 Laura Escoter-Torres1†
Laura Escoter-Torres1† Giorgio Caratti2†
Giorgio Caratti2† Aikaterini Mechtidou1†
Aikaterini Mechtidou1† Jan Tuckermann2
Jan Tuckermann2 Nina Henriette Uhlenhaut1,3*
Nina Henriette Uhlenhaut1,3* Sabine Vettorazzi2*
Sabine Vettorazzi2*- 1Molecular Endocrinology, Helmholtz Zentrum München (HMGU), German Center for Diabetes Research (DZD), Institute for Diabetes and Cancer IDC, Munich, Germany
- 2Department of Biology, Institute for Comparative Molecular Endocrinology, University of Ulm, Ulm, Germany
- 3Gene Center, Ludwig-Maximilians-Universität (LMU), Munich, Germany
For many decades, glucocorticoids have been widely used as the gold standard treatment for inflammatory conditions. Unfortunately, their clinical use is limited by severe adverse effects such as insulin resistance, cardiometabolic diseases, muscle and skin atrophies, osteoporosis, and depression. Glucocorticoids exert their effects by binding to the Glucocorticoid Receptor (GR), a ligand-activated transcription factor which both positively, and negatively regulates gene expression. Extensive research during the past several years has uncovered novel mechanisms by which the GR activates and represses its target genes. Genome-wide studies and mouse models have provided valuable insight into the molecular mechanisms of inflammatory gene regulation by GR. This review focusses on newly identified target genes and GR co-regulators that are important for its anti-inflammatory effects in innate immune cells, as well as mutations within the GR itself that shed light on its transcriptional activity. This research progress will hopefully serve as the basis for the development of safer immune suppressants with reduced side effect profiles.
Introduction
Glucocorticoids as Immunomodulators
Glucocorticoids (GCs) are steroid hormones secreted in a diurnal and stress responsive manner, under the control of the hypothalamic-pituitary-adrenal (HPA) axis (1).GCs regulate numerous essential physiological and developmental processes, ranging from lung maturation to glucose metabolism and immune responses. This is clearly demonstrated in mice with abrogated GC signaling, which die perinatally due to pulmonary atelectasis (2). The effect on lung maturation is not merely limited to mice: in clinical practice, pre-term neonates are given GCs to accelerate pulmonary development (3). In adult mammals, endogenous GCs play important homeostatic roles. For instance, GCs increase glucose production through glycogenolysis and gluconeogenesis in the liver upon fasting, and as part of daily rhythmic energy mobilization (4, 5).
Pharmacologically, GCs are widely used to treat acute and chronic inflammatory diseases, such as asthma, allergies, rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis etc., due to their potent anti-inflammatory actions. In addition, GCs are commonly prescribed to prevent graft-vs.-host immune responses after organ transplantation and for certain cancer types, such as lymphoma (6, 7). Currently, it is estimated that 1–3% of the adult Western population are receiving GCs, demonstrating their broad applications (8). GCs have been used for over 70 years as anti-inflammatory drugs, despite their adverse effects on systemic metabolism, which were noted soon after their first clinical use (9). Long term exposure to GCs induces adipocyte hypertrophy, glucose intolerance and insulin resistance, hypertension, muscle and skin atrophy, osteoporosis, glaucoma, impaired wound healing and psychological effects such as mood changes, insomnia, and depression (4, 10). Long term GC exposure due to increased secretion from endocrine tumors or chronic exogenous administration, often causes a pathological condition known as Cushing's syndrome (11). Cushing's manifests as debilitating muscle wasting, fat accumulation, and susceptibility to infection and can be fatal if left untreated.
Separating beneficial therapeutic properties from detrimental side effects based on a molecular understanding of GC action is a long-term goal of biomedical research. Furthermore, the glucocorticoid receptor (GR) has been key to understanding the basic molecular concepts of GC action. There have been several paradigm shifts of the molecular understanding of GC/GR mechanisms since cloning of the receptor more than 30 years ago (12). The generation of GR mutants that interfere with specific functions of the receptor, the introduction of several mutants into preclinical models and the characterization of genome wide profiles all revolutionized our view of GC action. In this review, we summarize recent insights into the anti-inflammatory effects of GR, focusing on mechanisms of macrophage gene regulation, GR co-regulators, novel GR target genes, and mouse models of inflammation. We also summarize the current understanding of immune modulatory mechanism in the innate immune system based on mouse mutants. These might explain why, despite much progress, developing novel immune modulators that match the efficacy of GCs but avoid the adverse effects remains a major challenge for the field.
The Glucocorticoid Receptor
The endogenous GC, cortisol in humans and corticosterone in rodents, binds to the GR, encoded by the NR3C1 gene. GR belongs to the nuclear receptor superfamily of ligand activated transcription factors. It consists of three major domains, the central DNA binding domain (DBD), the N-terminal transactivation domain (NTD), and the C-terminal ligand binding domain (LBD) [(12); Figure 1].
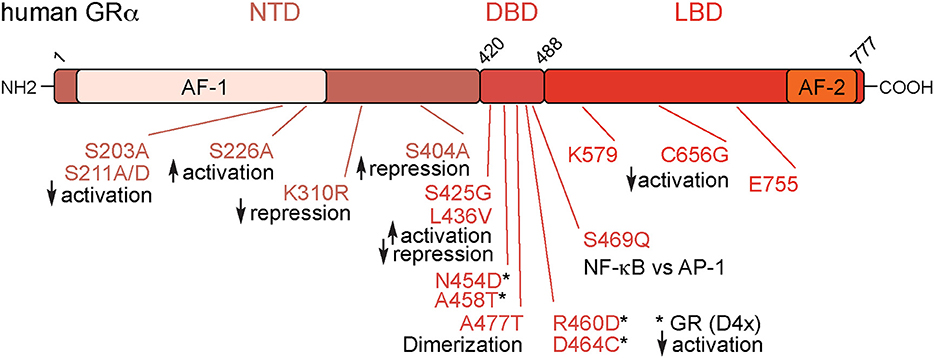
Figure 1. Overview of the glucocorticoid receptor protein. The Glucocorticoid Receptor (GR) is organized into three main domains: the N-terminal Transactivation Domain (NTD), the DNA-Binding Domain (DBD), and the Ligand Binding Domain (LBD). In addition, there are the transactivation domains 1 and 2 (AF-1 and AF-2). These mutations numbered above are relevant for GR's immunomodulatory effects. Numbers are amino acids of the human protein.
The NR3C1 gene encodes several isoforms that are generated by alternative splicing and alternative initiation of translation (10, 13). The full-length isoform GRα-A is the focus of this review. GRβ, a second splice variant, and other GR isoforms, are known to modify GC sensitivity, but are discussed in detail elsewhere (14).
In the absence of ligand, GR resides in the cytoplasm, bound to heat shock proteins 70 and 90 (Hsp70 and Hsp90) together with other chaperones and immunophilins (15). Upon binding of GCs, GR translocates to the nucleus where it binds to DNA sequences. In this way, GR is recruited to target gene enhancers and promoters where it can both activate and repress transcription (16, 17). Canonical binding sites for the GR are called glucocorticoid response elements (GREs) and are composed of two 6bp palindromes (half sites) separated by a 3bp spacer, with the consensus AGAACAnnnTGTTCT. However, GR binding sites (GBS) in the genome vary to a certain degree of motif mismatch, expanding the number of possible target sequences. Furthermore, the context of neighboring transcription factor binding sites and the ensuing crosstalk is relevant for the regulation of inflammatory genes by the GR. The beauty of using GR as a model transcription factor is that its ability to regulate genes can be easily controlled in vitro and in vivo by the absence or presence of the GC ligand.
Chromatin Residence Time and Multimerization of the Glucocorticoid Receptor
GR, along with other transcription factors, was assumed to bind DNA in a relatively static manner, “sitting down” for long periods of time to regulate gene expression. However, visualization of the dynamics of fluorescent-tagged GR in living cells led to the insight that occupancy of dimeric GR molecules at GREs is rather in the order of seconds and less (18). Only a small portion of available molecules are specifically bound to chromatin at a given time, suggesting that transcription factors and co-factors have a transient rather than stable interaction at genomic response elements (19).
GR acts as a monomer (20), dimer (21, 22), and even tetramer (23–25) depending on the subcellular localization, presence of ligand, GREs, or artificial response elements such as the MMTV array. Interestingly, DNA binding was proposed to trigger allosteric regulation of GR, followed by a change in its oligomeric state (24). Ligand bound GR is mainly nuclear and dimeric. Interestingly, upon DNA binding, the structural LBD rearrangement promotes the formation of higher order oligomers, predominantly tetramers, through unstudied LBD surfaces (25). The physiological relevance and implications of a tetrameric GR, however, are still open for debate and further investigation.
In general, chromatin binding and gene regulation by GR appear to be much more dynamic than previously thought, and the residence time of GR on chromatin may have differential effects. The LBD seems to regulate the number of GR molecules bound at a specific genomic region, which may also affect the transcription of target genes.
Glucocorticoid Receptor Co-regulators
All nuclear receptors (NRs), including GR, require a host of co-activators and co-repressors to ultimately control the transcriptional apparatus.
Steroid receptor coactivator-1 (SRC-1, also known as nuclear receptor co-activator 1, NCOA1) was one of the first identified (26), followed by glucocorticoid receptor interacting protein (GRIP1, SRC-2, and NCOA2) (27). Originally found to be a co-activator of the progesterone receptor (PR), SRC-1, and GRIP1 were shown to directly interact with GR and other steroid receptors. This direct co-activator interaction with GR depends on the evolutionarily conserved LXXLL motif, or NR-box, and without this motif, GR loses transcriptional activity (28). SRC-1 directly activates genes with its histone acetyltransferase (HAT) domain that decondenses chromatin [(29); Figure 2].
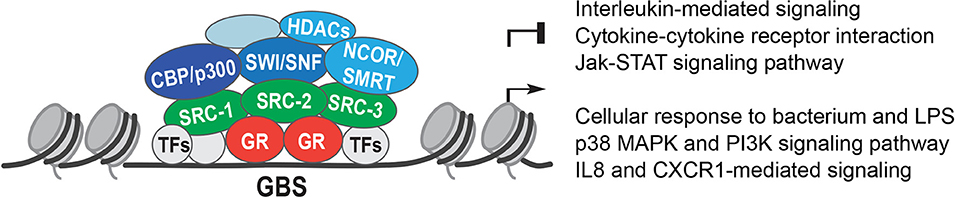
Figure 2. Glucocorticoid receptor co-regulators. The Glucocorticoid Receptor (GR) binds to Glucocorticoid Receptor Binding Sites (GBS) in open chromatin. GR interacts with other transcription factors (TFs) and recruits co-activators or co-repressors, such as: the Steroid Receptor co-activators 1, 2, and 3 (SRC-1, SRC-2, and SRC-3); the histone acetyl transferases CREB binding protein (CBP) and p300; the Nuclear Receptor co-repressors NCOR1 and NCOR2 (NCOR, SMRT), which recruit histone deacetylases 1 and 3 (HDACs); and the SWItch/Sucrose-Non Fermentable (SWI/SNF) chromatin remodeling complex.
