 Carlos Penha-Gonçalves
Carlos Penha-Gonçalves
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 July 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01771
Despite significant progress in combating malaria in recent years the burden of severe disease and death due to Plasmodium infections remains a global public health concern. Only a fraction of infected people develops severe clinical syndromes motivating a longstanding search for genetic determinants of malaria severity. Strong genetic effects have been repeatedly ascribed to mutations and allelic variants of proteins expressed in red blood cells but the role of inflammatory response genes in disease pathogenesis has been difficult to discern. We revisited genetic evidence provided by inflammatory response genes that have been repeatedly associated to malaria, namely TNF, NOS2, IFNAR1, HMOX1, TLRs, CD36, and CD40LG. This highlighted specific genetic variants having opposing roles in the development of distinct malaria clinical outcomes and unveiled diverse levels of genetic heterogeneity that shaped the complex association landscape of inflammatory response genes with malaria. However, scrutinizing genetic effects of individual variants corroborates a pathogenesis model where pro-inflammatory genetic variants acting in early infection stages contribute to resolve infection but at later stages confer increased vulnerability to severe organ dysfunction driven by tissue inflammation. Human genetics studies are an invaluable tool to find genes and molecular pathways involved in the inflammatory response to malaria but their precise roles in disease pathogenesis are still unexploited. Genome editing in malaria experimental models and novel genotyping-by-sequencing techniques are promising approaches to delineate the relevance of inflammatory response gene variants in the natural history of infection thereby will offer new rational angles on adjuvant therapeutics for prevention and clinical management of severe malaria.
During the last decade intensive efforts have succeeded in implementing malaria epidemiological control measures and in deploying new anti-malaria drugs that have significantly decreased the disease burden across the world (1). Nevertheless, malaria claims close to half a million deaths each year (2) demanding deeper understanding of severe malaria pathogenesis (3). Malaria is caused by mosquito-transmitted Plasmodium protozoan parasites that develop in multiple stages within the vertebrate host. The chronology of infection exposes the host to distinctive intracellular and extracellular forms that emerge sequentially during the natural history of infection (1, 4) (Figure 1). Free parasite forms- sporozoites and merozoites- are non-replicative and obligatory for life cycle progression. Although directly exposed to the immune system free parasites represent brief phases in infection that do not elicit protective responses in naive subjects (5). In contrast, intracellular parasite forms hidden in hepatocytes or in erythrocytes, undergo significant expansion, damaging, and ultimately destroying infected cells while offering a large number of inflammatory triggers and immunogenic targets (4, 6, 7).
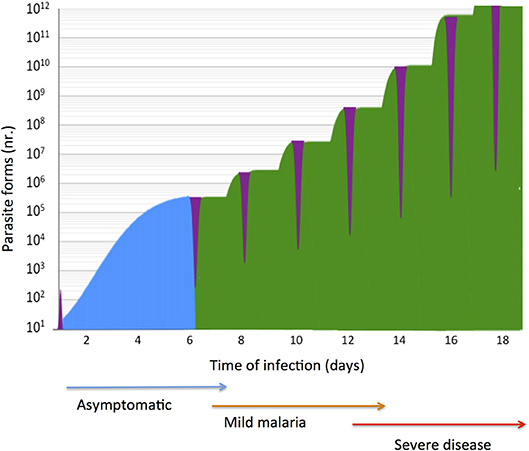
Figure 1. Natural history of malaria infection in a naïve susceptible host. P. falciparum expansion in the liver and in blood stage infection is represented in a logarithmic scale. Exposure of parasite extra-cellular forms (purple) to the host immune system is brief in case of sporozoites (<1 h) and momentaneous in the case of free merozoites in the blood (few minutes). In a typical infection liver stage parasites (blue) expand in few dozens of hepatocytes in circa 5.5 days, a clinically unapparent phase (blue arrow). Synchronous blood stage parasite expansion is represented (green), considering six rounds of trophozoite division during erythrocytic schizogony. In naïve individuals blood stage infection causes tertian fever (one brood infection) and mild malaria (brown arrow) but the clinical severity (red arrow) is co-determined by parasite pathogenicity factors and by the host genetic make-up.
Nevertheless, the effectiveness of anti-parasite responses is defied both by the dynamics of infection (e.g., high parasite growth rate) and by variations in the parasite antigenic complexity represented by stage-specific sets of antigens or by mechanisms of antigenic diversification and clonal antigenic variation (7–9). As a result the host immune system engages in multiple ineffective responses, a scenario that allows parasite cell-cycle progression and transmission to the next mosquito host. Clinical outcomes of blood-stage infection range from unapparent infection to life-threatening conditions depending on the caliber and effectiveness of anti-parasite responses (10). The severity of clinical manifestations result from direct parasite effects on infected cells (e.g., hemolysis), systemic effects of parasite activity and growth (e.g., malaria paroxysms and metabolic imbalances) as well as organ lesions (mainly in brain, lungs, liver, spleen, kidney, and placenta) generated by massive parasite clearance reactions and mal-adapted inflammatory responses (11). Thus, exacerbation of inflammatory responses during infection is a key determinant in development of immunopathology and organ dysfunction associated with severe malaria syndromes (12–14). This review scrutinizes genetic association evidence supporting that pro-inflammatory genetic variants have dual roles in disease pathogenesis.
The host response to malaria includes a wide range of both cell-intrinsic and systemic mechanisms but the initial responses in a naive host are mostly driven by non-specific reactions (1). Liver stage infection is clinically silent as the number of infected hepatocytes is relatively low (15). Liver infection progression is counteracted by hepatocyte killing mechanisms, including activation of hepatocyte-intrinsic apoptosis pathways (16–20) and innate responses of liver non-parenchymal cells (21–24). Nevertheless, completion of liver stage infection is usually secured by parasite-driven mechanisms that inhibit apoptosis of infected hepatocytes (19, 25), inhibit hepatocyte autophagy (26) or suppress liver macrophages responses (27, 28). As a result, a small number of infected hepatocytes sustain parasite growth and generate a relatively high number of merozoites that are freed in the bloodstream initiating blood stage infection.
An archetypal infection in a naive host leads to exponential increase blood stage parasites due to the cyclic expansion of asexual parasite forms (Figure 1) but in natural infection parasite growth is many times counteracted by strong host responses. Parasite molecules expressed in the surface of infected erythrocytes (IE) induce powerful innate responses leading to detection, engulfment, and destruction of large numbers of IE mainly in the spleen and liver (29–31). Pattern recognition receptors (e.g., TLRs) expressed by professional antigen presenting cells contribute to IE recognition and engulfment (32, 33). Phagocytosis triggers a respiratory burst response involving reactive oxygen species in intracellular parasite killing (34) but also leads to production of cytokines and chemokines that engage and polarize subsequent antibody- and cytotoxicity-mediated adaptive immune responses (35).
In this pro-inflammatory environment the mounting of adaptive responses in secondary lymphoid organs, mainly in the spleen white pulp (31) favors the generation of cytophilic or opsonizing antibodies that recognize Plasmodium antigens expressed in the surface of infected erythrocytes (36). Similarly, opsonizing antibody responses target free merozoites for destruction and correlate with disease protection (37, 38) and infection resolution (39–41). Nevertheless, variation and diversification of parasite antigens expressed in the erythrocyte surface subvert adaptive responses thereby sustaining infection progression (42). Furthermore, parasite virulence factors such as higher intrinsic growth rate, broad erythroid tropism, and cyto-adherence (43–46) are determinants of increased blood-stage parasite loads that in turn exacerbate the pro-inflammatory responses associated with severe disease (10, 47).
Several immunologic mechanisms triggered by infection in non-immune individuals paradoxically favor disease development as they reveal inefficiencies in eliminating the parasite. The host immune system reacts to parasite molecules released during IE rupture (e.g., hemozoin pigment and lipid-associated GPI anchors, so-called “malaria toxins”) through innate immunity receptor signaling (e.g., TLRs) leading to release of pro-inflammatory mediators, namely TNF-alpha, IL-1β, IL-6, and IFN-gamma (48–50). In overt blood-stage infection this inflammatory environment is amplified by reciprocal activation loops involving monocytes, NKT cells, T cells, and endothelial cells (11). Systemic microvascular endothelial activation by inflammatory mediators, IE and parasite components (e.g., nucleosomes and microvesicles) plays a decisive role in organ inflammation (51, 52) with marked up-regulation of adhesion molecules (e.g., ICAM1) and imbalances in vasoactive mediators (angiotensins, endothelin-1, and nitric oxide) (53–55). Often, adhesion of activated monocytes, T cells and platelets (56–58) leads to altered vascular permeability and coagulation disturbances with induction of endothelial cell damage and death. In counterbalance, tissue damage cues such as alarmins and cytokines released by parenchymal cells engage macrophages and lymphoid cells in inflammation resolution and tissue repairing responses (59). These general tissue protective responses that counteract inflammation may operate in malaria by induction of regulatory T cells (60, 61), production of anti-inflammatory cytokines [e.g., TGF-β (62, 63)] and stimulation of oxidative-stress protection systems [e.g., heme oxigenase 1 and nitric oxide (59)]. The inflammatory mechanisms and tissue protective responses elicited by infection interplay with parasite factors and microanatomy components in different organs to determine tissue-specific vulnerability to immunopathology (64), ultimately allowing development of specific malaria clinical syndromes.
The epidemiology of severe malaria suggests that cumulative exposure to the malaria parasite confers resilience against life-threatening disease (1). Nevertheless, malaria epidemiology alone does not explain why only a relatively small fraction of primary Plasmodium infections develop severe clinical forms. Compelling genetic evidence collected in recent years shows that malaria contributed to shape the human genome and it is established that variants in genes coding for proteins expressed in erythrocytes are major genetic determinants of resistance to infection and resilience to severe disease (65–71).
Interestingly, a growing number of genetic association studies also report that inflammation-related genes have a role in determining the course and clinical outcomes of infection. Yet, the action of individual genetic variants has been difficult to resolve and in some cases genetic variants appear to have pleiotropic effects (72). This is in part due to disparities in study design, regional differences in intensity of malaria transmission, diversity of Plasmodium biotypes, and relatively small sample sizes with unknown genetic substructure, particularly in Africa (70, 73). Usage of larger sample sizes in multicentric studies do not overcome the possibility that functional consequences of a given genetic variant may vary among geographic locations (74). Moreover, the diversity of malaria phenotypes and Plasmodium species used in case definition of different studies may preclude the identification of genetic variants as general malaria risk factors. Nevertheless, cumulative evidence corroborating the association of specific inflammatory genes with malaria phenotypes is instrumental to value their contribution to malaria pathogenesis and to scrutinize their roles during infection.
Here we attempted to integrate evidence obtained in genetic association studies in light of a malaria pathogenesis model informed by experimental and epidemiological evidence. This model attributes a dual role to inflammatory responsiveness during the natural history of infection and takes in consideration the intra-host infection dynamics and the history of exposure to malaria (Figure 2). Accordingly, pro-inflammatory alleles promoting strong anti-parasite responses are predicted to increase resistance against infection and uncomplicated disease but also contribute to mal-adapted inflammatory responses that underlie progression to severe malaria syndromes. Conversely, alleles that mediate low inflammatory responsiveness may increase susceptibility to unapparent infection and favor mild malaria but would confer resilience to inflammation-driven severe organ damage. In this context, low inflammatory responsiveness offers an explanation for why individuals surviving initial infections resisting to severe malaria are inefficient in clearing the parasite and show long periods of asymptomatic parasitemia. This implies, that genetic control of malaria clinical outcomes afforded by genetic variance in inflammatory genes is modified by cumulative exposure to infection translating into efficient infection resolution or asymptomatic infection (Figure 2).
