 Borros Arneth
Borros Arneth
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol., 07 August 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01697
This article is part of the Research TopicSkin AutoimmunityView all 69 articles
Introduction: Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that is characterized by the production of autoantibodies specific for components of the cell nucleus and that causes damage to body tissues and organs. The pathogenesis of SLE remains unclear, with numerous studies pointing to a combination of genetic and environmental factors. A critical stage in SLE development is cell necrosis, in which undegraded chromatin and nucleoproteins are released into the blood, resulting in circulating cell-free DNA and serum nucleoproteins that trigger anti-dsDNA autoantibody production. This systematic literature review aimed to examine whether SLE stems from a DNA degradation and elimination defect.
Materials and Methods: An advanced literature search was conducted in PubMed using the following keywords: [(“SLE” OR “Systemic Lupus Erythematosus” OR “Lupus”)] AND [(“DNA” OR “DNA Degradation”)] AND [(“Defect Elimination”)]. More articles were obtained from the references of the identified articles and basic Google searches. Twenty-five peer-reviewed articles published within the past 10 years (2007–2018) were included for review.
Results: The findings of each study are summarized in Tables 1, 2.
Discussion and Conclusion: The etiopathogenesis of SLE remains controversial, which limits therapeutic inventions for this disease. However, SLE is a DNA degradation and elimination disorder caused by uncleared histones and nuclear material that leak into the extracellular space and form cell-free DNA, triggering an immune response that destroys tissues and organs. Under normal conditions, apoptosis allows DNA and other nuclear material to be efficiently cleared through degradation and additional complex mechanisms such that this material does not trigger the immune system to produce nuclear autoantibodies.
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by the production of antinuclear antibodies (ANAs) specific for nuclear antigens originating from uncleared apoptotic cells. The disease affects several organs of the body. Globally, ~5 million people suffer from SLE, which has no cure to date (1). Evidence from previous studies shows that environmentally induced defects and genetic abnormalities in immune cells, mutations in regulatory components involved in cellular apoptosis and defects in mechanisms of cellular debris clearance are key contributors to SLE pathogenesis (2). More precise evidence from genome-wide studies shows that SLE patients have mutations in the three prime repair exonuclease 1 (TREX1), autophagy-related 5 (ATG5), RAD518, and deoxyribonuclease I (DNASE1) genes, which are involved in the degradation of DNA, cell apoptosis and the elimination of cellular debris (3).
Abnormalities in cell death processes have been implicated in the production of nuclear autoantigens, which result in a systemic autoimmune response if imperfectly cleared. This effect has been observed in SLE and many other chronic inflammatory diseases (4). These cell death processes include apoptosis, necrosis (primary and secondary), NETosis (a neutrophil-specific type of cell death), autophagy, necroptosis, and pyroptosis (5). Through a complex series of events, the production of autoantigens stimulates the immune system to produce autoantibodies that combine with uncleared cellular debris in the blood or tissues, leading to the formation of immune complexes (ICs). When dendritic cells, macrophages, and phagocytes clear ICs from the blood, proinflammatory cytokines are secreted, which subsequently leads to inflammation (4). The continued production of antibodies due to uncleared tissue debris in the blood and tissues results in perpetual inflammation and tissue damage, a characteristic of SLE (4).
Mitochondria hyperpolarization, ROS production, lysosomal membrane disintegration, organelle and cellular swelling, and plasma membrane rupture characterize necrotic cell death, which is often regarded as inflammatory since the loss of plasma membrane integrity stimulates the release of autoantigens and damage-associated molecular patterns (DAMPs), which act as chemoattractants for inflammatory cells (5).
Conversely, deoxyribonucleic acid (DNA) is an essential molecule for life; it carries hereditary information and is necessary for all organisms (6). Various events can trigger the degradation of DNA into nucleotides by DNase. Apoptosis is a classic example of such an event. This process has been well-known for years, but recent developments show that improper DNA degradation can result in various diseases, such as autoinflammation, cancer, and cataracts (7). SLE is characterized by the production of ANAs, which are autoantibodies specific for a variety of nuclear autoantigens. Anti-dsDNA autoantibodies are among the ANAs used to diagnose SLE (8). Normally, dsDNA is present in the nucleus and mitochondria, where it is shielded from recognition by the immune system. DNase I destroys DNA in the extracellular space, whereas DNAse II and III degrade endosomal and cytoplasmic DNA. Defective cell apoptosis, NETosis, and elimination processes result in elevated levels of cell-free dsDNA (cfDNA) in SLE patients (8). Therefore, understanding the complex pathways and processes involved in the formation of autoantigens and autoantibodies can help in the diagnosis and treatment of various diseases, such as cancer and SLE. Furthermore, these defects can be prevented effectively if proper DNA degradation is facilitated through extrinsic factors, such as drugs. In this paper, we review the connection between SLE and defective DNA degradation.
An advanced literature search was conducted to retrieve the most relevant articles from the evidence base. Additional methods of identifying relevant literature were also employed, including looking up sources in the reference lists of other research reports, manual searching of relevant journals, and searching by author.
The advanced search was done using PubMed only because PubMed features top resources in chemicals and bioassays, DNA and RNA, and genes and expression, among other topics. Boolean connectors (“AND” and “OR”) were used to connect keywords in the search boxes of PubMed. The following search terms were used: [(“SLE” OR “Systemic Lupus Erythematosus” OR “Lupus”)] AND [(“DNA” OR “DNA Degradation”)] AND [(“Defect Elimination”)]. When the search terms were applied, 2,728 results were retrieved, including 1,047 articles published within the last 10 years. Only articles with free full texts were selected (490 articles). After screening the relevance of these articles using the titles, 42 articles were selected. The selected articles were then emailed to the researcher directly from PubMed, and their abstracts were further screened for relevance; 23 articles were retrieved and included in this review.
However, before implementing the advanced search strategy, a basic Google search was conducted as a prescreen search, which yielded 144,000 results when the following keywords were used: “Systemic Lupus Erythematosus,” “SLE,” “DNA degradation,” and “Defect elimination.” Four articles were retrieved, of which two were excluded; one of the excluded articles discussed how circulating free DNA may signal SLE severity and be used to monitor therapy (9), and the other article was excluded because it was outdated despite being relevant (10). Regarding the articles that were retrieved and included, one was about the relationship between the presence of SLE and the failure of DNA degradation (11), whereas the other was about the important role of neutrophil extracellular traps (NETs) in SLE (12). Therefore, 25 articles were included in this review.
