 Manfred Kunz
Manfred Kunz Jan C. Simon
Jan C. Simon Anja Saalbach
Anja Saalbach- Department of Dermatology, Venereology and Allergology, University of Leipzig, Leipzig, Germany
Psoriasis is chronic inflammatory skin disease affecting skin, joints, cardiovascular system, brain, and metabolism. The pathogenesis of psoriasis is mediated by a complex interplay between the immune system, inflammatory mediators of different pathways, e.g., TNF-alpha and the IL-23/IL-17 pathways, psoriasis-associated susceptibility loci, autoantigens, and multiple environmental factors. Psoriasis is triggered by the combination of genetic and environmental factors. A novel environmental risk factor with rising importance is obesity. Several studies proved that obesity is an independent risk factor for the onset and severity of psoriasis. Due to the dramatic increase of obesity worldwide this minireview focuses on obesity as a major environmental risk factor for psoriasis and the mechanisms of obesity-mediated exacerbation of psoriasis.
Introduction
Psoriasis is a polygenic chronic inflammatory skin disease (1–3). A large proportion (20–30%) of the psoriasis patients suffer from additional joint involvement mainly affecting the distal extremities but also larger joints (3). Plaque-type psoriasis, the most common disease variant, which is seen in ~85% of cases, commonly manifests as dull-red, erythematous, scaly plaques particularly on the extensor surfaces of elbows, knees, and on the scalp. Less common psoriasis subtypes include pustular, guttate, inverse, erythrodermic, and palmoplantar psoriasis (4).
Psoriasis has a significant genetic background as shown by the enhanced risk for the development of the disease in offsprings and siblings of psoriasis patients and familial occurrence (5, 6). Genetic associations in psoriasis vulgaris were mainly described for the major histocompatibility complex (MHC) locus on chromosome 6 carrying the human leukocyte antigen (HLA) genes and other immune-regulatory genes such as complement factors and TNF-α (6). The strongest association was observed for the HLA-C allele Cw6, a classical HLA class I allele that was found in 46% of psoriasis patients but only in 7% of a control population (7). Subsequent genome-wide linkage studies by microsatellite analysis provided a further set of possible genomic regions with linkage to psoriasis such as the PSORS1 locus and other, non-MHC loci such as PSORS2-5 loci (8). More recently performed genome-wide association (GWAS) studies on psoriasis vulgaris have identified several additional psoriasis risk factors that comprise genes associated with chronic inflammation including IL12B (9, 10), IL23A and IL23R (9), IL2/IL21 (7), TNFAIP3 and TNIP1 (9), ZNF313 (11), and epidermal/antimicrobial genes such as SLC12A8 and HBD (human β-defensin gene) (12) and the LCE (late cornified envelope) gene cluster (10).
In contrast to psoriasis vulgaris, pustular psoriasis shows genetic associations with mutations in the IL36RN gene with the strongest association for generalized psoriasis pustulosa and a weaker association for palmoplantar pustulosis and acrodermatitis continua of Hallopeau (13). Palmoplantar pustulosis shows higher prevalence in female subjects and smokers. Guttate psoriasis is associated with environmental factors such as stress and infections, but no distinct genetic background has been defined so far. The different pathogenic mechanisms may also impact on treatment response, e.g., guttate psoriasis is less responsive to treatment with anti-TNF antibodies than plaque-type psoriasis (14).
Psoriasis is currently regarded as an auto-immune disease because it shares many features with other autoimmune diseases such as chronicity of the clinical symptoms and chronic inflammation, involvement of TNF-α and a genetic background with overlapping gene loci with other auto-immune diseases (15, 16). Potential autoantigens such as keratin 17 with sequence homologies to streptococcal M-proteins, the antimicrobial peptide LL37 and the melanocytic autoantigen ADAMTSL5 have been identified recently. LL37 and ADAMTSL5 are recognized by T-cells after binding to HLA-C*06:02 underlining the role of distinct HLA genotypes in the pathogenesis of psoriasis (17–19).
The central pathogenic cell types in psoriasis are epidermal keratinocytes, antigen presenting cells, and inflammatory T cells with complex feedback mechanisms (1, 2, 20–22).
Dysregulation of this complex interplay of cells of the innate and adaptive immune system promotes the proliferation and attenuates the differentiation of epidermal keratinocytes resulting in the distinctive thickened, scaly plaques seen in psoriasis vulgaris. Psoriasis is mediated by a plethora of cytokines and chemokines where TNF-α and the IL-23/IL-17 axis play an outstanding role (20, 23). IL-23 which is produced by antigen-presenting cells supports the development of IL-17-secreting CD4+ memory T cells (Th17 cells). Differential expression of both components of IL-23 (IL-23p19 and IL-12p40) was observed in psoriatic skin lesions in contrast to non-involved skin. However, there were no significant differences for the IL-12p35 subunit, suggestive for a particular role of IL-23 and not IL-12 in psoriasis (24). Th17 cells, neutrophils and mast cells produce IL-17A which exerts a feedforward inflammatory response in keratinocytes by triggering chemokine and cytokine production in keratinocytes. IL17 also activates neutrophils, B cells, monocytes, and macrophages (25, 26). The feedforward keratinocyte responses are self-amplifying, resulting in sustained pathogenic immune infiltration and the development of mature psoriatic plaques. Interestingly, inflammatory bowel diseases, which show a significant association with psoriasis vulgaris, and where IL-17 cytokines play a major pathogenic role, do not respond to anti-IL-17 treatment, while psoriasis does. This may be due to the fact that IL-17A can also play a protective role in the intestinal tract under inflammatory conditions as shown in an experimental mouse model of colitis (25).