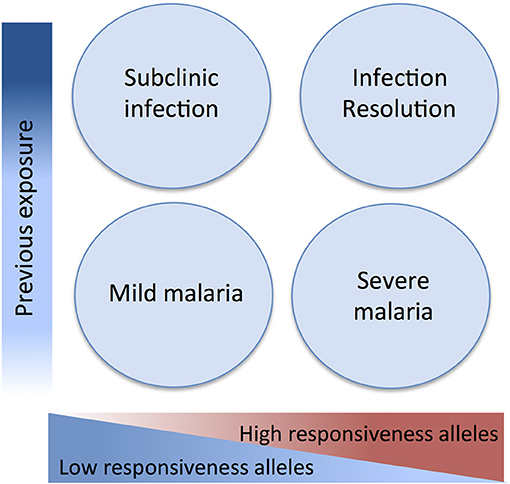
Figure 2. Genetic variants in inflammatory response genes and infection outcomes. Genetic variants controlling inflammatory responsiveness are proposed determinants of malaria clinical outcomes dependent on history of exposure to infection. Under the framework of a polygenic model of malaria susceptibility the additive effects of genetic variants conferring strong inflammatory responsiveness to infection concur to severe disease but may favor efficiency of anti-parasite responses in repeatedly exposed individuals. On the other hand, joint effects of alleles conferring low inflammatory responsiveness results in increased resilience to severe malaria but are associated to asymptomatic parasitemia in exposed individuals possibly due to inefficient parasite clearance.
We followed a gene-centered approach to screen malaria association studies encompassing different clinical phenotypes, geographic regions, and Plasmodium species aiming to identify gene variants that have repeatedly produced evidence for positive association, namely in TNF, NOS2, IFNAR1, HMOX1, TLRs, CD36, and CD40LG genes (Supplementary Table 1). Subsequently, genetic effects of these individual variants were scrutinized across different case-control studies using a terminology that takes in account the malaria status of the groups of individuals that are compared in each study (Table 1). The analysis of the malaria association landscape in individual genes reconciled apparently incongruent data and offers a perspective on the roles of specific inflammatory mediators in malaria pathogenesis.
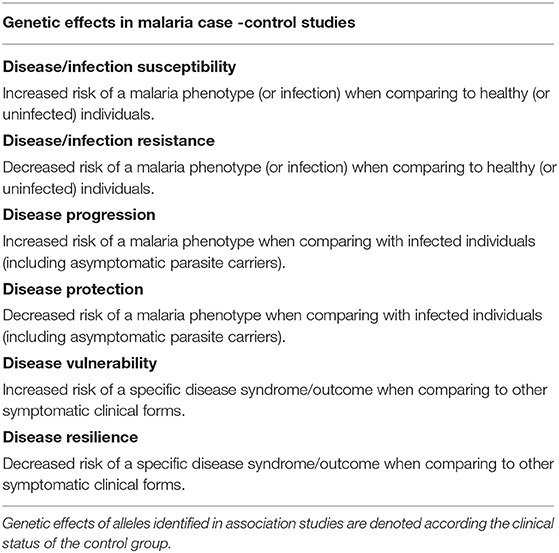
Table 1. Terminology of malaria genetic effects.
Tumor necrosis factor (TNF) is a potent pro-inflammatory mediator involved in various steps of immune responses. It has long been recognized that TNF takes part in anti-Plasmodium responses that lead to intra-erythrocytic parasite killing and parasitemia reduction (75). Yet, TNF was also implicated as a causative factor in development of malaria-associated organ pathology (76–78). Elevation of TNF serum levels is a common finding in malaria episodes (79) and was noted that high TNF production capacity correlates with faster parasite clearance and with resolution of malaria attacks (80). The TNF response appears to follow the parasitemia kinetics with increased serum levels in children with high-density parasitemia and decaying with parasite clearance (81, 82). Nevertheless, TNF serum levels were repeatedly found increased in children with severe malaria (82–87) implying an intricate role in malaria pathogenesis (88).
Several TNF-α promoter single nucleotide polymorphisms (SNPs) that control gene expression or TNF production (89–92) are associated with control of parasitemia levels (92–95) and with increased anti-P. falciparum IgG levels (96). This suggests that TNF variants may play a role in the effectiveness of anti-parasite responses. Furthermore, the role of promoter variants in clinical malaria (e.g., rs1800629) is highlighted by reports associating TNF with vulnerability to severe disease, including cerebral malaria (97–102) and malaria in pregnancy (103, 104) (Figure 3 and Supplementary Table 1). Likely, control of TNF gene expression by these polymorphisms is context-dependent regarding secreting cell types, cell activation status, and action of other inflammatory mediators (105). Accordingly, it was reported that TNF2 allele (-308, aka rs1800629) controlled elevation of serum TNF in severe malaria patients but not in patients with uncomplicated malaria suggesting that its capacity to control gene expression may be modified in the course of infection (106). Also, TNF-238 G allele (rs361525) shows opposing effects on vulnerability to cerebral malaria and severe malaria anemia (107) suggesting differential roles of TNF in the spectrum of severe malaria syndromes. It should be noted that some of these association signals could result from genotypic combinations with neighboring genes, namely lymphotoxin-alpha (LTA) (88) and more detailed analysis of this region is needed to discern the involvement of TNF in human severe malaria (108).
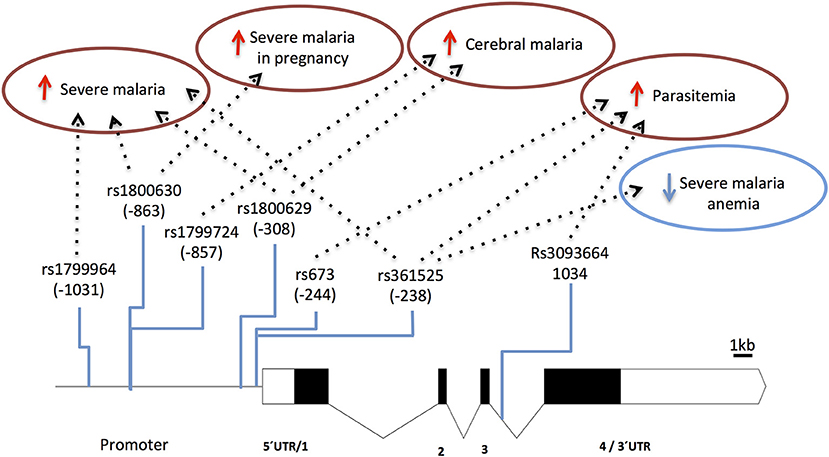
Figure 3. Genetic effects of TNF SNPs associated to clinical malaria. The scaled diagram of exon-intron structure of TNF gene shows malaria associated SNPs in the promoter region and one SNP in intron 3 (bar 1 Kb). Detrimental effects in malaria severity and parasitemia are represented by red arrows while beneficial effects are represented by blue arrows. TNF alleles associated with malaria are conferring several severe phenotypes with the exception of rs361525 that concomitantly is associated to protection from severe malaria anemia. For allele description and reported genetic effects (Supplementary Table 1).
Nevertheless, the data collectively suggest that TNF response capacity in malaria is controlled by TNF polymorphisms and that TNF represent a prototype of a pro-inflammatory factor in the natural course of infection; controlling parasite expansion in early stages of infection but acting in a context-dependent manner to increase the risk of specific severe malaria syndromes. The exact mechanisms by which TNF is involved in severe malaria remains unknown and requires a detailed functional testing of the network of TNF-TNFR family (88). However, it is noteworthy that other genetic modifiers of malaria susceptibility, namely NOS2, act as downstream effectors of TNF signaling encouraging further investigations on the intertwining of TNF and nitric oxide in disease development.
Nitric oxide (NO) is a free radical involved in multiple biological processes, including inflammatory responses. Decreased NO bioavailability in severe malaria patients is a well-established pathogenesis factor (53, 109, 110) possibly impacting on endothelial activation (111) and macrophage polarization (112). Experimental mouse models of cerebral malaria demonstrated that increasing nitric oxide bioavailability protects against cerebral malaria by deterring systemic inflammation and the activation of immunocytes and endothelial cells (113–115). However, the therapeutic value of NO in severe malaria still lacks solid supportive evidence (116).
The NOS2 gene codes for the inducible NO synthase (iNOS) and has been probed in several genetic studies that collectively uncovered common genetic variants functionally correlated with increased NO bioavailability in infected individuals (117–119). Meaningfully, these variants have beneficial effects in clinical malaria, namely lower incidence of symptomatic re-infection (119) and uncomplicated malaria (117, 118, 120–123), decreased risk of severe malaria (121, 124, 125), including severe anemia and cerebral malaria (117, 118). Nonetheless, the same NOS2 alleles (e.g., rs6505469) were also associated with higher incidence of asymptomatic infection among apparently healthy individuals (117) (Figure 4 and Supplementary Table 1). This suggests that increased NO bioavailability in infected individuals counteracts detrimental effects of malaria inflammatory responses but is also impairing parasite clearance responses in early stages of infection. In contrast, other NOS2 variants in the promoter region that favor reduced NO bioavailability in absence of infection (e.g., rs8078340) were underrepresented in individuals with lower markers of exposure to pre-erythrocytic infection (e.g., anti-CSP antibodies) suggesting they contribute to resistance to infection (117).
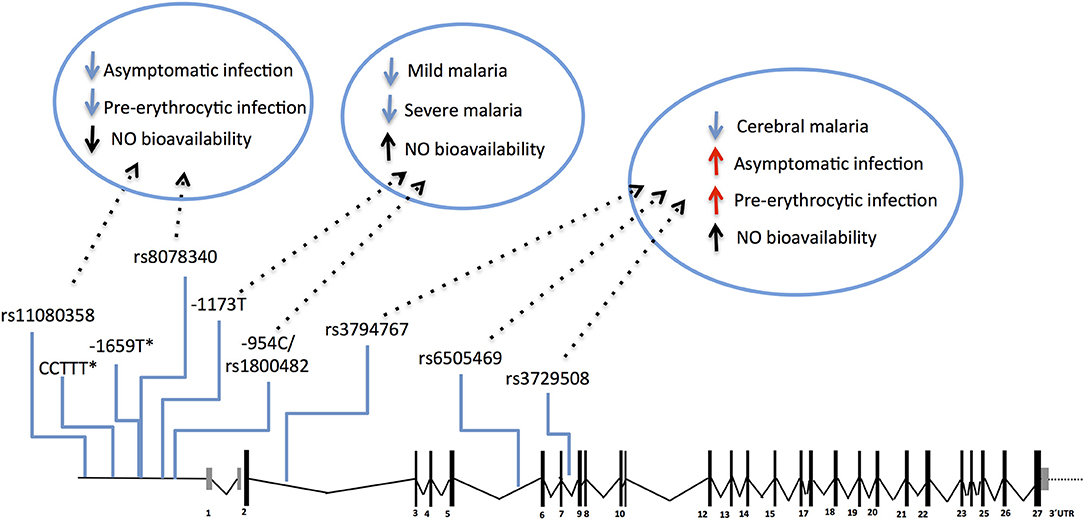
Figure 4. Genetic effects of NOS2 SNPs associated to malaria and nitric oxide bioavailability. The scaled diagram of NOS2 exon-intron structure depicts SNPs in the 5′region (rs11080358–rs1800482) and in the cistronic region (rs3794767–rs3729508) associated with malaria and with control of nitric oxide (NO) bioavailability. Genetic variants that decrease NO bioavailability in the 5′region have beneficial effects (blue arrows) in early stages of infection, possibly by favoring anti-parasite responses. In contrast, SNPs that increase NO bioavailability (mapping in the 5′ region or in the cistronic region) show an anti-inflammatory action favoring infection when parasite burden is low or when infection is unapparent (red arrows) but protect from mild malaria and increase resilience against severe malaria (blue arrows). (*) CCTTT and−1659 polymorphisms showed association with malaria severity and cerebral malaria but their role on NO bioavailability is not known. For allele description and reported genetic effects (Supplementary Table 1).
