
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 June 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01478
This article is part of the Research Topic Platelets and Immune Responses during Thromboinflammation View all 17 articles
 Fabrice Cognasse1,2*
Fabrice Cognasse1,2* Sandrine Laradi1,2
Sandrine Laradi1,2 Philippe Berthelot2,3Thomas Bourlet2,3
Philippe Berthelot2,3Thomas Bourlet2,3 Hubert Marotte4,5Patrick Mismetti4,6
Hubert Marotte4,5Patrick Mismetti4,6 Olivier Garraud2,7Hind Hamzeh-Cognasse2
Olivier Garraud2,7Hind Hamzeh-Cognasse2Blood platelets play a central hemostatic role, (i) as they repair vascular epithelial damage, and (ii) they play immune defense roles, as they have the capacity to produce and secrete various cytokines, chemokines, and related products. Platelets sense and respond to local dangers (infectious or not). Platelets, therefore, mediate inflammation, express and use receptors to bind infectious pathogen moieties and endogenous ligands, among other components. Platelets contribute to effective pathogen clearance. Damage-associated molecular patterns (DAMPs) are danger signals released during inflammatory stress, such as burns, trauma and infection. Each pathogen is recognized by its specific molecular signature or pathogen-associated molecular pattern (PAMP). Recent data demonstrate that platelets have the capacity to sense external danger signals (DAMPs or PAMPs) differentially through a distinct type of pathogen recognition receptor (such as Toll-like receptors). Platelets regulate the innate immune response to pathogens and/or endogenous molecules, presenting several types of “danger” signals using a complete signalosome. Platelets, therefore, use complex tools to mediate a wide range of functions from danger sensing to tissue repair. Moreover, we noted that the secretory capacity of stored platelets over time and the development of stress lesions by platelets upon collection, processing, and storage are considered stress signals. The key message of this review is the “inflammatory response to stress” function of platelets in an infectious or non-infectious context.
Several reviews have recently been published discussing the role of the interaction between platelets and both vascular endothelial cells and leukocytes during hemostasis and the initiation of the vascular repair process (1–3). This review focuses on the interactions between platelets and their environment beyond hemostasis, particularly in inflammation. It has been suggested that platelets detect and respond to local dangers such as infectious pathogens accidentally introduced into the bloodstream at the site of wounds. To achieve both goals (hemostasis /vascular repair and danger sensing), platelets use both membrane-bound and secreted products that interact with other cell types, including leukocytes.
Although platelets are regarded primarily as cells associated with hemostasis, it has now become clear that platelets play a wide variety of roles. Endothelial wall alteration or disruption exposes the sub-endothelial matrix rich in prohemostatic proteins. The engagement of platelet surface receptors with these matrix proteins leads to (i) platelet adhesion at the sites of lesions and (ii) the initiation of a complex intracellular signaling process (4). This process results in the formation and release of transcellular mediators, the exocytosis of adhesive and inflammatory proteins and the expression of both additional adhesive receptors and a procoagulant surface (5). Developing platelets retain cytoplasmic granules from their precursors during megacaryocyte differentiation and platelet production, the contents of which are secreted during platelet activation. Exocytosis of the platelet granule contents requires granule membranes to be fused with plasma membranes or open canalicular system (OCS) membranes. The OCS provides a transportation pathway for the release of platelet granule contents (6, 7). In general, these contents fall into three types. First, dense (δ) granules, which are rich in ADP, ATP, calcium, and serotonin, play an important role during hemostasis. Second are alpha (α) granules, which contain a variety of proteins, including adhesive proteins such as thrombospondin, von Willebrand factor, and fibronectin; growth factors such as insulin-like growth factor (IGF), transforming growth factor beta (TGF-β), and platelet-derived growth factor (PDGF); platelet factor 4 (PF4); and a variety of pro-inflammatory/modulatory chemokines and cytokines (1). In addition, α-granule exocytosis results in the expression of P-selectin (CD62P) on the external surface of platelets (5). Interestingly, there is now sound evidence to support the fact that α-granules are heterogeneous, in terms of both their contents and their exocytotic regulation (8–10). Third are platelet-enclosed lysosomes, which secrete hydrolases after activation (11). More recent reports have described a possible new type of granule termed a T-granule (11, 12). Resting platelets express basal surface TLR9 levels that increase significantly after thrombin activation, suggesting that although the majority of TLR9 is expressed intracellularly, some is relocalized to the plasma membrane upon agonist exposure (11–13).
The influence of the cytoskeleton on granule secretion has been a matter of discussion, with studies suggesting that reorganization of microtubules does not affect granule secretion (14, 15). Other studies suggest that the cytoskeleton does not facilitate the secretion of granules and that F-actin disassembly may be required for normal secretion of granules (16). In addition, the function of α-granules is dependent on inflammation, atherosclerosis, angiogenesis, wound healing, antimicrobial host defense, and platelet function in malignant hematological disorders. However, there is little knowledge about the cellular processes that help platelets release α-granule contents. Kamykowski et al. attributed the segregation of α-granule contents previously observed by investigators to the compartmentalization of cargo within single α-granules (17), while other reports showed that angiogenic factors are localized to different α-granules and released by different agonists upon stimulation (18, 19). Lastly, van Nispen tot Pannerden et al. showed that high spatial protein gradients exist within platelet α-granules and propose that tubular α-granules have different secretory capacities than conventional spherical granules and that the spatial segregation of cargo within tubular subtypes may result in differential release of their contents (20). A greater understanding of the dynamics of the fusion pore may illuminate the ways in which platelets drive the release of granule content with disparate platelet functions.
It has been known for more than three decades that platelets release arachidonic acid from membrane phospholipids (21). Arachidonic acid is converted into thromboxane A2 (TxA2), which has both prothrombotic and vasoconstrictive properties. This pathway serves as the target for aspirin, the primary antithrombotic currently in use. As outlined above, if an injured vessel is exposed to subendothelial structures (e.g., collagen), circulating platelets respond quickly, convert to an activated state, adhere and start to form the characteristic hemostatic clot (3). Among other processes, repair of the damaged vessel involves activated endothelial cells. Several reports describe the involvement of various cell adhesion molecules (e.g., P-Selectin, GPIb, GPVI, GPIaβ1, GPIIbβ3, CD40L, TNSF14; JAM-A, PSGL-1, P-Selectin, αvβ3, ICAM-1, CD40, TNSF14R, and JAM-A) acting at the interface between platelets and endothelial cells (19, 22, 23). P-selectin is a well-characterized endothelial and platelet adhesion receptor mediating the interaction of activated platelets and endothelial cells with leukocytes. Platelets release various growth factors (e.g., TGF-β, PDGF, and EGF) that influence endothelial cell physiology and vice versa and activate endothelial signalosome signaling (24).