The strength of GR's interaction with SRC-1 and GRIP1 might determine the steroid responsiveness of cancer cells, suggesting that the loss of GC-induced apoptosis or growth arrest is due to, at least in part, co-activator recruitment (30). However, GR seems to preferably interact with GRIP1 over SRC-1, while the opposite is true for PR, which confers selectivity of GR activation and PR activation on chromatin modifications (31).
Importantly, the co-activator GRIP1 can also act as a co-repressor. Depending on the individual GR target gene, GRIP1 functions as either an activator or repressor by using its co-repressor domain. For example, GRIP1 was described to act as a co-repressor at the osteocalcin promoter (32). Moreover, the functionality of GRIP1 is modulated by post-translational modifications. CDK9 mediated phosphorylation of GRIP1 was shown to increase GR dependent activation, but had no effect on repression (33).
SRC-3 (NCOA3), another member of the SRC family, was originally identified through interaction with the estrogen receptor (ER) (34). Similar to SRC-1 and GRIP1, SRC-3 is recruited in a locus-specific manner (35).
In the mid-1990s, the discovery of two nuclear receptor co-repressors (NCOR)—NCOR1 (36), and NCOR2 (otherwise known as SMRT, silencing mediator co-repressor) drove further research into the field of NR co-regulators (37). The NCOR family interacts with nuclear receptors via the coRNR-box, consisting of the consensus sequence LXX I/H I XXX I/L, which contacts the AF-2 domain of NRs (38, 39). This is analogous to the LXXLL sequence in co-activators and occupies a similar location on the receptors.
While the NCOAs display intrinsic HAT activity, the co-repressors NCOR/SMRT were described to interact with the histone deacetylase HDAC3 (40). Both NCOR1 and SMRT were able to recruit HDAC3 to condense chromatin as part of their repressive mechanism (41).
SUMOylation of mouse GR at K310 was shown to be essential for repression, and in point mutant mice, neither NCOR1, SMRT nor the associated HDAC3 complex were recruited (42, 43). GCs down-regulate expression of GR itself, through a negative feedback loop. This occurs by recruitment of a GR-NCOR1-HDAC3 complex to an nGRE in exon 6 of the NR3C1 gene (44). GC-mediated suppression of natural killer cells activity however, was described to be mediated by HDAC1 and SMRT specifically (45). The differential control of GR action by recruitment of alternative co-activators and co-repressors, in tissue or signal specific contexts, is still an open area of investigation. Different GR ligands selectively recruit alternate co-factors (46), suggesting that ligand induced conformational changes might have discrete effects on GR target genes, adding another level of complexity to GR mediated gene regulation.
Two major proteins that are recruited by co-activators are CBP (CREB binding protein) and p300. Both CBP and p300 are histone acetyl transferases (HATs), and induce chromatin relaxation (47) (Figure 2). SRC-1 was shown to recruit p300 into a complex with nuclear receptors to activate transcription (48). Part of GR's repressive action might involve competition for CBP and p300, as GR repression of an AP-1 (Activator Protein 1) reporter was abolished by overexpression of CBP and p300 (49). Moreover, enhanced engraftment of hematopoietic stem cells in response to GCs was described to be controlled by SRC-1 and p300 recruitment to the CXCR4 gene, with acetylation of histones H4K5 and H4K16 upregulating CXCR4 (50).
GR and the tumor suppressor protein 53 (p53) were shown to interact in a ligand dependent manner via Hd2m (a transcription factor), which enhanced the GC-induced degradation of both GR and p53 (51). In fact, the interaction between GR and p53 is important for the repression of NF-κB (nuclear factor-κB) responsive genes. Without p53, GR did not repress inflammation in a mouse model of endotoxic shock (52).
Finally, GR interacts with components of the SWI/SNF complex (SWItch/Sucrose-Non Fermentable). These highly evolutionarily conserved ATP-dependent chromatin remodelers use energy from ATP hydrolysis to alter nucleosome positioning. GR was shown to directly interact with the Baf250, Baf57, and Baf60a subunits of SWI/SNF complexes, further demonstrating the ability of GR to modify the chromatin architecture [(53–56); Figure 2].
In summary, GR recruits co-activators such as SRC family members, which in turn assemble a transcriptional complex containing histone modifying enzymes and chromatin remodelers to control the transcriptional machinery and RNA Pol II activity. These interactions are crucial for its anti-inflammatory actions and might present novel therapeutic targets in the future.
Mechanistic Insights Into Immunomodulation From GR Point Mutations in vitro
Introducing point mutations into the NR3C1 gene significantly contributed to our understanding of the molecular mechanisms of GR action. Here, we briefly address the insights gained from specific residues that revealed certain GR functions essential to suppress inflammation in cultured cells.
Besides promoter/enhancer occupancy, post-translational modifications of GR play a major role for transcriptional control. Three key phosphorylation sites were identified in the human GR: S203, S211, and S226 (57–59). All of them are located in the AF-1 domain, which is crucial for protein-protein interactions with TATA-box binding protein and others (60). By using phospho-deficient (S211A) or phospho-mimetic (S211D) mutations, it was shown that phosphorylation of GR at S211 increases association with the MED14 subunit of the mediator complex, a key bridge to the transcriptional machinery (59). In confirmation, the S211A mutant displays reduced expression of the GR targets GILZ and IRF8. S226A mutation however, had the opposite effect. The phosphorylation-deficient mutant S226A showed increased expression of GILZ and IRF8, suggesting an inhibitory role (59). In addition, S404, a site for GSK3β phosphorylation, regulates GR transcriptional activity. Mutation to S404A rewired the GR-regulated transcriptome, interestingly increasing its repressive capacity (61). Moreover, the SUMOylation-deficient murine GR K310R was shown to affect repression and the recruitment of co-regulators [(42, 43); Figure 1].
The AF-2 domain, located within the LBD (62), has additional sites modulating GR function. The mutation C656G within the AF-2 domain of the rat GR (C638 in human) reduced the ligand concentration required for activation of the PEPCK promoter (63). Mutations within the “charge-clamp”—that is the co-activator interaction site of K579 and E755—resulted in loss of transcriptional activation, but had no effect on repression (64).
Applying a random mutagenesis approach in yeast, Yamamoto and colleagues showed that multiple mutations within the zinc finger of the DBD impede GR binding to GREs in vitro, demonstrating the importance of this particular domain (65). Further mutagenesis studies in the 1990s identified a multitude of important amino acids involved in activation and repression. For example, the mutations S425G and L436V in the DBD could double the activation in a reporter assay, but almost completely abolished repression by GR (66).
Mutations in the dimer interface are also central for the understanding of GR biology. The GRdim (human A458T), corresponding to rat A477T (67), and GRmon (mouse A465T/I634A) (68) mutations disrupt the dimer interface. Further mutation of A458T outside the D-loop to the double N454D/A458T further increased the capacity of GR to repress a reporter in vitro (66). Generation of the GR(D4X), a quadruple mutant GR with the residues N454D, A458T, R460D, and D464C in the dimerization region of mouse GR provided deeper insight into the monomer/dimer action of GR. The GR (D4X) had equivalent repressive activity to wild type, while activation capacity as measured in reporter assays was near zero. This mutant demonstrated that opposition of TNF-α involved both activation of IKKB and repression, since mutant GR was unable to induce IKKB, but repressed the production of TNF-α (69). There is significant work on the GRdim mutation in vivo, covered in the next section. Early in vitro work however, showed that the A477T mutation induced loss of the dimer interface and reduced DNA residence time, making target gene regulation by A477T rather difficult to interpret (70). Both wild type GR and GRmon bound GRE half sites, but A447T was incapable of binding classic, full length GREs, which are occupied by receptor dimers [(67); Figure 1].
Another mutation in the second zinc finger of the DBD in rat GR R488Q (R469 in the human GR) was designed to discriminate between interactions with NF-κB and AP-1. Overexpressing GR R488Q in activated CV-1 cells under inflammatory conditions failed to suppress NF-κB reporter activity, whereas AP-1 inhibition was preserved (71). Additional GR mutations with less impact on inflammation are reviewed in more detail elsewhere (72).
Taken together, these GR point mutants show the importance and complexity of GR interactions with transcription factors and chromatin modifiers. In fact, several discrete mutations within the GR AF-1, AF-2 domains and the dimer interface alter its activity in a gene-specific manner, indicating that different parts of the receptor are dispensable for certain gene regulatory events, but essential for others (32). Differentially interfering with GR function therefore affects multiple physiological processes, and distinct types of inflammatory responses.
Lessons Learned From Genome-Wide Studies
Chromatin as a key determinant of GR function has been highlighted in multiple genome-wide ChIP-sequencing studies since the early 2010s. For instance, GR gene regulation is determined by the chromatin architecture of the responsive cell. GR does not act as its own pioneer factor, but rather cell-type-specific gene regulation is dependent on pre-existing available binding sites, determined by chromatin accessibility (73). The pro-inflammatory transcription factor AP-1 governs a large subset of GR regulatory sites, making areas of DNA accessible to GR (74). As GR is largely dependent on pre-existing open chromatin for binding, it cemented the possibility that stimuli which are known for chromatin remodeling, for example inflammation, alters GR binding. Indeed, treatment with TNF-α amends the transcriptional response to GCs, as well as chromatin occupancy of GR, and surprisingly GR activation also transformed the occupancy of NF-κB (75). Recent data showed that GR could indeed act as a pioneer factor for other transcription factors, such as FOXA1, but only at a minority of genomic sites, and thus far this effect has not been demonstrated in immune cells (76).
When assessing GR activity in a more relevant cell-type, macrophages treated with LPS, GR, p65 (part of the NF-κB complex), and c-Jun (one of the members of the AP-1 dimer) binding overlapped significantly (see below). However, the directionality of the gene regulatory response did not correlate well with the type of interaction. That is, contrary to established models, GR binding to NF-κB loci did not only result in repression of target genes, but either repression or activation depending on the particular locus. The inverse is also true, that GR binding to canonical GREs did not only result in up-regulation of transcription at the assigned gene. Rather than the presence or absence of GR as the determining factor, the recruitment of different chromatin modifiers, such as GRIP1, were the prime measure of whether the particular gene would be activated or repressed (77).
Moreover, GR effects can be dependent on the timing of the inflammatory signal. Pre-treatment of macrophages with GCs before LPS stimulation resulted in differential gene regulation compared to treatment with GCs after LPS stimulation. In addition, a large part of GR's anti-inflammatory action can be accounted for by the induction of negative regulators of inflammation such as Mkp1, GILZ, and A20, see below (78). GRdim macrophages treated with LPS and Dex also showed that the dimerization impaired GR preferentially occupied GR-half sites (16), a phenomenon also observed in cells overexpressing GR A477T (67).
