 Stefania Canè1Stefano Ugel1
Stefania Canè1Stefano Ugel1 Rosalinda Trovato1
Rosalinda Trovato1 Ilaria Marigo2
Ilaria Marigo2 Francesco De Sanctis1Silvia Sartoris1
Francesco De Sanctis1Silvia Sartoris1 Vincenzo Bronte1*
Vincenzo Bronte1*- 1Section of Immunology, Department of Medicine, University of Verona, Verona, Italy
- 2Veneto Institute of Oncology IOV-IRCCS, Padua, Italy
Cancer immunotherapy relies on either restoring or activating the function of adaptive immune cells, mainly CD8+ T lymphocytes. Despite impressive clinical success, cancer immunotherapy remains ineffective in many patients due to the establishment of tumor resistance, largely dependent on the nature of tumor microenvironment. There are several cellular and molecular mechanisms at play, and the goal is to identify those that are clinically significant. Among the hematopoietic-derived cells, monocytes are endowed with high plasticity, responsible for their pro- and anti-tumoral function. Indeed, monocytes are involved in several cancer-associated processes such as immune-tolerance, metastatic spread, neoangiogenesis, and chemotherapy resistance; on the other hand, by presenting cancer-associated antigens, they can also promote and sustain anti-tumoral T cell response. Recently, by high throughput technologies, new findings have revealed previously underappreciated, profound transcriptional, epigenetic, and metabolic differences among monocyte subsets, which complement and expand our knowledge on the monocyte ontogeny, recruitment during steady state, and emergency hematopoiesis, as seen in cancer. The subdivision into discrete monocytes subsets, both in mice and humans, appears an oversimplification, whereas continuum subsets development is best for depicting the real condition. In this review, we examine the evidences sustaining the existence of a monocyte heterogeneity along with functional activities, at the primary tumor and at the metastatic niche. In particular, we describe how tumor-derived soluble factors and cell-cell contact reprogram monocyte function. Finally, we point out the role of monocytes in preparing and shaping the metastatic niche and describe relevant targetable molecules altering monocyte activities. We think that exploiting monocyte complexity can help identifying key pathways important for the treatment of cancer and several conditions where these cells are involved.
Introduction
Monocyte diversity is well-recognized but the biologic and clinical significance of the different monocyte subtypes is far from being completely elucidated. The main hallmark of monocytes is their plastic nature, whereby they can exert multiple roles during the course of the immune response including cytokine production, pathogen clearance, antigen presentation, wound healing, and pro/anti-tumoral response (1–3). The original classification of monocytes into classical (in humans: CD14high, CD16−; in mice: Ly6Chigh), intermediate (in humans: CD14high, CD16low), and non-classical (in humans CD14low, CD16high; in mice: Ly6Clow) is currently being replaced by evidences supporting the existence of a “monocyte continuum” rather than stepwise differences between the different subtypes (4). Indeed, in mice under steady state, circulating classical monocyte subsets have been shown to switch into non-classical monocytes over time (5–7). However, it remains to be shown what relationship exists among the human monocytic subsets, and whether and how pathological conditions, like inflammation and cancer, impact this process.
Circulating monocytes have been viewed for many years, as precursor cells that provide tissue macrophages and dendritic cell (DCs) populations (8, 9); however, mounting evidence suggests that monocytes have their own effector functions in the blood and at peripheral sites throughout the body (10). The emerging data that distinct monocyte subsets, carrying different genetic, epigenetic, transcriptional, and metabolic arrangements, are committed to become macrophages and DCs seems to contradict the general accepted view of monocytes responding to a particular environmental stimuli and then differentiating into multifaceted macrophages and DCs. The intriguing evidence, both in mice and humans (11–13), of trained-monocytes, both present as mature and precursor cells, seems to strongly support the former hypothesis and reinvigorate the idea that monocytes have specific functions beyond being precursor cells. In this review, by combining the most recent advances in the field of monocytes' genetic, epigenetic, transcriptomic, and metabolomic, we outline and evaluate the changes occurring in monocyte subsets that underlie the aforementioned plasticity and heterogeneity. Secondly, we discuss new concepts in the monocyte field, like trained immunity and reprogramming and highlight the targetable pathways controlling monocyte fate and function.
We think that combining the information of single-cell transcriptome profiling, metabolomics array and epigenetic studies will elucidate complex relationships between cell types, thus solving limitations in the existing classification that relies on a relatively small number of markers.
Monocyte Phenotypical and Transcriptional Plasticity
Inflammatory and Patrolling Monocytes
Studies over the past two decades have delineated two major subsets of monocytes in mice and humans. Inflammatory monocytes (iMo), characterized by the high expression of the chemokine receptor CCR2, are repeatedly released from the bone marrow into the circulation. These cells, alternatively known as classical monocytes, are Ly6Chi in mice and correspond to the CD14hiCD16lo monocyte subset in humans. The fate of these cells is strictly dependent on the state of the body. Under steady state conditions, extravasated iMo and their derived-cells enrich in nearly all tissues throughout the body, where they form a small yet significant group of the so called local tissue-resident macrophages (7). The gradual accumulation of monocyte-derived macrophages in tissues is generally associated with the slow but progressive replacement of embryonic macrophages, in both quantitative and qualitative fashion (14). Monocyte-derived macrophages present sustained gene modifications as compared to their circulating counterparts, as they shape to the tissue microenvironment. They acquire transcriptional signatures resembling resident macrophages of embryonic origin, even though a certain level of differences remains at the epigenetic, transcriptional and functional levels (15–17). Whether monocyte-derived macrophages that infiltrate tissues under steady-state condition gain a self-renewal ability, comparable to their embryonic counterparts, is still a matter of debate and seems to strictly depend on the type of infiltrated tissue. On the other hand, iMo can also conserve their monocyte-like state inside the tissues without differentiating into macrophages, thereby acting as a local monocyte reservoir (18) (Table 1).
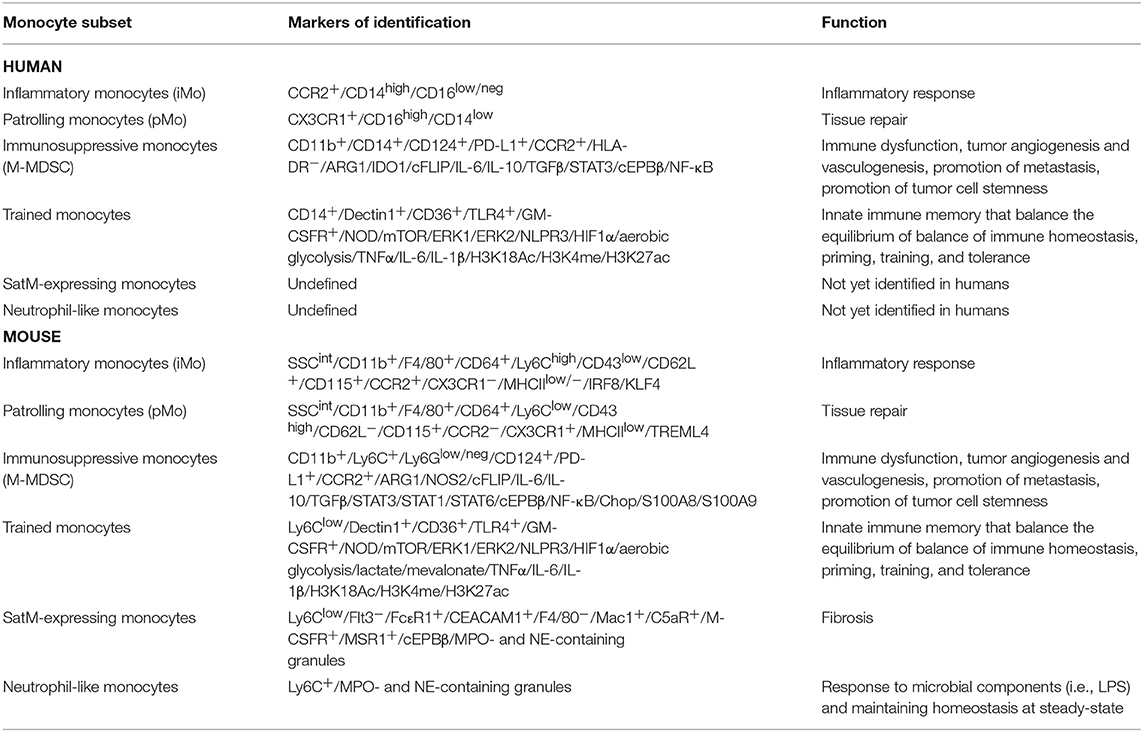
Table 1. Summary of monocyte subsets presented in this review, highlighting their markers and function in both humans (top part of the table) and mice (bottom part of the table).
Besides the aforementioned pathway of maturation, iMo can either remain in the blood, or transition into patrolling monocytes (pMo) by the setting up of de novo enhancers and activation of “frosted” enhancers (19, 20) (Table 1). The mechanisms driving the conversion of iMo into pMo are just beginning to be elucidated. It appears that Delta-like 1 (Dll1) signal from endothelial cells by interacting with NOTCH2 only iMo favors their switch into pMo cells (21). These data clearly indicate that iMo and pMo monocytes are biologically intertwined, corroborating observation obtained at the epigenetic level, which indicated that both monocyte subsets use the same promoter repertoire and minimally differ in their chromatin organization (19). Of course, this scenario raises several questions: are the iMo infiltrating the tissues able to reprogram into pMo? Is this switch tissue dependent? Can we interfere with this reprogramming to control the transition? Is there any factor maintaining iMo reservoir? Are pMo thus originated able to re-enter the blood stream? Are monocyte-derived macrophages transcriptionally similar to pMo? In mice, iMo can give rise to pMo, even though this does not rule out the presence of an alternative route for pMo development, independent from the iMo subset (8). Indeed, genetic evidence for this transition do exist. Two myeloid-determining transcription factors, like interferon regulatory factor 8 (IRF8), and the downstream Kruppel-like factor (KLF4), have been shown to regulate iMo generation without affecting the pMo numbers. Moreover, studies conducted on either global IRF8−/−mice, or fetal liver transplant of KLF4−/−cells into irradiated wild type mice, indicate a drastic reduced numbers of iMo in the bone marrow, while maintaining relatively normal pMo numbers (22). These findings suggest a pathway for pMo development untied from the iMo subset, probably originating directly from the common monocyte progenitor (cMoP). The identification of the transcription factors nerve growth factor IB (NR4A1) has helped to withstand the hypothesis that pMo can arise independently from iMo monocytes. On the other hand, recent single-cell RNA sequencing of murine and human monocytes indicate that circulating iMo and pMo represent, under physiological conditions, a nearly homogenous populations (19). Interestingly, data recently published (23) combining single profile and functional and phenotypic characterization, showed that monocytes subsets (defined as classical, intermediate, and non-classical) isolated from peripheral blood of both healthy mice and humans, can be further divided into two additional populations: one group expressing classical monocyte genes and also cytotoxic genes and the other one with undefined activity. Other studies conducted in human and mouse lung cancer samples (24) have showed that several tumor-infiltrating myeloid populations (TIM) and among those monocytes are uniquely associated with the disease and with clinical progress, highlighting the potential to use TIM as immunotherapeutic targets. We think that the multiple cell subsets identified in the aforementioned manuscripts, should be tested for their functional relevance in tumor progression, in disease progression and their abundance should be correlated with therapeutic response. Of course, the correlation between human and mouse TIM will help to achieve these goals with the ultimate purpose of gaining more insight into monocytes and monocyte-dependent therapies.