The role of IL-23 as a master regulator in psoriasis was highlighted by the induction of psoriasis-like ear swelling, epidermal hyperplasia and acanthosis upon injection of IL-23 into mouse ears, which was dependent on IL17- and IL-22 (27, 28). These findings were supportive for a role of IL-23, IL-17, and IL-22 in psoriasis. The role of these cytokines in psoriasis pathogenesis is further emphasized by the currently used highly effective treatment modalities for psoriasis and psoriasis arthritis using antibodies directed against TNF-α, IL-23p19, and IL-17 (4, 29, 30). The analysis of gene expression patterns in psoriasis lesional skin under treatment with biological agents showed that gene expression patterns of IL-23- and IL-17-induced genes were indeed reduced by treatment with an anti-IL-12/23 antibody in healing skin lesions (31).
The detrimental feedforward inflammatory process in psoriasis is not restricted to the skin. The uncontrolled inflammatory response contributes to a number of comorbid conditions in psoriasis including cardiometabolic disease, stroke, and metabolic syndrome (obesity, hypertension, dyslipidemia, and diabetes) (32–36).
In general, psoriasis is believed to be triggered by the combination of genetic and environmental factors. It has been accepted that the interplay between environmental and genetic factors contributes to the onset, development and clinical symptoms of psoriasis. A significant number of studies identified ultraviolet light, drugs, smoking, alcohol, and infections as well as mental and biomechanical stress as environmental risk factors affecting psoriasis by interfering with its genetic predisposition and immune response (37).
A novel risk factor for psoriasis of high socioeconomic importance is adiposity. Several studies have shown that obesity is an independent risk factor for the onset and severity of psoriasis (38, 39). Due to the dramatic increase of obesity worldwide, this minireview focuses on obesity as one environmental risk factors for psoriasis and the mechanisms of obesity-mediated exacerbation of psoriasis.
Psoriasis and Obesity
The incidence of psoriasis among adults had almost doubled between the 1970s and 2000 (40). Since the genetic basis should have not significantly changed, environmental factors including the Western lifestyle might have played a role in this growing prevalence (41). The dietary habits in industrialized nations often support high-fat, high-salt, and high-sugar diets with excess caloric intake resulting in obesity and metabolic syndrome (42). In a current large population-based Norwegian study including close to 35,000 subjects, an association of metabolic syndrome with an increased risk to develop psoriasis has been described. The analysis of metabolic factors indicated that adiposity is a central factor in this association (43). Similar findings were reported by others [reviewed in (38, 39)]. It is difficult to show what comes first, psoriasis or obesity. Pronounced social isolation, poor eating habits, depression, increased alcohol consumption, and decreased physical activity in patients with psoriasis might explain how psoriasis might lead to obesity (38).
However, epidemiological studies provide strong evidence that obesity predisposes patients to psoriasis and amplifies psoriatic inflammation. A study of Setty and co-workers including 78,626 women (of whom 892 reported having psoriasis) indicated that adiposity and weight gain were risk factors for the development of psoriasis (44). Patients with a body mass index (BMI) of 35 or more had a relative increased risk for development of psoriasis of 2.69 compared to lean patients (44). A recent prospective study indicated that obesity and high abdominal fat mass doubled the risk of psoriasis (45). These studies suggest that preventing weight gain, promoting maintenance of a normal body weight, and reduction of body mass may reduce incidence of psoriasis. Indeed, several studies showed a positive impact of weight loss on the severity of psoriasis (46). Thus, dietary weight reduction with a hypocaloric diet is recommended in overweight and obese patients with psoriasis (47). An open question is whether differences in the type of diet (low carbohydrate, ketogenic, or vegan/vegetarian diets) have an effect on psoriasis improvement. Understanding the epidemiological relationship between obesity/nutrition and psoriasis is important to assess the relevance of the environmental factors as modifiable risk factors in psoriasis pathogenesis and to develop new strategies to support anti-psoriatic treatments (48).
Since adipose tissue is an important endocrine organ secreting soluble factors involved in inflammation and immunity, it has been postulated that adipose tissue expansion and its secretion of pro-inflammatory mediators might worsen psoriasis. High levels of resistin and leptin have been found in obese psoriasis patients (39). A recent meta-analysis showed that patients with psoriasis have higher levels of leptin compared to persons without psoriasis (49).