Importantly, all these studies showed that GR not only binds to GREs, but occupies motifs near lineage determining factors, such as PU.1 in macrophages. Again this underscores the idea that GR requires open, pre-programmed chromatin for finding its genomic target sites (16, 74, 77–79). The chromatin landscape is cell-specific and depends on pioneer factors, cell lineage transcription factors and epigenetic marks that all predetermine GR binding. Only a minority of GR peaks are found in inaccessible chromatin and trigger chromatin remodeling upon hormone treatment (16, 73, 79–82). These findings strongly suggest that other DNA-binding proteins prime the chromatin landscape prior to GR arrival. The collaborative binding of lineage-determining transcription factors results in nucleosome remodeling, which generates open regions of chromatin. This provides access to signal-dependent transcription factors to bind open regions and modulate gene transcription in a cell-specific manner (83). In the context of macrophages, PU.1 and C/EBP are essential for the development of the myeloid lineage and have been shown to establish the monocyte-specific enhancer landscape (83, 84). PU.1 deletion results in loss of macrophages, neutrophils and B cells (85, 86). Importantly, PU.1 and C/EBP transcription factors often co-localize with GR in macrophages (16).
This new methodology has given deeper insights into the mechanisms by with GR regulates gene expression, identifying chromatin remodeling, and cooperation with other transcription factors, as a key determinants of GR activity. Importantly, GR's reliance on other factors to define its binding sites underscores the necessity of studying GC responses in a tissue-specific manner, rather than extrapolating effects from one cell-type to another.
Molecular Mechanisms of Immunomodulation by the Glucocorticoid Receptor
Non-genomic Actions of GR
Some therapeutic GC effects, such as bronchodilation, resolution of airway irritation or suppression of inflammation, occur almost too rapidly to result from transcription, raising the possibility of non-genomic GR actions (87, 88). These could be GR-unspecific interactions with cellular membranes, functions of membrane-bound GR or specific interactions with cytosolic GR, thereby altering posttranslational modifications like phosphorylation, or other mechanisms (89).
Membrane-bound GR was described in human monocytes and B cells (90, 91), and non-genomic functions have been found in macrophages (92), lung epithelial cells (93), and T-cells (94).
Downstream of inflammatory MAPK signaling, mitogen- and stress-activated protein kinase-1 (MSK1) is an essential kinase for NF-κB p65 S275 phosphorylation (95).Interestingly, GC-mediated repression of NF-κB targets involves loss of MSK1 kinase recruitment at inflammatory promoters and nuclear export of MSK1 via cytosolic GR (96). Putatively, GR can also crosstalk with AKT, GSK-3β, and mTOR signaling (93).
These non-genomic effects might be very interesting for the development of novel therapeutics, and will benefit from future studies, for example with novel cell lines or mouse models to dissect these complex interactions.
Genomic Actions of GR
Lipopolysaccharide (LPS) is a molecular component of the cell wall of Gram-negative bacteria commonly used to study inflammation (97, 98). On macrophages, LPS binds to Toll-Like Receptor 4 (TLR4) and activates a signaling cascade that results in NF-κB and AP-1 nuclear translocation. Together with other inflammatory transcription factors, these two protein complexes then activate pro-inflammatory gene expression (99, 100). TLR4 activates AP-1 via the MAPK signaling pathway and NF-κB via degradation of the cytosolic IKK complex that frees the NF-κB transcription factor (Figure 3).
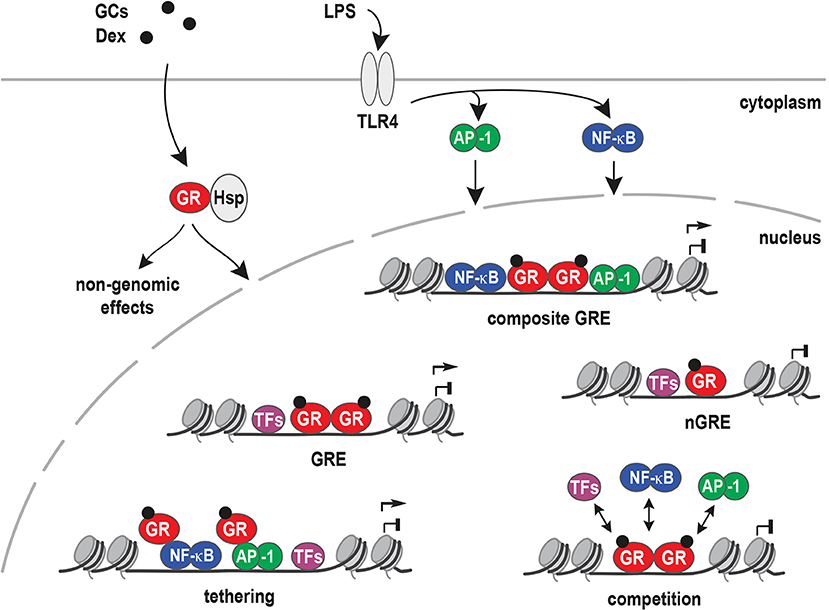
Figure 3. Models for inflammatory gene regulation by the glucocorticoid receptor. Upon ligand binding (GCs), the glucocorticoid receptor (GR) is released from heat shock proteins (Hsp) and translocates to the nucleus. Inflammation can be activated by lipopolysaccharide (LPS) binding to Toll-like receptor 4 (TLR4). TLR4 signaling results in the activation of NF-κB, AP-1, and other inflammatory transcription factors that bind and regulate pro-inflammatory target genes. Different mechanisms have been proposed for GR's potent anti-inflammatory actions, i.e., binding to Glucocorticoid Response Elements (GREs), to composite GREs together with other transcription factors, to negative GREs (nGRE), by tethering to DNA-bound transcription factors, by competing with other factors for DNA binding sites or by non-genomic actions.
GR can antagonize or synergize with pro-inflammatory signaling, depending on the context of promoters or enhancers. For antagonism of pro-inflammatory signaling, several mechanisms are proposed. These include the direct interference with MAPK or JNK signaling (101, 102), leading to repressive actions at the gene regulatory level. Conversely, repression of GR-target genes might be explained by tethering to other transcription factors or trans-repression, negative GREs (nGREs, with a different sequence), composite GREs, non-canonical novel GREs, DNA as a modulator of GR, and consensus classical GREs.
Most frequently, GR tethering to AP-1 or NF-κB via protein-protein interactions (trans-repression), instead of direct DNA binding, was suggested to underlie its repression of inflammatory responses (103, 104). In other words, GR has been shown to represses genes via protein-protein interactions with AP-1 (105), NF-κB (106), STAT3 (107), and other DNA-bound transcription factors (Figure 3). Interestingly, STAT3 tethering to GR resulted in synergistic gene regulation, and increased target gene expression in AtT-20 cells. On the other hand, GR tethering to DNA-bound STAT3 resulted in transcriptional repression (107).
Negative GREs (nGREs) were originally described as GREs motifs in the promoters of repressed target genes. nGREs can be found in very different cell types and genes involved in various processes, for example: HPA axis (POMC and CRH) (108, 109), lactation (PRL3) (110, 111), bone homeostasis (osteocalcin) (112), skin structure (keratins) (113), and inflammation (IL-1β) (114).
However, the definition of nGREs has not yet reached consensus in the literature, and subsequently, GBS with non-classical consensus sequences, near repressed targets, are also named nGREs. One study described a variation of nGREs, termed “inverted repeat (IR) nGRE.” IR nGRE is a complex GBS with the following consensus motif: CTCC(n)0−2GGAGA, which differs from the classical GRE (AGAACAnnnTGTTCT) or nGRE (115). These elements however, have not been identified by ChIP-seq, questioning how relevant they are to GR responses.
Similar to nGREs, composite elements, such as degenerate GREs overlapping with other transcription factor consensus motifs, may also affect the transcription of inflammatory targets. For example, a 25-base pair composite element (plfG element) in the promoter of the proliferin gene, is regulated by GR and AP-1 (116, 117). Furthermore, the GR DNA-binding domain (DBD) can bind a newly identified motif inside NF-κB consensus sequences. Crystal structures of the GR DBD demonstrated direct binding of GR to the AATTT nucleotides within the NF-κB motif from the promoter regions of CCL2, IL-8, PLAU, RELB, and ICAM1. This cryptic GR-binding site overlapping the NF-κB response element was named κBRE and was highly conserved between species (118).
An important aspect is the concept of DNA being an allosteric modulator of the GR. Here, the precise nucleotide sequence in a GBS is proposed to function as a shaping ligand that specifies GR's transcriptional activity. X-ray crystallography of GR DBD dimers bound to different GBSs showed that conformation of the lever arm in the DBD appeared to be influenced by the DNA sequence (24, 119). Furthermore, the addition of a single GR-binding site was sufficient to convert a gene, which was normally not regulated by GR, into a target gene, such as IL-1β and IL1R2 in U2OS cells (120). The presence of classical GREs in GR-bound enhancers near both activated and repressed genes in murine bone marrow-derived macrophages (BMDM) stimulated with LPS and Dexamethasone (Dex) challenge these models. These findings suggest that first, direct GR:GRE binding is relevant for repression of inflammatory genes. Secondly, that the classical models described above are not sufficient for prediction of GR mediated activation or repression. Therefore, the presence of a different combination of cofactors in activated vs. repressed sites could explain or contribute to the up- or down-regulation of GR target genes (77, 118, 121, 122).
Taken together, how GR activates one set of target genes while repressing another is still an open question, and the molecular mechanisms specifying the repression of inflammatory genes remain unknown. Repression by GR is a complex process which likely involves different determinant factors. One factor is GR itself (phosphorylation, post-translational modifications and ligand-specific conformations), another factor is the DNA sequence, the cell type-specific chromatin landscape and the cooperation with co-regulators and other transcription factors. All of these, together with potentially unknown factors, ultimately determine which target genes are up- or down-regulated.
Mechanistic Insights Into Immunomodulation From GR Point Mutations in vivo
As described above, one particular class of point mutations, which interfere with GR dimerization, caught considerable attention. In tissue culture experiments expressing these GRdim mutants (human GR A458T, mouse GR A465T, and rat A477T), the concept was developed that abrogation of dimerization could be beneficial to limit side effects of anti-inflammatory treatments. Therefore, pharmaceutical companies directed their research to develop dissociated ligands favoring GR monomer dependent favorable effects and reducing unwanted GR dimer action (123, 124).
Various selective GR agonists (SEGRAs), such as RU24858, RU24782, and non-steroidal ligands (LDG552, ZK216348, Compound A), were examined for desired anti-inflammatory effects with the hope that there would be minimal metabolic actions (124, 125). Only a few of these compounds, however, showed promise in preclinical trials (126). Their limited success arose from the generalized and oversimplified view that the GR monomer mediates trans-repression (anti-inflammatory) and the GR dimer regulates only unwanted effects (127). The disappointing conclusion of these programs for SEGRAs and non-steroidal ligands and their translation to the clinic called for new perspectives in the context of pathophysiology (10, 16, 104, 127–129). With knowledge gained from the GRdim mouse and others, the development of selective monomerizing GRagonists or modulators (SEMOGRAMs) and selective dimerizing GRagonists or modulators (SEDIGRAMs) has begun to make progress (130). To find SEDIGRAMs, a screening identified Cortivazol and AZD2906 as compounds that increase GR dimerization and enhance the transactivation capacity. Both chemicals, however, still have GR monomer activity, indicating that these are not yet the ideal SEDIGRAMS (129). Efforts are still ongoing to identify perfect GR modulators separating dimer from monomer.
