, Zhang HH, Hwang ST, Farber JM. IL-23- and imiquimod-induced models of experimental psoriasis in mice. Curr Protoc Immunol. (2019) 125:e71. doi: 10.1002/cpim.71
108. Cai YH, Shen XY, Ding CL, Qi CJ, Li KJ, Li X, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. (2011) 35:596–610. doi: 10.1016/j.immuni.2011.10.006
109. Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, et al. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat Immunol. (2018) 19:1286–98. doi: 10.1038/s41590-018-0256-2
110. Rendon A, Schakel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. (2019) 20:E1475. doi: 10.3390/ijms20061475
111. Singh S, Pradhan D, Puri P, Ramesh V, Aggarwal S, Nayek A, et al. Genomic alterations driving psoriasis pathogenesis. Gene. (2019) 683:61–71. doi: 10.1016/j.gene.2018.09.042
112. Ishizaki M, Muromoto R, Akimoto T, Sekine Y, Kon S, Diwan M, et al. Tyk2 is a therapeutic target for psoriasis-like skin inflammation. Int Immunol. (2014) 26:257–67. doi: 10.1093/intimm/dxt062
113. Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. (2005) 11:43–9. doi: 10.1038/nm1162
114. Works MG, Yin F, Yin CC, Yiu Y, Shew K, Tran TT, et al. Inhibition of TYK2 and JAK1 ameliorates imiquimod-induced psoriasis-like dermatitis by inhibiting IL-22 and the IL-23/IL-17 axis. J Immunol. (2014) 193:3278–87. doi: 10.4049/jimmunol.1400205
115. Krueger J, Clark JD, Suarez-Farinas M, Fuentes-Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: a randomized phase 2 study. J Allergy Clin Immunol. (2016) 137:1079–90. doi: 10.1016/j.jaci.2015.12.1318
116. Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol. (2012) 167:668–77. doi: 10.1111/j.1365-2133.2012.11168.x
117. Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. (2015) 386:552–61. doi: 10.1016/S0140-6736(14)62113-9
118. Asahina A, Etoh T, Igarashi A, Imafuku S, Saeki H, Shibasaki Y, et al. Oral tofacitinib efficacy, safety and tolerability in Japanese patients with moderate to severe plaque psoriasis and psoriatic arthritis: a randomized, double-blind, phase 3 study. J Dermatol. (2016) 43:869–80. doi: 10.1111/1346-8138.13258
119. Bissonnette R, Iversen L, Sofen H, Griffiths CE, Foley P, Romiti R, et al. Tofacitinib withdrawal and retreatment in moderate-to-severe chronic plaque psoriasis: a randomized controlled trial. Br J Dermatol. (2015) 172:1395–406. doi: 10.1111/bjd.13551
120. Papp KA, Menter MA, Abe M, Elewski B, Feldman SR, Gottlieb AB, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. (2015) 173:949–61. doi: 10.1111/bjd.14018
121. Zhang J, Tsai TF, Lee MG, Zheng M, Wang G, Jin H, et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: a Phase 3, randomized, double-blind, placebo-controlled study. J Dermatol Sci. (2017) 88:36–45. doi: 10.1016/j.jdermsci.2017.05.004
122. Valenzuela F, Korman NJ, Bissonnette R, Bakos N, Tsai TF, Harper MK, et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: long-term safety and efficacy in an open-label extension study. Br J Dermatol. (2018) 179:853–62. doi: 10.1111/bjd.16798
123. Papp KA, Bissonnette R, Gooderham M, Feldman SR, Iversen L, Soung J, et al. Treatment of plaque psoriasis with an ointment formulation of the Janus kinase inhibitor, tofacitinib: a Phase 2b randomized clinical trial. BMC Dermatol. (2016) 16:15. doi: 10.1186/s12895-016-0051-4
124. Punwani N, Scherle P, Flores R, Shi J, Liang J, Yeleswaram S, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. (2012) 67:658–64. doi: 10.1016/j.jaad.2011.12.018
125. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2016) 174:1266–76. doi: 10.1111/bjd.14403
126. Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br J Dermatol. (2018) 179:54–62. doi: 10.1111/bjd.16004
127. Bissonnette R, Luchi M, Fidelus-Gort R, Jackson S, Zhang H, Flores R, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J Dermatolog Treat. (2016) 27:332–8. doi: 10.3109/09546634.2015.1115819
128. Papp K, Gordon K, Thaci D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. (2018) 379:1313–21. doi: 10.1056/NEJMoa1806382
129. Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br J Dermatol. (2016) 174:985–95. doi: 10.1111/bjd.14399
130. Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol. (2016) 74:841–50. doi: 10.1016/j.jaad.2016.01.013
131. Muro R, Nitta T, Nakano K, Okamura T, Takayanagi H, Suzuki H. γδ TCR recruits the Syk/PI3K axis to drive proinflammatory differentiation program. J Clin Invest. (2018) 128:415–26. doi: 10.1172/JCI95837
132. Cook PW, Piepkorn M, Clegg CH, Plowman GD, DeMay JM, Brown JR, et al. Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J Clin Invest. (1997) 100:2286–94. doi: 10.1172/JCI119766
133. Cook PW, Brown JR, Cornell KA, Pittelkow MR. Suprabasal expression of human amphiregulin in the epidermis of transgenic mice induces a severe, early-onset, psoriasis-like skin pathology: expression of amphiregulin in the basal epidermis is also associated with synovitis. Exp Dermatol. (2004) 13:347–56. doi: 10.1111/j.0906-6705.2004.00183.x
134. Bhagavathula N, Nerusu KC, Fisher GJ, Liu G, Thakur AB, Gemmell L, et al. Amphiregulin and epidermal hyperplasia: amphiregulin is required to maintain the psoriatic phenotype of human skin grafts on severe combined immunodeficient mice. Am J Pathol. (2005) 166:1009–16. doi: 10.1016/S0002-9440(10)62322-X
135. Pastore S, Mascia F, Mariotti F, Dattilo C, Mariani V, Girolomoni G. ERK1/2 regulates epidermal chemokine expression and skin inflammation. J Immunol. (2005) 174:5047–56. doi: 10.4049/jimmunol.174.8.5047
136. Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med. (2013) 5:199ra111. doi: 10.1126/scitranslmed.3005886
137. Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. (2003) 163:303–12. doi: 10.1016/S0002-9440(10)63654-1
138. Tanida S, Joh T, Itoh K, Kataoka H, Sasaki M, Ohara H, et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology. (2004) 127:559–69. doi: 10.1053/j.gastro.2004.05.017
139. Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer. (2006) 6:803–12. doi: 10.1038/nrc1970
140. Varricchi G, Granata F, Loffredo S, Genovese A, Marone G. Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J Am Acad Dermatol. (2015) 73:144–53. doi: 10.1016/j.jaad.2015.03.041
141. Xia YP, Li B, Hylton D, Detmar M, Yancopoulos GD, Rudge JS. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood. (2003) 102:161–8. doi: 10.1182/blood-2002-12-3793
142. Halin C, Fahrngruber H, Meingassner JG, Bold G, Littlewood-Evans A, Stuetz A, et al. Inhibition of chronic and acute skin inflammation by treatment with a vascular endothelial growth factor receptor tyrosine kinase inhibitor. Am J Pathol. (2008) 173:265–77. doi: 10.2353/ajpath.2008.071074
143. Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, et al. An important role of lymphatic vessel activation in limiting acute inflammation. Blood. (2011) 117:4667–78. doi: 10.1182/blood-2010-10-316356
144. Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med. (2010) 207:2255–69. doi: 10.1084/jem.20100559
145. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
146. Harel S, Higgins CA, Cerise JE, Dai Z, Chen JC, Clynes R, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. (2015) 1:e1500973. doi: 10.1126/sciadv.1500973
147. Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. (2016) 1:e89776. doi: 10.1172/jci.insight.89776
148. Mackay-Wiggan J, Jabbari A, Nguyen N, Cerise JE, Clark C, Ulerio G, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. (2016) 1:e89790. doi: 10.1172/jci.insight.89790
149. Liu LY, Craiglow BG, King BA. Tofacitinib 2% ointment, a topical Janus kinase inhibitor, for the treatment of alopecia areata: a pilot study of 10 patients. J Am Acad Dermatol. (2018) 78:403–4.e1. doi: 10.1016/j.jaad.2017.10.043
150. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra23. doi: 10.1126/scitranslmed.3007811
151. Wang S, Zhou M, Lin F, Liu D, Hong W, Lu L, et al. Interferon-γ induces senescence in normal human melanocytes. PLoS ONE. (2014) 9:e93232. doi: 10.1371/journal.pone.0093232
152. Craiglow BG, King BA. Tofacitinib Citrate for the treatment of vitiligo: a pathogenesis-directed therapy. JAMA Dermatol. (2015) 151:1110–2. doi: 10.1001/jamadermatol.2015.1520
153. Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J Am Acad Dermatol. (2017) 77:675–82.e1. doi: 10.1016/j.jaad.2017.05.043
154. Rothstein B, Joshipura D, Saraiya A, Abdat R, Ashkar H, Turkowski Y, et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J Am Acad Dermatol. (2017) 76:1054–60.e1. doi: 10.1016/j.jaad.2017.02.049
155. Rock B, Labib RS, Diaz LA. Monovalent Fab' immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest. (1990) 85:296–9. doi: 10.1172/JCI114426
156. Anhalt GJ, Till GO, Diaz LA, Labib RS, Patel HP, Eaglstein NF. Defining the role of complement in experimental pemphigus vulgaris in mice. J Immunol. (1986) 137:2835–40.
157. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. (2006) 281:7623–34. doi: 10.1074/jbc.M512447200
158. Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. (2009) 129:908–18. doi: 10.1038/jid.2008.339
159. Spindler V, Waschke J. Pemphigus-A disease of desmosome dysfunction caused by multiple mechanisms. Front Immunol. (2018) 9:136. doi: 10.3389/fimmu.2018.00136
160. Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight. (2017) 2:92253. doi: 10.1172/jci.insight.92253
161. Satyam A, Khandpur S, Sharma VK, Sharma A. Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-Pemphigus vulgaris. Immunol Invest. (2009) 38:498–509. doi: 10.1080/08820130902943097
162. Tavakolpour S, Tavakolpour V. Interleukin 4 inhibition as a potential therapeutic in pemphigus. Cytokine. (2016) 77:189–95. doi: 10.1016/j.cyto.2015.09.017
163. Tavakolpour S. Tofacitinib as the potent treatment for refractory pemphigus: a possible alternative treatment for pemphigus. Dermatol Ther. (2018) 31:e12696. doi: 10.1111/dth.12696
164. Vodo D, Sarig O, Geller S, Ben-Asher E, Olender T, Bochner R, et al. Identification of a functional risk variant for pemphigus vulgaris in the ST18 gene. PLoS Genet. (2016) 12:e1006008. doi: 10.1371/journal.pgen.1006008
165. Gil MP, Modol T, Espana A, Lopez-Zabalza MJ. Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol. (2012) 21:254–9. doi: 10.1111/j.1600-0625.2012.01441.x