In contrast, patrolling monocytes (pMo) represent a more differentiated subset and are marked by the higher surface expression of CX3CR1. pMo express low levels of Ly6C in mice and are CD14loCD16hi in humans; they routinely check the vessels under physiological conditions through the engagement of an LFA/ICAM-dependent crawling mechanism with resting endothelial cells (25, 26). This patrolling behavior of pMo can be observed throughout the interdigitated system of capillaries, arterioles, and venules. Similarly, human CD14lo CD16hi pMo show patrolling behavior when adoptively transferred into immuno-compromised mice (27). The crawling features of pMo allows them to efficiently sense particles, on the one hand, and on the other hand to monitor of endothelial cell integrity. However, these cells are not restricted to the vessels as pMo also undergo diapedesis and can be identified within the parenchyma of multiple tissues (7). pMo display a longer lifespan at the steady state compared to iMo and they are, also for this reason, found in the blood, at any given time, more abundant than their counterpart (5, 28). Interestingly, pMo cells are strongly susceptible of the physiological status of the organism and therefore they might represent a potential diagnostic tool (29). Nevertheless, how the fine balance between iMo and pMo levels and differentiation capacity is maintained/regulated during severe inflammation, autoimmune diseases, and cancer is just began to be elucidated.
Under pathological conditions, such inflammation and cancer, the rapid recruitment of myeloid cells to sites of injury stimulates a constant development and mobilization of cells from the bone marrow, causing a state of “emergency” that might generate monocytes from different ways or precursors. These include monocytes that have circumvented the canonical MDP-cMoP-monocyte developmental axes and resemble neutrophil-like iMo derived from GMP precursors (30). An additional example of a recently described monocyte subset that appears under inflammatory conditions is the segregated nucleus-containing atypical pMo (SatM) (31). SatM and neutrophil-like monocytes represent a minor pool of monocyte subsets under steady-state (19, 30, 31) (Table 1), yet become conspicuous during inflammation (30, 31). At present, the lack of reliable surface markers, associated with a deep epigenetic and transcriptional profile unable to make a clear distinguish between neutrophil-like iMo identified using GFI1/IRF8-reporter mice (30) from classical iMo and SatM (identified as Ly6ClowCeacam1hiF4/80−Mac1hi), from non-classical pMo (31). The limited whole-cell proteomic data available so far showed, for example, that SatM cells contain granules expressing granulocyte-related protein, like myeloperoxidase (MPO) and neutrophil elastase (NE) (31) (Figure 1). These cells are related to fibrosic responses; in fact, adoptive transfer of SatM monocytes into bleomycin-treated mice exacerbates fibrosis. Furthermore, it was shown that chimeric mice, lacking the CCAAT/enhancer-binding protein beta (Cebpb) in the hematopoietic progenitors were resistant to fibrosis when exposed to bleomycin but they had unaltered inflammatory response, further supporting the role of SatM in sustaining the mechanism of fibrosis (31). Nevertheless, more studies will be required to uncover the origin of these subsets and their involvement in different pathologies. To add more complexity, a recent work by Hanna et al. (32), showed that a subset of circulating pMo, but not iMo, accumulate at the site of tumor where they display an anti-tumoral role, by directly engulfing cancer cells and by releasing factors which in turn activate cytotoxic natural killer cells (NK). Are these extravasating pMo similar to their blood counterpart? How the findings from Hanna et al. (32) correlate with observations that extravasating pMo can differentiate into macrophages during cancer? Are pMo “corrupted” by tumor cells once they have extravasated within tissues and switched to pro-tumoral cells? Are iMo and pMo competing for the access to the tumor site? Are iMo suppressing the anti-tumor function of pMo? Indeed, this mechanism is also consistent with the data from the pMo adoptive transfer experiments since pMo appear to act early during seeding but not after establishment of metastatic foci despite their continued accumulation. Therefore, under pathological conditions it still remains possible that pMo derive from either blood iMo, via the formation of an intermediate Ly6Cint monocyte, or from bone marrow Ly6C+ monocytes, from an independent bone marrow monocyte progenitor, or from a combination of all these pathways (Figure 1). In order to solve all these issues a detailed understanding of the factors and pathways regulating the development and survival of both iMo and pMo populations in specific inflammatory settings is necessary.
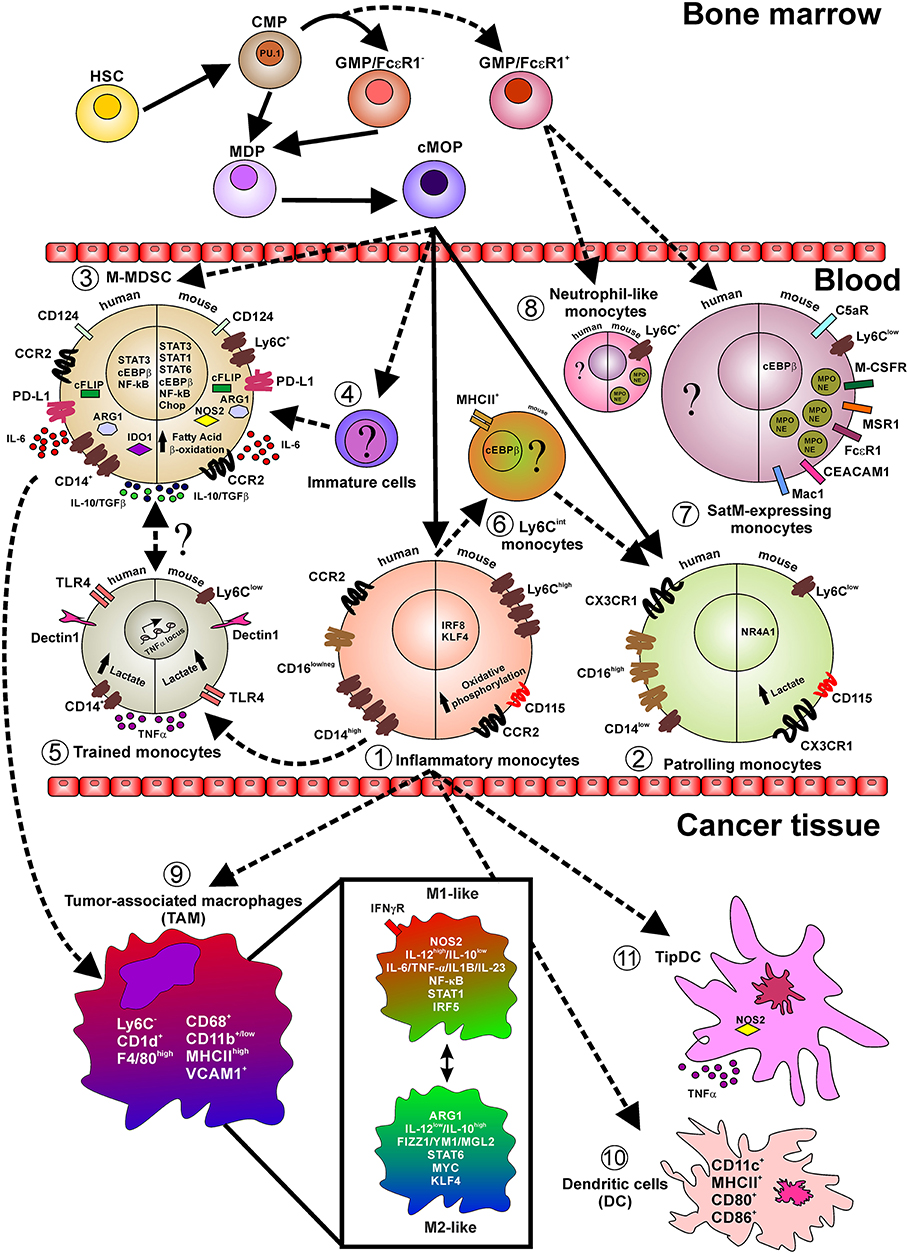
Figure 1. Layout depicting the monocyte lineage precursors (on top), the monocyte subsets in the peripheral blood (center), and monocyte fate in cancer tissues. Indicated are relevant surface markers, transcription factors, secreted cytokines, intracellular mediators, and relevant metabolic pathways. Continuous lines indicate events occurring during normal myelopoiesis while shaded lines indicate events in emergency myelopiesis (e.g., cancer and inflammation). Briefly, under steady state cMop precursors originate both inflammatory (1) and patrolling (2) monocytes, both in humans and mice. However, it has been reported that during emergency myelopoiesis cMop precursors can also differentiate into M-MDSC (3) and into not yet defined immature cells (4). Particularly, during infection, inflammatory monocytes acquire a trained phenotype (5) and also switch into Ly6Cint cells (6) only identified in mice and with not fully defined function, transcriptional regulators, and markers. During fibrosis a novel subset of monocytes, so called SatM (8), have been characterized, in mice, defined as Ly6C+ and expressing proteins typical of the neutrophil granules (MPO and NE). These cells, together with neutrophil-like monocytes (7), found in peripheral blood of mice during microbial infection and in the bone morrow in steady-state condition, originate from GMP/FcεRI+ precursors cells in the bone marrow. In pathological conditions, like cancer, inflammatory monocytes infiltrating the tissue give rise to TAM (9) which in turn represent a fultifaced population of macrophages. Additionally, inflammatory monocytes can also differentiate into classical DC (10) expressing the costimulatory molecules CD80 and CD86 and TipDC (11) expressing high level of NOS2 and TNFα.
As mentioned before, adoptive transfer and fate-mapping studies support the hypothesis that monocytes develop along a differentiation continuum in which inflammatory monocytes give rise to the patrolling subset in the circulation (3, 5). Development of monocytes from bone marrow progenitors combines the regulated expression of numerous transcription factors, with the contribution of growth factors and cytokines (33). Monocytes and neutrophils are both derived from hematopoietic stem cells (HSCs) via common myeloid progenitors (CMPs), which can originate from granulocyte-monocyte progenitor (GMP) and monocytes/macrophages, DC precursors (MDPs) (34, 35) (Figure 1). The commitment toward monocytes is characterized by three major lineage-determining factors (LDTF): PU.1, CCAAT/enhancer-binding protein beta (Cebpb) and Cebpa. PU.1 is a master regulator of myeloid and lymphoid cell development (36). The genetic ablation in mice of PU.1 promotes a lethal embryonic phenotype and the transfer of PU.1 mutated stem cells favors an altered myelopoiesis characterized by a robust contraction in both monocytes and DCs (37, 38). By binding closed chromatin through its C-terminal DNA-binding domain (39), PU.1 acts as a coordinator for the activation of selected genomic regions in collaboration with monocyte-associated transcriptional factors such as the IRF8 and KLF4 (40). Moreover, PU.1 synergistically cooperates with C/EBP-δ to activate the promoters of interleukin-6 (IL-6) and CCL5 (41) and transactivates the human macrophage colony-stimulating factor receptor (M-CSFR) promoter via the c-Jun pathway (42). Proteins of the C/EBP family radically impact the myeloid cells development. Since in Cebpa-deficient mice the transition from CMPs to GMPs is completely abrogated, these mice lack the granulocytic compartment (43, 44) indicating that C/EBPα is the master regulator of steady-state granulopoiesis.