In 1998, the GR A465T mutation was introduced into mice (131, 132). Intriguingly, mice born with this mutation survived in certain backgrounds (131), and simple inflammatory models, such as phorbol ester induced skin irritation, responded to GC treatment in these animals. This indicated that GR monomer and thus transrepression by tethering might be sufficient to reduce inflammation. However, for most other inflammatory models, GCs failed to have an effect in these GRdim mice (Figure 4A).
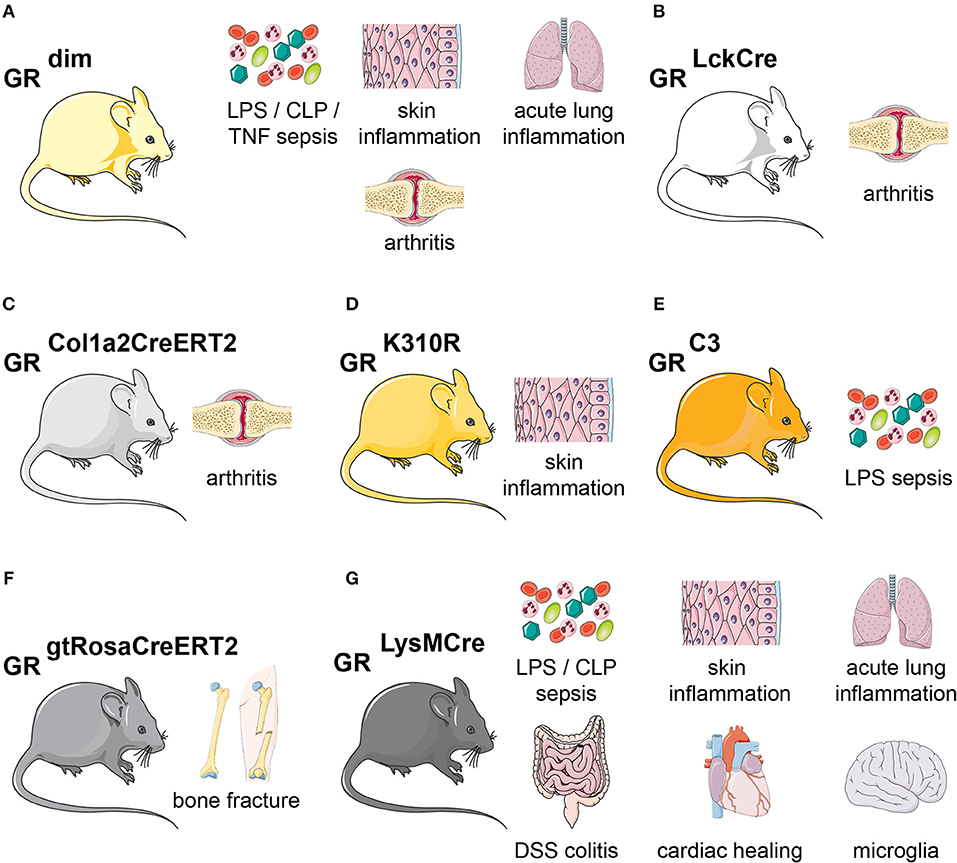
Figure 4. Glucocorticoid receptor mutant mouse models of inflammation. Overview of the mouse lines discussed in this article. (A) GRdim mice are more sensitive during LPS-, CLP-, or TNF inflammation. GRdim mice are refractory to GC treatment in models of skin inflammation, acute lung injury and arthritis. (B) In GRLckCre mice, GR is lacking in T-cells, making them refractory to GC treatment during arthritis. (C) GRCol1a2CreERT2 (lacking GR in fibroblasts) show delayed GC-induced suppression in arthritis. (D) GR K310R mutant mice lack GR SUMOylation and show impaired control of skin inflammation. (E) GR-C3 mice, lacking the most active GR isoform C3, are more sensitive to LPS-induced endotoxic shock. (F) During fracture, GR is necessary in all cells, as shown by GRgtRosaCreERT2 (tamoxifen-induced ubiquitous Cre-mediated recombination) for fracture healing. (G) GRLysMCre mice (GR is deleted in myeloid cells) show no proper healing in LPS-or CLP-sepsis, skin inflammation, acute lung injury, DSS colitis, cardiac healing, and Parkinson disease. The skin, lungs, bones, intestine, heart and brain cartoons were obtained from Servier Medical Art.
For instance, during LPS, CLP (cecal ligation and puncture), and TNF-α induced shock, GRdim mice were highly susceptible to inflammation and cytokine production, impaired thermoregulation and metabolic alterations (133–135). Furthermore, macrophages from GRdim mice were unable to efficiently repress cytokines in response to LPS (135). Moreover, GRdim mice treated with exogenous GCs showed impairment of anti-inflammatory responses in models of acute lung injury (ALI), arthritis, contact allergy, and allergic airway inflammation (136–139). During ALI, this was partially due to diminished expression of the GR-dimer target gene Sphk1 (138) (see above). In models of allergic airway inflammation, contact hypersensitivity, antigen-induced arthritis (AIA) or serum transfer-induced arthritis (STIA), GRdim mice failed to repress inflammation when given GC therapy (136, 137, 139, 140). In the model of AIA, GR dimerization was shown to be essential in T cells (GRLckCre mice) to reduce inflammation [(137); Figures 4A,B]. More recently, GRdim mice reconstituted with wild type hematopoietic stem cells failed to induce non-classical (CD11b+, F4/80+, Ly6C−), non-activated (CD11b+, F4/80+ MHCII−), anti-inflammatory (CD163, CD36, AnxA1, Axl, and MertK) macrophages during STIA, while cytokines were repressed normally (140). This strongly indicated that intact dimerization in stromal non-immune cells could contribute to the suppression of inflammation. More precise, the GR in fibroblast-like synoviocytes (GRCol1a2CreERT2) was crucial to reduce STIA (140) (Figure 4C). GRdim mice were also resistant to GC treatment during TNF-induced inflammation, and exhibited increased gut barrier leakiness, cell death of intestinal epithelial cells and cell death. An increased STAT1-responsive interferon-stimulated gene signature was observed in the gut of GRdim mice (141).
Whereas, the GRdim knock-in mice were intensively studied, less is known about other point mutations. The GRK310R mutation, which abrogates SUMOylation of the GR, failed to respond to GCs during skin inflammation. This was in part due to reduced SMRT/NCoR-co-repressor recruitment to GR/NF-κB/AP-1 repressive complexes [(42, 43); Figure 4D].
Finally, Cidlowski and colleagues published a knock-in mouse of the most active GR isoform C3. The lethality of these mice could be overcome by antenatal GC administration, and adult mice were hypersensitive to LPS administration. This indicated that either the absence of other isoforms like the most abundant GR-A, or indeed the specific overexpression of GR-C3 might confer anti-inflammatory actions [(142); Figure 4E]. However, further studies are warranted to dissect these observations in more detail.
Taken together, GR point mutations introduced in vivo, namely the GRdim mutation, but also the more recent mutations, have yield valuable insight into the molecular features of GR. With the emergence of CRISPR/Cas9 gene editing technology, more in vivo models for specific GR functions will help our understanding of GR in physiological processes in the future.
Glucocorticoid Action on Macrophages
GCs exert their immunosuppressive effects through many cells of the innate immune system, including dendritic cells, mast cells, neutrophils, and eosinophils (143, 144). GCs also play a major role in the regulation of adaptive immunity. For example, GCs decrease the proliferation of early B cell progenitors (145) and induce apoptosis in B cells and T cells (145–149). In this review, we will focus mainly on the effects of GCs in macrophages, since these innate immune cells are essential mediators of defense responses, beyond the mere removal of pathogens, and regulate tissue homeostasis in a myriad of ways (150).
Macrophages reside in many different tissues and are the first line of defense against pathogens (151). Depending on the activating stimulus, they can be categorized as M1-like and M2-like macrophages. The M1-like macrophages (classically activated macrophages) mediate pro-inflammatory actions. They are activated by exposure to LPS, INFγ, TNF-α, or pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) (151–153). GCs suppress inflammatory responses downstream of TLRs, in part by interfering with the NF-κB- and AP-1-activated transcription of pro-inflammatory cytokines and chemokines (154, 155).
The M2-like macrophages on the other hand, are characterized by their anti-inflammatory potential and are activated by cytokines involved in inflammatory resolution, like IL-4, IL-10, and IL-13 (151, 153, 156). GCs can also polarize macrophages to an M2-like phenotype by regulating the expression of anti-inflammatory proteins (153, 156). A major, yet undervalued aspect of GC control of anti-inflammatory macrophage polarization is the regulation of efferocytosis. GCs enhance the clearance of apoptotic cells, which in itself can augment the development of an anti-inflammatory macrophage phenotype (157, 158).
In sum, GCs can modulate macrophage activity in a number of different and intricate ways, which include suppressing the production of pro-inflammatory proteins and inducing anti-inflammatory mediators.
Glucocorticoid Receptor Target Genes Mediating Immune Modulation
GC stimulated macrophages shift to an M2-like anti-inflammatory and inflammation-resolving phenotype (156). These effects are achieved by the repression of pro-inflammatory genes, the induction of gene products antagonizing pro-inflammatory signaling, and by synergism with pro-inflammatory signaling pathways to activate genes resolving inflammation.
While the mechanisms of gene repression have been extensively discussed [referring to interleukins, chemokines, matrix metalloproteinases, inducible nitric oxide synthase (iNOS), and other mediators], the activated anti-inflammatory genes have only recently received attention (Table 1).
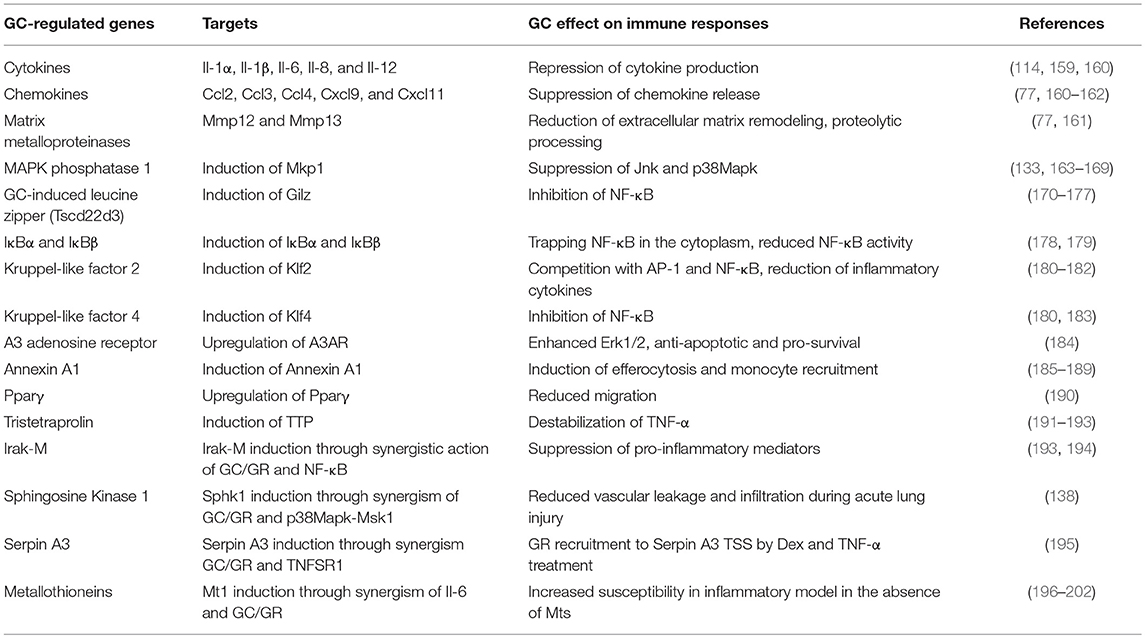
Table 1. GR target genes relevant for (anti-) inflammatory action.
