 Mark R. Zielinski
Mark R. Zielinski David M. Systrom3,4
David M. Systrom3,4
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 August 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01827
Profound and debilitating fatigue is the most common complaint reported among individuals with autoimmune disease, such as systemic lupus erythematosus, multiple sclerosis, type 1 diabetes, celiac disease, chronic fatigue syndrome, and rheumatoid arthritis. Fatigue is multi-faceted and broadly defined, which makes understanding the cause of its manifestations especially difficult in conditions with diverse pathology including autoimmune diseases. In general, fatigue is defined by debilitating periods of exhaustion that interfere with normal activities. The severity and duration of fatigue episodes vary, but fatigue can cause difficulty for even simple tasks like climbing stairs or crossing the room. The exact mechanisms of fatigue are not well-understood, perhaps due to its broad definition. Nevertheless, physiological processes known to play a role in fatigue include oxygen/nutrient supply, metabolism, mood, motivation, and sleepiness—all which are affected by inflammation. Additionally, an important contributing element to fatigue is the central nervous system—a region impacted either directly or indirectly in numerous autoimmune and related disorders. This review describes how inflammation and the central nervous system contribute to fatigue and suggests potential mechanisms involved in fatigue that are likely exhibited in autoimmune and related diseases.
According to the National Institutes of Health, autoimmune diseases are estimated to afflict over 20 million individuals in the United States (1, 2). Currently, there are over 100 recognized autoimmune diseases (3), and the prevalence of many autoimmune diseases continues to rise (4, 5). A recent self-reported survey of individuals with autoimmune and related disorders by the American Autoimmune and Related Disorders Association indicated that this population's primary concern is fatigue (6). Over two-thirds of respondents reported that their fatigue was profound, debilitating, and prevented them from completing simple everyday tasks. Indeed, a growing literature indicates that fatigue is common in most autoimmune-related diseases, as well as among individuals with related immunodeficiency disorders (7–12). Furthermore, it is estimated that 7–45% of people in the general population exhibit persistent fatigue (13), while almost 98% of individuals with autoimmune disease report that they suffer from fatigue (6). Fatigue can cause dramatic impairments in mood (14), diminish social aspects of life (15), lead to an inability to perform routine daily activities (16), and limit physical activity and work (17). Consequently, fatigue can severely affect well-being and has a financial burden on the individual, family, and society (18–21).
Fatigue is multifaceted and typically broadly defined making it difficult to decipher the causes in specific autoimmune diseases (19). Fatigue is generally described as a condition with prolonged periods of exhaustion accompanied by the inability to perform activities to an expected capacity. There are numerous aspects of fatigue that can be assessed that define the type of fatigue including the severity of functional impairment; time-of-day/circadian patterns of fatigue; length of the persistence of the fatigue from seconds to days; time between fatigue periods; duration of time necessary for the fatigue to dissipate; influence of sleep loss or disturbances in sleep; impact of depression or anxiety; degree of distress concurrently occurring with the fatigue; type of impairment, such as is cognition, motivation, attention, or physical abilities; and the type of physical performance or activity that is impaired, such as in walking, climbing stairs, socialization, chores, cooking, bathing, work, and sex. The type of fatigue experienced in autoimmune disease is variable These differences are likely related to the particular tissues/organs, cell types, brain areas, and molecular and physiological mechanisms affected by the condition (19).
Currently, there is a lack of efficacious long-lasting treatments for individuals experiencing fatigue in autoimmune disease. This is due, in part, to the limitations in our understanding of the multiple mechanisms responsible for fatigue. Evidence suggests several physiological functions can contribute to fatigue including oxygen/nutrient supply (22, 23), metabolism (24), mood (14), motivation (25), and daytime sleepiness (26, 27). Interestingly, inflammation is altered by many of the factors that modulate fatigue and vice versa (25, 28–30). Growing evidence indicates that neuroinflammation is a primary factor contributing to fatigue (25, 31). Since inflammation plays a large part in inducing fatigue, it is plausible that inflammatory pathways and the subsequent physiological alterations modulated by the inflammation are treatable targets for fatigue in patients with autoimmune disease. Indeed, evidence in autoimmune and related conditions, such as neurosarcoidosis, which is associated with increased lung inflammation, sleep disturbances, and fatigue, exhibit reduced fatigue from anti-inflammatory treatment (32). Herein, we discuss the role of factors contributing to fatigue in autoimmune disease including inflammatory-related mechanisms, relationships between peripheral and central nervous system (CNS) inflammation, particular brain areas and neurotransmitters, and cerebral vasohemodynamics (Table 1).
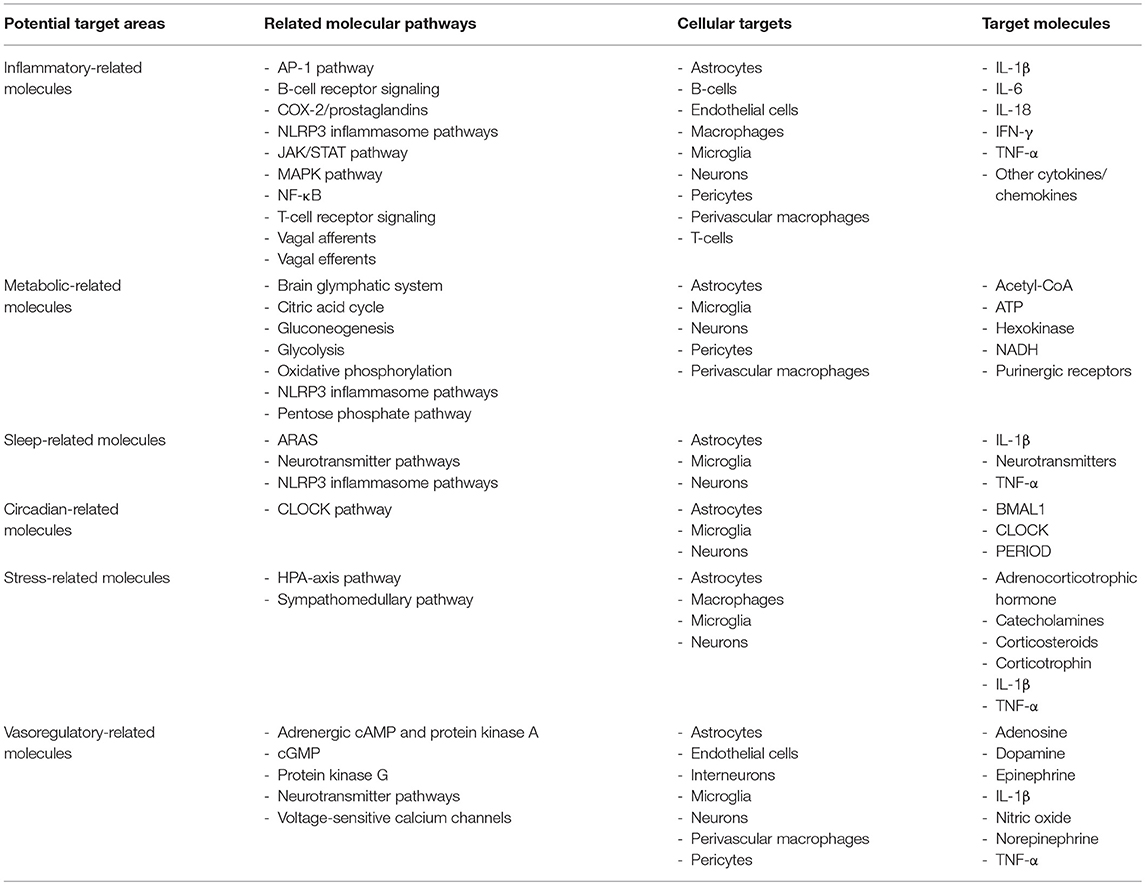
Table 1. Potential target areas, molecular pathways, cellular targets, and target molecules for understanding fatigue in individuals with autoimmune and related disorders.
Autoimmune diseases are associated with enhanced pro-inflammatory signals in the periphery and CNS (33–37). The location of the enhanced inflammation varies with the type of autoimmune disease and the progression of the condition. Fatigue is observed in non-autoimmune disease and related conditions that have increased inflammation in the periphery and/or CNS, including cancer (38), sleep disorders (31, 39, 40), stroke (41, 42), and traumatic brain injury (43, 44). Several non-inflammatory factors are known to be affect fatigue including impairments in hydration status (45), pain (46), interactions from pharmaceuticals (47), muscle/exercise (48), hypothyroidism (49), radiation therapy (50), lung function, and cardiovascular characteristics such as blood pressure, heart rate, cardiac output, and stroke volume (51, 52). Many of the non-inflammatory components contributing to fatigue are modulated by or modulate inflammatory processes. Regardless, inflammatory mediators are reported to affect different aspects that contribute to fatigue including motivation (53), sleepiness (54, 55), cognition (56), anxiety (57), depression (58, 59), and stress (60).
Cytokines are small proteins molecules involved in cell signaling allowing cells to communicate through autocrine, paracrine, or endocrine mechanisms (61). Cytokines modulate immune responses, inflammation, cell growth and maturation, and normal physiological functions. They are highly conserved among species ranging from invertebrates to rodents and humans. Inflammatory cytokines are produced by nucleated cell types including lymphocytes and macrophages, as well as microglia, astrocytes, and neurons in the CNS (62). Inflammatory mechanisms involved in the etiology of fatigue implicate a significant involvement of cytokines. Interleukin (IL)-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), IL-6, and interferon-gamma (IFN-γ) are cytokines that all have pro-inflammatory properties, and their enhancement are the most well-characterized in inducing fatigue and/or altering aspects contributing to fatigue (25, 63).
Autoimmune disease induces enhancement in cytokines such as IL-1β, TNF-α, IL-6, IL-12, IL-23, and IFN-γ, especially by T helper cells and macrophages (64, 65). Consequently, treatments targeting aspects of lymphocyte regulatory processes benefit the treatment of autoimmune diseases including Sjörgen's syndrome, rheumatoid arthritis, and inflammatory bowel disease. Macrophage activity is intertwined with T-cell functions (66), and macrophages are involved in the pathogenesis of autoimmune diseases (65). Macrophages in an M1 classical activated state tend to induce a pro-inflammatory response including IL-1β, TNF-α, IL-6, IFN-γ (67). Macrophages in a M2 alternatively activated state tend to have anti-inflammatory properties expressing IL-10, transforming growth factor-beta (TGF-β), and IL-1 receptor antagonist (IL-1RA). Pro- and anti-inflammatory cytokines regulate a balance in inflammatory status and the production of other cytokines. In addition, the overall level of localized cytokine secretion or its persistent enhancement or attenuation can lead to the upregulation or downregulation of associated receptors to modulate the downstream mechanisms or production of cytokines. Cytokines regulate normal physiological functions including mood, cognition, and sleep, and their expression varies over the course of the day and in response to local activity (30). Consequently, it is likely that dysregulation in inflammatory cytokines and their receptors in autoimmune disease serves to disrupt the normal physiological homeostasis of cytokines and contribute to fatigue.
Inflammation in the periphery can induce inflammation in the CNS and sickness behaviors (68, 69), which are behaviors associated with different aspects of fatigue (70). Experimental studies in rodents applying IL-1β, TNF-α, or IL-6 or substances that enhance their activity, such as the gram-negative bacterial cell wall component lipopolysaccharide (LPS), to the periphery or CNS result in alterations in behaviors affecting fatigue (30, 71, 72). Such applications can impair behaviors associated with fatigue including cognition, mood, pain, sleep, and motivation. Herein, we give several examples of how IL-1β modulates behaviors that can alter fatigue. Dysregulation of the IL-1 cytokine family member IL-1β or its receptor is implicated in fatigue in many autoimmune diseases (73). Enhanced IL-1β and its receptors are also implicated in fatigue (74). Nevertheless, evidence also suggests that IL-1β signaling is not necessary for other types of fatigue, including conditions like tumor-associated fatigue (75). Collectively, these findings from the animal literature indicate redundancy in how inflammatory cytokine affects fatigue-related behavior, although differences likely relate to the brain area and cells affected, amount of cytokine activity, timing of the cytokine activity, and interactions with normal physiological activities.
IL-1β binds to IL-1 receptor type I (IL-1R1) to induce inflammatory effects (76). IL-1β can also bind to IL-1 receptor type II, which acts as a decoy to inhibit IL-1β activity. Additionally, IL-1RA can inhibit IL-1β functioning by binding the IL-1R, thus preventing subsequent signaling processes. IL-1RI is functional in the presence of IL-1 receptor accessory protein (IL-1RAP). Activation of the IL-1RI promotes myeloid differentiation primary response 88 (MYD88), which subsequently activates the interleukin-1 receptor associated kinase 4 (IRAK4) (77). This activation ultimately leads to an enhancement of inhibitor of kappa B kinase (IKK) and mitogen-activated protein kinase (MAPK) kinase (MAPKK). IKK then can function to induce the inhibitory kappa B (IκB) to be released from nuclear factor kappa B (NF-κB), which allows NF-κB to translocate the nucleus and induce the transcription of pro-inflammatory molecules and processes. MAPKK induces c-Jun N-terminal nucleated kinase (JNK) and p38 to translocate the nucleus inducing activator protein-1 (AP-1) transcription of pro-inflammatory molecules and processes (78, 79). Interestingly, an IL-1 receptor accessor protein that is primarily expressed in the CNS (IL-1RAPb) was identified (80). IL-1RAPb binds to IL-1RI and inhibits the responses of the downstream adaptor molecules MYD88 and kinase IRAK4 (80, 81). Evidence suggests that the IL-1RAP is involved in modulating behavior under conditions of enhanced inflammation (82). Thus, although speculative, it is plausible that IL-1RAPb is involved in modulating cytokines involved in fatigue.
Cognitive fatigue involves declines in alertness, orientation, and mental performance on cognitive tasks and is associated with feelings of exhaustion that follow sustained cognitive demands (83). Individuals with autoimmune diseases, such as multiple sclerosis, often experience cognitive deficits and increased perceived cognitive fatigue associated with impaired cortical brain activity as determined using near-infrared spectroscopy (83). While there is a lack of specific tests in animal models regarding cognitive fatigue, the relationship between pro-inflammatory cytokines, especially IL-1β, and cognition is described (84, 85). Increased neuronal activity is observed with cognitive activities or whisker stimulation (86, 87), which enhance the expression of IL-1β or TNF-α in corresponding brain areas or barrel cortices (88, 89), respectively. In rodents, intracerebroventricular (ICV) or intraperitoneal (IP) injections of IL-1β prior to memory training impairs subsequent cognitive performance using the Morris water maze (90), a test of spatial memory that utilizes the hippocampus (91), or the eight-arm radial maze which tests working memory (92). The area of the brain where the inflammation occurs mediates the effect of the behavioral impairments. This is observed, for example, by the infusion of IL-1β locally into the hippocampus, which impairs working memory in a hippocampal-dependent memory task such as the three-panel runaway task administered to rats (93). Disease-specific aspects of autoimmune disease can also influence types of performance decrements. For instance, ICV injection of Human Immunodeficiency Virus-1 (HIV-1) envelope glycoprotein 120, a molecule that enhances IL-1β and TNF-α in individuals with HIV, increases IL-1β levels in the hippocampus and impairs contextual memory performance in rats (94). Inhibition of ligands for pro-inflammatory cytokines including IL-1β, TNF-α, IL-6, and IFN-γ using transgenic knockout mice, siRNA, or more cutting-edge technologies such as optogenetics or chemogenetics further implicate the role of inflammation in altering components related to fatigue (70, 95, 96). IL-1R1 knockout mice or an IL-1RA applied to the circulation given peripheral LPS demonstrate cognitive dysfunction associated with a fear conditioning test suggesting that reduced activation of peripheral inflammatory activity can inhibit central mediated behavior (97). However, evidence in animals also indicates that pro-inflammatory cytokines are required for normal behavioral functions and that an optimal zone exists for proper functioning (70, 84). This effect is seen in mice lacking IL-1R1 or mice given an IL-1RA to the periphery, which demonstrate reduced cognitive responses in the Morris water maze (90, 98, 99). Nevertheless, other studies demonstrate mice lacking IL-1R1 demonstrate normal learning in cognitive tests including the Y-maze, T-maze, and Morris water maze (100).
Autoimmune diseases including inflammatory bowel disease, multiple sclerosis, and rheumatoid arthritis have a high comorbidity with anxiety, depression, and pain (14), which can serve to induce fatigue. A study involving ICV injections of IL-1β demonstrated increased anxiety-like behavior in mice determined by shorter time spent in the center area of the open field test and increased time spent in the closed arms of the elevated plus maze (101). Additionally, central administration of IL-1β increases immobilization periods in the tail suspension test (102), which assesses depressive-like behavior in mice. Evidence also suggests that social aspects are impaired with enhanced CNS inflammation. For example, ICV injections of IL-1β reduces social interactions as evidenced by reductions in social exploration behavior including active investigation, anogenital sniffing, and wrestling in rats (103). Furthermore, mice lacking the IL-1R1 demonstrate anxiogenic-like behavior with increased time spent in the open arms of the elevated-plus maze (100). However, IL-1R1 knockout mice are reported to not exhibit reduced mobility in a forced swim test, which assesses depressive like behavior in rodents (100).
The perception of effort and motivation can modify fatigue and are affected in autoimmune disease. Animal studies indicate that effort expenditure is influenced by inflammation (104–106). In general, inflammation increases averseness toward negative stimuli and positive stimuli (107). Animal models of motivation indicate that both approach and avoidance motivation are affected by pro-inflammatory cytokines. For example, IP injections of IL-1β impairs motivation in rats as observed with reduced lever pressing in a chow feeding choice procedure (105). IL-1β applied centrally in rats that are food-deprived impairs rodent choice toward favoring a low-effort, low-reward option using a two-choice high vs. low effort/reward task (105). These findings suggest that IL-1β can influence motivation away from high effort/high award tasks, although in this study the use of operant responding for food pellets could be interpreted as IL-1β influencing consummatory behavior. However, in a study where mice received IP injections of LPS with free-feeding, the mice exhibited a reduction in nose pokes for the low-effort, low-reward stimuli of grain resulting in an increase in percentage of high-effort, high-reward stimuli for earned chocolate (108). These data suggest that inflammation affects incentive motivation, in part, though altering willingness to exert effort for reward instead of reducing sensitivity to the reward, although the differences in effects could also be due to species differences or additional cytokines and inflammatory pathways that LPS activates.
A primary mechanism that activates IL-1β occurs through the activation of inflammasomes (109). Inflammasomes are large intracellular signaling protein complexes found within the cytoplasm of most nucleated cells including neurons, astrocytes, microglia, and perivascular macrophages in the brain (Figure 1) (44, 109, 110). Inflammasome activation involves a priming step and a secondary step that induces the formation of the complex to activate caspase-1 to cleave the pro-forms of IL-1β and other IL-1 family members, such as IL-18 and IL-33, into their mature active forms (111). The priming signal involves the activation of transcriptional processes such as NF-κB or AP-1 to produce the components of the inflammasome as well as the pro-forms of the cytokines that will be activated upon inflammasome formation and subsequent caspase-1 release (112, 113). Inflammasome priming can occur by the activation of different types of receptors including the IL-1RI by IL-1β, TNF receptor I by TNF-α, or the toll-like receptor 4 (TLR4) by LPS to activate NF-κB (114). JNKs and MAPK/extracellular signal regulated kinase (ERK) pathways, which can activate AP-1 mediated transcription, also are implicated in the activation of inflammasomes (115). Most inflammasomes contain a nucleotide-binding oligomerization domain-like receptor or an absent in melanoma 2 (AIM2)-like receptor (109). The nucleotide leucine-rich protein-3 (NLRP3) inflammasome is the most widely characterized inflammasome, although multiple types of inflammasomes exist with unique recognition abilities in response to specific pathogen-associated molecular patterns (PAMPS) or danger-associated molecular patterns (DAMPS) (111). PAMPs and DAMPs include components of pathogens, energy-related molecules, double-stranded or single-stranded deoxyribonucleic acid (DNA), ribonucleic acid (RNA), or chemical substances. PAMPs and DAMPs are recognized by their specific associated pattern recognition receptor (PRR). These processes lead to the recruitment of the adapter apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC; also known as pycard1) and pro-caspase-1, which conglomerate to activate caspase-1.