The protective effects of NOS2 variants have not been consistently observed in different studies possibly owing to differences in the genetic variants analyzed, in the ethnic groups studied or in regional epidemiologic patterns of clinical malaria (126, 127). Nevertheless, the available genetic evidence cohesively supports the hypothesis that NO bioavailability holds an anti-inflammatory role in malaria pathogenesis that translates into decreased ability to clear the parasite in early stages of infection but enhancing protection against clinical inflammatory manifestations. The pathophysiological impact of NOS2 genetic variance is still not resolved but it should be noted that the NOS2 polymorphisms controlling NO bioavailability in absence of clinical malaria (e.g. rs8078340) are distinct from those associated with alterations of NO bioavailability in overt clinical disease (e.g. rs1800482) (117) (Figure 4). This implies that a distinct regimen of NOS2 genetic control is operating in infected individuals that should be taken into account when comparing groups of infected and non-infected individuals.
Type 1 interferons (IFN-1) are secreted by immune and non-immune cells including lymphocytes, macrophages, dendritic cells, fibroblasts, and endothelial cells. IFN-1 production is induced upon innate recognition of exogenous nucleic acids and proteins by innate receptors such as membrane-bound Toll-like receptors (TLRs) and cytosolic RNA helicases. Studies in vitro and in mice converge at the notion that sensing malaria parasites via innate immune receptors is linked to a IFN-1 response (128–130). A multi-level role of IFN-I signaling during Plasmodium infection in experimental models has been uncovered by the use of mice deficient in Ifnar1 (a subunit of the IFN-1 receptor) or in components of the IFN-I induction pathways (e.g., MDA5, MAVs, cGAS, STING, Irf7, and Irf3) (129–131). Mouse studies identified distinct mechanisms that recruit the IFN-1/IFNAR1 axis to the response against malaria, including: (i) stimulation of hepatocyte-intrinsic interferon responses that counteract liver stage parasite expansion (22, 24); (ii) exacerbated IFN-1 production by plasmocytoid dendritic cells (DCs) leading to enhanced IFNAR1 signaling in conventional DCs that in turn drive responses against blood-stage parasite (131); (iii) IFNAR1-mediated effects in the CD4T helper-antibody axis that dampen the response against blood stage parasite allowing hyperparasitemia development (132–135); and (iv) the requirement for IFNAR1 expression in CD8T cells to license the cytotoxic effector functions that lead to cerebral malaria development in mice (136, 137).
Together these findings raise the possibility that IFN-1/IFNAR1 signaling exerts beneficial effects in early stages of infection when innate immune responses dominate. In contrast, detrimental effects are expected at later stages if IFN-1 signaling intrudes in the mounting of adaptive immune responses by impairing anti-parasite responses driven by CD4T cells or exacerbating cytotoxic CD8T cell-mediated reactions leading to tissue damage.
Accordingly, several studies identified Type 1 Interferon Receptor 1 (IFNAR1) gene polymorphisms associated with mild malaria, disease severity and cerebral malaria in children (136, 138–141) (Figure 5 and Supplementary Table 1). Interestingly, IFNAR1 SNP alleles that are associated with CM resilience also show to confer increased risk of mild malaria development (Figure 6). This is exemplified by the rs2843710 derived allele which showed higher frequency in mild malaria cases as compared to uninfected controls but had lower frequency in CM patients when compared to patients with mild malaria (136) (Figure 6). The duality of this pathway in human malaria is paralleled in experimental models where increased amounts of IFN-1 improve anti-parasite responses by increasing IFNAR1 signaling in early stages of infection (131) while exacerbated IFN-1/IFNAR1 signaling later in infection increases vulnerability to severe disease (136, 137).These genetic association signals would be undetectable if CM patients were directly compared with healthy individuals providing a word of caution in the design of studies investigating genetic factors that concomitantly confer susceptibility to mild malaria and resilience to severe malaria syndromes.
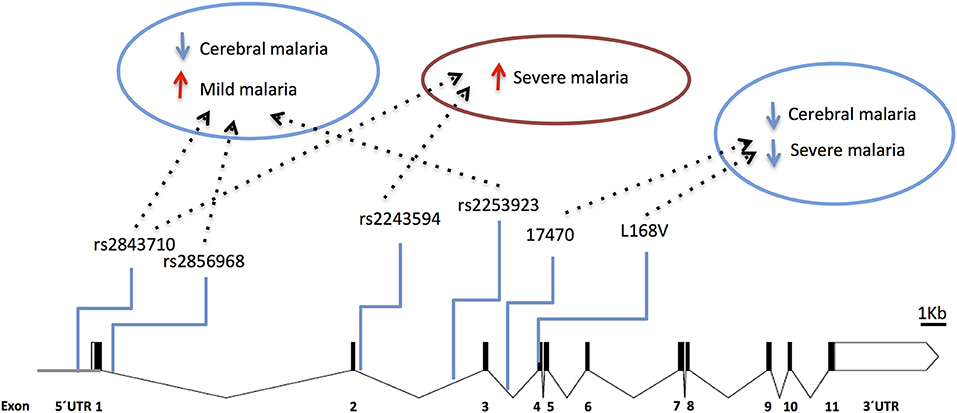
Figure 5. Genetic effects of IFNAR1 SNPs associated to clinical malaria. The scaled diagram of IFNAR1 exon-intron structure represents SNPs that have shown association with different malaria clinical outcomes (bar = 1 Kb). Experimental data suggest that detrimental IFNAR1 genetic effects (red arrows) could be attributed to alleles mediating decreased IFNAR1 signaling that impairs innate anti-parasite responses while beneficial effects (blue arrows) would be conferred by alleles that also decrease IFNAR1 signaling in the context of adaptive effector responses, and in particular deter cerebral immunopathology. For allele description and reported genetic effects (Supplementary Table 1).
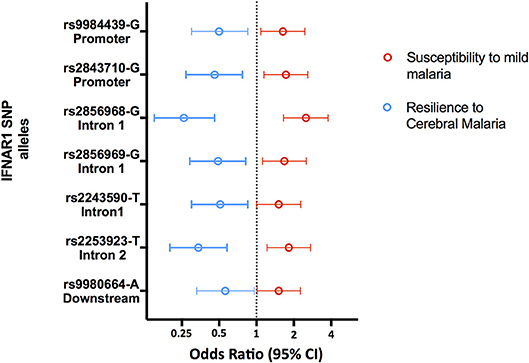
Figure 6. Opposing genetic effects of IFNAR1 SNP alleles in cerebral malaria and mild malaria. Genetic effects of minor frequency alleles at seven IFNAR1 SNPs in an Angolan dataset (136). Blue symbols represent the Odds Ratio and confidence intervals obtained by comparing 110 cases of CM and 129 mild malaria patients (OR< 1 corresponds to increased CM resilience). Red symbols represent the Odds Ratio and confidence intervals obtained by comparing 129 patients with mild malaria and 305 uninfected controls for the same alleles (OR > 1 corresponds to increased mild malaria susceptibility).
Heme oxygenase-1 (HO-1) is a catabolic enzyme that cleaves heme to generate biliverdin, carbon monoxide, and ferrous iron (142). HO-1 activity has anti-inflammatory effects and has been shown to be protective against severe forms of malaria in mice (143–145). In particular, vulnerability to experimental cerebral malaria (ECM) was unearthed by the deletion of Hmox1 (the gene encoding HO-1) in otherwise resilient mice and was abrogated by HO-1 induction or carbon monoxide administration (144). Tissue damage elicited in experimental infection is counteracted by HO-1 through reducing exposure to oxidative stress and inhibiting inflammatory action of immune cells without affecting parasite load; this represents a prototypic case of the disease tolerance phenomenon (59). In sharp contrast, modulation of Hmox1 expression and HO-1 enzymatic activity during experimental liver stage infection revealed that HO-1 is a down-modulator of the inflammatory response against intra-hepatocytic parasite forms and acts to promote liver stage infection (146).
Human studies focusing in the HMOX1 gene showed that polymorphisms functionally controlling HO-1 (promoter repeat, SS) expression or activity (147) were also associated with CM vulnerability in Asia (Myanmar) (148) and Africa (Angola) (149). On the other hand, the alternative alleles in the gene promoter (repeat, L) showed protection from respiratory distress in The Gambia (150) and mild malaria in Brazil (151) (Figure 7 and Supplementary Table 1), but not in Ghana (152). It was reported that HOMX1 promoter alleles associated with disease severity in children also confer higher levels of HO-1 and enhance neutrophil respiratory burst (150). Interestingly, it was demonstrated in the mouse that expression of Hmox1 induced by NO leads to protection from experimental cerebral malaria (114). Further research is needed to clarify how HMOX1 gene variants act in cerebral malaria pathogenesis (150) but its tempting to speculate that genetic interaction with NOS2 functional variants may help to explain the contribution of HMOX1 to the genetic complexity of human cerebral malaria.
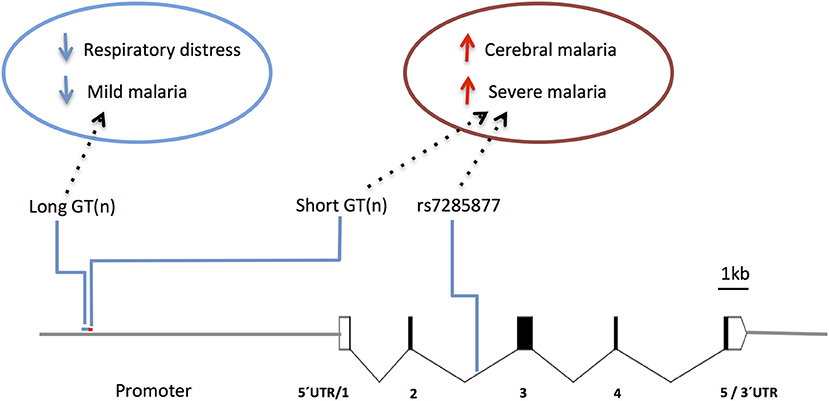
Figure 7. Genetic effects of HMOX1 gene polymorphisms associated to clinical malaria. Scaled diagram of HMOX1 exon-intron structure depicting a microsatellite (GT)n repeat in the HMOX1 gene promoter region and a malaria associated SNP in intron 2. Genetic effects of long alleles (GT)n repeat (blue) and short repeats and intronic SNP (red) have distinct association in occurrence of clinical malaria and are involved in the control of HOMX1 gene expression. For reported genetic associations (Supplementary Table 1).