166. Frusic-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y. Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis–possible involvement of the EGF receptor. Autoimmunity. (2006) 39:563–75. doi: 10.1080/08916930600971836
167. Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem. (2007) 282:13804–12. doi: 10.1074/jbc.M611365200
168. Rubenstein DS, Diaz LA. Pemphigus antibody induced phosphorylation of keratinocyte proteins. Autoimmunity. (2006) 39:577–86. doi: 10.1080/08916930600971885
169. Sanchez-Carpintero I, Espana A, Pelacho B, Lopez Moratalla N, Rubenstein DS, Diaz LA, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol. (2004) 151:565–70. doi: 10.1111/j.1365-2133.2004.06147.x
170. Kugelmann D, Rotzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of Src and cortactin in pemphigus skin blistering. Front Immunol. (2019) 10:626. doi: 10.3389/fimmu.2019.00626
171. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
172. Bagci IS, Horvath ON, Ruzicka T, Sardy M. Bullous pemphigoid. Autoimmun Rev. (2017) 16:445–55. doi: 10.1016/j.autrev.2017.03.010
173. Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun. (2008) 31:331–8. doi: 10.1016/j.jaut.2008.08.009
174. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. (1993) 92:2480–8. doi: 10.1172/JCI116856
175. Woodley DT, Ram R, Doostan A, Bandyopadhyay P, Huang Y, Remington J, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol. (2006) 126:1323–30. doi: 10.1038/sj.jid.5700254
176. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. (2005) 115:870–8. doi: 10.1172/JCI200521386
177. Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest. (1995) 95:1539–44. doi: 10.1172/JCI117826
178. Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. (1997) 100:1256–63. doi: 10.1172/JCI119639
179. Li Q, Ujiie H, Shibaki A, Wang G, Moriuchi R, Qiao HJ, et al. Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J Immunol. (2010) 185:7746–55. doi: 10.4049/jimmunol.1000667
180. Sitaru C, Chiriac MT, Mihai S, Buning J, Gebert A, Ishiko A, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. (2006) 177:3461–8. doi: 10.4049/jimmunol.177.5.3461
181. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. (2007) 178:6514–21. doi: 10.4049/jimmunol.178.10.6514
182. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med. (2018) 5:201. doi: 10.3389/fmed.2018.00201
183. Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. (2001) 108:1151–8. doi: 10.1172/JCI11494
184. Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem. (2011) 286:15003–9. doi: 10.1074/jbc.M111.221036
185. Kasprick A, Yu X, Scholten J, Hartmann K, Pas HH, Zillikens D, et al. Conditional depletion of mast cells has no impact on the severity of experimental epidermolysis bullosa acquisita. Eur J Immunol. (2015) 45:1462–70. doi: 10.1002/eji.201444769
186. Hiroyasu S, Ozawa T, Kobayashi H, Ishii M, Aoyama Y, Kitajima Y, et al. Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathol. (2013) 182:828–40. doi: 10.1016/j.ajpath.2012.11.029
187. Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol. (2014) 193:4415–28. doi: 10.4049/jimmunol.1400095
188. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol. (2009) 129:919–26. doi: 10.1038/jid.2008.305
189. Tie D, Da X, Natsuga K, Yamada N, Yamamoto O, Morita E. Bullous pemphigoid IgG induces cell dysfunction and enhances the motility of epidermal keratinocytes via Rac1/proteasome activation. Front Immunol. (2019) 10:200. doi: 10.3389/fimmu.2019.00200