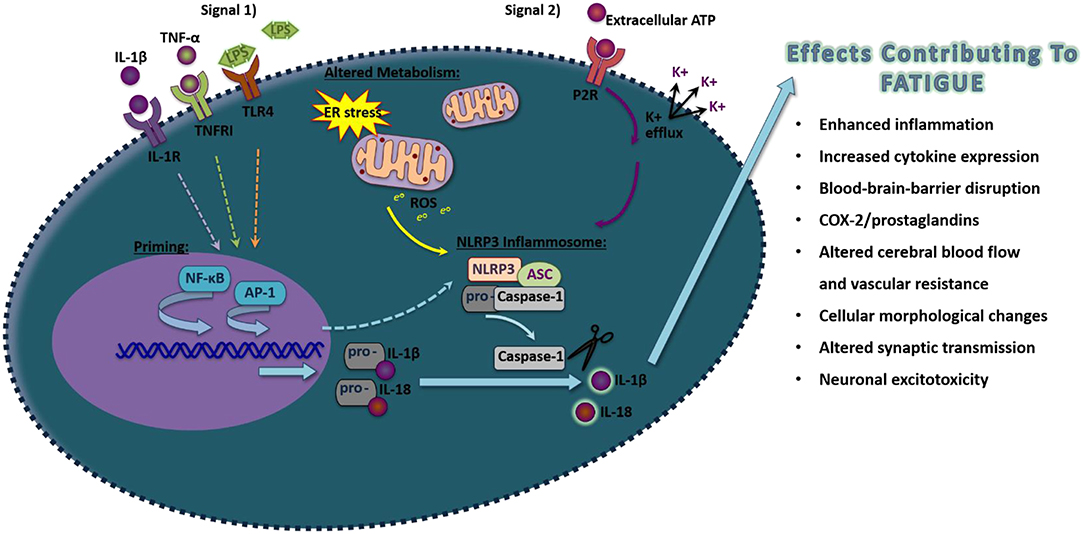
Figure 1. Schematic of proposed NLRP3 inflammasome activation in inducing pro-inflammatory molecules that induce fatigue. Exposure to pathogen-associated molecular pattern or danger-associated molecular pattern that act on their pattern recognition receptors (Signal 1), such as LPS, TNF-α, or IL-1β acting through Toll-like receptor 4, TNFR1, and IL-1R, respectively, will activate the transcriptional factors NF-κB that will prime components of the NLRP3 inflammasome. Additionally, transcription of inflammasome components can also occur through the activation of AP-1. A secondary signal is then required to activate inflammasome formation, including by alterations in metabolism that induce ROS or the activation of the purine type 2X receptors (P2XRs). Upon formation of NLRP3, ASC, and pro-caspase-1, caspase-1 will be allowed to disassociate and cleave the inactive pro-forms of IL-1β and IL-18 into their mature active forms. Upon release, these pro-inflammatory cytokines can alter surrounding cells leading to effects contributing to fatigue.
Inflammasomes are involved in modulating behavior including sleep (116), cognition (117), anxiety (118–120), and depression (120). Evidence also suggests that inflammasomes are hyper-activated in individuals with autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, spondyloarthritis, Sjogren's syndrome, and Crohn's disease (121, 122). Fatigue-related tests in mice including the repeated forced swim test, locomotor activity, and Rota-rod test have shown impaired performance associated with enhanced activation of inflammasome-related molecules, and mice lacking NLRP3 have demonstrated reduced fatigue-like behavior (123). TNF-α and IFN-γ have unique inflammatory pathways involved in their activation and are indirectly involved in mechanisms that can induce inflammasome activation (124, 125). Evidence also suggests that IL-1β can enhance reactive oxygen species (ROS), in part, through increasing the phosphatidylinositol 3-kinase (PI3-K)/protein kinase B (AKT)/mammalian target of the rapamycin (mTOR) pathway—a pathway implicated in inflammasome activation (125–127). Hexokinase-1 is also implicated in mTOR and NLRP3 inflammasome activation (128, 129).
IFN-γ is involved in innate and adaptive immunity. IFN-γ and its ligands (the IFN-γ receptor 1 and IFN-γ receptor 2) are found throughout tissues in the periphery and CNS, especially in macrophages, microglia, and lymphocytes (130). IFN-γ receptors stimulate the Janus kinase (JAK)-signal transducer and activator of transcription protein (STAT) pathway (130). The JAK-STAT pathway can interconnect with other inflammatory pathways including the PI3K/AKT/mTOR pathway and the MAPK/ERK pathway, which can serve to ultimately enhance NLRP3 inflammasome activation (130). Additional evidence suggests that IFN-γ effects on IL-1β are related to the enhancement of suppressor of cytokine signaling 1 (SOCS1) (131). Direct evidence also indicates that IFN-γ upregulates NLRP3, ASC and pro-caspase 1 expression (125).
Metabolism involves the conversion of fuel sources to energy-related molecules—such as adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate (NADPH)—enabling cellular processes; synthesis of components for proteins, lipids, nucleic acids, and carbohydrates; and the removal of ROS such as peroxides, superoxide, and hydroxyl radicals (132). Metabolism is involved in mechanisms that enhance pro-inflammatory cytokines including IL-1β, TNF-α, IL-6, and IFN-γ (133, 134). Alterations in metabolism have been implicated in sleep regulation and fatigue (135, 136). Metabolism also is tied to glycogen metabolism, glycogen synthesis, and energy substrates and their derivatives (132, 135), which could potentially alter fatigue. Alterations in metabolism are also implicated in the pathogenesis of autoimmune disease (63).
ATP is an energy molecule involved in the catabolism of macronutrients including carbohydrates, proteins, and lipids (137). ATP is modulated by intracellular glucose availability and mitochondria production (137). ATP can function as a neurotransmitter, in part, by acting on purine type 2 receptors (138, 139). Also, ATP can be co-released in to the extracellular space with other neurotransmitters such as norepinephrine or acetylcholine (138, 139). A major mechanism in the activation of NLRP3 inflammasomes involves extracellular ATP acting through purine type 2 (P2) receptors, including the P2X7 receptor, to induce potassium efflux leading to the formation of the NLRP3 inflammasome (140). Nevertheless, ATP is rapidly metabolized to adenosine diphosphate and adenosine monophosphate by ectonucleoside diphosphohydrolase (i.e., CD39). AMP is then metabolized to adenosine by ecto-5′ nucleotidase (i.e., CD73) (141), which is a molecule involved in modulating sleep (142). Adenosine acts on purine type 1 receptors, is involved in sleep regulation, and inhibits arousal (143), which could potentially lead to fatigue. Inhibition of purine type 1 receptors with adenosine 2A receptor antagonists or caffeine can enhance wakefulness and reduce some types of fatigue (144), although the persistent use of caffeine can be detrimental to sleep (145).
ROS are involved in cell signaling, homeostasis, and normal physiological processes including inflammatory processes (146). Evidence in animal models and patients indicates that increased ROS activity is involved in the pathogenesis of autoimmune diseases (147). ROS are formed from the metabolism of oxygen during cellular respiration (148). ROS production—done with the aid of enzymes including cyclooxygenase, lipoxygenase, NAPDH oxidases, and xanthine oxidase—can lead to enhanced pro-inflammatory cytokine production (149). Within the CNS, neurons largely rely on oxidative phosphorylation (OXPHOS) for ATP requirements (150). Neurons work synergistically with astrocytes—cells that utilize glycolysis to convert lipids and glucose to pyruvate, which is ultimately converted to lactate (151). Glycolysis and oxidative phosphorylation generate two ATP molecules per glucose molecule and 36 ATP molecules for OXPHOS, respectively (152). However, glycolysis also produces substrates for pentose phosphate pathway (PPP) to make ribose 5-phosphate for the generation of NADPH (153). NADPH oxidase aids in the production of ROS in microglia, astrocytes and neurons (110). Major areas of activation in the mitochondrial respiratory chain are at complex I and complex III. Electrons from NADH are accepted in complex I where they move through an electrochemical gradient to complex II through ubiquinone (154). Thereafter, the electrons are moved to cytochrome C and complex IV where oxygen is converted into water, which can lead to pro-inflammatory cytokine production (155, 156).
Evidence indicates that NADPH oxidases are activators of NLRP3 inflammasomes (157). In particular, cells undergoing ROS that are primed for NLRP3 activation enhance redox-dependent transcription factors including NF-κB (158). Other studies indicate that IL-1β and caspase-1 are ROS-dependent (159). IL-1β also enhances intracellular ROS by uncoupling superoxide dismutase, catalase, and glutathione peroxidase, which have anti-oxidant functions, thus inducing a feedforward production in inflammasome activation (115). ROS inhibitors also can reduce the secretion of IL-1β. NADPH oxidase appears to be a primary contributor for the ROS activation of inflammasomes involving extracellular ATP, although other contributors are also likely. This is consistent with studies indicating that the enhancement of ROS is dependent on P2X7 receptor activation (160). Moreover, the NADPH inhibitor diphenyleneiodonium attenuates caspase-1 activation by ATP further suggesting energy- and ROS-related mechanisms in inflammasome activation (115).
Mitochondrial DNA dysregulation has also been observed in inflammatory conditions including autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and granulomatosis with polyangiitis (161). Mitochondrial stress can induce the release of mitochondrial DNA into the cytosol, resulting in increased NLRP3 inflammasome activation (162). Mitochondrial DNA can be altered by the transcription factor A mitochondrial to impair OXPHOS (163). In addition, evidence indicates that the escape of mitochondrial DNA into the cytosol involves the DNA sensor cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), which promotes stimulator of interferon genes (STING)-interferon regulatory factor 3 (IRF3) dependent signaling to increase the expression of interferon-stimulated genes (164). STINGs are cytosolic proteins that are attached to the endoplasmic reticulum. STING activates IRF3 which then translocates to the nucleus to transcribe type I IFN genes and NF-κB. Interestingly, recent evidence suggests that IRF3 in antigen presenting cells and T-cells that is necessary for maximized IFN-γ responses (165).
Thioredoxin (TRX) and its endogenous inhibitor thioredoxin-interactive protein (TXNIP) are involved in the activation of inflammasomes (111). TXNIP, when released from the oxidized TRX, will bind directly to leucine-rich areas of NLRP3 inducing the inflammasomes to form (111). Interestingly, multiple sclerosis patients that were not exposed to interferon and immunosuppressive treatments exhibited significant enhancements in TRX expression in peripheral blood mononuclear cells compared to healthy controls while multiple sclerosis patients that did undergo treatments did not show significant differences (166). Moreover, all multiple sclerosis patients regardless of treatment exhibited greater TXNIP expression values (166).
The mechanistic target of rapamycin (mTORC) is a major sensor of metabolic stress and the pro-inflammatory response (167, 168). Evidence indicates that the mTORC1 complex is activated by mitochondrial ROS. Interestingly, mTORC1 and mitochondrial ROS are known to activate NLRP3 inflammasomes in a mouse model of lupus (127). The mTORC1 complex is also coupled with the metabolic depletion of glutathione (169). The depletion of glutathione can be reversed with N-acetylcysteine (NAC) by reducing cysteine and NADPH and inhibiting mTORC1 (169). NAC has been shown to be beneficial in blocking mTOR in systemic lupus erythematosus patients (170).
Cyclooxygenase (COX) converts prostaglandins from arachidonic acid (171). COX-2 is inducible and involved in modulating inflammatory mediators including IL-1β, while COX-1 is constitutively expressed (171). COX-2 is found in most cells including neurons, perivascular cells, and endothelial cells (171). The initiation and resolution of inflammation and alteration of autoimmune-related immunity is modulated by prostaglandin E2, in part, by IL-22 production, T helper 1 cell differentiation and Th17 cell proliferation (172). Evidence suggests that prostaglandin E2 is involved with inflammatory stimuli-induced fatigue (172). These findings collectively suggest that COX-prostaglandin E2 could be involved in fatigue occurring with autoimmune disease. Nevertheless, COX-2 inhibitors tend to have modest effects on fatigue, which could be related to its local cellular effects.
A bidirectional relationship appears to exist between sleep and circadian disturbances with autoimmune disease (173–175). Chronic insomnia is associated with an increased incidence of developing autoimmune disease (176). Findings in animal model of systemic lupus erythematosus also suggest that sleep deprivation could be involved in the etiology of the disease (177). Short sleep duration of <7 h of sleep per night is also associated with transitioning to systemic lupus erythematosus (178). Evidence also suggests that the sleep disorder narcolepsy has an autoimmune origin (179). Individuals with autoimmune disease often report disturbed sleep (180–185). In a mouse model of systemic lupus erythematosus, it was reported that an increased disturbances in sleep corresponded with the progression of the disease including increased sleep fragmentation and impaired sleep-stage transitions (186). The comorbidity of autoimmune disease and sleep disorders is documented for a small number of autoimmune diseases, although this area of research is widely under investigated. Rheumatoid arthritis, ankylosing spondylitis, systemic lupus erythematosus, Sjörgen's syndrome, pemphigus, and systemic sclerosis are associated with increased occurrence of sleep apnea (174, 187, 188). Multiple sclerosis is associated with an increased risk of rapid-eye movement (REM) sleep behavior disorder (189). Increased risk of restless legs syndrome is associated with autoimmune disorders including multiple sclerosis, psoriasis, and rheumatoid arthritis (190–192). Clinical diagnosis of chronic fatigue syndrome/myalgic encephalomyelitis is also partially based upon non-restorative sleep suggesting relationships between poor sleep and fatigue (193). In addition, secondary effects of autoimmune disease or sleep disorder pathology might serve to affect sleep. For example, chronic pain is found in individuals with autoimmune disease (194). Up to 88% of individuals with chronic pain also report disturbed sleep (195). Conversely, it is reported that up to 50% of individuals with insomnia also indicate enhanced pain (195), which could contribute to fatigue.
Sleepiness, often referred to as drowsiness, can be loosely defined as the inability to remain awake or the enhanced occurrence or compulsion to sleep. Sleep disorders and disturbed sleep are associated with sleepiness (196). Sleepiness can impact vigilance, cognition, mood, and attention as well as induce fatigue (197). Many sleep-related pathologies, including insomnia and sleep apnea, are associated with impairments in vigilance, cognition, mood, motivation, and attention (198–201). Sleepiness can be induced by acutely staying awake for extended periods of time or by the fragmented sleep that occurs chronically with many health conditions including sleep apnea and autoimmune diseases (202, 203). Evidence suggests that there is a dose-dependent impact whereby greater amounts of sleep loss correlate with enhanced sleepiness and poor performance (204). Nevertheless, sleepiness varies over the time of day (205), which can impair an individual's ability to sleep or the effectiveness of naps in preventing detriments of sleepiness in functional tasks such as cognition (206). Individuals experiencing fatigue often experience waves of periods of times when their fatigue is worse (207). Furthermore, individuals with multiple sclerosis may experience increased incidence of fatigue in the morning while others do so in the evening (207). Although speculative, prior sleep or circadian factors are likely contributors to these time-of-day fluctuations.
Sleep disorders—such as sleep apnea, insomnia, and REM sleep behavior disorder—are associated with enhanced levels of inflammatory molecules (208–210). Since inflammation can induce fatigue, it is plausible that neuroinflammation occurring with disturbed sleep or sleep loss could exacerbate fatigue in individuals with autoimmune disease. In a variety of species—including rats, mice, and rabbits—pro-inflammatory cytokines can enhance non-rapid-eye movement sleep when applied to the periphery or CNS (30, 211), although IL-1β and TNF-α are the most thoroughly investigated. When IL-1β or TNF-α are applied to the CNS they also enhance electroencephalogram delta activity (~0.5–4 Hz frequency range) (30, 211), a frequency of brain electrical activity that is associated with an increased pressure to sleep after sleep loss. Additionally, conditions that induce sleepiness—including sleep loss, pathogens, and related components such as influenza and LPS—enhance IL-1β and TNF-α in the brain (30, 211). However, inflammatory cytokines, such as IL-1β do not remain in a steady state throughout the course of a day (211). Inhibition of pro-inflammatory cytokines or their receptors via pharmaceuticals, siRNA, or knockout animals can inhibit enhanced sleepiness by pro-somnogenic substances (30, 211). Collectively, these findings supporting the idea that dysregulated homeostatic cytokine expression in autoimmune disease contributes, in part, to sleep disturbances in individuals with autoimmune disorders. Moreover, it is reasonable that sleep loss, which enhances pro-inflammatory cytokines, could exacerbate sleep disturbances in autoimmune disease. Indeed, prolonged sleep deprivation using a multiple platform method in an animal model of systemic lupus erythematosus using New Zealand Black/New Zealand White F1 mice were shown to exhibit earlier onset of disease like symptoms (177).
Recently, the NLRP3 inflammasome was found to be critical to sleep and electroencephalogram delta power fluctuations throughout the day as well as responses to sleep loss and inflammatory stimuli in mice (212). Additionally, infusing a caspase-1 inhibitor ICV into rats attenuated sleep and electroencephalogram delta power responses to LPS (213), further suggesting the involvement of the NLRP3 inflammasome in sleep regulation. Moreover, evidence indicates that there is an upregulation of NLRP3 inflammasome related components with sleep deprivation and sleep fragmentation (214–216). In the cortex, there are diurnal variations in NLRP3 expression, IL-1β protein, and caspase-1 activity occurring at times of day following high neuronal activity when sleep propensity is the greatest indicating a relationship between activity and inflammation in the CNS (212).
The transcriptional factors brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) and CLOCK (CLK) form heterodimers in the cytoplasm that regulate gene expression oscillator properties of cells that regulate both the persistence and duration of circadian rhythms (217). BMAL1 and CLK recruit the co-activator cyclic AMP response element-binding protein (CREB)-binding protein and, upon phosphorylation, form a CLK/CYCLE (CYC) complex that binds to the E-box of the promoter PERIOD (PER) (218), which enhances the expression of these promoters and modulates the circadian system. The CLK gene is a major transcription factor that serves as a circadian pacemaker and is a co-activator of CREB and modulates PER protein (219). CLK also induces histone acetyl transferase activity (220), which enhances the dimerization of BMAL1 and is associated with binding to another cytokine implicated in fatigue, IL-1alpha (221, 222). Clock gene expression is enhanced in a dosage-dependent manor with increased oxygen concentrations (223). Furthermore, mice lacking NLRP3 can inhibit CLK gene in hyperoxia-induced lung inflammation (223). An animal model of autoimmune encephalomyelitis found that within the CNS, the enhanced infiltration of IL-1β secreting CD11b/Ly6Chi monocytes enhanced IL17+/IFN-γ+ T cells with the loss of myeloid BMAL1 at midday instead of midnight (224). These data suggest that IL-1β and inflammasome activation or dysregulation could alter T cell cytokine responses to further dysregulate sleep and/or fatigue.
Melatonin is secreted by the pineal gland, regulates circadian rhythms, and has anti-inflammatory and anti-oxidant properties (225). Melatonin receptors are expressed on CD4 and CD8 T-cells and B-cells. Additional evidence suggests that melatonin attenuates the expression of IL-1β, TNF-α, IL-6, and IFN-γ (225). The anti-inflammatory properties of melatonin occur, in part, through the inhibition of NF-κB (225). An association between multiple sclerosis and melatonin has been observed clinically (225). Melatonin therapy has been shown to attenuate inflammatory cytokines and related pathways in both animal models and human studies of multiple sclerosis, type 1 diabetes, inflammatory bowel disease, and systemic lupus erythematosus (225), and thus could potentially be beneficial in combating fatigue.
The hypothalamic-pituitary-adrenal (HPA) axis involves interactions between the hypothalamus and pituitary glands and is involved in hormonal responses to stress that modulate fatigue (226, 227). Individuals with autoimmune disease are reported to have increased stress levels compared to the general population. Evidence indicates that stress can modulate brain inflammation in autoimmune diseases such as multiple scleorsis (228). Sympathomedullary system activation will release norepinephrine (also referred to as noradrenaline) from nerve terminals and epinephrine (also called adrenaline) from the adrenal medulla (229). These catecholamines can increase heart rate and blood pressure.
Microglia, macrophages, and astrocytes in the central nervous system express adrenaline receptors (230). Adrenaline receptors are altered by complement-induced innate immune responses. Interestingly, several autoimmune and related disorders are associated with dysregulated complement including Sjögren's syndrome, rheumatoid arthritis, and systemic lupus erythematosus (231). Evidence indicates that activated microglia are selectively inhibited by norepinephrine (232). Furthermore, norepinephrine applied with ATP attenuates the baseline rate and increased ATP-induced process extension and migration of microglia in vitro suggesting relationships exist between the activation of purinergic and adrenergic systems (233). Beta-adrenergic receptors also exhibit an anti-inflammatory influence on astrocytes by reducing TNF-α-related genes including IL-6, CXCL2, CXCL3, VCAM1, and ICAM1 expression (234).