Toll-like receptors (TLRs) are ancient components of the immune system that recognize pathogen associated molecular patterns (PAMPs) and are infection sensors at the onset of innate immune responses. Conceivably, TLR signaling could be involved both in resolving early infection and in heightening inflammation and immunopathology. It has been shown that binding to IE and recognition of Plasmodium moieties such as the glycosylphosphatidylinositol anchors (GPI) or hemozoin-DNA complexes can be mediated by several TLRs, providing sensor redundancy in the immuno-detection of blood stage parasites (48, 49, 153). A number of reports suggest that variants in different TLRs and their adaptor molecules are associated with control of parasite load (TLR9, rs187084), susceptibility to severe disease (TLR1, rs4833095), resilience to fatal disease (TIRAP S180L) and susceptibility to severe disease, including poor pregnancy outcomes (TLR4, rs4986790) (136, 154–160) (Supplementary Table 1). Together these studies convey the notion that recognition of Plasmodium-derived molecules by innate receptors contribute to the host response in different steps of the natural course of malaria likely by linking with pro-inflammatory responses, such as interferon production (131). Downstream effectors of TLR signaling include a variety of pro-inflammatory cytokines and chemokines such as IFN-γ, IL-6, TNF, IL-12, IFN-1, MCP-1, and IL-8 that take part in the amplification of anti-parasite responses during acute blood-stage infection (32, 161). Furthermore, it has been proposed that polymorphisms in TLRs influence circulating cytokines levels during Plasmodium vivax malaria (162). The association of different TLRs to malaria clinical outcomes suggests that Plasmodium components of diverse nature trigger genetically controlled innate responses that in turn determine the course of infection.
Several lines of evidence suggest that adhesion molecules expressed in endothelial cells are involved in pathogenesis of severe malaria by promoting cytoadhesion and possibly the sequestration of IE in microvessels (163, 164). CD36 is a scavenger receptor that was also identified as a receptor for P. falciparum-infected red blood cells (165). It has been noted that CD36 gene variants showing association to cerebral malaria are not associated to other severe malaria syndromes (74, 149). In particular, the low frequency allele (rs3211938; 1264G) in the exon 10 of CD36 was associated to cerebral malaria (166) and to higher malaria incidence [165) but favored resistance to severe anemia (167) (Supplementary Table 1). Individuals homozygous for this mutation which ablates CD36 protein surface expression showed reduced antibody response to malaria blood stage antigens (168, 169) suggesting that CD36 takes part in the initial steps of the adaptive immune response possibly by mediating phagocytosis of infected red blood cells. Although the role of CD36 in malaria is unsettled (164) these intriguing findings may be related with the multifunctional properties of CD36 namely as a co-receptor that together with TLR4-TLR6 heterodimers acts in the initiation of macrophage inflammatory responses triggered by microbial diacylated lipopeptides (170, 171). Nevertheless, it is possible that CD36 heterozygosity at rs3211938 interferes with the adhesion of IE to endothelial cells, a proposed cerebral malaria pathogenesis mechanism that involves other surface molecules expressed in endothelial cells, including ICAM-1, PECAM-1 and EPCR (164, 172–175).
CD40 ligand is expressed in the surface of activated T cells and binds to CD40 in B cells, regulating B cell proliferation, activation of antigen presentation cell, and immunoglobulin class switching. The CD40LG gene maps to chromosome X and hemizygous males or homozygous females for the minor allele at rs3092945 in the promoter region showed increased resistance to severe malaria (176) an effect that was detected in a multicentric study but was subjected to modifiers in different populations (70). This interesting finding raises the possibility that the genetic control of adaptive immune responses and in particular antibody responses could play a role in controlling parasitemia and thereby govern vulnerability to severe disease in exposed individuals. Accordingly, a recent study found that rs6682413 in the IL23R-IL12RB2 intergenic region was associated with severe malaria anemia (177), supporting observations that IL12 and IL23 pro-inflammatory effects are involved in pathogenesis of severe malaria syndromes (178, 179). In turn, Interferon gamma (IFN-γ) is a key mediator of inflammatory and immune responses induced primarily by interleukin-12 (IL-12). Besides its role in controlling early stages of infection, IFN-γ production by CD4T cells is a hallmark of Th1 polarization enhancing activation of CD8T cells, B cells, and macrophages that may lead to severe immunopathology (180). This is corroborated by genetic studies associating IFN-γ and IFNGR1 gene variants with malaria infection and cerebral malaria (140, 181).
The course of infection by the malaria parasite is contingent upon multiple factors including pathogenicity of parasite biotypes, transmission dynamics and host genetic make-up, that determine (im)balances between parasite expansion and host responses that in turn drive different clinical outcomes. The search for host genetic factors governing the development of clinical malaria phenotypes has attracted significant research efforts in the recent years. Under the selective pressure imposed by severe malaria, genetic variants with strong effects surfaced in endemic regions and were revealed in genes coding for proteins expressed in erythrocytes, prominently hemoglobin (74).
In contrast, inflammatory response genes associated with malaria show smaller genetic effects but unveiled key steps of the host response in the course of infection namely, innate recognition, phagocytosis, exacerbated inflammation, endothelial activation, and immune adaptive responses (Figure 8). Thus, innate receptors recognizing parasite moieties, including TLRs appear to be critical triggers of anti-parasite responses and together with other molecules that are also expressed in phagocytes such as CD36 highlight the central role of phagocytosis in combating infection. A number of pro-inflammatory cytokines and cytokine receptors have been identified in different association studies, including TNF and IFNAR1 but also interferon response genes and TGF beta 2 (Supplementary Table 1). These genetic associations collectively implicate amplification of the inflammatory response as a relevant determinant of infection outcomes. The role of exacerbated inflammatory responses in endothelial activation that underlie severe organ damage is further highlighted by the action of anti-inflammatory mediators such as NO and HO-1 and also by cell surface receptors promoting IE adhesion (Figure 8). Moreover, malaria genetic association with molecules that engage and polarize adaptive immune responses, including CD40L, IL4 (Supplementary Table 1), IFNGR1 (181), and TIM1 (182) underline a role for antibodies in infection resolution. Accumulation of genetic evidence revealing the involvement of inflammatory response genes in malaria provided an invaluable contribution to uncover nodes of the immune host responses that determine deleterious infection trajectories (Figure 8). The biological and physiological relevance of specific human genetic variants and their impact in disease pathogenesis is now experimentally approachable by usage of genome editing methodologies that allow precise introduction of defined mutations in malaria animal models.
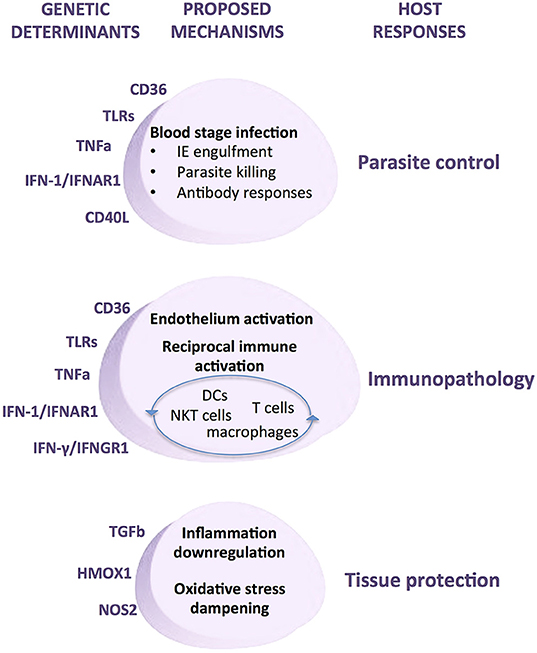
Figure 8. Mechanisms of genetic determination of malaria infection outcomes by inflammatory response genes. Genetic modifiers of human malaria infection have been repeatedly identified in a number of inflammatory response genes. Experimental evidence supports the involvement of these genetic determinants in multiple cellular and molecular mechanisms that underlie the host responses to malaria infection, namely control of parasite load, immunopathology, and tissue damage protection. Under this pathogenesis model specific genetic modifiers of inflammatory responsiveness may play disparate roles in the host responses to infection depending on contextual variables, including disease transmission dynamics, parasite virulence factors, infection stage, tissue-specific cues, and the overall inflammatory mellieu.
The scrutiny of inflammatory genes genetic variants associated with malaria reveals a complex genetic architecture with multiple levels of genetic heterogeneity, namely:
(1) Alternative alleles in one gene either favor or counteract severe disease development (e.g., HMOX1), offering direct interpretation of their genetic effects;
(2) Different genetic variants within a gene promoter are associated with different severe malaria syndromes (e.g., TNF) suggesting differential gene regulation in context of different inflammatory milieus;
(3) Alleles conferring both resilience to severe disease and susceptibility to asymptomatic infection/mild malaria (e.g., IFNAR1) suggesting that advantages of selecting for anti-inflammatory alleles carry fitness costs in infection susceptibility and chronic infection;
(4) Regulatory variants acting prior infection or during infection map in different gene regions (e.g., NOS2), suggesting that infection status alters gene regulatory regimens.
(5) Genetic variants that show disparate roles in malaria pathogenesis depending on their homo/heterozygous state (e.g., CD36).
In this scenario, evaluation of genetic effects requires that the overall allelic variation within an entire gene is taken into account. New genotyping by sequencing methodologies that generate long reads in single DNA molecules enables access to real-haplotype data across entire genes. Data on entire gene variants will allow a comprehensive analysis of their genetic effects in context of genotype combinations with other genes. These data will open new approaches to study non-allelic genetic interactions (e.g., TNF-NOS2 or NOS2-HMOX1 interactions) and to dissect the genetic architecture of inflammatory responses to malaria.
