 Lea Monteran
Lea Monteran Neta Erez
Neta Erez- Department of Pathology, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
Cancer-associated fibroblasts (CAFs) are prominent components of the microenvironment in most types of solid tumors, and were shown to facilitate cancer progression by supporting tumor cell growth, extracellular matrix remodeling, promoting angiogenesis, and by mediating tumor-promoting inflammation. In addition to an inflammatory microenvironment, tumors are characterized by immune evasion and an immunosuppressive milieu. In recent years, CAFs are emerging as central players in immune regulation that shapes the tumor microenvironment. CAFs contribute to immune escape of tumors via multiple mechanisms, including secretion of multiple cytokines and chemokines and reciprocal interactions that mediate the recruitment and functional differentiation of innate and adaptive immune cells. Moreover, CAFs directly abrogate the function of cytotoxic lymphocytes, thus inhibiting killing of tumor cells. In this review, we focus on recent advancements in our understanding of how CAFs drive the recruitment and functional fate of tumor-infiltrating immune cells toward an immunosuppressive microenvironment, and provide outlook on future therapeutic implications that may lead to integration of preclinical findings into the design of novel combination strategies, aimed at impairing the tumor-supportive function of CAFs.
Introduction
Tumors are complex multicellular systems, characterized by reciprocal interactions between cancer cells and the tumor microenvironment (TME). The non-cancerous components that comprise the TME are central to all stages of tumorigenesis, progression, and metastasis (1). The TME is composed of the extracellular matrix (ECM), as well as various cell types including immune cells, endothelial cells, pericytes, and fibroblasts.
In non-cancerous homeostatic conditions, resident tissue fibroblasts are important sentinels of tissue integrity (2). Fibroblasts can sense and respond to mechanical changes, as well as to various tissue damage signals and react by differentiating to myofibroblasts that orchestrate tissue repair and wound healing, mediated by their ECM synthesis and remodeling and by their crosstalk with innate immune cells (3–5). Dysregulation of the physiological wound healing response and chronicity of inflammatory responses lead to fibrosis and scarring, characterized by excess ECM production and deposition by activated fibroblasts. These physiological functions of tissue fibroblasts are hijacked in cancer-associated fibroblasts (CAFs) in the microenvironment of tumors, consistent with the description of tumors as “wounds that do not heal” (6).
Cancer-associated fibroblasts (CAFs) are a vastly heterogeneous stromal cell population and are prominent components of the microenvironment in solid tumors. In some cancer types, including breast and pancreatic carcinomas, CAFs are the most prominent stromal cell type. The presence and function of activated CAFs in the microenvironment are associated with worse prognosis in multiple cancers (7). Moreover, tumors with high stromal signatures have been found to be associated with therapy resistance and disease relapse (8, 9).
CAFs are composed of multiple subpopulations that were shown to have diverse origins, including reprogrammed resident tissue fibroblasts (10, 11), bone marrow-derived mesenchymal cells (MSCs) (12, 13), adipocytes (14), and endothelial cells (15). Functionally, CAFs were shown to enhance tumor growth by several mechanisms: directly promoting cancer cell proliferation via secretion of growth factors, by inducing angiogenesis and by remodeling the ECM, which supports tumor cell invasion (5, 10, 16–18). Importantly, CAFs were also implicated in mediating tumor-promoting inflammation in various cancer types via secretion of cytokines and chemokines that mediate the recruitment and activation of immune cells, and by their reciprocal interactions with immune cells in the TME (2, 19). Studies in recent years have elucidated that this plethora of tumor-promoting activities of CAFs is mediated by functionally distinct subpopulations of fibroblasts (12, 20–22). Analysis of CAFs at the single cell level in the coming years will undoubtedly add complexity to the emerging landscape of CAF functional heterogeneity.
The role of the immune system in cancer is multi-faceted: In addition to an inflammatory microenvironment, tumors are also characterized by immune evasion and an immunosuppressive milieu, that were acknowledged as hallmarks of cancer (23). In order to survive and proliferate in the primary tumor site and in distant organs, which may be initially hostile, tumor cells must escape immune surveillance and avoid killing by cytotoxic lymphocytes. This is achieved by shaping the immune microenvironment toward a tolerant and immunosuppressive milieu, characterized by the presence of immature myeloid cells, T regulatory cells, decreased levels of infiltrating killer cells (T cells and NK cells), and dysfunction of their cytotoxic activity (24, 25). These mechanisms of immune escape and suppression are achieved by tumor cell downregulation of antigen presentation, elevated expression of surface inhibitory molecules, and secretion of immunosuppressive factors (24).
In addition to tumor cell-mediated signaling that drives immune suppression, fibroblasts are emerging as central players in shaping the TME toward an immunosuppressive and growth-promoting phenotype (26). CAFs contribute to immune escape via upregulation of immunosuppressive cytokine production and immune checkpoint ligands, exclusion of anti-tumor CD8+ T cells from cancer cells, and by affecting the functional differentiation of tumor infiltrating inflammatory cells.
In this review, we focus on recent advancements in our understanding of how CAFs affect the recruitment and functional fate of tumor-infiltrating immune cells toward shaping an immunosuppressive tumor microenvironment, and examine future therapeutic implications.
CAFs Orchestrate Recruitment of Immune Cells
Recruitment of Myeloid Cells
Myeloid cells are the most abundant hematopoietic cells in the body and are critical components of the tumor microenvironment, that contribute to all aspects of tumor progression (1). Myeloid cells in the TME include various populations of tumor-associated macrophages (TAMs), neutrophils, eosinophils, basophils, dendritic cells (DC) and mast cells (1, 27, 28). In addition, immature myeloid cells that express CD11b, Ly6G and/or Ly6C are sometimes referred to as myeloid-derived suppressor cells (MDSCs), based on their ability to functionally suppress the proliferation and activity of T cells. MDSCs are characterized by their expression of Arginase (ARG1), TGF-β, Programmed death-ligand 1 (PD-L1)/ 2, IL-10, Prostaglandin E2 (PGE2), S100A8/A9 and Indoleamine-pyrrole 2,3-dioxygenase (IDO), and by their capacity to regulate dendritic and T cell functions. MDSCs are commonly divided into two subsets based on their expression of surface markers: monocytic MDSCs (CD11b+Ly6ChighLy6G−), and granulocytic (or polymorphonuclear) MDSCs (CD11b+Ly6CintLy6Ghigh) (29, 30). Notably, these surface markers are used to identify mouse MDSCs. Human tumor-associated MDSCs are identified by their expression of CD33+CD14+HLA-DRlow/− (monocytic MDSCs) or CD11b+CD14−CD15+/CD66b+ (29).
CAFs were shown to recruit macrophages into the TME in multiple mouse models of cancer, including squamous cell, prostate and breast carcinomas (19, 31, 32). In a mouse model of spontaneous lymphoma, tumor-educated CAFs (derived from MSCs) recruited CD11b+Ly6C+ monocytes, F4/80+ macrophages, and CD11b+Ly6G+ neutrophils via the CCL2–CCR2 axis, thus enhancing tumor growth (33). Moreover, in a mouse model of breast cancer lung metastasis, MSCs that were pre-conditioned with TNFα and co-injected with tumor cells were shown to recruit CXCR2+ neutrophils by secreting CXCR2 ligands (CXCL1, CXCL2, and CXCL5), resulting in enhanced lung metastasis (34). Expression of chemoattractants for myeloid cells was suggested to be mediated by enhanced expression of miR-1246 in breast cancer CAFs, in an NF-κB dependent manner (35). Notably, recent understanding of CAF heterogeneity implicates a distinct subpopulation of inflammatory CAFs (iCAFs) rather than myofibroblast-like CAFs (myCAFs) in the induction and maintenance of an inflammatory milieu via their expression of inflammatory mediators (IL-6, IL-11, CXCL1, CXCL2) (21, 36). While these findings were described in a mouse model of pancreatic cancer, they likely represent a general phenomenon whereby specific functions of CAFs in affecting immune cells are mediated by distinct subpopulations (12, 37).
Recruitment of tumor-promoting myeloid cells by CAFs is associated with shaping their functional differentiation toward an immunosuppressive phenotype: Secretion of Chitinase-like protein 3 (Chi3L1) by mammary CAFs was shown to drive an M2-like phenotype in recruited macrophages, associated with reduced infiltration of CD8+ T lymphocytes (19). Interestingly, the expression of fibroblast activation protein (FAP) in CAFs in multiple cancer types was shown to be associated with recruitment of immunosuppressive cells: In a mouse model of hepatic cancer, a subset of FAP+ fibroblasts had an inflammatory phenotype directed by STAT3 activation and increased CCL2 expression, resulting in enhanced recruitment of CCR2-expressing circulating MDSCs and enhanced tumor growth (38).
