 Manuel Alfredo Podestà
Manuel Alfredo Podestà Giuseppe Remuzzi
Giuseppe Remuzzi Federica Casiraghi
Federica Casiraghi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 June 2019
Sec. Alloimmunity and Transplantation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01287
This article is part of the Research Topic Safety, Efficacy and Mechanisms of Action of Mesenchymal Stem Cell Therapies View all 22 articles
In solid organ transplantation lifelong immunosuppression exposes transplant recipients to life-threatening complications, such as infections and malignancies, and to severe side effects. Cellular therapy with mesenchymal stromal cells (MSC) has recently emerged as a promising strategy to regulate anti-donor immune responses, allowing immunosuppressive drug minimization and tolerance induction. In this review we summarize preclinical data on MSC in solid organ transplant models, focusing on potential mechanisms of action of MSC, including down-regulation of effector T-cell response and activation of regulatory pathways. We will also provide an overview of available data on safety and feasibility of MSC therapy in solid organ transplant patients, highlighting the issues that still need to be addressed before establishing MSC as a safe and effective tolerogenic cell therapy in transplantation.
Transplantation represents the treatment of choice for end-stage solid organ failure. The dissection of the mechanisms regulating the interplay between the host immune system and the transplanted graft have led to the introduction into clinical practice of effective T-cell immunosuppressive agents, which have abated the risk of acute rejection in the peri-transplant period, increasing the 1-year graft survival above 90%. However, current immunosuppressive agents failed to significantly affect the long-term outcome of solid organ transplantation, because these drugs are less effective in preventing chronic allograft rejection (1). Moreover, the lifelong systemic immunosuppression exposes transplant recipients to a significant risk of side effects, infections and malignancy (2, 3).
Since acute T-cell mediated rejection can be successfully managed in most cases, transplant research should focus on the identification of innovative strategies to achieve allograft tolerance, i.e., long-term stable graft function in the absence of immunosuppression. Therefore, the ideal strategy should target not only T cells, which are the main players of alloimmunity, but regulate in a concerted action also B cells, dendritic cells and macrophages, which all contribute to both the acute and chronic alloimmune response.
In this scenario, mesenchymal stromal cells (MSC) seem a very promising cellular therapy in the pursuit of transplantation tolerance induction, allowing minimization or even discontinuation of life-long immunosuppression. Indeed, MSC have a unique capability to inhibit the immune alloresponse at different levels and to dampen the activation of cells of both the adaptive and innate immune systems, reprogramming them into regulatory cells. MSC are a heterogeneous subset of non-hematopoietic cells, currently defined by standard criteria that include the ability to differentiate into tissues of mesodermal lineages in vitro, plastic adherence under standard culture conditions, expression of CD73, CD90 and CD105, and lack of CD45, CD34, CD14, or CD11b, CD79α or CD19, and HLA-DR surface antigens (4). Although MSC can be obtained from several adult and fetal tissues (including umbilical cord, Wharton's jelly, amniotic fluid, adipose tissue and dental pulp) (5–9), the bone marrow has been traditionally considered as their main source, and thus bone marrow-derived MSC are the ones best characterized both in humans and animal models.
In this review we will provide a summary of the alloimmune response events that accompany solid organ transplantation, discussing the multiple immune modulatory effects that MSC have demonstrated on both the innate and adaptive arms of the immune system and highlighting their tolerance-inducing properties in preclinical transplant models. We will also provide an overview of available data from clinical trials on MSC infusion in solid organ transplant patients, discussing future perspectives and issues that still need to be addressed.
The crucial step of the adaptive response is represented by the recognition of donor alloantigens (mostly non-self MHC molecules) by recipient lymphocytes. These cells are activated in the presence of costimulatory molecules, leading to a cascade of events that ultimately precipitates into graft rejection (10).
Transplant rejection results in the generation of cells with either regulatory or effector functions, with the latter prevailing during the alloresponse. Effector functions are mainly mediated by CD8+ and CD4+ T cells: cytotoxic CD8+ T cells, activated by class I MHC-presented antigens, undergo clonal expansion, mature into effector cells and migrate into the graft, where they induce apoptosis and secrete cytotoxic molecules.
Class II MHC-restricted CD4+ T lymphocytes differentiate into distinct subsets of helper T cells, depending on the cytokine milieu: IL-12 prompts the differentiation in TH1 cells and promotes the secretion of IL-2 and IFNγ, which, respectively, sustain CD8+ T-cell proliferation and induce a delayed-type hypersensitivity reaction through macrophage activation (11). TH2 polarization is guided by IL-4, which results in the recruitment of graft-damaging eosinophils via IL-5 secretion (12). TH17 induction is mediated by IL-21, IL-23, IL-6, and TGFβ stimulation. TH17 cells play a role in both acute and chronic rejection through the recruitment of neutrophils into the transplanted graft (13). Finally, follicular helper T-cell (TFH) differentiation is primarily mediated by IL-12, IL-6, and TGF-β signaling; TFH cells promote B-cell differentiation into antibody-producing plasma cells (14, 15) and are the cells responsible for the development of donor-specific antibodies and antibody-mediated rejection (16, 17).
CD4+ T cells may also differentiate into several regulatory subsets, which actively inhibit the alloresponse and therefore constitute an essential part of peripheral tolerance. Regulatory T cells (TREG) express the transcription factor FoxP3 and are known to suppress the alloreaction through modulation of antigen presentation, production of anti-inflammatory cytokines, as well as competition and cytolysis of effector T cells (18, 19). T-regulatory type 1 cells (TR1) can be induced from naïve T cells upon TCR stimulation in the presence of IL-10; these cells downregulate the alloresponse by producing anti-inflammatory soluble mediators, including IL-10 and TGFβ (20).
B-cell mediated humoral immune responses also play an important role in alloimmunity, which is reflected by the elevated incidence of chronic antibody-mediated rejection in long-term graft recipients. B-cell differentiation into antibody-producing plasma cells and memory B cells after alloantigen uptake depend on cognate interactions with T cells. Conversely, following uptake through their B-cell receptor, B cells can mount alloantigens on class-II MHC and present them to T cells. Specific B-cell subsets have displayed immunoregulatory properties, and have been termed regulatory B cells (BREG). BREG are a heterogenous population that promotes the development of TREG, suppresses effector T cell differentiation and converts TH1 effectors into TR1 cells, mainly through secretion of the anti-inflammatory cytokines IL-10 and TGFβ (21, 22).
Despite the high specificity of the adaptive system for non-self-antigens, the set of allogeneic responses that ultimately lead to transplant rejection derives from a complex interplay with the innate immune system, an interaction that is much more intertwined than originally hypothesized.
Innate immunity relies on the activation of pattern recognition receptors (PRRs), which is induced by evolutionarily preserved molecular motifs from pathogens. However, also self-molecules released in response to cell stress can activate PRRs, triggering the innate immunity. These molecules are termed DAMPs (damage-associated molecular patterns) and are massively released in the context of organ transplantation in response to brain/cardiac death and ischemia-reperfusion injury (23, 24).
The best characterized cellular PRRs are toll-like receptors (TLRs), localized on the outer membrane and on the surface of intracellular vesicles, NOD-like receptors, which are part of the inflammasome complex, and C-type lectin receptors. DAMP-induced signaling through these receptors activates the transcription of specific genes, leading to the secretion of inflammatory cytokines and upregulation of adhesion and costimulatory molecules (25, 26).
DAMP-induced TLR signaling promotes the maturation of dendritic cells (DCs), which represents the pivotal link between innate and adaptive immunity. This process determines a critical switch in the biological functions of DCs in solid organ transplantation: donor DCs migrate from the graft to the lymphoid organs of the recipient and present intact donor MHC-antigen complexes to host T cells (“direct presentation”), inducing the intense activation and rapid proliferation of effector T cells that underlies early rejection episodes. In addition, mature recipient DCs infiltrating the graft can also present processed donor antigens to recipient T cells (“indirect presentation”), providing a weaker but long-lasting stimulation that can eventually result in both acute and chronic rejection (27, 28). In the setting of acute rejection, most of the DCs found in the graft derive from infiltrating monocytes that locally differentiate into antigen-presenting cells (29, 30).
Monocytes can also contribute to transplant rejection by differentiating into macrophages, which frequently represent the majority of graft-infiltrating cells during rejection (31). Similarly to other cell types, macrophages can damage the allograft or have immunoregulatory functions depending on their state of activation. Classically-activated M1 macrophages are pro-inflammatory cells that develop upon TLR engagement in the presence of IFNγ, damaging the graft via direct cytotoxicity or by inducing a delayed-type hypersensitivity reaction. Notably, these effector functions were shown to be triggered by alloantigens, and cytotoxicity proved to be allospecific after T-helper priming (32, 33). On the other end of the spectrum, alternatively-activated M2 macrophages, whose differentiation is guided by IL-4 and IL-13, produce anti-inflammatory cytokines (IL-10, TGFβ), induce regulatory T-cell differentiation and promote tissue repair, mitigating graft damage (34, 35).
MSC infusion has not only shown encouraging results in controlling autoimmunity in vivo (36), but has also consistently proven effective in prolonging allograft survival in multiple animal models of solid organ transplantation (37–39) (Table 1). Compelling evidence demonstrated that during the alloresponse these cells can tip the balance between effector and regulatory functions in favor of the latter. MSC reduce the host-vs.-graft response in part through contact-dependent regulation (49, 50) and, most importantly, by secreting soluble factors with paracrine immunomodulatory effects (51, 52). A constantly growing number of soluble mediators have been implicated in MSC-induced functions on the immune system, including indoleamine 2,3-dioxygenase (IDO) (53, 54), nitric oxide (55, 56), TGF-β (49, 57–59), prostaglandin-E2 (PGE2) (50, 60), heme-oxygenase 1 (HO-1) (61), galectins (62) and HLA-G5 (63). Indeed, MSC-conditioned medium and MSC-derived extracellular vesicles, which both contain these soluble bioproducts, have been increasingly studied as cell-free alternatives to MSC administration (64). In addition, a different approach has been explored by some researchers, who engineered MSC to overexpress and secrete several different soluble mediators, including IDO (65), TGFβ (66), IL-10 (67), and HO-1 (68–70). Preliminary evidence from animal models of solid organ transplantation showed a modest but consistent increase in the pro-tolerogenic activity of genetically modified MSC compared to their standard counterparts (Table 2). Soluble mediators act on multiple cell targets, resulting in dramatic changes in their phenotype and functions.