In conclusion, detailed analysis of genetic effects of inflammatory response gene variants is a key step in malaria research to motivate experimental investigations of the underlying pathogenesis mechanisms. This knowledge will be critical to identify rational adjuvant therapies to prevent fatality or undesired malaria complications and subsequent long-term sequels that represent a high burden in endemic regions.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported in part by research grant no. #6-FY15-191 from the March of Dimes and by a grant from ERA-NET NEURON grant (NEUINF), H2020 through the Fundação para a Ciência e Tecnologia (FCT).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I am grateful to Drs. Lars Hviid, Nádia Duarte, and Yash Pandya for reading the manuscript and for insightful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01771/full#supplementary-material
1. White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. (2013) 383:723–35. doi: 10.1016/S0140-6736(13)60024-0
3. Plassmeyer ML, Reiter K, Shimp RL, Kotova S, Smith PD, Hurt DE, et al. Structure of the Plasmodium falciparum circumsporozoite protein, a leading malaria vaccine candidate. J Biol Chem. (2009) 284:26951–63. doi: 10.1074/jbc.M109.013706
4. Farrar J, Hotez PJ, Junghanss T, Kang G, Lalloo D, White NJ. Manson's Tropical Diseases: Twenty-Third Edition. London, UK: Elsevier Health Sciences (2013). doi: 10.1016/C2010-0-66223-7
5. Cowman AF, Healer J, Marapana D, Marsh K. Malaria: biology and disease. Cell. (2016) 167:610–24. doi: 10.1016/j.cell.2016.07.055
6. Osier FH, Mackinnon MJ, Crosnier C, Fegan G, Kamuyu G, Wanaguru M, et al. Malaria: new antigens for a multicomponent blood-stage malaria vaccine. Sci Transl Med. (2014) 6:247ra102. doi: 10.1126/scitranslmed.3008705
7. Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol. (2017) 15:479–91. doi: 10.1038/nrmicro.2017.47
8. Abdi AI, Warimwe GM, Muthui MK, Kivisi CA, Kiragu EW, Fegan GW, et al. Global selection of Plasmodium falciparum virulence antigen expression by host antibodies. Sci Rep. (2016) 6:19882. doi: 10.1038/srep19882
9. Long CA, Zavala F. Malaria vaccines and human immune responses. Curr Opin Microbiol. (2016) 32:96–102. doi: 10.1016/j.mib.2016.04.006
10. Cunnington AJ, Walther M, Riley EM. Piecing together the puzzle of severe malaria. Sci Transl Med. (2009) 5:211ps18. doi: 10.1126/scitranslmed3007432
11. Deroost K, Pham TT, Opdenakker G, Van den Steen PE. The immunological balance between host and parasite in malaria. FEMS Microbiol Rev. (2016) 40:208–57. doi: 10.1093/femsre/fuv046
12. Artavanis-Tsakonas K, Tongren JE, Riley EM. The war between the malaria parasite and the immune system: immunity, immunoregulation and immunopathology. Clin Exp Immunol. (2003) 133:145–52. doi: 10.1046/j.1365-2249.2003.02174.x
13. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. (2005) 5:722–35. doi: 10.1038/nri1686
14. Yazdani SS, Mukherjee P, Chauhan VS, Chitnis CE. Immune responses to asexual blood-stages of malaria parasites. Curr Mol Med. (2006) 6:187–203. doi: 10.2174/156652406776055212
15. Borrmann S, Matuschewski K. Targeting Plasmodium liver stages: better late than never. Trends Mol Med. (2011) 17:527–36. doi: 10.1016/j.molmed.2011.05.008
16. Douglass AN, Kain HS, Abdullahi M, Arang N, Austin LS, Mikolajczak SA, et al. Host-based Prophylaxis Successfully Targets Liver Stage Malaria Parasites. Mol Ther. (2015) 23:857–65. doi: 10.1038/mt.2015.18
17. Gonçalves LA, Rodo J, Rodrigues-Duarte L, de Moraes LV, Penha-Gonçalves C. HGF secreted by activated kupffer cells induces apoptosis of Plasmodium-infected hepatocytes. Front Immunol. (2017) 8:90. doi: 10.3389/fimmu.2017.00090
18. Guha M, Kumar S, Choubey V, Maity P, Bandyopadhyay U. Apoptosis in liver during malaria: role of oxidative stress and implication of mitochondrial pathway. FASEB J. (2006) 20:1224–6. doi: 10.1096/fj.05-5338fje
19. Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, et al. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Rep. (2013). 3:630–7. doi: 10.1016/j.celrep.2013.02.010
20. Van De Sand C, Horstmann S, Schmidt A, Sturm A, Bolte S, Krueger A, et al. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol Microbiol. (2005) 58:731–42. doi: 10.1111/j.1365-2958.2005.04888.x
21. Gonçalves LA, Rodrigues-Duarte L, Rodo J, Vieira de Moraes L, Marques I, Penha-Gonçalves C. TREM2 governs Kupffer cell activation and explains belr1 genetic resistance to malaria liver stage infection. Proc Natl Acad Sci USA. (2013) 110:19531–6. doi: 10.1073/pnas.1306873110
22. Liehl P, Zuzarte-Luís V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med. (2014) 20:47–53. doi: 10.1038/nm.3424
23. Liehl P, Meireles P, Albuquerque IS, Pinkevych M, Baptista F, Mota MM, et al. Innate immunity induced by Plasmodium liver infection inhibits malaria reinfections. Infect Immun. (2015) 83:1172–80. doi: 10.1128/IAI.02796-14
24. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SHI. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep. (2014) 7:436–47. doi: 10.1016/j.celrep.2014.03.018
25. Rennenberg A, Lehmann C, Heitmann A, Witt T, Hansen G, Nagarajan K, et al. Exoerythrocytic Plasmodium parasites secrete a cysteine protease inhibitor involved in sporozoite invasion and capable of blocking cell death of host hepatocytes. PLoS Pathog. (2010) 6:e1000825. doi: 10.1371/journal.ppat.1000825
26. Real E, Rodrigues L, Cabal GG, Enguita FJ, Mancio-Silva L, Mello-Vieira J, et al. Plasmodium UIS3 sequesters host LC3 to avoid elimination by autophagy in hepatocytes. Nat Microbiol. (2017) 3:17–25. doi: 10.1038/s41564-017-0054-x
27. Klotz C, Frevert U. Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine Kupffer cells. Int J Parasitol. (2008) 38:1639–50. doi: 10.1016/j.ijpara.2008.05.018.Plasmodium
28. Usynin I, Klotz C, Frevert U. Malaria circumsporozoite protein inhibits the respiratory burst in Kupffer cells. Cell Microbiol. (2007) 9:2610–28. doi: 10.1111/j.1462-5822.2007.00982.x
29. Buffet PA, Safeukui I, Milon G, Mercereau-Puijalon O, David PH. Retention of erythrocytes in the spleen: A double-edged process in human malaria. Curr Opin Hematol. (2009) 16:157–64. doi: 10.1097/MOH.0b013e32832a1d4b
30. Buffet PA, Safeukui I, Deplaine G, Brousse V, Prendki V, Thellier M, et al. The pathogenesis of Plasmodium falciparum malaria in humans: Insights from splenic physiology. Blood. (2011) 117:381–92. doi: 10.1182/blood-2010-04-202911
31. del Portillo HA, Ferrer M, Brugat T, Martin-Jaular L, Langhorne J, Lacerda MVG. The role of the spleen in malaria. Cell Microbiol. (2012) 14:343–55. doi: 10.1111/j.1462-5822.2011.01741.x
32. Coban C, Ishii KJ, Horii T, Akira S. Manipulation of host innate immune responses by the malaria parasite. Trends Microbiol. (2007) 15:271–8. doi: 10.1016/j.tim.2007.04.003
33. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. (2014) 14:744–57. doi: 10.1038/nri3742
34. Ockenhouse CF, Schulman S, Shear HL. Induction of crisis forms in the human malaria parasite Plasmodium falciparum by gamma-interferon-activated, monocyte-derived macrophages. J Immunol. (1984) 133:1601–8.
35. Cordery DV, Urban BC. Immune recognition of Plasmodium-infected erythrocytes. In: Advances in Experimental Medicine and Biology. New York, NY: Springer (2009). p. 175–184. doi: 10.1007/978-1-4419-0901-5_12
36. Chan JA, Howell KB, Reiling L, Ataide R, Mackintosh CL, Fowkes FJI, et al. Targets of antibodies against Plasmodium falciparum-infected erythrocytes in malaria immunity. J Clin Invest. (2012) 122:3227–38. doi: 10.1172/JCI62182
37. Hill DL, Eriksson EM, Li Wai Suen CSN, Chiu CY, Ryg-Cornejo V, Robinson LJ, et al. Opsonising antibodies to P. falciparum merozoites associated with immunity to clinical malaria. PLoS ONE. (2013) 8:e74627. doi: 10.1371/journal.pone.0074627
38. Osier FHA, Feng G, Boyle MJ, Langer C, Zhou J, Richards JS, et al. Opsonic phagocytosis of Plasmodium falciparum merozoites: mechanism in human immunity and a correlate of protection against malaria. BMC Med. (2014) 12:108. doi: 10.1186/1741-7015-12-108
39. Cohen S, McGregor IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. (1961) 192:733–7. doi: 10.1038/192733a0
40. Langhorne J, Ndungu FMM, Sponaas A-M, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. (2008) 9:725–32. doi: 10.1038/ni.f.205
41. Sabchareon A, Burnouf T, Ouattara D, Attanath P, Bouharoun-Tayoun H, Chantavanich P, et al. Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am J Trop Med Hyg. (1991) 45:297–308. doi: 10.4269/ajtmh.1991.45.297
42. Hviid L, Jensen ATR. PfEMP1 - a parasite protein family of key importance in plasmodium falciparum malaria immunity and pathogenesis. Adv Parasitol. (2015) 88:51–84. doi: 10.1016/bs.apar.2015.02.004
43. Chan J-A, Fowkes FJI, Beeson JG. Surface antigens of Plasmodium falciparum-infected erythrocytes as immune targets and malaria vaccine candidates. Cell Mol Life Sci. (2014) 71:3633–57. doi: 10.1007/s00018-014-1614-3
44. Malleret B, Li A, Zhang R, Tan KSW, Suwanarusk R, Claser C, et al. Plasmodium vivax: restricted tropism and rapid remodeling of CD71-positive reticulocytes. Blood. (2015) 125:1314–24. doi: 10.1182/blood-2014-08-596015
45. Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev Mol Med. (2009) 11:e16. doi: 10.1017/S1462399409001082
46. Sherman IW, Eda S, Winograd E. Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes Infect. (2003) 5:897–909. doi: 10.1016/S1286-4579(03)00162-X
47. Barber BE, William T, Grigg MJ, Parameswaran U, Piera KA, Price RN, et al. Parasite biomass-related inflammation, endothelial activation, microvascular dysfunction and disease severity in vivax malaria. PLoS Pathog. (2015) 11:e1004558. doi: 10.1371/journal.ppat.1004558
48. Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol. (2007) 23:596–604. doi: 10.1016/j.pt.2007.09.003
49. Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. (2005) 280:8606–16. doi: 10.1074/jbc.M413541200
50. Tiemi Shio M, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. (2009) 5:e1000559. doi: 10.1371/journal.ppat.1000559
51. Gillrie MR, Lee K, Gowda DC, Davis SP, Monestier M, Cui L, et al. Plasmodium falciparum histones induce endothelial proinflammatory response and barrier dysfunction. Am J Pathol. (2012) 180:1028–39. doi: 10.1016/j.ajpath.2011.11.037
52. Sahu U, Sahoo PK, Kar SK, Mohapatra BN, Ranjit M. Association of TNF level with production of circulating cellular microparticles during clinical manifestation of human cerebral malaria. Hum Immunol. (2013) 74:713–21. doi: 10.1016/j.humimm.2013.02.006
53. Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA, et al. Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J Exp Med. (1996) 184:557–67. doi: 10.1084/jem.184.2.557
54. Gomes LT, Alves ER, Rodrigues-Jesus C, Nery AF, Gasquez-Martin TO, Fontes CJ. Angiopoietin-2 and angiopoietin-2/angiopoietin-1 ratio as indicators of potential severity of Plasmodium vivax malaria in patients with thrombocytopenia. PLoS ONE. (2014) 9:e109246. doi: 10.1371/journal.pone.0109246
55. Wenisch C, Wenisch H, Wilairatana P, Looareesuwan S, Vannaphan S, Wagner O, et al. Big endothelin in patients with complicated Plasmodium falciparum malaria. J Infect Dis. (1996) 173:1281–4. doi: 10.1093/infdis/173.5.1281
56. Bridges DJ, Bunn J, Van Mourik JA, Grau G, Preston RJS, Molyneux M, et al. Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet-decorated von Willebrand factor strings. Blood. (2010) 115:1472–4. doi: 10.1182/blood-2009-07-235150
57. Francischetti IMB, Seydel KB, Monteiro RQ, Whitten RO, Erexson CR, Noronha ALL, et al. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multimolecular coagulation complexes. J Thromb Haemost. (2007) 5:155–65. doi: 10.1111/j.1538-7836.2006.02232.x
58. Wassmer SC, de Souza JB, Frere C, Candal FJ, Juhan-Vague I, Grau GE. TGF- 1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis: a new mechanism for microvascular lesion during cerebral malaria. J Immunol. (2006) 176:1180–4. doi: 10.4049/jimmunol.176.2.1180
59. Soares MP, Teixeira L, Moita LF. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol. (2017) 17:83–96. doi: 10.1038/nri.2016.136
60. Scholzen A, Mittag D, Rogerson SJ, Cooke BM, Plebanski M. Plasmodium falciparum-mediated induction of human CD25 hiFoxp3hi CD4 T cells is independent of direct TCR stimulation and requires IL-2, IL-10, and TGFβ. PLoS Pathog. (2009) 5:e1000543. doi: 10.1371/journal.ppat.1000543
61. Scholzen A, Cooke BM, Plebanski M. Plasmodium falciparum induces Foxp3hi CD4 T cells independent of surface PfEMP1 expression via small soluble parasite components. Front Microbiol. (2014) 5:200. doi: 10.3389/fmicb.2014.00200
62. Omer FM, Riley EM. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J Exp Med. (1998) 188, 39–48. doi: 10.1084/jem.188.1.39
63. Wenisch C, Parschalk B, Burgmann H, Looareesuwan S, Graninger W. Decreased serum levels of TGF-beta in patients with acute Plasmodium falciparum malaria. J Clin Immunol. (1995) 15:69–73.