Importantly, recruitment of myeloid cells by CAFs was also shown to be associated with resistance to therapy: In a mouse model of transplantable colorectal carcinoma (CRC), FAPhigh fibroblasts were found to recruit myeloid cells via CCL2, leading to resistance to anti-PD-1 immune checkpoint therapy which was abrogated by targeting FAP. These findings were validated in human CRC tissue sections, where the abundance of FAPhigh fibroblasts was in correlation with increased infiltration of myeloid cells and inversely correlated with infiltrated T cells (39). Similarly, pharmacological targeting of FAP in a transplantable model of pancreatic cancer resulted in decreased macrophage recruitment and enhanced T cell infiltration (40). Moreover, targeting of FAP+ fibroblasts by immunization with an adenoviral vector in both transgenic and transplantable mouse models of melanoma abrogated the recruitment and function of immunosuppressive cells including monocytic and polymorphonuclear MDSCs within the TME (41).
CCL2-mediated recruitment of circulating monocytes by CAFs was also demonstrated in models of breast cancer, in vivo and in a 3D ex-vivo model (42, 43). Notably, while recruitment of macrophages into tumors by CAFs is operative in various cancer types, the molecular pathways are distinct: In primary in vitro cultures, CAFs isolated from human prostate tumors were found to recruit monocytes by secreting stromal cell-derived factor 1 (SDF1)/CXCL12. Moreover, these SDF1-producing CAFs enhanced M2-like polarization of circulating monocytes, reflected by high production of the immune suppressive cytokine IL-10 (44). These findings agree with the demonstrated functional role of CAF-derived SDF1 in promoting tumor growth and immunosuppression (45, 46).
Recruitment of myeloid cells into tumors by CAFs is not limited to monocytes: platelet-derived growth factor receptor A (PDGFRα)+ CAFs isolated from murine tumors were shown to be a major source of the granulocytic chemoattractant CXCL1, and to mediate the accumulation of Ly6C−Ly6G+ granulocytic cells (granulocytic MDSCs) with potent immune-suppressive activity, assessed by their ability to suppress T cell proliferation. Interestingly, this pathway may be an adaptive response to anti-CSF1R therapy, as it was induced in CAFs following treatment with CSF1R inhibitor in models of colon, lung, breast carcinomas and melanoma (47). These findings instructed the design of combination therapy, to block CSF1R signaling as well as CAFs: Combining CSF1R inhibitor with a CXCR2 antagonist blocked granulocyte infiltration and resulted in strong delay in tumor growth in models of lung carcinoma and melanoma (47). Interestingly, mast cells were also shown to be recruited by CAFs: CAFs isolated from hormone-dependent prostate tumors mediated the recruitment of CXCR4-expressing mast cells by secreting CXCL12 (48).
One of the suggested mechanisms for CAF-mediated recruitment of myeloid cells to the TME is the expression of a senescence-associated secretory phenotype (SASP) gene signature. Cellular senescence was originally thought to be a tumor-suppressive mechanism that limits malignant transformation by arresting cell proliferation. However, studies in recent years have shown that senescent fibroblasts acquire a senescence-associated secretory phenotype (SASP) that supports their pro-inflammatory and tumor-promoting functions (49, 50). Moreover, the acquisition of a senescent phenotype by CAFs was shown to contribute to recruitment of immunosuppressive cells: In a mouse model of stromal-specific induced senescence, senescent dermal fibroblasts were shown to mediate the formation of an immunosuppressive microenvironment by enhancing the recruitment of CD11b+Ly6C−Ly6Ghigh cells and T regulatory (CD3+CD4+FOXP3+) cells, and enhanced ECM deposition. Co-injection of senescent dermal fibroblasts with squamous cell carcinoma cells demonstrated that SASP-induced shaping of the immune microenvironment promotes tumor growth. SASP-mediated tumor promotion was inhibited by targeting SASP-derived IL-6 or by depleting Ly6G+ cells (51).
Thus, by employing multiple molecular pathways, CAFs recruit myeloid cells into tumors, that contribute to the formation of an immunosuppressive immune milieu (Figure 1).
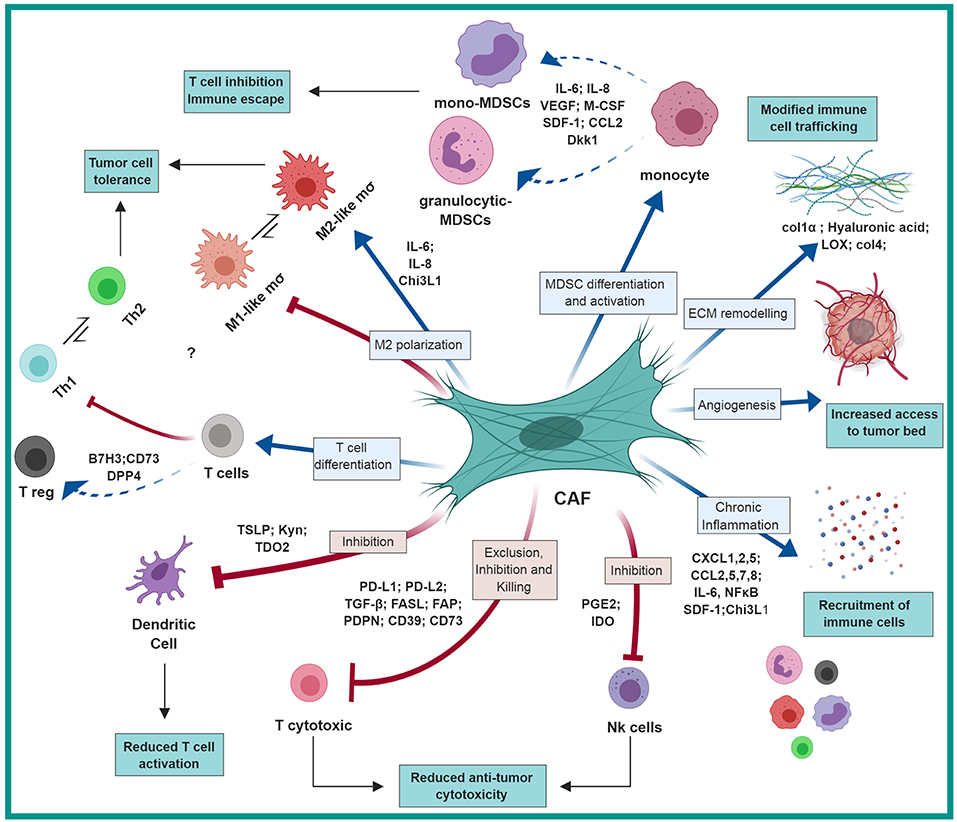
Figure 1. CAF-mediated immunosuppression: CAFs shape the immune microenvironment in tumors toward a pro-tumorigenic and immunosuppressive milieu by affecting the recruitment and function of various innate and adaptive immune cells. Red arrows represent negative regulation/inhibition and blue arrows represent positive regulation/induction. This figure was designed by using graphical elements from BioRender.
Recruitment of Regulatory T Cells
CAFs were found to potentiate the recruitment, differentiation and survival of T regulatory cells, contributing to the formation and maintenance of an immunosuppressive microenvironment. Treg cells are immunosuppressive T lymphocytes characterized by their expression of the IL-2 receptor α-chain (CD25) and the transcription factor forkhead box P3 (FOXP3). The mechanisms by which Treg mediate immunosuppressive function at tumor sites are not fully elucidated, but increased infiltration of Tregs within the tumor was shown to correlate with worse prognosis in multiple studies (52–55).
As the complexity of CAF populations is being gradually revealed, it is increasingly appreciated that mediating immunosuppression may be operative in a distinct subpopulation of CAFs: FACS-based analysis of CAFs in human breast tumors by using six surface markers identified four distinct CAF subsets which accumulated differently in different subtypes of human breast cancer (luminal A, Her2+, and triple-negative). Of these CAF populations, the subtype designated CAF-S1, characterized by expression of FAP, smooth muscle actin α (αSMA), PDGFRβ, and CD29, was found to be associated with recruitment, retention and differentiation of Treg cells: By secreting CXCL12, CAF-S1 promoted the attraction of CD4+CD25+ T cells, and mediated their retention via expression of OX40L, PD-L2, and Junctional adhesion molecule B (JAM2). Moreover, CAF-S1 were able to increase T cell survival, induce their differentiation to CD4+CD25+FOXP3+ regulatory lymphocytes and to enhance the capacity of Treg cells to inhibit the proliferation of CD4+CD25+ T cells in vitro (20). This mechanism is not restricted to breast cancer, as the presence and function of CAF-S1 in attraction, survival, and differentiation of CD25+FOXP3+ T lymphocytes was also demonstrated in human high-grade serous ovarian cancers (HGSOC), where it was shown to depend on differential regulation of CXCL12 by miR-200/141. Interestingly, upregulation of FOXP3 required direct cell-cell contact between CAF-S1 and CD4+ T cells (56). These findings are consistent with findings from immune competent mouse models which showed that targeting the CXCL12/CXCR4 axis inhibited the recruitment of Treg lymphocytes in ovarian and pancreatic cancer (45, 57).
Thus, by mediating the recruitment of various innate and adaptive immune cells, CAFs shape the immune composition of the TME and support tumor growth (Table 1; Figure 1).