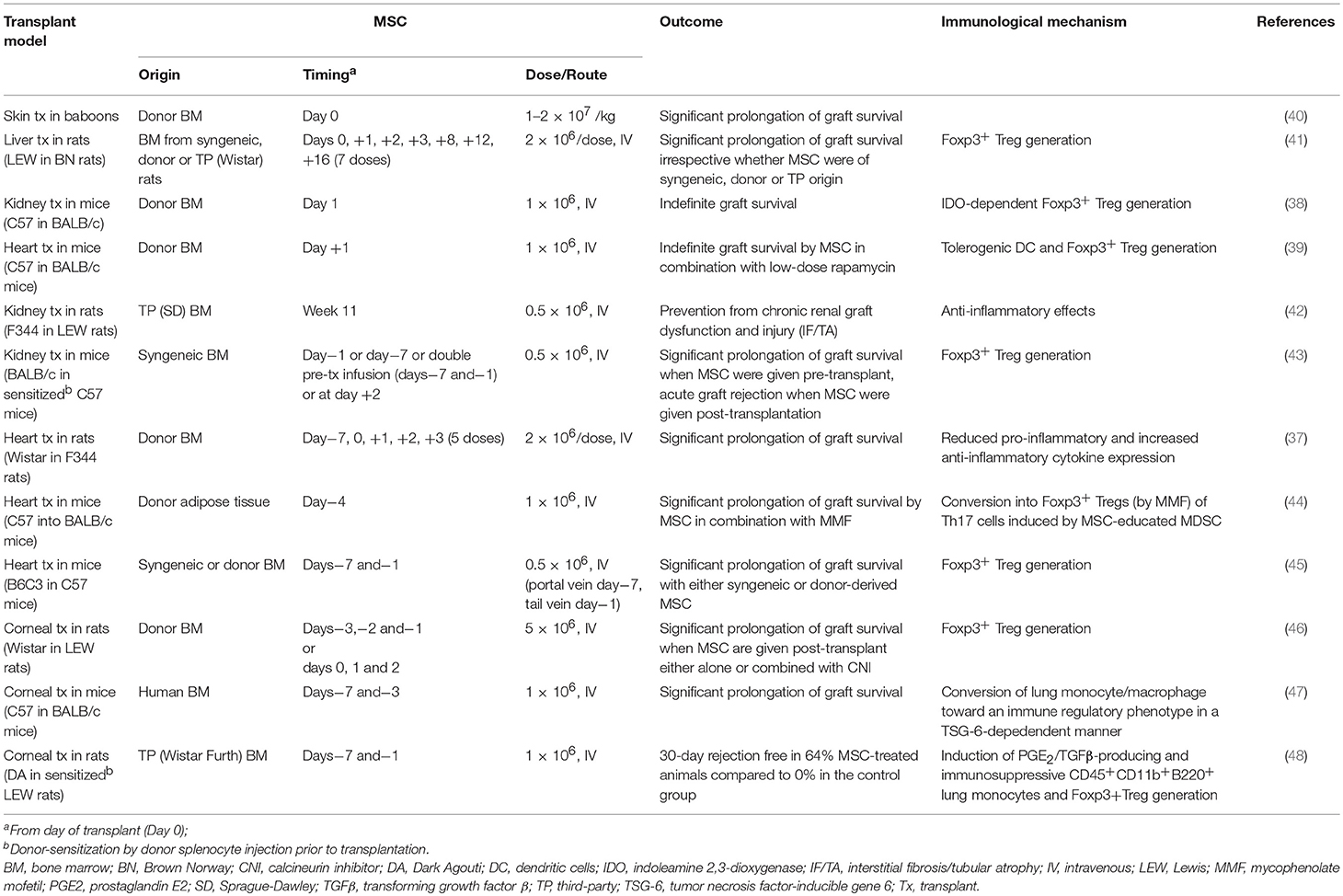
Table 1. MSC in preclinical transplant models.
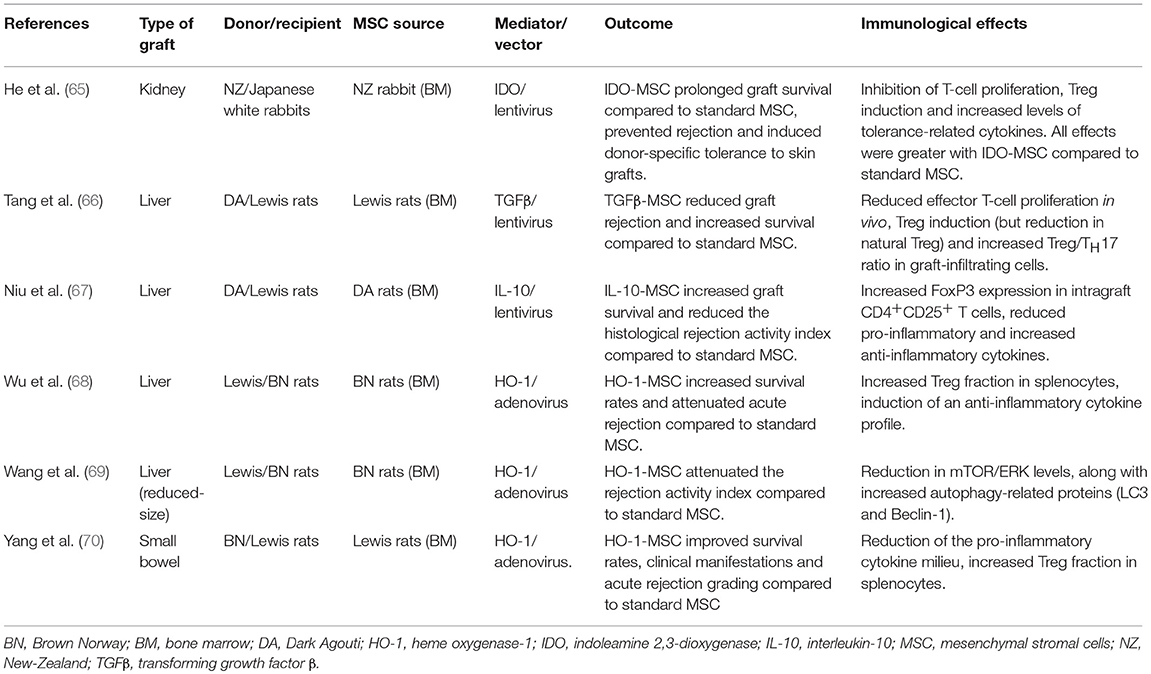
Table 2. Preclinical data on genetically-engineered MSC in solid organ transplantation.
Autologous and allogeneic MSC regulate the T-cell response to the graft via modulation of alloantigen presentation and through direct, antigen-independent effects on T cell. In their seminal work, Bartholomew et al. demonstrated for the first time that MSC can prevent not only T-cell alloreactivity, but also polyclonal activation and proliferation of baboon T cells induced by mitogens in vitro (40). This dose-dependent effect was also confirmed in mice with a series of elegant experiments assessing single-antigen reactivity (71, 72), which showed that MSC inhibition was not limited to antigen-specific T-cell clones, but determined a generalized division arrest anergy in both naïve and memory T cells, eventually resulting in reduced CD8+ T-cell proliferation and cytotoxicity. Similar results were obtained with human autologous and allogeneic MSC, which were able to suppress T-cell proliferation due to both antigen-priming and polyclonal activators (73–75). Findings that MSC are able to inhibit memory T cells, including CD8+ memory T cells, is of particular relevance, since a high frequency of alloreactive memory T cells before transplantation represents a barrier to tolerance induction (76–78) and threatens allograft survival, especially in the context of T-cell depleting induction therapy (79–82).
MSC can also modify the activity of helper T cells by rewiring their polarization. Addition of MSC to T-cell cultures stimulated by mitogens, primed by allogenic T cells, or under TH1-differentiating conditions, suppressed TH1-cell proliferation and activation, and promoted a strong TH2 polarization along with increased IL-10 production (74, 83, 84). The same effect was observed in a rat model of corneal transplantation, in which MSC injection shifted the balance between TH1- and TH2-specific cytokines in favor of the latter (46). Moreover, MSC displayed the capacity to inhibit TH17 generation by suppressing the transcription factor RORγt and to induce suppressive TREG from differentiated TH17 cells, both in vitro and in vivo (44, 50, 85). Although data regarding MSC effect on TFH cells in solid organ transplantation are currently lacking, MSC were shown to suppress the differentiation and proliferation of cultured TFH cells obtained from healthy human donors (86).
Alongside effector T-cell inhibition, MSC expand several subsets of regulatory cells, making them a unique tool to modulate the alloresponse. In murine models, MSC polarized T cells toward a FoxP3+ TREG phenotype both in vitro and in vivo (41, 45, 83), an effect that was evident also in human T-cell cultures (49, 63) and in transplant recipients undergoing MSC infusion (87). Recently, MSC were also reported to promote IL-10 secretion and to expand the TR1 regulatory subset in allogeneic mixed-lymphocyte reactions (MLRs), which mediated immunosuppression in vitro through the PGE2 and IDO pathways (61, 88).
Different reports have shown that MSC may block B-cell proliferation through cell-cycle arrest into the G0/G1 phase (89, 90). This alteration results in a strong inhibition of proliferation and maturation of B cells into plasmablasts, causing a steep reduction in antibody secretion (91, 92). Of particular interest for the transplantation field, the addition of MSC to standard allogeneic MLRs was shown to inhibit the formation of donor-specific antibodies (92). Interestingly, in vitro B-cell proliferation was not suppressed when MSC and B cells were cultured without T cells, suggesting that this effect is at least in part mediated by T-cell help (91). On the other hand, absence of T cells did not interfere with MSC-mediated inhibition of B-cell maturation and with BREG induction (93).
Inflammatory licensing by IFNγ significantly modifies MSC effect on B-cell subsets: MSC cultured in standard conditions were shown to increase the percentage of BREG along with IL-10 production, while MSC exposed to high IFNγ concentrations did not promote BREG expansion, but instead induced a greater inhibition of B-cell proliferation through IDO metabolic effects (94, 95).
Although, to date, there is a lack of studies exploring MSC effect on the B-cell compartment in animal models of solid organ transplantation, MSC infusion prevented the formation of circulating donor-specific antibodies in rats undergoing allogeneic kidney transplantation, suggesting that these cells are able to modulate humoral responses in vivo (42). Moreover, long term immunophenotyping of kidney allograft recipients that received MSC infusions revealed increased naïve and CD24HICD38HI (“transitional”) B-cell subsets, a phenotypic signature that was associated with spontaneous operational tolerance (87, 96). Additional characterization will be needed to clarify whether MSC may have increased the frequency of IL-10 producing B cells (i.e., those associated with BREG phenotype and function), especially in the “transitional” B-cell fraction.