Twenty-five articles were retrieved for review by using the above search strategy (see Table 1). Figure 1 shows a PRISMA diagram of the search strategy described above.
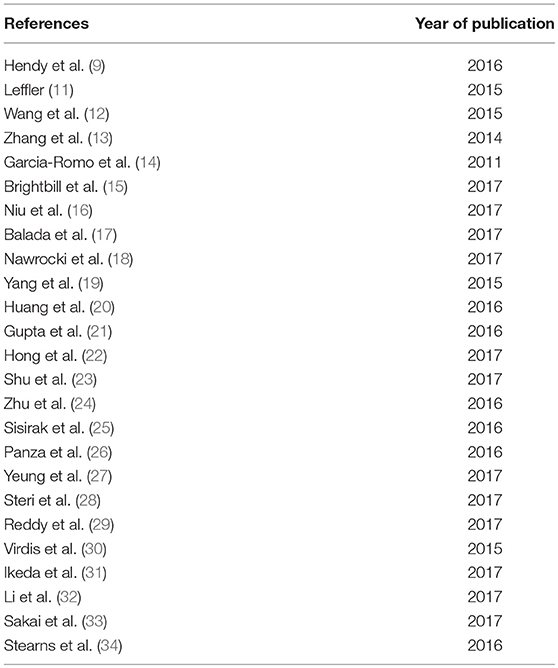
Table 1. Retrieved articles selected for review.
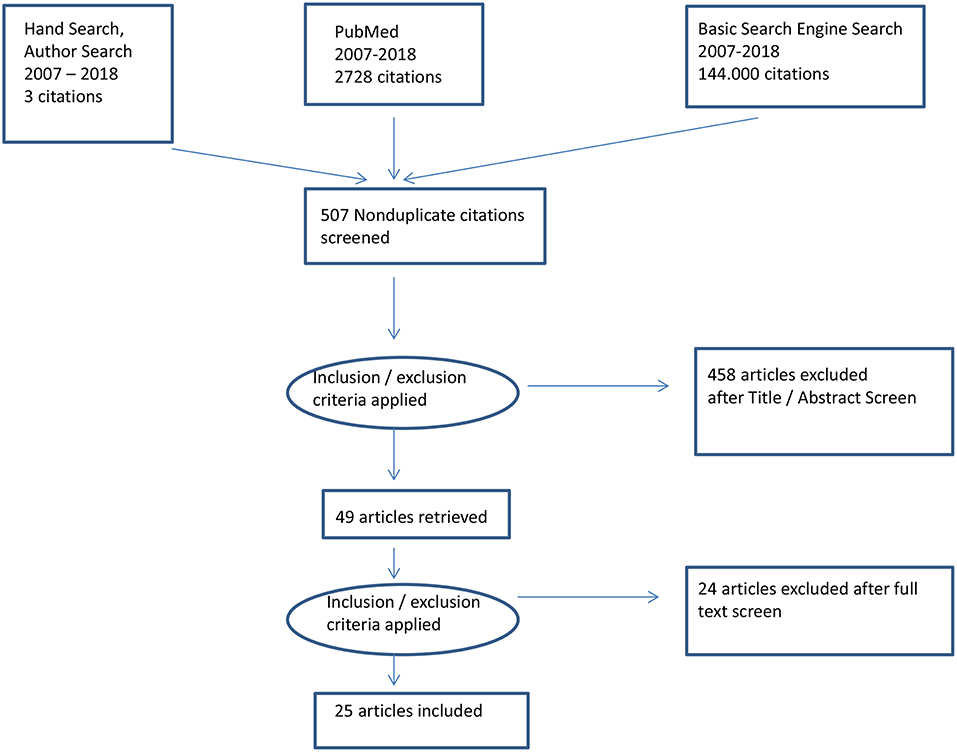
Figure 1. PRISMA diagram of the search strategy.
This subsection will summarize the findings of each study; the findings will then be coded and categorized in the Discussion section. Tables 1, 2 summarize the retrieved studies considered in the discussion.

Table 2. Findings from each study.
Several studies have been conducted in this field, with the most recent study reporting that NF-κB-inducing kinase (NIK) inhibition can improve outcomes in patients with SLE (35). Much remains unknown about SLE, and more studies are needed to explore more accurate and reliable ways of diagnosing and treating SLE. The following section of this paper will discuss these findings in the context of whether SLE is a DNA degradation defect.
Programmed cell death, termed apoptosis, is essential for homeostasis during both development and aging. Under normal conditions, apoptotic cells are engulfed into lysosomes within phagocytes and destroyed; this process occurs without inflammation (36). Many studies have maintained that SLE originates from the defective clearance of apoptotic remnants and NETs from the circulation and tissues (37–41). Thus, there is a consensus that SLE begins to develop when the clearance of dying cells is impaired. The clearance of cellular debris involves many ligands and receptors, and there must be sufficient bridging molecules between phagocytes and dying cells (42). This process is closely controlled and very intricate, but some studies have elucidated the details (43–45). Understanding this complex process has therapeutic potential, mainly in the timely prevention of SLE (45). Dying cells that are not quickly and efficiently removed go through secondary necrosis and subsequently burst, releasing nuclear material upon loss of an intact cell membrane (44).
The impaired clearance of apoptotic cells in SLE is due to their failed recognition by phagocytes because (a) the phagocytes are smaller (4), (b) the phagocytes have diminished and delayed phagocytic activity (46), (c) the phagocytes have reduced adherence (47), or (d) the differentiation of phagocytes from CD34-positive hematopoietic stem cells is reduced (48–50). Therefore, professional and non-professional phagocytes in patients with SLE have reduced phagocytic activity and are thus unable to clear NETs, and apoptosis remains incomplete (51).
During NETosis, NETs are generated by neutrophils upon pathogen stimulation to kill invading pathogens (40, 44). Cellular and nuclear components, including dsDNA, histones, neutrophil elastase, and myeloperoxidase, are released during NETosis. Proinflammatory components, including tumor necrosis factor (TNF)-α, interleukin (IL)-17, IL-8, and interferon (IFN)-γ, also trigger the production of NETs (13). Additionally, low-density granulocytes among peripheral blood mononuclear cells in SLE patients have been shown to form NETs (13). The formed NETs are normally degraded by DNase I, which hydrolyzes dsDNA through its endonuclease activity, thus breaking down chromatin during apoptosis (13).