Within the hypothalamus, stress induces the release of corticotrophin-releasing hormone (CRH) (229). CRH induces the release of adrenocorticotropic hormone (ACTH) from the pituitary, which enhances the release of glucocorticoids from the adrenal cortex (229). Glucocorticoids have an anti-inflammatory function that occurs, in part, by inhibiting IL-1β, IL-6, and IFN-γ, as well as cyclooxygenases and prostaglandins (229, 235). However, a relationship exists between cytokines and ACTH and glucocorticoids whereby IL-1β, IL-6 and TNF-α can stimulate the pituitary adrenal axis to increase serum levels of ACTH and glucocorticoids (229, 235), which will inhibit pro-inflammatory molecules. Nevertheless, glucocorticoids can also have pro-inflammatory influence on the immune system (235). Glucocorticoids can enhance the expression of NLRP3 to enhance IL-1β responses to ATP (236). Glucocorticoids are highly expressed in the hippocampus and prefrontal cortex and thus could modulate the inflammatory response for behaviors such as cognitive fatigue (229). Stress-related molecules including glucocorticoids exhibit diurnal patterns during which they tend to be greatest during the inactive period of the day (237). Sleep loss also can enhances levels of the glucocorticoid cortisol (238). Thus, the effects of stress, sleep, and fatigue are intertwining. Chronic stress, such as those undergoing chronic health conditions, can reduce glucocorticoid sensitivity to promote inflammatory signaling (239). This can occur, in part, the suppression of cortisol signaling sensitivity, the functional heterogeneity of monocytes, or the diminished ability of monocytes to transduce cortisol signals—all of which could serve to enhance inflammatory cytokines (239).
The weak androgen dehydroepiandrosterone (DHEA) produced in the adrenal cortex. The HPA-axis, in part, controls DHEA synthesis (240). The greatest levels of DHEA demonstrate a circadian pattern similar to the pattern of ACTH secretion (240). DHEA can act as a positive allosteric modulator for the NMDA receptor and a negative allosteric modulator of the GABAA receptor (240). DHEA is found to have reduced levels in autoimmune disease including multiple sclerosis, systemic lupus erythematosus, rheumatoid arthritis, and inflammatory bowel disease (241, 242). DHEA can function to attenuate the production of pro-inflammatory cytokines such as IL-1β and TNF-α though NF-κB (242). Collectively, stress can contribute to modulating brain inflammatory mediators, which could lead to exacerbation of molecules that can induce fatigue.
Several brain areas are associated with modulating fatigue (Table 2) (243), although this area of research remains poorly investigated. These brain areas function through neurons and glia to release neurotransmitters (243). Neurotransmitters, especially monoamines, are potent at producing arousal, wakefulness, and motivation to perform activities when desired (30, 144, 244). Dopamine, norepinephrine, epinephrine, glutamate, and histamine are excitatory transmitters. Serotonin and gamma-aminobutyric acid (GABA) are inhibitory neurotransmitters that prevent neuron signals from continuing (144). There are many redundancies in the actions of these neurotransmitters, although there are also unique characteristics for receptors on the cells and brain areas that neurotransmitters act upon, as well as functional differences that exist based on the prior state of the brain cells and the concentration of the neurotransmitters. Inflammatory molecules are affected by neurotransmitters and vice versa (245), which can lead to behavior changes including those affecting fatigue.
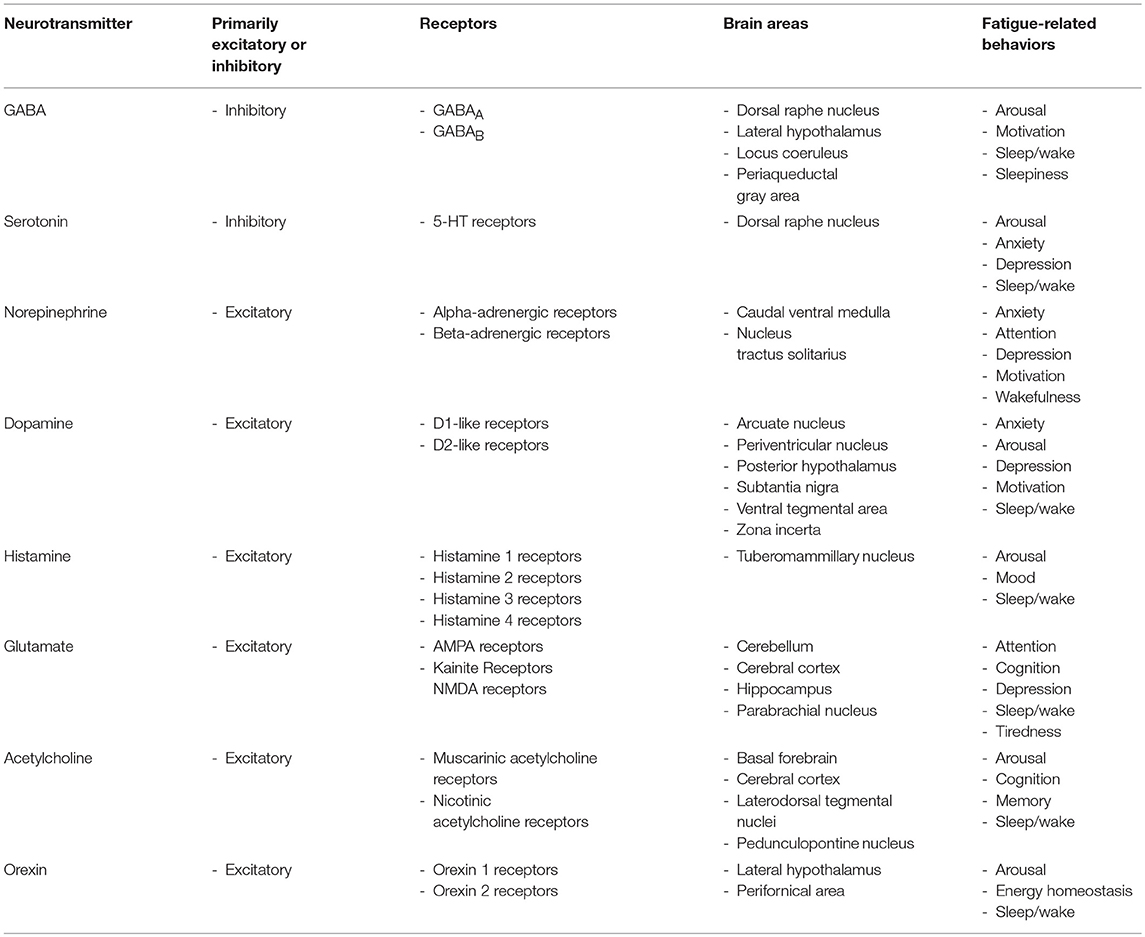
Table 2. Overview of neurotransmitters, their receptors, brain areas, and associated fatigue-related behaviors.
GABA is the major inhibitory neurotransmitter in the CNS (246). GABA ligands are GABAA receptors that are ligand-gated ion channels and GABAB, which are metabotropic receptors that are G protein-coupled receptors that open and close ion channels. GABA functions to help neurons recover after transmission. It has been shown to reduce anxiety and stress and is involved in sleep and sleepiness (246, 247). The dorsal raphe nucleus, periaqueductal gray area, locus coeruleus, and lateral hypothalamus have high concentrations of GABAergic neurons, which are activated during NREM sleep (30). GABAergic neurons in the ventrolateral preoptic nucleus area of the lateral hypothalamus are involved in modulating NREM sleep. The ventrolateral preoptic nucleus also projects to areas of the hypothalamus, including the median preoptic nucleus, which is involved in NREM sleep (248). Ventrolateral preoptic nucleus neurons are inhibited by the neurotransmitters acetylcholine, norepinephrine, serotonin, and dopamine. Moreover, the ventrolateral preoptic nucleus projects to the locus coeruleus, periaqueductal gray matter, parabrachial nucleus, histaminergic cells in the tuberomammillary nucleus, and the dorsal raphe nucleus where it can function to inhibit those arousal-producing brain areas. Evidence also indicates that IL-1β and IL-1RA, respectively, enhance and attenuate GABAergic neuron action potentials (249), which, in part, could contribute to the attenuation of arousal producing neurons resulting in fatigue and sleepiness.
Serotonin is involved with altering anxiety, depression, sleep, and fatigue (250–252). A main area of serotonin production in the CNS is the raphe nuclei, located in the brainstem (253). Serotonin acts on 5-hydroxytryptamine (5-HT) receptors located on cell membrane of nerve cells. These receptors have unique and repetitive functions including altering dopamine release into the mesocorticolimbic pathway, acetylcholine release in the prefrontal cortex, activation of GS signaling via activating adenylyl cyclase, and induction of vasoconstriction (254, 255). IL-1β is also associated with the release of serotonin, dopamine, and norepinephrine (256).
Norepinephrine is an excitatory neurotransmitter that binds to alpha- and beta-adrenergic receptors, which function as G protein-coupled receptors that act on secondary messenger systems (257). Norepinephrine is involved in anxiety and depression. However, low levels of norepinephrine are associated with fatigue and impaired motivation (258, 259). Neurons expressing norepinephrine are found in relatively small brain areas. The norepinephrine cell group A1 is located in the caudal ventrolateral part of the medulla within the brainstem (260). The norepinephrine cell group A2 is located in the brainstem area called the nucleus tractus solitarius (NTS) (260). Additionally, a major source of norepinephrine in the brain is the locus coeruleus, which is located in the pons of the brainstem (261). This area projects to all major parts of the brain including the cortex (262). Locus coeruleus activity is low during non-rapid-eye movement (NREM) sleep, with very little activity during REM sleep (262). Norepinephrine levels remain fairly constant during wakefulness but are enhanced when a stimulus is needed for reasons such as attention (262).
Dopamine is an excitatory neurotransmitter that binds to D1-like and D2-like receptors that increase or decrease intracellular amounts of cAMP by acting on adenylate cyclase and the trace amine-associated receptor 1 (263, 264). This increases intracellular amounts of cAMP and intracellular calcium levels (263). Dopamine is involved in motivation, interest, and drive (265). Low levels are found in individuals that experience difficulty completing tasks, poor concentration, no energy, and lack of motivation (266). Consequently, the dopamine imbalance hypothesis of fatigue suggests a U-shaped relationship between amounts of dopamine and levels of fatigue in which inadequate or excessive amounts of dopamine leads to fatigue (267). Main brain areas where dopaminergic neurons are found in the brain include the substantia nigra area of the basal ganglia, the ventral tegmental area (located near the midline of the midbrain of the brainstem), the posterior hypothalamus, the periventricular nucleus (located in the wall of the third ventricle within the hypothalamus), the arcuate nucleus area of the hypothalamus, the zona incerta (located below the thalamus), and the periventricular nucleus (268). The nigra-striatal pathway is heavily involved in motor functions. The ventral tegmental area dopaminergic neurons project to the prefrontal cortex in the mesocorticolimbic pathway while also projecting to the nucleus accumbens by the mesocorticolimbic pathway (268). The ventral tegmental area projects to the amygdala, cingulate gyrus (which is part of the cingulate cortex in the medial aspect of the cerebral cortex), hippocampus, and olfactory bulb (269, 270). The caudate is associated with cognitive fatigue in individuals with traumatic brain injury—a condition where inflammatory cytokines including TNF-α and IL-1β are enhanced (267, 271). Research suggests that individuals with multiple sclerosis have impaired dopamine function in the caudate (272).
Histamine is an excitatory neurotransmitter that acts on four G protein-coupled receptors (histamine 1, histamine 2, histamine 3, and histamine 4 receptors) (273). Histamine receptors function, in part, to activate phospholipase C, inhibit cAMP synthesis, and activate MAP kinases and the AKT/GSK3β pathway (274). Histamine can also affect sleep/wake, emotions, behavior, and fatigue (275, 276). Histamine is known to activate microglia leading to the production of pro-inflammatory cytokines including IL-6 and TNF-α (277). Histamine is also noted to be activated in chronic inflammatory diseases including autoimmune diseases such as multiple sclerosis (274). Histamine is involved in vasodilation and can alter blood pressure (273). Histamine also can function to release nitric oxide, which is a potent vasodilator (278). The tuberomammillary nucleus is the only known neuronal area to produce histamine (279). Histaminergic neurons project throughout the brain including the cortex and brainstem (280). However, histaminergic receptors are found on cells in other brain areas. Histamine 1 receptor is found on neurons in the tuberomammillary nucleus of the hypothalamus, which project to the dorsal raphe nucleus and locus coeruleus. The histamine 2 receptor is found in neurons in the dorsal striatum (caudate nucleus and putamen), external layers of the cerebral cortex, hippocampus, and dentate nucleus of the cerebellum. Histaminergic neurons within the tuberomammillary nucleus is involved in promoting arousal when activated (281). These neurons fire rapidly during wakefulness and stop during sleep (279). Histamine 1 receptor targeting drugs induce drowsiness, and over-the-counter anti-histamines are associated with increased sleepiness (282). Findings in experimental autoimmune encephalomyelitis (EAE), an animal model of enhanced brain inflammation, suggests that histamine acting through its histamine 1 and histamine 2 receptors can attenuate damage to the brain and inflammatory cytokines including IFN-γ (283, 284).
Glutamate is a major excitatory neurotransmitter found throughout the CNS that is produced by metabolism (285). Glutamate has affinity to bind to ionotropic and voltage-gated N-methyl-D-aspartate (NMDA) receptors that increase calcium membrane permeability; metabotropic glutamate receptors, which are G-coupled protein receptors that can activate phospholipase C or reduce intracellular levels of cAMP through adenylate cyclase; ionotropic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors; and kainate receptors that increase sodium and potassium membrane permeability (286). Low levels of glutamate can lead to tiredness and poor brain activity (287). Glutamatergic neurons located in the parabrachial nucleus located in the dorsolateral pons of the brainstem and the adjacent precoeruleus area induce arousal through their projections to areas of the ascending reticular activating system (ARAS) (30). Glutamatergic neurons are also found in synapses within additional brain areas including the hippocampus, cerebellum and cerebral cortex. Extracellular glutamate concentrations operate within a tight physiological range involving its release and uptake to protect neurons from excitotoxicity (288). Dysregulation of glutamate can lead to diminishing the sodium-dependent glial glutamate transporter and impairing the activity of volume sensitive anion channels and hemi-channels of astrocyte to alter astrocytic end-feet (289). Evidence suggests that mGluR5 receptors mediate NF-κB, which is suggested to alter chronic inflammation associated fatigue (290). Ketamine is a potent NMDA receptor antagonist, which recent evidence indicates has anti-fatigue effects (291). Studies indicate that IL-1β can modulate the glutamate receptor NMDA and vice versa. The IL-1R1 is co-localized with the NMDA receptor NR2B subunit (292). IL-1β is enhanced in microglia and astrocytes within the cortex after NMDA induced excitotoxicity. Furthermore, inhibiting NMDA receptors in rats attenuates IL-1β expression after cerebral ischemia and high potassium cortical spreading and depression (293). Collectively, these studies suggest that altered inflammatory response, neuronal activity, and cerebral blood flow to modulate glutamatergic neuronal activity and possibly fatigue.
Acetylcholine is an excitatory neurotransmitter that is synthesized by choline acetyltransferase from choline and acetyl-CoA (294). Acetylcholinesterase transforms acetylcholine into acetate and choline. Acetylcholine binds to nicotinic acetylcholine receptors and muscarinic acetylcholine receptors (mAChR). Nicotinic acetylcholine receptors are ligand-gated ion channels that modulate the state of neurons by the movement of cations to induce depolarization of the plasma membrane. This results in an excitatory postsynaptic potential or activation of voltage-gated ion channels or leads to the entry of calcium and to the activation of intracellular cascades that can alter gene expression or the release of neurotransmitters. mAChR exhibit longer lasting actions and function as G protein-coupled receptors to induce their effects by second messenger systems. Acetylcholine is involved in both CNS and peripheral function including the neuromuscular junction. Acetylcholine regulates sleep/wakefulness, arousal, cognition, memory, and fatigue (295–297). Myasthenia gravis disease is an autoimmune disease in which nicotinic acetylcholine receptors are affected and a condition that is associated with fatigue and feelings of weakness (298, 299). Acetylcholinesterase inhibitors have been used to treat symptoms in conditions including Alzheimer's disease, Parkinson's disease, and schizophrenia (300). Cholinergic neurons in the brainstem including the pedunculopontine and laterodorsal tegmental nuclei as well as the basal forebrain are involved with arousal and wakefulness (296). These brain areas project to the lateral hypothalamus, prefrontal cortex, and reticular thalamic nuclei. Acetylcholine also acts on cells within the basal ganglia, locus coeruleus, dorsal raphe nucleus, and hippocampus. Inflammatory molecules can inhibit acetylcholine and vice versa. For example, acetylcholine significantly attenuates LPS-induced TNF-α production in microglia in a dosage dependent manor (301). In human and rat monocytes, acetylcholine also is effective in suppressing other endotoxin-inducible pro-inflammatory cytokines—such as IL-1β, IL-6, and IL-18—by a post-transcriptional mechanism (302). IL-1β can also inhibit acetylcholine synthesis in the brains stem (303).
Arousal and wakefulness involve an ascending pathway that begins in brainstem monoaminergic and cholinergic neurons located between the pons and the midbrain in the mesopontine junction (30). Fatigue is associated with brain areas that regulate arousal, attention, and reaction time. These areas include the ARAS, limbic system, anterior cingulate, and basal ganglia (304). Increased self-reported fatigue is associated with poor sleep quality or sleep disturbances (305), and this type of fatigue likely involves the ARAS system that modulates arousal and wakefulness. Cholinergic neurons in the pedunculopontine and laterodorsal tegmental nuclei of the brainstem are active during wakefulness, and their output traverses the mesopontine junction to the thalamus, as well as project to the lateral hypothalamus, basal forebrain, prefrontal cortex, and reticular thalamic nuclei (144). Neurons in the brainstem project to the basal forebrain and the lateral hypothalamus affecting arousal. The basal forebrain is in particular a brain area involved in mediating arousal. Diverse populations of neurons including GABAergic, glutamatergic, and cholinergic neurons serve to activate cortical pyramidal neurons to increase cortical activation as evidenced by electroencephalogram desynchronization. Cortical GABAergic interneurons—cells that are often activated during sleep—are inhibited by neurons of the basal forebrain.
Orexin (also known as hypocretin) is a neuropeptide that binds to orexin 1 and orexin 2 receptors (306). These neurons release acetylcholine, serotonin, and norepinephrine and are sensitive to metabotropic glutamate receptors, adenosine A1 receptors, muscarinic M3 receptors, serotonin 5-HT1A receptors, neuropeptide Y receptors, cholecystokinin A receptors, and catecholamines, as well as ghrelin, leptin and glucose (307). Since orexin produces monoamines that all involved with arousal and wakefulness and links exist between energy homeostasis and orexins (144, 308), it is plausible that orexinergic cells could be involved in aspects of fatigue. Orexin neurons are located in the perifornical area and the lateral hypothalamus (307). These neurons project throughout the cortex, and to the brainstem, basal forebrain, tuberomammillary nucleus, locus coeruleus, and thalamus (309). The cerebral cortex, including the prefrontal cortex, also projects to the basal forebrain, hypothalamus, and brainstem to further modulate arousal, wakefulness, and sleep (144). In addition, evidence implicates the striatum, parietal cortex, basal ganglia, ventromedial prefrontal cortex, nucleus accumbens, and anterior cingulate cortex in cognitive fatigue with brain injury (310). Evidence indicates that IL-1β can block orexin neuron activity in the lateral hypothalamus and that inflammation-induced lethargy is mediated by the suppression of orexin neuron activity (311).
Two mechanisms whereby peripheral inflammation can enhance CNS inflammation are through leaky areas in the blood-brain-barrier and the vagus nerve (312). The vagus nerve, the tenth cranial nerve, is the longest nerve in the autonomic nervous system. This nerve has parasympathetic control of numerous organs that are involved in respiration—including the lungs, heart, and diaphragm—to mediate oxygen demand (313), which could contribute to fatigue. The vagal nerve afferents tend to relay pro-inflammatory responses cells from organs in the periphery to the CNS (Figure 2) (68). These vagal efferents tend to induce anti-inflammatory responses in peripheral tissue from CNS signals.
