 Kangsan Kim
Kangsan Kim Mauricio Marquez-Palencia
Mauricio Marquez-Palencia Srinivas Malladi
Srinivas Malladi- 1Department of Pathology, UT Southwestern Medical Center, Dallas, TX, United States
- 2Harold C. Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX, United States
Metastatic relapse is observed in cancer patients with no clinical evidence of disease for months to decades after initial diagnosis and treatment. Disseminated cancer cells that are capable of entering reversible cell cycle arrest are believed to be responsible for these late metastatic relapses. Dynamic interactions between the latent disseminated tumor cells and their surrounding microenvironment aid cancer cell survival and facilitate escape from immune surveillance. Here, we highlight findings from preclinical models that provide a conceptual framework to define and target the latent metastatic phase of tumor progression. The hope is by identifying patients harboring latent metastatic cells and providing therapeutic options to eliminate metastatic seeds prior to their emergence will result in long lasting cures.
Introduction
In a significant number of cancer patients considered disease free, metastatic relapses occur. If and when relapse will occur is a question that is both indeterminate and unanswerable. Depending on the tumor type, these relapses might occur within a few months or decades after initial diagnosis and treatment (1–3). Very late recurrences are reported in a subset of breast cancer, head and neck squamous cell carcinoma (HNSCC), prostate cancer, melanoma and renal cell carcinoma patients considered disease free, presenting a major treatment follow-up challenge (4–18) (Table 1). In comparison, small cell lung cancer patients with aggressive disease have no reported latency as they are diagnosed with metastatic disease and have very poor survival rate (19). Lung cancers have short metastatic latency spans, with majority of relapses occurring within a year (20). Breast cancers with high proliferative index, triple negative breast cancers (TNBCs), tend to have shorter latency periods compared to estrogen receptor (ER) positive breast cancer (21). The frequency of late recurrence after 5 years is greatly reduced in TNBCs compared to ER+ tumors, where disease recurrences have been reported in a significant number of patients as late as 20 years after primary diagnosis (22–24). Human autopsy and transplant studies report existence of disseminated tumor cells (DTCs; tumor cells that extravasate and reside in secondary organs) or metastatic lesions that persist as occult disease, highlighting the role of host immune system in limiting metastatic outgrowth (25, 26). Latency competent cancer cells (LCCs) are slow cycling or quiescent DTCs that persist in organs after surgery and initial therapy, and are the major source of disease relapse (2, 3, 27). LCCs reside next to the vasculature and are surrounded by extracellular matrix (ECM), soluble factors, stromal, and immune cells. LCCs remain unscathed in these sanctuaries, undergoing genetic/epigenomic adaptations that augment their ability to initiate metastasis and impede immune surveillance. Metastatic latency therefore depends on the oncogenomic status of the disseminated tumor cells, their proliferative capacity and the surrounding microenvironment.
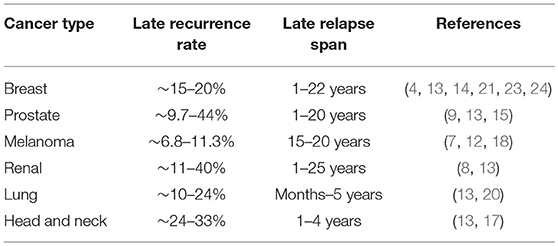
Table 1. Metastatic relapse rate and latency span in cancer patients.
Given that metastasis is the major cause for mortality in cancer patients, understanding how DTCs stay quiescent and remain viable for years before initiating metastasis is very critical (28). Assays to monitor the elusive LCCs and treatment strategies to effectively restrain or eliminate residual cancer cells is an unmet clinical need. Incorporating oncogenomic features of these cells along with tumor staging, presence of circulating/disseminated tumor cells or cell-free tumor DNA, will lead to better prediction of disease relapse in cancer patients with occult disease. Here, we summarize key determinants of metastatic latency, current concepts and proposed strategies to target and eliminate residual disease.
Dissemination: Get Out of Dodge
As solid tumors grow, tissue constraints and cellular energy needs drive genetic or epigenetic changes in cancer cells that facilitate epithelial to mesenchymal transition and acquisition of invasive and stem-cell like characteristics (29–32). Key concepts discussing dormancy and epithelial mesenchymal transition (EMT) have been recently reviewed (33). Invasive cancer cells within the primary tumors breach the basement membrane, permeate the surrounding tissue as single cells or clumps and migrate into vasculature or lymphatics (31, 34). How and when tumors become invasive in patients? Are the early or late disseminated tumor cells from the primary tumor responsible for initiating metastasis in patients (35, 36)? Do the early disseminators aid in developing pre-metastatic niche (37, 38)? And the role of stromal cells in driving invasion (39), are some of the open questions actively investigated.
Although cancer cells intravasate in large numbers, very few survive in circulation. Given their prognostic value, circulating tumor cell (CTC) counts have been used to predict relapse or metastatic disease in breast, colorectal, small cell lung, and prostate cancer patients (40–43). Efforts from several labs have been directed toward improving CTC capture and enrichment protocols to define surface biomarkers on these potential metastatic seeds and to predict metastatic incidence (44, 45). Many ultrasensitive devices that are able to segregate CTCs from patient blood using size, density, electrical and compressibility differences have been developed to address this clinical need (46–50). However, isolating viable CTCs and performing functional experiments has been a challenge. With improved protocols and devices, several groups are now able to isolate, culture and characterize CTCs from patients (51–53). Such models are indispensable to study and advance concepts in metastatic evolution (44). CTC clusters or aggregates have also been isolated from blood stream and are reported to have greater predisposition to form metastasis than single cells in animal models (54). How CTC clusters survive the shear stress in circulation and avoid entrapment in lung capillaries is unclear (55). Further research is needed to determine how CTC clusters are dispersed or assembled in circulation (56, 57); what are they composed of; and what aspect of clustering aids metastatic competence. It should be noted that reliable CTC isolation and characterization is feasible only in metastatic disease and may not be able to identify patients with minimal residual disease. Nonetheless, diagnostic leukapheresis may enable reliable detection of CTCs in non-metastatic patients (58, 59).