Incomplete degradation of NETs by DNase I results in the accumulation of the remaining NETs, which react with other proteins to form complexes that are involved in autoimmune SLE (9, 12–14, 35). Elevated levels of undegraded residual NETs are the major source of serum dsDNA, also called circulating cfDNA, in SLE patients (51). The clearance of NETs entirely depends on an efficient and active DNase I enzyme. Defective DNase I leads to undegraded chromatin, which is positively related to SLE disease activity (9, 12, 51). The presence of mitochondrial DNA (mtDNA) in NETs and its positive correlation with plasmacytoid dendritic cells (PDCs) and type 1 IFN-α imply that SLE is a disorder of defective DNA clearance. Studies have shown that NETs in SLE patients activate PDCs and the IFN pathway, triggering the production of autoantigens (14). Therefore, defective DNA degradation leads to defective chromatin breakdown and is thus etiopathogenically involved in SLE development.
NIK usually engages non-canonical NF-κB to signal downstream of several TNF family members, including TWEAK, BAF, OX40, and CD40, which are involved in SLE pathogenesis (15). In addition to type I IFN and Toll-like receptor (TLR), numerous members of the TNF receptor superfamily (TNFRSF) are involved in SLE pathophysiology. CD40 and B cell-activating factor (BAFF) are essential for B cell survival and differentiation into autoantibody-generating plasma cells. CD40 ligand (CD40L) blockade showed promise in initial clinical trials of lupus, although therapeutic development was halted because of thrombotic side effects. In contrast, blockade of BAFF via belimumab is moderately effective and is currently the only new approved treatment for lupus (15).
Autoantigens that trigger the production of autoantibodies in SLE originate from cell death by apoptosis and NETosis. Dying cells usually undergo morphological changes, such as DNA fragmentation, shrinking, and blebbing of the plasma membrane; the nuclear autoantigens that are targeted in SLE are usually stored in these plasma membrane blebs (52). On the other hand, NETosis results in rupture of the plasma membrane, releasing the nuclear autoantigens that are targeted in SLE (53). These autoantigens resulting from apoptosis and NETosis trigger the production of antibodies that bind uncleared apoptotic material to form ICs. The mechanism through which these autoantibodies are produced is well-documented; T cells trigger B cells to differentiate, proliferate, and finally mature. Then, the T cells stimulate class switching of B cells to produce different classes of autoantibodies (16, 54). Due to alterations in T cells that modulate the production of autoantibodies specific for lupus autoantigens by B cells, lupus T cells have been categorized as abnormal (54).
Lupus has also been shown to be associated with reduced expression of DNA cytosine-5-methyltransferases, which have hypomethylation activity that might be responsible for the production of autoreactive antibodies. Figure 2 gives an overview about the most important key-elements in the pathophysiology involved in systemic lupus erythematosus.
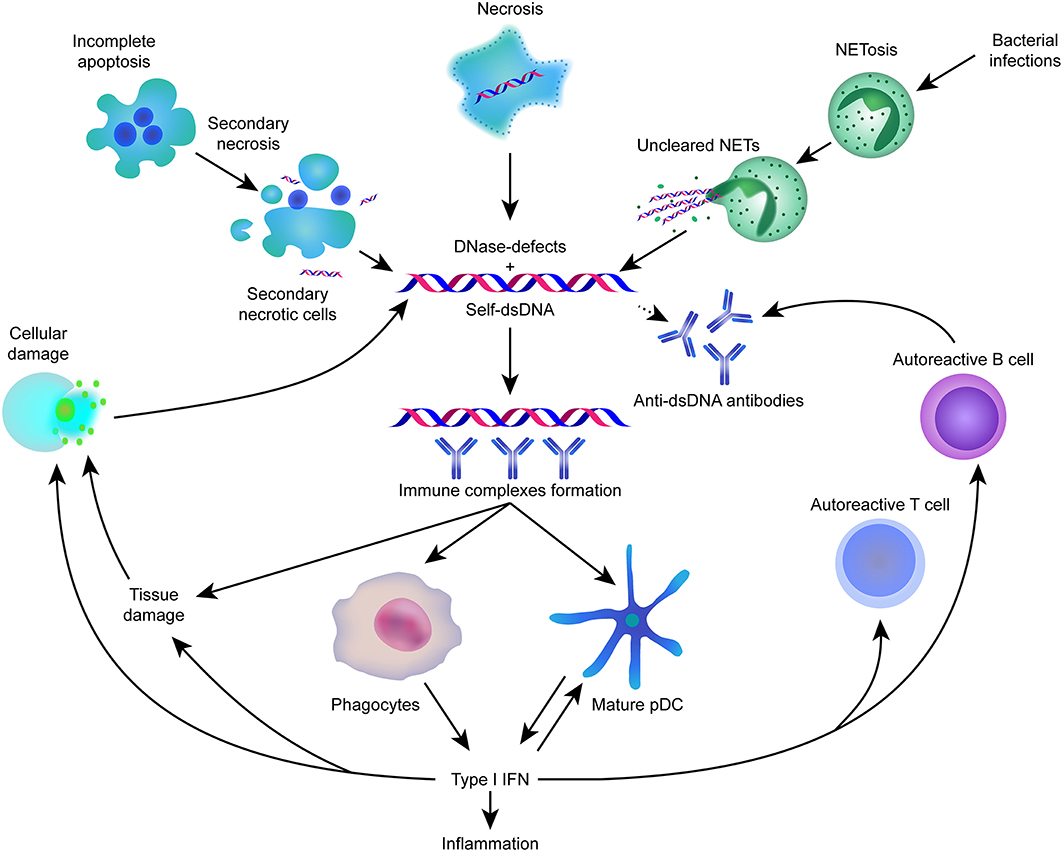
Figure 2. Self-dsDNA released during cell death plays an important role in the pathogenesis of SLE.
DNA methylation is essential for marking chromatin accessibility and regulating gene expression (55). DNA methylation involves the addition of a methyl group at the 5th carbon of cytosine residues in CG dinucleotides, and this process is usually involved in cell differentiation, X-chromosome inactivation, imprinting, and the suppression of transcriptional noise and parasitic DNA. Abnormalities in DNA methylation pathways have pathological consequences. For example, de novo mutation of the DNA methyltransferase DNMT3B leads to immunodeficiency-centromeric instability-facial anomalies syndrome (ICF syndrome). Complete DNMT1 deficiency is incompatible with life. Moreover, acquired defects in DNA methylation are related to diseases, including autoimmunity and cancer (55).
DNA methylation is principally a transcriptionally repressive epigenetic mark that renders chromatin inaccessible and promotes gene silencing through transcriptional repression; these events occur through different mechanisms, including the recruitment of methylcytosine binding domain-containing proteins that successively recruit histone deacetylases, which promote chromatin condensation (55).