MSC modulate DC phenotype and function at multiple levels, inducing pro-tolerogenic functions. Exposure to mouse MSC interferes with DC maturation, downregulating class II MHC and the costimulatory molecules CD40 and CD86 (97, 98). Moreover, MSC also impair DC homing to secondary lymphoid organs by reducing the expression of CCR7 and CD49dβ1 (99).
These effects have direct consequences on alloantigen presentation by both donor and recipient DCs: since MLR reactivity mostly depends on direct antigen presentation, the inhibition of proliferation observed in these cultures after MSC addition suggests an inhibitory effect on “donor” DCs (97). This was confirmed by the hypo-responsiveness displayed by human allogeneic T-cell responders to MSC-primed DCs compared to mature DCs (100–102). In vitro experiments with ovalbumin-specific TCR-transgenic mouse T cells demonstrated that MSC can also exert regulatory effects on ovalbumin-pulsed “recipient” DCs, leading to defective indirect alloantigen presentation to CD4+ T cells and reduced cross-presentation of allopeptides to CD8+ T cells (98, 99).
DC maturation block and inefficient antigen presentation, coupled with lack of co-stimulation, induces a regulatory phenotype from conventional T cells, thereby increasing the relative abundancy of TREG compared to effector T cells and inhibiting the alloresponse. In an allogeneic kidney transplant model, MSC infusion was associated with high frequency of immature, tolerogenic DCs, along with impaired donor-specific T-cell proliferation and enrichment of suppressive TREG in secondary lymphoid organs and into the graft (38). Similar results had been previously observed in mice receiving a cardiac allograft, in which tolerance was achieved by combining MSC infusion with low-dose rapamycin (39).
Several models have shown that MSC can increase macrophage proliferation and migration, while inducing a pro-tolerogenic polarization shift at the same time: MSC-reprogrammed M2 macrophages show reduced TNFα, IFNγ, and IL-12 secretion and increased IL-10 production, which inhibit effector T-cell responses and promote TREG proliferation (59, 103–105).
Several soluble mediators secreted by MSC have been implicated in macrophage reprogramming, including IDO and PGE2 (105, 106). Recent reports, however, have shown that also phagocytosis of apoptotic MSC or MSC microparticles by macrophages is sufficient to induce an M2 polarization shift, partly through IDO upregulation in these phagocytes (107–109). Moreover, macrophage uptake of the cell debris deriving from MSC-induced T-cell apoptosis also induced TGFβ secretion, which facilitated TREG expansion (110).
In vivo, MSC infusion before allogeneic corneal transplantation redirected macrophages toward an M2 phenotype, which conferred protection against allograft rejection (47). In this model, adoptive transfer and selective depletion experiments suggested that the monocyte lineage was responsible for tolerance induction (47). In pre-sensitized rats, allogeneic MSC administration before corneal transplant significantly increased rejection-free survival, which was associated with an early rise in alternatively-activated macrophages, followed by an increase in TREG at later time points (48).
Although MSC can act as potent immunomodulators of inflammation during the alloresponse, in resting conditions they mainly display homeostatic properties, supporting cells residing in their niche (111). Moreover, MSC can also acquire a completely opposite function, releasing pro-inflammatory cytokines and even acting as antigen-presenting cells following class II MHC upregulation (112–114). The acquisition of either of these specific functions strictly depends on the microenvironment MSC encounter: to become immunosuppressive, MSC need to “licensed” by a series of inflammatory stimuli that commonly occur in the setting of solid organ transplantation, deriving from both the innate and adaptive immune response.
Cytokines are the best studied mediators involved in MSC licensing: binding of IFNγ (chiefly secreted by T-cells after TH1 polarization) to its specific receptors on MSC represents a critical step of this process (115, 116). Other cytokines, including the macrophage-derived TNFα, IL-1α and IL-1β, have been shown to potentiate the licensing effect of IFNγ (55). Overall, this stimulation results in the production of chemokines (such as CXCR3 and CCR5) and in the induction of IDO, which are respectively necessary for T-cell recruitment and inhibition. However, below-threshold IFNγ and TNFα concentrations have been proven insufficient to induce IDO, therefore precluding the suppression of recruited T-cells and paradoxically increasing the inflammatory response (117).
MSC functional destiny is also influenced by the dynamics of DAMP-associated signaling that accompanies tissue injury. The presence of a wide array of TLRs has been confirmed on the surface of both human and mouse MSC (118–120), with variable expression influenced by environmental cues, including hypoxia and inflammatory cytokines (114, 121). TLR stimulation promotes MSC migration to the site of inflammation and enhances their survival (122, 123), but conflicting data have been reported regarding a DAMP-mediated licensing effect through TLR3 and TLR4 stimulation (124, 125). Recent evidence suggests that MSC can be differentially activated based on the type of TLR triggered: transient TLR4 engagement by low lipopolysaccharide concentrations polarizes MSC toward a proinflammatory phenotype, stimulating the secretion of IL-6 and IL-8, recruiting neutrophils and inducing T-cell activation in co-culture experiments (126, 127). On the other hand, sub-maximal triggering of TLR3 guides the differentiation into immunomodulating MSC, which upregulate IDO, secrete IL-10, CXCL10 and CCL5, and induce T-cell suppression in vitro (126).
In addition to its multiple pro-inflammatory effects, activation of the complement cascade also has direct effects on MSC activity. MSC are resistant to complement-mediated lysis due to expression and secretion of negative regulators (128, 129) but, at the same time, ligation of complement anaphylatoxins to their surface receptors enhances MSC resistance to oxidative stress and apoptosis (130). Although a direct licensing effect of complement has not been described, C3a and C5a act as potent chemotactic agents for MSC and recruit them at the site of inflammation, where functional polarization takes place (131, 132).
MSC survival is limited both in culture systems and following in vivo administration (45, 133). This peculiar feature seems antithetic to the development of long-term tolerance after a single MSC infusion, unless one takes into account that these cells may determine a multi-level protolerogenic polarization within a limited time frame. Therefore, the reduced lifespan of MSC raises by itself the issue of optimal infusion timing in allograft recipients. This concern was further corroborated by the first evidence that the timing of MSC infusion dictates their pro-tolerogenic effect: in an allogeneic rat model of heart transplantation, donor MSC infused 4 days before the transplant induced acceptance of the graft, as opposed to infusions on the same day or after 3 days from the procedure (134).
Building upon data indicating that systemically infused MSC tend to migrate to damaged tissues (such as those exposed to ischemia/reperfusion injury) (135–137), we hypothesized that timing of infusion may impact on MSC localization, and confirmed that administration of MSC after transplantation results in migration of these cells inside the graft, rather than toward secondary lymphoid organs (43). In addition, the site at which MSC are recruited tightly correlates with their immunomodulatory properties: indeed, migration of MSC into kidney allografts following post-transplant infusion was associated with a proinflammatory phenotype and resulted in neutrophilic infiltration, complement deposition and acute kidney injury, both in animal models and humans (43, 87). The events leading to this polarization are still ill-defined, but the inflammatory microenvironment and the massive release of DAMPs are likely key factors. On the other hand, administration of MSC before renal transplantation led to localization of these cells into secondary lymphoid organs, where they promoted the formation of a pro-tolerogenic environment and prolonged graft survival (43).
Localization by itself, however, does not fully explain the opposing characteristics that MSC may acquire in the setting of solid organ transplantation. We recently showed that experimental inhibition of C3a and C5a receptors on MSC infused after kidney transplantation prevents intra-graft migration and allows homing of these cells to secondary lymphoid organs. However, despite correct localization and reduction of donor-specific T-cell alloreactivity, MSC failed to induce TREG generation in this model and resulted only in limited prolongation of graft survival (132).
These experiments confirmed previous evidence suggesting that the effect of MSC on alloreactive T cells also depends on the degree of maturity these T cells display. Indeed, when MSC were added to MLRs after the primary stimulation step had already taken place, CD8+ T-cell mediated cytotoxicity was unhindered, suggesting that a complete polarization favors the escape from MSC-mediated immunoregulation (138). Similarly, the in vitro regulatory effect of MSC on TH1/TH17 proliferation, activation and differentiation was progressively lost when MSC were added with increasing delay from the start of the polarization process (83).
Consistent with effects observed in mice, MSC added at later time points in vitro were also unable to convert terminally differentiated conventional T cells into TREG (83, 139). Among mediators involved in TREG induction, the trophic cytokine IL-2 seems to be critical for the whole process to occur (140, 141): since neither MSC nor TREG secrete IL-2, the concentration of this cytokine relies on conventional T-cell production. Studies on the kinetics of IL-2 secretion during effector T-cell differentiation elucidated how terminal effectors progressively lose their ability to secrete this cytokine (142), resulting in a steep decline in IL-2 concentrations after 2 days in a conventional MLR (143). Thus, it is tempting to speculate that MSC may fail to induce TREG from terminally differentiated conventional T cells due to low IL-2 concentrations, but other concomitant factors are likely to play a role. For instance, timing seems also involved in MSC-mediated induction of an immature, tolerogenic DC phenotype: addition of lipopolysaccharide to MSC-DC co-cultures, mimicking the DAMP-induced TLR triggering that accompanies graft transplantation, reduced the inhibitory effect of MSC on DC maturation (144, 145). As previously described, the acquisition of a mature DC phenotype prevents TREG expansion, thus providing an additional plausible explanation of the reduced immunomodulating ability of MSC infused post-transplantation.
Failure to expand TREG has been frequently associated with graft rejection in animal models (38, 43), suggesting a central role for suppression in MSC-induced tolerance. Thus, infusion protocols aimed at expanding the TREG pool should have the highest potential of promoting a protolerogenic environment. Pre-transplant MSC administration has the best chances to achieve this aim, even though TREG expansion in the absence of antigenic pressure from the graft produces a broad repertoire, which includes both donor-specific and non-alloreactive regulatory T cells.
Indeed, antigen-pressure is not necessary to induce suppressive TREG from conventional T cells in MSC co-culture (146). However, FoxP3 expression in these cells is not stable, suggesting that polyclonal TREG induction without alloantigen presentation does not provide the required survival and proliferation stimuli for a stable long-term expansion (146). In vivo administration of MSC to healthy, non-transplanted mice expands the TREG pool (43), and adoptive transfer of splenocytes from these mice to syngeneic transplant recipients induces tolerance to the allograft (45), consistent with the notion that polyclonally-expanded TREG can modulate the alloresponse. However, after graft tolerance has been established, TREG-mediated suppression becomes donor-specific, without affecting the response to third-party alloantigen presentation in vivo (45). These findings suggest that MSC infused before the transplant and localizing to secondary lymphoid organs induce a polyclonal, antigen-independent expansion of TREG. Antigenic pressure from the graft then provides the required survival and expansion stimuli to donor-specific TREG, while non-alloreactive TREG clones are progressively lost over time (Figure 1).