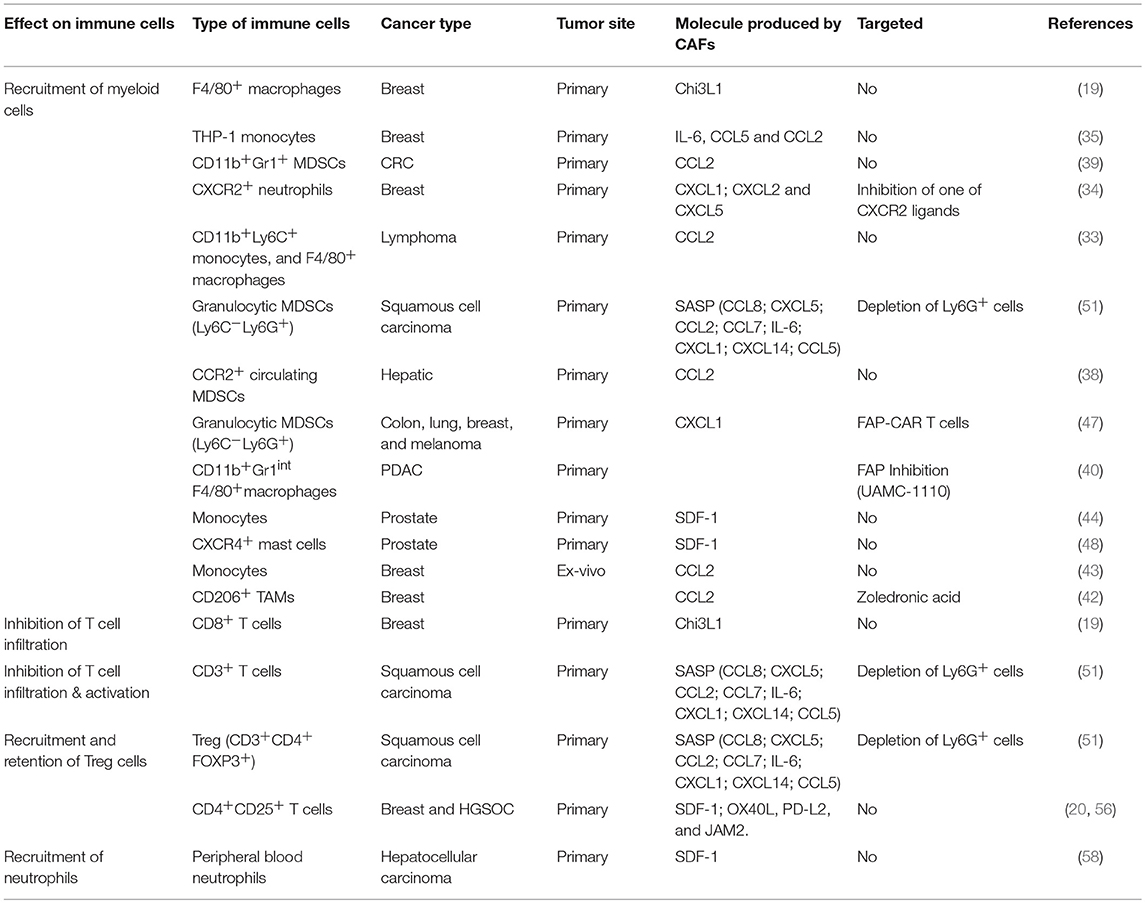
Table 1. Recruitment or exclusion of immune cells.
CAFs Drive an Immunosuppressive Function in Immune Cells
In addition to their capacity to recruit immune cells that foster tumor growth, CAFs were also implicated in affecting the function of various immune cells toward an immunosuppressive phenotype by multiple mechanisms. This may resonate the role of fibroblasts in wound healing, where their function favors M2-like and Th2 type immune reactions (59, 60). Notably, our recent understanding of CAF heterogeneity in origin and function suggests that driving immune suppression may be mediated by distinct subpopulations of CAFs. For example, bone marrow-derived mesenchymal stromal cells (MSCs) were shown to be a significant source of CAFs in breast cancer (12). Interestingly, in physiological wound healing MSCs were shown to mediate immunosuppression: In models of acute liver injury, the presence of pro-inflammatory cytokines (IFNγ, TNFα, or IL-1) induced MSCs to produce iNOS, which in turn suppressed the function of T cells (61, 62). These physiological functions of MSCs and fibroblasts may be hijacked in tumors, to elicit the formation of an immunosuppressive TME via the effect of CAFs on specific immune cell populations (Figure 1).
Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs) are a heterogeneous cell population arising from circulating monocytes or from tissue resident macrophages, and were implicated in various tumor-promoting tasks including pro-inflammatory signaling, enhancement of angiogenesis, invasion, metastasis and therapy resistance (63, 64). Macrophages can be classified according to their functional differentiation state and immunological responses: M1-like macrophages are involved in the response of type I T helper cells (Th1), they are activated by interferon gamma (IFNγ) and engagement of Toll-like receptors (TLRs) and characterized by production of pro-inflammatory molecules, nitric oxide (NO) and reactive oxygen species (ROS). M2-like macrophages, which are in general pro-tumorigenic, are involved in Th2-type immune responses, wound healing and tissue repair, activated by IL-4 and IL-13, and characterized by promotion of angiogenesis and secretion of immune suppressive factors that inhibit killing by cytotoxic T cells (63). This classification however, is not dichotomous, and different macrophage subtypes may share multiple features.
Functional differentiation of TAMs in the tumor microenvironment is affected by many factors. Recently, CAFs are emerging as novel effector cells in TAM differentiation toward an immunosuppressive phenotype, in addition to their role in monocyte recruitment. CAF-derived Chi3L1 was shown to be important for both recruitment and functional differentiation of bone marrow-derived macrophages in a mouse model of breast cancer: Genetic targeting of Chi3L1 expression in fibroblasts attenuated macrophage recruitment and their reprogramming to an M2-like phenotype, and promoted a Th1 phenotype in the tumor microenvironment (19). Prostate CAFs were shown to mediate both the recruitment and the M2-like differentiation of monocytes via SDF1 (44). A similar finding was demonstrated in an ex-vivo model of oral squamous cell carcinoma. CAFs isolated from human tumors, instigated an M2-like phenotype in patient-derived CD14+ myeloid cells (manifested by production of ARG1, IL-10 and TGF-β), which in turn potently suppressed the proliferation of autologous T cells (65). However, the underlying CAF-derived factors that mediated M2-like differentiation were not identified.
In this context, it is important to note that while TAMs and CAFs are both central players in the tumor microenvironment, their reciprocal interactions in cancer are not well characterized, and the main focus in the literature is on the effects of macrophages on fibroblasts. Future studies are required to further elucidate the contribution of CAF-derived signaling to the diverse functions of macrophages in the TME (Figure 1).
MDSCs
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature immune-suppressive myeloid cells. MDSCs are not found in healthy tissues and appear in pathologic conditions associated with chronic inflammation or stress, as well as in the microenvironment of tumors (29). The functional and phenotypic heterogeneity of MDSCs has been a source of confusion in their definition and terminology (66). Moreover, mouse and human MDSCs may be different in both surface markers (as detailed above) and in their immunosuppressive capacity: while in mice both granulocytic and monocytic MDSCs were shown to inhibit T cells in vitro, in human studies findings on the function of granulocytic vs. monocytic MDSCs depend on cancer type, underlying their diversity (30, 67). These differences should be considered when assessing murine studies.
Nevertheless, several studies in mouse and in human experimental systems demonstrated that CAFs are capable of reprogramming an immunosuppressive function in immature myeloid cells, typical of MDSCs. Primary pancreatic stellate cells (PSCs) isolated from human pancreatic tumors, but not normal PSCs, were demonstrated to induce an MDSC phenotype in peripheral blood mononuclear cells (PBMCs), manifested by inhibition of T cell proliferation in vitro. This reprogramming was dependent on IL-6 and STAT3 as their inhibition attenuated the induced immunosuppressive function (68). Moreover, IL-6 was found to be predominantly expressed in the stroma of human pancreatic tumors, and targeting it in transplantable and transgenic mouse models of pancreatic ductal adenocarcinoma (PDAC) in combination with PD-L1 blockade resulted in attenuated tumor growth, prolonged survival in a transgenic model of PDAC, and increased presence of intratumoral T cells (69). Importantly, this pathway may not be specific to pancreatic cancer: CAFs isolated from human hepatocellular carcinomas were capable of driving an immunosuppressive gene signature and functions in monocytes and in neutrophils via IL-6 mediated activation of STAT3 signaling (58, 70). Taken together, these findings suggest that combination of immune checkpoint therapeutics with targeting of stromal signaling may be beneficial in treatment of pancreatic and liver carcinomas.