Platelet interaction with leukocytes can facilitate the activation of adaptive immune responses. Platelets can promote dendritic cell maturation and NK cell and monocyte/macrophage responses, which themselves affect specific T and B cell responses. Furthermore, platelets can directly affect B cell isotype switching and CD8+ T cell proliferation (25). Czapiga et al. demonstrated that platelet-derived CD40L induces the maturation of immature dendritic cells, professional antigen-presenting cells, via the upregulation of co-stimulatory molecules and IL-12/p40 production (26). Kaneider et al. showed that platelets trigger dendritic cell maturation independently of cyclo-oxygenase-derived arachidonic acid metabolites by mechanisms involving CD40L (27). Lastly, our data indicate first that platelets secrete a soluble dendritic cell-activating factor that was shown not to be sCD40L, as was expected from previous in vivo and in vitro studies, but instead a nucleotide, and second, that cell-to-cell contact does not induce the maturation of dendritic cells, possibly since nucleotide release by platelets was prevented by direct contact with dendritic cells (28, 29).
Adhesive interactions between platelets and monocytes deliver specific signals that initiate inflammatory gene expression, as described by Dixon et al. (30), showing that activated platelets induce COX-2 synthesis in monocytes by signaling at the transcriptional and post-transcriptional levels (30). Moreover, the formation of platelet-monocyte complexes and the detection of platelet-bound CX(3)CL1 on inflamed smooth muscle cells suggest that the CX(3)CL1-CX(3)CR1 axis contributes significantly to platelet and monocyte concentration in atherosclerotic arterial injury (31). Wong et al. (32) showed that platelets interacted with Kupffer cells in the liver sinusoids and that those interactions quickly changed to firm adhesion after specific microbes were captured by Kupffer cells. Elzey et al. reported that platelet-derived sCD40L increases serum IgG levels and germinal center formation under conditions where antigen-specific CD4+ T lymphocyte amounts are limiting (33). Regarding T lymphocyte activation and platelets, platelet reduction was shown to decrease intrahepatic accumulation of virus-specific cytotoxic T lymphocytes (CTLs) and organ injury in mouse models of acute viral hepatitis. Moreover, activated platelets contribute independently of their procoagulant function to CTL-mediated liver immunopathology (34). Zamora et al. investigated the proliferation and cytokine release of CD36+ CD4+ lymphocytes. Flow cytometric analysis and immunofluorescence microscopy indicated that CD36+ platelets were responsible for CD36 recognition on CD4+ lymphocytes. Moreover, Zamora et al. described that IL-17 and IFN-γ production was reduced in CD4+ lymphocytes with bound platelets (35). Furthermore, CD40L-positive T lymphocytes stimulated platelet activation through a CD40-dependent interaction with RANTES release, which activated endothelial cells, and facilitated T cell recruitment (36). Chapman et al. in Craig Morrell's laboratory at the University of Rochester, provided evidence that murine and human platelets express MHC class I molecules and that platelets activate T cells in an MHC class I-dependent manner. This interesting report suggests a novel hypothesis that platelets participate in the initiation of the acquired immune response (37).
It is becoming increasingly clear that platelets have inflammatory functions and can influence both adaptive and innate immune responses. Below are discussed some of the mechanisms by which platelets contribute to the innate immune response.
Platelets express the transmembrane protein CD40 ligand (CD40L, CD154), a member of the TNF receptor family. CD40L engages CD40, a second member of the TNF receptor family also present on B cells, monocytes, macrophages, carcinoma cells, dendritic cells, Kupffer cells and vascular endothelial cells, as well as on non-hematopoietic cells such as endothelial cells, smooth muscle cells, fibroblasts and keratinocytes (38). Platelets also express CD40. CD40 and CD40L are instrumental in both innate and adaptive immunity, with complex functions. Platelet activation leads to the surface expression and secretion of a wide range of proteins. P-Selectin (CD62P) is present on the inner leaflet of platelet granules and, following exocytosis, is expressed on the external leaflet of the plasma membrane. The ligand for CD62P is P-selectin glycoprotein ligand-1, which is expressed on a variety of leukocytes, notably neutrophils, eosinophils, lymphocytes, and monocytes. The GPIIbIIIa complex is present on the surface of quiescent platelets in an inactive, closed configuration (36). However, upon activation, there is a structural change in GPIIbIIIa to an active open state, which allows its binding to ligands. Although the natural ligands for GPIIbIIIa are von Willebrand factor and fibrinogen, the HIV surface protein gp120 can be bound by GPIIIa (along with other receptors) on platelets.
TLR adapters and signaling proteins downstream of TLR activation are potential targets for therapeutic drugs in eukaryotic cells (2); however, a more complete understanding of the platelet signaling complex is necessary. An increasing number of studies report both that platelets participate in the inflammatory process and that they may have an impact on pathogen clearance and the pathogenesis of bacteraemia, sepsis and, potentially, severe sepsis (39–41). However, a recent study presents the opposite view, in which a dual-track clearance mechanism balances innate and adaptive immunity during bacteraemia. Liver macrophages mediate fast clearance of intravascular Listeria. monocytogenes via scavenger receptors, in contrast to platelets, whose binding shifts L. monocytogenes clearance from “fast” to CRIg-dependent “slow” clearance pathways (42). Of critical importance to immunity and inflammation are Toll-like receptors (TLRs). TLRs are sensors of pathogen-associated molecular patterns (PAMPs), molecular determinants generally expressed by pathogens, specifically infectious pathogens. Several groups have described the presence and functionality of TLRs in mice and humans on both the membrane (TLR2/TLR1/TLR6/TLR4 and TLR9) and within platelets (TLR9); and TLR3 and TLR7 have also been identified (11–13, 43–48). Several recent studies suggest that TLR2,-4 and -9 are targets for bacterial-platelet interactions during severe sepsis and that they provide interesting targets for pharmacological analysis. Clark et al. suggested platelet TLR4 to be a threshold switch for bacterial trapping in severe sepsis. LPS-activated neutrophils, in combination with TLR4-activated platelets, were found to lead to the formation of neutrophil extracellular traps (NETs), which were able to ensnare bacteria in the blood flow for targeting immune clearance events (46). The addition of septic plasma but not control plasma to healthy neutrophils and platelets in the presence of DNA dyes evidenced the formation of NETs. Moreover, the author showed that platelet TLR4 mediates NETosis by decreasing the neutrophil DNA release time from 2 to 3 h (generally observed when neutrophils are stimulated) to ~10 min (when platelets are present). Therefore, it has been suggested that inhibition of platelet activation with TLR4 inhibitors, such as eritoran, may reduce NET formation and limit tissue damage (46). Evidence is also emerging that certain TLRs play a major role in the pathogenesis of infectious and/or inflammatory diseases (49). Sabroe et al. reported that stimulation by natural ligands of TLR2 (Pam3CSK4) or TLR4 (LPS) did not cause any changes in platelet aggregation, the surface levels of CD62P or the intra-platelet calcium levels (44). These data are consistent with the absence of direct effects on platelet activation as a result of the engagement of TLR2 or TLR4; the authors therefore concluded that these receptors are non-functional residues from megakaryocytes. Similar data were obtained by Jayachandran et al. (50) who showed that LPS did not affect platelet responses. In contrast, other groups, including our own, have found that the TLRs on platelets are functional and that their engagement evokes a variety of platelet responses (1, 2, 36, 40). In contrast to