64. Coban C, Lee MSJ, Ishii KJ. Tissue-specific immunopathology during malaria infection. Nat Rev Immunol. (2018) 18:266–78. doi: 10.1038/nri.2017.138
65. Band G, Le QS, Jostins L, Pirinen M, Kivinen K, Jallow M, et al. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. (2013) 9:e1003509. doi: 10.1371/journal.pgen.1003509
66. Hedrick PW. Population genetics of malaria resistance in humans. Heredity. (2011) 107:283–304. doi: 10.1038/hdy.2011.16
67. Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. (2009) 41:657–65. doi: 10.1038/ng.388
68. Ma S, Cahalan S, LaMonte G, Grubaugh ND, Zeng W, Murthy SE, et al. Common PIEZO1 allele in African populations causes RBC dehydration and attenuates Plasmodium infection. Cell. (2018) 173:443–455.e12. doi: 10.1016/j.cell.2018.02.047
69. Malaria Genomic Epidemiology Network. A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature. (2015) 526:253–7. doi: 10.1038/nature15390
70. Rockett KA, Clarke GM, Fitzpatrick K, Hubbart C, Jeffreys AE, Rowlands K, et al. Reappraisal of known malaria resistance loci in a large multicenter study. Nat Genet. (2014) 46:1197–204. doi: 10.1038/ng.3107
71. Timmann C, Thye T, Vens M, Evans J, May J, Ehmen C, et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. (2012) 489:443–6. doi: 10.1038/nature11334
72. Driss A, Hibbert JM, Wilson NO, Iqbal SA, Adamkiewicz TV, Stiles JK. Genetic polymorphisms linked to susceptibility to malaria. Malar J. (2011) 10:271. doi: 10.1186/1475-2875-10-271
73. Marsh K, Snow RW. Host-parasite interaction and morbidity in malaria endemic areas. Philos Trans R Soc London B. (1997) 352:1385–94. doi: 10.1098/rstb.1997.0124
74. Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. (2005) 77:171–92. doi: 10.1086/432519
75. Clark IA, Virelizier JL, Carswell EA, Wood PR. Possible importance of macrophage-derived mediators in acute malaria. Infect Immun. (1981) 32:1058–1066
76. Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. (2007) 18:335–43. doi: 10.1016/j.cytogfr.2007.04.002
77. Clark IA. Along a TNF-paved road from dead parasites in red cells to cerebral malaria, and beyond. Parasitology. (2009) 136:1457–68. doi: 10.1017/S0031182009006088
78. Clark IA, Cowden WB, Butcher GA, Hunt NH. Possible roles of tumor necrosis factor in the pathology of malaria. Am J Pathol. (1987) 129:192–9.
79. McGuire W, D'Alessandro U, Stephens S, Olaleye BO, Langerock P, Greenwood BM, et al. Levels of tumour necrosis factor and soluble TNF receptors during malaria fever episodes in the community. Trans R Soc Trop Med Hyg. (1998) 92:50–3. doi: 10.1016/S0035-9203(98)90951-8
80. Mordmüller BG, Metzger WG, Juillard P, Brinkman BM, Verweij CL, Grau GE, et al. Tumor necrosis factor in Plasmodium falciparum malaria: high plasma level is associated with fever, but high production capacity is associated with rapid fever clearance. Eur Cytokine Netw. (1997) 8:29–35.
81. Othoro C, Lal AA, Nahlen B, Koech D, Orago ASS, Udhayakumar V. A low interleukin-10 tumor necrosis factor-α ratio is associated with malaria anemia in children residing in a holoendemic malaria region in Western Kenya. J Infect Dis. (1999) 179:279–82. doi: 10.1086/314548
82. Shaffer N, Grau GE, Hedberg K, Davachi F, Lyamba B, Hightower AW, et al. Tumor necrosis factor and severe malaria. J Infect Dis. (1991) 163:96–101.
83. Awandare GA, Goka B, Boeuf P, Tetteh JKA, Kurtzhals JAL, Behr C, et al. Increased levels of inflammatory mediators in children with severe Plasmodium falciparum malaria with respiratory distress. J Infect Dis. (2006) 194:1438–46. doi: 10.1086/508547
84. Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, et al. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med. (1989) 320:1586–91. doi: 10.1056/NEJM198906153202404
85. Kern P, Hemmer CJ, Van Damme J, Gruss HJ, Dietrich M. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med. (1989) 87:139–43. doi: 10.1016/S0002-9343(89)80688-6
86. Kwiatkowski D, Sambou I, Twumasi P, Greenwood BM, Hill AVS, Manogue KR, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. (1990) 336:1201–4. doi: 10.1016/0140-6736(90)92827-5
87. Tchinda VHM, Tadem AD, Tako EA, Tene G, Fogako J, Nyonglema P, et al. Severe malaria in Cameroonian children: correlation between plasma levels of three soluble inducible adhesion molecules and TNF-a. Acta Trop. (2007) 102:20–8. doi: 10.1016/j.actatropica.2007.02.011
88. Randall LM, Engwerda CR. TNF family members and malaria: old observations, new insights and future directions. Exp Parasitol. (2010) 126:326–31. doi: 10.1016/j.exppara.2010.04.016
89. Higuchi T, Seki N, Kamizono S, Yamada A, Kimura A, Kato H, et al. Polymorphism of the 5′-flanking region of the human tumor necrosis factor (TNF)-alpha gene in Japanese. Tissue Antigens. (1998) 51:605–12. doi: 10.1111/j.1399-0039.1998.tb03002.x
90. Huizinga TW, Westendorp RG, Bollen EL, Keijsers V, Brinkman BM, Langermans JA, et al. TNF-alpha promoter polymorphisms, production and susceptibility to multiple sclerosis in different groups of patients. J Neuroimmunol. (1997) 72:149–53.
91. Kaluza W, Reuss E, Grossmann S, Hug R, Schopf RE, Galle PR, et al. Different transcriptional activity and in vitro TNF-α production in psoriasis patients carrying the TNF-α 238A promoter polymorphism. J Invest Dermatol. (2000) 114:1180–3. doi: 10.1046/j.1523-1747.2000.00001.x
92. Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA. (1997) 94:3195–9. doi: 10.1073/pnas.94.7.3195
93. Basu M, Maji AK, Chakraborty A, Banerjee R, Mullick S, Saha P, et al. Genetic association of Toll-like-receptor 4 and tumor necrosis factor-α polymorphisms with Plasmodium falciparum blood infection levels. Infect Genet Evol. (2010) 10:686–96. doi: 10.1016/j.meegid.2010.03.008
94. Flori L, Delahaye NF, Iraqi FA, Hernandez-Valladares M, Fumoux F, Rihet P. TNF as a malaria candidate gene: polymorphism-screening and family-based association analysis of mild malaria attack and parasitemia in Burkina Faso. Genes Immun. (2005) 6:472–80. doi: 10.1038/sj.gene.6364231
95. Nguyen TN, Baaklini S, Koukouikila-Koussounda F, Ndounga M, Torres M, Pradel L, et al. Association of a functional TNF variant with Plasmodium falciparum parasitaemia in a congolese population. Genes Immun. (2017) 18:152–7. doi: 10.1038/gene.2017.13
96. Afridi S, Atkinson A, Garnier S, Fumoux F, Rihet P. Malaria resistance genes are associated with the levels of IgG subclasses directed against Plasmodium falciparum blood-stage antigens in Burkina Faso. Malar J. (2012) 11:308. doi: 10.1186/1475-2875-11-308
97. Dunstan SJ, Rockett KA, Quyen NTN, Teo YY, Thai CQ, Hang NT, et al. Variation in human genes encoding adhesion and proinflammatory molecules are associated with severe malaria in the Vietnamese. Genes Immun. (2012) 13:503–8. doi: 10.1038/gene.2012.25
98. Hananantachai H, Patarapotikul J, Ohashi J, Naka I, Krudsood S, Looareesuwan S, et al. Significant association between TNF-α (TNF) promoter allele (-1031C,−863C, and−857C) and cerebral malaria in Thailand. Tissue Antigens. (2007) 69:277–80. doi: 10.1111/j.1399-0039.2006.00756.x
99. McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. (1994) 371:508–10. doi: 10.1038/371508a0
100. Olaniyan SA, Amodu OK, Bakare AA, Troye-Blomberg M, Omotade OO, Rockett KA. Tumour necrosis factor alpha promoter polymorphism, TNF-238 is associated with severe clinical outcome of falciparum malaria in Ibadan southwest Nigeria. Acta Trop. (2016) 161:62–7. doi: 10.1016/j.actatropica.2016.05.006
101. Ubalee R, Suzuki F, Kikuchi M, Tasanor O, Wattanagoon Y, Ruangweerayut R, et al. Strong association of a tumor necrosis factor-?? promoter allele with cerebral malaria in Myanmar. Tissue Antigens. (2001) 58:407–10. doi: 10.1034/j.1399-0039.2001.580610.x
102. Wattavidanage J, Carter R, Perera KL, Munasingha A, Bandara S, McGuinness D, et al. TNFalpha*2 marks high risk of severe disease during Plasmodium falciparum malaria and other infections in Sri Lankans. Clin Exp Immunol. (1999) 115:350–5. doi: 10.1046/j.1365-2249.1999.00804.x
103. Nasr A, Allam G, Hamid O, Al-Ghamdi A. IFN-gamma and TNF associated with severe falciparum malaria infection in Saudi pregnant women. Malar J. (2014) 13:314. doi: 10.1186/1475-2875-13-314
104. Aidoo M, McElroy PD, Kolczak MS, Terlouw DJ, ter Kuile FO, Nahlen B, et al. Tumor necrosis factor-α promoter variant 2 (TNF2) is associated with pre-term delivery, infant mortality, and malaria morbidity in western Kenya: Asembo Bay Cohort Project IX. Genet Epidemiol. (2001) 21:201–11. doi: 10.1002/gepi.1029
105. El-Tahan RR, Ghoneim AM, El-Mashad N. TNF-α gene polymorphisms and expression. Springerplus. (2016) 5:1508. doi: 10.1186/s40064-016-3197-y
106. Perera MK, Herath NP, Pathirana SL, Phone-Kyaw M, Alles HK, Mendis KN, et al. Association of high plasma TNF-alpha levels and TNF-alpha/IL-10 ratios with TNF2 allele in severe P. falciparum malaria patients in Sri Lanka. Pathog Glob Health. (2013) 107:21–9. doi: 10.1179/2047773212Y.0000000069
107. McGuire W, Knight JC, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Severe malarial anemia and cerebral malaria are associated with different tumor necrosis factor promoter alleles. J Infect Dis. (1999) 179:287–90. doi: 10.1086/314533
108. Clark TG, Diakite M, Auburn S, Campino S, Fry AE, Green A, et al. Tumor necrosis factor and lymphotoxin-alpha polymorphisms and severe malaria in African populations. J Infect Dis. (2009) 199:569–75. doi: 10.1086/596320
109. Anstey NM, Granger DL, Hassanali MY, Mwaikambo ED, Duffy PE, Weinberg JB. Nitric oxide, malaria, and anemia: inverse relationship between nitric oxide production and hemoglobin concentration in asymptomatic, malaria-exposed children. Am J Trop Med Hyg. (1999) 61:249–52.