Metastatic Latency: It's not Kansas Anymore to Make Oneself at Home
Metastatic latency span is both variable and indeterminate as it is a function of the rate at which the disseminated cancer cells adapt to and alter the surrounding microenvironment to initiate a metastatic lesion that impairs organ function. The composition and architecture of metastatic microenvironment determines the likelihood of DTC colonization (60). Majority of CTCs that extravasate into the new cellular milieu face resistance and perish upon extravasation (61). Cancer cells therefore have the propensity to reside in precincts that resemble stroma of the primary tumor (62). Depending on the robustness of the perceived cues, cancer cells are likely to proliferate, apoptose, or enter into a quiescent-slow cycling state (Figure 1). Proliferating DTCs are also more likely to be eliminated by chemotherapies and adjuvant therapies (22, 63). Slow cycling and quiescent LCCs, that are adapting to the new microenvironment remain unaffected by therapies targeting dividing cells and are enriched for stem cell like characteristics, that are critical to initiate secondary or metastatic tumors (27, 64, 65). Hypoxic microenvironments in the primary tumors promotes activation of dormancy programs and DTCs with these features are likely to survive better post-extravasation (66). Overall, absence of proliferating signals or a self-imposed block to these cues may result in activation of dormancy programs (27).
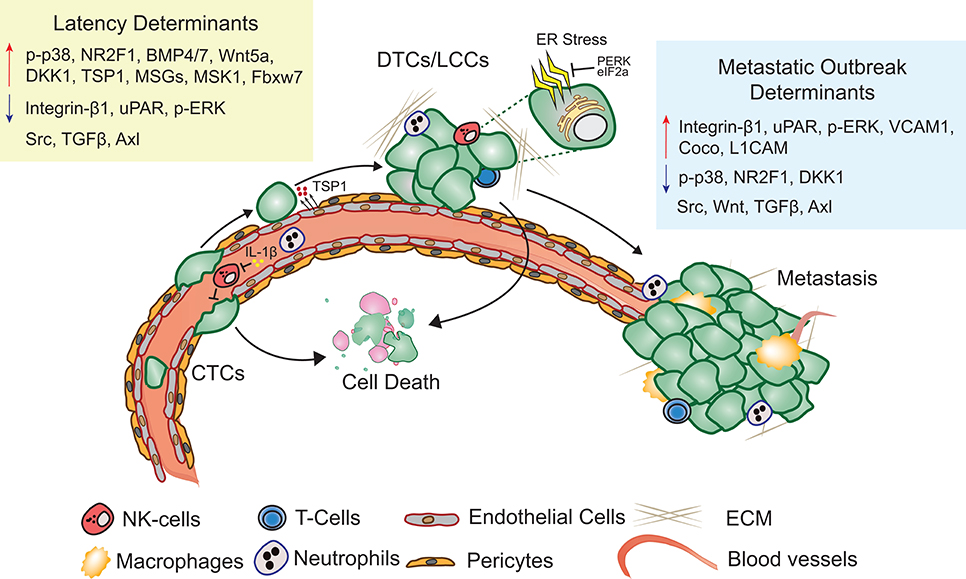
Figure 1. Metastatic latency. Upon extravasation, many DTCs perish, few surviving LCCs adapt to and modify the surrounding microenvironment, eventually giving rise to metastasis. Key molecular determinants of latency and metastatic outbreak are highlighted (Refer text for more details).
Extracellular Matrix
The distribution and availability of growth factors and cytokines is tightly regulated by the ECM surrounding DTCs (37, 67). Non-structural matrix protein such as Thrombospondin-1 (TSP-1) and Periostin through direct interaction with membrane receptors and fibrous ECM molecules modulate cancer cell proliferation status (68). Collagen enriched fibrotic environment leads to activation of myosin light chain kinase through integrin β1 signaling and promotes proliferation in cancer cells, while failure to engage proliferative signals results in dormancy (69, 70). Mitogen activated protein kinase (MAPK) activity has been clearly demonstrated to regulate proliferation status of human squamous carcinoma, melanoma, breast, and prostate cancer cells (71). Increased p38 and decreased ERK activity is observed in dormant cancer cells. Urokinase-type plasminogen activator receptor (uPAR) drives activation of ERK through FAK and Src kinases and promotes proliferation, while loss of either uPAR expression or FAK/Src activity leads to increased p38 kinase activity and unleashes downstream quiescence effectors DEC2, NR2F1, and CDK inhibitors (60, 64, 72–74). Src activation in response to CXCL12 and IGF1, potentiates PI3K/AKT activation, and aids survival of latent breast cancer DTCs independent of their hormone receptor status or cancer subtype (62). Expression of metastatic suppressor genes (MSGs: KISS1, KAI1, MKK4/6/7, and NM23) has also been reported to limit metastasis initiating capacity of DTCs by modulating the activity of MAPKs through G-protein coupled receptors and tyrosine receptor kinases (75, 76). Over-expression of KISS1 results in limiting metastatic outgrowth of aggressive human melanoma cell line (77). Similarly, NM23 and MKK4/6 activate p38 and inhibit ERK to induce dormancy in ovarian and breast cancer cells (76). Mitogen and stress-activated kinase 1 (MSK1) functions downstream of p38 and restrains breast DTCs into a steady micro-metastatic state by promoting luminal differentiation through GATA3 and FOXA1 in ER+ breast cancer (78). Along these lines, GATA6 and HOPX have been reported to limit lung adenocarcinoma metastasis by promoting alveolar differentiation (79). Integrin α5β3 signaling response can also promote differentiation of luminal A breast cancer cells and limit tumor progression (80). L1CAM and YAP signaling enable the outgrowth of metastasis-initiating cells through integrin-ILK both immediately following their infiltration of target organs and as they exit metastatic latency (81). Taken together, altered ECM and MAPK activity in response to microenvironmental cues influences the proliferation status of latent DTCs.