Pbx1 is a member of the TALE family of homeodomain-containing transcription factors that regulates the DNA binding of Hox proteins. Pbx1 plays an essential role during organogenesis and development by integrating several signals via interactions with various partners, including Prep1 TALE proteins and Meis, that control chromatin remodeling and co-activator access. During immune system development, Pbx1 is vital for maintaining hematopoietic stem cell self-renewal and limiting myeloid maturation to preserve the differentiation ability of lymphoid progenitors. The absence of Pbx1 in embryonic stem cells leads to the failure to produce common lymphoid progenitors, the absence of NK and B cells, and the development of impaired T cells (16). In addition, Pbx1 regulates chromatin accessibility to various genes and is conserved between humans and mice. The Pbx1-d dominant-negative isoform is more commonly expressed in CD4+ T cells from lupus patients than in those from healthy controls. Pbx1-d is related to autoreactive T cell production in mice with the Sle1a1 lupus vulnerability locus (56).
Dual-specificity protein phosphatase 23 (DUSP23) activates the IFN and IL pathways via dephosphorylation (17), and these pathways are responsible for the formation of NETs, which are sources of cfDNA (17, 18, 57). DUSP3, DUSP22, and VH1 participate in both the IL and IFN signaling pathways mainly by dephosphorylating signal transducer and activator of transcription (STAT) proteins (37, 38). High IFN-α activity is frequently detected in sera from SLE patients (39). Moreover, patients with SLE exhibit a particular mRNA expression profile of IFN-dependent genes in leucocytes known as the IFN signature (17, 40, 41).
The presence of hypomethylated DNA in SLE patients implies that the DNA will not be degraded; hence, SLE is a defective DNA degradation disorder. Additionally, simultaneously high levels of anti-dsDNA, anti-nucleosome, and anti-histone antibodies in serum suggest that defective DNA degradation marks the genesis of SLE (19) and can indicate severe nephropathy in SLE. In summary, these findings improve our understanding of the role of T cells in SLE (20). During SLE pathogenesis, autoreactive T cells trigger the differentiation, proliferation, and maturation of B cells, thus supporting the formation of autoantibodies. Moreover, attenuating T cells normally alleviates the manifestations of autoimmune diseases, such as clearing pathogenic autoreactive T cells in SLE.
The presence of pathogens, including lupus autoantigens, stimulates host cells to make and release a group of signaling proteins (cytokines) called IFNs through complex processes. There are three types of IFNs, namely, alpha, beta, and gamma IFNs. Gamma IFNs are known for the expansion of macrophages and inducing the class II major histocompatibility complex (MHC). Anti-IFN-γ autoantibodies are associated with amplified disease activity in patients with SLE (21). IFN-γ is commonly referred to as a type II IFN, whereas IFN-α and IFN-β are referred to as type I IFNs, a subtype of cytokines that help regulate the immune system.
Type I IFNs generated during a viral infection trigger the production of MCP-1, which is responsible for inducing the migration of inflammatory monocytes to the inflammation site. After recruitment, inflammatory monocytes are triggered by type I IFNs to generate IL-18, which then signals via IL-18R expressed by NK cells to induce the production of IFN-γ.
Abundant autoantibody production and IC formation cause tissue damage; hence, SLE is regarded as a B cell disorder. Conversely, the significance of helper T cells in inducing B cell immunity was identified based on defective CD3ζ function or expression. Reduced CD3ζ-chain expression has been identified in cancer patients and those with autoimmune illnesses. In contrast, decreased CD3ζ-chain levels are maintained during the course of SLE in most patients (22).
Moreover, IFN regulatory factor 5 (IRF5) plays a crucial role in the stimulation of type I IFNs, TNF-α, and the proinflammatory cytokines IL-6 and IL-12. It is also involved in adaptive and innate immunity. The first identified single-nucleotide polymorphism (SNP) in IRF5, Rs2004640, is closely related to the high expression of several IRF5 isoforms and is a valuable genetic risk factor for SLE (23).
Gene expression studies have identified hypomethylated upregulated genes and hypermethylated downregulated genes in SLE patients, indicating that DNA methylation plays a role in SLE development (24). Differentially expressed genes (differential gene co-expression) in SLE are involved in the IFN and TLR signaling pathways (24). The transcriptional repressor growth factor independence 1 (Gfi1) plays a critical role in myeloid cell regulation, as well as in the prevention of unprompted lupus autoimmunity, mainly by negatively regulating TLR7 signaling (58).
Before therapy with Gfi1, SLE mice showed high levels of serum autoantibodies specific for DNA and RNA, which are indicative of defective nucleic acid degradation in SLE. Hence, IFNs are involved in the etiopathogenesis of SLE by stimulating the formation of NETs, the main sources of cfDNA, and exploring their mechanisms of involvement has therapeutic potential (58).
Suurmond et al. (59) maintained that studying genetic risk alleles can improve our understanding of failed tolerance mechanisms for DNA-reactive B cells and B cell alterations in SLE. Understanding the complexity of interactions between immune disturbance in SLE and epigenetic mechanisms is a potential way to explore new therapeutic targets (60). In particular, the digestion of chromatin in apoptotic cell microparticles (25) marks a tolerance mechanism for SLE. The absence of DNASE1L3, a DNA degrading enzyme, results in the production of anti-dsDNA antibodies specific for serum dsDNA; hence, defective DNA degradation is associated with SLE (25). When this process reaches a point at which autoantibodies are produced because of defective clearance of NETs and apoptotic remnants, the person is said to have global loss of self-tolerance (25). This is the point at which impaired DNA degradation plays a role in the etiopathogenesis of SLE.
Nuclear autoantigens (e.g., antigens specific for DNA and histones) (61) and proteins from the cytoplasm of neutrophils (62) play a role in the etiopathogenesis of SLE when cellular material containing these components leaks to the extracellular space through apoptosis or NETosis, hence exposing these factors to the immune system. Histones, which are a group of chromatin proteins that are abundant in NETs and apoptotic blebs, are modified through various processes, such as methylation (63), acetylation (64), ubiquitination (65), and poly-ADP ribosylation (66), during apoptosis, and NETosis. When modified histones are exposed to the immune system in the extracellular space, they are recognized as foreign and dangerous by receptors of the innate immune system, such as TLRs. Therefore, the H4/PK assay is usually a reliable and simple test that is valuable for the differential diagnosis and evaluation of symptomatic activity in SLE patients (26).