Prominent examples are the induction of MAPK phosphatase 1 (Mkp1 or Dusp1), that interferes with the p38MAPK pathway; GC induced leucine zipper (GILZ/Tsc22d3), which binds to the NF-κB subunit p65; the induction of IκBα and β, which oppose NF-κB activity; the activation of kruppel like transcription factors (Klf), which are important for alternative macrophage polarization, and many others (Table 1). This upregulation of anti-inflammatory genes further emphasizes that both gene repression and activation are required for the immunomodulatory effects of GCs.
More recently, there were intriguing observations that GCs not only antagonize inflammatory signaling, but also synergize with pro-inflammatory signaling pathways (Table 1). GCs synergize with Haemophilus influenzae activated inflammatory pathways in macrophages, bronchial epithelial cells (BEAS-2B) and lung epithelial cells (A549) to induce IRAK-M, a negative regulator of TLR signaling (203). Mechanistically, this synergistic activation of Irak-M/Irak-3 transcription is dependent on binding of both GR and p65 to its promoter, showing a cooperative induction by NF-κB and GR that limits inflammation (203). Similarly, GCs activate TLR2 expression synergistically with H. influenza signaling in vitro (194).
In ALI models, GR was shown to cooperate with LPS-induced p38MAPK-Msk1 to induce Sphingosine Kinase 1 (SphK1) expression in macrophages (138). SphK1 produces the active mediator Sphingosine-1-phosphate (S1P), that binds to the S1P receptor 1 (S1PR1) on endothelial cells to reduce vascular leakage and infiltration during lung inflammation (138, 204–208). In ALI, mice lacking SphK1 in macrophages were resistant to GC treatment and showed reduced S1P levels. Additional examples of synergistically regulated genes important for modulation of inflammation are acute phase proteins like Serpin A3 (α1-antichymotrypsin) (195) and Metallothioneins (Mt1 and Mt2) (196, 197).
The synergistic regulation of immune-modulating genes by GCs and pro-inflammatory pathways is an important component of their mechanism, but the underlying dynamics and time windows are still poorly understood.
Loss of Function Models of GC Signaling in Macrophages
Strong evidence for the role of GR during homeostasis and inflammation was derived from conditional loss-of-function studies in mice. Applying the Cre/LoxP system, GR tamoxifen-inducible mice (GRgtROSACreERT2) could be used to determine the impact of GR deletion in adult animals, circumventing the lethality of global GR knockouts. For example, they have been useful to study GR during inflammation-dependent bone repair after fracture (209). Overall, the mice displayed a mild increase in inflammation, with elevated serum IL-6 levels and increased IL-1β levels at the fracture hematoma, accompanied by increased CD3+ and CD8+ cells. Consequently, the lack of GR and potentially the elevated inflammation, caused a delayed endochondral regeneration and maturation of callus and a decreased healing response [(209); Figure 4F].
Since the publications of conditional GR alleles in 1999 (210), 2003 (211), and 2012 (212), many cell types have been targeted with specific Cre lines to characterize specific functions of the GR in numerous cell types in the brain, muscle, heart, T lymphocytes, and others.
Insights into the function of GR in macrophages in vivo mainly stems from Lysozyme 2 (LysM)–Cre mice crossed to GR floxed alleles, which causes deletion in the myeloid cell lineage (monocytes, mature macrophages, and granulocytes) [(135, 136, 163, 213); Figure 4G].
In both the LPS-induced endotoxic shock model and during CLP, myeloid GR is crucial for the repression of inflammatory cytokines and for survival (135, 163). Not only in LPS-induced inflammation, but also in dextran sodium sulfate (DSS)-induced colitis, the action of endogenous GCs in macrophages was essential to reduce intestinal inflammation (214). Mice deficient for macrophage GR had a higher disease score, with increased infiltration of neutrophils, T cells and macrophages in the colon, which was associated with enhanced serum IL-6 (214). Moreover, macrophages were shown to play an essential role for cardiac healing, tissue repair and hence survival in myocardial infarction (215). Deletion of GR in macrophages delayed cardiac healing 7 days after myocardial infarct, with impaired cardiac function, collagen scar formation and neovascularization, and larger myofibroblasts. Consequently, targeting macrophage GR during myocardial infarction might be a potential pharmacological intervention for tissue repair (215). In contrast, in a mouse model of atherosclerosis, macrophage GR deletion was beneficial and showed reduced levels of vascular calcification, due to reduced RANKL, BMP2, and Mx2 expression (216).
During skin inflammation in a model of contact hypersensitivity, the anti-inflammatory effects of GCs required GR in myeloid cells (136). Additionally, in a model of ALI, GRLysMCre mice were resistant to GC therapy, did not reduce cellular infiltration in the lung and did not induce the endothelial barrier stabilizing sphingosine-1-phosphate [(138); Figure 4G].
GRLysMCre mice were shown to efficiently express Cre in microglia, knocking out GR in brain resident macrophages. Studies on the function of microglial GR during acute inflammation demonstrated more cellular lesions, damage, demyelination in the corpus callosum, and increased neuronal degeneration. It also significantly increased pro-inflammatory cytokines after LPS injections (217). The activation of microglia induces secretion of pro-inflammatory proteins that contribute to dopaminergic neuronal death, a major a hallmark of Parkinson's disease. The absence of GR in microglia revealed that increased death of dopaminergic neurons in Parkinson's may contribute to neurodegenerative processes (218). Additionally, recent studies suggest that the absence of microglia GR facilitates TLR9 activation of inflammatory processes and affects Parkinson's disease progression (219).
In summary, the genetic deletion of GR in myeloid cells in various inflammatory models demonstrated the pivotal role of this cell type for GC actions. However, one of the limitations of the LysMCre mouse is the recombination in other myeloid cells such as neutrophils, whose contribution cannot be excluded. Nonetheless, this wealth of data supports the concept that selective targeting of glucocorticoids to macrophages, while sparing other cell types, could be a promising approach to optimize therapy.
Conclusion
During the past decade, much has been learned about the immunomodulatory mechanisms employed by GR: analyzing various mouse models, creating distinct mutations, mapping GR target genes genome-wide, functionally characterizing individual proteins mediating GC responses, studying different inflammatory settings, identifying essential co-regulators, and applying novel molecular biology methods, have broadened our understanding of these steroids' intricate actions. Taken together, it becomes obvious how basic research is fundamental in enabling drug development. However, we now realize that GR's molecular mechanisms are very complex, cell-type, locus- and signal-specific, and much more sophisticated than we previously anticipated. Intra- and extra-cellular signals can control GR function on many levels, and these multi-layered machineries demand new interpretation of previous over-simplified models. In the future, the rapid advancement of high-throughput technologies such as machine learning, genomics, proteomics, genome engineering, etc. will be key to the development of safer immunomodulators or novel GR ligands.
Author Contributions
LE-T, GC, AM, and SV wrote the manuscript with supervision of JT and NU. SV, JT, and NU secured funding.
Funding
This work was supported by funding from the DFG (SFB 1064 Chromatin Dynamics) & the ERC (ERC-StG-2014-StG 638573 SILENCE) to NU; and from the DFG (SFB1149, B07) & Start-Up Funding Program of the Medical Faculty University Ulm (Bausteinprogramm Universität Ulm) to SV; and from the DFG (SFB1149, C02) & AMPK ANR (DFG Tu220/13-1) to JT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors JT.
Acknowledgments
We apologize to all authors who could not be cited due to space constraints. We sincerely thank Ivonne Guderian and Sybille Regn for assistance.
References
1. Biddie SC, Conway-Campbell BL, Lightman SL. Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology (Oxford). (2012) 51:403–12. doi: 10.1093/rheumatology/ker215
2. Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. (1995) 9:1608–21. doi: 10.1101/gad.9.13.1608
3. Olaloko O, Mohammed R, Ojha U. Evaluating the use of corticosteroids in preventing and treating bronchopulmonary dysplasia in preterm neonates. Int J Gen Med. (2018) 11: 265–74. doi: 10.2147/IJGM.S158184
4. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. (2013) 34:518–30. doi: 10.1016/j.tips.2013.07.003
5. Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. (2016) 6:603–21. doi: 10.1002/cphy.c150015
6. Ramamoorthy S, Cidlowski JA. Corticosteroids: mechanisms of action in health and disease. Rheum Dis Clin North Am. (2016) 42:15–31; vii. doi: 10.1016/j.rdc.2015.08.002
7. De Lucena DD, Rangel EB. Glucocorticoids use in kidney transplant setting. Expert Opin Drug Metab Toxicol. (2018) 14:1023–41. doi: 10.1080/17425255.2018.1530214
8. McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. (2008) 20:131–7. doi: 10.1097/BOR.0b013e3282f51031
9. Hench PS. The reversibility of certain rheumatic and nonrheumatic conditions by the use of cortisone or of the pituitary adrenocotropic hormone. Ann Intern Med. (1952) 36:1–38. doi: 10.7326/0003-4819-36-1-1
10. Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, et al. Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. (2016) 96:409–7. doi: 10.1152/physrev.00011.2015
11. Raff H, Carroll T. Cushing's syndrome: from physiological principles to diagnosis and clinical care. J Physiol. (2015) 593:493–506. doi: 10.1113/jphysiol.2014.282871
12. Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell. (1986) 46:645–52. doi: 10.1016/0092-8674(86)90339-9
13. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. (2005) 18:331–42. doi: 10.1016/j.molcel.2005.03.025
14. Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract Res Clin Endocrinol Metab. (2015) 29:545–6. doi: 10.1016/j.beem.2015.04.007
15. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood). (2003) 228:111–33. doi: 10.1177/153537020322800201
16. Lim HW, Uhlenhaut NH, Rauch A, Weiner J, Hubner S, Hubner N, et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. (2015) 25:836–44. doi: 10.1101/gr.188581.114
17. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. (2010) 10:365–76. doi: 10.1038/nri2748
18. Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, et al. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods. (2013) 10:421–6. doi: 10.1038/nmeth.2411
19. Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, et al. Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat Commun. (2017) 8: 15896. doi: 10.1038/ncomms15896
20. Wrange O, Carlstedt-Duke J, Gustafsson JA. Stoichiometric analysis of the specific interaction of the glucocorticoid receptor with DNA. JBiol Chem. (1986) 261:11770–8.
21. Tsai SY, Carlstedt-Duke J, Weigel NL, Dahlman K, Gustafsson JA, Tsai MJ, et al. Molecular interactions of steroid hormone receptor with its enhancer element: evidence for receptor dimer formation. Cell. (1988) 55:361–9. doi: 10.1016/0092-8674(88)90059-1
22. Wrange O, Eriksson P, Perlmann T. The purified activated glucocorticoid receptor is a homodimer. J Biol Chem. (1989) 264:5253–9.