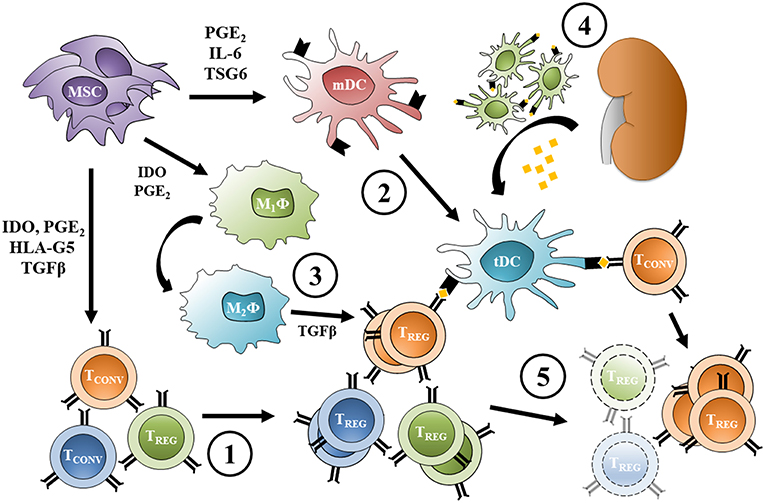
Figure 1. Expansion of donor-specific TREG following pre-transplant MSC infusion. (1) MSC infused before transplant induce TREG from conventional T cells and promote a polyclonal, antigen-independent expansion; at the same time, (2) MSC inhibit dendritic cell maturation increasing the frequency of protolerogenic DCs, and (3) reprogram macrophages toward an M2 phenotype. (4) Allogeneic transplantation causes migration of donor DCs from the graft to lymphoid organs and provides alloantigen for recipient DC uptake (other antigen presenting cells can act the same way). (5) Direct and indirect antigen presentation provide the survival stimuli necessary for donor-specific TREG proliferation, while the non-alloreactive TREG pool is progressively lost over time. Protolerogenic DCs can also induce TREG from conventional alloreactive T cells, while alternatively-activated macrophage TGFβ secretion promotes TREG expansion. MSC, mesenchymal stromal cell; TCONV, conventional T cell; TREG, regulatory T cell; mDC, mature dendritic cell; tDC, tolerogenic dendritic cell; M1ϕ, classically-activated macrophage; M2ϕ, alternatively-activated macrophage; PGE2, prostaglandin-E2; IDO, indoleamine 2,3-dioxygenase; TGFβ, transforming growth factor β; HLA-G5, human leukocyte antigen G5; IL-6, interleukin 6; TSG6, tumor necrosis factor-inducible gene 6 protein.
Cellular therapy with MSC has been tested so far in phase 1 clinical studies in kidney, liver and lung transplantation (Table 3) using either autologous bone marrow- (BM-)derived (87, 147–151), allogeneic BM-derived (152, 155–158) or umbilical cord-derived MSC (154). These studies have indicated the safety, feasibility and tolerability of the procedure in all settings, including the intravenous injection of MSC in lung transplant recipients with moderate obstructive chronic lung allograft rejection (158). All MSC-treated patients showed good graft function over the 1–2 years-follow-up.
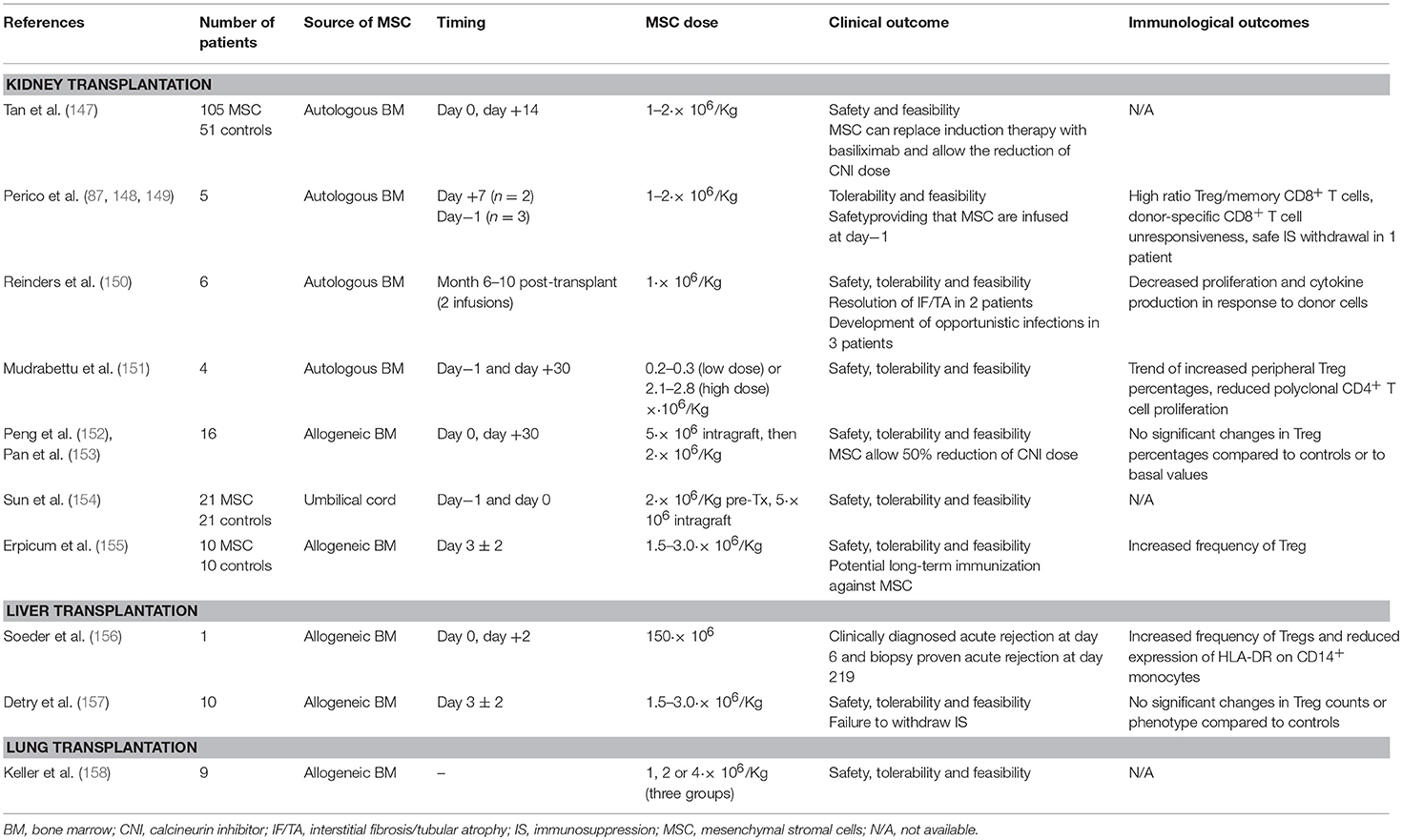
Table 3. Summary of clinical trials assessing MSC infusion in the setting of renal, liver and pulmonary transplantation.
Even though an initial study in kidney transplantation reported a high incidence of opportunistic infections in MSC-treated kidney transplant recipients (150), subsequent studies allayed this concern. In kidney and liver transplant recipients MSC infusion was associated with a reduced (147) or unchanged (87, 155) incidence of opportunistic infections in the first 1–2 years post-transplant compared to controls. In our kidney transplant patients given autologous BM-derived MSC we did not observe increased susceptibility to infection or neoplasm even in the long term (5–7-years follow-up) (87).
The hypothetical increased risk of malignancy following MSC administration constitutes an additional matter of debate (159). This concern originally stemmed from the observation that murine MSC may undergo spontaneous malignant transformation during ex vivo cultures (160). However, this feature of murine MSC is not shared by their human counterparts, which are not prone to maldifferentiation even after long-term in vitro expansion (159, 161). Despite the lack of malignant potential, human MSC may still theoretically promote the growth of pre-existing undiagnosed tumors in transplant recipients. Preclinical evidence in this context is not univocal, with MSC effects varying from neoplastic facilitation to anti-tumoral activity (159, 162–164). Even though studies carried out so far did not detect any association between MSC infusion and malignancy (165), long-term surveillance systems need to be established in order to gauge a precise estimate of adverse events related to MSC administration.
A major concern regarding the use of third-party MSC therapy refers to possible recipient sensitization due to development of antibodies against allogeneic MSC, a phenomenon demonstrated in animal models (166, 167). This fundamental issue, however, has been considered only by one recent study in kidney transplantation (155): ten kidney transplant recipients from deceased donor were given third-party BM-derived MSC on day 3 ± 2 post-transplant. During the 12 months-follow up, four MSC-treated patients developed de novo antibodies against MSC or shared kidney donor-MSC HLA, albeit at very low levels and with only one patient showing de novo MSC-donor-specific antibodies with MFI > 1,500. The clinical significance of these alloantibodies is uncertain and further studies with longer follow-up are needed to definitely address the issue of possible sensitization after allogeneic MSC injection.
The available studies in kidney transplantation also provide some evidence of potential efficacy of the MSC therapy in enabling minimization of induction (147) and maintenance (152, 153) immunosuppressive drugs, in inducing a pro-tolerogenic environment (87, 148, 149) and in repairing chronic allograft damage (150).
In a large clinical trial conducted in China, autologous BM-MSC infusions (at reperfusion and 2 weeks later, 1–2 × 106/Kg each) could safely replace induction therapy with the anti-IL-2R antibody basiliximab and enable 20% CNI dose reduction in living donor kidney transplantation (147). In a subsequent study from China, allogeneic BM-derived MSC allowed a 50% reduction of tacrolimus dose used as maintenance therapy. Patients given MSC showed graft survival, incidence of acute rejection and graft function similar to the control patients receiving a full tacrolimus dose (153).