Additionally, during myelopoiesis, C/EBPα controls and activates the myeloid-associated gene expression program by binding to either promoters or enhancers of myeloid-related genes, such as colony stimulating factor 3 receptor (CSF3R), growth factor independent-1 (GFI-1), interleukin 6 receptor (IL-6R), or C/EBPε, both in mice and in human stem cells (45). C/EBPα inhibits specific transcriptional factors attenuating the expression of non-myeloid lineage genes (46, 47). Moreover, genetically enforced inducible C/EBPα expression in GMPs by tamoxifen administration favors monocyte development demonstrating the critical role of this transcriptional factor during monopoiesis (48). By contrast, C/EBPβ is not necessary for steady-state granulopoiesis. However, C/EBPβ was recently identified as the key factor during the epigenetic default differentiation of iMo monocytes into pMo cells under steady state condition, highlighting the multifaceted role of this transcriptional factor during monocyte development (19). In this regard, a key contribution of C/EBPβ to mylopoiesis is highlighted by data showing that mice knockout for Cebpb have a dramatic reduction in circulating monocytes (19). C/EBPβ is also the master regulator of emergency myelopoiesis; in fact, inflammatory signals (i.e., cytokine stimulation) strongly induce the downregulation of all members of C/EBP family except for C/EBPβ (43). Under pathological condition, like fibrosis, a C/EBPβ-associated gene program in FcεR1+ GMPs progenitors promote the development of alternative monocytes, like SatM, previously described. As we will discuss below, C/EBPβ-driven programs are also activated in cancer-educated myeloid cells.
Myeloid-Derived Suppressor Cells
In cancer, tumor-derived soluble factors, such as growth factors, cytokines, chemokines, and tumor-derived exosomes, not only support an increased recruitment of monocytes from bone marrow to tumor-microenvironment bypassing the canonical monocyte development but, also, favor the acquisition of immunosuppressive features in myeloid cells. To highlight these acquired functional properties, myeloid cells comprising monocytes, neutrophils, and immature cells were named myeloid-derived suppressor cells (MDSCs) (49, 50). Although this terminology generated some controversies, it represents a useful ground for scientific researches on altered hematopoiesis. It relies on the concept that myelopoiesis in pathology might give rise to cellular subsets that can share some markers with the cells present under steady state but are functionally and molecularly distinct, sharing the property of negatively regulating effectors of adaptive and innate immunity. The monocytic-MDSC (M-MDSC) subset is broadly defined in mice as Ly6C+CD11b+ cells and in human as CD14+HLA-DR− or CD14+CD124+ cells and is endowed with a stronger ability to arrest T cell response, when compared to the granulocytic-MDSC (G-MDSC) counterpart (51, 52), in part dependent on the activation of two enzymes, arginase 1 (ARG1) and inducible nitric oxide synthases (NOS2/iNOS), which are directly regulated by C/EBPβ expression (53) (Figure 1; Table 1). We demonstrated that, in tumor-bearing mice, both the expansion and the immunosuppressive function of MDSCs are abrogated in the absence of C/EBPβ, resulting in restricted tumor spread (53). These data confirm the central role of C/EBPβ in tumor-associated inflammation, underscoring it as promising therapeutic target to develop new approach to limit cancer progression. Both human and mouse M-MDSCs secrete immunoregulatory cytokines, like IL-10, TGFβ, and IL-6 and present and array of molecules, such as ARG1, FADD-like IL-1β-converting enzyme-inhibitory protein (c-FLIP), indoleamine 2,3-dioxygenase 1 (IDO1), and nitric oxide synthase 2 (NOS2), which can contribute to the suppressive activity of these cells. Mechanistically, IL-6, for example, activates PI3Kγ, which stimulates mTOR, S6Kα, and C/EBPβ-mediated anti-inflammatory gene expression and inhibits NFkB-mediated pro-inflammatory gene expression, thereby promoting the immune suppressive function of these cells mediated by, but not limited to, IL-10, TGFβ, and ARG1 (54, 55). In particular, Arg1 gene in MDSCs is strictly controlled by several inducible transcriptional factors able to recognize sequences characterized by high content in GC that impacts the nucleosomal stability (56), such as signal transducer and activator of transcription 3 (STAT3), IRF8, as well as CHOP, PU.1, KLF4, and activator-protein 1 (AP-1) (57). Moreover, STAT3 promotes both expansion and survival of M-MDSCs through Bcl-XL, c-Myc, and Cyclin D1 expression (58) as well as the induction of several immune regulatory mediators like bFGF, HGF, VEGF, IL-1β, MMP9, CCL2, and CXCL2 (50). Interestingly, phosphorylated STAT3 binds to multiple sites in the Arg1 promoter, suggesting that STAT3 inhibitors, like Stattic, could reduce ARG1 dependent immunosuppression by dampening the expression of Arg1 mRNA (59). Within the tumor environment, ARG1 can cooperate with NOS2 to produce high levels of superoxide anion () that can react with either nitric oxide (NO) or H2O generating reactive–nitrogen species (RNS), such as peroxinitrites (ONOO−), which damage both the function and migration of T cells to tumor site (60), and reactive-oxygen species (ROS), such as H2O2 which decreases T cellular CD3ζ expression limiting the activation of T cells, respectively (61). However, ARG1 has a hierarchically dominant negative role compared to NOS2 in developing an immunosuppressive tumor microenvironment by limiting the activity of monocyte-derived NOS2-expressing and TNF-producing dendritic cells (defined as Tip-DCs) that can sustain and favor the anti-tumor effect of transferred T lymphocytes (62). An alternative way to reprogram MDSC differentiation and function is through the expression of p53. It was recently demonstrated that M-MDSC can be driven to differentiate into potent antigen-presenting, defined as Ly6C+CD103+ DCs by inflammation-induced activation of p53. In fact, mice with a targeted deletion of p53 in myeloid cells specifically loose the Ly6C+CD103+ population and became unresponsive to different forms of immunotherapy and immunogenic chemotherapy (63).
Recently, we demonstrated the ability of c-FLIP, which controls the extrinsic apoptotic pathway and caspase 8 activation (64), to re-program monocytes into MDSC-like cells (65). In fact, FLIP-expressing monocytes displayed impressive regulatory features both in vitro, constraining the activated T cell proliferation, and in vivo, controlling the development of graft vs. host disease in a xenogeneic mouse model. Indeed, enforced expression of c-FLIP in monocytes up-regulates MDSC-associated immunosuppressive genes, such as CD273, CD124, IL-6, IL-10, CFS3, PTGS2, and IDO1, as a result of a “steered” NF-kB activation induced by the nuclear co-localization of c-FLIP with NF-kB p50 (65). During the course of a disease, like cancer, MDSCs infiltrate the tumor, differentiating into tumor-associated macrophages (TAMs) which can sustain primary tumor growth and contribute to the pre-metastatic niche formation (Figure 1).
Trained Immunity and Metabolic Landscape of Monocyte Subsets
Recent studies have shown that during infection with some pathogens iMo can undergo extensive epigenetic, transcriptional, and metabolic reprogramming, with the functional consequence of an enhanced immune reactivity upon a second encounter, in other words they acquire an immunological memory. The existence of this innate immune memory was initially suggested by studies in mice deficient for functional T and B cells and exposed to mild C. albicans infection, which show protection against C. albicans reinfection by increased responsiveness of monocytes (66). Even though the requirements for monocyte training has been primarily investigated either in vitro or under in vivo steady state, trained monocytes seem to originate from iMo during emergency hematopoiesis by a profound epigenetic and metabolic rewiring (Table 1). Exposure of iMo to either C. albicans or β-glucan in vitro, induce profound genome-wide changes in epigenetic marks, including, but not restricted, histone H3 lysine 4 monomethylation (H3K4me1), trimethylation (H3K4me3), and H3 lysine 27 acetylation (H3K27ac) (67) as a consequence of Dectin-1/AKT/mTOR/HIF-1α signaling pathway activation and secretion of IL-6, IL-1β, and TNFα. Other studies instead, identified Bacille Calmette-Guérin (BCG) and peptidoglycan as potent inducers of the aforementioned trained-related epigenetic modifications, though a different mechanism dependent on nucleotide-binding oligomerization domain-containing protein 2 (NOD2) pathway and activation of NF-kB (68). Concomitantly to these epigenetic changes, a metabolic switch also occurs. Trained monocytes are mainly glycolytic (aerobic glycolysis) with impairment of the oxidative phosphorylation, production of lactate and disruption of the Krebs cycle at the level of both citrate, which is withdrawn for fatty acid biosynthesis, and succinate, which activates HIF-1α and consequently up regulates the expression of several pro-inflammatory cytokines, mainly IL-1β and TNF-α (69).
Beside microbial particles, products of the lipid metabolism were found to be activators of the trained immunity. Oxidized-low density lipoprotein (oxLDL), a damage-associated molecular pattern (DAMP), interacts with CD36 on myeloid cells leading to the activation of NLRP3 inflammasome and consequent production of IL-1β (70–72).
Although the role of epigenetic programing as a mechanism required to insure innate immune memory is becoming more clear, one crucial aspect still remains unanswered: what is the cellular process that induces and maintains such epigenetic changes? Initial evidences seem to suggest that metabolites might play a role since they can act as cofactors for the enzymes (mainly methylases, methyltransferases, histone deacetylases, and histone acetylases) involved in epigenetic modulation of gene transcription (67, 73, 74). Of course, more studies are required to fill the gap and also to deeply examine the role that different chromatin modifications have on the stability of the chromatin. It is expected that stable histone modifications (e.g., histone methylation) would be more suitable to maintain a functional modification than those with short half-life (e.g., histone acetylation). Thus, the long-lasting persistence of some histone modifications could reflect both the stability of such modifications or the persistent activation status of the signaling pathways and transcription factors upstream (75, 76). Understanding these regulations is a sine qua non for designing therapeutic intervention aimed at modulating trained immunity, to dampen it when in excess (e.g., organ rejection, autoimmunity, allergy, atherosclerosis) or enhance it when defective (e.g., cancer, infection).