23. Payvar F, DeFranco D, Firestone GL, Edgar B, Wrange O, Okret S, et al. Sequence-specific binding of glucocorticoid receptor to MTVDNA at sites within and upstream of the transcribed region. Cell. (1983) 35(2 Pt 1):381–92. doi: 10.1016/0092-8674(83)90171-X
24. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. (2009) 324:407–10. doi: 10.1126/science.1164265
25. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci USA. (2016) 113:8236–41. doi: 10.1073/pnas.1606774113
26. Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. (1995) 270:1354–7. doi: 10.1126/science.270.5240.1354
27. Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci USA. (1996) 93:4948–52. doi: 10.1073/pnas.93.10.4948
28. Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol Endocrinol. (1998) 12:302–13. doi: 10.1210/mend.12.2.0065
29. Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. (1997) 389:194–8. doi: 10.1038/38304
30. Clarisse D, Thommis J, Van Wesemael K, Houtman R, Ratman D, Tavernier J, et al. Coregulator profiling of the glucocorticoid receptor in lymphoid malignancies. Oncotarget. (2017) 8:109675–91. doi: 10.18632/oncotarget.22764
31. Li X, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol Cell Biol. (2003) 23:3763–73. doi: 10.1128/MCB.23.11.3763-3773.2003
32. Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, et al. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA. (2003) 100:13845–50. doi: 10.1073/pnas.2336092100
33. Rollins DA, Kharlyngdoh JB, Coppo M, Tharmalingam B, Mimouna S, Guo Z, et al. Glucocorticoid-induced phosphorylation by CDK9 modulates the coactivator functions of transcriptional cofactor GRIP1 in macrophages. Nat Commun. (2017) 8:1739. doi: 10.1038/s41467-017-01569-2
34. Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. (1997) 277:965–8. doi: 10.1126/science.277.5328.965
35. Grenier J, Trousson A, Chauchereau A, Amazit L, Lamirand A, Leclerc P, et al. Selective recruitment of p160 coactivators on glucocorticoid-regulated promoters in Schwann cells. Mol Endocrinol. (2004) 18:2866–79. doi: 10.1210/me.2004-0241
36. Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. (1995) 377:397–404. doi: 10.1038/377397a0
37. Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. (1995) 377:454–7. doi: 10.1038/377454a0
38. Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, et al. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. (1999) 13:3198–208. doi: 10.1101/gad.13.24.3198
39. Webb P, Anderson CM, Valentine C, Nguyen P, Marimuthu A, West BL, et al. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs). Mol Endocrinol. (2000) 14:1976–85. doi: 10.1210/mend.14.12.0566
40. Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. (2012) 481:335–40. doi: 10.1038/nature10728
41. Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. (2001) 21:6091–101. doi: 10.1128/MCB.21.18.6091-6101.2001
42. Hua G, Ganti KP, Chambon P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc Natl Acad Sci USA. (2016) 113:E635–43. doi: 10.1073/pnas.1522826113
43. Hua G, Paulen L, Chambon P. GRSUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci USA. (2016) 113:E626–34. doi: 10.1073/pnas.1522821113
44. Ramamoorthy S, Cidlowski JA. Ligand-induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long-range chromatin interactions with intragenic glucocorticoid response elements. Mol Cell Biol. (2013) 33:1711–22. doi: 10.1128/MCB.01151-12
45. Bush KA, Krukowski K, Eddy JL, Janusek LW, Mathews HL. Glucocorticoid receptor mediated suppression of natural killer cell activity: identification of associated deacetylase and corepressor molecules. Cell Immunol. (2012) 275:80–9. doi: 10.1016/j.cellimm.2012.02.014
46. Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS Jr, Louw A, et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. (2009) 299:219–31. doi: 10.1016/j.mce.2008.10.008
47. Turner BM. Histone acetylation and control of gene expression. J Cell Sci. (1991) 99 (Pt 1):13–20.
48. Yao TP, Ku G, Zhou N, Scully R, Livingston DM. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci USA. (1996) 93:10626–31. doi: 10.1073/pnas.93.20.10626
49. Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, et al. ACBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. (1996) 85:403–14. doi: 10.1016/S0092-8674(00)81118-6
50. Guo B, Huang X, Cooper S, Broxmeyer HE. Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat Med. (2017) 23:424–8. doi: 10.1038/nm.4298
51. Sengupta S, Wasylyk B. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. (2001) 15:2367–80. doi: 10.1101/gad.202201
52. Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ, et al. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci USA. (2011) 108:17117–22. doi: 10.1073/pnas.1114420108
53. Fryer CJ, Archer TK. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature. (1998) 393:88–91. doi: 10.1038/30032
54. Hsiao PW, Fryer CJ, Trotter KW, Wang W, Archer TK. BAF60a mediates critical interactions between nuclear receptors and the BRG1 chromatin-remodeling complex for transactivation. Mol Cell Biol. (2003) 23:6210–20. doi: 10.1128/MCB.23.17.6210-6220.2003
55. Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, et al. A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol Cell Biol. (2000) 20:8879–88. doi: 10.1128/MCB.20.23.8879-8888.2000
56. Hebbar PB, Archer TK. Chromatin remodeling by nuclear receptors. Chromosoma. (2003) 111:495–504. doi: 10.1007/s00412-003-0232-x
57. Bodwell JE, Orti E, Coull JM, Pappin DJ, Smith LI, Swift F. Identification of phosphorylated sites in the mouse glucocorticoid receptor. J Biol Chem. (1991) 266:7549–55.
58. Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. (1997) 272:9287–93. doi: 10.1074/jbc.272.14.9287
59. Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. (2008) 22:1754–66. doi: 10.1210/me.2007-0219
60. Copik AJ, Webb MS, Miller AL, Wang Y, Kumar R, Thompson EB. Activation function 1 of glucocorticoid receptor binds TATA-binding protein in vitro and in vivo. Mol Endocrinol. (2006) 20:1218–30. doi: 10.1210/me.2005-0257
61. Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. (2008) 28:7309–22. doi: 10.1128/MCB.00808-08
62. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. (2002) 110:93–105. doi: 10.1016/S0092-8674(02)00817-6
63. Kucera T, Waltner-Law M, Scott DK, Prasad R, Granner DK. A point mutation of the AF2 transactivation domain of the glucocorticoid receptor disrupts its interaction with steroid receptor coactivator 1. J Biol Chem. (2002) 277:26098–102. doi: 10.1074/jbc.M204013200
64. Wu J, Li Y, Dietz J, Lala DS. Repression of p65 transcriptional activation by the glucocorticoid receptor in the absence of receptor-coactivator interactions. Mol Endocrinol. (2004) 18:53–62. doi: 10.1210/me.2002-0373
65. Schena M, Freedman LP, Yamamoto KR. Mutations in the glucocorticoid receptor zinc finger region that distinguish interdigitated DNA binding and transcriptional enhancement activities. Genes Dev. (1989) 3:1590–601. doi: 10.1101/gad.3.10.1590
66. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. (1994) 13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x
67. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. (2014) 15:418. doi: 10.1186/s13059-014-0418-y
68. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. (2014) 12:e1001813. doi: 10.1371/journal.pbio.1001813
69. Heck S, Bender K, Kullmann M, Gottlicher M, Herrlich P, Cato AC. I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. (1997) 16:4698–707. doi: 10.1093/emboj/16.15.4698
70. Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. (2013) 20:876–3. doi: 10.1038/nsmb.2595
71. Bladh LG, Liden J, Dahlman-Wright K, Reimers M, Nilsson S, Okret S. Identification of endogenous glucocorticoid repressed genes differentially regulated by a glucocorticoid receptor mutant able to separate between nuclear factor-kappaB and activator protein-1 repression. Mol Pharmacol. (2005) 67:815–26. doi: 10.1124/mol.104.005801
72. Beck IM, De Bosscher K, Haegeman G. Glucocorticoid receptor mutants: man-made tools for functional research. Trends Endocrinol Metab. (2011) 22:295–310. doi: 10.1016/j.tem.2011.03.009
73. John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. (2011) 43:264–8. doi: 10.1038/ng.759
74. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. (2011) 43:145–55. doi: 10.1016/j.molcel.2011.06.016
75. Rao NA, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, Kolisis FN, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. (2011) 21:1404–6. doi: 10.1101/gr.118042.110
76. Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, et al. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell. (2016) 165:593–605. doi: 10.1016/j.cell.2016.02.067
77. Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. (2013) 49:158–71. doi: 10.1016/j.molcel.2012.10.013
78. Oh KS, Patel H, Gottschalk RA, Lee WS, Baek S, Fraser ID C, et al. Anti-inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity. (2017) 47:298–309 e5. doi: 10.1016/j.immuni.2017.07.012
79. John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell. (2008) 29:611–24. doi: 10.1016/j.molcel.2008.02.010
80. So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. (2007) 3:e94. doi: 10.1371/journal.pgen.0030094
81. Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. (2009) 19:2163–71. doi: 10.1101/gr.097022.109
82. Siersbaek R, Nielsen R, John S, Sung MH, Baek S, Loft A, et al. Extensive chromatin remodelling and establishment of transcription factor 'hotspots’ during early adipogenesis. EMBO J. (2011) 30:1459–72. doi: 10.1038/emboj.2011.65
83. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. (2010) 38:576–89. doi: 10.1016/j.molcel.2010.05.004
84. Jin F, Li Y, Ren B, Natarajan R. PU.1 and C/EBP(alpha) synergistically program distinct response to NF-kappaB activation through establishing monocyte specific enhancers. Proc Natl Acad Sci USA. (2011) 108:5290–5. doi: 10.1073/pnas.1017214108
85. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. (1994) 265:1573–7. doi: 10.1126/science.8079170
86. Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity. (1997) 6:437–7. doi: 10.1016/S1074-7613(00)80287-3
87. Buttgereit F, Scheffold A. Rapid glucocorticoid effects on immune cells. Steroids. (2002) 67:529–34. doi: 10.1016/S0039-128X(01)00171-4