In addition, obesity alters the cellular composition and activity of inflammatory cells in the skin. Nakamizo and co-workers described an accumulation of IL-17A-producing γδ T cells in psoriatic skin lesions of high fat diet (HFD)-induced obese mice, which resulted in an exacerbation of psoriatic dermatitis (50). Moreover, genetically engineered diabetic (db/db) mice showed an enhanced psoriatic skin inflammation with enhanced levels of IL-17A and IL-22 (51). Another study showed that long-term HFD over 9 months promoted the accumulation of specific CD11c+ macrophages in the skin, in an epidermal fatty acid binding protein (E-FABP)-dependent manner (52). In elegant studies, Christ and co-workers showed that Western diet (WD) induces a long-lasting trained immunity in myeloid cells. The authors induced systemic inflammation in Ldlr−/− mice by WD feeding that subsided after shifting mice to chow diet. WD induced long-lasting transcriptomic and epigenomic reprogramming of myeloid progenitor cells resulting in increased proliferation and innate immune responses (53).
Another important aspect is the fact that obesity and nutrition affect the microbiome (54, 55). Recently it has been shown that the microbiome -which stands for the entire microorganisms that live on human outer and inner body surfaces- exerts a strong influence on human autoimmune diseases (56). There is already some evidence that this might also be the case in psoriasis (57). The role of the microbiome for metabolic processes has also been emphasized in recent experimental studies (58). Interestingly, a correlation exists between the microbiome and IL-17 production in autoimmune diseases (59–61). Changes in the gut microbiome in psoriasis refer to a decrease in the Bacteroidetes phylum with an increase in the Faecalibacterium genus. It was suggested that bacteria shed their cell wall components, such as lipopolysaccharide and lipoteichoic acid into the blood stream thereby supporting a chronic inflammatory state. Along this line, pro-biotic substances have been shown to exert an influence on autoimmune diseases such as Crohn's disease, colitis ulcerosa, and rheumatoid arthritis but so far have not been tested for their impact on psoriasis (62).
Psoriasis and Fatty Acids
Interestingly, psoriatic patients on low energy diet showed a significant decrease of serum lipids in parallel to a reduction of skin involvement compared to a control group on a normal diet (63). However, body weight did not differ between both groups linking obesity and psoriasis independent of adipose tissue. In line with this, a recent study by our group using an imiquimod-induced psoriasis mouse model showed that specific dietary components, rather than obesity itself, may exacerbate psoriasis (64). In this study, a correlation of serum concentrations of free fatty acids (FFAs) with severity of psoriatic inflammation in HFD-induced obese mice was observed (64, 65). Interestingly, these data could be recapitulated in a human cohort of psoriasis patients where blood levels of FFAs correlated with the severity of psoriatic skin lesions (64). In accordance with these findings, in the above mentioned large population-based Norwegian study only blood fat levels and adiposity showed a positive association with psoriasis while glucose levels did not (43). To examine the causal relationship between FFAs and the HFD-induced exacerbation of psoriatic skin inflammation we fed mice for only 5 weeks (64). At this time point mice are lean and metabolically healthy while expressing elevated levels of serum FFAs. Psoriatic skin inflammation was strongly enhanced accompanied by an increased tissue infiltration of myeloid cells, epidermal thickening and expression of S100 proteins, chemokines, and pro-inflammatory cytokines such as IL-1β (64). Consistently, ob/ob mice, another model of murine obesity on normal diet, did not exhibit enhanced psoriatic skin inflammation (50). To further discriminate between the impact of adipose tissue and FFAs we used different dietary approaches (64). First, mice received standard HFD with high saturated FA (SFAs) for 2.5 weeks, which was then changed to a HFD with reduced SFAs but enriched polyunsaturated FAs (PUFA) content for additional 2.5 weeks. After 5 weeks, weight did not significantly differ between these groups but the modified HFD fed mice showed significantly lower SFA blood levels, and psoriatic inflammation was strongly reduced. Mice were then fed for 20 weeks with HFD to mimic the situation of obese patients. Then, the diet was switched to a low-fat chow diet without PUFA supplementation. Again, diet change decreased the serum concentration of SFAs without effect on weight. Importantly, these obese mice with reduced serum concentration of SFA exhibited a reduced psoriatic skin inflammation compared to obese mice on HFD (64). This study showed that dietary SFAs seem to be key amplifiers of psoriatic inflammation and suggest that restriction of SFAs may be beneficial for both lean and obese patients (Figure 1).