Endoplasmic Reticulum (ER) Stress
Transducers of unfolded protein response (PERK and eIF2α) are also activated in p38 active dormant cells and have been shown to be essential for cancer cell survival under chemotherapy induced genotoxic stress (74, 82). CK19 and MHC class I negative dormant pancreatic DTCs activate PERK and relieving ER stress pharmacologically or by expression of XBP1 in combination with T-cell depletion resulted in metastatic outgrowth (83). Administration of chemical chaperone 4-PBA to relieve ER stress in DTCs preoperatively has been proposed to drive DTCs out of quiescence and be cleared by active adaptive immune surveillance (83). Likewise, Fbxw7, a component of SCF-E3 ubiquitin ligase complex has been reported to maintain dormancy in breast DTCs and its ablation led to increased proliferation in this model system. A combination of depleting Fbxw7 and chemotherapy has been proposed to limit residual disease (84). Will these approaches result in reduced metastatic incidence or worsen survival outcome in patients by unleashing restrained heterogeneous metastatic clones needs to be further explored.
Supportive Niches
Specialized microenvironments surrounding LCCs limit proliferation and facilitate cancer cell survival and quiescence. For example, perivascular niche (PVN) supports survival of hematopoietic stem cells (HSCs) as well as disseminated lung, melanoma, breast and prostate cancer cells in the bone marrow (85–87). TSP1 secreted by the microvascular endothelium in the bone and lung induce growth arrest in breast DTCs, while high Periostin and TGF-β1 expression in the neovascular tip cells triggers metastatic relapse (68). Inhibition of integrin-mediated interactions between DTCs (either quiescent or proliferating) and PVN sensitizes them to chemotherapy (88). Several stromal derived factors have inhibitory effect on LCC proliferation. For example, leukemia inhibitory factor (LIF) secreted by osteoblasts and bone marrow stromal cells limit growth of breast cancer DTCs in bone by activating LIFR: STAT3 signaling (89). Prostate cancer DTCs and drug resistant dormant myeloma cells in the bone marrow respond to osteoblast derived growth arrest specific 6 (GAS6), through Axl, a receptor tyrosine kinase and remain dormant (73, 90, 91). HSC driving factors such as osteoblast secreted stromal cell-derived factor 1 (SDF-1/CXCL12) binds to CXCR4 on cancer cells and retains them in the HSC niche (87). SDF-1 CXCR4 interaction plays an important role in keeping chronic myeloid leukemia stem cells dormant. Depletion of CXCL12 in mesenchymal stromal cells led to increased proliferation of these dormant cells while deletion of CXCL12 in endothelial cells resulted in reduced proliferation (92). Dormant breast cancer cells are predominantly found in the E-selectin and SDF-1 rich perisinusoidal vascular regions. Simultaneous blockade of CXCR4 and E-selectin in patients could release dormant micro metastases from the protective bone microenvironment and also prevent adhesion in the first place (93).
TGF-β2 rich bone microenvironment promotes quiescence in HNSCC DTCs by inducing cell cycle inhibitor p27, metastatic suppressor DEC2 and SMAD1/5 activation, while the TGF-β2 low lung microenvironment permits metastatic outgrowth. Removing this break by inhibiting TGF-βRIII increased metastatic burden in mice (94). BMP7, another TGF-β family member secreted by bone stromal cells induces senescence in prostate DTCs through BMPR2 dependent activation of p38 and p21. Withdrawal of BMP7 in this mouse model of prostate cancer induces recurrent metastatic growth in the bone (95). Similarly, BMP4 supports breast cancer dormancy in the lung, while its antagonist, Coco, drives metastatic outgrowth (96). Cancer cells have also been reported to either cannibalize bone marrow derived mesenchymal stem cells or prime them to secrete microRNA packed exosomes that promote quiescence (97, 98). WNT5a from the osteoblastic niche induces dormancy in prostate cancer cells by activating non-canonical ROR/SIAH2 signaling and repressing canonical WNT/β-catenin signaling (99). In an autocrine fashion, breast and lung cancer DTCs can also enforce a slow cycling state by inhibiting WNT/β-catenin signaling (27).