We are currently conducting clinical studies of MSC therapy in living (NCT00752479 and NCT02012153) (87, 148, 149) and deceased donor kidney (NCT 02565459) and liver (NCT02260375) transplantation. In our experience with autologous BM-derived MSC infusion in living donor kidney transplant recipients we showed that timing of cell infusion was crucial in establishing the eventual effect of MSC. MSC infusion at day 7 after transplantation on top of induction therapy with low dose thymoglobulin and basiliximab, resulted in transient renal insufficiency in the first two patients (148), likely caused by MSC migration toward the inflamed graft with the consequent activation of their pro-inflammatory phenotype. We therefore modified the protocol and the subsequent two patients were given infusion of autologous MSC the day before kidney transplantation (149). In this setting, we removed basiliximab from the induction therapy to avoid any deleterious effect of the anti-IL-2R antibody on TREG possibly expanded by MSC. MSC infusion was no longer associated with acute renal insufficiency (149) but the basiliximab-free regimen exposed patients to an increased risk of acute rejection. The protocol was therefore further modified by re-introducing basiliximab induction therapy in the setting of pre-transplant (day−1) MSC infusion. Two additional patients have been treated so far with this protocol and results of 1 year follow up of the first patient have been recently published (87). The first four patients are now at 5–7-years follow-up with stable graft function and no major side effect (87). In these patients we performed extensive longitudinal immunological studies and were able to document a long lasting increase in the ratio between TREG and memory CD8+ T cells, along with a persistent reduction of donor-specific CD8+ T-cell cytotoxicity ex-vivo. These parameters were particularly evident in two patients and were associated with high levels of circulating naïve and transitional B cells, parameters proposed as the B-cell signature of spontaneous and induced tolerance to kidney transplantation (96). In one of these two patients, who was also showing normal histology at 1-year protocol biopsy and no evidence of de novo donor-specific antibodies, we attempted immunosuppressive drug withdrawal. Cyclosporin was successfully withdrawn, mycophenolate mofetil was gradually tapered, and the patient is currently free from immunosuppression (Perico et al. manuscript submitted).
Analogously to our approach, the group of Detry et al. attempted immunosuppression withdrawal in liver transplant patients given allogeneic MSC infusion, despite the lack of laboratory evidence of a pro-tolerogenic environment (unchanged percentages of total TREG or their naïve, resting, activated and proliferating subsets) in MSC-treated patients compared to controls. Immunosuppression withdrawal, attempted in nine MSC recipients, was unsuccessful and immunosuppressive drug discontinuation was not achieved (157). Ineffective MSC-mediated immunomodulation in these patients could be due, at least in part, to the absence of a peri-transplant T-cell depleting induction therapy, which would have likely enabled MSC to further expand TREG while restraining memory T cells at the same time.
Overall, the available studies indicate the safety and feasibility of MSC infusion of both autologous and allogeneic source in solid organ transplant recipients. Some suggestions of possible therapeutic efficacy derived from the evidence that MSC allowed minimization of immunosuppression and could promote a TREG-mediated pro-tolerogenic environment.
Many open issues such as source, number of infusions, extent of in vitro expansion and concomitant immunosuppression regimens still need to be addressed before MSC can become a standard treatment protocol for transplant recipients. In particular, large variations in the dose of MSC infusions are a common occurrence in clinical trials, since the choice is empirical. No direct comparison of MSC doses has been performed in solid organ transplantation so far, but a clinical trial on graft-vs.-host disease showed that amounts ranging from 0.4 to 9.0 × ·106/Kg were not only safe and well tolerated, but also had a similar immunomodulatory effect (168). On the other hand, lack of clinical trial uniformity regarding timing of MSC infusion likely reflects organizational issues rather than insufficient scientific evidence. In light of preclinical and clinical data available, MSC infusion should be always programmed before the transplantation procedure in kidney allograft recipients, in order to avoid potential graft damage resulting from infiltrating proinflammatory MSC. Similarly, the lack of tolerance induction in liver transplant recipients suggests that post-transplant MSC infusion does not efficiently modulate the alloresponse, an effect that might be obtained instead by a pre-transplant administration schedule.
Another fundamental question remains to be addressed, i.e., the final aim of using MSC cellular therapy in solid organ transplantation. We think that MSC should not be adopted to prevent acute allograft rejection, since this phenomenon is well controlled by conventional, less expensive and effective immunosuppressive drugs. Instead, MSC should be harnessed for complementing the tolerogenic potential of induction therapies, which should still be used for the prevention of acute graft rejection, allowing MSC to create a pro-tolerogenic environment at later stages. To this purpose, future efforts should focus on a deeper understanding of mechanism of action of MSC and their interplay with immune cell subsets, with the aim to identify biomarkers of response to MSC therapy that may allow to select patients amenable to safe immunosuppressive drug withdrawal.
MP and FC searched the literature and wrote the manuscript. GR critically revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the European Union's Horizon 2020 research and innovation programme under grant agreement No 634086. The materials presented and views expressed here are the responsibility of the authors only. The EU commission takes no responsibility for any use made of the information set out.
APC, antigen presenting cell; BREG, regulatory B cell; DAMP, damage-associated molecular pattern; DC, dendritic cell; HO-1, heme-oxygenase 1; IDO, indoleamine 2,3-dioxygenase; IFNγ, interferon gamma; MLR, mixed-lymphocyte reaction; MSC, mesenchymal stromal cell; NK, natural killer; PGE2, prostaglandin-E2; PRR, pattern recognition receptor; TFH, follicular helper T-cell; TFR, T-follicular regulatory cell; TGFβ, transforming growth factor β; TH, helper T cell; TLR, toll-like receptor; TNFα, tumor necrosis factor α; TR1, T-regulatory type 1 cell; TREG, regulatory T cell.
1. Lodhi SA, Lamb KE, Meier-Kriesche HU. Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant. (2011) 11:1226–35. doi: 10.1111/j.1600-6143.2011.03539.x
2. Kim SI. Bacterial infection after liver transplantation. World J Gastroenterol. (2014) 20:6211–20. doi: 10.3748/wjg.v20.i20.6211
3. Rama I, Grinyó JM. Malignancy after renal transplantation: the role of immunosuppression. Nat Rev Nephrol. (2010) 6:511–9. doi: 10.1038/nrneph.2010.102
4. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. (2006) 8:315–7. doi: 10.1080/14653240600855905
5. Sarugaser R, Lickorish D, Baksh D, Hosseini MM, Davies JE. Human umbilical cord perivascular (HUCPV) cells: a source of mesenchymal progenitors. Stem Cells. (2005) 23:220–9. doi: 10.1634/stemcells.2004-0166
6. Batsali AK, Kastrinaki M-C, Papadaki HA, Pontikoglou C. Mesenchymal stem cells derived from Wharton's Jelly of the umbilical cord: biological properties and emerging clinical applications. Curr Stem Cell Res Ther. (2013) 8:144–55. doi: 10.2174/1574888X11308020005
7. In't Anker PS, Scherjon SA, Kleijburg-van der Keur C, Noort WA, Claas FHJ, Willemze R, et al. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood. (2003) 102:1548–9. doi: 10.1182/blood-2003-04-1291
8. Orbay H, Tobita M, Mizuno H. Mesenchymal stem cells isolated from adipose and other tissues: basic biological properties and clinical applications. Stem Cells Int. (2012) 2012:461718. doi: 10.1155/2012/461718
9. La Noce M, Paino F, Spina A, Naddeo P, Montella R, Desiderio V, et al. Dental pulp stem cells: state of the art and suggestions for a true translation of research into therapy. J Dent. (2014) 42:761–8. doi: 10.1016/j.jdent.2014.02.018
10. Wood KJ, Goto R. Mechanisms of rejection: current perspectives. Transplantation. (2012) 93:1–10. doi: 10.1097/TP.0b013e31823cab44
11. Jiang S, Herrera O, Lechler RI. New spectrum of allorecognition pathways: implications for graft rejection and transplantation tolerance. Curr Opin Immunol. (2004) 16:550–7. doi: 10.1016/j.coi.2004.07.011
12. Goldman M, Le Moine A, Braun M, Flamand V, Abramowicz D. A role for eosinophils in transplant rejection. Trends Immunol. (2001) 22:247–51. doi: 10.1016/S1471-4906(01)01893-2
13. Liu Z, Fan H, Jiang S. CD4+ T-cell subsets in transplantation. Immunol Rev. (2013) 252:183–91. doi: 10.1111/imr.12038
14. Yan L, de Leur K, Hendriks RW, van der Laan LJW, Shi Y, Wang L, et al. T Follicular Helper Cells as a new target for immunosuppressive therapies. Front Immunol. (2017) 8:1510. doi: 10.3389/fimmu.2017.01510
15. Chen M, Lin X, Cheukfai L, Olsen N, He X, Zheng SG. Advances in T follicular helper and T follicular regulatory cells in transplantation immunity. Transplant Rev (Orlando). (2018) 32:187–93. doi: 10.1016/j.trre.2018.07.002
16. Walters GD, Vinuesa CG. T Follicular Helper Cells in transplantation. Transplantation. (2016) 100:1650–5. doi: 10.1097/TP.0000000000001217
17. Chenouard A, Chesneau M, Bui Nguyen L, Le Bot S, Cadoux M, Dugast E, et al. Renal operational tolerance is associated with a defect of blood Tfh cells that exhibit impaired B cell help. Am J Transplant. (2017) 17:1490–501. doi: 10.1111/ajt.14142
18. Caridade M, Graca L, Ribeiro RM. Mechanisms underlying CD4+ Treg immune regulation in the adult: from experiments to models. Front Immunol. (2013) 4:378. doi: 10.3389/fimmu.2013.00378
19. Waldmann H, Hilbrands R, Howie D, Cobbold S. Harnessing FOXP3+ regulatory T cells for transplantation tolerance. J Clin Invest. (2014) 124:1439–45. doi: 10.1172/JCI67226
20. Battaglia M, Gregori S, Bacchetta R, Roncarolo M-G. Tr1 cells: from discovery to their clinical application. Semin Immunol. (2006) 18:120–7. doi: 10.1016/j.smim.2006.01.007
21. Wortel CM, Heidt S. Regulatory B cells: phenotype, function and role in transplantation. Transpl Immunol. (2017) 41:1–9. doi: 10.1016/j.trim.2017.02.004
22. Najar M, Raicevic G, Fayyad-Kazan H, Bron D, Toungouz M, Lagneaux L. Mesenchymal stromal cells and immunomodulation: a gathering of regulatory immune cells. Cytotherapy. (2016) 18:160–71. doi: 10.1016/j.jcyt.2015.10.011
23. Fung A, Zhao H, Yang B, Lian Q, Ma D. Ischaemic and inflammatory injury in renal graft from brain death donation: an update review. J Anesth. (2016) 30:307–16. doi: 10.1007/s00540-015-2120-y