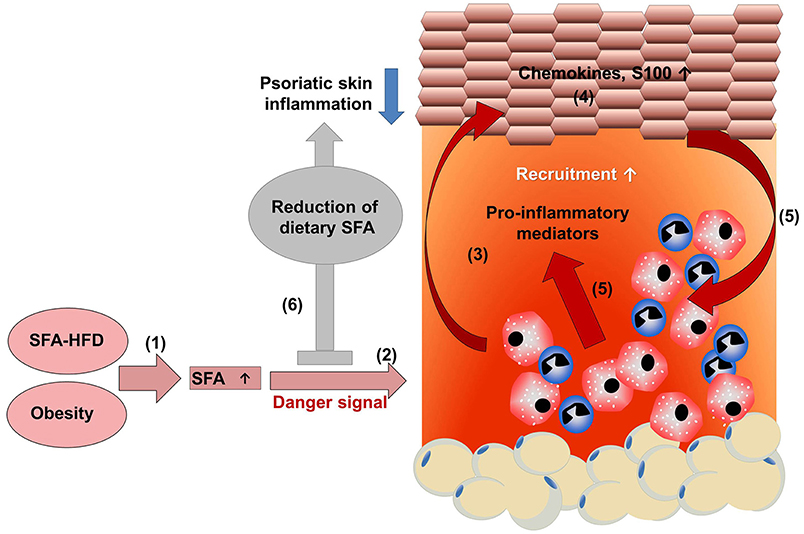
Figure 1. HFD-derived SFAs amplify psoriatic inflammation. (1) A diet rich in saturated fatty acids (SFAs) increases SFA serum concentration. (2) Chronic intake of a high fat diet increases adipose tissue, resulting in obesity with high SFA serum levels. (2) SFAs sensitize myeloid cells resulting in an amplified pro-inflammatory response with enhanced secretion of pro-inflammatory mediators in the presence of a danger signal. (3) The enhanced myeloid cell activation contributes to a disturbance of keratinocyte proliferation, differentiation, and (4) enhances the production of chemokines and S100 proteins. (5) Consequently, more myeloid immune cells are recruited into skin lesion and activated, further enhancing psoriatic skin inflammation. (6) Dietary reduction of SFAs dampens psoriatic skin inflammation, which might support treatment efficacy in psoriatic patients.
Due to limited evidence of a beneficial effect of fish oil for psoriasis, fish oil supplementation is not recommended for psoriasis treatment (47). Data from our study might explain the failure of PUFA supplementation as a therapeutic measure in psoriasis (66–69). It appears that a reduction of SFAs is more efficient than PUFA supplementation. At present, caloric restriction is recommended for overweight and obese psoriasis patients. Future clinical trials have to verify whether a specification of this recommendation—the reduction of SFAs as adjuvant dietary measure—might support conventional anti-inflammatory therapies.
Fatty Acids and Inflammation
Long chain SFAs such as palmitate are enriched in states of nutrient excess and obesity (65, 70). SFAs can produce insulin resistance, endoplasmatic reticulum stress, oxidative stress, and cell death, a phenomenon referred to as lipotoxicity (70). They can bind to cell surface molecules such as CD36, free fatty acid receptors (FFAR1-4) and intracellular receptors/sensors [E-FABP and Peroxisome Proliferator Activated Receptor (PPAR) γ] that control inflammatory cell signaling and gene expression (71). SFAs are able to induce pro-inflammatory cytokines in human macrophages via pathways involving de novo ceramide synthesis (72). SFAs, but not unsaturated FAs, bind to E-FABP which activates retinoid acid receptor (RAR) resulting in differentiation of CD11c+ macrophages and expression of proinflammatory cytokines (73). It has been suggested that SFAs, via binding to TLR2 and TLR4, stimulate the expression of pro-inflammatory signaling pathways (74, 75). Current findings indicate that SFAs are not TLR4 agonists, but instead provide a second hit of activation that is dependent on prior TLR4 activation (76). Consistently, several studies did not detect any direct FFA-mediated activation of myeloid cells (64, 77, 78). However, amplification of the pro-inflammatory response of myeloid cells in the presence of SFAs has been described in many studies (64, 65, 72, 77–79). How can SFAs amplify the pro-inflammatory response?
PPARs are specialized receptors detecting FFA-derived signal molecules. Loss of PPAR-γ dampens de novo sterol biosynthesis and augments IFN-β production, which in turn suppresses the transcription of IL-1α and IL-1β in LPS-stimulated macrophages (80). Uptake of SFAs leads to enhanced ceramide generation, which in turn activates PKC-ζ and MAPK, resulting in increased IL-6 and IL-8 secretion upon LPS stimulation (79).
Elevated FFAs caused by HFD or obesity activate the NLRP3 inflammasome in macrophages resulting in increased IL-1β and IL-18 secretion (78). In the presence of danger signals SFAs induce inflammasome activation by induction of mitochondrial reactive oxygen species, or by stimulation of AMP-activated protein kinase, autophagy, or lysosome- and calcineurin-dependent pathways (70, 78). Excess SFAs uptake induces intracellular SFAs crystallization that leads to NLRP3 inflammasome activation and subsequent IL-1β release via lysosomal dysfunction (81).
Taken together, SFAs can amplify the pro-inflammatory response via direct and indirect actions (Figure 2). Thus, restriction of dietary SFAs might be helpful to suppress psoriatic inflammation.