A growing body of evidence suggests that the development of immune cells and their different effector functions are the results of a dynamic changes occurring at the metabolic level (77, 78). In mouse models of cancer, myeloid cells are metabolically influenced by tumor-derived factors to become MDSCs, helping to protect tumor from the effects of chemotherapy (79). Specifically, mouse MDSCs undergo a major metabolic reprogramming by switching off glycolysis and enhancing fatty acid β oxidation (FAO) pathway. This metabolic reprogramming is generally characterized by an up-regulation of lipid uptake receptors CD36 and Mrs1, an increase in FAO enzymes, mainly carnitine palmitoyltransferase 1 (CPT1) and 3-hydroxyacyl-Coa dehydrogenase (HADHA), and an increase in oxygen consumption. These events are associated with the activation of immunosuppressive pathways, namely upregulation of ARG1 and NOS2 synthesis and production of ONOO−, contributing to dampen T cell proliferation and IFNγ secretion (80–82). Blockade of FAO, both in vitro and in vivo in different tumor models, decreased the incorporation of fatty acid and ATP production, holding up the development of suppressive MDSCs (82) and leading to increased efficacy of chemotherapy and adoptive T cell therapy. Interestingly, fatty acid oxidation also plays an important role in regulating the inflammatory properties of iMo. Increased intracellular level of unsaturated fatty acid (arachidonic acid) was shown to stimulate the secretion of pro-inflammatory IL-1α by uncoupling the mitochondrial respiration (83, 84) thus exacerbating the pathogenesis of atherosclerosis (85). The relevance of changes in the lipid metabolism occurring during myeloid cells differentiation, was also recently demonstrated by Mitroulis et al., in an vivo model of trained immunity (13). Treatment with β-glucan determines an increase in gene expression of several enzymes involved in cholesterol biosynthesis and decrease in expression of Abca1, a transporter regulating cholesterol efflux (86). Consistently, β-glucan administration in mice not only upregulates CD131, a subunit of the receptor for IL-3, IL-5, and GM-CSF, expression in myeloid precursors, but also activates downstream signaling, as demonstrated by STAT5 phosphorylation. The capacity of β-glucan to enhance the biosynthesis of cholesteryl esters and significantly decrease glycerophospholipid-containing arachidonic fatty acid chains highlight the capacity of cells to alter their lipidome and, thus, the physicochemical features of their membranes. This adaptive response has direct consequences in the composition of cellular membranes (87) and consequently in cell signaling (88). In this regard, alterations in the quantitative and qualitative cholesterol composition of the membrane can impact the localization of CD131, its signaling (86, 89) and consequently the differentiation of specific myeloid subsets.
Amino acids, besides being the building block of several molecules, serve as essential precursors of different metabolites. Different studies have shown that glutamine metabolism into glutamate, α-ketoglutarate and succinate semialdehyde can fuel the synthesis of fumarate and succinate for the tricarboxylic acid cycle (TCA) (90). Inhibition of glutaminolysis, in mice, down regulates the production of pro-inflammatory cytokines in monocytes exposed to C. albicans, dampening the development of efficient trained monocytes triggered by β-glucan. In line with these observations, the biochemical catheterization of β-glucan trained monocytes has revealed that upon induction of the signaling cascade Dectin-1-Akt-mTOR-HIF-1α, a metabolic shift occurs leading to an increase aerobic glycolysis, glucose usage, lactate production, and TNF-α secretion (67).
Thus, it appears that modulation of metabolic landscape represents a fundamental step to unravel the functional consequences of different monocytes subsets helping to identify new strategies of intervention for the treatment of several patho-physiological conditions. It remains instead undefined whether cancer-derived factors can also generate trained monocytes and if these cells contribute to dampen the anti-tumor response or favor metastatic spread.
Monocyte Functions
Monocytes at the Primary Tumor
Tumor derived factors (TDFs) are key mediators in the crosstalk between monocytes and tumor cells. They are involved in monocyte recruitment from the hematopoietic organs in adult life, i.e., bone marrow and in part the spleen, survival, and differentiation within the tumor site. Tumor-released monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) was identified as the major TDF involved in iMo recruitment, through the CCL2-CCR2 axis, into several mouse and human tumors (91). Indeed, inhibition of CCL2-CCR2 signaling in a mouse model of breast cancer significantly impair iMo infiltration and reduce tumor growth and metastases (92). Several studies have described the presence of other chemokines within the tumor microenvironment (TME), including CCL3, CCL4, CCL5, CXCL12, and growth factors such as colony stimulating factor-1 (CSF1), which may also contribute to monocyte recruitment to tumors (93). Indeed, in both mouse (94) and human (91) tumors, cells secrete high level of CSF1 that is involved in recruitment, survival, and differentiation of monocytes. The inhibition of CSF1 signaling in an experimental model of lung carcinoma significantly reduced the number of mature TAMs due to impaired recruitment, proliferation and maturation of iMo cells (95). Moreover, CSF1R signaling blockade can also reprogram immunosuppressive TAMs prompting the differentiation of iMo in anti-tumoral M1-like TAMs (96). However, abrogation of CSF1R signaling, by either small molecules (97) or monoclonal antibodies (98), even though appealing, have so far demonstrated a limited anti-tumor effects. Only a recent work done by Kumar et al. (99) highlighted that the CSF1-dependent cross-talk between tumor cells and cancer-associated fibroblasts (CAF) might explain the inefficacy of such a treatment. Thus, combinatorial therapy targeting both CSF1R and CXCR2 seems to have more chances to generate an effective anti-tumor T cell response. Thus, the therapeutic focus has shifted to combinations of CSF1R inhibitors with other agents. Indeed, treatment with CSF1 inhibitor in combination with either paclitaxel or radiotherapy, is showing to improve the survival of mouse model of breast or prostate cancer, respectively (79, 100) and improve the efficacy of ACT when combined with anti-PD-1 and anti-CTLA4 in a pancreatic mouse model (101). It is becoming clear that cytotoxic therapies, for example, induce mammary epithelial cells to produce monocyte/macrophage recruitment factors, including CSF1 and interleukin-34 (IL-34), which together enhance CSF1R-dependent monocytes/macrophage infiltration, making its inhibition more effective.
One of the strongest stimuli inducing the secretion of TDFs is hypoxia. Indeed, during tumor growth the level of available O2 is significantly reduced, especially in the inner part of the neoplastic mass. This hypoxic microenvironment triggers HIF-1α stabilization in tumor cells and the consequent release of pro-angiogenic factors, such as growth factors (VEGF, PDGF, PIGF, ANG-2), chemokines (CXCL8, CXCL12), cytokines (TNFα, IL1β, TGFβ), and metalloproteases (MMPs), which results in the sprouting of new vessels supporting cancer cells growth (102, 103). In addition, hypoxia is a powerful monocyte and macrophage attractant. Through the release of VEGFB and PIGF, tumor cells can enhance haematopoiesis and monocyte recruitment (104). In addition, angiopoietin-2 (ANG-2) is able to recruit circulating Tie2-expressing monocytes (TEMs) that inhibit apoptosis in both tumor and endothelial cells, by mechanisms depending on TNFα release (105), and exhibits an essential pro-angiogenetic role with a not completely clarified mechanism. Interestingly, it was recently discovered that a small subgroup of recruited iMo can be educated by VEGF, and exert their proangiogenic function, supporting the formation of capillaries and larger vessels, as short-lived monocytes without becoming macrophages (106). Secreted VEGF promotes the acquisition of immunosuppressive features in monocytes generating M-MDSCs by upregulating both ARG1 and iNOS through hypoxia response elements and NF-kB (107). In addition, MDSCs can fuel this circuit by releasing MMP-9, which induces VEGF release from ECM (108). Therefore, targeting VEGF/VEGFR has received attention as a strategy to interfere with monocyte-driven angiogenesis. Moreover, blocking this axis will affect monocytes recruitment to the tumor site (109), and favor the conversion from predominant suppressive to anti-tumoral monocytes (110). An additional way to interfere with MDSC differentiation is by interfering with p53 expression, as mentioned before (63).
In the last few years, extracellular vesicles (EVs) emerged among the TDFs as additional determinants in the formation of TME, both at primary and metastatic sites (111). EVs are a heterogeneous group of membrane vesicles mainly composed of exosomes and microvesicles. Interestingly, EVs released by tumor cells (tEVs) and tumor-derived exosomes (TEX) interact with immune cells inducing their switch toward a pro-tumoral phenotype. Particularly, exosomes target monocytes altering their normal function by several mechanisms. In melanoma and colon cancer, TEX block peripheral iMo differentiation into DCs, and favor the acquisition of a peculiar phenotype reminiscent of M-MDSCs and characterized by decreased expression of HLA-DR and costimulatory molecules (112). A similar modulation of monocytes has been described in many other malignancies, including pancreatic cancer, bladder carcinoma, glioblastoma, and multiple myeloma (113–115) and it is often associated with increased cytokine secretion, i.e., CCL2, CCL4, and IL-6, as well as programmed death-ligand 1 (PD-L1) expression (116). Moreover, glioblastoma-derived EVs may skew the differentiation of peripheral blood monocytes to alternatively activated M2 macrophages inducing the expression of elevated levels of VEGF, IL6, Cox2, ARG1, and PD-L1 through STAT3 activation (117). Interestingly, in gastric cancer, TEX effectively educated monocytes to differentiate into a peculiar type of M2 TAM expressing PD-1, which induce T cell dysfunction through IL-10 secretion by interacting directly with PD-L1+ cells and thereby promote tumor progression (118). TEX have been described to contain miRNA which can be transferred to target cells and modulate cellular function (119). These data strongly highlight the role of TEX as additional mediators of monocyte dysfunction in TME. In the last few years, significant advances in understanding the mechanisms associated with exosome biogenesis/release have been obtained identifying some possible targets to interfere with this cell-cell communication. Recent studies in mouse models demonstrated that RAB27A or RAB35 inhibition significantly impair TEX secretion in HeLa cervical carcinoma and Oli-Neu oligodendroglial precursor cell lines (120), respectively. Moreover, RAB27A deficient tumor cell lines displayed reduced growth due to impaired recruitment of bone-marrow derived, pro-tumoral immune cells (121).
In addition to be modulated by soluble factors released by the tumor cells, monocytes can shape their effector function also in a contact-dependent manner. For instance, breast cancer stem cells express CD90 and Ephrin A4 receptor (EphA4R) that interact with CD11b and EphA4 present on tumor-associated monocytes and macrophages, respectively, leading to the secretion of inflammatory cytokine (IL-6, IL-8, GM-CSF), which in turn sustain tumor stem cell fate (122).
Among factors that could shape monocyte plasticity, TGFβ, a key multifunctional cytokine, involved in both cancer and inflammation, appears to play a key role. Besides being targeted in a number of human diseases (123), TGFβ has a very well-recognized ability to regulate T cell responses (124), supporting Th9 (125, 126) and Th17 (127) differentiation, and promoting regulatory T cell function (128–130). However, how TGFβ regulates innate immune responses just began to be appreciated. Many TME-associated cells, and among those monocytes and macrophages, express high amount of latent LTGFβ. The recent work by Kelly et al. (131), demonstrates that iMo, beside tumor cells, express high levels of αvβ8 integrin responsible for the activation TGFβ from LTGFβ form (131, 132). Additionally, monocyte-derived macrophages, integrin expression and TGFβ signaling are generally maintained in anti-inflammatory macrophages but down-modulated in pro-inflammatory macrophages. To sustain the immunosuppressive microenvironment, tumor cells exploit this regulatory mechanism upregulating the expression of integrin αvβ8 and activating TGFβ from LTGFβ-expressing monocytes and macrophages (133).