Innate and Adaptive Leukocytes
Host immunity plays an important role in shaping and limiting tumor growth and progression (100–106). Neutrophils are the most abundant circulating immune cells and among the first ones to infiltrate the lung metastatic niche. Their role in either promoting or inhibiting metastasis is highly debated (107). MET expressing neutrophils secrete reactive oxygen species and are reported to be anti-metastatic (108, 109). In contrast, several studies identify a pro-metastatic function for neutrophils (110). Neutrophils inhibit natural killer (NK) cell function and facilitate extravasation of tumor cells by secreting IL-1β and matrix metalloproteinases (111). Neutrophil derived leukotrienes further support early colonization of breast cancer cells (112, 113). Depletion of neutrophils or genetic ablation of CXCR2, suppressed metastasis in pancreatic cancer models and lead to increased T-cell infiltration and extended survival (114). Recent reports highlight the role of neutrophils in metastatic outbreaks induced by sustained lung inflammation caused by tobacco smoke or bacterial derived lipopolysaccharide (115). Systemic inflammatory response induced after surgery can also promote the re-emergence of tumors that were kept in check by a tumor-specific T-cell response (116). Inflammation in lung, induced formation of neutrophil extracellular traps (NET) that resulted in cleavage and remodeling of laminin. Remodeled laminin activated integrin signaling and induced proliferation in otherwise dormant lung DTCs. This escape from latency is reported to be dependent on expression of Zeb1, a key modulator of EMT (117). Antibodies against NET-remodeled laminin prevented awakening of dormant cells and has been proposed as an approach to prevent metastatic outbreaks and prolong survival of cancer patients (115). Of note, obesity causes lung neutrophilia and the increase in neutrophils favors breast cancer metastasis to lung (118, 119).
Tissue resident macrophages or infiltrating monocytes are also reported to play an important role in either limiting or promoting early colonization of DTCs post extravasation (105, 120). Monocyte chemotactic and activating factor (CCL2) secreted by cancer cells and stroma recruits CXCR2+ positive monocytes and macrophages to enable seeding, colonization and outgrowth (121, 122). VCAM1 on breast cancer cells in leukocyte rich lung microenvironment binds to α4β1 integrin on macrophages and activates Ezrin-AKT survival pathway in cancer cells (123). In the bone, aberrant expression of VCAM1 promotes transition from indolent to overt metastasis in breast DTCs. VCAM1 expressing DTCs attract and tether to integrin α4β1 expressing osteoclast progenitors and give rise to osteolytic metastasis. Antibodies against α4 integrin block this prosurvival function of VCAM1 and metastatic burden (124). NR4A1 positive patrolling monocytes that are enriched in the microvasculature of the lung, engulf melanoma, and breast tumor cells and reduce lung colonization and metastasis (125, 126). They also promote recruitment and activation of NK cells. Administration of selective class IIa histone deacetylate (HDAC) inhibitor, in MMTV-pyMT mouse model, resulted in reduced tumor burden and spontaneous pulmonary metastasis. HDAC inhibition reverts the pro-tumorigenic phenotype of tumor associated macrophages, recruits anti-tumor phagocytic macrophages and stimulates the adaptive immune response (127). Selective inhibition of histone deacetylase may unleash the antitumor potential of macrophages and keep DTCs in check.
NK cells play an important role in surveilling and eradicating cancer cells in circulation and upon extravasation (106, 128). By releasing cytolytic granules and pro-apoptotic factors or cytokines, NK cells kill tumor cells. They also release chemokines that attract T-cells, dendritic cells, and monocytes promoting adaptive immune response (129, 130). NK cell cytotoxicity has been negatively correlated with metastatic burden in several cancer types (131, 132). Depletion of NK cells aid metastatic outbreaks in disseminated cancer cells from breast and lung cancers (27, 111, 133). As tumors become invasive and acquire mesenchymal traits, they upregulate expression of cell surface NK cell activating ligands and are more susceptible to elimination by NK cells (134). DTCs are therefore more susceptible to immune recognition in circulation and upon extravasation (106). Nonetheless, cancer cells evade NK mediated immune surveillance by either down regulating NK cell activating ligands and death inducing receptors (135, 136). For example, extravasated breast and lung cancer DTCs in lung, brain, liver, and kidneys evade immune attack by NK cells by entering into a slow cycling or quiescent state enforced by autocrine inhibition of WNT signaling pathway (27, 133). Through mechanisms yet to be defined, these slow cycling DTCs downregulate expression of several NK cell activating sensors (27).
In a spontaneous mouse model of melanoma, early dissemination of tumor cells to the lung was observed and the DTCs remained dormant for varying periods of time. Depletion of CD8+ T cells in these metastasis models resulted to increased metastatic out breaks (137). Similarly, depletion of CD4+ and CD8+ T cells 5 months after surgical removal of methylcholanthrene-induced fibro sarcoma tumor results in lung metastasis, highlighting the role of T cells in eliminating proliferative DTCs (138). In this model, intratumoral MHC-I heterogeneity dictates metastatic capacity and is proposed to predict response to immunotherapy (139). It is possible that the immune equilibrium at the metastatic site is maintained by the immune suppressive (MDSCs, Treg) and tumor inhibiting (T cells, NK cells) cells. Taken together, all these studies reinforce the role of innate and adaptive immune system in either delaying or limiting metastatic incidence. They also provide a framework to investigate the effect of host microenvironment on metastatic latency. Given that mouse and human immune systems are different, development of reliable preclinical models that replicate human immune surveillance are desired.
Target Residual Disease: How to Eliminate the Veiled Threat?
Tracking residual disease in patients with no obvious symptoms is challenging. In order to accurately predict relapse, genomic and epigenomic characteristics of divergent disseminated cancer cells at the metastatic site and their associated phenotypic information is needed. Disease predictions depend on preclinical models, that are imperfect as they are based on assumptions that change with novel insights and discoveries. Nonetheless, every model, in spite of its limitations, has advanced our understanding of this phase of tumor progression (140, 141) (Table 2).