110. Sobolewski P, Gramaglia I, Frangos J, Intaglietta M, Van Der Heyde HC. Nitric oxide bioavailability in malaria. Trends Parasitol. (2005) 21:415–22. doi: 10.1016/j.pt.2005.07.002
111. Weinberg JB, Lopansri BK, Mwaikambo E, Granger DL. Arginine, nitric oxide, carbon monoxide, and endothelial function in severe malaria. Curr Opin Infect Dis. (2008) 21:468–75. doi: 10.1097/QCO.0b013e32830ef5cf
112. Weinberg JB, Volkheimer AD, Rubach MP, Florence SM, Mukemba JP, Kalingonji AR, et al. Monocyte polarization in children with falciparum malaria: relationship to nitric oxide insufficiency and disease severity. Sci Rep. (2016) 6:29151. doi: 10.1038/srep29151
113. Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. (2007) 12:1417–22. doi: 10.1038/nm1499
114. Jeney V, Ramos S, Bergman M-L, Bechmann I, Tischer J, Ferreira A, et al. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep. (2014) 8:126–36. doi: 10.1016/j.celrep.2014.05.054
115. Serghides L, Kim H, Lu Z, Kain DC, Miller C, Francis RC, et al. Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS ONE. (2011) 6:e27714. doi: 10.1371/journal.pone.0027714
116. Hawkes MT, Conroy AL, Opoka RO, Hermann L, Thorpe KE, McDonald C, et al. Inhaled nitric oxide as adjunctive therapy for severe malaria: a randomized controlled trial. Malar J. (2015) 14:421. doi: 10.1186/s12936-015-0946-2
117. de Jesus Trovoada M, Martins M, Mansour R, Ben Do Rosário Sambo M, Fernandes AB, Gonçalves LA, et al. NOS2 variants reveal a dual genetic control of nitric oxide levels, susceptibility to Plasmodium infection, and cerebral malaria. Infect. Immun. (2014) 82:1287–95. doi: 10.1128/IAI.01070-13
118. Hobbs MR, Udhayakumar V, Levesque MC, Booth J, Roberts JM, Tkachuk AN, et al. A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet. (2002) 360:1468–75. doi: 10.1016/S0140-6736(02)11474-7
119. Kun JF, Mordmuller B, Perkins DJ, May J, Mercereau-Puijalon O, Alpers M, et al. Nitric oxide synthase 2(Lambarene) (G-954C), increased nitric oxide production, and protection against malaria. J Infect Dis. (2001) 184:330–6. doi: 10.1086/322037
120. Apinjoh TO, Anchang-Kimbi JK, Njua-Yafi C, Ngwai AN, Mugri RN, Clark TG, et al. Association of candidate gene polymorphisms and TGF-beta/IL-10 levels with malaria in three regions of Cameroon: a case–control study. Malar J. (2014) 13:236. doi: 10.1186/1475-2875-13-236
121. Kun JF, Mordmuller B, Lell B, Lehman LG, Luckner D, Kremsner PG. Polymorphism in promoter region of inducible nitric oxide synthase gene and protection against malaria. Lancet. (1998) 351:265–6. doi: 10.1016/S0140-6736(05)78273-8
122. Lwanira CN, Kironde F, Kaddumukasa M, Swedberg G. Prevalence of polymorphisms in glucose-6-phosphate dehydrogenase, sickle haemoglobin and nitric oxide synthase genes and their relationship with incidence of uncomplicated malaria in Iganga, Uganda. Malar J. (2017) 16:322. doi: 10.1186/s12936-017-1970-1
123. Parikh S, Dorsey G, Rosenthal PJ. Host polymorphisms and the incidence of malaria in Ugandan children. Am J Trop Med Hyg. (2004) 71:750–3. doi: 10.4269/ajtmh.2004.71.750
124. Burgner D, Xu W, Rockett K, Gravenor M, Charles IG, Hill AV, et al. Inducible nitric oxide synthase polymorphism and fatal cerebral malaria. Lancet. (1998) 352:1193–4. doi: 10.1016/S0140-6736(05)60531-4
125. Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, Tokunaga K. Significant association of longer forms of CCTTT Microsatellite repeat in the inducible nitric oxide synthase promoter with severe malaria in Thailand. J Infect Dis. (2002) 186:578–81. doi: 10.1086/341779
126. Cramer JP, Mockenhaupt FP, Ehrhardt S, Burkhardt J, Otchwemah RN, Dietz E, et al. iNOS promoter variants and severe malaria in Ghanaian children. Trop Med Int Heal. (2004) 9:1074–80. doi: 10.1111/j.1365-3156.2004.01312.x
127. Levesque MC, Hobbs MR, Anstey NM, Chancellor JA, Misukonis MAM, Granger DL, et al. A review of polymorphisms in the human gene for inducible nitric oxide synthase (NOS2) in patients with malaria. Sepsis. (2001) 4:217–31. doi: 10.1023/A:1012913023602
128. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. (2011) 35:194–207. doi: 10.1016/j.immuni.2011.05.016
129. Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B, et al. STING-licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe Plasmodium yoelii malaria. PLoS Pathog. (2016) 12:e1005975. doi: 10.1371/journal.ppat.1005975
130. Torre S, Polyak MJ, Langlais D, Fodil N, Kennedy JM, Radovanovic I, et al. USP15 regulates type I interferon response and is required for pathogenesis of neuroinflammation. Nat Immunol. (2016) 18:54–63. doi: 10.1038/ni.3581
131. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria Immunity and Host Mortality. Immunity. (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
132. Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH, et al. Type I interferons suppress CD4 + T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol. (2011) 41:2688–98. doi: 10.1002/eji.201141539
133. Haque A, Best SE, De Oca MM, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8-DCs impairs Th1-dependent malaria immunity. J Clin Invest. (2014) 124:2483–96. doi: 10.1172/JCI70698
134. Sebina I, James KR, Soon MSF, Fogg LG, Best SE, Labastida Rivera F, et al. IFNAR1-signalling obstructs ICOS-mediated humoral immunity during non-lethal blood-stage Plasmodium infection. PLoS Pathog. (2016) 12:e1005999. doi: 10.1371/journal.ppat.1005999
135. Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJJ, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog. (2016) 12:e1005945. doi: 10.1371/journal.ppat.1005945
136. Ball EA, Sambo MR, Martins M, Trovoada MJ, Benchimol C, Costa J, et al. IFNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in Plasmodium berghei-infected mice. J Immunol. (2013) 190:5118–27. doi: 10.4049/jimmunol.1300114
137. Palomo J, Fauconnier M, Coquard L, Gilles M, Meme S, Szeremeta F, et al. Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood-stage Plasmodium berghei ANKA. Eur J Immunol. (2013) 43:2683–95. doi: 10.1002/eji.201343327
138. Aucan C, Walley AJ, Hennig BJW, Fitness J, Frodsham A, Zhang L, et al. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun. (2003) 4:275–82. doi: 10.1038/sj.gene.6363962
139. Kanchan K, Jha P, Pati SS, Mohanty S, Mishra SK, Sharma SK, et al. Interferon-γ (IFNG) microsatellite repeat and single nucleotide polymorphism haplotypes of IFN-α receptor (IFNAR1) associated with enhanced malaria susceptibility in Indian populations. Infect Genet Evol. (2015) 29:6–14. doi: 10.1016/j.meegid.2014.10.030
140. Khor CC, Vannberg FO, Chapman SJ, Walley A, Aucan C, Loke H, et al. Positive replication and linkage disequilibrium mapping of the chromosome 21q22.1 malaria susceptibility locus. Genes Immun. (2007) 8:570–6. doi: 10.1038/sj.gene.6364417
141. Feintuch CM, Tare A, Cusumano L, Benayoun J, Ryu S, Sixpence A, et al. Type I interferon receptor variants in gene regulatory regions are associated with susceptibility to cerebral malaria in Malawi. Am J Trop Med Hyg. (2018) 98:1692–8. doi: 10.4269/ajtmh.17-0887
142. Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. (1997) 37:517–54. doi: 10.1146/annurev.pharmtox.37.1.517
143. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. (2007) 13:703–10. doi: 10.1038/nm1586
144. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci USA. (2009) 106:15837–42. doi: 10.1073/pnas.0903419106
145. Pereira MLM, Marinho CRF, Epiphanio S. Could heme oxygenase-1 be a new target for therapeutic intervention in malaria-associated acute lung injury/acute respiratory distress syndrome? Front Cell Infect Microbiol. (2018) 8:161. doi: 10.3389/fcimb.2018.00161
146. Epiphanio S, Mikolajczak SA, Gonçalves LA, Pamplona A, Portugal S, Albuquerque S, et al. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine Plasmodium liver infection. Cell Host Microbe. (2008) 3:331–8. doi: 10.1016/j.chom.2008.04.003
147. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. (2004) 37:1097–104. doi: 10.1016/j.freeradbiomed.2004.07.008
148. Takeda M, Kikuchi M, Ubalee R, Na-Bangchang K, Ruangweerayut R, Shibahara S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to cerebral malaria in Myanmar. Jpn J Infect Dis. (2005) 58:268–71.