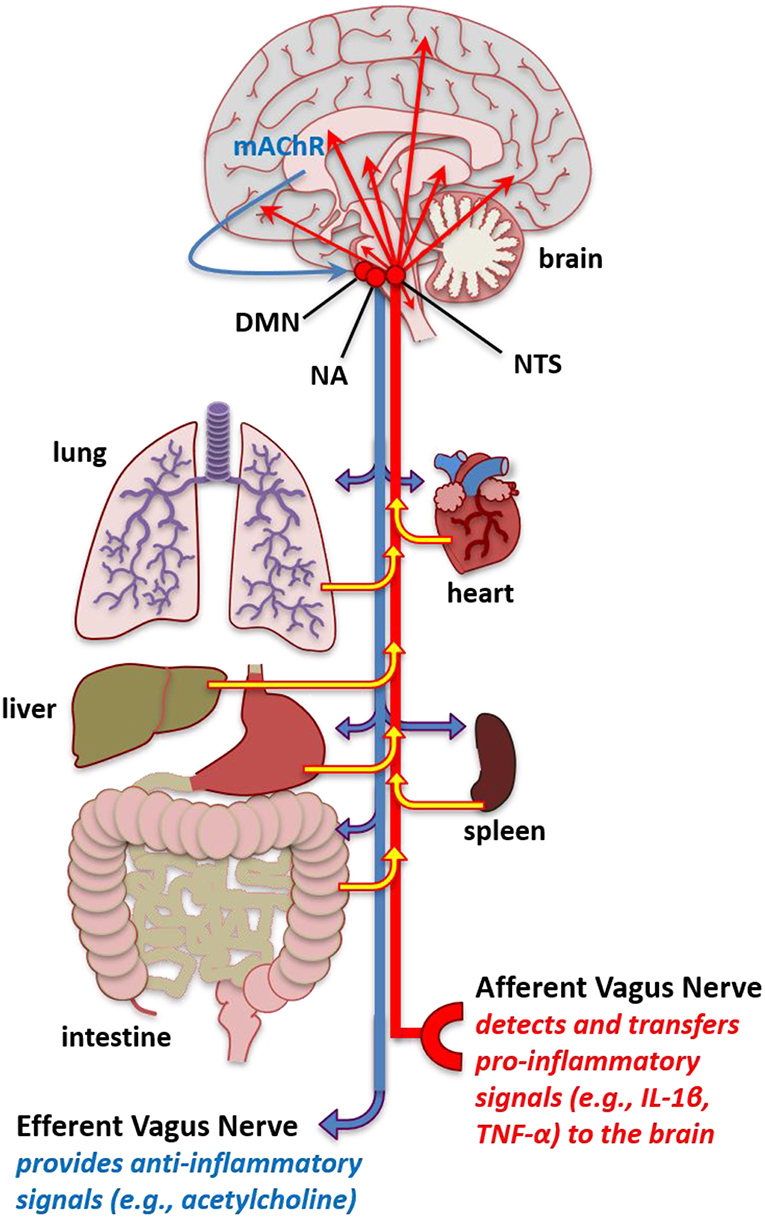
Figure 2. Increased pro-inflammatory molecules in the brain. Schematic of vagal afferent and efferent modulation of inflammation. The vagal afferents mediate pro-inflammatory signals, such as IL-1β and TNF-α from the periphery including the peritoneum and organs such as the lung intestine, hear, spleen, liver, and lung to stimulate inflammatory cytokines in the nucleus tractus solitarius (NTS). The NTS has projections to multiple brain areas where this pro-inflammatory signal that originates in the periphery leads to enhanced pro-inflammatory cytokine expression in brain areas that affect fatigue and sleep. Conversely, stimulation of the vagal efferents, such as that occurring from cholinergic mechanisms in the brain, such as muscarinic acetylcholine receptors (mAChR) acting through the, NTS, dorsal motor nucleus (DMN), and nucleus ambiguus (NA) can lead to anti-inflammatory reactions in peripheral tissues. The vagal afferents could serve to transfer enhanced inflammatory signals in the periphery occurring from autoimmune and related pathologies into dysregulated inflammatory regulation in the brain to induce fatigue-like behavior. Additionally, abnormal vagal efferent activity could server to affect physiological functions mediating fatigue-like behavior, such as heart rate, bronchoconstriction, and gluconeogenesis.
The vagal nerve afferents project to the dorsal vagal complex, which consists of the NTS, the dorsal motor nucleus (DMN) of the vagus, and the area postrema, located in the medulla area of the brainstem. However, the NTS is the primary area of vagal afferent stimulation. Neurons in the brainstem project toward many areas of the brain that can modulate fatigue including the amygdala, cortex, central nucleus of the amygdala, nucleus accumbens, paraventricular nucleus, and lateral hypothalamic areas of the hypothalamus, cerebellum, and other areas of the brainstem (314, 315). The NTS projects to areas of the brain that modulate the respiratory response (315), which could serve to alter oxygen and nutrient supply to affect fatigue. The NTS has projections to the other areas of the brain stem including the dorsal raphe nucleus and locus coeruleus. The NTS also projects to the parabrachial complex, which are nuclei located in the dorsolateral pons and surrounds the superior cerebellar peduncle. The parabrachial complex projects to areas of the brain involved in arousal including the thalamus, medial and lateral hypothalamus, and amygdala (316). Additionally, the NTS projects to the reticular formation, which are interconnected nuclei found thought the brainstem. The reticular formation has ascending pathways to the cortex in the ARAS (144).
Studies in rats and mice have demonstrated that the vagal afferent stimulation by IL-1β, TNF-α, or LPS delivered IP can enhance inflammatory IL-1β or TNF-α gene expression in the brain (69, 317, 318). Conversely, vagal efferents originating in the brainstem projecting to organs in the periphery can function to attenuate inflammation (68). Inhibiting the vagus nerve by vagotomy can demonstrate directional changes between the periphery and CNS. Inhibiting vagal afferents by a vagotomy and applying inflammatory substances to the periphery indicate that at lower dosages the vagal afferent nerves can be a major mechanism of translating peripheral inflammation to central inflammation and subsequently can affect behavior such as sleep (68, 69). Application of IL-1β, TNF-α, or LPS, which stimulates IL-1β and TNF-α production, into the peritoneum enhances sleep in mice and/or rats and is attenuated in animals that have their vagal nerves severed (69, 319, 320). Using vagotomy animal models, it has been shown that peripheral inflammation achieves this, in part, through stimulating the vagal afferents that enhance IL-1β and TNF-α in the NTS—a brain area that projects to brain areas involved in regulating sleep and are associated with modulating fatigue (69). In rats, the direct electrical stimulation of the afferent vagus nerve enhanced mRNA gene expression and protein levels in the hypothalamus and hippocampus (321). Nevertheless, the types of cytokines stimulating the vagal afferents from the periphery likely stimulate CNS cytokines over different time courses and affect different brain areas and cytokines. Thus, differences in peripheral inflammation associated with the type of autoimmune disease and progression of the disease could interact with circadian, sleep/wake states, activity-dependent, and pharmaceutical-related alterations in inflammatory processes to affect the severity or development of specific types of fatigue.
Vagotomy does not inhibit central inflammation induced by peripheral inflammation by large concentrations of inflammatory stimuli (69), suggesting that leaky areas of the blood-brain barrier also contribute to CNS inflammation. Leaky areas of the blood-brain-barrier exist in the circumventricular organs including the area postrema, and the subfornical organs, as well as the vascular organ of lamina terminalis (VOLT) (322). However, immunohistological evidence suggests that a diffusion barrier exists between the circumventricular organs and the NTS (323). VOLT, along with the subfornical organ, is interconnected with the mid-ventral hypothalamus and surrounds the third ventricle along with the median eminence. The VOLT capillaries do not have a blood-brain-barrier, and circulating factors present in the systemic circulation can enter the brain through this area (324). These areas are well-vascularized and respond to a wide variety of hormones and neurotransmitters. This area of cells heavily regulates osmoregulation, cardiovascular regulate, and energy homeostasis. Thus, leaky areas of the brainstem or the vagal afferents could be involved in peripheral inflammation enhancement in fatigue occurring in individuals with autoimmune disease that have enhanced peripheral inflammation.
The DMN and NTS are major sources of efferent motor vagal input. Longer preganglionic cholinergic neurons communicate with postganglionic neurons in closer proximity and within tissues of the viscera to induce anti-inflammatory signals (68). Acetylcholine released from neurons interacts with muscarinic acetylcholine receptors and utilizes the DMN, NTS, nucleus ambiguus (NA), and the vagal efferent nerve to alter heart rate, gluconeogenesis, and bronchial constriction (68). Consequently, these physiological effects could potentially affect fatigue-like behaviors.
Vagal efferent nerves project to the reticuloendothelial system, other peripheral organs, and the brain-derived motor output (325). Acetylcholine plays a large role in modulating the anti-inflammatory actions of the vagal efferent nerve on systemic and local peripheral inflammation (326). Muscarinic acetylcholine receptors in the CNS can induce anti-inflammatory effect on the periphery. This effect has been observed with the acetylcholinesterase inhibitor galantamine in the activation in the CNS. The nicotinic acetylcholine receptor alpha-7 (α7 nAChR) is a mechanism that signals the vagal efferent aspects of the anti-inflammatory effect. Cholinergic vagal efferent stimulation can downregulate CD14, TLR4, and NF-κB activation thus inhibiting pro-inflammatory processes within the periphery (68). This suppressor effect can be, in part, attributed to the activation of the JAK-STAT pathway. Additionally, the vagal efferents are involved in reducing hepatic glucose production and enhancement of glycogen synthesis and pancreas secretion of insulin, which could modulate metabolism and affect fatigue. Vagal nerve stimulation is used as an alternative therapy to inhibit TNF-α in patients with rheumatoid arthritis (327). Additional studies suggest that vagal nerve stimulation could benefit individuals with inflammatory bowel disease (327). The benefits of this anti-inflammatory treatment occur, in part, through the activation of cholinergic neurons to induce a suppression of peripheral inflammation, which then attenuates CNS inflammation. In fact, vagal nerve stimulation in individuals with Sjörgen's syndrome treated with a non-invasive method for 28 days were found to have reduced daytime sleepiness, improvements in fatigue, and reduced whole blood cells levels of TNF-α, IFN-γ, IL-6, and IL-1β (328).
Interestingly, evidence suggests relationships between peripheral muscle weakness and pain, inflammatory pathways, neuroinflammation, and fatigue in certain autoimmune disorders. A recent study found that serum TNF-related apoptosis-inducing ligand, which is a TNF-superfamily member, is enhanced in the serum and expressed in infiltrating inflammatory cells in patients with polymyostitis and dermatomyostitis (329)—conditions that are associated with increased fatigue and neuroinflammation. Chronic fatigue syndrome/myalgic encephalomyelitis is a condition that is defined by muscle pain and CNS inflammation, and fatigue (330) Thus, it is plausible that the vagus nerve is involved in muscle pain/CNS inflammation, and fatigue associated with these conditions. While much more research is needed to define the inflammatory pathways involved, targeting specific inflammatory pathways in the periphery could provide novel treatments to attenuate neuroinflammation and associated detriments.
Cerebral blood flow is an essential component involved in providing nutrients and oxygen supply to cells in the CNS (331). Additionally, the neurovasculature removes waste products including carbon dioxide (CO2), signaling molecules, and provides adequate supplies of energy reserves (332, 333). A reduction in blood supply within an area surrounding neurons in the CNS can lead to cellular and performance dysfunction (334). Autoimmune diseases, such as multiple sclerosis, type 1 diabetes, and Alzheimer's disease, and celiac disease are often found to be associated with hypoperfusion (335–337) Neural activity functions in synchrony with localized cerebral blood flow and is referred to as neurovascular coupling (338). This neural activity is associated with increases in the release of vasoactive substances, which can directly or indirectly alter vasohemodynamics (339). Pro-inflammatory cytokines including IL-1β and TNF-α are reported alter vasohemodynamics, although these relationships are not well-understood (340–343). In rats, IL-1β applied ICV induced global cerebral hypoperfusion (340). In rabbits, application of TNF-α into the cisterna magna induces a prolonged reduction in cerebral blood flow (343). It has been postulated under the hemo-neuro hypothesis that vasohemodynamics modulate the gain of local cortical circuits to alter the detection and discrimination of sensory stimuli, and thus, cerebral blood flow could affect neural activity (344). This process could involve alterations in diffusible or mechanical factors that lead to alterations in neuronal membranes, temperature, or indirect mechanisms such as altering astrocyte functions, which are tightly regulated with neurons. Indeed, neurovascular coupling depends not only on neurons but also numerous cells of the neurovascular unit such as neurons, astrocytes with end feet that surround the vasculature, endothelial cells, pericytes, and surrounding supporting cells including microglia and perivascular macrophages (Figure 3) (338, 345, 346). Interestingly, inhibiting perivascular macrophages and brain macrophages using liposomes with clodronate can reduce IL-1β in the brain and increase central mediated fatigue in an exhaustive exercise test in mice (347), which suggests that inflammation in the neurovascular unit is critical to CNS-mediated fatigue.
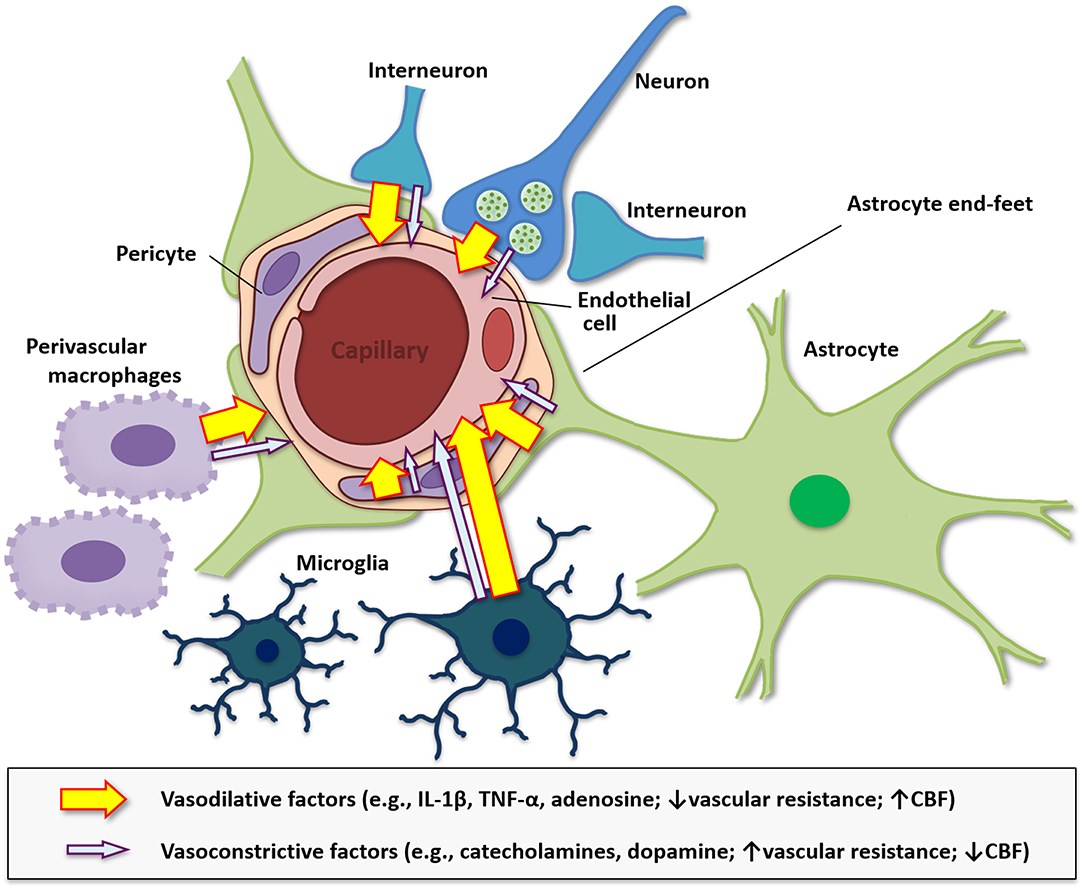
Figure 3. Diagram of the neurovascular unit in modulating vasohemodynamics. The neurovascular unit at the level of the cerebral microvasculature including the arterioles and capillaries is comprised of endothelial cells, smooth muscle, astrocytes, neurons, pericytes, and is modulated by surrounding microglia and perivascular macrophages. Additionally, alterations in metabolism and inflammation can modulate astrocyte end-feet to modulate cerebral blood flow (CBF). The neurovascular unit modulates blood flow throughout the brain and is regulated by energy needs of the surrounding cells and the vasoconstrictive, such as catecholamines and dopamine, and vasodilative factors, such as IL-1β, TNF-α, and adenosine, that are released by these cells. Pro-inflammatory molecules tend to be vasodilative, reduce vascular resistance, and increase cerebral CBF, while monoamines released by neurons have both vasodilative and vasoconstrictive properties, which can influence blood flow. Vasoconstrictive substances typically increase vascular resistance and reduce CBF.
The neurovascular unit functions to regulate cerebral blood flow, blood velocity, and blood volume in local areas of the brain (333, 338). Substances can pass through blood capillaries by osmosis, filtration, or diffusion. Oxygen and CO2 pass through the blood vessel walls by diffusion whereas fluid passes through by both hydrostatic and osmotic pressure (348). Blood travels from areas of high pressure to areas of lower pressure and flows in the direction of the lower pressure gradient (349). Blood velocity changes inversely with the cross-sectional area of the blood vessels, where vasodilation will increase blood flow and blood volume (350). Blood pressure can affect blood flow in the circulation including the cerebral vasculature (351). Blood pressure is affected by cardiac output, blood volume, peripheral resistance, and viscosity, which is dependent on the heart and the lung. Systemic blood pressure is expressed as the ratio of systolic pressure to diastolic pressure. Cerebral autoregulation is the process where cerebrovascular resistance, which is largely affected by the diameter of blood vessels, is adjusted to compensate for changes in perfusion pressure to maintain a constant blood flow. The mean arterial pressure is the average pressure of blood in the arteries driving blood into vessels and is dependent upon systolic and diastolic blood pressure values.
There is evidence that autoimmune conditions are associated with increased hypertension including systemic lupus erythematosus, psoriasis, multiple sclerosis, and rheumatoid arthritis (352, 353). Inflammation is a primary contributor to hypertension (354). Hypertension is associated with injured endothelium, which affects microvessel integrity. Astrocyte end feet interact with the endothelium to form the basal lamina matrix, which forms a blood-brain-barrier by forming tight junctions that inhibit larger molecules, including pro-inflammatory cytokines, from entering the CNS (355). Prolonged enhanced inflammation can make the vascular more rigid and less compliant (356). Vascular compliance is a necessary process that allows the vasculature to dilate or contract appropriately when required by an activity, and the lack of proper function could result in fatigue. A major component in modulating blood flow is vascular compliance, which allows increased blood volume to occur and resultant increases in blood flow. Impairments in vessel compliance are seen in inflammatory conditions such as cardiovascular disease and autoimmune diseases such as multiple sclerosis, type 1 diabetes, and systemic lupus erythematosus (357–360). Moreover, pro-inflammatory cytokines, such as TNF-α, are elevated with reductions in vascular compliance in patients with systemic lupus erthematosus (358).
Multiple autoimmune diseases, such as rheumatoid arthritis, Sjörgen's syndrome (SS), Wegener's granulomatosis, Churg-Strauss syndrome, Good pasture's syndrome, and ankylosing spondylitis are known to induce inflammation in the lung and alter lung functions (361). Macrophages in the lungs are located in the airways, alveoli, lung interstitium and can travel into the lung microvasculature (362). These cells are potent sources of cytokines, chemokines, and other pro-inflammatory molecules (363). Pulmonary hemodynamics with associated impairments in pulmonary vascular compliance and increased vascular resistance can persist following enhancements in exercise in individuals with pulmonary hypertension suggesting that lung function is important in fatigue (364).
Hyperventilation can induce fatigue and is implicated in perpetuating chronic fatigue syndrome/myalgic encephalomyelitis (365). A condition related to chronic fatigue syndrome/myalgic encephalomyelitis, postural osrthostatic tachycardia syndrome, is a conditions that induces cerebrovasoconstriction and hyperventilation affecting cerebral blood flow (366, 367). Interestingly, CNS related deficits, such as cognitive impairments are found in individuals with chronic fatigue syndrome and postural orthostatic tachycardia syndrome in support of the idea that central-mediated impairments could be, in part, induced from hyperventilation (368). Conditions of dysfunctional breathing, such as asthma and chronic obstructive pulmonary disease (369), are often associated with hyperventilation during exercise. Thus, it would be expected that individuals with dyspnea, such as those with chronic fatigue syndrome/myalgic encephalomyelitis, could have exercise-induced hyperventilation and associated fatigue.
A complex and not well-understood relationship exists between the brain vasculature, cerebrospinal fluid, and the perivascular spaces that act as lymphatics in the brain (30). In 1968, Foldi and colleagues alluded that perivascular spaces function as lymphatics in the brain (370), which has since been termed the glymphatic system (371). They glymphatic system is modulated by cerebrovascular pulsation and astrocyte water channels around the perivascular channels. The glymphatic system is altered during sleep/wake periods and is thought to clear the formation of ROS and other waste products in the brain. Persistent inflammation can alter astrocytic channels and end-feet morphology (372), which could modulate the clearance and lead to further inflammation affecting fatigue and/or sleep. Although speculative, it is plausible that an enhanced ROS content in the CNS might be unable to be cleared in autoimmune diseases and sleep related disorders leading to exacerbated inflammation and impaired vasohemodynamics leading to fatigue.