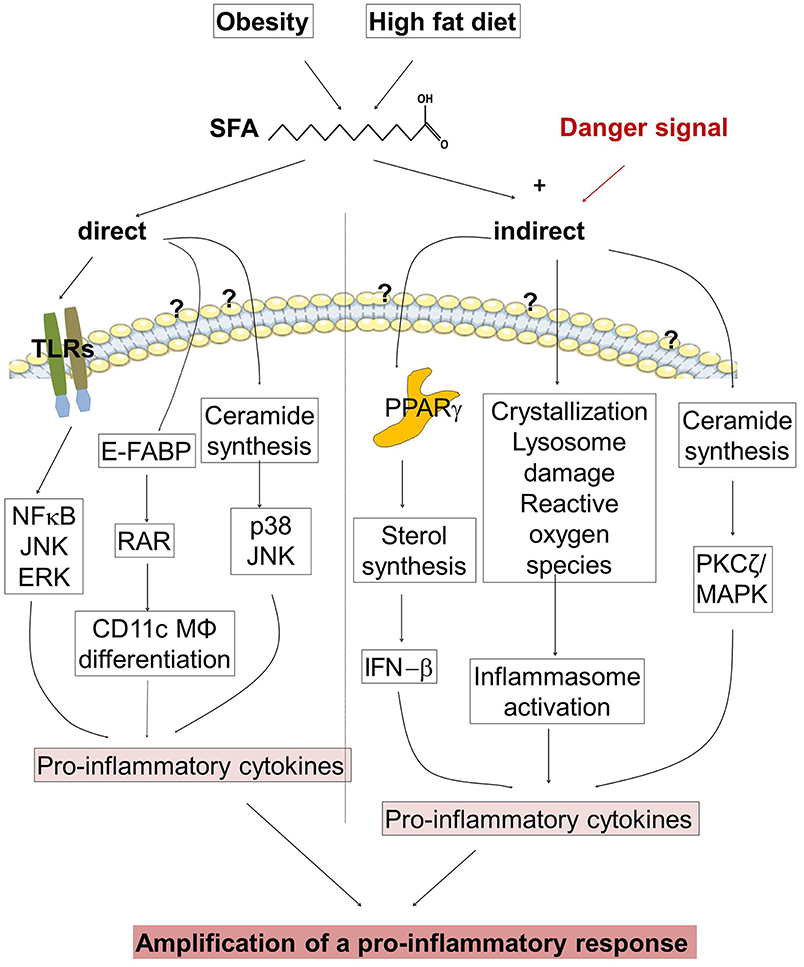
Figure 2. Regulation of the pro-inflammatory response in psoriasis. (Left half of the figure) Obesity and high fat diet (HFD) increase the concentration of saturated fatty acids (SFAs). SFAs are able to stimulate directly the expression of pro-inflammatory cytokines. SFAs activate toll-like receptors (TLR), and bind to cytoplasmic epidermal fatty acid binding proteins (E-FABPs) activating retinoid acid receptor (RAR) and stimulate the differentiation of CD11c+ macrophages (MØ). An Increase of SFAs modulates ceramide synthesis. (Right half of the figure) SFAs amplify the pro-inflammatory response in the presence of a danger signal. SFAs stimulate the expression of pro-inflammatory cytokines via binding to PPARs, by inflammasome activation, and by modulation of ceramide synthesis. Until now it is not clear which receptors are involved in FFA binding, translocation into the cell and subsequent pro-inflammatory activity.
Conclusions
Psoriasis is a chronic inflammatory skin disease mediated by a complex interplay between immune cells and tissue resident cells. Genetic and environmental factors contribute to psoriasis pathogenesis. Environmental factors such obesity and nutrition have an important impact on onset and severity of psoriasis. Recent studies suggest that dietary SFAs seem to be key amplifiers of psoriatic inflammation and suggest that restriction of SFAs may be beneficial for both lean and obese patients. The clinical relevance has to be proven in future clinical trials to improve psoriasis treatment responses and co-morbidities.
Author Contributions
AS and MK wrote the manuscript. JS edited and discussed the manuscript.
Funding
This work was supported by Deutscher Psoriasis Bund e.V. and PsoNet Leipzig/Westsachsen and by the Deutsche Forschungsgemeinschaft (grants to AS: SA863/2-3; SA863/4-1).
Conflict of Interest Statement
MK received travel grants from UCB Pharma, and is member of advisory boards of Novartis Pharma and LEO Pharma. JS received speakers honoraria and is member of advisory boards of Novartis Pharma, Janssen Pharma, UCB Pharma, and AbbVie Pharma and received travel grants from these companies and from LEO Pharma.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge support from the German Research Foundation (DFG), the PsoNet Leipzig, the Deutsche Psoriasisbund, and Universität Leipzig within the program of Open Access Publishing.