24. van Werkhoven MB, Damman J, van Dijk MCRF, Daha MR, de Jong IJ, Leliveld A, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant. (2013) 13:875–82. doi: 10.1111/ajt.12130
25. Todd JL, Palmer SM. Danger signals in regulating the immune response to solid organ transplantation. J Clin Invest. (2017) 127:2464–72. doi: 10.1172/JCI90594
26. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol. (2009) 21:242–53. doi: 10.1016/j.smim.2009.06.005
27. Hubo M, Trinschek B, Kryczanowsky F, Tüttenberg A, Steinbrink K, Jonuleit H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front Immunol. (2013) 4:82. doi: 10.3389/fimmu.2013.00082
28. Sagoo P, Lombardi G, Lechler RI. Relevance of regulatory T cell promotion of donor-specific tolerance in solid organ transplantation. Front Immunol. (2012) 3:184. doi: 10.3389/fimmu.2012.00184
29. Zuidwijk K, de Fijter JW, Mallat MJK, Eikmans M, van Groningen MC, Goemaere NN, et al. Increased influx of myeloid dendritic cells during acute rejection is associated with interstitial fibrosis and tubular atrophy and predicts poor outcome. Kidney Int. (2012) 81:64–75. doi: 10.1038/ki.2011.289
30. Zhuang Q, Liu Q, Divito SJ, Zeng Q, Yatim KM, Hughes AD, et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat Commun. (2016) 7:12623. doi: 10.1038/ncomms12623
31. Hancock WW, Thomson NM, Atkins RC. Composition of interstitial cellular infiltrate identified by monoclonal antibodies in renal biopsies of rejecting human renal allografts. Transplantation. (1983) 35:458–63. doi: 10.1097/00007890-198305000-00013
32. Liu W, Xiao X, Demirci G, Madsen J, Li XC. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol. (2012) 188:2703–11. doi: 10.4049/jimmunol.1102997
33. Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, Lakkis FG. An innate response to allogeneic nonself mediated by monocytes. J Immunol. (2009) 183:7810–6. doi: 10.4049/jimmunol.0902194
34. Salehi S, Reed E. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transpl. (2015) 20:446–53. doi: 10.1097/MOT.0000000000000209
35. Schmidt A, Zhang X-M, Joshi RN, Iqbal S, Wahlund C, Gabrielsson S, et al. Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-β. Immunol Cell Biol. (2016) 94:747–62. doi: 10.1038/icb.2016.34
36. Pistoia V, Raffaghello L. Mesenchymal stromal cells and autoimmunity. Int Immunol. (2017) 29:49–58. doi: 10.1093/intimm/dxx008
37. Zhou HP, Yi DH, Yu SQ, Sun GC, Cui Q, Zhu HL, et al. Administration of donor-derived mesenchymal stem cells can prolong the survival of rat cardiac allograft. Transplant Proc. (2006) 38:3046–51. doi: 10.1016/j.transproceed.2006.10.002
38. Ge W, Jiang J, Arp J, Liu W, Garcia B, Wang H. Regulatory T-cell generation and kidney allograft tolerance induced by mesenchymal stem cells associated with indoleamine 2,3-dioxygenase expression. Transplantation. (2010) 90:1312–20. doi: 10.1097/TP.0b013e3181fed001
39. Ge W, Jiang J, Baroja ML, Arp J, Zassoko R, Liu W, et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am J Transplant. (2009) 9:1760–72. doi: 10.1111/j.1600-6143.2009.02721.x
40. Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. (2002) 30:42–8. doi: 10.1016/S0301-472X(01)00769-X
41. Wang Y, Zhang A, Ye Z, Xie H, Zheng S. Bone marrow-derived mesenchymal stem cells inhibit acute rejection of rat liver allografts in association with regulatory T-cell expansion. Transplant Proc. (2009) 41:4352–6. doi: 10.1016/j.transproceed.2009.08.072
42. Franquesa M, Herrero E, Torras J, Ripoll E, Flaquer M, Gomà M, et al. Mesenchymal stem cell therapy prevents interstitial fibrosis and tubular atrophy in a rat kidney allograft model. Stem Cells Dev. (2012) 21:3125–35. doi: 10.1089/scd.2012.0096
43. Casiraghi F, Azzollini N, Todeschini M, Cavinato RA, Cassis P, Solini S, et al. Localization of mesenchymal stromal cells dictates their immune or proinflammatory effects in kidney transplantation. Am J Transplant. (2012) 12:2373–83. doi: 10.1111/j.1600-6143.2012.04115.x
44. Obermajer N, Popp FC, Soeder Y, Haarer J, Geissler EK, Schlitt HJ, et al. Conversion of Th17 into IL-17Aneg regulatory T cells: a novel mechanism in prolonged allograft survival promoted by mesenchymal stem cell-supported minimized immunosuppressive therapy. J Immunol. (2014) 193:4988–99. doi: 10.4049/jimmunol.1401776
45. Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, Cugini D, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. (2008) 181:3933–46. doi: 10.4049/jimmunol.181.6.3933
46. Jia Z, Jiao C, Zhao S, Li X, Ren X, Zhang L, et al. Immunomodulatory effects of mesenchymal stem cells in a rat corneal allograft rejection model. Exp Eye Res. (2012) 102:44–9. doi: 10.1016/j.exer.2012.06.008
47. Ko JH, Lee HJ, Jeong HJ, Kim MK, Wee WR, Yoon S-O, et al. Mesenchymal stem/stromal cells precondition lung monocytes/macrophages to produce tolerance against allo- and autoimmunity in the eye. Proc Natl Acad Sci USA. (2016) 113:158–63. doi: 10.1073/pnas.1522905113
48. Lohan P, Murphy N, Treacy O, Lynch K, Morcos M, Chen B, et al. Third-party allogeneic mesenchymal stromal cells prevent rejection in a pre-sensitized high-risk model of corneal transplantation. Front Immunol. (2018) 9:2666. doi: 10.3389/fimmu.2018.02666
49. English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, Mahon BP. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin Exp Immunol. (2009) 156:149–60. doi: 10.1111/j.1365-2249.2009.03874.x
50. Duffy MM, Pindjakova J, Hanley SA, McCarthy C, Weidhofer GA, Sweeney EM, et al. Mesenchymal stem cell inhibition of T-helper 17 cell- differentiation is triggered by cell-cell contact and mediated by prostaglandin E2 via the EP4 receptor. Eur J Immunol. (2011) 41:2840–51. doi: 10.1002/eji.201141499
51. Gao F, Chiu SM, Motan DAL, Zhang Z, Chen L, Ji H-L, et al. Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. (2016) 7:e2062. doi: 10.1038/cddis.2015.327
52. Burr SP, Dazzi F, Garden OA. Mesenchymal stromal cells and regulatory T cells: the Yin and Yang of peripheral tolerance? Immunol Cell Biol. (2013) 91:12–8. doi: 10.1038/icb.2012.60
53. Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. (2004) 103:4619–21. doi: 10.1182/blood-2003-11-3909
54. Reading JL, Yang JHM, Sabbah S, Skowera A, Knight RR, Pinxteren J, et al. Clinical-grade multipotent adult progenitor cells durably control pathogenic T cell responses in human models of transplantation and autoimmunity. J Immunol. (2013) 190:4542–52. doi: 10.4049/jimmunol.1202710
55. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
56. Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. (2007) 109:228–34. doi: 10.1182/blood-2006-02-002246
57. Nemeth K, Keane-Myers A, Brown JM, Metcalfe DD, Gorham JD, Gorham JD, et al. Bone marrow stromal cells use TGF-beta to suppress allergic responses in a mouse model of ragweed-induced asthma. Proc Natl Acad Sci USA. (2010) 107:5652–7. doi: 10.1073/pnas.0910720107
58. Zhao W, Wang Y, Wang D, Sun B, Wang G, Wang J, et al. TGF-beta expression by allogeneic bone marrow stromal cells ameliorates diabetes in NOD mice through modulating the distribution of CD4+ T cell subsets. Cell Immunol. (2008) 253:23–30. doi: 10.1016/j.cellimm.2008.06.009
59. Melief SM, Schrama E, Brugman MH, Tiemessen MM, Hoogduijn MJ, Fibbe WE, et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. (2013) 31:1980–91. doi: 10.1002/stem.1432
60. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. (2005) 105:1815–22. doi: 10.1182/blood-2004-04-1559
61. Mougiakakos D, Jitschin R, Johansson CC, Okita R, Kiessling R, Blanc KL. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood. (2011) 117:4826–35. doi: 10.1182/blood-2010-12-324038
62. Gieseke F, Böhringer J, Bussolari R, Dominici M, Handgretinger R, Müller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood. (2010) 116:3770–9. doi: 10.1182/blood-2010-02-270777
63. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte Antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells. (2008) 26:212–22. doi: 10.1634/stemcells.2007-0554
64. Ferreira JR, Teixeira GQ, Santos SG, Barbosa MA, Almeida-Porada G, Gonçalves RM. Mesenchymal stromal cell secretome: influencing therapeutic potential by cellular pre-conditioning. Front Immunol. (2018) 9:2837. doi: 10.3389/fimmu.2018.02837
65. He Y, Zhou S, Liu H, Shen B, Zhao H, Peng K, et al. Indoleamine 2, 3-Dioxgenase transfected mesenchymal stem cells induce kidney allograft tolerance by increasing the production and function of regulatory T cells. Transplantation. (2015) 99:1829–38. doi: 10.1097/TP.0000000000000856
66. Tang J, Yang R, Lv L, Yao A, Pu L, Yin A, et al. Transforming growth factor-β-expressing mesenchymal stem cells induce local tolerance in a rat liver transplantation model of acute rejection. Stem Cells. (2016) 34:2681–92. doi: 10.1002/stem.2437
67. Niu J, Yue W, Song Y, Zhang Y, Qi X, Wang Z, et al. Prevention of acute liver allograft rejection by IL-10-engineered mesenchymal stem cells. Clin Exp Immunol. (2014) 176:473–84. doi: 10.1111/cei.12283
68. Wu B, Song H-L, Yang Y, Yin M-L, Zhang B-Y, Cao Y, et al. Improvement of liver transplantation outcome by Heme Oxygenase-1-transduced bone marrow mesenchymal stem cells in rats. Stem Cells Int. (2016) 2016:9235073. doi: 10.1155/2016/9235073
69. Wang R, Shen Z, Yang L, Yin M, Zheng W, Wu B, et al. Protective effects of heme oxygenase-1-transduced bone marrow-derived mesenchymal stem cells on reduced-size liver transplantation: role of autophagy regulated by the ERK/mTOR signaling pathway. Int J Mol Med. (2017) 40:1537–48. doi: 10.3892/ijmm.2017.3121
70. Yang Y, Song HL, Zhang W, Wu BJ, Fu NN, Dong C, et al. Heme oxygenase-1-transduced bone marrow mesenchymal stem cells in reducing acute rejection and improving small bowel transplantation outcomes in rats. Stem Cell Res Ther. (2016) 7:164. doi: 10.1186/s13287-016-0427-8
71. Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. (2003) 101:3722–9. doi: 10.1182/blood-2002-07-2104
72. Glennie S, Soeiro I, Dyson PJ, Lam EW-F, Dazzi F. Bone marrow Mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. (2005) 105:2821–7. doi: 10.1182/blood-2004-09-3696
73. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. (2002) 99:3838–43. doi: 10.1182/blood.V99.10.3838
74. Mareschi K, Castiglia S, Sanavio F, Rustichelli D, Muraro M, Defedele D, et al. Immunoregulatory effects on T lymphocytes by human mesenchymal stromal cells isolated from bone marrow, amniotic fluid, and placenta. Exp Hematol. (2016) 44:138–50 e1. doi: 10.1016/j.exphem.2015.10.009
75. Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. (2003) 75:389–97. doi: 10.1097/01.TP.0000045055.63901.A9
76. Nadazdin O, Boskovic S, Murakami T, Tocco G, Smith R-N, Colvin RB, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med. (2011) 3:86ra51. doi: 10.1126/scitranslmed.3002093
77. Bestard O, Cruzado JM, Lucia M, Crespo E, Casis L, Sawitzki B, et al. Prospective assessment of antidonor cellular alloreactivity is a tool for guidance of immunosuppression in kidney transplantation. Kidney Int. (2013) 84:1226–36. doi: 10.1038/ki.2013.236
78. Heeger PS, Greenspan NS, Kuhlenschmidt S, Dejelo C, Hricik DE, Schulak JA, et al. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol. (1999) 163:2267–75.