Interestingly, stromal signaling that drives differentiation of peripheral MDSCs may be bone marrow-derived: In transplantable models of lung carcinoma and melanoma (Lewis lung carcinoma and B16 melanoma cell lines), tumor-bearing mice had elevated systemic levels of Dickkopf-related protein 1 (Dkk1), which were found to originate in the bone stromal compartment (osteoblasts and osteocytes) (71). Dkk1 effect on MDSCs was via inhibition of β-catenin, previously shown to be essential in mediating immunosuppressive functions of MDSCs (72). Thus, stromal signaling is central to shaping the functional differentiation of immature myeloid cells toward an immunosuppressive phenotype both locally and systemically in a variety of tumor types.
Importantly, the interactions of CAFs with recruited myeloid cells are reciprocal: Once monocytic and granulocytic cells are recruited to the TME they affect the activation of fibroblasts. For example, activated neutrophils secrete large amounts of reactive oxygen species which are known pro-fibrotic mediators (4, 18). Moreover, activated neutrophils release granules containing multiple proteases including MMPs, elastase, and cathepsins, capable of cleaving collagenous and non-collagenous connective tissue components. This release of ECM breakdown products further activates stromal fibroblasts, physiologically programmed to facilitate matrix remodeling during tissue repair. Similarly, macrophage secreted factors were demonstrated to facilitate reprogramming of resident dermal fibroblasts, or of mesenchymal stromal cells in an NF-κB dependent manner in skin and gastric carcinomas (32, 73).
T Cells
T cell-mediated immune response can be classified into Th1 or Th2-type immunity, based on their profile of cytokine production. In general, Th2-mediated immunity is considered tumor promoting, as it entails pro-angiogenic signaling, activation of M2-like macrophage function and inhibition of cell-mediated tumor cell killing (74).
Accumulating evidence suggest that signaling by CAFs may shape the T cell milieu in the TME toward a tumor-promoting function, either directly or via innate immune cells. Many of the findings emerge from murine models of cancer in which targeting of specific signaling molecules in CAFs resulted in attenuation of tumor growth and metastasis, accompanied by a shift in the T cell responses: In vivo elimination of FAP+ CAFs by vaccination lead to a switch from Th2 to Th1-type immunity, characterized by increased expression of the cytotoxic cytokines IL-2 and IL-7, increased CD8+ T cell tumor infiltration, and diminished recruitment of macrophages, MDSCs and T regulatory cells in a transplantable model of triple-negative breast cancer (75). Targeting of CAF-derived Chi3L1 had a similar effect in another transplantable model of breast cancer, and resulted in enhanced infiltration of CD8+ T cells and a shift in the tumor cytokine profile toward a Th1-type phenotype. However, in both studies these effects of CAFs on T cells may be indirect.
A direct effect of CAFs on T cell function was demonstrated in an in vitro study that utilized fibroblasts and tumor-infiltrating T lymphocytes (TILs) isolated from human lung tumors: CAF-derived IL-6 enhanced production of IFNγ and IL-17A in activated TILs, suggesting that fibroblasts may also have an immunostimulatory effect on T cells (76).
Notably, the crosstalk between CAFs and T cells is reciprocal. Secreted factors from activated T cells enhanced the production of IL-6 by lung CAFs (76). Activated lymphocytes were also shown to induce the expression of cyclooxygenase-2 (COX-2) and intercellular adhesion molecule-1 (ICAM-1) in normal human lung fibroblasts. These activated fibroblasts then induced a reduction in the expression of T cell activation/co-stimulation markers (CD69, LFA-1; CD3 and CD28) suggesting that fibroblasts are able to modulate effector functions of T cells recruited into sites of inflammation (77) (Figure 1).
Many of the pathways that are operative in tumors represent “hijacking” of physiological pathways. Indeed, the interactions between fibroblasts and T cells are probably not restricted to tumors and represent a physiological role of fibroblasts, as normal skin fibroblasts and autologous T cells showed similar interactions in vitro (76). The ability of fibroblasts to drive type-2 immunity may also be a physiological capability of fibroblasts: A recent study in normal lung tissue suggested that fibroblast-like adventitial stromal cells (ASCs) support the accumulation and activation of group 2 innate lymphoid cells (ILC2s), which are important mediators of type 2 immunity. Accumulation of ILC2s resulted in a formation of a tissue niche with Treg and dendritic cells, and depletion of ASCs abrogated these functions, suggesting that subpopulations of fibroblasts are required for optimal accumulation of ILC2s during type 2 immune responses (78). Future studies are required to demonstrate whether these fibroblast-mediated functions are also operative in tumors.
Dendritic Cells
Another mechanism by which CAFs hinder anti-tumor immune responses and impede on the function of T cells in the TME is by affecting the function of DCs, the most important population of antigen-presenting cells. Activated fibroblasts are a major source of transforming growth factor b (TGF-β), a pleotropic and immunosuppressive cytokine that functions in wound healing, ECM remodeling, and can affect the functional differentiation of multiple types of immune cells (79). TGF-β was shown to mediate downregulation of MHC class II molecules and the co-stimulatory molecules CD40, CD80, and CD86 in dendritic cells, thus inhibiting their antigen presentation capacity and their capability to activate cytotoxic T cell responses (80). CAF-mediated modulation of DC function was also shown to be mediated by their secretion of pro-inflammatory cytokines (Figure 1). CAFs isolated from human hepatic carcinomas were shown to secrete IL-6, which activated STAT3 in DCs, resulting in generation of regulatory DCs. These CAF-educated DCs exhibited lower expression of antigen presenting molecules and co-stimulatory molecules (CD1a, HLA-DR, CD80, CD86), and elevated expression of immunosuppressive cytokines (such as IL-10 and TGF-β). Moreover, hepatic CAF-educated DCs could affect T cells toward a suppressive phenotype, including induction of CD4+CD25+Foxp3+ Tregs, and decreased production of IFN-γ in CD8+ T cells (81). In lung cancer, inhibition of DCs differentiation and function was shown to be mediated via CAF-secreted tryptophan 2,3-dioxygenase (TDO2). Analysis of lung cancer surgical specimens revealed increased TDO2 expression in the fibroblasts adjacent to the cancer, and inhibition of TDO2 in a transplantable model of lung carcinoma resulted in improved DC function and T cell response, and decreased experimental metastasis (82).
The crosstalk between CAFs and dendritic cells was also shown to affect the ability of DCs to polarize the differentiation of T cells toward a Th2 phenotype in pancreatic cancer, via CAF secretion of Thymic stromal lymphopoietin (TSLP). CAFs that were activated by tumor-derived pro-inflammatory signaling (TNFα and IL-1β) secreted TSLP, which endowed them with the ability to drive the differentiation of naïve CD4+CD45RA+ T cells toward a Th2 phenotype. Human data from pancreatic cancer patients indicated that DCs with features of TSLP-treated DCs and Th2-attracting chemokines were present in pancreatic tumors, and the Th2/Th1 ratio in pancreatic tumors was an independent marker of poor survival (83).
Taken together, these studies demonstrate multiple mechanisms by which CAFs modulate the functional differentiation of immune cells in the TME toward an immunosuppressive function (Table 2, Figure 1).
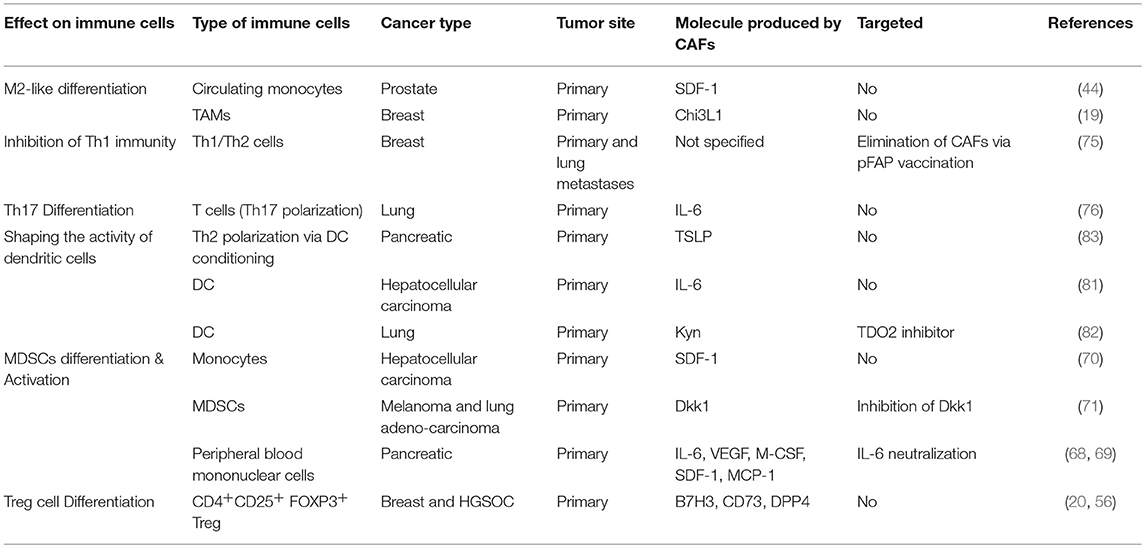
Table 2. CAF-mediated modulation of immune cell differentiation.