References
2. Kim J, Krueger JG. The immunopathogenesis of psoriasis. Dermatol Clin. (2015) 33:13–23. doi: 10.1016/j.det.2014.09.002
4. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. (2017) 140:645–53. doi: 10.1016/j.jaci.2017.07.004
5. Bowcock AM. Understanding the pathogenesis of psoriasis, psoriatic arthritis, and autoimmunity via a fusion of molecular genetics and immunology. Immunol Res. (2005) 32:45–56. doi: 10.1385/IR:32:1-3:045
6. Roberson ED, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet. (2010) 26:415–23. doi: 10.1016/j.tig.2010.06.006
7. Liu Y, Krueger JG, Bowcock AM. Psoriasis: genetic associations and immune system changes. Genes Immun. (2007) 8:1–12. doi: 10.1038/sj.gene.6364351
8. Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. (2004) 13:R43–R55. doi: 10.1093/hmg/ddh094
9. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. (2009) 41:199–204. doi: 10.1038/ng.311
10. Zhang XJ, Huang W, Yang S, Sun LD, Zhang FY, Zhu QX, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet. (2009) 41:205–10. doi: 10.1038/ng.310
11. Capon F, Bijlmakers MJ, Wolf N, Quaranta M, Huffmeier U, Allen M, et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet. (2008) 17:1938–45. doi: 10.1093/hmg/ddn091
12. Hollox EJ. Copy number variation of beta-defensins and relevance to disease. Cytogenet Genome Res. (2008) 123:148–55. doi: 10.1159/000184702
13. Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. (2019) 143:1021–6. doi: 10.1016/j.jaci.2018.06.038
14. Brummer GC, Hawkes JE, Duffin KC. Ustekinumab-induced remission of recalcitrant guttate psoriasis: a case series. JAAD Case Rep. (2017) 3:432–5. doi: 10.1016/j.jdcr.2017.06.015
15. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med. (2015) 21:730–8. doi: 10.1038/nm.3897
16. Hwang ST, Nijsten T, Elder JT. Recent highlights in psoriasis research. J Invest Dermatol. (2017) 137:550–6. doi: 10.1016/j.jid.2016.11.007
17. Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. (2015) 212:2203–12. doi: 10.1084/jem.20151093
18. Prinz JC. Human leukocyte antigen-class I alleles and the autoreactive T cell response in psoriasis pathogenesis. Front Immunol. (2018) 9:954. doi: 10.3389/fimmu.2018.00954
19. Schon MP, Erpenbeck L. The interleukin-23/interleukin-17 axis links adaptive and innate immunity in psoriasis. Front Immunol. (2018) 9:1323. doi: 10.3389/fimmu.2018.01323
20. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. (2018) 201:1605–13. doi: 10.4049/jimmunol.1800013
21. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
22. Nickoloff BJ, Qin JZ, Nestle FO. Immunopathogenesis of psoriasis. Clin Rev Allergy Immunol. (2007) 33:45–56. doi: 10.1007/s12016-007-0039-2
23. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A, beyond. Front Immunol. (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
24. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
25. Bianchi E, Rogge L. The IL-23/IL-17 pathway in human chronic inflammatory diseases-new insight from genetics and targeted therapies. Genes Immun. (2019) 20:415–25. doi: 10.1038/s41435-019-0067-y
26. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
27. Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. (2011) 186:1495–502. doi: 10.4049/jimmunol.1001001
28. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
29. Chan TC, Hawkes JE, Krueger JG. Interleukin 23 in the skin: role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther Adv Chronic Dis. (2018) 9:111–9. doi: 10.1177/2040622318759282
30. Kim J, Krueger JG. Highly effective new treatments for psoriasis target the IL-23/type 17 T cell autoimmune axis. Annu Rev Med. (2017) 68:255–69. doi: 10.1146/annurev-med-042915-103905
31. Brodmerkel C, Li K, Garcet S, Hayden K, Chiricozzi A, Novitskaya I, et al. Modulation of inflammatory gene transcripts in psoriasis vulgaris: differences between ustekinumab and etanercept. J Allergy Clin Immunol. (2019) 143:1965–9. doi: 10.1016/j.jaci.2019.01.017
32. Fernandez-Armenteros JM, Gomez-Arbones X, Buti-Soler M, Betriu-Bars A, Sanmartin-Novell V, Ortega-Bravo M, et al. Psoriasis, metabolic syndrome and cardiovascular risk factors. A population-based study. J Eur Acad Dermatol Venereol. (2019) 33:128–35. doi: 10.1111/jdv.15159
33. Gerdes S, Mrowietz U, Boehncke WH. Comorbidity in psoriasis. Hautarzt. (2016) 67:438–44. doi: 10.1007/s00105-016-3805-3
34. Husni ME, Wilson Tang WH, Lucke M, Chandrasekharan UM, Brennan DM, Hazen SL. Correlation of high-density lipoprotein-associated paraoxonase 1 activity with systemic inflammation, disease activity, and cardiovascular risk factors in psoriatic disease. Arthritis Rheumatol. (2018) 70:1240–50. doi: 10.1002/art.40499
35. Ryan C, Korman NJ, Gelfand JM, Lim HW, Elmets CA, Feldman SR, et al. Research gaps in psoriasis: opportunities for future studies. J Am Acad Dermatol. (2014) 70:146–67. doi: 10.1016/j.jaad.2013.08.042
36. Boehncke WH, Gladman DD, Chandran V. Cardiovascular comorbidities in psoriasis and psoriatic arthritis: pathogenesis, consequences for patient management, and future research agenda: a report from the GRAPPA 2009 annual meeting. J Rheumatol. (2011) 38:567–71. doi: 10.3899/jrheum.101124