79. Rosenblum JM, Kirk AD. Recollective homeostasis and the immune consequences of peritransplant depletional induction therapy. Immunol Rev. (2014) 258:167–82. doi: 10.1111/imr.12155
80. Moxham VF, Karegli J, Phillips RE, Brown KL, Tapmeier TT, Hangartner R, et al. Homeostatic proliferation of lymphocytes results in augmented memory-like function and accelerated allograft rejection. J Immunol. (2008) 180:3910–8. doi: 10.4049/jimmunol.180.6.3910
81. Ayasoufi K, Yu H, Fan R, Wang X, Williams J, Valujskikh A. Pretransplant antithymocyte globulin has increased efficacy in controlling donor-reactive memory T cells in mice. Am J Transplant. (2013) 13:589–99. doi: 10.1111/ajt.12068
82. Neujahr DC, Chen C, Huang X, Markmann JF, Cobbold S, Waldmann H, et al. Accelerated memory cell homeostasis during T cell depletion and approaches to overcome it. J Immunol. (2006) 176:4632–9. doi: 10.4049/jimmunol.176.8.4632
83. Luz-Crawford P, Kurte M, Bravo-Alegría J, Contreras R, Nova-Lamperti E, Tejedor G, et al. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther. (2013) 4:65. doi: 10.1186/scrt216
84. Aksu AE, Horibe E, Sacks J, Ikeguchi R, Breitinger J, Scozio M, et al. Co-infusion of donor bone marrow with host mesenchymal stem cells treats GVHD and promotes vascularized skin allograft survival in rats. Clin Immunol. (2008) 127:348–58. doi: 10.1016/j.clim.2008.02.003
85. Ghannam S, Pène J, Moquet-Torcy G, Torcy-Moquet G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol. (2010) 185:302–12. doi: 10.4049/jimmunol.0902007
86. Liu R, Li X, Zhang Z, Zhou M, Sun Y, Su D, et al. Allogeneic mesenchymal stem cells inhibited T follicular helper cell generation in rheumatoid arthritis. Sci Rep. (2015) 5:12777. doi: 10.1038/srep12777
87. Perico N, Casiraghi F, Todeschini M, Cortinovis M, Gotti E, Portalupi V, et al. Long-term clinical and immunological profile of kidney transplant patients given mesenchymal stromal cell immunotherapy. Front Immunol. (2018) 9:1359. doi: 10.3389/fimmu.2018.01359
88. Hsu W-T, Lin C-H, Chiang B-L, Jui H-Y, Wu KK-Y, Lee C-M. Prostaglandin E2 potentiates mesenchymal stem cell-induced IL-10+IFN-γ+CD4+ regulatory T cells to control transplant arteriosclerosis. J Immunol. (2013) 190:2372–80. doi: 10.4049/jimmunol.1202996
89. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. (2006) 107:367–72. doi: 10.1182/blood-2005-07-2657
90. Tabera S, Pérez-Simón JA, Díez-Campelo M, Sánchez-Abarca LI, Blanco B, López A, et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica. (2008) 93:1301–9. doi: 10.3324/haematol.12857
91. Rosado MM, Bernardo ME, Scarsella M, Conforti A, Giorda E, Biagini S, et al. Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev. (2015) 24:93–103. doi: 10.1089/scd.2014.0155
92. Comoli P, Ginevri F, Maccario R, Avanzini MA, Marconi M, Groff A, et al. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol Dial Transplant. (2008) 23:1196–202. doi: 10.1093/ndt/gfm740
93. Franquesa M, Mensah FK, Huizinga R, Strini T, Boon L, Lombardo E, et al. Human adipose tissue-derived mesenchymal stem cells abrogate plasmablast formation and induce regulatory B cells independently of T helper cells. Stem Cells. (2015) 33:880–91. doi: 10.1002/stem.1881
94. Luk F, Carreras-Planella L, Korevaar SS, de Witte SFH, Borràs FE, Betjes MGH, et al. Inflammatory conditions dictate the effect of mesenchymal stem or stromal cells on B cell function. Front Immunol. (2017) 8:1042. doi: 10.3389/fimmu.2017.01042
95. Hermankova B, Zajicova A, Javorkova E, Chudickova M, Trosan P, Hajkova M, et al. Suppression of IL-10 production by activated B cells via a cell contact-dependent cyclooxygenase-2 pathway upregulated in IFN-γ-treated mesenchymal stem cells. Immunobiology. (2016) 221:129–36. doi: 10.1016/j.imbio.2015.09.017
96. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest. (2010) 120:1836–47. doi: 10.1172/JCI39933
97. Djouad F, Charbonnier L-M, Bouffi C, Louis-Plence P, Bony C, Apparailly F, et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. (2007) 25:2025–32. doi: 10.1634/stemcells.2006-0548
98. English K, Barry FP, Mahon BP. Murine mesenchymal stem cells suppress dendritic cell migration, maturation and antigen presentation. Immunol Lett. (2008) 115:50–8. doi: 10.1016/j.imlet.2007.10.002
99. Chiesa S, Morbelli S, Morando S, Massollo M, Marini C, Bertoni A, et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. Proc Natl Acad Sci USA. (2011) 108:17384–9. doi: 10.1073/pnas.1103650108
100. Zhang W, Ge W, Li C, You S, Liao L, Han Q, et al. Effects of mesenchymal stem cells on differentiation, maturation, and function of human monocyte-derived dendritic cells. Stem Cells Dev. (2004) 13:263–71. doi: 10.1089/154732804323099190
101. Nauta AJ, Kruisselbrink AB, Lurvink E, Willemze R, Fibbe WE. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol. (2006) 177:2080–7. doi: 10.4049/jimmunol.177.4.2080
102. Zhao Z-G, Xu W, Sun L, You Y, Li F, Li Q-B, et al. Immunomodulatory function of regulatory dendritic cells induced by mesenchymal stem cells. Immunol Invest. (2012) 41:183–98. doi: 10.3109/08820139.2011.607877
103. Gao S, Mao F, Zhang B, Zhang L, Zhang X, Wang M, et al. Mouse bone marrow-derived mesenchymal stem cells induce macrophage M2 polarization through the nuclear factor-κB and signal transducer and activator of transcription 3 pathways. Exp Biol Med (Maywood). (2014) 239:366–75. doi: 10.1177/1535370213518169
104. Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. (2009) 37:1445–53. doi: 10.1016/j.exphem.2009.09.004
105. Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS ONE. (2010) 5:e9252. doi: 10.1371/journal.pone.0009252
106. François M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. (2012) 20:187–95. doi: 10.1038/mt.2011.189
107. Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med. (2017) 9:7828. doi: 10.1126/scitranslmed.aam7828
108. de Witte SFH, Luk F, Sierra Parraga JM, Gargesha M, Merino A, Korevaar SS, et al. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells. (2018) 36:602–15. doi: 10.1002/stem.2779
109. Gonçalves F, da C, Luk F, Korevaar SS, Bouzid R, Paz AH, et al. Membrane particles generated from mesenchymal stromal cells modulate immune responses by selective targeting of pro-inflammatory monocytes. Sci Rep. (2017) 7:12100. doi: 10.1038/s41598-017-12121-z
110. Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. (2012) 10:544–55. doi: 10.1016/j.stem.2012.03.007
111. Krampera M. Mesenchymal stromal cell “licensing”: a multistep process. Leukemia. (2011) 25:1408–14. doi: 10.1038/leu.2011.108
112. Chan JL, Tang KC, Patel AP, Bonilla LM, Pierobon N, Ponzio NM, et al. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood. (2006) 107:4817–24. doi: 10.1182/blood-2006-01-0057
113. François M, Romieu-Mourez R, Stock-Martineau S, Boivin M-N, Bramson JL, Galipeau J. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood. (2009) 114:2632–8. doi: 10.1182/blood-2009-02-207795
114. Raicevic G, Rouas R, Najar M, Stordeur P, Boufker HI, Bron D, et al. Inflammation modifies the pattern and the function of Toll-like receptors expressed by human mesenchymal stromal cells. Hum Immunol. (2010) 71:235–44. doi: 10.1016/j.humimm.2009.12.005
115. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. (2006) 24:386–98. doi: 10.1634/stemcells.2005-0008
116. English K, Barry FP, Field-Corbett CP, Mahon BP. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett. (2007) 110:91–100. doi: 10.1016/j.imlet.2007.04.001
117. Li W, Ren G, Huang Y, Su J, Han Y, Li J, et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ. (2012) 19:1505–13. doi: 10.1038/cdd.2012.26
118. Opitz CA, Litzenburger UM, Lutz C, Lanz TV, Tritschler I, Köppel A, et al. Toll-like receptor engagement enhances the immunosuppressive properties of human bone marrow-derived mesenchymal stem cells by inducing indoleamine-2,3-dioxygenase-1 via interferon-beta and protein kinase R. Stem Cells. (2009) 27:909–19. doi: 10.1002/stem.7
119. Pevsner-Fischer M, Morad V, Cohen-Sfady M, Rousso-Noori L, Zanin-Zhorov A, Cohen S, et al. Toll-like receptors and their ligands control mesenchymal stem cell functions. Blood. (2007) 109:1422–32. doi: 10.1182/blood-2006-06-028704
120. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. (2012) 12:383–96. doi: 10.1038/nri3209
121. Hwa Cho H, Bae YC, Jung JS. Role of toll-like receptors on human adipose-derived stromal cells. Stem Cells. (2006) 24:2744–52. doi: 10.1634/stemcells.2006-0189