CAF-mediated ECM Remodeling and Fibrosis Drives an Immunosuppressive Microenvironment
Fibrosis is a scarring process, characterized by excess deposition of collagenous and non-collagenous extracellular matrix (ECM) due to the accumulation, proliferation, and activation of fibroblasts. One of the hallmarks of CAFs is the excessive production/deposition of extracellular matrix components and degradation enzymes. This CAF-mediated deregulation of the ECM results in biomechanical and biochemical changes that facilitate tumor growth, invasion, and metastasis (84, 85). In addition to their effect on cancer cells, CAF-mediated deregulation of the ECM protein network modulates immune cells trafficking. Aberrant ECM protein composition and fragments of the ECM that are derived from tissue-remodeling processes can influence immune cell activation and survival, thereby actively contributing to immune responses at these sites (86). Various ECM components were shown to modulate macrophage polarization (toward an M2-like signature) and mediate the migration and maturation of monocytes and MDSCs (87). Moreover, stiffed collagen-rich matrix was found to induce CAFs production of monocytic chemoattractants like CCL2 and M-CSF (87), and CAFs in tumor-associated fibrosis produce high levels of cytokines and chemokines that favor tumor-promoting Th2 and Th17 responses (88). One example for an ECM component demonstrated to affect macrophage trafficking to tumors is hyaluronan (HA). Genetic targeting of the HA synthase gene in fibroblasts in a transplantable model of mammary carcinoma, leading to HA deficiency in the stroma, resulted in impaired macrophage recruitment and attenuated tumor angiogenesis and lymphangiogenesis (89).
While ECM components promote immune cell recruitment and activation, excessive deposition of collagen by fibroblasts in the TME leading to formation of scar-like tissue, was shown in pancreatic cancer to form a physical barrier that prevented cytotoxic T cell infiltration into the tumor, thus contributing to immune escape in pancreatic cancer. In vitro experiments demonstrated that while activated T cells migrated in low-density collagen matrices, migration was inhibited in dense collagen (90). Real-time imaging in viable slices of human lung tumors revealed that antigen-specific T cells within the tumor accumulate more in the stromal rich area than in the tumor islets. The density and the orientation of collagen and fibronectin fibers were suggested to play key roles in controlling T cells trafficking, as matrix loosening induced by collagenase treatment increased the ability of T cells to contact tumor cells (91, 92). Indeed, highly desmoplastic stroma, associated with activation of focal adhesion kinase (FAK) in pancreatic tumors, was shown to correlate with poor CD8+ cytotoxic T cell infiltration. FAK inhibition in a transgenic mouse model of PDAC resulted in attenuated tumor fibrosis, improved response to immune checkpoint therapy and prolonged survival. These findings suggest that targeting fibrosis may be beneficial for overcoming CAF-mediated immune suppression (93).
The composition of the ECM is an important factor in enabling tumor metastasis (94). CAF-mediated remodeling of the ECM was recently shown to have an important role in enabling melanoma metastasis, in association with aging. While young skin fibroblasts produced abundant ECM components, aged fibroblasts were shown to lose the expression of the hyaluronic and proteoglycan link protein (HAPLN1), resulting in enhanced alignment of ECM matrices that promoted metastasis of melanoma cells in a mouse model of transplantable melanoma. However, the effect of matrices produced by aged fibroblasts was inhibitory on the migration of T cells, which may contribute to impaired immune response in the TME (95). Interestingly, age-related changes in HAPLN1 were also shown to increase lymphatic permeability, which affected melanoma lymph node metastasis. Age-related loss of HAPLN1 was shown to be associated with loss of integrity in the lymphatic vasculature and with enhanced lymphatic endothelial permeability, which enabled the escape of melanoma cells from the lymphatic system to distant metastatic sites (96).
Thus, CAF-mediated ECM remodeling and fibrosis contribute to the formation of an immunosuppressed and growth promoting microenvironment by multiple mechanisms (Figure 1).
Direct Inhibition of Cell Mediated Killing: CAFs Abrogate the Function of Cytotoxic Lymphocytes
In addition to mediating the recruitment and functional differentiation of immune cells in the TME, findings from multiple studies implicate CAFs in affecting killing of tumor cells by cytotoxic lymphocytes.
CAFs isolated from human metastatic melanoma lesions or from hepatocellular carcinomas interfered in co-culture experiments with NK ability to kill melanoma cells. This inhibition was mediated by CAF-derived PGE2 and IDO, which abrogated NK cells expression of cytotoxic molecules (granzyme B and perforin) and cytokines, and impaired their cytotoxic activation and surface expression of NKp44 and NKp30 (97, 98). Another mechanism by which CAFs abrogate NK killing of tumor cells was demonstrated in melanoma-associated fibroblasts: CAFs isolated from tumors of melanoma patients decreased in vitro susceptibility of cancer cells to NK-mediated lysis. Mechanistically, CAFs secreted active matrix-metalloproteinases (MMPs) that were able to degrade two NKG2D ligands (MICA/B) on the surface of melanoma cells, resulting in an inhibition of NKG2-dependent cytotoxic activity of NK cells (99).
CAFs were also shown to directly abrogate the function of cytotoxic T cells by multiple mechanisms: Expression of immune checkpoint molecules is emerging as an important process by which CAFs directly suppress the function of cytotoxic T lymphocytes (CTL). Fibroblasts isolated from melanoma patient biopsies could directly suppress CD8+ T cells proliferation and function, via upregulating their expression of the PD-1 ligand PD-L1, mediated by IL-1α/β. These finding suggest that blockade of IL-1 may benefit melanoma patients and potentially synergize with immunotherapeutic interventions (100). CAFs isolated from resected human pancreatic tumors were recently shown to express PD-L1 and PD-L2. Furthermore, these CAFs were shown in vitro to inhibit the proliferation of T cells and to stimulate their expression of the inhibitory molecules TIM-3, PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and LAG-3, possibly via the activity of CAF-derived PGE2 (101).
Expression of immune checkpoint molecules is likely a physiological pathway in fibroblasts during inflammation: Normal colon fibroblasts expressing PD-L1 and/or PD-L2 were shown to be involved in the regulation of mucosal CD4+ T cell response (102). Moreover, expression of PD-L1 is upregulated in human dermal fibroblasts and in mesenchymal stromal cells via IFNγ, highly secreted by activated T cells (103, 104). Future studies are required to assess whether IFNγ also mediates the expression of PD-L1/2 in CAFs in tumors.
Another suggested mechanism for abrogating the function of cytotoxic T cells is by CAF-mediated metabolic effects. MSCs isolated from cervical tumors had elevated expression levels of CD39 and CD73 as compared with normal tissue fibroblasts. This feature was associated with the ability to strongly suppress the proliferation, activation and effector functions of cytotoxic T cells through generation of large amounts of adenosine from the hydrolysis of ATP, ADP, and AMP nucleotides (105). Similarly, glycolytic CAFs in prostate cancer were found to affect the polarization and function of effector T cells via their release of lactate (106).
Immunotherapy approaches are designed to unleash the cytotoxic T cell function of “dysfunctional” CD8+ T cells by blocking the immunosuppressive signaling restraining these T cells. This requires not only that activated cancer-specific T cells be present in the TME, but also that their location allows physical contact with tumor cells. Many tumors exhibit an “immune excluded” phenotype, in which T cells are restricted to a peritumoral zone rich in fibroblasts, with few lymphocytes within the epithelial tumor mass itself (107). In a murine model of pancreatic cancer, CAF-derived CXCL12 protected tumor cells from T cell accumulation. Depletion of FAP+ CAFs resulted in enhanced T cell infiltration and better response to anti-CTLA4 and anti-PD-L1 treatment in mice (45, 92, 108).
Indeed, CAFs were suggested to be important in affecting the non-effectiveness of immune therapy in multiple cancer types, partially via activation of TGF-β signaling. TGF-β signature in fibroblasts was shown to be associated with poor response to anti-PD-L1 treatment in metastatic urothelial cancer. Moreover, TGF-β signaling in fibroblasts was correlated with exclusion of CD8+ T cells within the tumor, which were instead found in the fibroblasts and collagen-rich peritumoral stroma. Therapeutic co-administration of TGF-β-blocking reagents and anti-PD-L1 antibodies reduced TGF-β signaling in stromal cells, facilitated T-cell tumor penetration, and promoted tumor regression (109).
Similarly, targeting of TGF-β in a transgenic mouse model of metastatic colorectal cancer unleashed a potent and enduring cytotoxic T cell response. Tumors in these mice were characterized by T cell exclusion, highly activated stromal TGF-β, and a limited response to anti-PD-1 and anti-PD-L1 treatment. Inhibition of TGF-β enabled T cell infiltration to tumors and metastatic lesions, facilitated the response to anti-PD-1 and anti-PD-L1 therapy, diminished tumor growth and reduced liver metastasis (110). Taken together, these studies suggest that CAFs play a central role in inhibiting tumor cell killing by T cells, and advocate the use of combination therapies that target immune checkpoint inhibitors together with abrogating the immunosuppressive ability of CAFs (111).