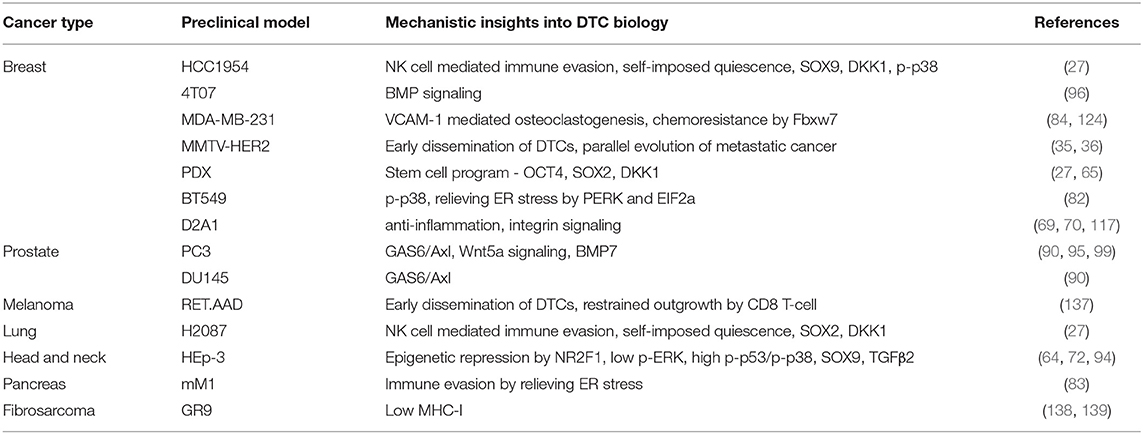
Table 2. Metastatic latency preclinical models.
Keeping DTCs in a quiescent non-proliferative state is an attractive viable approach to limit delayed metastatic incidence (115, 142, 143). Adjuvant anti-estrogen therapy with the ER antagonists is a standard of care for patients with ER+ breast cancers for years after initial diagnosis and this approach has significantly improved survival outcomes (22, 63). FDA approved CDK4/6 inhibitors for ER+ breast cancers, block cancer cell proliferation and induce dormancy or senescence in various models (144). Such inhibitors have potential to limit relapse in cancers with prolonged metastatic latency phase. Inhibition of integrin β1, uPAR, ERK, and Src driven signaling might prevent metastatic breakouts. Activation of p38, NR2F1, or administration of GAS6, BMP4/7, WNT inhibitors, and TGF-β2 might be effective in limiting relapse. The major challenge for this approach is identifying enforcers of quiescence that are effective in all tissues and specific for cancers with distinct oncogenomic features. Also unknown is how well tolerated these extended therapies will be in patients and how effective this approach will be on slow cycling DTCs. Nevertheless, the threat of disease relapse will still remain.
Removing the proliferative break or mobilizing DTCs from their niches and allowing anti-proliferative drugs or immune surveillance to target DTCs is an alternative strategy (83, 84, 94, 95, 145, 146). Unleashing the proliferative potential of quiescent population has disease management concerns. In order to be effective, this approach would have to drive all DTCs out of quiescence and the subsequent treatment has to effectively eliminate all proliferating cancer cells, which is unlikely. Moreover, this approach may result in selection of clones that don't respond to available therapies and can be detrimental to patient health. Eliminating quiescent DTCs by targeting intrinsic or extrinsic enforcers of this state is an attractive approach that needs to be further explored in clinic (142, 143).
Concluding Remarks
Early detection of disseminated disease with improved understanding of cellular and molecular mechanisms driving metastatic latency in an organ with distinct tissue architecture is critical to provide effective therapeutic interventions. Designing a clinical trial to assess the benefit of proposed strategies is a major challenge. Some obvious questions apart from the cost being: how to define patients with likelihood of disease relapse, trial duration and endpoint criteria. Further research with preclinical models that faithfully represent this phase of tumor progression will provide risk prognostication tools, novel targets and treatment strategies to eliminate minimal residual disease.
Author Contributions
SM conception, design, and wrote the manuscript. KK, MM-P, and SM literature search. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by grants from CPRIT (RR170003), KCP SPORE DRP (P50CA196516) grants to SM, and NSF grant (2019281049) to MM-P. We apologize to colleagues whose work was not cited due to space limitations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Pollard JW. Defining metastatic cell latency. N Engl J Med. (2016) 375:280–2. doi: 10.1056/NEJMcibr1606716
2. Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. (2010) 10:871–7. doi: 10.1038/nrc2933
3. Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Cancer Res. (2009) 102:67–101. doi: 10.1016/S0065-230X(09)02003-X
4. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. (2004) 10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110
5. Cote RJ, Rosen PP, Hakes TB, Sedira M, Bazinet M, Kinne DW, et al. Monoclonal antibodies detect occult breast carcinoma metastases in the bone marrow of patients with early stage disease. Am J Surg Pathol. (1988) 12:333–40. doi: 10.1097/00000478-198805000-00001
6. Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. (2010) 2:25ra3. doi: 10.1126/scitranslmed.3000403
7. Tsao H, Cosimi AB, Sober AJ. Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer. (1997) 79:2361–70. doi: 10.1002/(SICI)1097-0142(19970615)79:12<2361::AID-CNCR10>3.0.CO;2-P
8. McNichols DW, Segura JW, DeWeerd JH. Renal cell carcinoma: long-term survival and late recurrence. J Urol. (1981) 126:17–23. doi: 10.1016/S0022-5347(17)54359-1
9. Liesenfeld L, Kron M, Gschwend JE, Herkommer K. Prognostic factors for biochemical recurrence more than 10 years after radical prostatectomy. J Urol. (2017) 197:143–8. doi: 10.1016/j.juro.2016.07.004
10. Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev. (2011) 21:42–9. doi: 10.1016/j.gde.2010.10.011
12. Leman JA, Mac Kie RM. Late (>10 years) recurrence of melanoma: the Scottish experience. Br J Dermatol. (2003) 148:372–3. doi: 10.1046/j.1365-2133.2003.05097_8.x
13. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. (2015) 6:10697–711. doi: 10.18632/oncotarget.4037
14. Janni W, Vogl FD, Wiedswang G, Synnestvedt M, Fehm T, Jückstock J, et al. Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse–a European pooled analysis. Clin Cancer Res. (2011) 17:2967–76. doi: 10.1158/1078-0432.CCR-10-2515
15. Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res. (2009) 15:677–83. doi: 10.1158/1078-0432.CCR-08-1754
16. Chin AI, Lam JS, Figlin RA, Belldegrun AS. Surveillance strategies for renal cell carcinoma patients following nephrectomy. Rev Urol. (2006) 8:1–7. doi: 10.1007/s11934-005-0062-x