On the other hand, DNA methylation, an epigenetic mechanism for controlling gene expression (67), plays a significant role in the etiopathogenesis of SLE. Abnormal DNA methylation in immune-related cells is associated with SLE and can be detected in unfractionated white blood cells, which has diagnostic implications (27). This abnormal DNA methylation in immune-related cells involves altered DNA methylation of cytokine genes, which makes the chromatin inaccessible for destruction (60).
Moreover, SLE is associated with decreased DNA methyltransferase mRNA expression levels (18), which is the exact mechanism through which abnormal DNA methylation occurs in immune-related cells. Therefore, DNA hypomethylation of immune-related cells plays a key role in the etiopathogenesis of SLE; however, little is known about hypermethylation in SLE.
Only one study examined the role of CD3Z (a T cell surface glycoprotein and component of the T cell antigen receptor) hypermethylation in SLE, and it was determined that this modification is a potential risk factor associated with severe SLE manifestations (28).
Hypermethylation of CD3Z could be an environmentally induced epigenetic modification (22). In particular, CD3Z hypermethylation could provide an important mechanism for CD3ζ-chain downregulation in SLE T cells. Furthermore, CD3Z DNA hypermethylation was connected to more severe clinical signs in persons with SLE; thus, CD3Z hypermethylation is a marker of disease severity (22).
Many other factors have recently been associated with the etiopathogenesis, diagnosis, and treatment of SLE. There is extensive evidence for the association of IFNs and ILs with SLE; however, little is known about the role of TNFs (additional cytokines) in the etiopathogenesis of SLE. Variants of TNF ligand superfamily member 13B (TNFSF13B) are associated with defective DNA degradation in SLE disease development. Elevated serum levels of TNFSF13B correlate with serum dsDNA antigen levels, suggesting that TNF is associated with abnormal DNA degradation in SLE patients (28).
Moreover, recent studies have shown that some types of drugs can treat or contribute to the etiopathogenesis of SLE. Rituximab, which has proved efficacious in the treatment of diffuse alveolar hemorrhage in SLE, can cause IgM hypogammaglobulinemia, which can be predicted by lower baseline serum IgM levels and sequential therapy with mycophenolate mofetil (MMF) (29). This finding shows that high SLE disease activity is associated with dsDNA antigens in serum, suggesting defective DNA degradation. Early treatment with hydroxychloroquine can curb the development of endothelial dysfunction in SLE patients (30). Bortezomib, an anticancer drug, increases proinflammatory cytokine levels in mice, hence worsening SLE (31). Aconitine, a C19 norditerpenoid alkaloid, can inhibit disease progression and ameliorate pathologic lesions of SLE (32). This alkaloid, which is the key active component of Aconitum, has immunomodulatory properties and may be valuable for treating autoimmune diseases such as SLE (32).
TREX1 frame-shift mutations, such as V235fs and D272fs, trigger autoantibodies chiefly against non-nuclear antigens. Thus, patients who have such mutations are possibly ANA negative. High serum ANA levels are very rare among patients who have retinal vasculopathy with cerebral leukodystrophy (RVCL). Thus, diseases associated with such mutations may be under- or misdiagnosed. The oligosaccharyltransferase (OST) inhibitor aclacinomycin effectively suppressed the production of autoantibodies, confirming the possible therapeutic value of such medicines for treating phenotypes associated with the V235fs mutation. Moreover, the D272fs and V235fs mutations usually affect the DNase-independent roles of TREX1 and trigger serologic autoimmunity, which likely contributes to the development of autoimmune disease (33, 34).
In addition, IFN-α expression is positively related to IRF5 expression. Childhood-onset SLE normally follows a more aggressive course, with higher mortality and morbidity and a greater occurrence of serological and immunological abnormalities, than does adult-onset SLE, perhaps implicating dissimilar mechanisms in both clusters. Nonetheless, IRF5 plays an important role in childhood-onset SLE pathogenesis. Sp1 can increase IRF5 promoter activity and mRNA expression. Sp1 expression is increased in childhood-onset SLE and is positively related to IRF5 levels; thus, high Sp1 expression might contribute to high IRF5 levels, and the subsequent high IFN-α levels are a fundamental feature of SLE pathology (68).
DNase I, an endonuclease, is usually secreted to cleave extracellular DNA to tetranucleotides with 3′ hydroxyl DNA ends and 5′ monophosphate (68). Moreover, during apoptosis, DNase I is the specific endonuclease that facilitates chromatin breakdown. Together with the plasminogen system, DNase I enables prompt and efficient chromatin breakdown through the instantaneous degradation of DNA and DNA-binding proteins (68).
DNase I mutations have been associated with a familiar form of SLE (69).
DNase II is vital for self-dsDNA clearance in lysosomes to circumvent an immune response. Moreover, DNase II knockout is usually embryonically lethal in mice due to uncontrolled inflammation, whereas cGAS deletion in DNase II-deficient mice salvages this lethal phenotype. DNASEII defects are associated with autoinflammtion, but however have so far not been linked to the classical lupus phenotype.
TREX1, also known as DNase III, is a DNA exonuclease that engages in cytoplasmic dsDNA and ssDNA clearance. Loss-of-function mutations of TREX1 result in the accumulation of self-DNA and autoimmune diseases, such as SLE. Mutations in DNase III/TREX1 have also been associated with a familiar form of lupus erythematosus (70–72).
TREX1-deficient mice display markedly reduced survival due to unprompted inflammatory myocarditis, increased circulatory failure and cardiomyopathy Trex1 prevents the cell-intrinsic initiation of autoimmunity in the mice model of TREX1-deficient mice (73–76).
Depletion of STING, IRF3, cGAS, or IFN-I receptor in TREX1-deficient mice protects against autoimmune disorders and death, indicating that cGAS–STING–IRF3 axis-regulated IFN-I production is responsible for autoimmune disorder development in TREX1-deficient mice. Nevertheless, in STING-deficient lupus mice, serum cytokine levels, autoantibody production, and lymphoid hypertrophy increase significantly in comparison to STING-adequate littermates.
Mutations leading to STING activation cause autoinflammation and can lead to an inflammatory vascular and pulmonary syndrome (77) and has also been linked to a familiar form of lupus erythematosus (78, 79).
But on the other hand STING deficiency can also lead to autoimmunity mediated by enhanced TLR activation in lupus prone mice (80). In this case STING potently suppresses inflammation in a model of SLE. This controversial behavior points out the complexity of regulation and is important to be considered for the development of targeted therapies directed against STING.