There are several areas of research and logistics that need to be established to understand the exact mechanisms of fatigue and sleep in autoimmune diseases. First, a detailed description of particular types of fatigue needs to be established in the clinic. This can be achieved, in part, with more precise medical coding. Second, there needs to be standardized questionnaires and diagnostic tests that can more precisely indicate the determents observed from fatigue. This will provide insight into the types of fatigue that are observed with the pathogenesis of the autoimmune disorders. Understanding the neurocircuitry of fatigue and its relationship between inflammation and autoimmune diseases is also needed. This area can aid in understanding the manifestation of types and severity of fatigue found in autoimmune patients. Since inflammation and metabolism are implicated in fatigue and impairments are found in these in autoimmune disorders, a detailed understating of the mechanisms of these systems, unique cells, and brain areas are lacking in autoimmune research. More information on interactions between circadian timing and sleep/wake state or sleep loss, alterations in neuronal activity, and normal daily functioning that affect fatigue is required. And there needs to be a better understanding of the relationship between the vagal afferents in modulating brain inflammation to induce fatigue is needed. More research is also necessary for understanding the relationship between vasohemodynamics and fatigue in autoimmune disease. Additionally, several autoimmune diseases are associated with disproportionately greater incidence and disease severity in particular genders and little information is known regarding the relationship of fatigue with these gender differences (373). The relationship of gender to fatigue in autoimmune disease should be a topic for future research.
An autoimmune disease involves multiple interactions between genetics and environmental factors. Most autoimmune-related disorders are more prevalent in monozygotic twins vs. dizygotic twins or siblings, indicating the involvement of genetics in disease development. Furthermore, genome wide association studies (GWAS) have found several genetic loci and small nucleotide polymorphisms that are associated with specific autoimmune disease prevalence. Interestingly, proteins encoded by genes that are involved in inflammatory mechanisms including NF-κB, apoptosis, Toll-like receptor, and immune complexes are described in autoimmune and related disorders. Nevertheless, a number of autoimmune diseases exhibit similar genetic modifications, which likely contributes to the higher incidence of multiple autoimmune diseases found in individuals with autoimmune disorders and potentially similar disease characteristics including fatigue. Indeed, larger data sets (i.e., >10,000 individuals) are finding new associations of inflammatory mechanism I with impairments in respiratory function and sleep (374). Several inflammatory gene small nucleotide polymorphisms have been implicated in fatigue including TNF-α, IL-1β, IL-6, and IFN-γ (375). Nevertheless, a high amount of the heritability of the genetics behind autoimmune and related disorders remains unexplained. In recent years, researchers, agencies, and governments, such as the British Biobank, Trans-Omics for Precision Medicine (TOPMED), and the Million Veteran Program have begun assembling very large sample populations that are giving enough statistical power to unmask genetic links between diseases.
In summary, fatigue is a major early finding in individuals with autoimmune diseases, and inflammation is a contributing factor relating to this impairment, one that affects the ability of people to perform daily activities, work, and thus their overall well-being. Recent research reveals a relationship between types of fatigue and certain brain areas, cell types, and phenotypes that mediate the symptoms observed. In addition, inflammatory molecules that are enhanced in the periphery with specific types of autoimmune disease can alter brain inflammation and neurocircuitry affecting fatigue. Consequently, immunomodulatory agents and drugs targeting inflammatory pathways could serve to treat fatigue occurring in autoimmune and related diseases. Understanding the mechanisms behind fatigue will not only aid individuals with autoimmune diseases but could also benefit transplant recipients, cancer patients, and infectious disease patients who experience debilitating fatigue.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The preparation of the report was supported by the Department of Veterans Affairs grant IBX002823 (MZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review was based on a colloquium supported by the American Autoimmune Related Diseases Association entitled Sleep, Fatigue, and Autoimmune Disease, which was organized by NR, DS, and MZ. We also thank Marissa Anderson for taking notes during the colloquium and Thea Anderson for assistance with the figures and references.
5-HT, 5-hydroxytryptamine; ACTH, adrenocorticotropic hormone; AIM2, absent in melanoma 2; AKT, protein kinase B; AMPA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AP-1, activator protein-1; ARAS, ascending reticular activating system; ASC, adapter apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; ATP, adenosine triphosphate; BMAL1, brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1; CBF, cerebral blood flow; CD39, adenosine monophosphate by ectonucleoside diphosphohydrolase; CD73, adenosine by ecto-5' nucleotidase; cGAS, cyclic guanosine monophosphate-adenosine monophosphate synthase; CLK, CLOCK; CNS, central nervous system; CO2, carbon dioxide; CREB, cyclic AMP response element-binding protein; CRH, corticotrophin-releasing hormone; CYC, CYCLE; DAMPS, danger-associated molecular patterns; DNA, single-stranded deoxyribonucleic acid; EAE, experimental autoimmune encephalomyelitis; ERK, extracellular signal regulated kinase; GABA, gamma-aminobutyric acid; GWAS, genome wide association studies; HIV-1, Human Immunodeficiency Virus-1; HPA, hypothalamic-pituitary-adrenal; ICV, intracerebroventricular; IFN-γ, interferon-gamma; IκB, inhibitory kappa B; IKK, inhibitor of kappa B kinase; IL-1, interleukin; IL-1β, Interleukin-1 beta; IL-1R1, IL-1 receptor type I; IL-1RA, IL-1 receptor antagonist; IL-1RAP, IL-1 receptor accessory protein; IP, intraperitoneal; IRAK4, interleukin-1 receptor associated kinase 4; IRF3, interferon regulatory factor 3; JAK, Janus kinase; JNK, c-Jun N-terminal nucleated kinase; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MAPKK, mitogen-activated protein kinase kinase; mTOR, mammalian target of the rapamycin; mTORC, mechanistic target of rapamycin; MYD88, myeloid differentiation primary response 88; NAC, N-acetylcysteine; NADPH, nicotinamide adenine dinucleotide phosphate; α7 nAChR, nicotinic acetylcholine receptor alpha-7; NF-κB, nuclear factor kappa B; NLRP3, nucleotide leucine-rich protein-3; NREM, non-rapid-eye movement; NTS, nucleus tractus solitarii; OXPHOS, oxidative phosphorylation; P2, purine type 2; P2X7, purine type 2 X7; PAMPS, pathogen-associated molecular patterns; PER, PERIOD; PI3-K, phosphatidylinositol 3-kinase; PPP, pentose phosphate pathway; PRR, pattern recognition receptor; REM, rapid-eye movement; RNA, ribonucleic acid; ROS, reactive oxygen species; SOCS1, suppressor of cytokine signaling 1; SS, Sjörgen's syndrome; STAT, signal transducer and activator of transcription protein; STING, stimulator of interferon genes; TGF-β, transforming growth factor-beta; TLR4, toll-like receptor 4; TNF-α, tumor necrosis factor-alpha; TOPMED, trans-Omics for Precision Medicine; TRX, Thioredoxin; TXNIP, thioredoxin-interactive protein; VOLT, vascular organ of lamina terminalis.
1. American Autoimmune Related Disease Association. Autoimmune Disease Statistics AARDA. Available online at: https://www.aarda.org/news-information/statistics/ (accessed March 17, 2019).
2. Diseases, & Conditions|NIH: National Institute of Allergy and Infectious Diseases. Available online at: https://www.niaid.nih.gov/diseases-conditions/all (accessed March 24, 2019).
3. Autoimmune Disease List • AARDA. Available online at: https://www.aarda.org/diseaselist/ (accessed March 17, 2019).
4. Bakker JP, Weaver TE, Parthasarathy S, Aloia MS. Adherence to CPAP: what should we be aiming for, and how can we get there? Chest. (2019) 155:1272–87. doi: 10.1016/j.chest.2019.01.012
5. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis. (2016) 3:151–5. doi: 10.12691/ijcd-3-4-8
6. Fatigue Survey Results Released • AARDA. Available online at: https://www.aarda.org/fatigue-survey-results-released/ (accessed March 17, 2019).
7. Chalah MA, Ayache SS. Is there a link between inflammation and fatigue in multiple sclerosis? J Inflamm Res. (2018) 11:253–64. doi: 10.2147/JIR.S167199
8. Griggs S, Morris NS. Fatigue among adults with type 1 diabetes mellitus and implications for self-management: an integrative review. Diabetes Educ. (2018) 44:325–39. doi: 10.1177/0145721718782148
9. Torres J, Mehandru S, Colombel J-F, Peyrin-Biroulet L. Crohn's disease. Lancet. (2017) 389:1741–55. doi: 10.1016/S0140-6736(16)31711-1
10. Nguyen MH, Bryant K, O'Neill SG. Vitamin D in SLE: a role in pathogenesis and fatigue? A review of the literature. Lupus. (2018) 27:2003–11. doi: 10.1177/0961203318796293
11. Hajjar J, Kutac C, Rider NL, Seeborg FO, Scalchunes C, Orange J. Fatigue and the wear-off effect in adult patients with common variable immunodeficiency. Clin Exp Immunol. (2018) 194:327–38. doi: 10.1111/cei.13210
12. Hajjar J, Guffey D, Minard CG, Orange JS. Increased incidence of fatigue in patients with primary immunodeficiency disorders: prevalence and associations within the US immunodeficiency network registry. J Clin Immunol. (2017) 37:153–65. doi: 10.1007/s10875-016-0367-1
13. Junghaenel DU, Christodoulou C, Lai J-S, Stone AA. Demographic correlates of fatigue in the US general population: results from the patient-reported outcomes measurement information system (PROMIS) initiative. J Psychosom Res. (2011) 71:117–23. doi: 10.1016/j.jpsychores.2011.04.007
14. Enns MW, Bernstein CN, Kroeker K, Graff L, Walker JR, Lix LM, et al. The association of fatigue, pain, depression and anxiety with work and activity impairment in immune mediated inflammatory diseases. PLoS ONE. (2018) 13:e0198975. doi: 10.1371/journal.pone.0198975
15. Aghaei N, Karbandi S, Gorji MAH, Golkhatmi MB, Alizadeh B. Social support in relation to fatigue symptoms among patients with multiple sclerosis. Indian J Palliat Care. (2016) 22:163–7. doi: 10.4103/0973-1075.179610
16. Flachenecker P. Autoimmune diseases and rehabilitation. Autoimmun Rev. (2012) 11:219–25. doi: 10.1016/j.autrev.2011.05.016
17. Cook KF, Molton IR, Jensen MP. Fatigue and aging with a disability. Arch Phys Med Rehabil. (2011) 92:1126–33. doi: 10.1016/j.apmr.2011.02.017
18. Agarwal N, Kumar V. Burden of lupus on work: issues in the employment of individuals with lupus. Work. (2016) 55:429–39. doi: 10.3233/WOR-162398
19. Finsterer J, Mahjoub SZ. Fatigue in healthy and diseased individuals. Am J Hosp Palliat Care. (2014) 31:562–75. doi: 10.1177/1049909113494748
20. Taylor PC, Moore A, Vasilescu R, Alvir J, Tarallo M. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol Int. (2016) 36:685–95. doi: 10.1007/s00296-015-3415-x
21. Sabes-Figuera R, McCrone P, Hurley M, King M, Donaldson AN, Ridsdale L. The hidden cost of chronic fatigue to patients and their families. BMC Health Serv Res. (2010) 10:56. doi: 10.1186/1472-6963-10-56
22. Boissoneault J, Letzen J, Robinson M, Staud R. Cerebral blood flow and heart rate variability predict fatigue severity in patients with chronic fatigue syndrome. Brain Imaging Behav. (2018) 13:789–97. doi: 10.1007/s11682-018-9897-x
23. Trinity JD, Broxterman RM, Richardson RS. Regulation of exercise blood flow: role of free radicals. Free Radic Biol Med. (2016) 98:90–102. doi: 10.1016/j.freeradbiomed.2016.01.017
24. Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. (2013) 48:190–204. doi: 10.1007/s12035-013-8425-7
25. Dantzer R, Heijnen CJ, Kavelaars A, Laye S, Capuron L. The neuroimmune basis of fatigue. Trends Neurosci. (2014) 37:39–46. doi: 10.1016/j.tins.2013.10.003
26. Popp RFJ, Fierlbeck AK, Knüttel H, König N, Rupprecht R, Weissert R, et al. Daytime sleepiness versus fatigue in patients with multiple sclerosis: a systematic review on the Epworth sleepiness scale as an assessment tool. Sleep Med Rev. (2017) 32:95–108. doi: 10.1016/j.smrv.2016.03.004
27. Nociti V, Losavio FA, Gnoni V, Losurdo A, Testani E, Vollono C, et al. Sleep and fatigue in multiple sclerosis: a questionnaire-based, cross-sectional, cohort study. J Neurol Sci. (2017) 372:387–92. doi: 10.1016/j.jns.2016.10.040
28. Moulton CD, Pavlidis P, Norton C, Norton S, Pariante C, Hayee B, et al. Depressive symptoms in inflammatory bowel disease: an extraintestinal manifestation of inflammation? Clin Exp Immunol. (2019). doi: 10.1111/cei.13276. [Epub ahead of print].
29. Katarina V, Gordana T, Svetlana MD, Milica B. Oxidative stress and neuroinflammation should be both considered in the occurrence of fatigue and depression in multiple sclerosis. Acta Neurol Belg. (2018). doi: 10.1007/s13760-018-1015-8. [Epub ahead of print].
30. Zielinski MR, McKenna JT, McCarley RW. Functions and mechanisms of sleep. AIMS Neurosci. (2016) 3:67–104. doi: 10.3934/Neuroscience.2016.1.67
31. Lacourt TE, Vichaya EG, Chiu GS, Dantzer R, Heijnen CJ. The high costs of low-grade inflammation: persistent fatigue as a consequence of reduced cellular-energy availability and non-adaptive energy expenditure. Front Behav Neurosci. (2018) 12:78. doi: 10.3389/fnbeh.2018.00078
32. Atkins C, Wilson AM. Managing fatigue in sarcoidosis - A systematic review of the evidence. Chron Respir Dis. (2017) 14:161–73. doi: 10.1177/1479972316661926
33. Duarte-Delgado NP, Vásquez G, Ortiz-Reyes BL. Blood-brain barrier disruption and neuroinflammation as pathophysiological mechanisms of the diffuse manifestations of neuropsychiatric systemic lupus erythematosus. Autoimmun Rev. (2019) 18:426–32. doi: 10.1016/j.autrev.2018.12.004
34. Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. (2019) 25:112–23. doi: 10.1016/j.molmed.2018.11.005
35. Luty J, Ruckemann-Dziurdzinska K, Witkowski JM, Bryl E. Immunological aspects of autoimmune thyroid disease - Complex interplay between cells and cytokines. Cytokine. (2019) 116:128–33. doi: 10.1016/j.cyto.2019.01.003
36. Toubi E, Vadasz Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev. (2019) 18:306–11. doi: 10.1016/j.autrev.2018.10.005
37. Salvi V, Gianello V, Tiberio L, Sozzani S, Bosisio D. Cytokine targeting by miRNAs in autoimmune diseases. Front Immunol. (2019) 10:15. doi: 10.3389/fimmu.2019.00015
38. Santos JC, Pyter LM. Neuroimmunology of behavioral comorbidities associated with cancer and cancer treatments. Front Immunol. (2018) 9:1195. doi: 10.3389/fimmu.2018.01195
39. Vinnikov D, Blanc PD, Alilin A, Zutler M, Holty J-EC. Fatigue and sleepiness determine respiratory quality of life among veterans evaluated for sleep apnea. Health Qual Life Outcomes. (2017) 15:48. doi: 10.1186/s12955-017-0624-x
40. Rosenzweig I, Williams SCR, Morrell MJ. The impact of sleep and hypoxia on the brain: potential mechanisms for the effects of obstructive sleep apnea. Curr Opin Pulm Med. (2014) 20:565–71. doi: 10.1097/MCP.0000000000000099
41. Cumming TB, Packer M, Kramer SF, English C. The prevalence of fatigue after stroke: a systematic review and meta-analysis. Int J Stroke. (2016) 11:968–77. doi: 10.1177/1747493016669861
42. Barrington J, Lemarchand E, Allan SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol. (2017) 27:205–12. doi: 10.1111/bpa.12476
43. Cronin H, O'Loughlin E. Sleep and fatigue after TBI. NeuroRehabilitation. (2018) 43:307–17. doi: 10.3233/NRE-182484
44. Mortezaee K, Khanlarkhani N, Beyer C, Zendedel A. Inflammasome: its role in traumatic brain and spinal cord injury. J Cell Physiol. (2018) 233:5160–9. doi: 10.1002/jcp.26287
45. Cincotta MC, Engelhard MM, Stankey M, Goldman MD. Fatigue and fluid hydration status in multiple sclerosis: a hypothesis. Mult Scler. (2016) 22:1438–43. doi: 10.1177/1352458516663854
46. Louati K, Berenbaum F. Fatigue in chronic inflammation - a link to pain pathways. Arthritis Res Ther. (2015) 17:254. doi: 10.1186/s13075-015-0784-1
47. Katz P. Causes and consequences of fatigue in rheumatoid arthritis. Curr Opin Rheumatol. (2017) 29:269–76. doi: 10.1097/BOR.0000000000000376
48. Theofilidis G, Bogdanis GC, Koutedakis Y, Karatzaferi C. Monitoring exercise-induced muscle fatigue and adaptations: making sense of popular or emerging indices and biomarkers. Sport. (2018) 6:153. doi: 10.3390/sports6040153
49. Antonelli A, Fallahi P, Di Bari F, Giuggioli D, Ferrari SM, Ferri C. Fatigue in patients with systemic sclerosis and hypothyroidism. A review of the literature and report of our experience. Clin Exp Rheumatol. (2019). 35(Suppl. 106):193–7.
50. Wang XS, Woodruff JF. Cancer-related and treatment-related fatigue. Gynecol Oncol. (2015) 136:446–52. doi: 10.1016/j.ygyno.2014.10.013
51. Burnley M, Jones AM. Power-duration relationship: physiology, fatigue, and the limits of human performance. Eur J Sport Sci. (2018) 18:1–12. doi: 10.1080/17461391.2016.1249524
52. Boccia G, Dardanello D, Rinaldo N, Coratella G, Schena F, Rainoldi A. Electromyographic manifestations of fatigue correlate with pulmonary function, 6-minute walk test, and time to exhaustion in COPD. Respir Care. (2015) 60:1295–302. doi: 10.4187/respcare.04138
53. Felger JC, Treadway MT. Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology. (2017) 42:216–41. doi: 10.1038/npp.2016.143
54. Pollmächer T, Schuld A, Kraus T, Haack M, Hinze-Selch D, Mullington J. Experimental immunomodulation, sleep, and sleepiness in humans. Ann NY Acad Sci. (2000) 917:488–99. doi: 10.1111/j.1749-6632.2000.tb05413.x
55. Mullington JM, Hinze-Selch D, Pollmächer T. Mediators of inflammation and their interaction with sleep: relevance for chronic fatigue syndrome and related conditions. Ann NY Acad Sci. (2001) 933:201–10. doi: 10.1111/j.1749-6632.2001.tb05825.x
56. Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, et al. Tumor necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast. (2018) 2018:8430123. doi: 10.1155/2018/8430123
57. Renna ME, O'Toole MS, Spaeth PE, Lekander M, Mennin DS. The association between anxiety, traumatic stress, and obsessive-compulsive disorders and chronic inflammation: a systematic review and meta-analysis. Depress Anxiety. (2018) 35:1081–94. doi: 10.1002/da.22790
58. Jeon SW, Kim Y-K. Inflammation-induced depression: Its pathophysiology and therapeutic implications. J Neuroimmunol. (2017) 313:92–8. doi: 10.1016/j.jneuroim.2017.10.016
59. Konsman JP. Inflammation and depression: a nervous plea for psychiatry to not become immune to interpretation. Pharmaceuticals. (2019) 12:29. doi: 10.3390/ph12010029
60. Deak T, Quinn M, Cidlowski JA, Victoria NC, Murphy AZ, Sheridan JF. Neuroimmune mechanisms of stress: sex differences, developmental plasticity, and implications for pharmacotherapy of stress-related disease. Stress. (2015) 18:367–80. doi: 10.3109/10253890.2015.1053451