other cell types involved in immune responses (e.g., macrophages and dendritic cells), any possible link formed by platelets between innate immunity and adaptive immunity has yet to be proven (1–3, 33, 36, 40). Recently, Panigrahi et al. showed that physiological platelet agonists, primed by either suboptimal concentrations of thrombin receptor-activating peptide (TRAP) or the weak agonist ADP, act synergistically with TLR9 ligands by inducing TLR9 expression on the platelet surface and that the platelet TLR9 receptor is a functional receptor linking oxidative stress, innate immunity, and thrombosis (12). Thon et al. further demonstrated that TLR9 is located in a newly identified intracellular compartment in platelets and described a new organizational and signaling mechanism for TLR9 in human platelets (11). Finally, Hally et al. observed that platelet TLR9 expression was significantly elevated in subjects with acute coronary syndromes (ACSs) compared to that in healthy subjects, which may result in increased sensitivity to TLR9 agonists. Platelet activation caused increased expression of TLR9 in healthy platelets. We suggest that platelet activation, which occurs as part of ACSs, is a potential mechanism explaining the increased expression of platelet TLR9 observed in ACS patients (51). We provided evidence for differential signaling in platelets exposed to various TLR ligands leading to cytokine and chemokine secretion (52, 53). This difference indicates that platelet TLRs are functional, as they not only engage intracellular signaling pathways but also select among distinct adaptors (MyD88 vs. TRIF) to terminate NF-kB phosphorylation. The correlation between platelet TLR2 and TLR4 stimulation in vitro and the NF-kB/TRIF/Myd88 adaptor and signaling molecules is currently under investigation. In addition, studies showing the expression of TLR3 (54) and TLR7 (48) in human platelets have been published recently. Human platelets express TLR3 and are able to respond to poly I:C, indicating that these cells influence the innate immune response after exposure to viral dsRNA (54). Encephalomyocarditis virus (EMCV) infection rapidly reduces platelet count, and this phenomenon is credited to platelet Toll-like receptor 7 (TLR7) (48). Interestingly, Koupenova et al. demonstrated that platelets express all TLR transcripts and that these transcripts are more important in women with regard to cardiovascular risk and inflammatory markers (55).
Platelets are the major physiological contributor of sCD40L in plasma (56, 57). As outlined above, these soluble molecules can interact with the epithelium lining cells or mononuclear circulating cells that constitutively express CD40 counter receptors. It has been suggested that platelets can alter the binding of CD40/sCD40L, which is essential to inflammation. Platelets are cells that co-express surface CD40 and sCD40L molecules (58–63) in a platelet activation-dependent manner (CD40L is expressed and secreted only after activation, unlike CD40, which is constitutively expressed and not upregulated). However, while sCD40L is characteristic because of its quantitative and qualitative importance, this molecule is just one of the many secretory platelet molecules that contribute significantly to both hemostasis and immune modulation. In addition to releasing molecules that alter immune responses, platelets are involved in antimicrobial responses; indeed, platelets aggregate when exposed to certain bacteria (such as Staphylococcus aureus) and viruses (such as HIV), which may trigger responses to danger signals (39, 40, 64). In support of this finding, platelet degranulation, endocytic bacterial, and viral engulfment, and the release of antibacterial/antifungal proteins have been observed in conjunction with platelet aggregation events. Several studies have attempted to determine the involvement of platelets in immune responses dependent on CD40/CD40L and to determine the interactions between platelets and peripheral B cells. Platelets and B cells (in an in vitro co-culture model) were mutually activated, as validated by the increased expression of membrane platelet CD62P and B cell CD86. Platelet and B cell interactions were accompanied by changes in the membrane expression of CD40 and CD40L by both platelets and B cells. Differentiated B lymphocytes increased their production of IgG1, IgG2 and IgG3 but not IgG4, IgA, or IgM after a 3-days incubation with platelets in vitro (65). Another example of indirect interactions is at the interactions between platelets and macrophages in innate immunity and inflammation (66).
Platelets respond rapidly to changes in their environment, as they express surface receptors for a variety of ligands, such as the subendothelial proteins von Willebrand factor and collagen, as well as soluble agonists, such as thrombin, ADP and TxA2. This activation process leads to a variety of changes in platelets, including the extension of pseudopodia, the secretion of granule contents and PMPs, the synthesis and secretion of TxA2 and IL-1β, the formation of a procoagulant surface and the surface expression of a range of adhesive proteins, either by the exposure of granular membrane proteins on the plasma membrane or by structural changes in surface proteins from inert to active conformations (3).
While it is not yet generally accepted that platelets are also stimulators of immunity and inflammation, several recent reports argue in favor of acknowledging platelets as sensors in innate immunity and players in inflammation (2, 67–71). Platelets exert immune functions by acting to remove pathogen-infected host cells by binding directly to bacteria, viruses, and fungi (1, 39) and by mediating interactions between target cells and these infectious agents to potentiate the immune response (3, 33, 72). As a consequence, platelets have been linked with various inflammatory pathologies, such as cardiovascular disease (73), sepsis (72), and arthritis (36). A variety of mechanisms are involved in the contribution of platelets to the inflammatory process, including the increased expression of receptors for various immune mediators such as cytokines and chemokines and the exocytosis of a range of soluble factors, immunomodulatory factors, growth factors, biological response modifiers, etc., from α-granules (74–79). Several proteins are associated with the inflammasome family. These proteins are divided into two groups depending on the domains they contain: NLRPs contain pyrin domains and NLRCs contain caspase recruitment domains (CARDs). Hottz et al. noted that platelets constitutively express the inflammasome components NLRP3 and ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) and can use them to assemble functional inflammasomes, activate caspase-1, and process IL-1β (80).
In addition, in patients with dengue or after platelet exposure to dengue virus in vitro, increased expression of IL-1β in platelets and platelet-derived microparticles was observed. Infection with dengue virus results in NLRP3 inflammation, caspase-1 activation, and caspase-1-dependent IL-1β secretion. IL-1β derived from platelets is released mainly as microparticles through mechanisms dependent on inflammatory NLRP3 triggered by mitochondrial ROS. Activation of IL-1β-rich microparticles by the inflammasome and platelet shedding is correlated with increased vascular permeability. These findings show that platelets contribute to the increased vascular permeability in dengue virus infection by the inflammation-dependent release of IL-1β (80).
Moreover, Dr. Craig Jenne group are interested by the infections mediated by multidrug-resistant S. aureus. Recently, Surewaard et al. demonstrated (81), using an elegantly intravital imaging, that alpha toxin targets platelets directly, resulting in circulation detrimental aggregation. Moreover, neutralizing alpha toxin during infection of S. aureus, while escaping microvascular damage, does not interfere with beneficial platelet responses. In this context, Platelets are always able to recruit macrophages and participate to the eradication of S. aureus. Considered platelets as sensors in innate immunity in response to infectious stresses (1, 2, 36, 39, 82–94) contributes to the understanding of the interrelationship between infection, inflammation, and coagulation.