Monocytes at the Metastatic Niche
Cancer metastasis is a multi-step process of the neoplastic progression termed “invasion-metastasis cascade” (134, 135). Monocytes are, among other myeloid cells (e.g., neutrophils), corrupted to foster tumor progression and metastasis. Accumulating evidence indicates that monocytes (primarily iMo) are essential pre-metastatic promoters being rapidly recruited from the bone marrow to the pre-metastatic niche, mainly by CCL2/CCR2 axis (136, 137), where they promote tumor colonization by secreting angiogenic factors, like VEGFA (92, 138). Indeed, in the MMTV-PyMT mouse model of spontaneous breast cancer, CCL2, released by either tumor cells or stromal cells at the metastatic lung niche, induces the recruitment of CCR2-expressing iMo, which in turn favor the extravasation of tumor cells, through the release of VEGFA (92). Consequently, the inhibition of CCL2-CCR2 signaling axis abrogated the recruitment of monocytes thus reducing metastasis formation. Similarly, in a mouse model of metastatic melanoma (B16F10 model), accumulation of CXCR3+monocytes/macrophages in the lung was a prerequisite to mediate melanoma engraftment and metastatic disease (139). However, how this process takes place remains undefined. In a different set of experiments employing B16F10 melanoma model, it was shown that M-MDSCs were recruited to the pre-metastatic niche, mainly by CCL12 expression. By releasing IL-1β, these cells promoted the expression of E-selectin on endothelial cells thus promoting the adhesion of tumor cells to the vascular endothelium (140).
At the metastatic targeted-organs, monocytes can offer survival stimuli for cancer cells. Metastatic cells in the lung, from either mouse or human breast cancer, overexpress vascular cell adhesion molecule-1 (VCAM-1) and shRNA-mediated depletion of VCAM-1 inhibited metastasis formation. Moreover, monocytic cells expressing α4-integrin can bind VCAM-1 present on the surface of tumor cells. Thus, upon α4-integrin engagement on monocytes, VCAM-1 delivers anti-apoptotic signals into breast cancer cells through the PI3K/Akt pathway favoring tumor cell survival (141).
EVs can cross the basal lamina of alveolar capillaries in the lung. In the lungs, alveolar and interstitial macrophages upon taking up these EVs start to secret CCL2 favoring the recruitment of iMo, which in turn differentiate into macrophages, mostly M2-like cells, promoting tumor growth by the secretion of IL-6 and deposition of fibrin (142). Similarly, in colorectal cancer (CRC) patients the high expression of serum exosomal-derived miR-203 was associated with increased probability to develop distant metastases. It was shown that TEX-derived miR-203 uptaken by monocytes promoted their differentiation into M2-like macrophages, in vitro. Furthermore, mice injected with CRC cells transfected with miR-203 developed significantly more liver metastases than the control group (143).
We have previously underlined the plasticity of monocytes and how iMo and pMo play different roles in cancer progression and surveillance. In line with these observations, it has been demonstrated that TEX from poorly-metastatic melanoma cells are taken up by bone-marrow monocytes, promoting their differentiation into pMo, which in turn migrate at the metastatic niche clearing tumor cells by direct engulfment or by activating cytotoxic NK cells (144). Interestingly, TEX from poorly-metastatic tumors caused macrophage alteration toward M1-like cells expressing TRAIL, which competed with NK cells for tumor killing. These findings suggest that prior to the acquisition of the metastatic capacity, tumors continuously alert host immune system by producing vesicles that affect innate immune responses and support the concept of developing new cancer immunotherapeutic approach based on TEX to deliver specifically immune triggers.
Future Prospective: Targeting of Monocytes as the New Frontier for Cancer Immunotherapy
Myeloid cells are extremely plastic and can develop specialized functions in response to micro-environmental pathologic conditions such as infections, autoimmunity or cancer (145). Myeloid cell polarization into either tumor-suppressive or tumor-promoting phenotypes is fundamental for shaping TME. Once at the tumor site, these myeloid cells generally acquire a pro-tumor phenotype (146). Thus, one of the major goals of contemporary tumor immunotherapy is targeting tumor-associated myeloid cells by depletion, recruitment inhibition or reprogramming their polarization/activation status.
As mentioned above, inhibition of the CCL2-CCR2 axis, used to prevent the egression of monocytes from bone marrow, improved the efficacy of chemo-, radio- and immune-therapy in several preclinical models. Nevertheless, the use of either CCL2 or CCR2 inhibitors, in clinical trials, gave disappointing results, indicating the need of supplementary studies considering the presence of potential TME-dependent compensatory mechanisms acting on tumor-resident myeloid cells (146, 147). Moreover, although the continuous blockade of macrophages constrains tumor progression, cessation of the CCL2 blocking therapy stimulates them to a rapid rebound, leading to accelerated metastatic disease via a mechanism dependent on VEGF-A and IL-6 production monocyte-derived by macrophages (102).
Targeting Trained Immunity
Trained immunity inducing factors were tested for their anti-tumor activity, both in vitro and in vivo, and some of them reached the clinical application. A β-glucan PAMP, Imprime PGG (Imprime), is currently in clinical development in combination with immune checkpoint inhibitors, tumor-targeting antibodies, and anti-angiogenic antibodies. The results from a randomized phase 2 clinical trial of Imprime in combination with bevacizumab and carboplatin/paclitaxel vs. bevacizumab and chemotherapy alone in the 1st-line treatment of stage IV non-small cell lung cancer showed promising efficacy in terms of both objective tumor response and survival (ClinicalTrials.gov NCT 00874107, EudraCT 2008-006780-37). Earlier results have shown that both the M2 macrophages and DCs derived from Imprime-trained monocytes have higher expression of PD-L1 and CD86, rendering these cells suitable for treatment with anti-PD-1 antibody. Ex vivo treatment of T cells with Nivolumab, an anti-PD-1 antibody, enhanced proliferation in response to αCD3/αCD28 stimulation and co-culture with Imprime-trained monocytes-derived M2 macrophages or DCs further improved T cell expansion and increased production of several cytokines, including IFNγ, IL-2, TNF-α, and GM-CSF. Results were further validated in syngeneic mouse model, like the CT26 colon carcinoma. Bacillus Calmette-Guerin (BCG), another trained immunity inducer, is currently the only agent approved by the US Food and Drug Administration for first line treatment of carcinoma in situ of the bladder. BCG therapy reduces the risk of recurrence and maintenance therapy with BCG decreases the risk of progression in patients with high-grade, non–muscle invasive bladder cancer (148, 149). It has been speculated that the mechanism of action involves the autophagy. In fact, pharmacologic or genetic inhibition of autophagy blocks the epigenetic reprogramming of monocytes at the level of H3K4 trimethylation, arresting the mechanism of trained immunity induced in vitro by BCG. Single nucleotide polymorphisms associated with bladder cancer progression and recurrence, in the autophagy genes ATG2B (rs3759601) and ATG5 (rs2245214), affected both the in vitro and in vivo training effect of BCG (150).
Muramyl dipeptide (MDP), a synthetic peptide of N-acetyl muramic acid attached to a short amino acid chain of L-Ala-D-isoGln, is a bacterial cell wall peptidoglycan active as NOD2 agonist and contributing to the generation of trained monocytes (151). Interestingly, Paclitaxel conjugated to MDP showed not only antitumor activity, but also immune enhancement capacity. In fact, compared with either paclitaxel or MDP alone, the combination significantly increased the expression and secretion of TNFα and IL-12 from mouse peritoneal monocytes (152). Moreover, it was shown that MDP can upregulate PD-L1 in healthy monocytes, but in patients with Crohn's disease, carrying the Leu1007 frameshift mutation of the NOD2 gene, such effect was completely lost (153) (Table 2).
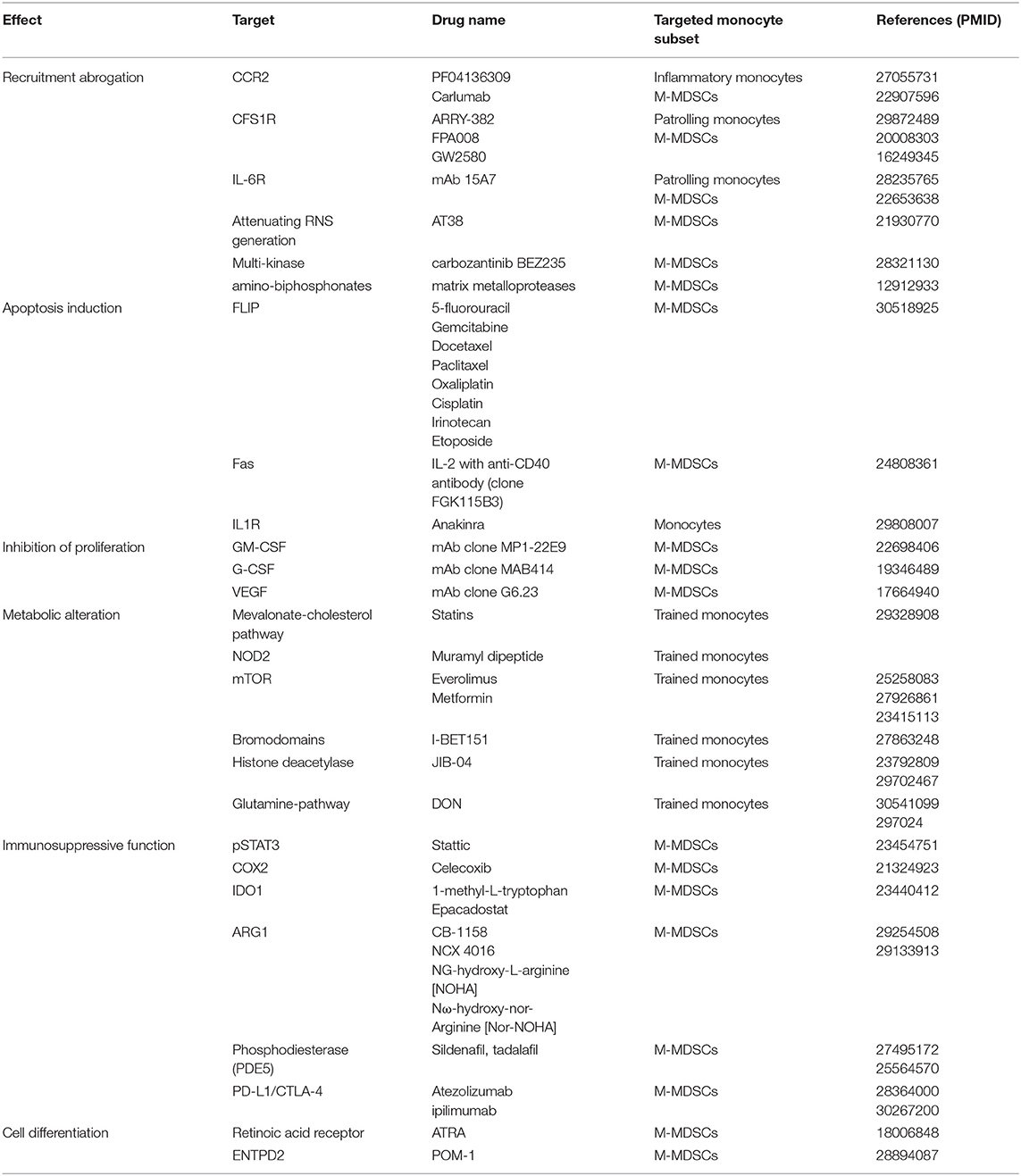
Table 2. Inhibitors and their corresponding targets found to impact pathways regulating different aspects of monocyte biology.