61. Murphy K, Kenneth M, Weaver C. Janeway's Immunobiology. 9th Edn. New York, NY: Garland Science (2017).
62. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. (2012) 33:116–25. doi: 10.1016/j.yfrne.2011.12.002
63. Morris G, Berk M, Walder K, Maes M. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med. (2015) 13:28. doi: 10.1186/s12916-014-0259-2
64. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm. (2017) 2017:3908061. doi: 10.1155/2017/3908061
65. Taniyama K, Kuno T, Tanaka C. Distribution of beta-adrenoceptors associated with cAMP-generating system in cat colon. Am J Physiol. (1987) 253(3 Pt 1):G378–82. doi: 10.1152/ajpgi.1987.253.3.G378
66. Hilhorst M, Shirai T, Berry G, Goronzy JJ, Weyand CM. T cell-macrophage interactions and granuloma formation in vasculitis. Front Immunol. (2014) 5:432. doi: 10.3389/fimmu.2014.00432
67. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
68. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol. (2012) 8:743–54. doi: 10.1038/nrendo.2012.189
69. Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-α and lipopolysaccharide in mice. Sleep. (2013) 36:1227–38. doi: 10.5665/sleep.2892
70. Zielinski MR, Krueger JM. Sleep and innate immunity. Front Biosci. (2011) 3:632–42. doi: 10.2741/s176
71. Hodes GE, Ménard C, Russo SJ. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. (2016) 4:15–22. doi: 10.1016/j.ynstr.2016.03.003
72. Morris G, Berk M, Galecki P, Walder K, Maes M. The neuro-immune pathophysiology of central and peripheral fatigue in systemic immune-inflammatory and neuro-immune diseases. Mol Neurobiol. (2016) 53:1195–219. doi: 10.1007/s12035-015-9090-9
73. Zhao R, Zhou H, Su SB. A critical role for interleukin-1β in the progression of autoimmune diseases. Int Immunopharmacol. (2013) 17:658–69. doi: 10.1016/j.intimp.2013.08.012
74. Yadlapati S, Efthimiou P. Impact of IL-1 inhibition on fatigue associated with autoinflammatory syndromes. Mod Rheumatol. (2016) 26:3–8. doi: 10.3109/14397595.2015.1069459
75. Grossberg AJ, Vichaya EG, Christian DL, Molkentine JM, Vermeer DW, Gross PS, et al. Tumor-associated fatigue in cancer patients develops independently of IL1 signaling. Cancer Res. (2018) 78:695–705. doi: 10.1158/0008-5472.CAN-17-2168
76. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
77. Patra MC, Choi S. Recent progress in the molecular recognition and therapeutic importance of interleukin-1 receptor-associated kinase 4. Molecules. (2016) 21:1529. doi: 10.3390/molecules21111529
79. Kappelmann M, Bosserhoff A, Kuphal S. AP-1/c-Jun transcription factors: regulation and function in malignant melanoma. Eur J Cell Biol. (2014) 93:76–81. doi: 10.1016/j.ejcb.2013.10.003
80. Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev. (2018) 281:197–232. doi: 10.1111/imr.12606
81. Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity. (2009) 30:817–31. doi: 10.1016/j.immuni.2009.03.020
82. Taishi P, Davis CJ, Bayomy O, Zielinski MR, Liao F, Clinton JM, et al. Brain-specific interleukin-1 receptor accessory protein in sleep regulation. J Appl Physiol. (2012) 112:1015–22. doi: 10.1152/japplphysiol.01307.2011
83. Borragán G, Gilson M, Atas A, Slama H, Lysandropoulos A, De Schepper M, et al. Cognitive fatigue, sleep and cortical activity in multiple sclerosis disease. A behavioral, polysomnographic and functional near-infrared spectroscopy investigation. Front Hum Neurosci. (2018) 12:378. doi: 10.3389/fnhum.2018.00378
84. Huang Z-B, Sheng G-Q. Interleukin-1β with learning and memory. Neurosci Bull. (2010) 26:455–68. doi: 10.1007/s12264-010-6023-5
85. O'Léime CS, Cryan JF, Nolan YM. Nuclear deterrents: Intrinsic regulators of IL-1β-induced effects on hippocampal neurogenesis. Brain Behav Immun. (2017) 66:394–412. doi: 10.1016/j.bbi.2017.07.153
86. Ferezou I, Bolea S, Petersen CCH. Visualizing the cortical representation of whisker touch: voltage-sensitive dye imaging in freely moving mice. Neuron. (2006) 50:617–29. doi: 10.1016/j.neuron.2006.03.043
87. Shine JM, Breakspear M, Bell PT, Ehgoetz Martens KA, Shine R, Koyejo O, et al. Human cognition involves the dynamic integration of neural activity and neuromodulatory systems. Nat Neurosci. (2019) 22:289–96. doi: 10.1038/s41593-018-0312-0
88. Hallett H, Churchill L, Taishi P, De A, Krueger JM. Whisker stimulation increases expression of nerve growth factor- and interleukin-1beta-immunoreactivity in the rat somatosensory cortex. Brain Res. (2010) 1333:48–56. doi: 10.1016/j.brainres.2010.03.048
89. Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, et al. Tumor necrosis factor α: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience. (2008) 156:71–80. doi: 10.1016/j.neuroscience.2008.06.066
90. Takemiya T, Fumizawa K, Yamagata K, Iwakura Y, Kawakami M. Brain interleukin-1 facilitates learning of a water maze spatial memory task in young mice. Front Behav Neurosci. (2017) 11:202. doi: 10.3389/fnbeh.2017.00202
91. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. (2006) 1:848–58. doi: 10.1038/nprot.2006.116
92. Song C, Phillips AG, Leonard BE, Horrobin DF. Ethyl-eicosapentaenoic acid ingestion prevents corticosterone-mediated memory impairment induced by central administration of interleukin-1beta in rats. Mol Psychiatry. (2004) 9:630–8. doi: 10.1038/sj.mp.4001462
93. Matsumoto Y, Yamaguchi T, Watanabe S, Yamamoto T. Involvement of arachidonic acid cascade in working memory impairment induced by interleukin-1 beta. Neuropharmacology. (2004) 46:1195–200. doi: 10.1016/j.neuropharm.2004.02.012
94. Pugh CR, Johnson JD, Martin D, Rudy JW, Maier SF, Watkins LR. Human immunodeficiency virus-1 coat protein gp120 impairs contextual fear conditioning: a potential role in AIDS related learning and memory impairments. Brain Res. (2000) 861:8–15. doi: 10.1016/S0006-8993(99)02445-2
95. Rohleder N, Aringer M, Boentert M. Role of interleukin-6 in stress, sleep, and fatigue. Ann NY Acad Sci. (2012) 1261:88–96. doi: 10.1111/j.1749-6632.2012.06634.x
96. Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev. (2018) 98:477–504. doi: 10.1152/physrev.00039.2016
97. Terrando N, Rei Fidalgo A, Vizcaychipi M, Cibelli M, Ma D, Monaco C, et al. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. (2010) 14:R88. doi: 10.1186/cc9019
98. Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. (2002) 78:379–89. doi: 10.1006/nlme.2002.4072
99. Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. (2003) 13:826–34. doi: 10.1002/hipo.10135
100. Murray CL, Obiang P, Bannerman D, Cunningham C. Endogenous IL-1 in cognitive function and anxiety: a study in IL-1RI−/− mice. PLoS ONE. (2013) 8:e78385. doi: 10.1371/journal.pone.0078385
101. Rossi S, Sacchetti L, Napolitano F, De Chiara V, Motta C, Studer V, et al. Interleukin-1β causes anxiety by interacting with the endocannabinoid system. J Neurosci. (2012) 32:13896–905. doi: 10.1523/JNEUROSCI.1515-12.2012
102. Dunn AJ, Swiergiel AH. Effects of interleukin-1 and endotoxin in the forced swim and tail suspension tests in mice. Pharmacol Biochem Behav. (2005) 81:688–93. doi: 10.1016/j.pbb.2005.04.019
103. Park H-J, Shim H-S, An K, Starkweather A, Kim KS, Shim I. IL-4 inhibits IL-1β-induced depressive-like behavior and central neurotransmitter alterations. Mediators Inflamm. (2015) 2015:941413. doi: 10.1155/2015/941413
104. Larson SJ. Behavioral and motivational effects of immune-system activation. J Gen Psychol. (2002) 129:401–14. doi: 10.1080/00221300209602104
105. Nunes EJ, Randall PA, Estrada A, Epling B, Hart EE, Lee CA, et al. Effort-related motivational effects of the pro-inflammatory cytokine interleukin 1-beta: studies with the concurrent fixed ratio 5/chow feeding choice task. Psychopharmacology. (2014) 231:727–36. doi: 10.1007/s00213-013-3285-4
106. Yohn SE, Arif Y, Haley A, Tripodi G, Baqi Y, Müller CE, et al. Effort-related motivational effects of the pro-inflammatory cytokine interleukin-6: pharmacological and neurochemical characterization. Psychopharmacology. (2016) 233:3575–86. doi: 10.1007/s00213-016-4392-9
107. Vichaya EG, Dantzer R. Inflammation-induced motivational changes: perspective gained by evaluating positive and negative valence systems. Curr Opin Behav Sci. (2018) 22:90–5. doi: 10.1016/j.cobeha.2018.01.008
108. Vichaya EG, Hunt SC, Dantzer R. Lipopolysaccharide reduces incentive motivation while boosting preference for high reward in mice. Neuropsychopharmacology. (2014) 39:2884–90. doi: 10.1038/npp.2014.141
109. Yang Q, Liu R, Yu Q, Bi Y, Liu G. Metabolic regulation of inflammasomes in inflammation. Immunology. (2019) 157: 95–109. doi: 10.1111/imm.13056
110. Belarbi K, Cuvelier E, Destée A, Gressier B, Chartier-Harlin M-C. NADPH oxidases in Parkinson's disease: a systematic review. Mol Neurodegener. (2017) 12:84. doi: 10.1186/s13024-017-0225-5
111. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. (2015) 265:35–52. doi: 10.1111/imr.12286
112. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
113. Barker BR, Taxman DJ, Ting JP-Y. Cross-regulation between the IL-1β/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol. (2011) 23:591–7. doi: 10.1016/j.coi.2011.07.005
114. Groslambert M, Py BF. Spotlight on the NLRP3 inflammasome pathway. J Inflamm Res. (2018) 11:359–74. doi: 10.2147/JIR.S141220
115. Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. (2014) 5:352. doi: 10.3389/fphys.2014.00352
116. Zielinski MR, Gerashchenko D, Karpova SA, Konanki V, McCarley RW, Sutterwala FS, et al. The NLRP3 inflammasome modulates sleep and NREM sleep delta power induced by spontaneous wakefulness, sleep deprivation and lipopolysaccharide. Brain Behav Immun. (2017) 62:137–50. doi: 10.1016/j.bbi.2017.01.012
117. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
118. Lei Y, Chen C-J, Yan X-X, Li Z, Deng X-H. Early-life lipopolysaccharide exposure potentiates forebrain expression of NLRP3 inflammasome proteins and anxiety-like behavior in adolescent rats. Brain Res. (2017) 1671:43–54. doi: 10.1016/j.brainres.2017.06.014
119. Wu P-J, Liu H-Y, Huang T-N, Hsueh Y-P. AIM 2 inflammasomes regulate neuronal morphology and influence anxiety and memory in mice. Sci Rep. (2016) 6:32405. doi: 10.1038/srep32405
120. Wong M-L, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J, et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry. (2016) 21:797–805. doi: 10.1038/mp.2016.46
121. Yi Y-S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol Pharmacol. (2018) 22:1–15. doi: 10.4196/kjpp.2018.22.1.1
122. Opipari A, Franchi L. Role of inflammasomes in intestinal inflammation and Crohn's disease. Inflamm Bowel Dis. (2015) 21:173–81. doi: 10.1097/MIB.0000000000000230
123. Zhang Z, Ma X, Xia Z, Chen J, Liu Y, Chen Y, et al. NLRP3 inflammasome activation mediates fatigue-like behaviors in mice via neuroinflammation. Neuroscience. (2017) 358:115–23. doi: 10.1016/j.neuroscience.2017.06.048
124. McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, Peña CA, et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J Clin Invest. (2017) 127:4488–97. doi: 10.1172/JCI90699
125. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol. (2017) 8:873. doi: 10.3389/fimmu.2017.00873
126. Labzin LI, Lauterbach MAR, Latz E. Interferons and inflammasomes: cooperation and counterregulation in disease. J Allergy Clin Immunol. (2016) 138:37–46. doi: 10.1016/j.jaci.2016.05.010
127. Li X, Zhang X, Pan Y, Shi G, Ren J, Fan H, et al. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim Biophys Sin. (2018) 50:888–96. doi: 10.1093/abbs/gmy088
128. Cade BE, Chen H, Stilp AM, Louie T, Ancoli-Israel S, Arens R, et al. Associations of variants In the hexokinase 1 and interleukin 18 receptor regions with oxyhemoglobin saturation during sleep. PLoS Genet. (2019) 15:e1007739. doi: 10.1371/journal.pgen.1007739
129. Moon J-S, Hisata S, Park M-A, DeNicola GM, Ryter SW, Nakahira K, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. (2015) 12:102–15. doi: 10.1016/j.celrep.2015.05.046
130. Monteiro S, Roque S, Marques F, Correia-Neves M, Cerqueira JJ. Brain interference: revisiting the role of IFNγ in the central nervous system. Prog Neurobiol. (2017) 156:149–63. doi: 10.1016/j.pneurobio.2017.05.003
131. Masters SL, Mielke LA, Cornish AL, Sutton CE, O'Donnell J, Cengia LH, Roberts AW, et al. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. (2010) 11:640–6. doi: 10.1038/embor.2010.93
132. Griffiths HR, Gao D, Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. (2017) 12:50–7. doi: 10.1016/j.redox.2017.01.023
133. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. (2011) 32:373–9. doi: 10.1016/j.it.2011.05.004
134. Wen H, Ting JP-Y, O'Neill LAJ. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat Immunol. (2012) 13:352–7. doi: 10.1038/ni.2228
135. Petit J-M, Burlet-Godinot S, Magistretti PJ, Allaman I. Glycogen metabolism and the homeostatic regulation of sleep. Metab Brain Dis. (2015) 30:263–79. doi: 10.1007/s11011-014-9629-x
136. He F, Li J, Liu Z, Chuang C-C, Yang W, Zuo L. Redox mechanism of reactive oxygen species in exercise. Front Physiol. (2016) 7:486. doi: 10.3389/fphys.2016.00486
137. Owen L, Sunram-Lea SI. Metabolic agents that enhance ATP can improve cognitive functioning: a review of the evidence for glucose, oxygen, pyruvate, creatine, and L-carnitine. Nutrients. (2011) 3:735–55. doi: 10.3390/nu3080735
138. Kennedy C. ATP as a cotransmitter in the autonomic nervous system. Auton Neurosci. (2015) 191:2–15. doi: 10.1016/j.autneu.2015.04.004
139. Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol. (2014) 99:16–34. doi: 10.1113/expphysiol.2013.071951
140. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. (2012) 3:414. doi: 10.3389/fimmu.2012.00414
141. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
142. Zielinski MR, Taishi P, Clinton JM, Krueger JM. 5'-Ectonucleotidase-knockout mice lack non-REM sleep responses to sleep deprivation. Eur J Neurosci. (2012) 35:1789–98. doi: 10.1111/j.1460-9568.2012.08112.x
143. Urry E, Landolt H-P. Adenosine, caffeine, and performance: from cognitive neuroscience of sleep to sleep pharmacogenetics. Curr Top Behav Neurosci. (2015) 25:331–66. doi: 10.1007/7854_2014_274
144. Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. (2012) 92:1087–187. doi: 10.1152/physrev.00032.2011
145. Snel J, Lorist MM. Effects of caffeine on sleep and cognition. Prog Brain Res. (2011) 190:105–17. doi: 10.1016/B978-0-444-53817-8.00006-2
146. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
147. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights. (2016) 7:11. doi: 10.1007/s13317-016-0083-0
148. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
149. Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/cr.2010.178
150. Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. (2016) 5:e13374. doi: 10.7554/eLife.13374
151. Jha MK, Morrison BM. Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp Neurol. (2018) 309:23–31. doi: 10.1016/j.expneurol.2018.07.009
152. Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. (2015) 86:883–901. doi: 10.1016/j.neuron.2015.03.035
153. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
154. Lukyanova LD, Kirova YI. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front Neurosci. (2015) 9:320. doi: 10.3389/fnins.2015.00320
155. Gouveia A, Bajwa E, Klegeris A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim Biophys Acta Gen Subj. (2017) 1861:2274–81. doi: 10.1016/j.bbagen.2017.06.017
156. Eleftheriadis T, Pissas G, Liakopoulos V, Stefanidis I. Cytochrome C as a potentially clinical useful marker of mitochondrial and cellular damage. Front Immunol. (2016) 7:279. doi: 10.3389/fimmu.2016.00279
157. Ma MW, Wang J, Dhandapani KM, Brann DW. NADPH oxidase 2 regulates NLRP3 inflammasome activation in the brain after traumatic brain injury. Oxid Med Cell Longev. (2017) 2017:6057609. doi: 10.1155/2017/6057609
158. Dolunay A, Senol SP, Temiz-Resitoglu M, Guden DS, Sari AN, Sahan-Firat S, et al. Inhibition of NLRP3 inflammasome prevents LPS-induced inflammatory hyperalgesia in mice: contribution of NF-κB, Caspase-1/11, ASC, NOX, and NOS isoforms. Inflammation. (2017) 40:366–86. doi: 10.1007/s10753-016-0483-3
159. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
160. Munoz FM, Gao R, Tian Y, Henstenburg BA, Barrett JE, Hu H. Neuronal P2X7 receptor-induced reactive oxygen species production contributes to nociceptive behavior in mice. Sci Rep. (2017) 7:3539. doi: 10.1038/s41598-017-03813-7
161. Boyapati RK, Tamborska A, Dorward DA, Ho G-T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Research. (2017) 6:169. doi: 10.12688/f1000research.10397.1
162. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
163. Kang I, Chu CT, Kaufman BA. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. (2018) 592:793–811. doi: 10.1002/1873-3468.12989
164. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
165. Guinn Z, Lampe AT, Brown DM, Petro TM. Significant role for IRF3 in both T cell and APC effector functions during T cell responses. Cell Immunol. (2016) 310:141–9. doi: 10.1016/j.cellimm.2016.08.015
166. Mahmoudian E, Khalilnezhad A, Gharagozli K, Amani D. Thioredoxin-1, redox factor-1 and thioredoxin-interacting protein, mRNAs are differentially expressed in Multiple Sclerosis patients exposed and non-exposed to interferon and immunosuppressive treatments. Gene. (2017) 634:29–36. doi: 10.1016/j.gene.2017.08.021
167. Perez-Alvarez MJ, Villa Gonzalez M, Benito-Cuesta I, Wandosell FG. Role of mTORC1 controlling proteostasis after brain ischemia. Front Neurosci. (2018) 12:60. doi: 10.3389/fnins.2018.00060
168. Arbore G, Kemper C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur J Immunol. (2016) 46:1563–73. doi: 10.1002/eji.201546131
169. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. (2016) 12:169–82. doi: 10.1038/nrrheum.2015.172
170. Lai Z-W, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2012) 64:2937–46. doi: 10.1002/art.34502
171. Saper CB, Romanovsky AA, Scammell TE. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci. (2012) 15:1088–95. doi: 10.1038/nn.3159
172. Tsuge K, Inazumi T, Shimamoto A, Sugimoto Y. Molecular mechanisms underlying prostaglandin E2-exacerbated inflammation and immune diseases. Int Immunol. (2019) dxz021. doi: 10.1093/intimm/dxz021. [Epub ahead of print].