190. Schmidt E, Reimer S, Kruse N, Jainta S, Brocker EB, Marinkovich MP, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol. (2000) 115:842–8. doi: 10.1046/j.1523-1747.2000.00141.x
191. Zhao ML, Trimbeger ME, Li N, Diaz LA, Shapiro SD, Liu Z. Role of FcRs in animal model of autoimmune bullous pemphigoid. J Immunol. (2006) 177:3398–405. doi: 10.4049/jimmunol.177.5.3398
192. Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Kohl J, et al. Fcγ receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol. (2014) 184:2185–96. doi: 10.1016/j.ajpath.2014.05.007
193. Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol. (2002) 161:301–11. doi: 10.1016/S0002-9440(10)64182-X
194. Dimson OG, Giudice GJ, Fu CL, Van den Bergh F, Warren SJ, Janson MM, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol. (2003) 120:784–8. doi: 10.1046/j.1523-1747.2003.12146.x
195. Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. (2017) 177:1644–53. doi: 10.1111/bjd.15924
196. Yu KK, Crew AB, Messingham KA, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol. (2014) 71:468–74. doi: 10.1016/j.jaad.2014.04.053
197. Liu Z, Zhao M, Li N, Diaz LA, Mayadas TN. Differential roles for beta2 integrins in experimental autoimmune bullous pemphigoid. Blood. (2006) 107:1063–9. doi: 10.1182/blood-2005-08-3123
198. Deng FY, Chen Y, Zheng JF, Huang QNA, Cao XT, Zillikens D, et al. CD11b-deficient mice exhibit an increased severity in the late phase of antibody transfer-induced experimental epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1175–8. doi: 10.1111/exd.13434
199. Nemeth T, Virtic O, Sitaru C, Mocsai A. The Syk tyrosine kinase is required for skin inflammation in an in vivo mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:2131–9. doi: 10.1016/j.jid.2017.05.017
200. Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathol. (2007) 212:56–65. doi: 10.1002/path.2157
201. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-genome expression profiling in skin reveals sYK as a key regulator of inflammation in experimental epidermolysis Bullosa acquisita. Front Immunol. (2018) 9:249. doi: 10.3389/fimmu.2018.00249
202. Nemeth T, Futosi K, Sitaru C, Ruland J, Mocsai A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun. (2016) 7:11004. doi: 10.1038/ncomms11004
203. Schmidt E, Bastian B, Dummer R, Tony HP, Brocker EB, Zillikens D. Detection of elevated levels of IL-4, IL-6, and IL-10 in blister fluid of bullous pemphigoid. Arch Dermatol Res. (1996) 288:353–7. doi: 10.1007/BF02507102
204. Schmidt E, Mittnacht A, Schomig H, Dummer R, Brocker EB, Zillikens D. Detection of IL-1 alpha, IL-1 beta and IL-1 receptor antagonist in blister fluid of bullous pemphigoid. J Dermatol Sci. (1996) 11:142–7. doi: 10.1016/0923-1811(95)00435-1
205. Schmidt E, Ambach A, Bastian B, Brocker EB, Zillikens D. Elevated levels of interleukin-8 in blister fluid of bullous pemphigoid compared with suction blisters of healthy control subjects. J Am Acad Dermatol. (1996) 3:310–2. doi: 10.1016/S0190-9622(96)80146-0
206. Juczynska K, Wozniacka A, Waszczykowska E, Danilewicz M, Wagrowska-Danilewicz M, Wieczfinska J, et al. Expression of the JAK/STAT signaling pathway in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflamm. (2017) 2017:6716419. doi: 10.1155/2017/6716419
207. Samavedam U, Scheuber J, Seavey M, Koga H, Witte M, Schulze F, et al. Therapeutic efficacy of a novel selective JAK2 inhibitor (CEP-33779) in organ-specific, autoantibody-induced tissue injury. J Invest Dermatol. (2014) 134:S16.