149. Sambo MR, Trovoada MJ, Benchimol C, Quinhentos V, Gonçalves L, Velosa R, et al. Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS ONE. (2010) 5:e11141. doi: 10.1371/journal.pone.0011141
150. Walther M, De Caul A, Aka P, Njie M, Amambua-Ngwa A, Walther B, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. (2012) 8:e1002579. doi: 10.1371/journal.ppat.1002579
151. Mendonça VRR, Luza NF, Santos NJG, Borges VM, Gonçalves MS, Andrade BB, et al. Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect Immun. (2012) 80:1445–54. doi: 10.1128/IAI.05933-11
152. Hansson HH, Maretty L, Balle C, Goka BQ, Luzon E, Nkrumah FN, et al. Polymorphisms in the Haem Oxygenase-1 promoter are not associated with severity of Plasmodium falciparum malaria in Ghanaian children. Malar J. (2015) 14:153. doi: 10.1186/s12936-015-0668-5
153. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. PNAS. (2007) 104:1919–24. doi: 10.1073/pnas.0608745104
154. Costa AG, Ramasawmy R, Ibiapina HNS, Sampaio VS, Xábregas LA, Brasil LW, et al. Association of TLR variants with susceptibility to Plasmodium vivax malaria and parasitemia in the Amazon region of Brazil. PLoS ONE. (2017) 12:e0183840. doi: 10.1371/journal.pone.0183840
155. Hamann L, Bedu-Addo G, Eggelte TA, Schumann RR, Mockenhaupt FP. The toll-like receptor 1 variant S248N influences placental malaria. Infect Genet Evol. (2010) 10:785–9. doi: 10.1016/j.meegid.2010.05.005
156. Leoratti FMS, Farias L, Alves FP, Suarez-Mútis MC, Coura JR, Kalil J, et al. Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. J Infect Dis. (2008) 198:772–80. doi: 10.1086/590440
157. Manning L, Cutts J, Stanisic DI, Laman M, Carmagnac A, Allen S, et al. A Toll-like receptor-1 variant and its characteristic cellular phenotype is associated with severe malaria in Papua New Guinean children. Genes Immun. (2016) 17:52–9. doi: 10.1038/gene.2015.50
158. Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, et al. Toll-Like Receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. J Commun Dis. (2006) 38:230–45. doi: 10.1073/pnas.0506803102
159. Mockenhaupt FP, Hamann L, von Gaertner C, Bedu-Addo G, von Kleinsorgen C, Schumann RR, et al. Common polymorphisms of toll-like receptors 4 and 9 are associated with the clinical manifestation of malaria during pregnancy. J Infect Dis. (2006) 194:184–8. doi: 10.1086/505152
160. Panda AK, Das BK, Panda A, Tripathy R, Pattnaik SS, Mahto H, et al. Heterozygous mutants of TIRAP (S180L) polymorphism protect adult patients with Plasmodium falciparum infection against severe disease and mortality. Infect Genet Evol. (2016) 43:146–50. doi: 10.1016/j.meegid.2016.04.035
161. Erdman L, Finney C, Liles W, Kain K. Inflammatory pathways in malaria infection: TLRs share the stage with other components of innate immunity. Mol Biochem Parasitol. (2008) 162:105–11. doi: 10.1016/j.molbiopara.2008.08.006
162. Costa AG, Ramasawmy R, Val FFA, Ibiapina HNS, Oliveira AC, Tarragô AM, et al. Polymorphisms in TLRs influence circulating cytokines production in Plasmodium vivax malaria: TLR polymorphisms influence cytokine productions in malaria-vivax. Cytokine. (2018) 110:374–80. doi: 10.1016/j.cyto.2018.04.008
163. Berendt AR, Ferguson DJP, Newbold CI. Sequestration in Plasmodium falciparum malaria: sticky cells and sticky problems. Parasitol Today. (1990) 6:247–54. doi: 10.1016/0169-4758(90)90184-6
164. Cabrera A, Neculai D, Kain KC. CD36 and malaria: friends or foes? A decade of data provides some answers. Trends Parasitol. (2014) 30:436–44. doi: 10.1016/j.pt.2014.07.006
165. Barnwell JW, Asch AS, Nachman RL, Yamaya M, Aikawa M, Ingravallo P. A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. J Clin Invest. (1989) 84:765–72. doi: 10.1172/JCI114234
166. Aitman TJ, Cooper LD, Norsworthy PJ, Wahid FN, Gray JK, Curtis BR, et al. Malaria susceptibility and CD36 mutation. Nature. (2000) 405:1015–6. doi: 10.1038/35016636
167. Pain A, Urban BC, Kai O, Casals-Pascual C, Shafi J, Marsh K, et al. A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet. (2001) 357:1502–3. doi: 10.1016/S0140-6736(00)04662-6
168. Kajeguka D, Mwanziva C, Daou M, Ndaro A, Matondo S, Mbugi E, et al. CD36 c.1264 T>G null mutation impairs acquisition of IgG antibodies to Plasmodium falciparum MSP1-19 antigen and is associated with higher malaria incidences in Tanzanian children. Scand J Immunol. 75:355–60. doi: 10.1111/j.1365-3083.2011.02661.x
169. Shelton JMG, Corran P, Risley P, Silva N, Hubbart C, Jeffreys A, et al. Genetic determinants of anti-malarial acquired immunity in a large multi-centre study. Malar J. (2015) 14:333. doi: 10.1186/s12936-015-0833-x
170. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature. (2005) 433:523–7. doi: 10.1038/nature03253
171. Stewart CR, Stuart LM, Wilkinson K, Van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
172. Bernabeu M, Smith JD. EPCR and Malaria Severity: The Center of a Perfect Storm. Trends Parasitol. (2017) 33:295–308. doi: 10.1016/j.pt.2016.11.004
173. Chakravorty SJ, Craig A. The role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur J Cell Biol. (2005) 84:15–27. doi: 10.1016/j.ejcb.2004.09.002
174. Kessler A, Dankwa S, Bernabeu M, Harawa V, Danziger SA, Duffy F, et al. Linking EPCR-Binding PfEMP1 to Brain Swelling in Pediatric Cerebral Malaria. Cell Host Microbe. (2017) 22:601–14.e5. doi: 10.1016/j.chom.2017.09.009
175. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JEV, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. (2013) 498:502–5. doi: 10.1038/nature12216
176. Sabeti P, Usen S, Farhadian S, Jallow M, Doherty T, Newport M, et al. CD40L association with protection from severe malaria. Genes Immun. (2002) 3:286–91. doi: 10.1038/sj.gene.6363877
177. Ravenhall M, Campino S, Sepúlveda N, Manjurano A, Nadjm B, Mtove G, et al. Novel genetic polymorphisms associated with severe malaria and under selective pressure in North-eastern Tanzania. PLoS Genet. (2018) 14:e1007172. doi: 10.1371/journal.pgen.1007172
178. Zhang L, Prather D, Eng J, Vanden Eng J, Crawford S, Kariuki S, et al. Polymorphisms in genes of interleukin 12 and its receptors and their association with protection against severe malarial anaemia in children in western Kenya. Malar J. (2010) 9:87. doi: 10.1186/1475-2875-9-87
179. Marquet S, Conte I, Poudiougou B, Argiro L, Cabantous S, Dessein H, et al. The IL17F and IL17RA genetic variants increase risk of cerebral malaria in two African populations. Infect Immun. (2016) 84:590–7. doi: 10.1128/IAI.00671-15
180. King T, Lamb T. Interferon-γ: The Jekyll and Hyde of Malaria. PLoS Pathog. (2015) 11:e1005118. doi: 10.1371/journal.ppat.1005118
181. Koch O, Awomoyi A, Usen S, Jallow M, Richardson A, Hull J, et al. IFNGR1 gene promoter polymorphisms and susceptibility to cerebral malaria. J Infect Dis. (2002) 185:1684–7. doi: 10.1086/340516
182. Nuchnoi P, Ohashi J, Kimura R, Hananantachai H, Naka I, Krudsood S, et al. Significant association between TIM1 promoter polymorphisms and protection against cerebral malaria in Thailand. Ann Hum Genet. (2008) 72:327–36. doi: 10.1111/j.1469-1809.2007.00424.x
183. Opi DH, Swann O, Macharia A, Uyoga S, Band G, Ndila CM, et al. Two complement receptor one alleles have opposing associations with cerebral malaria and interact with α+thalassaemia. Elife. (2018) 7:1–27. doi: 10.7554/eLife.31579
184. Sinha S, Mishra SK, Sharma S, Patibandla PK, Mallick PK, Sharma SK, et al. Polymorphisms of TNF-enhancer and gene for FcγRIIa correlate with the severity of falciparum malaria in the ethnically diverse Indian population. Malar J. (2008) 7:13. doi: 10.1186/1475-2875-7-13
185. Meyer CG, May J, Luty AJ, Lell B, Kremsner PG. TNFalpha-308A associated with shorter intervals of Plasmodium falciparum reinfections. Tissue Antigens. (2002) 59:287–92. doi: 10.1034/j.1399-0039.2002.590406.x
186. Purohit P, Mohanty PK, Patel S, Das P, Das K, Panigrahi J. Association of CD40L gene polymorphism with severe Plasmodium falciparum malaria in Indian population. J Vector Borne Dis. (2017) 54:74–9.
187. Luoni G, Verra F, Arcà B, Sirima BS, Troye-Blomberg M, Coluzzi M, et al. Antimalarial antibody levels and IL4 polymorphism in the Fulani of West Africa. Genes Immun. (2001) 2:411–4. doi: 10.1038/sj.gene.6363797
188. Jha AN, Singh VK, Singh R, Pati SS, Patra PK, Singh L, et al. A rare non-synonymous c.102C>G SNP in the IFNB1 gene might be a risk factor for cerebral malaria in Indian populations. Infect Genet Evol. (2013) 14:369–74. doi: 10.1016/j.meegid.2012.12.029
189. Cabantous S, Poudiougou B, Oumar AA, Traore A, Barry A, Vitte J, et al. Genetic evidence for the aggravation of Plasmodium falciparum malaria by interleukin 4. J Infect Dis. (2009) 200:1530–9. doi: 10.1086/644600
190. Ojurongbe O, Funwei RI, Snyder TJ, Farid I, Aziz N, Li Y, et al. Genetic variants of tumor necrosis factor-α−308G/A (rs1800629) but not Toll-interacting proteins or vitamin D receptor genes enhances susceptibility and severity of malaria infection. Immunogenetics. (2018) 70:135–40. doi: 10.1007/s00251-017-1032-4
191. Burgner D, Rockett K, Ackerman H, Hull J, Usen S, Pinder M, et al. Haplotypic relationship between SNP and microsatellite markers at the NOS2A locus in two populations. Genes Immun. (2003) 4:506–14. doi: 10.1038/sj.gene.6364022
Keywords: malaria, NOS2 gene, type 1 interferon receptor 1 (IFNAR1), genetics, TNFA gene, CD40LG gene, HMOX1 gene, TLRs (toll-like receptors)
Citation: Penha-Gonçalves C (2019) Genetics of Malaria Inflammatory Responses: A Pathogenesis Perspective. Front. Immunol. 10:1771. doi: 10.3389/fimmu.2019.01771
Received: 11 July 2018; Accepted: 15 July 2019;
Published: 30 July 2019.
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Beatrix Schumak, University of Bonn, GermanyCopyright © 2019 Penha-Gonçalves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Penha-Gonçalves, Y3BlbmhhQGlnYy5ndWxiZW5raWFuLnB0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.