173. Shaver JL, Iacovides S. Sleep in women with chronic pain and autoimmune conditions: a narrative review. Sleep Med Clin. (2018) 13:375–94. doi: 10.1016/j.jsmc.2018.04.008
174. Hsiao YH, Chen YTYM, Tseng CM, Wu LA, Lin WC, Su VY, et al. Sleep disorders and increased risk of autoimmune diseases in individuals without sleep apnea. Sleep. (2015) 38:581–6. doi: 10.5665/sleep.4574
175. Mirrakhimov AE. Obstructive sleep apnea and autoimmune rheumatic disease: is there any link? Inflamm Allergy Drug Targets. (2013) 12:362–7. doi: 10.2174/18715281113129990051
176. Kok VC, Horng JT, Hung GD, Xu JL, Hung TW, Chen YC, et al. Risk of autoimmune disease in adults with chronic insomnia requiring sleep-inducing pills: a population-based longitudinal study. J Gen Intern Med. (2016) 31:1019–26. doi: 10.1007/s11606-016-3717-z
177. Palma BD, Gabriel A, Colugnati FAB, Tufik S. Effects of sleep deprivation on the development of autoimmune disease in an experimental model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. (2006) 291:R1527–32. doi: 10.1152/ajpregu.00186.2006
178. Young KA, Munroe ME, Harley JB, Guthridge JM, Kamen DL, Gilkensen GS, et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus. (2018) 27:1524–31. doi: 10.1177/0961203318778368
179. Mahoney CE, Cogswell A, Koralnik IJ, Scammell TE. The neurobiological basis of narcolepsy. Nat Rev Neurosci. (2019) 20:83–93. doi: 10.1038/s41583-018-0097-x
180. Blattner MS, de Bruin GS, Bucelli RC, Day GS. Sleep disturbances are common in patients with autoimmune encephalitis. J Neurol. (2019) 266:1007–15. doi: 10.1007/s00415-019-09230-2
181. Grabovac I, Haider S, Berner C, Lamprecht T, Fenzl KH, Erlacher L, et al. Sleep quality in patients with rheumatoid arthritis and associations with pain, disability, disease duration, and activity. J Clin Med. (2018) 7:336. doi: 10.3390/jcm7100336
182. Lamis DA, Hirsch JK, Pugh KC, Topciu R, Nsamenang SA, Goodman A, et al. Perceived cognitive deficits and depressive symptoms in patients with multiple sclerosis: perceived stress and sleep quality as mediators. Mult Scler Relat Disord. (2018) 25:150–5. doi: 10.1016/j.msard.2018.07.019
183. Benn BS, Lehman Z, Kidd SA, Miaskowski C, Sunwoo BY, Ho M, et al. Sleep disturbance and symptom burden in sarcoidosis. Respir Med. (2018) 144S:S35–40. doi: 10.1016/j.rmed.2018.03.021
184. Griggs S, Redeker NS, Grey M. Sleep characteristics in young adults with type 1 diabetes. Diabetes Res Clin Pract. (2019) 150:17–26. doi: 10.1016/j.diabres.2019.02.012
185. van Langenberg DR, Yelland GW, Robinson SR, Gibson PR. Cognitive impairment in Crohn's disease is associated with systemic inflammation, symptom burden and sleep disturbance. United Eur Gastroenterol J. (2017) 5:579–87. doi: 10.1177/2050640616663397
186. Palma BD, Tufik S. Increased disease activity is associated with altered sleep architecture in an experimental model of systemic lupus erythematosus. Sleep. (2010) 33:1244–8. doi: 10.1093/sleep/33.9.1244
187. Shen TC, Hang LW, Liang SJ, Huang CC, Lin CL, Tu CY, et al. Risk of obstructive sleep apnoea in patients with rheumatoid arthritis: a nationwide population-based retrospective cohort study. BMJ Open. (2016) 6:e013151. doi: 10.1136/bmjopen-2016-013151
188. Kang J-H, Lin H-C. Obstructive sleep apnea and the risk of autoimmune diseases: a longitudinal population-based study. Sleep Med. (2012) 13:583–8. doi: 10.1016/j.sleep.2012.03.002
189. Marrie RA, Reider N, Cohen J, Trojano M, Sorensen PS, Cutter G, et al. A systematic review of the incidence and prevalence of sleep disorders and seizure disorders in multiple sclerosis. Mult Scler. (2015) 21:342–9. doi: 10.1177/1352458514564486
190. Schell C, Schleich R, Walker F, Yazdi AS, Lerche H, Röcken M, et al. Restless legs syndrome in psoriasis: an unexpected comorbidity. Eur J Dermatol. (2015) 25:255–60. doi: 10.1684/ejd.2015.252
191. Gjevre JA, Taylor Gjevre RM. Restless legs syndrome as a comorbidity in rheumatoid arthritis. Autoimmune Dis. (2013) 2013:352782. doi: 10.1155/2013/352782
192. Ning P, Hu F, Yang B, Shen Q, Zhao Q, Huang H, et al. Systematic review and meta-analysis of observational studies to understand the prevalence of restless legs syndrome in multiple sclerosis: an update. Sleep Med. (2018) 50:97–104. doi: 10.1016/j.sleep.2018.05.039
193. Jackson ML, Bruck D. Sleep abnormalities in chronic fatigue syndrome/myalgic encephalomyelitis: a review. J Clin Sleep Med. (2012) 8:719–28. doi: 10.5664/jcsm.2276
194. Mifflin KA, Kerr BJ. Pain in autoimmune disorders. J Neurosci Res. (2017) 95:1282–94. doi: 10.1002/jnr.23844
195. McBeth J, Wilkie R, Bedson J, Chew-Graham C, Lacey RJ. Sleep disturbance and chronic widespread pain. Curr Rheumatol Rep. (2015) 17:469. doi: 10.1007/s11926-014-0469-9
196. Slater G, Steier J. Excessive daytime sleepiness in sleep disorders. J Thorac Dis. (2012) 4:608–16. doi: 10.3978/j.issn.2072-1439.2012.10.07
197. Mullins HM, Cortina JM, Drake CL, Dalal RS. Sleepiness at work: a review and framework of how the physiology of sleepiness impacts the workplace. J Appl Psychol. (2014) 99:1096–12. doi: 10.1037/a0037885
198. Cellini N. Memory consolidation in sleep disorders. Sleep Med Rev. (2017) 35:101–12. doi: 10.1016/j.smrv.2016.09.003
199. Batool-Anwar S, Kales SN, Patel SR, Varvarigou V, DeYoung PN, Malhotra A. Obstructive sleep apnea and psychomotor vigilance task performance. Nat Sci Sleep. (2014) 6:65–71. doi: 10.2147/NSS.S53721
200. Altena E, Van Der Werf YD, Strijers RLM, Van Someren EJW. Sleep loss affects vigilance: effects of chronic insomnia and sleep therapy. J Sleep Res. (2008) 17:335–43. doi: 10.1111/j.1365-2869.2008.00671.x
201. Spiegelhalder K, Regen W, Nanovska S, Baglioni C, Riemann D. Comorbid sleep disorders in neuropsychiatric disorders across the life cycle. Curr Psychiatry Rep. (2013) 15:364. doi: 10.1007/s11920-013-0364-5
202. Gaspar LS, Álvaro AR, Moita J, Cavadas C. Obstructive sleep apnea and hallmarks of aging. Trends Mol Med. (2017) 23:675–92. doi: 10.1016/j.molmed.2017.06.006
203. Ranjbaran Z, Keefer L, Stepanski E, Farhadi A, Keshavarzian A. The relevance of sleep abnormalities to chronic inflammatory conditions. Inflamm Res. (2007) 56:51–7. doi: 10.1007/s00011-006-6067-1
204. Van Dongen HPA, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. (2003) 26:117–26. doi: 10.1093/sleep/26.2.117
205. Goel N, Basner M, Rao H, Dinges DF. Circadian rhythms, sleep deprivation, and human performance. Prog Mol Biol Transl Sci. (2013) 119:155–90. doi: 10.1016/B978-0-12-396971-2.00007-5
206. Mantua J, Spencer RMC. Exploring the nap paradox: are mid-day sleep bouts a friend or foe? Sleep Med. (2017) 37:88–97. doi: 10.1016/j.sleep.2017.01.019
207. Powell DJH, Liossi C, Schlotz W, Moss-Morris R. Tracking daily fatigue fluctuations in multiple sclerosis: ecological momentary assessment provides unique insights. J Behav Med. (2017) 40:772–83. doi: 10.1007/s10865-017-9840-4
208. Unnikrishnan D, Jun J, Polotsky V. Inflammation in sleep apnea: an update. Rev Endocr Metab Disord. (2015) 16:25–34. doi: 10.1007/s11154-014-9304-x
209. Mondello S, Kobeissy F, Mechref Y, Zhao J, Talih FR, Cosentino F, et al. Novel biomarker signatures for idiopathic REM sleep behavior disorder: a proteomic and system biology approach. Neurology. (2018) 91:e1710–5. doi: 10.1212/WNL.0000000000006439
210. Pejovic S, Vgontzas AN. Neurobiological Disturbances in Insomnia: Clinical Utility of Objective Measures of Sleep. Insomnia: Diagnosis and Treatment. London: CRC Press (2016). p. 65–76.
211. Pals DT, Lawson JA, Couch SJ. Rat model for evaluating inhibitors of human renin. J Pharmacol Methods. (1990) 23:239–45. doi: 10.1016/0160-5402(90)90052-M
212. Gerashchenko D, Schmidt MA, Zielinski MR, Moore ME, Wisor JP. Sleep state dependence of optogenetically evoked responses in neuronal nitric oxide synthase-positive cells of the cerebral cortex. Neuroscience. (2018) 379:189–201. doi: 10.1016/j.neuroscience.2018.02.006
213. Imeri L, Bianchi S, Opp MR. Inhibition of caspase-1 in rat brain reduces spontaneous nonrapid eye movement sleep and nonrapid eye movement sleep enhancement induced by lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol. (2006) 291:R197–204. doi: 10.1152/ajpregu.00828.2005
214. Xia M, Li X, Yang L, Ren J, Sun G, Qi S, et al. The ameliorative effect of fluoxetine on neuroinflammation induced by sleep deprivation. J Neurochem. (2017) 146:63–75. doi: 10.1111/jnc.14272
215. Li X, Liang S, Li Z, Li S, Xia M, Verkhratsky A, et al. Leptin increases expression of 5-HT2B receptors in astrocytes thus enhancing action of fluoxetine on the depressive behavior induced by sleep deprivation. Front psychiatry. (2018) 9:734. doi: 10.3389/fpsyt.2018.00734
216. Yoon DW, Shin S, Choi J, Shin C, Lee C, Lee J, et al. Sleep fragmentation induces activation of NOD-like receptor protein-3 inflammasome in rat hippocampus. Sleep Med Res. (2017) 8:26–32. doi: 10.17241/smr.2017.00017
217. Menet JS, Pescatore S, Rosbash M. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. (2014) 28:8–13. doi: 10.1101/gad.228536.113
218. Dubowy C, Sehgal A. Circadian rhythms and sleep in Drosophila melanogaster. Genetics. (2017) 205:1373–97. doi: 10.1534/genetics.115.185157
219. Lee B, Li A, Hansen KF, Cao R, Yoon JH, Obrietan K. CREB influences timing and entrainment of the SCN circadian clock. J Biol Rhythms. (2010) 25:410–20. doi: 10.1177/0748730410381229
220. Hardin PE, Yu W. Circadian transcription: passing the HAT to CLOCK. Cell. (2006) 125:424–6. doi: 10.1016/j.cell.2006.04.010
221. Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun. (2018) 9:1156. doi: 10.1038/s41467-018-03362-1
222. Roerink ME, van der Schaaf ME, Dinarello CA, Knoop H, van der Meer JWM. Interleukin-1 as a mediator of fatigue in disease: a narrative review. J Neuroinflammation. (2017) 14:16. doi: 10.1186/s12974-017-0796-7
223. Lagishetty V, Parthasarathy PT, Phillips O, Fukumoto J, Cho Y, Fukumoto I, et al. Dysregulation of CLOCK gene expression in hyperoxia-induced lung injury. Am J Physiol Cell Physiol. (2014) 306:C999–1007. doi: 10.1152/ajpcell.00064.2013
224. Sutton CE, Finlay CM, Raverdeau M, Early JO, DeCourcey J, Zaslona Z, et al. Loss of the molecular clock in myeloid cells exacerbates T cell-mediated CNS autoimmune disease. Nat Commun. (2017) 8:1923. doi: 10.1038/s41467-017-02111-0
225. Lin G-J, Huang S-H, Chen S-J, Wang C-H, Chang D-M, Sytwu H-K. Modulation by melatonin of the pathogenesis of inflammatory autoimmune diseases. Int J Mol Sci. (2013) 14:11742–66. doi: 10.3390/ijms140611742
226. Matura LA, Malone S, Jaime-Lara R, Riegel B. A systematic review of biological mechanisms of fatigue in chronic illness. Biol Res Nurs. (2018) 20:410–21. doi: 10.1177/1099800418764326
227. Silverman MN, Heim CM, Nater UM, Marques AH, Sternberg EM. Neuroendocrine and immune contributors to fatigue. PM R. (2010) 2:338–46. doi: 10.1016/j.pmrj.2010.04.008
228. Karagkouni A, Alevizos M, Theoharides TC. Effect of stress on brain inflammation and multiple sclerosis. Autoimmun Rev. (2013) 12:947–53. doi: 10.1016/j.autrev.2013.02.006
229. Bellavance M-A, Rivest S. The HPA - immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front Immunol. (2014) 5:136. doi: 10.3389/fimmu.2014.00136
230. Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. (2015) 6:171. doi: 10.3389/fphar.2015.00171
231. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res. (2013) 56:477–91. doi: 10.1007/s12026-013-8422-y
232. Barnes MA, Carson MJ, Nair MG. Non-traditional cytokines: how catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine. (2015) 72:210–9. doi: 10.1016/j.cyto.2015.01.008
233. Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. (2013) 288:15291–302. doi: 10.1074/jbc.M113.458901
234. Laureys G, Gerlo S, Spooren A, Demol F, De Keyser J, Aerts JL. β-adrenergic agonists modulate TNF-α induced astrocytic inflammatory gene expression and brain inflammatory cell populations. J Neuroinflammation. (2014) 11:21. doi: 10.1186/1742-2094-11-21
235. Liu Y-Z, Wang Y-X, Jiang C-L. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. (2017) 11:316. doi: 10.3389/fnhum.2017.00316
236. Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. (2011) 286:38703–13. doi: 10.1074/jbc.M111.275370
237. Dickmeis T. Glucocorticoids and the circadian clock. J Endocrinol. (2009) 200:3–22. doi: 10.1677/JOE-08-0415
238. Leproult R, Copinschi G, Buxton O, Van Cauter E. Sleep loss results in an elevation of cortisol levels the next evening. Sleep. (1997) 20:865–70.
239. Miller GE, Murphy MLM, Cashman R, Ma R, Ma J, Arevalo JM, et al. Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun. (2014) 41:191–9. doi: 10.1016/j.bbi.2014.05.016
240. Prough RA, Clark BJ, Klinge CM. Novel mechanisms for DHEA action. J Mol Endocrinol. (2016) 56:R139–55. doi: 10.1530/JME-16-0013
241. Téllez N, Comabella M, Julià E, Río J, Tintoré M, Brieva L, et al. Fatigue in progressive multiple sclerosis is associated with low levels of dehydroepiandrosterone. Mult Scler. (2006) 12:487–94. doi: 10.1191/135248505ms1322oa
242. Sawalha AH, Kovats S. Dehydroepiandrosterone in systemic lupus erythematosus. Curr Rheumatol Rep. (2008) 10:286–91. doi: 10.1007/s11926-008-0046-1
243. Harrington ME. Neurobiological studies of fatigue. Prog Neurobiol. (2012) 99:93–105. doi: 10.1016/j.pneurobio.2012.07.004
244. Galtress T, Marshall AT, Kirkpatrick K. Motivation and timing: clues for modeling the reward system. Behav Process. (2012) 90:142–53. doi: 10.1016/j.beproc.2012.02.014
245. Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. (2013) 30:297–306. doi: 10.1002/da.22084
246. Gottesmann C. GABA mechanisms and sleep. Neuroscience. (2002) 111:231–9. doi: 10.1016/S0306-4522(02)00034-9
247. Goddard AW. Cortical and subcortical gamma amino acid butyric acid deficits in anxiety and stress disorders: clinical implications. World J Psychiatry. (2016) 6:43–53. doi: 10.5498/wjp.v6.i1.43
248. Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann NY Acad Sci. (2008) 1129:275–86. doi: 10.1196/annals.1417.027
249. Patel RR, Khom S, Steinman MQ, Varodayan FP, Kiosses WB, Hedges DM, et al. IL-1β expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain Behav Immun. (2019) 75:208–19. doi: 10.1016/j.bbi.2018.10.009
250. Cordeiro LMS, Rabelo PCR, Moraes MM, et al. Physical exercise-induced fatigue: the role of serotonergic and dopaminergic systems. Braz J Med Biol Res. (2017) 50:e6432. doi: 10.1590/1414-431x20176432
251. Liu Y, Zhao J, Guo W. Emotional roles of mono-aminergic neurotransmitters in major depressive disorder and anxiety disorders. Front Psychol. (2018) 9:2201. doi: 10.3389/fpsyg.2018.02201
252. Holst SC, Landolt H-P. Sleep-wake neurochemistry. Sleep Med Clin. (2018) 13:137–46. doi: 10.1016/j.jsmc.2018.03.002
253. Quentin E, Belmer A, Maroteaux L. Somato-dendritic regulation of raphe serotonin neurons; a key to antidepressant action. Front Neurosci. (2018) 12:982. doi: 10.3389/fnins.2018.00982
254. Nero D, Bowditch N, Pickert S, MacIntyre RJ. A genetic and molecular analysis of P-induced mutations at the glucose-6-phosphate dehydrogenase locus in Drosophila melanogaster. Mol Gen Genet. (1989) 219:429–38. doi: 10.1007/BF00259616
255. Watts SW, Morrison SF, Davis RP, Barman SM. Serotonin and blood pressure regulation. Pharmacol Rev. (2012) 64:359–88. doi: 10.1124/pr.111.004697
256. Shintani F, Kanba S, Nakaki T, Nibuya M, Kinoshita N, Suzuki E, et al. Interleukin-1 beta augments release of norepinephrine, dopamine, and serotonin in the rat anterior hypothalamus. J Neurosci. (1993) 13:3574–81. doi: 10.1523/JNEUROSCI.13-08-03574.1993
257. Cotecchia S, Stanasila L, Diviani D. Protein-protein interactions at the adrenergic receptors. Curr Drug Targets. (2012) 13:15–27. doi: 10.2174/138945012798868489
258. McMorris T, Barwood M, Corbett J. Central fatigue theory and endurance exercise: Toward an interoceptive model. Neurosci Biobehav Rev. (2018) 93:93–107. doi: 10.1016/j.neubiorev.2018.03.024
259. Vitrac C, Benoit-Marand M. Monoaminergic modulation of motor cortex function. Front Neural Circuits. (2017) 11:72. doi: 10.3389/fncir.2017.00072