Strikingly, recent evidence suggested that CAFs are capable of antigen presentation, leading to antigen-specific deletion of CD8+ T cells to protect tumor cells. CAFs isolated from lung adenocarcinomas and melanoma tumors were shown to process and present antigens, and directly interact with activated CD8+ T cells, thus inducing T cell death via PD-L2 and FAS ligand (FASL) engagement. Moreover, the capacity of antigen-specific T cells to kill their target tumor cells was dramatically impaired when conditioned by antigen-loaded CAFs, indicating that CAFs are capable of driving dysfunction and death of tumor-specific T cells, leading to enhanced tumor cell survival (112).
Interestingly, this function of CAFs is similar to the physiological role of lymph node fibroblastic reticular cells (FRCs) that display specific immunological properties to maintain peripheral tolerance. Lymph node structure consists of defined niches for B and T lymphocytes. This structure is provided by FRCs, which also provide the lymphocytes with a scaffold upon which to migrate. FRCs produce collagen rich reticular fibers that form a dense network within the lymphoid tissue. The network of fibers supports and guides the movement of dendritic cells (DCs), T lymphocytes and B lymphocytes (113). Moreover, FRCs express and present peripheral tissue antigens to T cells in lymph nodes (LNs), receive peptide-MHC II loaded exosomes from DCs, induce CD4+ T cell hyporesponsiveness, and dampen T cell proliferation through the production of nitric oxide (114). These physiological functions may be hijacked in tumors, granting CAFs the capacity to actively regulate T cell function within the TME. For example, in lymph nodes, T cell migration is guided by FRCs that secrete the CCR7 ligands CCL21 and CCL19, which guide the interactions between CCR7+ T cells and antigen-presenting cells (APCs) needed for T cell education and priming (115). These events are central for maintaining peripheral tolerance, as Treg cells require LN occupancy and CCR7 signaling for their activation and function, and the loss of CCR7 signaling is associated with spontaneous autoimmunity. Interestingly, similar pathways of immune tolerance and T cell exclusion are operative in tumors: Expression of CCL21 in melanoma tumors in mice was associated with the induction of stromal zones that were reminiscent of lymph node paracortex stroma, recruitment of regulatory immune cells, an altered cytokine milieu, and an immunotolerant microenvironment, which depended on host expression of CCR7 (116).
In human and mouse breast tumors, this direct immunosuppressive capacity of CAFs was attributed to a distinct subpopulation of fibroblasts expressing FAP and Podoplanin (PDPN). FAP+PDPN+ CAFs expressed a TGF-β and fibrosis-related gene signature, and were in direct contact with T cells in the peritumoral dense ECM of mammary tumors. Moreover, FAP+PDPN+ CAFs were shown to suppress T cell proliferation in a nitric oxide-dependent manner (117). This function is reminiscent of the immune suppressive function of FRCs in the lymph nodes: Under inflammatory conditions, FRCs acquire immunosuppressive potential, and attenuate T cell expansion by producing nitric oxide (118, 119).
Thus, tumor cells co-opt tissue fibroblasts to generate stromal architecture and function that restrains tumor-infiltrating immune cells and impedes proper function of cytotoxic lymphocytes (Table 3, Figure 1). Taken together, these studies suggest that better understanding of CAF interactions with T cells and with regulation of immune checkpoint pathways may be beneficial for better design of immunotherapy treatments.
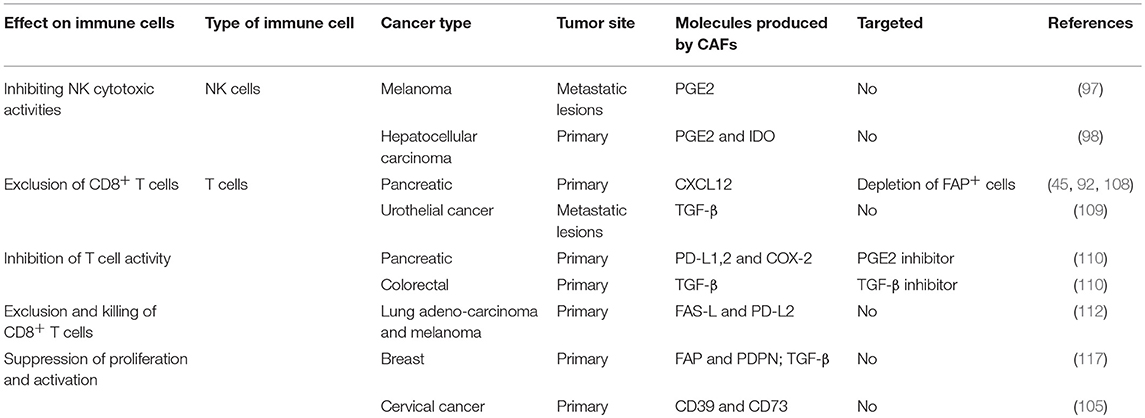
Table 3. CAF-mediated inhibition of anti-tumor cytotoxicity.
Therapeutic Perspectives: CAF Targeting Approaches
Therapeutic approaches of treating cancer are increasingly moving toward combinatorial strategies that target key operative pathways and mediators in the TME, rather than solely targeting cancer cell-intrinsic pathways. This is a result of improved understanding of the complexity of tumor eco-systems, as well as improved capacity of precision diagnostics that enable tailored therapeutic approaches. As our understanding of the important role of CAFs in mediating multiple tumor-promoting functions increases, it becomes clear that targeting CAFs in combination with other therapeutics may be beneficial. Based on the role of CAFs in mediating an immunosuppressed microenvironment that was reviewed herein, co-targeting of CAFs in combination with immunotherapeutics is an attractive option.
Pre-clinical trials targeting CAFs indicated that targeting a subpopulation of FAP+ CAFs was beneficial in transplantable models of Lewis lung carcinoma and pancreatic adenocarcinomas. Depletion of FAP+ CAFs using transgenic mice with FAP promoter- driven diphtheria toxin receptor (DTR) resulted in tumor regression in an IFNγ and TNFα dependent manner (120). Moreover, depletion of FAP+ CAFs in a mouse model of PDAC enabled the therapeutic effects of anti-CTLA4 and anti-PD-1 (45). In an effort to design more applicable ways to target FAP, multiple other approaches were developed for the targeting of FAP+ CAFs, including pharmacological inhibitors (e.g., PT630) (121), monoclonal antibodies (FAP5-DM1) (122), a FAP- targeting immunotoxin (αFAP- PE38) (123), and an oral DNA FAP vaccine (124, 125), which showed efficacy in mouse models of breast, pancreatic, lung and colon carcinomas. Moreover, a chimeric antigen receptor (CAR) T-cell specific for FAP was demonstrated to inhibit the growth of various subcutaneously transplanted tumors in mice by augmenting CD8+ T cell antitumor responses (126). However, depletion of FAP+ cells using the DTR system had severe systemic toxicity, including cachexia and anemia in mouse models of transgenic PDAC (KPC mice) and in transplantable colon carcinoma (C26 cells), likely reflecting the importance of FAP+ stromal cells in maintaining normal muscle mass and hematopoiesis (127). Thus, caution should be taken when designing FAP-targeting approaches for clinical testing in cancer patients.
Depletion of αSMA+ myofibroblasts in a mouse model of PDAC, utilizing thymidine kinase-Ganciclovir-mediated ablation, resulted unexpectedly in more invasive tumors, increased presence of CD4+Foxp3+ Treg cells and reduced survival (128). These findings suggest that the effect of targeting CAFs may depend on tumor type and on the experimental systems that were used, and requires careful consideration. In this context, it is important to note that depleting entire fibroblast populations is highly problematic in human patients, as fibroblasts have many critical physiologic functions. Moreover, both αSMA and FAP are not expressed exclusively by CAFs, which adds to the complexity of targeting cell populations based on these markers.
Therefore, targeting of molecules or pathways that are essential for the tumor-promoting functions of CAFs is likely a more clinically relevant approach. For example, targeting the pro-fibrotic function of CAFs with Pirfenidone (PFD—an anti-fibrotic agent as well as a TGF–β antagonist) was shown to be efficient in combination with doxorubicin in a mouse model of triple-negative breast cancer (4T1) (129). Similarly, targeting the fibrotic activity of CAFs with tranilast, an anti-fibrotic agent, in transplantable tumor models (lymphoma, Lewis lung carcinomas and melanoma) resulted in decreased presence of Treg cells and MDSCs, and enhanced cytotoxic CD8+ T cell response. These beneficial tumor-inhibitory effects were enhanced when CAFs were targeted in combination with effector-stimulatory immunotherapy such as dendritic cell-based vaccines (130). Importantly, CAF-mediated fibrosis contributes not only to enhanced tumor growth and invasiveness, but also to the immunosuppressive role of CAFs, as the increased matrix stiffness forms a physical barrier that limits the access of anti-tumor immune cells (87). Thus, targeting the pro-fibrotic functions of CAFs is beneficial for multiple reasons.