208. Kaye A, Gordon SC, Deverapalli SC, Her MJ, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. (2018) 154:1225–6. doi: 10.1001/jamadermatol.2018.2526
209. Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum. (2010) 62:2086–92. doi: 10.1002/art.27452
210. Chalmers SA, Wen J, Doerner J, Stock A, Cuda CM, Makinde HM, et al. Highly selective inhibition of Bruton's tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res Ther. (2018) 20:10. doi: 10.1186/s13075-017-1500-0
211. Yu C, Chang C, Zhang J. Immunologic and genetic considerations of cutaneous lupus erythematosus: a comprehensive review. J Autoimmun. (2013) 41:34–45. doi: 10.1016/j.jaut.2013.01.007
212. Deng GM, Tsokos GC. Pathogenesis and targeted treatment of skin injury in SLE. Nat Rev Rheumatol. (2015) 11:663–9. doi: 10.1038/nrrheum.2015.106
213. Robinson ES, Werth VP. The role of cytokines in the pathogenesis of cutaneous lupus erythematosus. Cytokine. (2015) 73:326–34. doi: 10.1016/j.cyto.2015.01.031
214. Chasset F, Arnaud L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev. (2018) 17:44–52. doi: 10.1016/j.autrev.2017.11.009
215. Deng GM. Pathogenesis of skin injury of systemic lupus erythematosus. Curr Rheumatol Rep. (2018) 20:5. doi: 10.1007/s11926-018-0713-9
216. Chan ES, Herlitz LC, Jabbari A. Ruxolitinib attenuates cutaneous lupus development in a mouse lupus model. J Invest Dermatol. (2015) 135:1912–5. doi: 10.1038/jid.2015.107
217. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
218. Satoh TK, Mellett M, Contassot E, French LE. Are neutrophilic dermatoses autoinflammatory disorders? Br J Dermatol. (2018) 178:603–13. doi: 10.1111/bjd.15105
219. Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol. (2019) 10:1059. doi: 10.3389/fimmu.2019.01059
220. Gurung P, Fan G, Lukens JR, Vogel P, Tonks NK, Kanneganti TD. Tyrosine kinase SYK licenses MyD88 adaptor protein to instigate IL-1α-mediated inflammatory disease. Immunity. (2017) 46:635–48. doi: 10.1016/j.immuni.2017.03.014
221. Tartey S, Gurung P, Samir P, Burton A, Kanneganti TD. Cutting edge: dysregulated CARD9 signaling in neutrophils drives inflammation in a mouse model of neutrophilic dermatoses. J Immunol. (2018) 201:1639–44. doi: 10.4049/jimmunol.1800760
222. Kawakami T, Ohashi S, Kawa Y, Takahama H, Ito M, Soma Y, et al. Elevated serum granulocyte colony-stimulating factor levels in patients with active phase of sweet syndrome and patients with active behcet disease: implication in neutrophil apoptosis dysfunction. Arch Dermatol. (2004) 140:570–4. doi: 10.1001/archderm.140.5.570
223. White JM, Mufti GJ, Salisbury JR, du Vivier AW. Cutaneous manifestations of granulocyte colony-stimulating factor. Clin Exp Dermatol. (2006) 31:206–7. doi: 10.1111/j.1365-2230.2005.01996.x
224. Boyadzhiev M, Marinov L, Boyadzhiev V, Iotova V, Aksentijevich I, Hambleton S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr Rheumatol Online J. (2019) 17:19. doi: 10.1186/s12969-019-0322-9
225. Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Curr Opin Chem Biol. (2016) 32:29–33. doi: 10.1016/j.cbpa.2016.03.006
226. Strober BE, Gottlieb AB, van de Kerkhof PCM, Puig L, Bachelez H, Chouela E, et al. Benefit-risk profile of tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: pooled analysis across six clinical trials. Br J Dermatol. (2019) 180:67–75. doi: 10.1111/bjd.17149
227. Wolk R, Armstrong EJ, Hansen PR, Thiers B, Lan S, Tallman AM, et al. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J Clin Lipidol. (2017) 11:1243–56. doi: 10.1016/j.jacl.2017.06.012
Keywords: tyrosine kinases, signaling, Syk, Jak, Src-family, dermatitis, psoriasis, autoimmune blistering diseases
Citation: Szilveszter KP, Németh T and Mócsai A (2019) Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 10:1862. doi: 10.3389/fimmu.2019.01862
Received: 24 April 2019; Accepted: 23 July 2019;
Published: 09 August 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Zhi Liu, University of North Carolina at Chapel Hill, United StatesFrank Petersen, Forschungszentrum Borstel (LG), Germany
Copyright © 2019 Szilveszter, Németh and Mócsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kata P. Szilveszter, c3ppbHZlc3p0ZXIua2F0YUBtZWQuc2VtbWVsd2Vpcy11bml2Lmh1; Attila Mócsai, bW9jc2FpLmF0dGlsYUBtZWQuc2VtbWVsd2Vpcy11bml2Lmh1