88. Panettieri RA, Schaafsma D, Amrani Y, Koziol-White C, Ostrom R, Tliba O. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci. (2019) 40:38–49. doi: 10.1016/j.tips.2018.11.002
89. Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. (2003) 49:703–8. doi: 10.1002/art.11388
90. Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. (2004) 18:70–80. doi: 10.1096/fj.03-0328com
91. Spies CM, Schaumann DH, Berki T, Mayer K, Jakstadt M, Huscher D, et al. Membrane glucocorticoid receptors are down regulated by glucocorticoids in patients with systemic lupus erythematosus and use a caveolin-1-independent expression pathway. Ann Rheum Dis. (2006) 65:1139–46. doi: 10.1136/ard.2005.048272
92. Long F, Wang YX, Liu L, Zhou J, Cui RY, Jiang CL. Rapid nongenomic inhibitory effects of glucocorticoids on phagocytosis and superoxide anion production by macrophages. Steroids. (2005) 70:55–61. doi: 10.1016/j.steroids.2004.10.004
93. Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D. Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol Endocrinol. (2008) 22:1320–30. doi: 10.1210/me.2007-0154
94. Lowenberg M, Verhaar AP, Bilderbeek J, Marle J, Buttgereit F, Peppelenbosch MP, et al. Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep. (2006) 7:1023–9. doi: 10.1038/sj.embor.7400775
95. Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. (2003) 22:1313–24. doi: 10.1093/emboj/cdg139
96. Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, et al. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J. (2008) 27:1682–93. doi: 10.1038/emboj.2008.95
97. Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 Signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines (Basel). (2017) 5:E34. doi: 10.3390/vaccines5040034
98. Nijland R, Hofland T, van Strijp JA. Recognition of LPS by TLR4: potential for anti-inflammatory therapies. Mar Drugs. (2014) 12:4260–73. doi: 10.3390/md12074260
99. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
100. Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, et al. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther. (2008) 10:201. doi: 10.1186/ar2338
101. Caelles C, Gonzalez-Sancho JM, Munoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. (1997) 11:3351–64. doi: 10.1101/gad.11.24.3351
102. Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, et al. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest. (2002) 110:1729–38. doi: 10.1172/JCI0215481
103. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front Endocrinol (Lausanne). (2017) 8:16. doi: 10.3389/fendo.2017.00016
104. Desmet SJ, De Bosscher K. Glucocorticoid receptors: finding the middle ground. J Clin Invest. (2017) 127:1136–45. doi: 10.1172/JCI88886
105. Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, et al. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. (1990) 62:1205–15. doi: 10.1016/0092-8674(90)90396-V
106. Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. (1994) 91:752–6. doi: 10.1073/pnas.91.2.752
107. Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell. (2012) 47:38–49. doi: 10.1016/j.molcel.2012.04.021
108. Drouin J, Trifiro MA, Plante RK, Nemer M, Eriksson P, Wrange O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol Cell Biol. (1989) 9:5305–14. doi: 10.1128/MCB.9.12.5305
109. Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol. (1999) 13:1629–44. doi: 10.1210/mend.13.10.0351
110. Subramaniam N, Cairns W, Okret S. Studies on the mechanism of glucocorticoid-mediated repression from a negative glucocorticoid response element from the bovine prolactin gene. DNA Cell Biol. (1997) 16:153–63. doi: 10.1089/dna.1997.16.153
111. Sakai DD, Helms S, Carlstedt-Duke J, Gustafsson JA, Rottman FM, Yamamoto KR. Hormone-mediated repression: a negative glucocorticoid response element from the bovine prolactin gene. Genes Dev. (1988) 2:1144–54. doi: 10.1101/gad.2.9.1144
112. Aslam F, Shalhoub V, van Wijnen AJ, Banerjee C, Bortell R, Shakoori AR, et al. Contributions of distal and proximal promoter elements to glucocorticoid regulation of osteocalcin gene transcription. Mol Endocrinol. (1995) 9:679–90. doi: 10.1210/mend.9.6.8592514
113. Radoja N, Komine M, Jho SH, Blumenberg M, Tomic-Canic M. Novel mechanism of steroid action in skin through glucocorticoid receptor monomers. Mol Cell Biol. (2000) 20:4328–39. doi: 10.1128/MCB.20.12.4328-4339.2000
114. Zhang G, Zhang L, Duff GW. A negative regulatory region containing a glucocorticosteroid response element (nGRE) in the human interleukin-1beta gene. DNA Cell Biol. (1997) 16:145–52. doi: 10.1089/dna.1997.16.145
115. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. (2011) 145:224–41. doi: 10.1016/j.cell.2011.03.027
116. Mordacq JC, Linzer DI. Co-localization of elements required for phorbol ester stimulation and glucocorticoid repression of proliferin gene expression. Genes Dev. (1989) 3:760–9. doi: 10.1101/gad.3.6.760
117. Miner JN, Yamamoto KR. The basic region of AP-1 specifies glucocorticoid receptor activity at a composite response element. Genes Dev. (1992) 6:2491–501. doi: 10.1101/gad.6.12b.2491
118. Hudson WH, Vera IMS, Nwachukwu JC, Weikum ER, Herbst AG, Yang Q, et al. Cryptic glucocorticoid receptor-binding sites pervade genomic NF-kappaB response elements. Nat Commun. (2018) 9:1337. doi: 10.1038/s41467-018-03780-1
119. Schone S, Bothe M, Einfeldt E, Borschiwer M, Benner P, Vingron M, et al. Synthetic STARR-seq reveals how DNA shape and sequence modulate transcriptional output and noise. PLoS Genet. (2018) 14:e1007793. doi: 10.1371/journal.pgen.1007793
120. Thormann V, Glaser LV, Rothkegel MC, Borschiwer M, Bothe M, Fuchs A, et al. Expanding the repertoire of glucocorticoid receptor target genes by engineering genomic response elements. Life Sci Alliance. (2019) 2:e201800283. doi: 10.26508/lsa.201800283
121. Hoffman JA, Trotter KW, Ward JM, Archer TK. BRG1 governs glucocorticoid receptor interactions with chromatin and pioneer factors across the genome. Elife. (2018) 7:e35073. doi: 10.7554/eLife.35073
122. Hemmer MC, Wierer M, Schachtrup K, Downes M, Hubner N, Evans RM, et al. E47 modulates hepatic glucocorticoid action. Nat Commun. (2019) 10:306. doi: 10.1038/s41467-018-08196-5
123. Schacke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. (2007) 275:109–7. doi: 10.1016/j.mce.2007.05.014
124. De Bosscher K, Beck IM, Haegeman G. Classic glucocorticoids versus non-steroidal glucocorticoid receptor modulators: survival of the fittest regulator of the immune system? Brain Behav Immun. (2010) 24:1035–42. doi: 10.1016/j.bbi.2010.06.010
125. Hubner S, Dejager L, Libert C, Tuckermann JP. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. (2015) 396:1223–31. doi: 10.1515/hsz-2015-0106
126. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. (2013) 154:993–1007. doi: 10.1210/en.2012-2045
127. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. (2012) 134:54–67. doi: 10.1016/j.pharmthera.2011.12.004
128. Schone S, Jurk M, Helabad MB, Dror I, Lebars I, Kieffer B, et al. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat Commun. (2016) 7:12621. doi: 10.1038/ncomms12621
129. Souffriau J, Eggermont M, Van Ryckeghem S, Van Looveren K, Van Wyngene L, Van Hamme E, et al. A screening assay for selective dimerizing glucocorticoid receptor agonists and modulators (SEDIGRAM) that are effective against acute inflammation. Sci Rep. (2018) 8:12894. doi: 10.1038/s41598-018-31150-w
130. De Bosscher K, Beck IM, Ratman D, Berghe WV, Libert C. Activation of the Glucocorticoid Receptor in Acute Inflammation: the SEDIGRAMConcept. Trends Pharmacol Sci. (2016) 37:4–16. doi: 10.1016/j.tips.2015.09.002
131. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
132. Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. (2001) 20:7168–3. doi: 10.1093/emboj/20.24.7168
133. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. (2012) 122:2130–40. doi: 10.1172/JCI60006
134. Silverman MN, Mukhopadhyay P, Belyavskaya E, Tonelli LH, Revenis BD, Doran JH, et al. Glucocorticoid receptor dimerization is required for proper recovery of LPS-induced inflammation, sickness behavior and metabolism in mice. Mol Psychiatry. (2013) 18:1006–7. doi: 10.1038/mp.2012.131
135. Kleiman A, Hubner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. (2012) 26:722–9. doi: 10.1096/fj.11-192112
136. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. (2007) 117:1381–90. doi: 10.1172/JCI28034
137. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci USA. (2011) 108:19317–22. doi: 10.1073/pnas.1105857108
138. Vettorazzi S, Bode C, Dejager L, Frappart L, Shelest E, Klassen C, et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat Commun. (2015) 6:7796. doi: 10.1038/ncomms8796
139. Klassen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Luhder F, et al. Airway epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. J Immunol. (2017) 199:48–61. doi: 10.4049/jimmunol.1601691
140. Koenen M, Culemann S, Vettorazzi S, Caratti G, Frappart L, Baum W, et al. Glucocorticoid receptor in stromal cells is essential for glucocorticoid-mediated suppression of inflammation in arthritis. Ann Rheum Dis. (2018) 77:1610–8. doi: 10.1136/annrheumdis-2017-212762
141. Ballegeer M, Van Looveren K, Timmermans S, Eggermont M, Vandevyver S, Thery F, et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J Clin Invest. (2018) 128:3265–79. doi: 10.1172/JCI96636
142. Oakley RH, Ramamoorthy S, Foley JF, Busada JT, Lu NZ, Cidlowski JA. Glucocorticoid receptor isoform-specific regulation of development, circadian rhythm, and inflammation in mice. FASEB J. (2018) 32:5258–71. doi: 10.1096/fj.201701153R
143. Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol. (2000) 30:1233–42. doi: 10.1002/(SICI)1521-4141(200004)30:4<1233::AID-IMMU1233>3.0.CO;2-F
144. Tuckermann JP, Kleiman A, McPherson KG, Reichardt HM. Molecular mechanisms of glucocorticoids in the control of inflammation and lymphocyte apoptosis. Crit Rev Clin Lab Sci. (2005) 42:71–104. doi: 10.1080/10408360590888983
145. Alnemri ES, Fernandes TF, Haldar S, Croce CM, Litwack G. Involvement of BCL-2 in glucocorticoid-induced apoptosis of human pre-B-leukemias. Cancer Res. (1992) 52:491–5.