ANAs are considered the serological characteristic of SLE. These antibodies often bind various nuclear antigens, including DNA, histones, non-histone proteins and protein complexes with RNA and DNA. Due to the frequent manifestation of ANA abnormalities in SLE, ANA analysis is a vital element of clinical assessments and determinations of suitability for clinical tests or the use of particular therapies (34).
DNase I, an endonuclease, is usually secreted to cleave extracellular DNA to tetranucleotides with 3′ hydroxyl DNA ends and 5′ monophosphate (68). Moreover, during apoptosis, DNase I is the specific endonuclease that facilitates chromatin breakdown. Together with the plasminogen system, DNase I enables prompt and efficient chromatin breakdown by the instantaneous degradation of DNA and DNA-binding proteins (68).
Self-dsDNA is involved in SLE pathogenesis; these molecules are normally cleared in apoptosis, necrosis and NETs, but these processes are defective in SLE patients, thereby triggering the production of autoantibodies through unknown mechanisms. In addition, anti-dsDNA antibodies are formed by autoreactive B cells and autoreactive T cells. dsDNA ICs and autoantibodies stimulate robust IFN-I production via several intercellular DNA sensors, primarily in PDCs and phagocytes (81).
The etiopathogenesis of SLE is still controversial, and there are limited therapeutic inventions for this disease. However, this paper has demonstrated that SLE is a DNA degradation disorder. In normal apoptosis, DNA and other nucleic materials are efficiently cleared through degradation and other complex mechanisms, blocking the formation of nuclear autoantigens. Autoantibodies produced against cfDNA are responsible for tissue and organ damage in SLE patients.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/pubmed/.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ANAs, Antinuclear antibodies; ATG5, Autophagy related 5; BAF, Barrier-to-autointegration factor; CD3Z, Cluster of differentiation 3Z; CD40, Cluster of differentiation 40; CD40L, Cluster of differentiation 40 ligand (CD40L); cfDNA, Circulating free DNA; DAMPs, Damage associated molecular patterns; DNA, Deoxyribonucleic acid; DNASE II, Deoxyribonuclease II; DNASE III, Deoxyribonuclease III; DNASE I, Deoxyribonuclease I; DNMT1, DNA methyltransferase 1; DNMT3B, DNA methyltransferase 3 beta; DUSP23, Dual-specificity protein phosphatase 23; Gfi1, Growth factor independence 1; ICF, Immunodeficiency-centromeric instability-facial anomalies; ICs, Immune complexes; IFN-γ, Interferon gamma; IRF5, Interferon regulatory factor 5; MHC, Major histocompatibility complex; MMF, Mycophenolate mofetil; mtDNA, Mitochondrial DNA; NETs, Neutrophil extracellular traps; NIC, NF-κB-inducing kinase; NK, Natural killer; PDCs, Plasmacytoid dendritic cells; RNA, Ribonucleic acid; RVCL, Retinal vasculopathy with cerebral leukodystrophy; SLE, Systemic lupus erythematosus; SNP, Single-nucleotide polymorphism; STAT, Signal transducer and activator of transcription; STING, Stimulator of interferon genes; TLR, Toll-like receptor; TNFRSF, Tumor necrosis factor receptor superfamily; TNF-α, Tumor necrosis factor alpha; TREX1, Three prime repair exonuclease 1.
1. Meas R, Burak M, Sweasy J. DNA repair and systemic lupus erythematosus. DNA Repair. (2017) 56:174–82. doi: 10.1016/j.dnarep.2017.06.020
2. Wu H, Fu S, Zhao M, Lu L, Lu Q. Dysregulation of cell death and its epigenetic mechanisms in systemic lupus erythematosus. Molecules. (2017) 2:30. doi: 10.3390/molecules22010030
3. Teruel M, Alarcón-Riquelme M. The genetic basis of lupus erythematosus: what are the risk factors and what we have learned. J Autoimmun. (2016) 74:161–75. doi: 10.1016/j.jaut.2016.08.001
4. Mahajan A, Herrmann M, Muñoz L. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. (2016) 7:35. doi: 10.3389/fimmu.2016.00035
5. Mistry P, Kaplan M. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. (2016) 185:59–73. doi: 10.1016/j.clim.2016.08.010
6. Kawane K, Motani K, Nagata S. DNA degradation and its defects. Cold Spring HarborPerspect Biol. (2014) 6:a016394. doi: 10.1101/cshperspect.a016394
7. Jackson S, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
8. Rekvig OP. Anti-dsDNA antibodies as a classification criterion and a diagnostic markers for systemic lupus erythematosus: critical remarks. Clin Exp Immunol. (2015) 179:5–10. doi: 10.1111/cei.12296
9. Hendy O, Motalib T, El Shafie M, Khalaf F, Kotb S, Khalil A, et al. Circulating cell free DNA as a predictor of systemic lupus erythematosus severity and monitoring of therapy. Egypt J Med Hum Genet. (2016) 17:79–85. doi: 10.1016/j.ejmhg.2015.07.001
10. Lunec J, Herbert K, Blount S, Griffiths H, Emery P. 8-hydroxydeoxyguanosine: a marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. (1994) 348:131–8. doi: 10.1016/0014-5793(94)00583-4
11. Leffler J, Ciacma K, Gullstrand B, Bengtsson A, Martin M, Blom A. A subset of patients with systemic lupus erythematosus fails to degrade DNA from multiple clinically relevant sources. Arthr Res Ther. (2015) 17:1–10. doi: 10.1186/s13075-015-0726-y
12. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthr Rheumatol. (2015) 67:3190–200. doi: 10.1002/art.39296
13. Zhang S, Lu X, Shu X, Tian X, Yang H, Yang W, et al. Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Inter Med. (2014) 53:2763–71. doi: 10.2169/internalmedicine.53.2570
14. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
15. Brightbill H, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol E, et al. NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat Commun. (2018) 9:179. doi: 10.1038/s41467-017-02672-0
16. Niu Y, Sengupta M, Titov A, Choi S, Morel L. The PBX1 lupus susceptibility gene regulates CD44 expression. Mol Immunol. (2017) 85:148–54. doi: 10.1016/j.molimm.2017.02.016