Platelet concentrates for transfusion are living cell products with a certain life span that degrade in a physiological mechanism-dependent manner via mechanisms that may be accelerated by mechanical production and storage mechanisms (95). Platelets prepared for transfusion are subject to stress injury upon collection, preparation and storage (96). Under these types of stress, platelets undergo morphologic/metabolic changes likely to lead to platelet activation and an increase in the concentration of BRMs (82). Ex vivo platelet processing can have an effect on BRM secretion (97). These BRM-promoting events lead to negative changes and a gradual deterioration in platelet viability, structure, and function.
When stored as PCs, platelets can undergo changes that are mainly related to the storage solutions and conditions (platelet agitation and storage temperature and time).
In general, PCs—especially those prepared for prophylactic usage—are stored for an average of 5 days at a maximum temperature of 22 ± 2°C under constant, gentle agitation to prevent platelet aggregation. Additionally, buffy coat-derived pooled platelet concentrates (PPCs) and single-donor apheresis platelet concentrates (SDA-PCs) are stored in suspension in 35% donor plasma and 65% platelet additive solution (PAS). Compared to platelet storage in autologous plasma, platelet storage in an additive solution has satisfactorily improved platelet function preservation. PASs are generally used as plasma replacements to (i) reduce the quantity of plasma transfused; (ii) avoid the transfusion of large volumes of plasma to reduce the incidence of adverse reactions and circulatory overload; (iii) enable certain photochemical treatments for pathogen inactivation; and (iv) maintain storage conditions (98). Platelet storage lesions include the appearance of platelet morphological changes, activation markers, GPIbα expression loss, α granule secretion, and mitochondrial dysfunction (99). Platelet concentrate storage can lead to the secretion of several BRMs, such as sCD40L, PDGFAA, RANTES, IL1β, IL6, IL7, IL8, PF4, IL13, OX40L, IL27, and TGFβ (2, 82). Generally, extended PC storage is accompanied by increased BRM production, which may be related to an increase in the percentage of adverse events (AEs) observed according to PC storage time. To minimize AEs, it would be preferable to transfuse PCs as early as possible. It is fitting, however, to consider this conclusion in light of the PC production and issuing constraints on blood establishments according to the demand for the product in hospital banks. In particular, it has been demonstrated that from the 3rd day of PC storage, there is a significant increase in the concentration of BRMs, especially sCD40L (96). These observations suggest that storage lesions play a role in the inflammation caused by PC. sCD40L induces the production of reactive oxygen species (ROS) during PC storage, leading to an increase in the production and release of pro-inflammatory substances (100).
Additionally, the type of PC processing used during the preparation and storage process can have an effect on platelet activation. Leitner et al. showed that platelets stored in an InterSol™ solution exhibited significantly higher initial activation levels, as indicated by CD62P expression, than platelets stores in other additive solutions (Composol® and SSP+®) (101). However, platelet storage in an additive solution has demonstrated a certain number of benefits, especially a reduction in serious adverse reactions (102). Although the different types of PCs are of comparable quality, there is debate about their safety. Daurat et al. showed that AEs were less commonly related to PPCs than to SDA-PCs (103). These results challenge the widespread use of SDA-PCs and suggest that these concentrated should be prescribed for specific indications. Evaluation of the risk/benefit balance of transfusing different types of PCs would enable the prescription of the optimal product according to the medical indication.
BRMs contained in PC are also transfused. It has been shown that BRMs can induce immune responses (33) and post-transfusion reactions (104) and can affect hemostasis (105) and inflammation in the recipient (106). Storage lesions triggered by extrinsic factors (preparation methods) or intrinsic mechanisms (plasma and platelet factors, residual leukocytes) could be largely responsible for both reducing the therapeutic efficacy of PC transfusion and inducing AEs (76). In addition to blood platelets, BRMs contained in PC are also transfused to the recipient. Among these molecules, sCD40L is described as being partly responsible for febrile non-hemolytic transfusion reactions (FNHTRs) after platelet transfusions (58, 107). In addition to its role in inflammation, CD40L seems to play a role in AEs. sCD40L is found in PCs, and its concentration increases during storage (96). Numerous studies have shown that sCD40L is involved in PC transfusion reactions (106, 108). In addition, we showed that other soluble factors, such as IL27 and sOX40L, are involved in FNHTR (104). Several soluble factors with high predictive value for the occurrence of AEs, such as sCD40L, IL13, and MIP1α, have been identified, primarily via machine learning algorithms (109). Indeed, this study shows a correlation between the concentrations of sCD40L and IL13 and the onset of AEs. Additionally, the concentration of MIP1α found in the supernatants that induced AEs seems to be able to differentiate the type of AEs, FNHTR or allergies. PCs also contain mitochondrial DNA (mtDNA), which is associated with adverse effects (96, 110). Boudreau et al. showed that activated platelets release mitochondria, in both encapsulated microparticles and membrane-free organelles. Extracellular mitochondria are found at higher levels in transfused PCs that caused acute reactions (FNHTR, cutaneous, and cardiovascular signs) in transfused patients than in those that did not (99, 110, 111).
It is clearly acknowledged that the increased levels of cytokines and chemokines in platelet concentrates developed during storage, in the absence of detectable exogenous stimuli, can contribute to AEs (96, 112). In addition to cytokines/chemokines, platelet Extracellular vesicles (EVs), and platelet microparticles (PMs), which are important mediators of inflammation and immune response regulation also seem to be involved in the onset of AEs (113, 114). EVs are a heterogeneous group of structures and comprise a large group of particles, including exosomes and microvesicles, and are released from virtually all cell types. EVs can be divided according to their size into microparticles (MPs) or microvesicles (MVs) that vary in size between 0.1 and 1 μM, and exosomes in size of 30–100 nm (115). Platelet exosomes strongly expressed tetraspanin CD63, CD9, CD63, TSG101, ALIX, CD31, CD41, CD42a, P-selectin, PF4, and GPIIb/IIIa. Platelet exosomes might play a lesser role in procoagulant activity than PMPs. Platelet exosomes can directly stimulate target cells by providing ligands that increase the secretion of various signaling molecules, e.g., growth factors or cytokines. They can also transfer membrane receptors and molecules of adhesion. In addition, Platelet exosomes provide proteins, mRNA, and transcription factors that cause target cell epigenetic reprogramming (116).
PMPs containing microRNA can also be involved in a pathophysiologic response and AE induction following PC transfusion. Additionally, studies have shown that pathogen reduction technologies aimed at reducing the potential risk of transfusion-transmitted infections induce platelet activation and a reduction in the mRNA (117) and microRNA levels (118). These RNA changes are correlated with an increase in the PMP concentration. As a result, it seems likely that pathogenic agent reduction technologies can increase PMP formation in PCs (118). Given the pro-inflammatory properties of PMPs, it is reasonable to presume that they can aggravate acute and chronic inflammatory reactions in blood vessels, such as those associated with platelet transfusion and atherosclerosis (119).