Another attractive option is targeting the transcriptional reprogramming of CAFs, which contributes to their activation. Vitamin D receptor (VDR) in pancreatic stellate cells was shown to be a central transcriptional repressor of their inflammatory and fibrotic functions, and treatment of mice with the VDR ligand calcipotriol induced stromal reprogramming that inhibited inflammation and fibrosis, enabled gemcitabine delivery into tumors, and improved survival in a PDAC model, suggesting that vitamin D may be utilized therapeutically in the treatment of pancreatic cancer (131). In addition, targeting central cytokines and chemokines that contribute to the pro-inflammatory, immunosuppressive and matrix remodeling function of CAFs may also be beneficial. For example, targeting IL-6 was suggested as a stromal-targeting therapeutic approach in cancer (132). In addition, CAFs are the main source of SDF-1/CXCL12 and blockade of the SDF-1/CXCL12-CXCR4 signaling pathway was shown to be beneficial in alleviating CAF-mediated immunosuppression (45, 92).
Importantly, the functional complexity and heterogeneity of CAF populations that may be specific to tumor type, specific organ and physiological context warrants careful consideration of CAF-targeted therapeutic strategies in patients.
Future Challenges and Outlook
The central role of fibroblasts in all stages of tumorigenesis and metastasis has emerged in recent years, as part of our growing understanding of tumors as multicellular organs. In addition to their “classical” functions in matrix remodeling and secretion of ECM components, accumulating evidence from many studies implicate CAFs in immunoregulatory functions that shape the immune milieu of tumors toward a pro-inflammatory and immunosuppressive function. These functions are mediated by CAF secretion of multiple cytokines and chemokines, and by reciprocal interactions with innate and adaptive immune cells. However, the heterogeneity and plasticity of CAFs are still poorly understood. This is partially a result of limited experimental tools: much of our knowledge relies on in vitro studies, or studies of CAF co-injection with tumor cells, which may not faithfully recapitulate the physiological function of CAFs. A major limitation to our ability to elucidate the role of specific CAF-derived factors is the sparsity of reliable CAF-specific Cre mice which will enable conditional ablation of candidate factors in CAFs, in order to identify potential therapeutic targets.
Another future challenge is the use of reliable pre-clinical models of spontaneous metastasis (133) that will enable better understanding of the role of CAFs in the formation of a pre-metastatic niche, and in facilitating the early stages of metastasis.
In the coming years, we will likely see multiple studies that will profile CAF populations at the single cell level, enabling better identification of their functional heterogeneity. Such understanding will provide us with both context-specific understanding of unique CAF functions, and unifying mechanisms that are common to CAF tasks in various cancer types. While knowledge from preclinical studies on immunoregulatory functions of CAFs is emerging, clinical data on CAF targeting is still limited. Hopefully, we will see in the future integration of the preclinical findings described in this review (Figure 1) into the design of novel therapeutic combination strategies aimed at impairing the tumor-supportive and immunosuppressive responses of CAFs.
Author Contributions
LM analyzed the relevant literature, prepared the tables and figures, and participated in writing the manuscript. NE wrote the manuscript.
Funding
NE was supported by funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 637069 MetCAF), The Israel Science Foundation and the Israel Cancer Research Fund (ICRF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank members of the Erez lab for critical reading of the manuscript.
References
1. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
2. Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol. (2013) 229:198–207. doi: 10.1002/path.4103
3. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. (2014) 102:258–69. doi: 10.1093/cvr/cvu062
4. Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. (2011) 79:944–56. doi: 10.1038/ki.2010.516
5. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. (2018) 11:dmm029447. doi: 10.1242/dmm.029447
6. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
7. Liu L, Liu L, Yao HH, Zhu ZQ, Ning ZL, Huang Q. Stromal myofibroblasts are associated with poor prognosis in solid cancers: a meta-analysis of published studies. PLoS ONE. (2016) 11:e0159947. doi: 10.1371/journal.pone.0159947
8. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. (2015) 47:320–9. doi: 10.1038/ng.3225
9. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. (2019) 18:70. doi: 10.1186/s12943-019-0994-2
10. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
11. Sharon Y, Raz Y, Cohen N, Ben-Shmuel A, Schwartz H, Geiger T, et al. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. (2015) 75:963–73. doi: 10.1158/0008-5472.CAN-14-1990
12. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med. (2018) 215:3075–93. doi: 10.1084/jem.20180818
13. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
14. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE. (2012) 7:e30563. doi: 10.1371/journal.pone.0030563
15. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
16. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. (2016) 30:1002–19. doi: 10.1101/gad.279737.116
17. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. (2006) 6:392–401. doi: 10.1038/nrc1877
18. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. (2014) 211:1503–23. doi: 10.1084/jem.20140692
19. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene. (2017) 36:4457–68. doi: 10.1038/onc.2017.65
20. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
21. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
22. Bartoschek M, Oskolkov N, Bocci M, Lovrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun. (2018) 9:5150. doi: 10.1038/s41467-018-07582-3
23. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
24. Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of immune evasion in breast cancer. BMC Cancer. (2018) 18:556. doi: 10.1186/s12885-018-4441-3
25. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
26. Barnas JL, Simpson-Abelson MR, Yokota SJ, Kelleher RJ, Bankert RB. T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron. (2010) 3:29–47. doi: 10.1007/s12307-010-0044-5
27. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
28. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
29. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
30. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425–6. doi: 10.1158/0008-5472.CAN-06-3037
31. Augsten M, Sjoberg E, Frings O, Vorrink SU, Frijhoff J, Olsson E, et al. Cancer-associated fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide signaling for their tumor-supporting properties. Cancer Res. (2014) 74:2999–3010. doi: 10.1158/0008-5472.CAN-13-2740
32. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
33. Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell. (2012) 11:812–24. doi: 10.1016/j.stem.2012.08.013
34. Yu PF, Huang Y, Han YY, Lin LY, Sun WH, Rabson AB, et al. TNFalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene. (2017) 36:482–90. doi: 10.1038/onc.2016.217
35. Bott A, Erdem N, Lerrer S, Hotz-Wagenblatt A, Breunig C, Abnaof K, et al. miRNA-1246 induces pro-inflammatory responses in mesenchymal stem/stromal cells by regulating PKA and PP2A. Oncotarget. (2017) 8:43897–914. doi: 10.18632/oncotarget.14915
36. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. (2019) 9:282–301. doi: 10.1158/2159-8290.CD-18-0710
37. Biffi G, Tuveson DA. Deciphering cancer fibroblasts. J Exp Med. (2018) 215:2967–8. doi: 10.1084/jem.20182069
38. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. (2016) 76:4124–35. doi: 10.1158/0008-5472.CAN-15-2973
39. Chen L, Qiu X, Wang X, He J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem Biophys Res Commun. (2017) 487:8–14. doi: 10.1016/j.bbrc.2017.03.039
40. Gunderson AJ, Yamazaki T, McCarty K, Phillips M, Alice A, Bambina S, et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE. (2019) 14:e0211117. doi: 10.1371/journal.pone.0211117
41. Zhang Y, Ertl HC. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget. (2016) 7:23282–99. doi: 10.18632/oncotarget.7818
42. Jia XH, Du Y, Mao D, Wang ZL, He ZQ, Qiu JD, et al. Zoledronic acid prevents the tumor-promoting effects of mesenchymal stem cells via MCP-1 dependent recruitment of macrophages. Oncotarget. (2015) 6:26018–28. doi: 10.18632/oncotarget.4658
43. Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart LA. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology. (2010) 215:737–47. doi: 10.1016/j.imbio.2010.05.019
44. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. (2014) 33:2423–31. doi: 10.1038/onc.2013.191
45. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
46. Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. (2014) 158:564–78. doi: 10.1016/j.cell.2014.05.045
47. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. (2017) 32:654–68.e5. doi: 10.1016/j.ccell.2017.10.005
48. Ellem SJ, Taylor RA, Furic L, Larsson O, Frydenberg M, Pook D, et al. A pro-tumourigenic loop at the human prostate tumour interface orchestrated by oestrogen, CXCL12 and mast cell recruitment. J Pathol. (2014) 234:86–98. doi: 10.1002/path.4386
49. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. (2010) 29:273–83. doi: 10.1007/s10555-010-9220-9
50. Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. (2012) 72:2251–61. doi: 10.1158/0008-5472.CAN-11-3386
51. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. (2016) 7:11762. doi: 10.1038/ncomms11762
52. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. (2009) 182:1746–55. doi: 10.4049/jimmunol.182.3.1746
53. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell. (2012) 22:291–303. doi: 10.1016/j.ccr.2012.07.023
54. Karavitis J, Hix LM, Shi YH, Schultz RF, Khazaie K, Zhang M. Regulation of COX2 expression in mouse mammary tumor cells controls bone metastasis and PGE2-induction of regulatory T cell migration. PLoS ONE. (2012) 7:e46342. doi: 10.1371/journal.pone.0046342
55. Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. (2009) 69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619
56. Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun. (2018) 9:1056. doi: 10.1038/s41467-018-03348-z
57. Gil M, Komorowski MP, Seshadri M, Rokita H, McGray AJ, Opyrchal M, et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol. (2014) 193:5327–37. doi: 10.4049/jimmunol.1400201
58. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. (2018) 9:422. doi: 10.1038/s41419-018-0458-4
59. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci. (2017) 18:1545. doi: 10.3390/ijms18071545
60. Krzyszczyk P, Schloss R, Palmer A, Berthiaume F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front Physiol. (2018) 9:419. doi: 10.3389/fphys.2018.00419
61. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
62. Han X, Yang Q, Lin L, Xu C, Zheng C, Chen X, et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. (2014) 21:1758–68. doi: 10.1038/cdd.2014.85
63. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
64. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009
65. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget. (2017) 8:8633–47. doi: 10.18632/oncotarget.14374
66. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
67. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
68. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. (2013) 73:3007–18. doi: 10.1158/0008-5472.CAN-12-4601
69. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. (2018) 67:320–32. doi: 10.1136/gutjnl-2016-311585
70. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. (2017) 36:1090–101. doi: 10.1038/onc.2016.273
71. D'Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. (2016) 213:827–40. doi: 10.1084/jem.20150950
72. Capietto AH, Kim S, Sanford DE, Linehan DC, Hikida M, Kumosaki T, et al. Down-regulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. (2013) 210:2257–71. doi: 10.1084/jem.20130281
73. Yang T, Zhang X, Wang M, Zhang J, Huang F, Cai J, et al. Activation of mesenchymal stem cells by macrophages prompts human gastric cancer growth through NF-kappaB pathway. PLoS ONE. (2014) 9:e97569. doi: 10.1371/journal.pone.0097569
74. DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. (2007) 9:212. doi: 10.1186/bcr1746
75. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE. (2009) 4:e7965. doi: 10.1371/journal.pone.0007965
76. Barnas JL, Simpson-Abelson MR, Brooks SP, Kelleher RJ Jr, Bankert RB, Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol. (2010) 185:2681–92. doi: 10.4049/jimmunol.1000896
77. Vancheri C, Mastruzzo C, Trovato-Salinaro E, Gili E, Lo Furno D, Pistorio MP, et al. Interaction between human lung fibroblasts and T-lymphocytes prevents activation of CD4+ cells. Respir Res. (2005) 6:103. doi: 10.1186/1465-9921-6-103
78. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity. (2019) 50:707–22.e6. doi: 10.1016/j.immuni.2019.02.002
79. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
80. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. (2010) 10:554–67. doi: 10.1038/nri2808
81. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. (2016) 5:e198. doi: 10.1038/oncsis.2016.7
82. Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS, et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget. (2016) 7:27584–98. doi: 10.18632/oncotarget.8488
83. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. (2011) 208:469–78. doi: 10.1084/jem.20101876
84. Eble JA, Niland S. The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis. (2019) 36:171–98. doi: 10.1007/s10585-019-09966-1
85. Venning FA, Wullkopf L, Erler JT, Targeting ECM disrupts cancer progression. Front Oncol. (2015) 5:224. doi: 10.3389/fonc.2015.00224
86. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
87. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. (2017) 66:1037–1048. doi: 10.1007/s00262-017-2003-1
88. Protti MP, De Monte L. Cross-talk within the tumor microenvironment mediates Th2-type inflammation in pancreatic cancer. Oncoimmunology. (2012) 1:89–91. doi: 10.4161/onci.1.1.17939
89. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. (2010) 70:7073–83. doi: 10.1158/0008-5472.CAN-09-4687
90. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res. (2014) 20:3422–33. doi: 10.1158/1078-0432.CCR-13-2972
91. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/JCI45817
92. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
93. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. (2016) 22:851–60. doi: 10.1038/nm.4123
94. Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell. (2019) 49:332–346. doi: 10.1016/j.devcel.2019.03.026
95. Kaur A, Ecker BL, Douglass SM, Kugel CH III, Webster MR, Almeida FV, et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. (2019) 9:64–81. doi: 10.1158/2159-8290.CD-18-0193
96. Ecker BL, Kaur A, Douglass SM, Webster MR, Almeida FV, Marino GE, et al. Age-related changes in HAPLN1 increase lymphatic permeability and affect routes of melanoma metastasis. Cancer Discov. (2019) 9:82–95. doi: 10.1158/2159-8290.CD-18-0168
97. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:20847–52. doi: 10.1073/pnas.0906481106
98. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. (2012) 318:154–61. doi: 10.1016/j.canlet.2011.12.020
99. Ziani L, Safta-Saadoun TB, Gourbeix J, Cavalcanti A, Robert C, Favre G, et al. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget. (2017) 8:19780–94. doi: 10.18632/oncotarget.15540
100. Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. (2012) 18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632
101. Gorchs L, Fernandez Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4(+) and CD8(+) T-Cells. Front Immunol. (2019) 10:847. doi: 10.3389/fimmu.2019.00847
102. Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology. (2008) 135:1228–37, 1237.e1–2. doi: 10.1053/j.gastro.2008.07.016
103. Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. (2005) 40:95–103. doi: 10.1016/j.jdermsci.2005.06.008
104. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells. (2017) 35:766–76. doi: 10.1002/stem.2509
105. de Lourdes Mora-Garcia M, Garcia-Rocha R, Morales-Ramirez O, Montesinos JJ, Weiss-Steider B, Hernandez-Montes J, et al. Mesenchymal stromal cells derived from cervical cancer produce high amounts of adenosine to suppress cytotoxic T lymphocyte functions. J Transl Med. (2016) 14:302. doi: 10.1186/s12967-016-1057-8
106. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene. (2019) 38:3681–95. doi: 10.1038/s41388-019-0688-7
107. Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. (2015) 42:663–71. doi: 10.1053/j.seminoncol.2015.05.011
108. Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. (2014) 2:187–93. doi: 10.1158/2326-6066.CIR-14-0002
109. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
110. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
111. Ganesh K, Massague J. TGF-beta inhibition and immunotherapy: checkmate. Immunity. (2018) 48:626–628. doi: 10.1016/j.immuni.2018.03.037
112. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. (2018) 9:948. doi: 10.1038/s41467-018-03347-0
113. Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J Immunol. (2015) 194:1389–94. doi: 10.4049/jimmunol.1402520
114. Lane RS, Lund AW. Non-hematopoietic control of peripheral tissue T cell responses: implications for solid tumors. Front Immunol. (2018) 9:2662. doi: 10.3389/fimmu.2018.02662
115. Mrass P, Petravic J, Davenport MP, Weninger W. Cell-autonomous and environmental contributions to the interstitial migration of T cells. Semin Immunopathol. (2010) 32:257–74. doi: 10.1007/s00281-010-0212-1
116. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. (2010) 328:749–52. doi: 10.1126/science.1185837
117. Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. (2018) 6:1472–85. doi: 10.1158/2326-6066.CIR-18-0098
118. Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS ONE. (2011) 6:e27618. doi: 10.1371/journal.pone.0027618
119. Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. (2011) 12:1096–104. doi: 10.1038/ni.2112
120. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. (2010) 330:827–30. doi: 10.1126/science.1195300
121. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. (2009) 119:3613–25. doi: 10.1172/JCI38988
122. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res. (2008) 14:4584–92. doi: 10.1158/1078-0432.CCR-07-5211
123. Fang J, Xiao L, Joo KI, Liu Y, Zhang C, Liu S, et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int J Cancer. (2016) 138:1013–23. doi: 10.1002/ijc.29831
124. Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. (2006) 116:1955–62. doi: 10.1172/JCI26532
125. Duperret EK, Trautz A, Ammons D, Perales-Puchalt A, Wise MC, Yan J, et al. Alteration of the tumor stroma using a consensus DNA vaccine targeting fibroblast activation protein (FAP) synergizes with antitumor vaccine therapy in mice. Clin Cancer Res. (2018) 24:1190–201. doi: 10.1158/1078-0432.CCR-17-2033
126. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
127. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. (2013) 210:1137–51. doi: 10.1084/jem.20122344
128. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
129. Takai K, Le A, Weaver VM, Werb Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget. (2016) 7:82889–901. doi: 10.18632/oncotarget.12658
130. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. (2015) 106:134–42. doi: 10.1111/cas.12584
131. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. (2014) 159:80–93. doi: 10.1016/j.cell.2014.08.007
132. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
Keywords: CAFs, immunosuppression, immune modulation, inflammation, tumor microenvironment
Citation: Monteran L and Erez N (2019) The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 10:1835. doi: 10.3389/fimmu.2019.01835
Received: 29 May 2019; Accepted: 22 July 2019;
Published: 02 August 2019.
Edited by:
Leila Akkari, The Netherlands Cancer Institute (NKI), NetherlandsReviewed by:
Claudia Cantoni, University of Genoa, ItalyKawaljit Kaur, University of California, Los Angeles, United States
Copyright © 2019 Monteran and Erez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neta Erez, bmV0YWVyZXpAdGF1ZXgudGF1LmFjLmls