17. Balada E, Felip L, Ordi-Ros J, Vilardell-Tarrés M. DUSP23is over-expressed and linked to the expression ofDNMTsin CD4+T cells from systemic lupus erythematosus patients. Clin Exp Immunol. (2016) 187:242–50. doi: 10.1111/cei.12883
18. Nawrocki M, Majewski D, Puszczewicz M, Jagodzinski P. Decreased mRNA expression levels of DNA methyltransferases type 1 and 3A in systemic lupus erythematosus. Rheumatol Int. (2017) 37:775–83. doi: 10.1007/s00296-017-3711-8
19. Yang J, Xu Z, Sui M, Han J, Sun L, Jia X, et al. Co-positivity for Anti-dsDNA, -nucleosome and -histone antibodies in lupus nephritis is indicative of high serum levels and severe nephropathy. PLoS ONE. (2015) 10:e0140441. doi: 10.1371/journal.pone.0140441
20. Huang L, Yang Y, Kuang Y, Wei D, Li W, Yin Q, et al. The impact of T cell vaccination in alleviating and regulating systemic lupus erythematosus manifestation. J Immunol Res. (2016) 2016:1–9. doi: 10.1155/2016/5183686
21. Gupta S, Tatouli I, Rosen L, Hasni S, Alevizos I, Manna Z, et al. Distinct functions of autoantibodies against interferon in systemic lupus erythematosus: a comprehensive analysis of anticytokine autoantibodies in common rheumatic diseases. Arthr Rheumatol. (2016) 68:1677–87. doi: 10.1002/art.39607
22. Hong K, Kim H, Park S, Poojan S, Kim M, Sung J, et al. CD3Zhypermethylation is associated with severe clinical manifestations in systemic lupus erythematosus and reduces CD3ζ-chain expression in T cells. Rheumatology. (2016) 56:kew405. doi: 10.1093/rheumatology/kew405
23. Shu J, Li L, Zhou L, Qian J, Fan Z, Zhuang L, et al. IRF5 is elevated in childhood-onset SLE and regulated by histone acetyltransferase and histone deacetylase inhibitors. Oncotarget. (2017) 8:47184–94. doi: 10.18632/oncotarget.17586
24. Zhu H, Mi W, Luo H, Chen T, Liu S, Raman I, et al. Whole-genome transcription and DNA methylation analysis of peripheral blood mononuclear cells identified aberrant gene regulation pathways in systemic lupus erythematosus. Arthr Res Ther. (2016) 18:162. doi: 10.1186/s13075-016-1050-x
25. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Özçakar Z, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
26. Panza F, Alcaro M, Petrelli F, Angelotti F, Pratesi F, Rovero P, et al. A novel DNA/histone H4 peptide complex detects autoantibodies in systemic lupus erythematosus sera. Arthr Res Ther. (2016) 18:220. doi: 10.1186/s13075-016-1117-8
27. Yeung K, Chung B, Choufani S, Mok M, Wong W, Mak C, et al. Genome-wide DNA methylation analysis of chinese patients with systemic lupus erythematosus identified hypomethylation in genes related to the type I interferon pathway. PLoS ONE. (2017) 12:e0169553. doi: 10.1371/journal.pone.0169553
28. Steri M, Orrù V, Idda M, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med. (2017) 376:1615–26. doi: 10.1056/NEJMoa1610528
29. Reddy V, Martinez L, Isenberg D, Leandro M, Cambridge G. Pragmatic treatment of patients with systemic lupus erythematosus with rituximab: long-term effects on serum immunoglobulins. Arthr Care Res. (2017) 69:857–66. doi: 10.1002/acr.22993
30. Virdis A, Tani C, Duranti E, Vagnani S, Carli L, Kühl A, et al. Early treatment with hydroxychloroquine prevents the development of endothelial dysfunction in a murine model of systemic lupus erythematosus. Arthr Res Ther. (2015) 17:277. doi: 10.1186/s13075-015-0790-3
31. Ikeda T, Fujii H, Nose M, Kamogawa Y, Shirai T, Shirota Y, et al. Bortezomib treatment induces a higher mortality rate in lupus model mice with a higher disease activity. Arthr Res Ther. (2017) 19:187. doi: 10.1186/s13075-017-1397-7
32. Li X, Gu L, Yang L, Zhang D, Shen J. Aconitine: a potential novel treatment for systemic lupus erythematosus. J Pharmacol Sci. (2017) 133:115–21. doi: 10.1016/j.jphs.2017.01.007
33. Sakai T, Miyazaki T, Shin D, Kim Y, Qi C, Fariss R, et al. DNase-active TREX1 frame- shift mutants induce serologic autoimmunity in mice. J Autoimmun. (2017) 81:13–23. doi: 10.1016/j.jaut.2017.03.001
34. Stearns N, Zhou S, Petri M, Binder S, Pisetsky D. The use of poly-L-lysine as a capture agent to enhance the detection of antinuclear antibodies by ELISA. PLoS ONE. (2016) 11:e0161818. doi: 10.1371/journal.pone.0161818
35. Elliott M, Ravichandran K. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. (2010) 189:1059–70. doi: 10.1083/jcb.201004096
36. Biermann M, Maueroder C, Brauner JM, Chaurio R, Janko C, Herrmann M, et al. Surface code –biophysical signals for apoptotic cell clearance. Phys Biol. (2013) 10:065007. doi: 10.1088/1478-3975/10/6/065007
37. Shao W, Cohen P. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthr Res Ther. (2010) 13:202. doi: 10.1186/ar3206
38. Muñoz L, Lauber K, Schiller M, Manfredi A, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. (2010) 6:280–9. doi: 10.1038/nrrheum.2010.46
39. Fadeel B, Xue D, Kagan V. Programmed cell clearance: molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem Biophy Res Commun. (2010) 396:7–10. doi: 10.1016/j.bbrc.2010.02.106
40. Elliott M, Chekeni F, Trampont P, Lazarowski E, Kadl A, Walk S, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
41. Muñoz L, Peter C, Herrmann M, Wesselborg S, Lauber K. Scent of dying cells: the role of attraction signals in the clearance of apoptotic cells and its immunological consequences. Autoimmun Rev. (2010) 9:425–30. doi: 10.1016/j.autrev.2009.11.016
42. Rahman Z, Shao W, Khan T, Zhen Y, Cohen P. Impaired apoptotic cell clearance in the germinal center by mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J Immunol. (2010) 185:5859–68. doi: 10.4049/jimmunol.1001187
43. Ravichandran K. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. (2010) 207:1807–17. doi: 10.1084/jem.20101157
44. Röszer T. Transcriptional control of apoptotic cell clearance by macrophage nuclear receptors. Apoptosis. (2016) 22:284–94. doi: 10.1007/s10495-016-1310-x
45. Poon I, Lucas C, Rossi A, Ravichandran K. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
46. Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, et al. Clearance deficiency and systemic lupus erythematosus (SLE). J Autoimmun. (2007) 28:114–21. doi: 10.1016/j.jaut.2007.02.005