Platelets contain and secrete numerous cytokines and other immunomodulatory proteins, which may be candidates for innovative targeted therapeutic approaches. For instance, there is now sound evidence that the platelet-activating factor (PAF)/PAF-receptor pathway is a promising target for pharmacological involvement in acute coronary syndrome (120), central nervous system diseases (121), autoimmune diseases (122), and rheumatoid arthritis (123). Platelet-related CD40L, IL-1β, PF4, and RANTES are currently under consideration as molecular targets against inflammation and hypercholesterolemia syndromes (124). Platelets continue to motivate much biological research. To date, a substantial amount of data has been collected over several decades on the hemostatic properties and immunogenic competence of platelets, yet much about the roles of platelets in physiological and pathological processes remains to be clarified. Furthermore, if platelets are immune sentinels with the capacity to bridge innate and adaptive immunity, preformed products from platelets, and products resulting from platelet neosynthesis can act on cell-cell interactions (platelet-dendritic cell, platelet-B cell and platelet-T cell) and play a major role in immune responses. Several reports have investigated “platelet physiology” as an immune cell concept (2, 71), and a notable number of papers (67–70, 90, 125, 126) have recently detailed this new concept. Indeed, future directions for research concern the critical role of platelets as an immune cell in the host immune response. It is now clear, therefore, that in addition to their roles in hemostasis and thrombosis, platelets have a large range of other functions, notably playing key roles in the inflammatory process, immune responses (2, 82, 84), regenerative medicine and host defense against pathogens (Figure 1). The challenge for therapeutic intervention in pathological processes will be to identify drugs that block specific targets involved in the complex contribution of platelets to inflammation/immunity without affecting their hemostatic function.
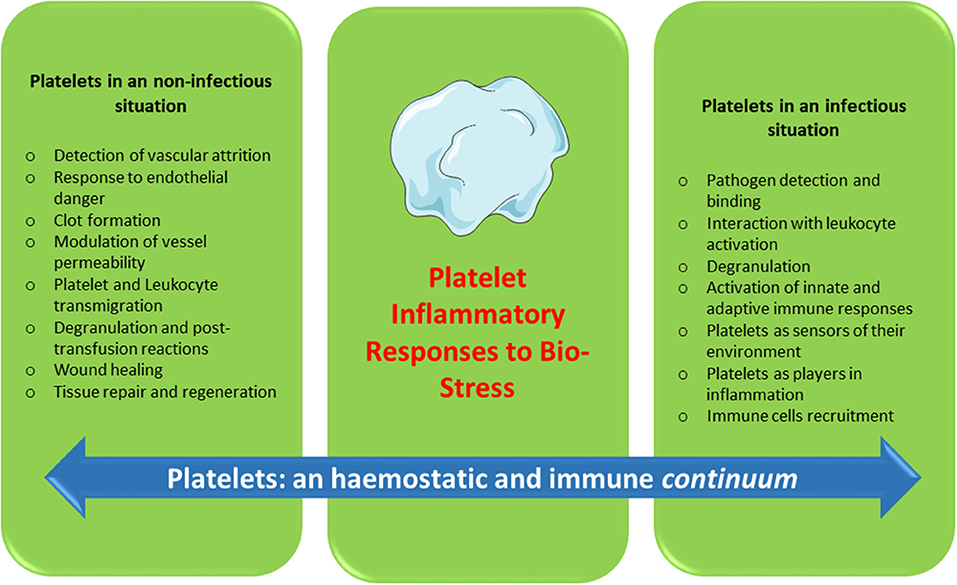
Figure 1. Platelets: an hemostatic and immune continuum.
FC, SL, PB, TB, HM, PM, OG, and HH-C have made a substantial, direct and intellectual contribution to the work, wrote and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Charles Antoine Arthaud, Marie Ange Eyraud, and Jocelyne Fagand for their contribution of original data. We would also like to thank the medical staff of the Etablissement Français du Sang Rhone-Alpes-Auvergne, Saint-Etienne, France for technical support throughout our studies. This work was supported by grants from the Etablissement Français du Sang (EFS) [French National Blood Service] (Grant APR), France; the Association Recherche et Transfusion (ART) [Association for Research and Transfusion], Paris, France; the Agence Nationale de la Sécurité et du Médicament et des Produits de Santé (ANSM) [French National Agency for Medicines and Health Products Safety] (AAP-2012-011, Reference 2012S055), the Association Les Amis de Rémi Savigneux, France, the ADSB Boisset-Saint-Pal-Tiranges, France, the Academic Research Community-1 of the Rhône-Alpes Region, and the Agence Nationale de la Recherche (ANR) [French National Research Agency], reference ANR-12-JSV1-0012-01.
1. Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and siglec receptors. Front Immunol. (2015) 6:83. doi: 10.3389/fimmu.2015.00083
2. Cognasse F, Garraud O, Pozzetto B, Laradi S, Hamzeh-Cognasse H. How can non-nucleated platelets be so smart? J Thromb Haemost. (2016) 14:794–6. doi: 10.1111/jth.13262
3. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
4. Bennett JS, Berger BW, Billings PC. The structure and function of platelet integrins. JTH. (2009) 7 (Suppl. 1):200–5. doi: 10.1111/j.1538-7836.2009.03378.x
6. Thon JN, Italiano JE. Platelets: production, morphology, and ultrastructure. Handb Exp Pharmacol. (2012) 3–22. doi: 10.1007/978-3-642-29423-5_1
7. Flaumenhaft R, Dilks JR, Rozenvayn N, Monahan-Earley RA, Feng D, Dvorak AM. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. (2005) 105:3879–87. doi: 10.1182/blood-2004-04-1392
8. Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. (2002) 415:175–9. doi: 10.1038/415175a
9. Peters CG, Michelson AD, Flaumenhaft R. Granule exocytosis is required for platelet spreading: differential sorting of alpha-granules expressing VAMP-7. Blood. (2012) 120:199–206. doi: 10.1182/blood-2011-10-389247
10. Ravindran R, Krishnan LK. A biochemical study on the effect of proteolysis of beta-thromboglobulin proteins released from activated platelets on fibroblast proliferation. Pathophysiol Haemost Thromb. (2007) 36:285–9. doi: 10.1159/000296282
11. Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, et al. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. (2012) 198:561–74. doi: 10.1083/jcb.201111136
12. Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, et al. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyper-reactivity and thrombosis. Circul Res. (2012) 112:103–12. doi: 10.1161/CIRCRESAHA.112.274241
13. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll-like receptor molecules on human platelets. Immunol Cell Biol. (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
14. van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. (2019) 16:166–79. doi: 10.1038/s41569-018-0110-0
15. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules or not? JTH. (2015) 13:2141–51. doi: 10.1111/jth.13147
16. Marcu MG, Zhang L, Nau-Staudt K, Trifaro JM. Recombinant scinderin, an F-actin severing protein, increases calcium-induced release of serotonin from permeabilized platelets, an effect blocked by two scinderin-derived actin-binding peptides and phosphatidylinositol 4,5-bisphosphate. Blood. (1996) 87:20–4.
17. Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood. (2011) 118:1370–3. doi: 10.1182/blood-2011-01-330910
18. Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, et al. Distinct platelet packaging, release, surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood. (2011) 117:3907–11. doi: 10.1182/blood-2010-12-327007
19. Battinelli EM, Markens BA, Italiano JE Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood. (2011) 118:1359–69. doi: 10.1182/blood-2011-02-334524
20. van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HF. The platelet interior revisited: electron tomography reveals tubular alpha-granule subtypes. Blood. (2010) 116:1147–56. doi: 10.1182/blood-2010-02-268680
21. Bills TK, Smith JB, Silver MJ. Selective release of archidonic acid from the phospholipids of human platelets in response to thrombin. J Clin Invest. (1977) 60:1–6. doi: 10.1172/JCI108745
22. Mannel DN, Grau GE. Role of platelet adhesion in homeostasis and immunopathology. Mol Pathol. (1997) 50:175–85. doi: 10.1136/mp.50.4.175
23. Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. (2004) 114:447–53. doi: 10.1016/j.thromres.2004.07.020
24. van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. (2009) 85:195–204. doi: 10.1189/jlb.0708400
25. Li N. Platelet-lymphocyte cross-talk. J Leukoc Biol. (2008) 83:1069–78. doi: 10.1189/jlb.0907615
26. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: a potential bridge between injury and immune activation. Exp Hematol. (2004) 32:135–9. doi: 10.1016/j.exphem.2003.11.004
27. Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-dependent maturation of human monocyte-derived dendritic cells by activated platelets. Int J Immunopathol Pharmacol. (2003) 16:225–31. doi: 10.1177/039463200301600307
28. Hamzeh-Cognasse H, Cognasse F, Palle S, Chavarin P, Olivier T, Delezay O, et al. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol. (2008) 9:54–69. doi: 10.1186/1471-2172-9-54
29. Perros AJ, Christensen AM, Flower RL, Dean MM. Soluble mediators in platelet concentrates modulate dendritic cell inflammatory responses in an experimental model of transfusion. J Interf Cytok Res. (2015) 35:821–30. doi: 10.1089/jir.2015.0029
30. Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest. (2006) 116:2727–38. doi: 10.1172/JCI27209
31. Postea O, Vasina EM, Cauwenberghs S, Projahn D, Liehn EA, Lievens D, et al. Contribution of platelet CX(3)CR1 to platelet-monocyte complex formation and vascular recruitment during hyperlipidemia. Arterioscl Thromb Vasc Biol. (2012) 32:1186–93. doi: 10.1161/ATVBAHA.111.243485
32. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
33. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity. (2003) 19:9–19. doi: 10.1016/S1074-7613(03)00177-8
34. Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med. (2005) 11:1167–9. doi: 10.1038/nm1317
35. Zamora C, Canto E, Nieto JC, Ortiz MA, Diaz-Torne C, Diaz-Lopez C, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol. (2013) 94:521–29. doi: 10.1189/jlb.0213074
36. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol. (2015) 194:5579–87. doi: 10.4049/jimmunol.1500259
37. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, et al. Platelets present antigen in the context of MHC class I. J Immunol. (2012) 189: 916–23. doi: 10.4049/jimmunol.1200580
38. van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. (2000) 67:2–17. doi: 10.1002/jlb.67.1.2
39. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections - complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
40. Garraud O, Cognasse F, Hamzeh-Cognasse H, Pozzetto B. Platelets and their immune role in anti-infective immunity. Future Microbiol. (2016) 11:167–70. doi: 10.2217/fmb.15.146
41. Vincent JL, Yagushi A, Pradier O. Platelet function in sepsis. Crit Care Med. (2002) 30 (5 Suppl):S313–7. doi: 10.1097/00003246-200205001-00022
42. Steven Kooten P, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe. (2016) 20:36–48. doi: 10.1016/j.chom.2016.05.023
43. Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y. Expression of toll-like receptors on human platelets. Thromb Res. (2004) 113:379–85. doi: 10.1016/j.thromres.2004.03.023
44. Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. (2005) 94:831–8. doi: 10.1160/TH05-01-0009
45. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
46. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
47. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. (2008) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
48. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
49. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity. update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
50. Jayachandran M, Brunn GJ, Karnicki K, Miller RS, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J Appl Physiol. (2007) 102:429–33. doi: 10.1152/japplphysiol.01576.2005
51. Hally KE, La Flamme AC, Larsen PD, Harding SA. Toll-like receptor 9 expression and activation in acute coronary syndrome patients on dual anti-platelet therapy. Thromb Res. (2016) 148:89–95. doi: 10.1016/j.thromres.2016.10.026
52. Berthet J, Damien P, Hamzeh-Cognasse H, Arthaud CA, Eyraud MA, Zeni F, et al. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin Immunol. (2012) 145:189–200. doi: 10.1016/j.clim.2012.09.004
53. Kappelmayer J, Beke Debreceni I, Vida A, Antal-Szalmas P, Clemetson KJ, Nagy B Jr. Distinct effects of Re- and S-forms of LPS on modulating platelet activation. J Thromb Haemost. (2013) 11:775–8. doi: 10.1111/jth.12151
54. Anabel AS, Eduardo PC, Antonio HC, Carlos SM, Juana NM, Honorio TA, et al. Human platelets express toll-like receptor 3 and respond to poly I:C. Hum Immunol. (2014) 75:1244–51. doi: 10.1016/j.humimm.2014.09.013
55. Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscl Thromb Vascu Biol. (2015) 35:1030–7. doi: 10.1161/ATVBAHA.114.304954
56. Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circuluation. (2002) 106:896–9. doi: 10.1161/01.CIR.0000028962.04520.01
57. Aloui C, Prigent A, Tariket S, Sut C, Fagan J, Cognasse F, et al. Levels of human platelet-derived soluble CD40 ligand depend on haplotypes of CD40LG-CD40-ITGA2. Scient Rep. (2016) 6:24715. doi: 10.1038/srep24715
58. Phipps RP, Kaufman J, Blumberg N. Platelet derived CD154 (CD40 ligand) and febrile responses to transfusion. Lancet. (2001) 357:2023–4. doi: 10.1016/S0140-6736(00)05108-4
59. Freedman JE. CD40-CD40L and platelet function: beyond hemostasis. Circ Res. (2003) 92:944–6. doi: 10.1161/01.RES.0000074030.98009.FF