146. Goossens S, Van Vlierberghe P. Overcoming steroid resistance in T cell acute lymphoblastic leukemia. PLoS Med. (2016) 13:e1002208. doi: 10.1371/journal.pmed.1002208
147. Wang Z, Malone MH, He H, McColl KS, Distelhorst CW. Microarray analysis uncovers the induction of the proapoptotic BH3-only protein Bim in multiple models of glucocorticoid-induced apoptosis. J Biol Chem. (2003) 278:23861–7. doi: 10.1186/1475-4924-3-4
148. Gruver-Yates AL, Cidlowski JA. Tissue-specific actions of glucocorticoids on apoptosis: a double-edged sword. Cells. (2013) 2:202–23. doi: 10.3390/cells2020202
149. Cao Y, Bender IK, Konstantinidis AK, Shin SC, Jewell CM, Cidlowski JA, et al. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood. (2013) 121:1553–62. doi: 10.1182/blood-2012-05-432336
150. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci. (2017) 19:E92. doi: 10.3390/ijms19010092
151. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
152. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
153. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
154. Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. (2009) 9:692–703. doi: 10.1038/nri2634
155. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. (2004) 18:2195–224. doi: 10.1101/gad.1228704
156. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
157. McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C, et al. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol. (2009) 183:2167–75. doi: 10.4049/jimmunol.0803503
158. Lauber K, Keppeler H, Munoz LE, Koppe U, Schroder K, Yamaguchi H, et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ. (2013) 20:1230–40. doi: 10.1038/cdd.2013.82
159. Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. (2005) 19:1116–27. doi: 10.1101/gad.1297105
160. Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol. (2010) 30:4564–74. doi: 10.1128/MCB.00146-10
161. Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci USA. (2012) 109:11776–81. doi: 10.1073/pnas.1206059109
162. Gupte R, Muse GW, Chinenov Y, Adelman K, Rogatsky I. Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proc Natl Acad Sci USA. (2013) 110:14616–21. doi: 10.1073/pnas.1309898110
163. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. (2007) 109:4313–9. doi: 10.1182/blood-2006-10-048215
164. Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBOJ. (2001) 20:7108–16. doi: 10.1093/emboj/20.24.7108
165. Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. (2002) 22:7802–11. doi: 10.1128/MCB.22.22.7802-7811.2002
166. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. (2006) 203:1883–9. doi: 10.1084/jem.20060336
167. Vattakuzhi Y, Abraham SM, Freidin A, Clark AR, Horwood NJ. Dual-specificity phosphatase 1-null mice exhibit spontaneous osteolytic disease and enhanced inflammatory osteolysis in experimental arthritis. Arthritis Rheum. (2012) 64:2201–10. doi: 10.1002/art.34403
168. Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. (2006) 176:1899–907. doi: 10.4049/jimmunol.176.3.1899
169. Hammer M, Echtenachter B, Weighardt H, Jozefowski K, Rose-John S, Mannel DN, et al. Increased inflammation and lethality of Dusp1-/- mice in polymicrobial peritonitis models. Immunology. (2010) 131:395–404. doi: 10.1111/j.1365-2567.2010.03313.x
170. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. (2009) 23:3649–58. doi: 10.1096/fj.09-134684
171. Riccardi C, Bruscoli S, Ayroldi E, Agostini M, Migliorati G. GILZ, a glucocorticoid hormone induced gene, modulates T lymphocytes activation and death through interaction with NF-κB. Adv Exp Med Biol. (2001) 495:31–9. doi: 10.1007/978-1-4615-0685-0_5
172. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimege L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med. (2013) 5:456–70. doi: 10.1002/emmm.201201683
173. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology. (2009) 136:530–41. doi: 10.1053/j.gastro.2008.09.024
174. Hahn RT, Hoppstadter J, Hirschfelder K, Hachenthal N, Diesel B, Kessler SM, et al. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis. (2014) 234:391–400. doi: 10.1016/j.atherosclerosis.2014.03.028
175. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum. (2010) 62:2651–61. doi: 10.1002/art.27566
176. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H, et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum. (2013) 65:1203–12. doi: 10.1002/art.37858
177. Robert O, Boujedidi H, Bigorgne A, Ferrere G, Voican CS, Vettorazzi S, et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J Hepatol. (2016) 64:916–24. doi: 10.1016/j.jhep.2015.11.023
178. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. (1995) 270:286–90. doi: 10.1126/science.270.5234.286
179. Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS Jr. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. (1995) 270:283–6. doi: 10.1126/science.270.5234.283
180. Chinenov Y, Coppo M, Gupte R, Sacta MA, Rogatsky I. Glucocorticoid receptor coordinates transcription factor-dominated regulatory network in macrophages. BMC Genomics. (2014) 15:656. doi: 10.1186/1471-2164-15-656
181. Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, et al. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci USA. (2006) 103:6653–8. doi: 10.1073/pnas.0508235103
182. Das M, Lu J, Joseph M, Aggarwal R, Kanji S, McMichael BK, et al. Kruppel-like factor 2 (KLF2) regulates monocyte differentiation and functions in mBSA and IL-1beta-induced arthritis. Curr Mol Med. (2012) 12:113–25. doi: 10.2174/156652412798889090
183. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. (2011) 121:2736–49. doi: 10.1172/JCI45444
184. Barczyk K, Ehrchen J, Tenbrock K, Ahlmann M, Kneidl J, Viemann D, et al. Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood. (2010) 116:446–55. doi: 10.1182/blood-2009-10-247106
185. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. (2009) 9:62–70. doi: 10.1038/nri2470
186. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of Inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res. (2016) 2016:8239258. doi: 10.1155/2016/8239258
187. Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1-/- mouse. FASEB J. (2003) 17:253–5. doi: 10.1096/fj.02-0239fje
188. Damazo AS, Yona S, Flower RJ, Perretti M, Oliani SM. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J Immunol. (2006) 176:4410–8. doi: 10.4049/jimmunol.176.7.4410
189. Patel HB, Kornerup KN, Sampaio AL, D'Acquisto F, Seed MP, Girol AP, et al. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann Rheum Dis. (2012) 71:1872–80. doi: 10.1136/annrheumdis-2011-201180
190. Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, et al. Peroxisome proliferator-activated receptor-gamma modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front Immunol. (2018) 9:893. doi: 10.3389/fimmu.2018.00893
191. Smoak K, Cidlowski JA. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol. (2006) 26:9126–35. doi: 10.1128/MCB.00679-06
192. Ishmael FT, Fang X, Galdiero MR, Atasoy U, Rigby WF, Gorospe M, et al. Role of the RNA-binding protein tristetraprolin in glucocorticoid-mediated gene regulation. J Immunol. (2008) 180:8342–53. doi: 10.4049/jimmunol.180.12.8342
193. Qiu LQ, Stumpo DJ, Blackshear PJ. Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J Immunol. (2012) 188:5150–9. doi: 10.4049/jimmunol.1103700
194. Shuto T, Imasato A, Jono H, Sakai A, Xu H, Watanabe T, et al. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J Biol Chem. (2002) 277:17263–70. doi: 10.1074/jbc.M112190200
195. Lannan EA, Galliher-Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFalpha coregulate gene expression in vitro and in vivo. Endocrinology. (2012) 153:3701–12. doi: 10.1210/en.2012-1020
196. Itoh N, Kasutani K, Muto N, Otaki N, Kimura M, Tanaka K. Blocking effect of anti-mouse interleukin-6 monoclonal antibody and glucocorticoid receptor antagonist, RU38486, on metallothionein-inducing activity of serum from lipopolysaccharide-treated mice. Toxicology. (1996) 112:29–36. doi: 10.1016/0300-483X(96)03345-8
197. Kasutani K, Itoh N, Kanekiyo M, Muto N, Tanaka K. Requirement for cooperative interaction of interleukin-6 responsive element type 2 and glucocorticoid responsive element in the synergistic activation of mouse metallothionein-I gene by interleukin-6 and glucocorticoid. Toxicol Appl Pharmacol. (1998) 151:143–51. doi: 10.1006/taap.1998.8452
198. Kelly EJ, Sandgren EP, Brinster RL, Palmiter RD. A pair of adjacent glucocorticoid response elements regulate expression of two mouse metallothionein genes. Proc Natl Acad Sci USA. (1997) 94:10045–50. doi: 10.1073/pnas.94.19.10045
199. Waelput W, Broekaert D, Vandekerckhove J, Brouckaert P, Tavernier J, Libert C. A mediator role for metallothionein in tumor necrosis factor-induced lethal shock. J Exp Med. (2001) 194:1617–24. doi: 10.1084/jem.194.11.1617
200. Takano H, Inoue K, Yanagisawa R, Sato M, Shimada A, Morita T, et al. Protective role of metallothionein in acute lung injury induced by bacterial endotoxin. Thorax. (2004) 59:1057–62. doi: 10.1136/thx.2004.024232
201. Inoue K, Takano H, Shimada A, Wada E, Yanagisawa R, Sakurai M, et al. Role of metallothionein in coagulatory disturbance and systemic inflammation induced by lipopolysaccharide in mice. FASEB J. (2006) 20:533–5. doi: 10.1096/fj.05-3864fje
202. Subramanian Vignesh K, Deepe GS Jr. Metallothioneins: emerging modulators in immunity and infection. Int J Mol Sci. (2017) 18:E2197. doi: 10.3390/ijms18102197
203. Miyata M, Lee JY, Susuki-Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun. (2015) 6:6062. doi: 10.1038/ncomms7062
204. Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. (2001) 108:689–701. doi: 10.1172/JCI12450
205. McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med. (2004) 170:987–93. doi: 10.1164/rccm.200405-684OC
206. Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. (2004) 169:1245–51. doi: 10.1164/rccm.200309-1258OC
207. Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. (2008) 103:1164–72. doi: 10.1161/01.RES.0000338501.84810.51
208. Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. (2009) 119:1871–9. doi: 10.1172/JCI38575
209. Rapp AE, Hachemi Y, Kemmler J, Koenen M, Tuckermann J, Ignatius A. Induced global deletion of glucocorticoid receptor impairs fracture healing. FASEB J. (2018) 32:2235–45. doi: 10.1096/fj.201700459RR
210. Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. (1999) 23:99–103. doi: 10.1038/12703
211. Brewer JA, Khor B, Vogt SK, Muglia LM, Fujiwara H, Haegele KE, et al. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. (2003) 9:1318–22. doi: 10.1038/nm895
212. Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest. (2012) 122:2384–94. doi: 10.1172/JCI63067
213. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. (1999) 8:265–77. doi: 10.1023/A:1008942828960
214. Meers GK, Bohnenberger H, Reichardt HM, Luhder F, Reichardt SD. Impaired resolution of DSS-induced colitis in mice lacking the glucocorticoid receptor in myeloid cells. PLoS ONE. (2018) 13:e0190846. doi: 10.1371/journal.pone.0190846
215. Galuppo P, Vettorazzi S, Hovelmann J, Scholz CJ, Tuckermann JP, Bauersachs J, et al. The glucocorticoid receptor in monocyte-derived macrophages is critical for cardiac infarct repair and remodeling. FASEB J. (2017) 31:5122–32. doi: 10.1096/fj.201700317R
216. Preusch MR, Rattazzi M, Albrecht C, Merle U, Tuckermann J, Schutz G, et al. Critical role of macrophages in glucocorticoid driven vascular calcification in a mouse-model of atherosclerosis. Arterioscler Thromb Vasc Biol. (2008) 28:2158–64. doi: 10.1161/ATVBAHA.108.174128
217. Carrillo-de Sauvage MA, Maatouk L, Arnoux I, Pasco M, Sanz Diez A, Delahaye M, et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ. (2013) 20:1546–57. doi: 10.1038/cdd.2013.108
218. Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci USA. (2011) 108:6632–7. doi: 10.1073/pnas.1017820108
Keywords: glucocorticoid receptor, inflammation, macrophages, mouse models, gene regulation
Citation: Escoter-Torres L, Caratti G, Mechtidou A, Tuckermann J, Uhlenhaut NH and Vettorazzi S (2019) Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 10:1859. doi: 10.3389/fimmu.2019.01859
Received: 15 May 2019; Accepted: 23 July 2019;
Published: 07 August 2019.
Edited by:
Claude Libert, Flanders Institute for Biotechnology, BelgiumReviewed by:
Zsuzsa Szondy, University of Debrecen, HungaryMarcel Schaaf, Leiden University, Netherlands
Copyright © 2019 Escoter-Torres, Caratti, Mechtidou, Tuckermann, Uhlenhaut and Vettorazzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Henriette Uhlenhaut, aGVucmlldHRlLnVobGVuaGF1dEBoZWxtaG9sdHotbXVlbmNoZW4uZGU=; Sabine Vettorazzi, c2FiaW5lLnZldHRvcmF6emlAdW5pLXVsbS5kZQ==
†These authors have contributed equally to this work