Targeting Signaling Pathways
An alternative approach to target trained immunity is to inhibit the pathway Dectin-1-Akt-mTOR-HIF-1α. To this end, the beneficial effects of Metformin and Everolimus, an mTOR activator rapamycin analog, administration to patients with type 2 diabetes and cancer, respectively, were linked with the modulation of trained monocytes (67, 90, 154). Interestingly, inhibitors of other kinases, such as Raf-1, PI3K, and ERK are of particular interest in modulating trained monocytes because they represent downstream effectors of Dectin-1 and NOD2 activation (Table 2). In particular the knockout of PI3Kγ was reported to break tumor tolerance by MDSC reduction (54) as well as, the combination targeting of PI3Kδ in association with PD-L1- based immunotherapy better limited tumor progression (155, 156). Moreover, the pharmacological treatment using multi-kinase inhibitors carbozantinib and BEZ235, which limit MDSC accumulation, in combination with immune checkpoint therapy controlled more efficiently tumor growth in a castration-resistant prostate tumor model than the single agents (157) underlying the possibility to overcame de novo resistance to antibody blockade based therapy by limiting MDSCs.
The development of epigenetic modulators is acquiring increased interest due to the relevance of epigenetic changed in several diseases. The broad jumonji histone demethylase inhibitor JIB-04 decreased trained immunity response by modulating of the methylation status of H3K9 (158). Interestingly, a clinically relevant small molecule of the BET family of bromodomains, I-BET151, was shown to prevent monocyte tolerance when administered concomitantly with LPS, but it was ineffective when administered after LPS stimulation. These results suggest that I-BET151 is not an effective treatment in monocytes that have already experienced an inflammatory response (159) (Table 2).
Targeting Metabolic Pathways
Metabolic modulation also represents another interesting approach to target both trained monocytes and M-MDSCs. Some metabolites and metabolic enzymes function as either substrates or cofactors for chromatin modifying enzymes, thereby influencing the epigenetic landscape of target cells. Accordingly, fumarate can increase trained immunity by increasing H3K4me3 and H3K27ac and inhibiting the degradation of HIF-1α (90). Moreover, the decreased expression of lysine demethylase 5 family of HDAC (KDM5), responsible for H3K4 demethylation, is also inhibited by fumarate, maintaining then the accessibility of the chromatin. On the other hand, it was shown that tolerant monocytes lack the activity of KDM5, whose function can be restored by its cofactor, α-ketoglutarate (90). Mevalonate, intermediate of the cholesterol pathway has been shown to induce trained immunity (74), consequently, statins can be used to prevent this process under conditions in which accumulation of trained monocytes is detrimental, like patient with hyper-IgD syndrome or inflammatory conditions. Additionally, given the enhanced glutamynolysis associated to trained immunity (90), administration of 6-diazo-5-oxo-L-norleucine (DON), which inhibits glutamine uptake and metabolism, was shown to have favorable effects after organ transplant preventing rejection (160) (Table 2). Even though targeting metabolic pathways, to modulate trained monocyte function, is feasible, toxicity, and side effects represent the main drawbacks of metabolic drugs. Overcoming this limitation, for example, by delivering drugs trough nanoparticles/nanocarriers, could open the access to a variety of molecules that have already demonstrated their efficacy in vitro.
Targeting Immunosuppressive Monocytes (M-MDSC)
Together with trained monocytes, MDSCs represent the other group of targetable cells. Proliferating T cells need a large supply of amino acid like L-arginine and L-tryptophan. MDSCs have developed a strategy to modulate local concentrations of these amino acids via the up regulation of enzymes involved in their degradation like ARG1, NOS2, and IDO. Developing inhibitors of these enzymes represent a field of intense research. To this end, nitroaspirin, consisting of a nitric oxide group covalently linked to aspirin, was shown to restore L-arginine levels in T cells, by suppressing the production of ROS and iNOS (161). Moreover, our laboratory showed that treatment with AT38 [3-(aminocarbonyl) furoxan-4-yl]methyl salicylate], decreased MDSC-induced nitration within the tumor environment, increasing CCL2 binding and T cell tumor infiltration in mice (60). N-hydroxy-L-arginine (NOHA) is an intermediate in the conversion of arginine to citrulline and NO by iNOS (162) (Table 2). It is a potent physiologic inhibitor of ARG1. Mice exposed to NOHA demonstrated inhibition of MDSC function, and mice with B cell lymphoma treated with NOHA had decreased numbers of circulating Treg cells and improved immune responses to the cancer (163). We also demonstrated that the production of polyamine by ARG1 activity promotes IDO1 activation through Src kinase signaling (164). Therefore, a combined targeting of ARG1 and IDO1 using pharmacological compounds (165) could be an effective treatment to constrain tumor-associated immunosuppression improving cancer immunotherapy. MDSCs generate an immunosuppressive environment also by producing prostaglandin E2 (PGE2), which levels are regulated through the enzyme Cox2 (166). PGE2 activates PGE2-R on MDSCs altering the differentiation of MDSCs. In the bone marrow, activation of PGE2-R hampers the differentiation of monocytes into antigen presenting cells, while increased PGE2 switch monocytes into MDSCs via increased expression of IDO, IL-4Rα, iNOS, and IL-10 (167, 168). In line with these findings, blocking the production of PGE2 in mice bearing lung carcinoma, with Cox2 inhibitors, decreased the expression of ARG1 in MDSC and tumor growth (169). Cox2 inhibitors may also provide other antitumor effects (Table 2). Celecoxib, a Cox2 inhibitors, was shown to decrease MDSC recruitment and increased CD8+ T cell tumor infiltration in gliomas and colon carcinoma by decreasing CCL2 production (170).
Methionine, an essential amino acid for normal T cells function, is generally supplied by antigen presenting cells. DCs and macrophages import cysteine to create methionine, which is then secreted; they additionally release thioredoxin converting cysteine to methionine. However, in the TME, MDSCs transport cysteine intracellularly thus depleting T cells of methionine (171, 172). Consequently, blocking thioredoxin could prevent T cells proliferation arrest. In this situation small molecules or neutralizing antibodies targeting extracellularly released enzymes could be beneficial to restore T cells function and used in combination with ACT of tumor specific T cells or with check point inhibitors.
Constitutive activation of the JAK-STAT pathway has been implicated in the proliferation of MDSCs via anti-apoptotic and pro-proliferative genes (173). Moreover, ARG1 and iNOS, immunosuppressive enzymes in MDSCs, are controlled via STAT1 and STAT3 (174). Consequently, inhibition of the JAK-STAT pathway has been of great interest. Many inhibitors of STAT1/STAT3 have been discovered and several of them already enter clinical trials. Among those one, Stattic, an inhibitor of pSTAT3, reduces the suppressive activity of MDSC in vitro both in mice and in humans (175, 176) (Table 2).
In the last 10 years, several approaches to target immunosuppressive monocyte referred as M-MDSC were developed using a large spectrum of pharmacological compounds and immunotherapeutic approaches with the aim to limit MDSC proliferation, function, and recruitment to tumor site. For instance, non-therapeutic low doses of conventional chemotherapeutic drugs such as gemcitabine and 5-fluorouracil [which does not induce immunogenic cancer-cell death of tumor cells (177)] were able to limit Ly6C+MDSC number and activity by inducing c-FLIP down-regulation (65). The M-MDSC elimination is essential to restore the immune response in tumor-bearing mice by rescuing the frequency of circulating anti-tumor T cells (51). These data obtained in preclinical cancer models were also confirmed in tumor patients. In fact, renal cell cancer (RCC) patients with low frequency of M-MDSCs showed a better disease control after anti-tumor peptide-based vaccination in combination with chemotherapy (178); moreover, the HPV16 long-peptide-based vaccination during CarboTaxol treatment was able to reduce circulating MDSCs and generated a strong immune response, confirming the relevance of M-MDSC elimination as a valid approach to enforce anti-tumor immune response (179). Based on these data, several chemotherapeutic compounds with different mechanisms of action were listed as M-MDSC-targeting drugs (173). M-MDSC elimination can be also achieved using immune compounds such cytokines and antibodies. In this line, the combination of IL-2 with anti-CD40 antibody was effective on cancer growth control in two different mouse tumor models by inducing M-MDSC elimination trough Fas-mediated apoptosis (180). Similarly, to improve the efficacy of CAR T cells immunotherapy in leukemia setting, by limiting transferred T cells-associated life-threatening cytokine-release syndrome (CRS) and neurotoxicity, either monocyte-depletion or infusion of IL-1 receptor antagonist anakinra were reported as effective strategies (181). Other therapeutic approaches aim at favoring MDSC differentiation into anti-tumoral cell subsets such as macrophages and DCs. For example, all-trans-retinoic acid (ATRA) treatment was reported to promote the differentiation of MDSC into mature anti-tumoral myeloid cells via the activation of the ERK1/2 signaling pathway (182) as well as the treatment with 25-hydroxyvitamin D3 was reported to reduce the frequency of immature immune suppressive cells in peripheral blood of HNSCC patients (183). Moreover, M-MDSC differentiation toward mature anti-tumoral monocytes-derived DCs can be achieved using pharmacological inhibitor of ATP-converting ectoenzyme ENTPD2, thus mitigating cancer growth and enhancing the efficiency of immune checkpoint inhibitors (184). Finally, some targeting approaches aim at blocking M-MDSC migration from bone marrow to tumor site. For example, amino-biphosphonates were able to prevent the activation of matrix metalloproteases (MMPs) limiting cancer aggressiveness and distal spread (185). Along the same line, pharmacological antagonist of chemokine receptors (i.e., S-265610, CXCR2-specific antagonist) were able to drastically reduce M-MDSC in tumor-bearing mice (186).
Concluding Remarks
Monocytes are relevant immunological cells connecting the innate and adaptive immune compartments. Here, we have attempted to integrate recent advances in the molecular, metabolic, and functional aspects of monocyte biology with the current state of understanding about the role of these cells in cancer growth and metastatic spread. Although a large body of evidence supports the notion that circulating monocytes serve only as precursor cells that replenish tissue macrophages and DC populations, the overwhelming complexity underlined by high throughput technologies, is supporting a direct contribution of monocyte to cancer development. Targeting monocytes at the immunological, metabolic, epigenetic, and transcriptional level is a promising strategy to treat both disease with impaired immune function, like cancer, or with over-reactive immune response, like autoimmune diseases. The advances in our knowledge on monocyte development, response, and reprogramming, particularly during cancer evolution and metastatic spread, will pave the way for the development of new therapeutic strategy with specificity and limited toxicity.
Author Contributions
SC, SU, RT, IM, FD, and SS wrote parts of the manuscript. SC and VB edited and finalized the manuscript.