60. Davi G, Ferroni P. CD40-CD40L interactions in platelet activation. Thromb Haemost. (2005) 93:1011–2. doi: 10.1160/TH05-04-0270
61. Corash L, Cognasse F, Osselaer C-J, Messe N, Hooydonk MV, Garraud O. Cytokines in platelet components associated with acute transfusion reactions: the role of sCD40Blood L. ASH Ann Meeting Abstracts. (2006) 108:952. Available online at: http://www.bloodjournal.org/content/108/11
62. Elzey BD, Ratliff TL, Sowa JM, Crist SA. Platelet CD40L at the interface of adaptive immunity. Thromb Res. (2011) 127:180–3. doi: 10.1016/j.thromres.2010.10.011
63. Aloui C, Prigent A, Sut C, Tariket S, Hamzeh-Cognasse H, Pozzetto B, et al. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int J Mol Sci. (2014) 15:22342–64. doi: 10.3390/ijms151222342
64. Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, et al. Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol. (2015) 16:26. doi: 10.1186/s12865-015-0092-1
65. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogne M, Richard Y, et al. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp Hematol. (2007) 35:1376–87. doi: 10.1016/j.exphem.2007.05.021
66. Mantovani A, Garlanda C. Platelet-macrophage partnership in innate immunity and inflammation. Nat Immunol. (2013) 14:768–70. doi: 10.1038/ni.2666ni.2666
67. Gaertner F, Massberg S. Blood coagulation in immunothrombosis—At the frontline of intravascular immunity. Sem Immunol. (2016) 28:561–69. doi: 10.1016/j.smim.2016.10.010
68. Iannacone M. Platelet-mediated modulation of adaptive immunity. Sem Immunol. (2016) 28:555–60. doi: 10.1016/j.smim.2016.10.008
69. Kim S-J, Jenne CN. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Sem Immunol. (2016) 28:546–54. doi: 10.1016/j.smim.2016.10.013
70. Kubes P. The versatile platelet contributes to inflammation, infection, hemostasis, coagulation and cancer. Sem Immunol. (2016) 28:535. doi: 10.1016/j.smim.2016.11.002
71. Garraud O, Cognasse F. Are platelets cells? and if yes, are they immune cells? Front Immunol. (2015) 6:70. doi: 10.3389/fimmu.2015.00070
72. Schubert S, Weyrich AS, Rowley JW. A tour through the transcriptional landscape of platelets. Blood. (2014) 124:493–502. doi: 10.1182/blood-2014-04-512756
73. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. (2005) 115:3378–84. doi: 10.1172/JCI27196
74. Garraud O, Cognasse F. Platelet immunology and the immune response. Transfus Clin Biol. (2009) 16:106–17. doi: 10.1016/j.tracli.2009.03.002
75. Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, et al. Platelet functions beyond haemostasis. J Thromb Haemost. (2009) 7:1759–66. doi: 10.1111/j.1538-7836.2009.03586.x
76. Morrell CN. Immunomodulatory mediators in platelet transfusion reactions. Hematol Am Soc Hematol Educ Program. (2011) 2011:470–4. doi: 10.1182/asheducation-2011.1.470
77. Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. (2014) 29:153–62. doi: 10.1016/j.blre.2014.10.003
78. Lannan KL, Sahler J, Kim N, Spinelli SL, Maggirwar SB, Garraud O, et al. Breaking the mold: transcription factors in the anucleate platelet and platelet-derived microparticles. Front Immunol. (2015) 6:48. doi: 10.3389/fimmu.2015.00048
79. Boilard E, Duchez AC, Brisson A. The diversity of platelet microparticles. Curr Opin Hematol. (2015) 22:437–44. doi: 10.1097/MOH.0000000000000166
80. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
81. Surewaard BGJ, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, et al. Alpha-toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microbe. (2018) 24:271–84 e3. doi: 10.1016/j.chom.2018.06.017
82. Sut C, Tariket S, Aubron C, Aloui C, Hamzeh-Cognasse H, Berthelot P, et al. The non-hemostatic aspects of transfused platelets. Front Med. (2018) 5:42. doi: 10.3389/fmed.2018.00042
83. Vardon Bounes F, Mujalli A, Cenac C, Severin S, Le Faouder P, Chicanne G, et al. The importance of blood platelet lipid signaling in thrombosis and in sepsis. Adv Biol Regul. (2018) 67:66–73. doi: 10.1016/j.jbior.2017.09.011
84. Kim SJ, Davis RP, Jenne CN. Platelets as modulators of inflammation. Semin Thromb Hemost. (2018) 44:91–101. doi: 10.1055/s-0037-1607432
85. Jenne CN. Pathogen-induced coagulation: a new angle? Blood. (2018) 132:771–3. doi: 10.1182/blood-2018-07-859967
86. Hamzeh-Cognasse H, Berthelot P, Tardy B, Pozzetto B, Bourlet T, Laradi S, et al. Platelet toll-like receptors are crucial sensors of infectious danger moieties. Platelets. (2018) 29:533–40. doi: 10.1080/09537104.2018.1445842
87. Manne BK, Xiang SC, Rondina MT. Platelet secretion in inflammatory and infectious diseases. Platelets. (2017) 28:155–64. doi: 10.1080/09537104.2016.1240766
88. D' Atri LP, Schattner M. Platelet toll-like receptors in thromboinflammation. Front Biosci. (2017) 22:1867–83. doi: 10.2741/4576
89. Blumberg N, Cholette JM, Schmidt AE, Phipps RP, Spinelli SL, Heal JM, et al. Management of platelet disorders and platelet transfusions in ICU patients. Transfus Med Rev. (2017) 31:252–27. doi: 10.1016/j.tmrv.2017.04.002
90. Deppermann C, Kubes P. Platelets and infection. Semin Immunol. (2016) 28:536–45. doi: 10.1016/j.smim.2016.10.005
91. Nording HM, Seizer P, Langer HF. Platelets in inflammation and atherogenesis. Front Immunol. (2015) 6:98. doi: 10.3389/fimmu.2015.00098
92. Morrell CN. Understanding platelets in malaria infection. Curr Opin Hematol. (2014) 21:445–9. doi: 10.1097/MOH.0000000000000073
93. de Stoppelaar SF, van 't Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost. (2014) 112:666–77. doi: 10.1160/TH14-02-0126
94. Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: toll-like receptors in platelets and megakaryocytes. Thromb Res. (2010) 125:205–9. doi: 10.1016/j.thromres.2009.11.004
95. Devine DV, Serrano K. The platelet storage lesion. Clin Lab Med. (2010) 30:475–87. doi: 10.1016/j.cll.2010.02.002
96. Cognasse F, Boussoulade F, Chavarin P, Acquart S, Fabrigli P, Lamy B, et al. Release of potential immunomodulatory factors during platelet storage. Transfusion. (2006) 46:1184–9. doi: 10.1111/j.1537-2995.2006.00869.x
97. Garraud O, Hamzeh-Cognasse H, Cognasse F. Platelets and cytokines: how and why? Transfus Clin Biol. (2012) 19:104–8. doi: 10.1016/j.tracli.2012.02.004
99. Ng MSY, Tung JP, Fraser JF. Platelet storage lesions: what more do we know now? Transfus Med Rev. (2018) 32:144–54. doi: 10.1016/j.tmrv.2018.04.001