47. Munoz LE, Chaurio RA, Gaipl US, Schett G, Kern P. MoMa from patients with systemic lupus erythematosus show altered adhesive activity. Autoimmunity. (2009) 42:269–71. doi: 10.1080/08916930902827983
48. Bradley S, Suarez-Fueyo A, Moss D, Kyttaris V, Tsokos G. T cell transcriptomes describe patient subtypes in systemic lupus erythematosus. PLoS ONE. (2015) 10:e0141171. doi: 10.1371/journal.pone.0141171
49. Munoz LE, Frey B, Appelt U, Janko C, Sarter K, Voll R, et al. Peripheral blood stem cells of patients with systemic lupus erythematosus show altered differentiation into macrophages. Open Autoimmun J. (2010) 2:11–6. doi: 10.2174/1876894601002010011
50. Szondy Z, Garabuczi Ã, JoÃs G, Tsay G, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol. (2014) 5:1–8. doi: 10.3389/fimmu.2014.00354
51. Fernado MV, Eva B, Joseph O, MIquel V. DNase 1 and systemic lupus erythematosus. Sci Direct. (2008) 7:359–63. doi: 10.1016/j.autrev.2008.02.002
52. Pieterse E, van der Vlag J. Breaking immunological tolerance in systemic lupus erythematosus. Front Immunol. (2014) 5:1–8. doi: 10.3389/fimmu.2014.00164
53. Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Front Immunol. (2012) 3:420. doi: 10.3389/fimmu.2012.00420
54. Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. (2014) 2014:419029. doi: 10.1155/2014/419029
55. Hughes T, Webb R, Fei Y, Wren JD, Sawalha AH. DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks. Genes Immun. (2010) 11:554–60. doi: 10.1038/gene.2010.24
56. Choi SC, Hutchinson TE, Titov AA, Seay HR, Li S, Brusko TM, et al. The lupus susceptibility gene Pbx1 regulates the balance between follicular helper T cell and regulatory T cell differentiation. J Immunol. (2016) 197:458–69. doi: 10.4049/jimmunol.1502283
57. Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. (1979) 301:5–8 doi: 10.1056/NEJM197907053010102
58. Desnues B, Macedo A, Ordoñez-Rueda D, Roussel-Queval A, Malissen B, Bruhns P, et al. The transcriptional repressor Gfi1 prevents lupus autoimmunity by restraining TLR7 signaling. Eur J Immunol. (2016) 46:2801–11. doi: 10.1002/eji.201646573
59. Suurmond J, Calise J, Malkiel S, Diamond B. DNA-reactive B cells in lupus. Curr Opin Immunol. (2016) 43:1–7. doi: 10.1016/j.coi.2016.07.002
60. Wang Z, Chang C, Peng M, Lu Q. Translating epigenetics into clinic: focus on lupus. Clin Epigen. (2017) 9:78. doi: 10.1186/s13148-017-0378-7
61. Herrmann M, Podolska M, Biermann M, Maueröder C, Hahn J. Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflamm Res. (2015) 8:161–71. doi: 10.2147/JIR.S70325
62. Bouts Y, Wolthuis D, Dirkx M, Pieterse E, Simons E, Van Boekel A, et al. Apoptosis and NET formation in the pathogenesis of SLE. Autoimmunity. (2012) 45:597–601. doi: 10.3109/08916934.2012.719953
63. Liu C, Tangsombatvisit S, Rosenberg J, Mandelbaum G, Gillespie E, Gozani O, et al. Correction: specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthr Res Ther. (2012) 14:403. doi: 10.1186/ar3933
64. Long H, Huang W, Yin H, Zhao S, Zhao M, Lu Q. Abnormal expression pattern of histone demethylases in CD4 + T cells of MRL/lpr lupus-like mice. Lupus. (2009) 18:1327–8. doi: 10.1177/0961203309104869
65. Kawasaki A, Ito I, Ito S, Hayashi T, Goto D, Matsumoto I, et al. Association of TNFAIP3 polymorphism with susceptibility to systemic lupus erythematosus in a Japanese population. J Biomed Biotechnol. (2010) 2010:1–5. doi: 10.1155/2010/207578
66. Krukenberg K, Kim S, Tan E, Maliga Z, Mitchison T. Extracellular poly(ADP-Ribose) is a pro-inflammatory signal for macrophages. Chem Biol. (2015) 22:446–52. doi: 10.1016/j.chembiol.2015.03.007
67. Matzke M, Mosher R. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. (2014) 15:394–408. doi: 10.1038/nrg3683
68. Baranovskii AG, Buneva VN, Nevinsky GA. Human deoxyribonucleases. Biochemistry. (2004) 9:587–601. doi: 10.1023/B:BIRY.0000033731.50496.01
69. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
70. Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. (2007) 85:531–7. doi: 10.1007/s00109-007-0199-9
71. Günther C, Berndt N, Wolf C, Lee-Kirsch MA. Familial chilblain lupus due to a novel mutation in the exonuclease III domain of 3' repair exonuclease 1 (TREX1). JAMA Dermatol. (2015) 151:426–31. doi: 10.1001/jamadermatol.2014.3438
72. Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun. (2016) 7:11752. doi: 10.1038/ncomms11752
73. Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. (2007) 131:873–86. doi: 10.1016/j.cell.2007.10.017
74. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
75. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. (2008)22:587–98. doi: 10.1016/j.cell.2008.06.032
76. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. (2012) 36:120–31. doi: 10.1016/j.immuni.2011.11.018
77. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014)371:507–18. doi: 10.1056/NEJMoa1312625
78. König N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tüngler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. (2017)76:468–72. doi: 10.1136/annrheumdis-2016-209841
79. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. (2014)124:5516–20. doi: 10.1172/JCI79100
80. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci USA. (2015)112:E710–7. doi: 10.1073/pnas.1420217112
Keywords: SLE, DNA-degradation, DNA-elimination, SLE-Systemic Lupus Nephritis, DNA-anti DNA
Citation: Arneth B (2019) Systemic Lupus Erythematosus and DNA Degradation and Elimination Defects. Front. Immunol. 10:1697. doi: 10.3389/fimmu.2019.01697
Received: 16 April 2019; Accepted: 08 July 2019;
Published: 07 August 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Claudia Günther, University Hospital Carl Gustav Carus, GermanyCopyright © 2019 Arneth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Borros Arneth, Ym9ycm9zLmFybmV0aEBrbGluY2hlbWllLm1lZC51bmktZ2llc3Nlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.