Funding
This work was supported by the Italian Association for Cancer Research (18603 and 12182 that refers to Special Program Molecular Clinical Oncology 5 per mille), by the Cariverona Fondation (Project call, 2017), by the Cancer Research Institute (Clinic and Laboratory Integration Program, CLIP-2017), and by Euronanomed III (Joint Translational Call 2017, Project resolve) to VB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. (2009) 27:669–92. doi: 10.1146/annurev.immunol.021908.132557
2. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. (2005) 5:953–64. doi: 10.1038/nri1733
3. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
4. Cignarella A, Tedesco S, Cappellari R, Fadini GP. The continuum of monocyte phenotypes: experimental evidence and prognostic utility in assessing cardiovascular risk. J Leukoc Biol. (2018) 103:1021–8. doi: 10.1002/JLB.5RU1217-477RR
5. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
6. Thomas GD, Hanna RN, Vasudevan NT, Hamers AA, Romanoski CE, McArdle S, et al. Deleting an Nr4a1 super-enhancer subdomain ablates Ly6C(low) monocytes while preserving macrophage gene function. Immunity. (2016) 45:975–87. doi: 10.1016/j.immuni.2016.10.011
7. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
8. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol. (2013) 34:440–5. doi: 10.1016/j.it.2013.06.001
9. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
10. Mildner A, Yona S, Jung S. A close encounter of the third kind: monocyte-derived cells. Adv Immunol. (2013) 120:69–103. doi: 10.1016/B978-0-12-417028-5.00003-X
11. de Bree C, Marijnissen RJ, Kel JM, Rosendahl Huber SK, Aaby P, Benn CS, et al. Bacillus calmette-guerin-induced trained immunity is not protective for experimental influenza A/Anhui/1/2013 (H7N9) infection in mice. Front Immunol. (2018) 9:869. doi: 10.3389/fimmu.2018.02471
12. Saz-Leal P, Del Fresno C, Brandi P, Martinez-Cano S, Dungan OM, Chisholm JD, et al. Targeting SHIP-1 in myeloid cells enhances trained immunity and boosts response to infection. Cell Rep. (2018) 25:1118–26. doi: 10.1016/j.celrep.2018.09.092
13. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61 e12. doi: 10.1016/j.cell.2017.11.034
14. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
15. Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med. (2018) 215:1627–47. doi: 10.1084/jem.20180247
16. T'Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol. (2018) 330:43–53. doi: 10.1016/j.cellimm.2018.02.005
17. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
18. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
19. Mildner A, Schonheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, et al. Genomic characterization of murine monocytes reveals C/EBPbeta transcription factor dependence of Ly6C(-) cells. Immunity. (2017) 46:849–62 e7. doi: 10.1016/j.immuni.2017.04.018
20. Polletti S, Natoli G. Understanding spontaneous conversion: the case of the Ly6C(-) monocyte. Immunity. (2017) 46:764–6. doi: 10.1016/j.immuni.2017.04.010
21. Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseno CG, et al. Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat Commun. (2016) 7:12597. doi: 10.1038/ncomms12597
22. Kurotaki D, Osato N, Nishiyama A, Yamamoto M, Ban T, Sato H, et al. Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood. (2013) 121:1839–49. doi: 10.1182/blood-2012-06-437863
23. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
24. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. (2019) 50:1317–34 e10. doi: 10.1016/j.immuni.2019.03.009
25. Shen JJ, Lee PH, Holden JJ, Shatkay H. Using cluster ensemble and validation to identify subtypes of pervasive developmental disorders. AMIA Annu Symp Proc. (2007) 666–70.
26. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
27. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. (2010) 33:375–86. doi: 10.1016/j.immuni.2010.08.012
28. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
29. Selimoglu-Buet D, Wagner-Ballon O, Saada V, Bardet V, Itzykson R, Bencheikh L, et al. Characteristic repartition of monocyte subsets as a diagnostic signature of chronic myelomonocytic leukemia. Blood. (2015) 125:3618–26. doi: 10.1182/blood-2015-01-620781
30. Yanez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, et al. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity. (2017) 47:890–902 e4. doi: 10.1016/j.immuni.2017.10.021
31. Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature. (2017) 541:96–101. doi: 10.1038/nature20611
32. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. (2011) 12:778–85. doi: 10.1038/ni.2063
33. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
34. Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. (2006) 211:609–18. doi: 10.1016/j.imbio.2006.05.025
35. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
36. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. (1994) 265:1573–7. doi: 10.1126/science.8079170
37. Nutt SL, Metcalf D, D'Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. (2005) 201:221–31. doi: 10.1084/jem.20041535
38. McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. (1996) 15:5647–58. doi: 10.1002/j.1460-2075.1996.tb00949.x
39. Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. (2010) 32:317–28. doi: 10.1016/j.immuni.2010.02.008
40. Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol. (2016) 17:26–33. doi: 10.1038/ni.3306
41. Nagulapalli S, Pongubala JM, Atchison ML. Multiple proteins physically interact with PU.1. Transcriptional synergy with NF-IL6 beta (C/EBP delta, CRP3). J Immunol. (1995) 155:4330–8.
42. Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. (1994) 14:373–81. doi: 10.1128/MCB.14.1.373
43. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is required for 'emergency' granulopoiesis. Nat Immunol. (2006) 7:732–9. doi: 10.1038/ni1354
44. Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. (1998) 18:4301–14. doi: 10.1128/MCB.18.7.4301
45. Avellino R, Havermans M, Erpelinck C, Sanders MA, Hoogenboezem R, van de Werken HJ, et al. An autonomous CEBPA enhancer specific for myeloid-lineage priming and neutrophilic differentiation. Blood. (2016) 127:2991–3003. doi: 10.1182/blood-2016-01-695759
46. Ma O, Hong S, Guo H, Ghiaur G, Friedman AD. Granulopoiesis requires increased C/EBPalpha compared to monopoiesis, correlated with elevated Cebpa in immature G-CSF receptor versus M-CSF receptor expressing cells. PLoS ONE. (2014) 9:e95784. doi: 10.1371/journal.pone.0095784
47. Rossi A, Sebastiani A. Photocoagulation treatment of diabetic retinopathy. Minerva Endocrinol. (1988) 13:63–6.
48. Wang D, D'Costa J, Civin CI, Friedman AD. C/EBPalpha directs monocytic commitment of primary myeloid progenitors. Blood. (2006) 108:1223–9. doi: 10.1182/blood-2005-12-008763
49. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
50. De Sanctis F, Bronte V, Ugel S. Tumor-induced myeloid-derived suppressor cells. Microbiol Spectr. (2016) 4:1–22. doi: 10.1128/microbiolspec.MCHD-0016-2015
51. Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. (2012) 2:628–39. doi: 10.1016/j.celrep.2012.08.006
52. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
53. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
54. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
55. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. (2016) 6:870–85. doi: 10.1158/2159-8290.CD-15-1346
56. Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. (2009) 138:114–28. doi: 10.1016/j.cell.2009.04.020
57. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
58. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
59. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. (2013) 123:1580–9. doi: 10.1172/JCI60083
60. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
61. De Sanctis F, Sandri S, Ferrarini G, Pagliarello I, Sartoris S, Ugel S, et al. The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front Immunol. (2014) 5:69. doi: 10.3389/fimmu.2014.00069
62. Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AHR, Ugel S, et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell. (2016) 30:377–90. doi: 10.1016/j.ccell.2016.08.004
63. Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, et al. Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen-presenting cells in tumors. Immunity. (2018) 48:91–106 e6. doi: 10.1016/j.immuni.2017.12.014
65. Fiore A, Ugel S, De Sanctis F, Sandri S, Fracasso G, Trovato R, et al. Induction of immunosuppressive functions and NF-kappaB by FLIP in monocytes. Nat Commun. (2018) 9:5193. doi: 10.1038/s41467-018-07654-4
66. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
67. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
68. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille calmette-guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
69. Mourits VP, Wijkmans JC, Joosten LA, Netea MG. Trained immunity as a novel therapeutic strategy. Curr Opin Pharmacol. (2018) 41:52–58. doi: 10.1016/j.coph.2018.04.007
70. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75 e14. doi: 10.1016/j.cell.2017.12.013
71. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
72. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
73. Dominguez-Andres J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab. (2019) 29:211–20 e5. doi: 10.1016/j.cmet.2018.09.003
74. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46 e9. doi: 10.1016/j.cell.2017.11.025
75. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. (2013) 152:157–71. doi: 10.1016/j.cell.2012.12.018
76. Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. (2013) 51:310–25. doi: 10.1016/j.molcel.2013.07.010
77. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
78. Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, Netea MG. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. (2017) 26:142–56. doi: 10.1016/j.cmet.2017.06.001
79. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
80. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
81. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
82. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
83. Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat Immunol. (2013) 14:1045–53. doi: 10.1038/ni.2704
84. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
85. Christ A, Bekkering S, Latz E, Riksen NP. Long-term activation of the innate immune system in atherosclerosis. Semin Immunol. (2016) 28:384–93. doi: 10.1016/j.smim.2016.04.004
86. Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. (2010) 328:1689–93. doi: 10.1126/science.1189731
87. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
88. Coskun U, Simons K. Cell membranes: the lipid perspective. Structure. (2011) 19:1543–8. doi: 10.1016/j.str.2011.10.010
89. Kaul S, Xu H, Zabalawi M, Maruko E, Fulp BE, Bluemn T, et al. Lipid-free apolipoprotein A-I reduces progression of atherosclerosis by mobilizing microdomain cholesterol and attenuating the number of CD131 expressing cells: monitoring cholesterol homeostasis using the cellular ester to total cholesterol ratio. J Am Heart Assoc. (2016) 5:e004401. doi: 10.1161/JAHA.116.004401
90. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
91. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. (2016) 7:28697–710. doi: 10.18632/oncotarget.7376
92. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
93. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
94. Scholl SM, Lidereau R, de la Rochefordiere A, Le-Nir CC, Mosseri V, Nogues C, et al. Circulating levels of the macrophage colony stimulating factor CSF-1 in primary and metastatic breast cancer patients. A pilot study. Breast Cancer Res Treat. (1996) 39:275–83. doi: 10.1007/BF01806155
95. Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, et al. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res. (2009) 15:778–87. doi: 10.1158/1078-0432.CCR-08-1283
96. Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Cancer Res. (2016) 76:35–42. doi: 10.1158/0008-5472.CAN-15-0869
97. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
98. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
99. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. (2017) 32:654–68 e5. doi: 10.1016/j.ccell.2017.10.005
100. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. (2013) 73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981
101. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
102. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
103. Bernardini G, Ribatti D, Spinetti G, Morbidelli L, Ziche M, Santoni A, et al. Analysis of the role of chemokines in angiogenesis. J Immunol Methods. (2003) 273:83–101. doi: 10.1016/S0022-1759(02)00420-9
104. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. (2016) 18:356–65. doi: 10.1038/ncb3330
105. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. (2016) 17:611–25. doi: 10.1038/nrm.2016.87
106. Scavelli C, Nico B, Cirulli T, Ria R, Di Pietro G, Mangieri D, et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene. (2008) 27:663–74. doi: 10.1038/sj.onc.1210691
107. Avraham-Davidi I, Yona S, Grunewald M, Landsman L, Cochain C, Silvestre JS, et al. On-site education of VEGF-recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells. J Exp Med. (2013) 210:2611–25. doi: 10.1084/jem.20120690
108. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. (2010) 70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439
109. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
110. Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. (1999) 5:2963–70.