37. Zeng J, Luo S, Huang Y, Lu Q. Critical role of environmental factors in the pathogenesis of psoriasis. J Dermatol. (2017) 44:863–72. doi: 10.1111/1346-8138.13806
38. Carrascosa JM, Rocamora V, Fernandez-Torres RM, Jimenez-Puya R, Moreno JC, Coll-Puigserver N, et al. Obesity and psoriasis: inflammatory nature of obesity, relationship between psoriasis and obesity, and therapeutic implications. Actas Dermosifiliogr. (2014) 105:31–44. doi: 10.1016/j.ad.2012.08.003
40. Icen M, Crowson CS, McEvoy MT, Dann FJ, Gabriel SE, Maradit KH. Trends in incidence of adult-onset psoriasis over three decades: a population-based study. J Am Acad Dermatol. (2009) 60:394–401. doi: 10.1016/j.jaad.2008.10.062
41. Moroni L, Bianchi I, Lleo A. Geoepidemiology, gender and autoimmune disease. Autoimmun Rev. (2012) 11:A386–92. doi: 10.1016/j.autrev.2011.11.012
42. Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in obesity among adults in the United States, 2005 to 2014. JAMA. (2016) 315:2284–91. doi: 10.1001/jama.2016.6458
43. Snekvik I, Nilsen TIL, Romundstad PR, Saunes M. Metabolic syndrome and risk of incident psoriasis: prospective data from the HUNT Study, Norway. Br J Dermatol. (2019) 180:94–9. doi: 10.1111/bjd.16885
44. Setty AR, Curhan G, Choi HK. Obesity, waist circumference, weight change, and the risk of psoriasis in women: nurses' health study II. Arch Intern Med. (2007) 167:1670–5. doi: 10.1001/archinte.167.15.1670
45. Snekvik I, Smith CH, Nilsen TIL, Langan SM, Modalsli EH, Romundstad PR, et al. Obesity, waist circumference, weight change, and risk of incident psoriasis: prospective data from the HUNT study. J Invest Dermatol. (2017) 137:2484–90. doi: 10.1016/j.jid.2017.07.822
46. Alotaibi HA. Effects of weight loss on psoriasis: a review of clinical trials. Cureus. (2018) 10:e3491. doi: 10.7759/cureus.3491
47. Ford AR, Siegel M, Bagel J, Cordoro KM, Garg A, Gottlieb A, et al. Dietary recommendations for adults with psoriasis or psoriatic arthritis from the medical board of the national psoriasis foundation: a systematic review. JAMA Dermatol. (2018) 154:934–50. doi: 10.1001/jamadermatol.2018.1412
48. Barrea L, Nappi F, Di SC, Savanelli MC, Falco A, Balato A, et al. Environmental risk factors in psoriasis: the point of view of the nutritionist. Int J Environ Res Public Health. (2016) 13:743. doi: 10.3390/ijerph13070743
49. Zhu KJ, Zhang C, Li M, Zhu CY, Shi G, Fan YM. Leptin levels in patients with psoriasis: a meta-analysis. Clin Exp Dermatol. (2013) 38:478–83. doi: 10.1111/ced.12171
50. Nakamizo S, Honda T, Adachi A, Nagatake T, Kunisawa J, Kitoh A, et al. High fat diet exacerbates murine psoriatic dermatitis by increasing the number of IL-17-producing gammadelta T cells. Sci Rep. (2017) 7:14076. doi: 10.1038/s41598-017-14292-1
51. Kanemaru K, Matsuyuki A, Nakamura Y, Fukami K. Obesity exacerbates imiquimod-induced psoriasis-like epidermal hyperplasia and interleukin-17 and interleukin-22 production in mice. Exp Dermatol. (2015) 24:436–42. doi: 10.1111/exd.12691
52. Zhang Y, Li Q, Rao E, Sun Y, Grossmann ME, Morris RJ, et al. Epidermal fatty acid binding protein promotes skin inflammation induced by high-fat diet. Immunity. (2015) 42:953–64. doi: 10.1016/j.immuni.2015.04.016
53. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75. doi: 10.1016/j.cell.2017.12.013
54. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. (2011) 474:327–36. doi: 10.1038/nature10213
55. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
56. Woo V, Alenghat T. Host-microbiota interactions: epigenomic regulation. Curr Opin Immunol. (2017) 44:52–60. doi: 10.1016/j.coi.2016.12.001
57. Statnikov A, Alekseyenko AV, Li Z, Henaff M, Perez-Perez GI, Blaser MJ, et al. Microbiomic signatures of psoriasis: feasibility and methodology comparison. Sci Rep. (2013) 3:2620. doi: 10.1038/srep02620
58. Zhou D, Pan Q, Shen F, Cao HX, Ding WJ, Chen YW, et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep. (2017) 7:1529. doi: 10.1038/s41598-017-01751-y
59. Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, et al. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity. (2016) 44:659–71. doi: 10.1016/j.immuni.2016.02.007
60. Speeckaert R, Lambert J, Grine L, Van GM, De SS, van GN. The many faces of interleukin-17 in inflammatory skin diseases. Br J Dermatol. (2016) 175:892–901. doi: 10.1111/bjd.14703
61. Zakostelska Z, Malkova J, Klimesova K, Rossmann P, Hornova M, Novosadova I, et al. Intestinal microbiota promotes psoriasis-like skin inflammation by enhancing Th17 response. PLoS ONE. (2016) 11:e0159539. doi: 10.1371/journal.pone.0159539
62. Forbes JD, Van Domselaar G, Bernstein CN. The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol. (2016) 7:1081. doi: 10.3389/fmicb.2016.01081
63. Rucevic I, Perl A, Barisic-Drusko V, Adam-Perl M. The role of the low energy diet in psoriasis vulgaris treatment. Coll Antropol. (2003) 27 (Suppl 1):41–8.