17. Denaro N, Merlano MC, Russi EG. Follow-up in head and neck cancer: do more does it mean do better? A systematic review and our proposal based on our experience. Clin Exp Otorhinolaryngol. (2016) 9:287–97. doi: 10.21053/ceo.2015.00976
18. Faries MB, Steen S, Ye X, Sim M, Morton DL. Late recurrence in melanoma: clinical implications of lost dormancy. J Am Coll Surg. (2013) 217:27–34; discussion−6. doi: 10.1016/j.jamcollsurg.2013.03.007
19. Gazdar AF, Minna JD. Small cell lung cancers made from scratch. J Exp Med. (2019) 216:476–8. doi: 10.1084/jem.20182216
20. Gomis RR, Gawrzak S. Tumor cell dormancy. Mol Oncol. (2017) 11:62–78. doi: 10.1016/j.molonc.2016.09.009
21. Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. (2010) 28:3271–7. doi: 10.1200/JCO.2009.25.9820
22. Early Breast Cancer Trialists' Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. (2005) 365:1687–717. doi: 10.1016/S0140-6736(05)66544-0
23. Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. (2013) 381:805–16. doi: 10.1016/S0140-6736(12)61963-1
24. Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. (2017) 377:1836–46. doi: 10.1056/NEJMoa1701830
26. Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol. (2010) 11:790–6. doi: 10.1016/S1470-2045(10)70024-3
27. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
28. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. (2016) 529:298–306. doi: 10.1038/nature17038
29. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
30. Wei SC, Yang J. Forcing through tumor metastasis: the interplay between tissue rigidity and epithelial-mesenchymal transition. Trends Cell Biol. (2016) 26:111–20. doi: 10.1016/j.tcb.2015.09.009
31. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
32. Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. (2006) 26:87–98. doi: 10.3233/BD-2007-26108
33. Weidenfeld K, Barkan D. EMT and stemness in tumor dormancy and outgrowth: are they intertwined processes? Front Oncol. (2018) 8:381. doi: 10.3389/fonc.2018.00381
34. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
35. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Corrigendum: mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. (2018) 553:366. doi: 10.1038/nature24666
36. Hosseini H, Obradović MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. (2016) 540:552–8. doi: 10.1038/nature20785
37. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
38. Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Res. (2006) 66:11089–93. doi: 10.1158/0008-5472.CAN-06-2407
39. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. (2015) 5:932–43. doi: 10.1158/2159-8290.CD-15-0012
40. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. (2004) 351:781–91. doi: 10.1056/NEJMoa040766
41. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. (2004) 10:6897–904. doi: 10.1158/1078-0432.CCR-04-0378
42. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. (2008) 14:6302–9. doi: 10.1158/1078-0432.CCR-08-0872
43. Aggarwal C, Wang X, Ranganathan A, Torigian D, Troxel A, Evans T, et al. Circulating tumor cells as a predictive biomarker in patients with small cell lung cancer undergoing chemotherapy. Lung Cancer. (2017) 112:118–25. doi: 10.1016/j.lungcan.2017.08.008
44. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. (2017) 14:155–67. doi: 10.1038/nrclinonc.2016.144
45. Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. (2016) 35:1216–24. doi: 10.1038/onc.2015.192
46. Zhou L, Dicker DT, Matthew E, El-Deiry WS, Alpaugh RK. Circulating tumor cells: silent predictors of metastasis. F1000Res. (2017) 6:1445. doi: 10.12688/f1000research.11313.1
47. Zheng S, Lin H, Liu JQ, Balic M, Datar R, Cote RJ, et al. Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J Chromatogr A. (2007) 1162:154–61. doi: 10.1016/j.chroma.2007.05.064
48. Hosokawa M, Hayata T, Fukuda Y, Arakaki A, Yoshino T, Tanaka T, et al. Size-selective microcavity array for rapid and efficient detection of circulating tumor cells. Anal Chem. (2010) 82:6629–35. doi: 10.1021/ac101222x
49. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci USA. (2010) 107:18392–7. doi: 10.1073/pnas.1012539107
50. Song Y, Tian T, Shi Y, Liu W, Zou Y, Khajvand T, et al. Enrichment and single-cell analysis of circulating tumor cells. Chem Sci. (2017) 8:1736–51. doi: 10.1039/C6SC04671A
51. Hamza B, Ng SR, Prakadan SM, Delgado FF, Chin CR, King EM, et al. Optofluidic real-time cell sorter for longitudinal CTC studies in mouse models of cancer. Proc Natl Acad Sci USA. (2019) 116:2232–6. doi: 10.1073/pnas.1814102116
52. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. (2014) 20:897–903. doi: 10.1038/nm.3600
53. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. (2017) 23:114–9. doi: 10.1038/nm.4239
54. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. (2014) 158:1110–22. doi: 10.1016/j.cell.2014.07.013
55. Aceto N, Toner M, Maheswaran S, Haber DA. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer. (2015) 1:44–52. doi: 10.1016/j.trecan.2015.07.006
56. Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. (2019) 9:96–113. doi: 10.1158/2159-8290.CD-18-0065
57. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
58. Fischer JC, Niederacher D, Topp SA, Honisch E, Schumacher S, Schmitz N, et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc Natl Acad Sci USA. (2013) 110:16580–5. doi: 10.1073/pnas.1313594110
59. Fehm TN, Meier-Stiegen F, Driemel C, Jäger B, Reinhardt F, Naskou J, et al. Diagnostic leukapheresis for CTC analysis in breast cancer patients: CTC frequency, clinical experiences and recommendations for standardized reporting. Cytometry A. (2018) 93:1213–9. doi: 10.1002/cyto.a.23669
60. Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. (2012) 195:25–39. doi: 10.1007/978-3-642-28160-0_3
61. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. (1998) 153:865–73. doi: 10.1016/S0002-9440(10)65628-3
62. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. (2013) 154:1060–73. doi: 10.1016/j.cell.2013.07.036
63. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
64. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun. (2015) 6:6170. doi: 10.1038/ncomms7170
65. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. (2015) 526:131–5. doi: 10.1038/nature15260
66. Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. (2017) 19:120–32. doi: 10.1038/ncb3465
67. Hynes RO. The extracellular matrix: not just pretty fibrils. Science. (2009) 326:1216–9. doi: 10.1126/science.1176009
68. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. (2013) 15:807–17. doi: 10.1038/ncb2767
69. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. (2010) 70:5706–16. doi: 10.1158/0008-5472.CAN-09-2356
70. Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci USA. (2009) 106:10290–5. doi: 10.1073/pnas.0904227106
71. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. (2003) 63:1684–95. doi: 10.1016/j.urolonc.2003.12.012
72. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. (2001) 12:863–79. doi: 10.1091/mbc.12.4.863
73. Cackowski FC, Eber MR, Rhee J, Decker AM, Yumoto K, Berry JE, et al. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J Cell Biochem. (2017) 118:891–902. doi: 10.1002/jcb.25768
74. Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther. (2006) 5:729–35. doi: 10.4161/cbt.5.7.2968
75. Khan I, Steeg PS. Metastasis suppressors: functional pathways. Lab Invest. (2018) 98:198–210. doi: 10.1038/labinvest.2017.104
76. Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. (2003) 3:55–63. doi: 10.1038/nrc967
77. Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. (1996) 88:1731–7. doi: 10.1093/jnci/88.23.1731
78. Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat Cell Biol. (2018) 20:211–21. doi: 10.1038/s41556-017-0021-z
79. Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE, et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell. (2013) 23:725–38. doi: 10.1016/j.ccr.2013.04.009
80. Abu-Tayeh H, Weidenfeld K, Zhilin-Roth A, Schif-Zuck S, Thaler S, Cotarelo C, et al. 'Normalizing' the malignant phenotype of luminal breast cancer cells via alpha(v)beta(3)-integrin. Cell Death Dis. (2016) 7:e2491. doi: 10.1038/cddis.2016.387
81. Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol. (2018) 20:966–78. doi: 10.1038/s41556-018-0138-8
82. Bambang IF, Lu D, Li H, Chiu LL, Lau QC, Koay E, et al. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp Cell Res. (2009) 315:1964–74. doi: 10.1016/j.yexcr.2009.02.017
83. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. (2018) 360:eaao4908. doi: 10.1126/science.aao4908
84. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight. (2019) 4:125138. doi: 10.1172/jci.insight.125138
85. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. (2010) 16:116–22. doi: 10.1038/nm.2072
86. Cao Z, Scandura JM, Inghirami GG, Shido K, Ding BS, Rafii S. Molecular checkpoint decisions made by subverted vascular niche transform indolent tumor cells into chemoresistant cancer stem cells. Cancer Cell. (2017) 31:110–26. doi: 10.1016/j.ccell.2016.11.010
87. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. (2011) 121:1298–312. doi: 10.1172/JCI43414
88. Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol. (2019) 21:238–50. doi: 10.1038/s41556-018-0267-0
89. Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. (2016) 18:1078–89. doi: 10.1038/ncb3408
90. Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE. (2013) 8:e61873. doi: 10.1371/journal.pone.0061873
91. Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood. (2019) 134:30–43. doi: 10.1182/blood.2018880930
92. Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz Á, et al. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell. (2019) 24:769–84.e6. doi: 10.1016/j.stem.2019.02.018
93. Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. (2016) 8:340ra73. doi: 10.1126/scitranslmed.aad4059
94. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. (2013) 15:1351–61. doi: 10.1038/ncb2861
95. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. (2011) 208:2641–55. doi: 10.1084/jem.20110840
96. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. (2012) 150:764–79. doi: 10.1016/j.cell.2012.06.035
97. Bartosh TJ, Ullah M, Zeitouni S, Beaver J, Prockop DJ. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc Natl Acad Sci USA. (2016) 113:E6447–E56. doi: 10.1073/pnas.1612290113
98. Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K, et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. (2016) 76:5832–44. doi: 10.1158/0008-5472.CAN-16-1092
99. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med. (2019) 216:428–49. doi: 10.1084/jem.20180661
100. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. (1970) 13:1–27. doi: 10.1159/000386035
101. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
102. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. (2007) 450:903–7. doi: 10.1038/nature06309
103. Teng MW, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. (2008) 84:988–93. doi: 10.1189/jlb.1107774
104. Teng MW, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, et al. Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res. (2012) 72:3987–96. doi: 10.1158/0008-5472.CAN-12-1337
105. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. (2015) 15:73–86. doi: 10.1038/nri3789
106. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
107. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. (2016) 16:431–46. doi: 10.1038/nrc.2016.52
108. Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. (2015) 522:349–53. doi: 10.1038/nature14407
109. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. (2011) 20:300–14. doi: 10.1016/j.ccr.2011.08.012
110. Acharyya S, Massague J. Arresting supporters: targeting neutrophils in metastasis. Cell Res. (2016) 26:273–4. doi: 10.1038/cr.2016.17
111. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. (2016) 6:630–49. doi: 10.1158/2159-8290.CD-15-1157
112. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. (2015) 522:345–8. doi: 10.1038/nature14282
113. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. (2015) 528:413–7. doi: 10.1038/nature16140
114. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
115. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
116. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med. (2018) 10:eaan3464. doi: 10.1126/scitranslmed.aan3464
117. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. (2016) 76:6778–84. doi: 10.1158/0008-5472.CAN-16-0608
118. Olson OC, Quail DF, Joyce JA. Obesity and the tumor microenvironment. Science. (2017) 358:1130–1. doi: 10.1126/science.aao5801
119. Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. (2017) 19:974–87. doi: 10.1038/ncb3578
120. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
121. Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. (2013) 73:662–71. doi: 10.1158/0008-5472.CAN-12-0653
122. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
123. Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. (2011) 20:538–49. doi: 10.1016/j.ccr.2011.08.025
124. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
125. Hanna BS, McClanahan F, Yazdanparast H, Zaborsky N, Kalter V, Rößner PM, et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia. (2016) 30:570–9. doi: 10.1038/leu.2015.305
126. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science. (2015) 350:985–90. doi: 10.1126/science.aac9407
127. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
128. Roato I, Vitale M. The uncovered role of immune cells and NK cells in the regulation of bone metastasis. Front Endocrinol (Lausanne). (2019) 10:145. doi: 10.3389/fendo.2019.00145
129. Sun JC, Lanier LL. Natural killer cells remember: an evolutionary bridge between innate and adaptive immunity? Eur J Immunol. (2009) 39:2059–64. doi: 10.1002/eji.200939435
130. Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res. (2003) 90:127–56. doi: 10.1016/S0065-230X(03)90004-2
131. Donskov F, von der Maase H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J Clin Oncol. (2006) 24:1997–2005. doi: 10.1200/JCO.2005.03.9594
132. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. (1997) 79:2320–8. doi: 10.1002/(SICI)1097-0142(19970615)79:12<2320::AID-CNCR5>3.0.CO;2-P
133. Nair VR, Malladi S. Mouse models to study natural killer cell-mediated immunosurveillance and metastatic latency. Methods Mol Biol. (2019) 1884:141–50. doi: 10.1007/978-1-4939-8885-3_9
134. López-Soto A, Huergo-Zapico L, Galván JA, Rodrigo L, de Herreros AG, Astudillo A, et al. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J Immunol. (2013) 190:4408–19. doi: 10.4049/jimmunol.1202950
135. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
136. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) 35:S185–98. doi: 10.1016/j.semcancer.2015.03.004
137. Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. (2010) 120:2030–9. doi: 10.1172/JCI42002
138. Romero I, Garrido C, Algarra I, Collado A, Garrido F, Garcia-Lora AM. T lymphocytes restrain spontaneous metastases in permanent dormancy. Cancer Res. (2014) 74:1958–68. doi: 10.1158/0008-5472.CAN-13-2084
139. Romero I, Garrido C, Algarra I, Chamorro V, Collado A, Garrido F, et al. MHC intratumoral heterogeneity may predict cancer progression and response to immunotherapy. Front Immunol. (2018) 9:102. doi: 10.3389/fimmu.2018.00102
140. Linde N, Fluegen G, Aguirre-Ghiso JA. The relationship between dormant cancer cells and their microenvironment. Adv Cancer Res. (2016) 132:45–71. doi: 10.1016/bs.acr.2016.07.002
141. Carpenter RA, Kwak JG, Peyton SR, Lee J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat Biomed Eng. (2018) 2:915–29. doi: 10.1038/s41551-018-0307-x
142. Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. (2019) 40:128–41. doi: 10.1016/j.tips.2018.12.004
143. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. (2015) 15:238–47. doi: 10.1038/nrc3910
144. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417–30. doi: 10.1038/nrclinonc.2016.26
145. Morrissey C, Vessella RL, Lange PH, Lam HM. The biology and clinical implications of prostate cancer dormancy and metastasis. J Mol Med. (2016) 94:259–65. doi: 10.1007/s00109-015-1353-4
Keywords: metastasis, dormancy, immune-surveillance, microenvironment, minimal residual disease, latency
Citation: Kim K, Marquez-Palencia M and Malladi S (2019) Metastatic Latency, a Veiled Threat. Front. Immunol. 10:1836. doi: 10.3389/fimmu.2019.01836
Received: 15 May 2019; Accepted: 22 July 2019;
Published: 06 August 2019.
Edited by:
Khashayarsha Khazaie, Mayo Clinic College of Medicine and Science, United StatesReviewed by:
Robert J. Canter, University of California, Davis, United StatesEkaterina Jordanova, Center for Gynaecologic Oncology Amsterdam, Netherlands
Melanie Werner-Klein, Universität Regensburg, Germany
Copyright © 2019 Kim, Marquez-Palencia and Malladi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Srinivas Malladi, c3Jpbml2YXMubWFsbGFkaUB1dHNvdXRod2VzdGVybi5lZHU=