111. Du Four S, Maenhout SK, De Pierre K, Renmans D, Niclou SP, Thielemans K, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology. (2015) 4:e998107. doi: 10.1080/2162402X.2014.998107
112. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. (2016) 30:836–48. doi: 10.1016/j.ccell.2016.10.009
113. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. (2006) 66:9290–8. doi: 10.1158/0008-5472.CAN-06-1819
114. Javeed N, Gustafson MP, Dutta SK, Lin Y, Bamlet WR, Oberg AL, et al. Immunosuppressive CD14(+)HLA-DR(lo/neg) monocytes are elevated in pancreatic cancer and “primed” by tumor-derived exosomes. Oncoimmunology. (2017) 6:e1252013. doi: 10.1080/2162402X.2016.1252013
115. Yuan XK, Zhao XK, Xia YC, Zhu X, Xiao P. Increased circulating immunosuppressive CD14(+)HLA-DR(-/low) cells correlate with clinical cancer stage and pathological grade in patients with bladder carcinoma. J Int Med Res. (2011) 39:1381–91. doi: 10.1177/147323001103900424
116. Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, et al. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(-)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol. (2010) 72:540–7. doi: 10.1111/j.1365-3083.2010.02463.x
117. de Vrij J, Maas SL, Kwappenberg KM, Schnoor R, Kleijn A, Dekker L, et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer. (2015) 137:1630–42. doi: 10.1002/ijc.29521
118. Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. (2018) 7:e1412909. doi: 10.1080/2162402X.2017.1412909
119. Gershbein LL, Lieberman HL. Lactate dehydrogenase of concentric zones of human senile cataracts and calf lenses produced by ultrasound. Biotechnol Appl Biochem. (1989) 11:602–9.
120. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. (2010) 12:19–30. doi: 10.1038/ncb2000
121. Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA, et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol. (2010) 189:223–32. doi: 10.1083/jcb.200911018
122. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. (2014) 16:1105–17. doi: 10.1038/ncb3041
123. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. (2012) 11:790–811. doi: 10.1038/nrd3810
124. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
125. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. (2008) 9:1347–55. doi: 10.1038/ni.1677
126. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta 'reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. (2008) 9:1341–6. doi: 10.1038/ni.1659
127. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
128. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
129. Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. (2007) 178:4022–6. doi: 10.4049/jimmunol.178.7.4022
130. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. (2008) 9:632–40. doi: 10.1038/ni.1607
131. Kelly A, Gunaltay S, McEntee CP, Shuttleworth EE, Smedley C, Houston SA, et al. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J Exp Med. (2018) 215:2725–36. doi: 10.1084/jem.20171491
132. Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. (2002) 157:493–507. doi: 10.1083/jcb.200109100
133. Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, et al. Integrin alphavbeta8-expressing tumor cells evade host immunity by regulating TGF-beta activation in immune cells. JCI Insight. (2018) 3:122591. doi: 10.1172/jci.insight.122591
134. Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. (2008) 359:2814–23. doi: 10.1056/NEJMra0805239
135. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
136. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. (2006) 354:610–21. doi: 10.1056/NEJMra052723
137. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7:311–7. doi: 10.1038/ni1309
138. Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol. (2013) 15:284–94. doi: 10.1038/ncb2690
139. Butler KL, Clancy-Thompson E, Mullins DW. CXCR3(+) monocytes/macrophages are required for establishment of pulmonary metastases. Sci Rep. (2017) 7:45593. doi: 10.1038/srep45593
140. Shi H, Zhang J, Han X, Li H, Xie M, Sun Y, et al. Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1beta-mediated increase in E-selectin expression. Int J Cancer. (2017) 140:1370–83. doi: 10.1002/ijc.30538
141. Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. (2011) 20:538–49. doi: 10.1016/j.ccr.2011.08.025
142. Zhang H, Yu Y, Zhou L, Ma J, Tang K, Xu P, et al. Circulating tumor microparticles promote lung metastasis by reprogramming inflammatory and mechanical niches via a macrophage-dependent pathway. Cancer Immunol Res. (2018) 6:1046–56. doi: 10.1158/2326-6066.CIR-17-0574
143. Takano Y, Masuda T, Iinuma H, Yamaguchi R, Sato K, Tobo T, et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget. (2017) 8:78598–613. doi: 10.18632/oncotarget.20009
144. Plebanek MP, Angeloni NL, Vinokour E, Li J, Henkin A, Martinez-Marin D, et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun. (2017) 8:1319. doi: 10.1038/s41467-017-01433-3
145. Murray PJ. Immune regulation by monocytes. Semin Immunol. (2018) 35:12–18. doi: 10.1016/j.smim.2017.12.005
146. Bednar M, Kraemer R, Abraham NG, Mullane KM. Arachidonic acid monooxygenase and lipoxygenase activities in polymorphonuclear leukocytes. Biochem Pharmacol. (1987) 36:1741–7. doi: 10.1016/0006-2952(87)90063-3
147. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
148. Kawai K, Miyazaki J, Joraku A, Nishiyama H, Akaza H. Bacillus Calmette-Guerin (BCG) immunotherapy for bladder cancer: current understanding and perspectives on engineered BCG vaccine. Cancer Sci. (2013) 104:22–7. doi: 10.1111/cas.12075
149. Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer–a current perspective. Nat Rev Urol. (2014) 11:153–62. doi: 10.1038/nrurol.2014.15
150. Buffen K, Oosting M, Quintin J, Ng A, Kleinnijenhuis J, Kumar V, et al. Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLoS Pathog. (2014) 10:e1004485. doi: 10.1371/journal.ppat.1004485
151. Ifrim DC, Quintin J, Joosten LA, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. (2014) 21:534–45. doi: 10.1128/CVI.00688-13
152. Li X, Yu J, Xu S, Wang N, Yang H, Yan Z, et al. Chemical conjugation of muramyl dipeptide and paclitaxel to explore the combination of immunotherapy and chemotherapy for cancer. Glycoconj J. (2008) 25:415–25. doi: 10.1007/s10719-007-9095-3
153. Hewitt RE, Pele LC, Tremelling M, Metz A, Parkes M, Powell JJ. Immuno-inhibitory PD-L1 can be induced by a peptidoglycan/NOD2 mediated pathway in primary monocytic cells and is deficient in Crohn's patients with homozygous NOD2 mutations. Clin Immunol. (2012) 143:162–9. doi: 10.1016/j.clim.2012.01.016
154. Rojas LB, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndr. (2013) 5:6. doi: 10.1186/1758-5996-5-6
155. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res. (2017) 77:2607–19. doi: 10.1158/0008-5472.CAN-16-2534
156. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. (2014) 510:407–11. doi: 10.1038/nature13444
157. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. (2017) 543:728–32. doi: 10.1038/nature21676
158. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. (2013) 4:2035. doi: 10.1038/ncomms3035
159. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167:1354–68 e14. doi: 10.1016/j.cell.2016.09.034
160. Bettencourt IA, Powell JD. Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation, and transplantation. J Immunol. (2017) 198:999–1005. doi: 10.4049/jimmunol.1601318
161. Fiorucci S, Santucci L, Cirino G, Mencarelli A, Familiari L, Soldato PD, et al. IL-1 beta converting enzyme is a target for nitric oxide-releasing aspirin: new insights in the antiinflammatory mechanism of nitric oxide-releasing nonsteroidal antiinflammatory drugs. J Immunol. (2000) 165:5245–54. doi: 10.4049/jimmunol.165.9.5245
162. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
163. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. (2004) 64:6337–43. doi: 10.1158/0008-5472.CAN-04-0757
164. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A Relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. (2017) 46:233–44. doi: 10.1016/j.immuni.2017.01.005
165. Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol. (2017) 35:30–9. doi: 10.1016/j.coph.2017.05.002
166. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
167. Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal advance: tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J Leukoc Biol. (2010) 88:839–48. doi: 10.1189/jlb.1209821
168. Obermajer N, Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant Res. (2012) 1:15. doi: 10.1186/2047-1440-1-15
169. Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA, et al. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol. (2013) 190:2403–14. doi: 10.4049/jimmunol.1202369
170. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
171. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. (2010) 70:68–77. doi: 10.1158/0008-5472.CAN-09-2587
172. Gmunder H, Eck HP, Droge W. Low membrane transport activity for cystine in resting and mitogenically stimulated human lymphocyte preparations and human T cell clones. Eur J Biochem. (1991) 201:113–7. doi: 10.1111/j.1432-1033.1991.tb16263.x
173. Ugel S, Delpozzo F, Desantis G, Papalini F, Simonato F, Sonda N, et al. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol. (2009) 9:470–81. doi: 10.1016/j.coph.2009.06.014
174. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
175. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. (2014) 63:513–28. doi: 10.1007/s00262-014-1527-x
176. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. (2016) 7:1168–84. doi: 10.18632/oncotarget.6662
177. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
178. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. (2012) 18:1254–61. doi: 10.1038/nm.2883
179. Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. (2016) 8:334ra52. doi: 10.1126/scitranslmed.aad8307
180. Weiss JM, Subleski JJ, Back T, Chen X, Watkins SK, Yagita H, et al. Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/alphaCD40 therapy. J Immunol. (2014) 192:5821–9. doi: 10.4049/jimmunol.1400404
181. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
182. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. (2007) 67:11021–8. doi: 10.1158/0008-5472.CAN-07-2593
183. Lathers DM, Clark JI, Achille NJ, Young MR. Phase IB study of 25-hydroxyvitamin D(3) treatment to diminish suppressor cells in head and neck cancer patients. Hum Immunol. (2001) 62:1282–93. doi: 10.1016/S0198-8859(01)00317-2
184. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
185. Santini D, Vincenzi B, Dicuonzo G, Avvisati G, Massacesi C, Battistoni F, et al. Zoledronic acid induces significant and long-lasting modifications of circulating angiogenic factors in cancer patients. Clin Cancer Res. (2003) 9:2893–7.
Keywords: monocytes heterogeneity, monocyte continuum, primary tumor, metastatic niche, targeting of monocytes
Citation: Canè S, Ugel S, Trovato R, Marigo I, De Sanctis F, Sartoris S and Bronte V (2019) The Endless Saga of Monocyte Diversity. Front. Immunol. 10:1786. doi: 10.3389/fimmu.2019.01786
Received: 17 May 2019; Accepted: 16 July 2019;
Published: 06 August 2019.
Edited by:
Wei Zheng, Albert Einstein College of Medicine, United StatesReviewed by:
Robin Parihar, Baylor College of Medicine, United StatesShentong Fang, Wihuri Research Institute, Finland
Copyright © 2019 Canè, Ugel, Trovato, Marigo, De Sanctis, Sartoris and Bronte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Bronte, dmluY2Vuem8uYnJvbnRlQHVuaXZyLml0