64. Herbert D, Franz S, Popkova Y, Anderegg U, Schiller J, Schwede K, et al. High-fat diet exacerbates early psoriatic skin inflammation independent of obesity: saturated fatty acids as key players. J Invest Dermatol. (2018) 138:1999–2009. doi: 10.1016/j.jid.2018.03.1522
65. Stelzner K, Herbert D, Popkova Y, Lorz A, Schiller J, Gericke M, et al. Free fatty acids sensitize dendritic cells to amplify TH1/TH17-immune responses. Eur J Immunol. (2016) 46:2043–53. doi: 10.1002/eji.201546263
66. Mayser P, Grimm H, Grimminger F. n-3 fatty acids in psoriasis. Br J Nutr. (2002) 87 (Suppl 1):S77–S82. doi: 10.1079/BJN2001459
67. Park MK, Li W, Paek SY, Li X, Wu S, Li T, et al. Consumption of polyunsaturated fatty acids and risk of incident psoriasis and psoriatic arthritis from the Nurses' Health Study II. Br J Dermatol. (2017) 177:302–6. doi: 10.1111/bjd.15059
68. Qin S, Wen J, Bai XC, Chen TY, Zheng RC, Zhou GB, et al. Endogenous n-3 polyunsaturated fatty acids protect against imiquimod-induced psoriasis-like inflammation via the IL-17/IL-23 axis. Mol Med Rep. (2014) 9:2097–104. doi: 10.3892/mmr.2014.2136
69. Teng KT, Chang CY, Chang LF, Nesaretnam K. Modulation of obesity-induced inflammation by dietary fats: mechanisms and clinical evidence. Nutr J. (2014) 13:12. doi: 10.1186/1475-2891-13-12
70. Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem. (2014) 289:9158–71. doi: 10.1074/jbc.M113.531202
71. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol. (2016) 114:3–13. doi: 10.1016/j.bcp.2016.03.017
72. Haversen L, Danielsson KN, Fogelstrand L, Wiklund O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis. (2009) 202:382–93. doi: 10.1016/j.atherosclerosis.2008.05.033
73. Zeng J, Zhang Y, Hao J, Sun Y, Liu S, Bernlohr DA, et al. Stearic acid induces CD11c expression in proinflammatory macrophages via epidermal fatty acid binding protein. J Immunol. (2018) 200:3407–19. doi: 10.4049/jimmunol.1701416
74. Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. (2013) 191:4337–47. doi: 10.4049/jimmunol.1300298
75. Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. (2012) 53:2002–13. doi: 10.1194/jlr.D029546
76. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. (2018) 27:1096–110. doi: 10.1016/j.cmet.2018.03.014
77. Chang CF, Chau YP, Kung HN, Lu KS. The lipopolysaccharide-induced pro-inflammatory response in RAW264.7 cells is attenuated by an unsaturated fatty acid-bovine serum albumin complex and enhanced by a saturated fatty acid-bovine serum albumin complex. Inflamm Res. (2012) 61:151–60. doi: 10.1007/s00011-011-0399-1
78. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
79. Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, et al. Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler Thromb Vasc Biol. (2010) 30:802–8. doi: 10.1161/ATVBAHA.109.201681
80. Weber KJ, Sauer M, He L, Tycksen E, Kalugotla G, Razani B, et al. PPARgamma deficiency suppresses the release of IL-1beta and IL-1alpha in macrophages via a type 1 IFN-dependent mechanism. J Immunol. (2018) 201:2054–69. doi: 10.4049/jimmunol.1800224
Keywords: psoriasis, obesity, fatty acids, metabolism, genetics
Citation: Kunz M, Simon JC and Saalbach A (2019) Psoriasis: Obesity and Fatty Acids. Front. Immunol. 10:1807. doi: 10.3389/fimmu.2019.01807
Received: 29 March 2019; Accepted: 17 July 2019;
Published: 31 July 2019.
Edited by:
Michael P. Schön, University Medical Center Göttingen, GermanyReviewed by:
Matthew Stoll, University of Alabama at Birmingham, United StatesAlexandros A. Drosos, University of Ioannina, Greece
Copyright © 2019 Kunz, Simon and Saalbach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manfred Kunz, bWFuZnJlZC5rdW56QG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=