260. Dahlstroem A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand Suppl. (1964) 232:1–55.
261. Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. (1981) 1:876–86. doi: 10.1523/JNEUROSCI.01-08-00876.1981
262. Rho H-J, Kim J-H, Lee S-H. Function of selective neuromodulatory projections in the mammalian cerebral cortex: comparison between cholinergic and noradrenergic systems. Front Neural Circuits. (2018) 12:47. doi: 10.3389/fncir.2018.00047
263. Hasbi A, O'Dowd BF, George SR. Dopamine D1-D2 receptor heteromer signaling pathway in the brain: emerging physiological relevance. Mol Brain. (2011) 4:26. doi: 10.1186/1756-6606-4-26
264. Miller GM. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J Neurochem. (2011) 116:164–76. doi: 10.1111/j.1471-4159.2010.07109.x
265. Liu C, Kaeser PS. Mechanisms and regulation of dopamine release. Curr Opin Neurobiol. (2019) 57:46–53. doi: 10.1016/j.conb.2019.01.001
266. Sadeghniiat-Haghighi K, Yazdi Z. Fatigue management in the workplace. Ind Psychiatry J. (2015) 24:12–7. doi: 10.4103/0972-6748.160915
267. Wylie GR, Dobryakova E, DeLuca J, Chiaravalloti N, Essad K, Genova H. Cognitive fatigue in individuals with traumatic brain injury is associated with caudate activation. Sci Rep. (2017) 7:8973. doi: 10.1038/s41598-017-08846-6
268. Taber KH, Black DN, Porrino LJ, Hurley RA. Neuroanatomy of dopamine: reward and addiction. J Neuropsychiatry Clin Neurosci. (2012) 24:1–4. doi: 10.1176/appi.neuropsych.24.1.1
269. Ranaldi R. Dopamine and reward seeking: the role of ventral tegmental area. Rev Neurosci. (2014) 25:621–30. doi: 10.1515/revneuro-2014-0019
270. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. (2013) 14:609–25. doi: 10.1038/nrn3381
271. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. (2010) 7:22–30. doi: 10.1016/j.nurt.2009.10.016
272. Dobryakova E, Genova HM, DeLuca J, Wylie GR. The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front Neurol. (2015) 6:52. doi: 10.3389/fneur.2015.00052
273. Parsons ME, Ganellin CR. Histamine and its receptors. Br J Pharmacol. (2006) 147Suppl(S1):S127–35. doi: 10.1038/sj.bjp.0706440
274. Passani MB, Ballerini C. Histamine and neuroinflammation: insights from murine experimental autoimmune encephalomyelitis. Front Syst Neurosci. (2012) 6:32. doi: 10.3389/fnsys.2012.00032
275. Shan L, Dauvilliers Y, Siegel JM. Interactions of the histamine and hypocretin systems in CNS disorders. Nat Rev Neurol. (2015) 11:401–13. doi: 10.1038/nrneurol.2015.99
276. Loy BD, O'Connor PJ. The effect of histamine on changes in mental energy and fatigue after a single bout of exercise. Physiol Behav. (2016) 153:7–18. doi: 10.1016/j.physbeh.2015.10.016
277. Branco ACCC, Yoshikawa FSY, Pietrobon AJ, Sato MN. Role of histamine in modulating the immune response and inflammation. Mediators Inflamm. (2018) 2018:9524075. doi: 10.1155/2018/9524075
278. Akerman S, Williamson DJ, Kaube H, Goadsby PJ. The role of histamine in dural vessel dilation. Brain Res. (2002) 956:96–102. doi: 10.1016/S0006-8993(02)03485-6
279. Scammell TE, Arrigoni E, Lipton JO. Neural circuitry of wakefulness and sleep. Neuron. (2017) 93:747–65. doi: 10.1016/j.neuron.2017.01.014
280. Panula P, Chazot PL, Cowart M, Gutzmer R, Leurs R, Liu WL, et al. International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol Rev. (2015) 67:601–55. doi: 10.1124/pr.114.010249
281. Thakkar MM. Histamine in the regulation of wakefulness. Sleep Med Rev. (2011) 15:65–74. doi: 10.1016/j.smrv.2010.06.004
282. Church MK, Church DS. Pharmacology of antihistamines. Indian J Dermatol. (2013) 58:219–24. doi: 10.4103/0019-5154.110832
283. Musio S, Gallo B, Scabeni S, Lapilla M, Poliani PL, Matarese G, et al. A key regulatory role for histamine in experimental autoimmune encephalomyelitis: disease exacerbation in histidine decarboxylase-deficient mice. J Immunol. (2006) 176:17–26. doi: 10.4049/jimmunol.176.1.17
284. Saligrama N, Noubade R, Case LK, del Rio R, Teuscher C. Combinatorial roles for histamine H1-H2 and H3-H4 receptors in autoimmune inflammatory disease of the central nervous system. Eur J Immunol. (2012) 42:1536–46. doi: 10.1002/eji.201141859
285. Walker MC, van der Donk WA. The many roles of glutamate in metabolism. J Ind Microbiol Biotechnol. (2016) 43:419–30. doi: 10.1007/s10295-015-1665-y
286. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. (2010) 460:525–42. doi: 10.1007/s00424-010-0809-1
287. Rönnbäck L, Hansson E. On the potential role of glutamate transport in mental fatigue. J Neuroinflammation. (2004) 1:22. doi: 10.1186/1742-2094-1-22
288. Moussawi K, Riegel A, Nair S, Kalivas PW. Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci. (2011) 5:94. doi: 10.3389/fnsys.2011.00094
289. Langer J, Gerkau NJ, Derouiche A, Kleinhans C, Moshrefi-Ravasdjani B, Fredrich M, et al. Rapid sodium signaling couples glutamate uptake to breakdown of ATP in perivascular astrocyte endfeet. Glia. (2017) 65:293–308. doi: 10.1002/glia.23092
290. Feng LR, Fernández-Martínez JL, Zaal KJM, deAndrés-Galiana EJ, Wolff BS, Saligan LN. mGluR5 mediates post-radiotherapy fatigue development in cancer patients. Transl Psychiatry. (2018) 8:110. doi: 10.1038/s41398-018-0161-3
291. Saligan LN, Farmer C, Ballard ED, Kadriu B, Zarate CA. Disentangling the association of depression on the anti-fatigue effects of ketamine. J Affect Disord. (2019) 244:42–5. doi: 10.1016/j.jad.2018.10.089
292. Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J Neuroinflammation. (2011) 8:14. doi: 10.1186/1742-2094-8-14
293. Pearson VL, Rothwell NJ, Toulmond S. Excitotoxic brain damage in the rat induces interleukin-1beta protein in microglia and astrocytes: correlation with the progression of cell death. Glia. (1999) 25:311–23. doi: 10.1002/(SICI)1098-1136(19990215)25:4<311::AID-GLIA1>3.3.CO;2-5
294. Szutowicz A, Bielarczyk H, Jankowska-Kulawy A, Pawełczyk T, Ronowska A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem Res. (2013) 38:1523–42. doi: 10.1007/s11064-013-1060-x
295. Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. (2012) 76:116–29. doi: 10.1016/j.neuron.2012.08.036
296. Watson CJ, Baghdoyan HA, Lydic R. Neuropharmacology of sleep and wakefulness. Sleep Med Clin. (2010) 5:513–28. doi: 10.1016/j.jsmc.2010.08.003
297. Meeusen R, Roelands B. Central fatigue and neurotransmitters, can thermoregulation be manipulated? Scand J Med Sci Sports. (2010) 20(Suppl. 3):19–28. doi: 10.1111/j.1600-0838.2010.01205.x
298. Jordan B, Mehl T, Schweden TLK, Menge U, Zierz S. Assessment of physical fatigability and fatigue perception in myasthenia gravis. Muscle Nerve. (2017) 55:657–63. doi: 10.1002/mus.25386
299. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. (2015) 14:1023–36. doi: 10.1016/S1474-4422(15)00145-3
300. Hsu W-Y, Lane H-Y, Lin C-H. Medications used for cognitive enhancement in patients with schizophrenia, bipolar disorder, Alzheimer's disease, and Parkinson's disease. Front Psychiatry. (2018) 9:91. doi: 10.3389/fpsyt.2018.00091
301. Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. (2004) 89:337–43. doi: 10.1046/j.1471-4159.2004.02347.x
302. Hecker A, Küllmar M, Wilker S, Richter K, Zakrzewicz A, Atanasova S, et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1β release. J Immunol. (2015) 195:2325–34. doi: 10.4049/jimmunol.1400974
303. Brambilla D, Barajon I, Bianchi S, Opp MR, Imeri L. Interleukin-1 inhibits putative cholinergic neurons in vitro and REM sleep when microinjected into the rat laterodorsal tegmental nucleus. Sleep. (2010) 33:919–29. doi: 10.1093/sleep/33.7.919
304. Chaudhuri A, Behan PO. Fatigue in neurological disorders. Lancet. (2004) 363:978–88. doi: 10.1016/S0140-6736(04)15794-2
305. Christie AD, Seery E, Kent JA. Physical activity, sleep quality, and self-reported fatigue across the adult lifespan. Exp Gerontol. (2016) 77:7–11. doi: 10.1016/j.exger.2016.02.001
306. Sakurai T. The role of orexin in motivated behaviours. Nat Rev Neurosci. (2014) 15:719–31. doi: 10.1038/nrn3837
307. Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. (2009) 61:162–76. doi: 10.1124/pr.109.001321
308. Sakurai T. Orexin: a link between energy homeostasis and adaptive behaviour. Curr Opin Clin Nutr Metab Care. (2003) 6:353–60. doi: 10.1097/00075197-200307000-00001
309. Ohno K, Sakurai T. Orexin neuronal circuitry: role in the regulation of sleep and wakefulness. Front Neuroendocrinol. (2008) 29:70–87. doi: 10.1016/j.yfrne.2007.08.001
310. Ryan NP, Beauchamp MH, Beare R, Coleman L, Ditchfield M, Kean M, et al. Uncovering cortico-striatal correlates of cognitive fatigue in pediatric acquired brain disorder: evidence from traumatic brain injury. Cortex. (2016) 83:222–30. doi: 10.1016/j.cortex.2016.07.020
311. Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG Jr, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci. (2011) 31:11376–86. doi: 10.1523/JNEUROSCI.2311-11.2011
312. Erickson MA, Banks WA. Neuroimmune axes of the blood-brain barriers and blood-brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. (2018) 70:278–314. doi: 10.1124/pr.117.014647
313. Bordoni B, Purgol S, Bizzarri A, Modica M, Morabito B. The influence of breathing on the central nervous system. Cureus. (2018) 10:e2724. doi: 10.7759/cureus.2724
314. García-Medina NE, Miranda MI. Nucleus of the solitary tract chemical stimulation induces extracellular norepinephrine release in the lateral and basolateral amygdala. Brain Stimul. (2013) 6:198–201. doi: 10.1016/j.brs.2012.03.020
315. Zeinvand-Lorestani M, Kalantari H, Khodayar MJ, Teimoori A, Saki N, Ahangarpour A, et al. Autophagy upregulation as a possible mechanism of arsenic induced diabetes. Sci Rep. (2018) 8:11960. doi: 10.1038/s41598-018-30439-0
316. Luo T, Yu S, Cai S, Zhang Y, Jiao Y, Yu T, et al. Parabrachial neurons promote behavior and electroencephalographic arousal from general anesthesia. Front Mol Neurosci. (2018) 11:420. doi: 10.3389/fnmol.2018.00420
317. Layé S, Bluthé RM, Kent S, Combe C, Médina C, Parnet P, et al. Subdiaphragmatic vagotomy blocks induction of IL-1 beta mRNA in mice brain in response to peripheral LPS. Am J Physiol. (1995) 268(5 Pt 2):R1327–31. doi: 10.1152/ajpregu.1995.268.5.R1327
318. Hansen MK, Taishi P, Chen Z, Krueger JM. Vagotomy blocks the induction of interleukin-1beta (IL-1beta) mRNA in the brain of rats in response to systemic IL-1beta. J Neurosci. (1998) 18:2247–53. doi: 10.1523/JNEUROSCI.18-06-02247.1998
319. Kubota T, Fang J, Guan Z, Brown RA, Krueger JM. Vagotomy attenuates tumor necrosis factor-alpha-induced sleep and EEG delta-activity in rats. Am J Physiol Regul Integr Comp Physiol. (2001) 280:R1213–20. doi: 10.1152/ajpregu.2001.280.4.R1213
320. Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol. (1997) 273(4 Pt 2):R1246–53. doi: 10.1152/ajpregu.1997.273.4.R1246
321. Hosoi T, Okuma Y, Nomura Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am J Physiol Regul Integr Comp Physiol. (2000) 279:R141–7. doi: 10.1152/ajpregu.2000.279.1.R141
322. Kaur C, Ling EA. The circumventricular organs. Histol Histopathol. (2017) 32:879–92. doi: 10.14670/HH-11-881
323. Wang QP, Guan JL, Pan W, Kastin AJ, Shioda S. A diffusion barrier between the area postrema and nucleus tractus solitarius. Neurochem Res. (2008) 33:2035–43. doi: 10.1007/s11064-008-9676-y
324. Miyata S. New aspects in fenestrated capillary and tissue dynamics in the sensory circumventricular organs of adult brains. Front Neurosci. (2015) 9:390. doi: 10.3389/fnins.2015.00390
325. Chavan SS, Tracey KJ. Essential Neuroscience in Immunology. J Immunol. (2017) 198:3389–97. doi: 10.4049/jimmunol.1601613
326. Corsi-Zuelli FM das G, Brognara F, Quirino GFDS, Hiroki CH, Fais RS, Del-Ben CM, et al. Neuroimmune interactions in schizophrenia: focus on vagus nerve stimulation and activation of the alpha-7 nicotinic acetylcholine receptor. Front Immunol. (2017) 8:618. doi: 10.3389/fimmu.2017.00618
327. Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Natl Acad Sci USA. (2016) 113:8284–9. doi: 10.1073/pnas.1605635113
328. Tarn J, Legg S, Mitchell S, Simon B, Ng WF. The effects of noninvasive vagus nerve stimulation on fatigue and immune responses in patients with primary sjögren's syndrome. Neuromodulation. (2019) 22:580–5. doi: 10.1111/ner.12879
329. Zhou H, Wang Y, Bi K, Qi H, Song S, Zhou M, et al. Serum-soluble TRAIL: a potential biomarker for disease activity in myositis patients. Clin Rheumatol. (2019) 38:1425–31. doi: 10.1007/s10067-018-04418-9
330. Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an 11C-(R)-PK11195 PET study. J Nucl Med. (2014) 55:945–50. doi: 10.2967/jnumed.113.131045
331. Joris PJ, Mensink RP, Adam TC, Liu TT. Cerebral blood flow measurements in adults: a review on the effects of dietary factors and exercise. Nutrients. (2018) 10:530. doi: 10.3390/nu10050530
332. Sidorenko I, Turova V, Botkin N, Eckardt L, Alves-Pinto A, Felderhoff-Müser U, et al. Modeling cerebral blood flow dependence on carbon dioxide and mean arterial blood pressure in the immature brain with accounting for the germinal matrix. Front Neurol. (2018) 9:812. doi: 10.3389/fneur.2018.00812
333. Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. (2017) 18:419–34. doi: 10.1038/nrn.2017.48
334. Bordeleau M, ElAli A, Rivest S. Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice. Oncotarget. (2016) 7:11864–80. doi: 10.18632/oncotarget.7689
335. D'haeseleer M, Hostenbach S, Peeters I, Sankari SE, Nagels G, De Keyser J, et al. Cerebral hypoperfusion: a new pathophysiologic concept in multiple sclerosis? J Cereb Blood Flow Metab. (2015) 35:1406–10. doi: 10.1038/jcbfm.2015.131
336. Keymeulen B, Jacobs A, de Metz K, de Sadeleer C, Bossuyt A, Somers G. Regional cerebral hypoperfusion in long-term type 1 (insulin-dependent) diabetic patients: relation to hypoglycaemic events. Nucl Med Commun. (1995) 16:10–6. doi: 10.1097/00006231-199501000-00004
337. Love S, Miners JS. Cerebral hypoperfusion and the energy deficit in Alzheimer's disease. Brain Pathol. (2016) 26:607–17. doi: 10.1111/bpa.12401
338. Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. (2017) 96:17–42. doi: 10.1016/j.neuron.2017.07.030
339. Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. (2014) 15:43–53. doi: 10.1038/nrn3617
340. Maher CO, Anderson RE, Martin HS, McClelland RL, Meyer FB. Interleukin-1beta and adverse effects on cerebral blood flow during long-term global hypoperfusion. J Neurosurg. (2003) 99:907–12. doi: 10.3171/jns.2003.99.5.0907
341. Farkas E, Süle Z, Tóth-Szuki V, Mátyás A, Antal P, Farkas IG, et al. Tumor necrosis factor-alpha increases cerebral blood flow and ultrastructural capillary damage through the release of nitric oxide in the rat brain. Microvasc Res. (2006) 72:113–9. doi: 10.1016/j.mvr.2006.05.007
342. Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-alpha reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain. (2002) 125(Pt 11):2446–59. doi: 10.1093/brain/awf256
343. Tureen J. Effect of recombinant human tumor necrosis factor-alpha on cerebral oxygen uptake, cerebrospinal fluid lactate, and cerebral blood flow in the rabbit: role of nitric oxide. J Clin Invest. (1995) 95:1086–91. doi: 10.1172/JCI117755
344. Moore CI, Cao R. The hemo-neural hypothesis: on the role of blood flow in information processing. J Neurophysiol. (2008) 99:2035–47. doi: 10.1152/jn.01366.2006
345. Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. (2016) 19:771–83. doi: 10.1038/nn.4288
346. McConnell HL, Kersch CN, Woltjer RL, Neuwelt EA. The translational significance of the neurovascular unit. J Biol Chem. (2017) 292:762–70. doi: 10.1074/jbc.R116.760215
347. Carmichael MD, Davis JM, Murphy EA, Carson JA, Van Rooijen N, Mayer E, et al. Role of brain macrophages on IL-1beta and fatigue following eccentric exercise-induced muscle damage. Brain Behav Immun. (2010) 24:564–8. doi: 10.1016/j.bbi.2009.12.011
348. Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. (2014) 11:26. doi: 10.1186/2045-8118-11-26
349. Fantini S, Sassaroli A, Tgavalekos KT, Kornbluth J. Cerebral blood flow and autoregulation: current measurement techniques and prospects for noninvasive optical methods. Neurophotonics. (2016) 3:031411. doi: 10.1117/1.NPh.3.3.031411
351. Castro P, Azevedo E, Sorond F. Cerebral autoregulation in stroke. Curr Atheroscler Rep. (2018) 20:37. doi: 10.1007/s11883-018-0739-5
352. Mösges R, Kaatz V, Schmalz P, Meiser P, Eschmann K. Glycerol lidocaine eardrops for the treatment of acute abacterial otitis externa. Arzneimittelforschung. (2010) 60:427–31. doi: 10.1055/s-0031-1296307
353. Marrie RA, Yu BN, Leung S, Elliott L, Caetano P, Warren S, et al. Rising prevalence of vascular comorbidities in multiple sclerosis: validation of administrative definitions for diabetes, hypertension, and hyperlipidemia. Mult Scler. (2012) 18:1310–9. doi: 10.1177/1352458512437814
354. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertens. (2017) 70:660–7. doi: 10.1161/HYPERTENSIONAHA.117.07802
355. Quintana FJ. Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation. J Clin Invest. (2017) 127:2897–9. doi: 10.1172/JCI95769
356. Park S, Lakatta EG. Role of inflammation in the pathogenesis of arterial stiffness. Yonsei Med J. (2012) 53:258–61. doi: 10.3349/ymj.2012.53.2.258
357. Thenappan T, Prins KW, Pritzker MR, Scandurra J, Volmers K, Weir EK. The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann Am Thorac Soc. (2016) 13:276–84. doi: 10.1513/AnnalsATS.201509-599FR
358. Barnes JN, Nualnim N, Sugawara J, Sommerlad SM, Renzi CP, Tanaka H. Arterial stiffening, wave reflection, and inflammation in habitually exercising systemic lupus erythematosus patients. Am J Hypertens. (2011) 24:1194–200. doi: 10.1038/ajh.2011.143
359. Tryfonopoulos D, Anastasiou E, Protogerou A, Papaioannou T, Lily K, Dagre A, et al. Arterial stiffness in type 1 diabetes mellitus is aggravated by autoimmune thyroid disease. J Endocrinol Invest. (2005) 28:616–22. doi: 10.1007/BF03347260
360. Fjeldstad C, Frederiksen C, Fjeldstad AS, Bemben M, Pardo G. Arterial compliance in multiple sclerosis: a pilot study. Angiology. (2010) 61:31–6. doi: 10.1177/0003319709334120
361. Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Pulmonary manifestations of systemic autoimmune diseases. Maedica. (2011) 6:224–9.
362. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, et al. Inflammatory mechanisms in the lung. J Inflamm Res. (2009) 2:1–11.
363. Losa García JE, Rodríguez FM, Martín de Cabo MR, García Salgado MJ, Losada JP, Villarón LG, et al. Evaluation of inflammatory cytokine secretion by human alveolar macrophages. Mediators Inflamm. (1999) 8:43–51. doi: 10.1080/09629359990711
364. Oliveira RKF, Agarwal M, Tracy JA, Karin AL, Opotowsky AR, Waxman AB, et al. Age-related upper limits of normal for maximum upright exercise pulmonary haemodynamics. Eur Respir J. (2016) 47:1179–88. doi: 10.1183/13993003.01307-2015
365. Bazelmans E, Bleijenberg G, Vercoulen JH, van der Meer JW, Folgering H. The chronic fatigue syndrome and hyperventilation. J Psychosom Res. (1997) 43:371–7. doi: 10.1016/S0022-3999(97)00169-4
366. Stewart JM, Medow MS, Cherniack NS, Natelson BH. Postural hypocapnic hyperventilation is associated with enhanced peripheral vasoconstriction in postural tachycardia syndrome with normal supine blood flow. Am J Physiol Heart Circ Physiol. (2006) 291:H904–13. doi: 10.1152/ajpheart.01359.2005
367. Coelho R, Hughes AM, da Fonseca AF, Bond MR. Essential hypertension: the relationship of psychological factors to the severity of hypertension. J Psychosom Res. (1989) 33:187–96. doi: 10.1016/0022-3999(89)90046-9
368. Shanks L, Jason LA, Evans M, Brown A. Cognitive impairments associated with CFS and POTS. Front Physiol. (2013) 4:113. doi: 10.3389/fphys.2013.00113
369. Depiazzi J, Everard ML. Dysfunctional breathing and reaching one's physiological limit as causes of exercise-induced dyspnoea. Breathe. (2016) 12:120–9. doi: 10.1183/20734735.007216
370. Földi M, Csillik B, Zoltán OT. Lymphatic drainage of the brain. Experientia. (1968) 24:1283–7. doi: 10.1007/BF02146675
371. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. (2013) 342:373–7. doi: 10.1126/science.1241224
372. Schiweck J, Eickholt BJ, Murk K. Important shapeshifter: mechanisms allowing astrocytes to respond to the changing nervous system during development, injury and disease. Front Cell Neurosci. (2018) 12:261. doi: 10.3389/fncel.2018.00261
373. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. (2014) 35:347–69. doi: 10.1016/j.yfrne.2014.04.004
374. Cade BE, Chen H, Stilp AM, Gleason KJ, Sofer T, Ancoli-Israel S, et al. Genetic associations with obstructive sleep apnea traits in hispanic/latino Americans. Am J Respir Crit Care Med. (2016) 194:886–97. doi: 10.1164/rccm.201512-2431OC
Keywords: autoimmune, cytokines, fatigue, inflammasome, inflammation, sleep, neurovascular unit
Citation: Zielinski MR, Systrom DM and Rose NR (2019) Fatigue, Sleep, and Autoimmune and Related Disorders. Front. Immunol. 10:1827. doi: 10.3389/fimmu.2019.01827
Received: 21 May 2019; Accepted: 18 July 2019;
Published: 06 August 2019.
Edited by:
Attila Mócsai, Semmelweis University, HungaryReviewed by:
Robert Weissert, University of Regensburg, GermanyCopyright © 2019 Zielinski, Systrom and Rose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark R. Zielinski, bWFya196aWVsaW5za2lAaG1zLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.