100. Ghasemzadeh M, Hosseini E. Platelet granule release is associated with reactive oxygen species generation during platelet storage: a direct link between platelet pro-inflammatory and oxidation states. Thromb Res. (2017) 156:101–4. doi: 10.1016/j.thromres.2017.06.016
101. Leitner GC, List J, Horvath M, Eichelberger B, Panzer S, Jilma-Stohlawetz P. Additive solutions differentially affect metabolic and functional parameters of platelet concentrates. Vox sanguinis. (2016) 110:20–6. doi: 10.1111/vox.12317
102. van der Meer PF. PAS or plasma for storage of platelets? A concise review. Transfus Med. (2016) 26:339–42. doi: 10.1111/tme.12325
103. Daurat A, Roger C, Gris J, Daurat G, Feissel M, Le Manach Y, et al. Apheresis platelets are more frequently associated with adverse reactions than pooled platelets both in recipients and in donors: a study from French hemovigilance data. Transfusion. (2016) 56:1295–303. doi: 10.1111/trf.13475
104. Hamzeh-Cognasse H, Damien P, Nguyen KA, Arthaud CA, Eyraud MA, Chavarin P, et al. Immune-reactive soluble OX40 ligand, soluble CD40 ligand, interleukin-27 are simultaneously oversecreted in platelet components associated with acute transfusion reactions. Transfusion. (2014) 54:613–25. doi: 10.1111/trf.12378
105. Refaai MA, Phipps RP, Spinelli SL, Blumberg N. Platelet transfusions: impact on hemostasis, thrombosis, inflammation, and clinical outcomes. Thromb Res. (2011) 127:287–91. doi: 10.1016/j.thromres.2010.10.012
106. Sahler J, Spinelli S, Phipps R, Blumberg N. CD40 ligand (CD154) involvement in platelet transfusion reactions. Transfus Clin Biol. (2012) 19:98–103. doi: 10.1016/j.tracli.2012.02.003
107. Cognasse F, Payrat JM, Corash L, Osselaer JC, Garraud O. Platelet components associated with acute transfusion reactions: the role of platelet-derived soluble CD40 ligand. Blood. (2008) 112:4779–80. doi: 10.1182/blood-2008-05-157578
108. Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP. The platelet as an immune cell-CD40 ligand and transfusion immunomodulation. Immunol Res. (2009) 45:251–60. doi: 10.1007/s12026-009-8106-9
109. Nguyen KA, Hamzeh-Cognasse H, Sebban M, Fromont E, Chavarin P, Absi L, et al. A computerized prediction model of hazardous inflammatory platelet transfusion outcomes. PLoS ONE. (2014) 9:e97082. doi: 10.1371/journal.pone.0097082
110. Yasui K, Matsuyama N, Kuroishi A, Tani Y, Furuta RA, Hirayama F. Mitochondrial damage-associated molecular patterns as potential proinflammatory mediators in post-platelet transfusion adverse effects. Transfusion. (2016) 56:1201–12. doi: 10.1111/trf.13535
111. Cognasse F, Aloui C, Anh Manach K, Hamzeh-Cognasse H, Fagan J, Arthaud CA, et al. Platelet components associated with adverse reactions: predictive value of mitochondrial DNA relative to biological response modifiers. Transfusion. (2016) 56:497–504. doi: 10.1111/trf.13373
112. Muylle L. The role of cytokines in blood transfusion reactions. Blood Rev. (1995) 9:77–83. doi: 10.1016/S0268-960X(95)90028-4
113. Kriebardis A, Antonelou M, Stamoulis K, Papassideri I. Cell-derived microparticles in stored blood products: innocent-bystanders or effective mediators of post-transfusion reactions? Blood Transfus. (2012) 10 (Suppl. 2):s25–38. doi: 10.2450/2012.006S
114. Burnouf T, Chou ML, Goubran H, Cognasse F, Garraud O, Seghatchian J. An overview of the role of microparticles/microvesicles in blood components: are they clinically beneficial or harmful? Transfus Apher Sci. (2015) 53:137–45. doi: 10.1016/j.transci.2015.10.010
115. Zmigrodzka M, Guzera M, Miskiewicz A, Jagielski D, Winnicka A. The biology of extracellular vesicles with focus on platelet microparticles and their role in cancer development and progression. Tumour Biol. (2016) 37:14391–401. doi: 10.1007/s13277-016-5358-6
116. Tao SC, Guo SC, Zhang CQ. Platelet-derived extracellular vesicles: an emerging therapeutic approach. Int J Biol Sci. (2017) 13:828–34. doi: 10.7150/ijbs.19776
117. Osman A, Hitzler WE, Ameur A, Provost P. Differential expression analysis by RNA-Seq reveals perturbations in the platelet mRNA transcriptome triggered by pathogen reduction systems. PLoS ONE. (2015) 10:e0133070. doi: 10.1371/journal.pone.0133070
118. Osman A, Hitzler WE, Meyer CU, Landry P, Corduan A, Laffont B, et al. Effects of pathogen reduction systems on platelet microRNAs, mRNAs, activation, function. Platelets. (2015) 26:154–63. doi: 10.3109/09537104.2014.898178
119. Boilard E, Blanco P, Nigrovic PA. Platelets: active players in the pathogenesis of arthritis and SLE. Nat Rev Rheumatol. (2012) 8:534–42. doi: 10.1038/nrrheum.2012.118
120. Palur Ramakrishnan AV, Varghese TP, Vanapalli S, Nair NK, Mingate MD. Platelet activating factor: a potential biomarker in acute coronary syndrome? Cardiovasc Therap. (2017) 35:64–70. doi: 10.1111/1755-5922.12233
121. Liu Y, Shields LB, Gao Z, Wang Y, Zhang YP, Chu T, et al. Current understanding of platelet-activating factor signaling in central nervous system diseases. Mol Neurobiol. (2016) 54:5563–72. doi: 10.1007/s12035-016-0062-5
122. Onat A, Can G. Enhanced proinflammatory state and autoimmune activation. a breakthrough to understanding chronic diseases. Curr Pharmaceut Design. (2014) 20:575–84. doi: 10.2174/138161282004140213145551
123. Cognasse F, Garraud O, Marotte H. Unlike tocilizumab, etanercept slightly increases experimental thrombin-induced aggregation in healthy individuals. Joint, Bone, Spine. (2017) 84:373–5. doi: 10.1016/j.jbspin.2016.04.010
124. Alexandru N, Popov D, Georgescu A. Platelet dysfunction in vascular pathologies and how can it be treated. Thromb Res. (2012) 129:116–26. doi: 10.1016/j.thromres.2011.09.026
125. Doni A, Garlanda C, Mantovani A. Innate immunity, hemostasis, and matrix remodeling. PTX3 as a link. Sem Immunol. (2016) 28:570–7. doi: 10.1016/j.smim.2016.10.012
Keywords: platelets, innate immunity, transfusion, cytokine/chemokine, inflammation
Citation: Cognasse F, Laradi S, Berthelot P, Bourlet T, Marotte H, Mismetti P, Garraud O and Hamzeh-Cognasse H (2019) Platelet Inflammatory Response to Stress. Front. Immunol. 10:1478. doi: 10.3389/fimmu.2019.01478
Received: 14 March 2019; Accepted: 13 June 2019;
Published: 28 June 2019.
Edited by:
Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Beate E. Kehrel, University Hospital Münster, GermanyCopyright © 2019 Cognasse, Laradi, Berthelot, Bourlet, Marotte, Mismetti, Garraud and Hamzeh-Cognasse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrice Cognasse, ZmFicmljZS5jb2duYXNzZUBlZnMuc2FudGUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.