122. Tomchuck SL, Zwezdaryk KJ, Coffelt SB, Waterman RS, Danka ES, Scandurro AB. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells. (2008) 26:99–107. doi: 10.1634/stemcells.2007-0563
123. Wang Z, Zhang F, Wang L, Yao Y, Zhao Q, Gao X. Lipopolysaccharides can protect mesenchymal stem cells (MSCs) from oxidative stress-induced apoptosis and enhance proliferation of MSCs via Toll-like receptor(TLR)-4 and PI3K/Akt. Cell Biol Int. (2009) 33:665–74. doi: 10.1016/j.cellbi.2009.03.006
124. Liotta F, Angeli R, Cosmi L, Filì L, Manuelli C, Frosali F, et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells. (2008) 26:279–89. doi: 10.1634/stemcells.2007-0454
125. Rashedi I, Gómez-Aristizábal A, Wang X-H, Viswanathan S, Keating A. TLR3 or TLR4 activation enhances mesenchymal stromal cell-mediated Treg induction via notch signaling. Stem Cells. (2017) 35:265–75. doi: 10.1002/stem.2485
126. Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLOS ONE. (2010) 5:e10088. doi: 10.1371/journal.pone.0010088
127. Brandau S, Jakob M, Hemeda H, Bruderek K, Janeschik S, Bootz F, et al. Tissue-resident mesenchymal stem cells attract peripheral blood neutrophils and enhance their inflammatory activity in response to microbial challenge. J Leukoc Biol. (2010) 88:1005–15. doi: 10.1189/jlb.0410207
128. Tu Z, Li Q, Bu H, Lin F. Mesenchymal stem cells inhibit complement activation by secreting factor H. Stem Cells Dev. (2010) 19:1803–9. doi: 10.1089/scd.2009.0418
129. Moll G, Jitschin R, von Bahr L, Rasmusson-Duprez I, Sundberg B, Lönnies L, et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS ONE. (2011) 6:e21703. doi: 10.1371/journal.pone.0021703
130. Le Blanc K, Davies LC. Mesenchymal stromal cells and the innate immune response. Immunol Lett. (2015) 168:140–6. doi: 10.1016/j.imlet.2015.05.004
131. Schraufstatter IU, DiScipio RG, Zhao M, Khaldoyanidi SK. C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. J Immunol. (2009) 182:3827–36. doi: 10.4049/jimmunol.0803055
132. Casiraghi F, Todeschini M, Azzollini N, Cravedi P, Cassis P, Solini S, et al. Effect of timing and complement receptor antagonism on intragraft recruitment and pro-tolerogenic effects of mesenchymal stromal cells in murine kidney transplantation. Transplantation. (2019) doi: 10.1097/TP.0000000000002611. [Epub ahead of print].
133. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol. (2012) 3:297. doi: 10.3389/fimmu.2012.00297
134. Popp FC, Eggenhofer E, Renner P, Slowik P, Lang SA, Kaspar H, et al. Mesenchymal stem cells can induce long-term acceptance of solid organ allografts in synergy with low-dose mycophenolate. Transpl Immunol. (2008) 20:55–60. doi: 10.1016/j.trim.2008.08.004
135. Li Q, Zhou X, Shi Y, Li J, Zheng L, Cui L, et al. In vivo tracking and comparison of the therapeutic effects of MSCs and HSCs for liver injury. PLoS ONE. (2013) 8:e62363. doi: 10.1371/journal.pone.0062363
136. Herrera MB, Bussolati B, Bruno S, Morando L, Mauriello-Romanazzi G, Sanavio F, et al. Exogenous mesenchymal stem cells localize to the kidney by means of CD44 following acute tubular injury. Kidney Int. (2007) 72:430–41. doi: 10.1038/sj.ki.5002334
137. Dong F, Harvey J, Finan A, Weber K, Agarwal U, Penn MS. Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation. (2012) 126:314–24. doi: 10.1161/CIRCULATIONAHA.111.082453
138. Rasmusson I, Ringdén O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit the formation of cytotoxic T lymphocytes, but not activated cytotoxic T lymphocytes or natural killer cells. Transplantation. (2003) 76:1208–13. doi: 10.1097/01.TP.0000082540.43730.80
139. Carrión F, Nova E, Luz P, Apablaza F, Figueroa F. Opposing effect of mesenchymal stem cells on Th1 and Th17 cell polarization according to the state of CD4+ T cell activation. Immunol Lett. (2011) 135:10–6. doi: 10.1016/j.imlet.2010.09.006
140. Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal period. J Exp Med. (2005) 201:769–77. doi: 10.1084/jem.20041179
141. Amado IF, Berges J, Luther RJ, Mailhé M-P, Garcia S, Bandeira A, et al. IL-2 coordinates IL-2-producing and regulatory T cell interplay. J Exp Med. (2013) 210:2707–20. doi: 10.1084/jem.20122759
142. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. (2008) 8:247–58. doi: 10.1038/nri2274
143. Cayeux S, Meuer S, Pezzutto A, Körbling M, Haas R, Schulz R, et al. Allogeneic mixed lymphocyte reactions during a second round of ontogeny: normal accessory cells did not restore defective interleukin-2 (IL-2) synthesis in T cells but induced responsiveness to exogeneous IL-2. Blood. (1989) 74:2278–84.
144. Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. (2005) 105:2214–9. doi: 10.1182/blood-2004-07-2921
145. Spaggiari GM, Abdelrazik H, Becchetti F, Moretta L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood. (2009) 113:6576–83. doi: 10.1182/blood-2009-02-203943
146. Khosravi M, Bidmeshkipour A, Cohen JL, Moravej A, Hojjat-Assari S, Naserian S, et al. Induction of CD4+CD25+FOXP3+ regulatory T cells by mesenchymal stem cells is associated with modulation of ubiquitination factors and TSDR demethylation. Stem Cell Res Ther. (2018) 9:273. doi: 10.1186/s13287-018-0991-1
147. Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. (2012) 307:1169–77. doi: 10.1001/jama.2012.316
148. Perico N, Casiraghi F, Introna M, Gotti E, Todeschini M, Cavinato RA, et al. Autologous mesenchymal stromal cells and kidney transplantation: a pilot study of safety and clinical feasibility. Clin J Am Soc Nephrol. (2011) 6:412–22. doi: 10.2215/CJN.04950610
149. Perico N, Casiraghi F, Gotti E, Introna M, Todeschini M, Cavinato RA, et al. Mesenchymal stromal cells and kidney transplantation: pretransplant infusion protects from graft dysfunction while fostering immunoregulation. Transpl Int. (2013) 26:867–78. doi: 10.1111/tri.12132
150. Reinders MEJ, de Fijter JW, Roelofs H, Bajema IM, de Vries DK, Schaapherder AF, et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl Med. (2013) 2:107–11. doi: 10.5966/sctm.2012-0114
151. Mudrabettu C, Kumar V, Rakha A, Yadav AK, Ramachandran R, Kanwar DB, et al. Safety and efficacy of autologous mesenchymal stromal cells transplantation in patients undergoing living donor kidney transplantation: a pilot study. Nephrology (Carlton). (2015) 20:25–33. doi: 10.1111/nep.12338
152. Peng Y, Ke M, Xu L, Liu L, Chen X, Xia W, et al. Donor-derived mesenchymal stem cells combined with low-dose tacrolimus prevent acute rejection after renal transplantation: a clinical pilot study. Transplantation. (2013) 95:161–8. doi: 10.1097/TP.0b013e3182754c53
153. Pan G-H, Chen Z, Xu L, Zhu J-H, Xiang P, Ma J-J, et al. Low-dose tacrolimus combined with donor-derived mesenchymal stem cells after renal transplantation: a prospective, non-randomized study. Oncotarget. (2016) 7:12089–101. doi: 10.18632/oncotarget.7725
154. Sun Q, Huang Z, Han F, Zhao M, Cao R, Zhao D, et al. Allogeneic mesenchymal stem cells as induction therapy are safe and feasible in renal allografts: pilot results of a multicenter randomized controlled trial. J Transl Med. (2018) 16:52. doi: 10.1186/s12967-018-1422-x
155. Erpicum P, Weekers L, Detry O, Bonvoisin C, Delbouille M-H, Grégoire C, et al. Infusion of third-party mesenchymal stromal cells after kidney transplantation: a phase I-II, open-label, clinical study. Kidney Int. (2019) 95:693–707. doi: 10.1016/j.kint.2018.08.046
156. Soeder Y, Loss M, Johnson CL, Hutchinson JA, Haarer J, Ahrens N, et al. First-in-human case study: multipotent adult progenitor cells for immunomodulation after liver transplantation. Stem Cells Transl Med. (2015) 4:899–904. doi: 10.5966/sctm.2015-0002
157. Detry O, Vandermeulen M, Delbouille M-H, Somja J, Bletard N, Briquet A, et al. Infusion of mesenchymal stromal cells after deceased liver transplantation: a phase I-II, open-label, clinical study. J Hepatol. (2017) 67:47–55. doi: 10.1016/j.jhep.2017.03.001
158. Keller CA, Gonwa TA, Hodge DO, Hei DJ, Centanni JM, Zubair AC. Feasibility, safety, and tolerance of mesenchymal stem cell therapy for obstructive chronic lung allograft dysfunction. Stem Cells Transl Med. (2018) 7:161–7. doi: 10.1002/sctm.17-0198
159. Casiraghi F, Remuzzi G, Abbate M, Perico N. Multipotent mesenchymal stromal cell therapy and risk of malignancies. Stem Cell Rev. (2013) 9:65–79. doi: 10.1007/s12015-011-9345-4
160. Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V, et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells. (2006) 24:1095–103. doi: 10.1634/stemcells.2005-0403
161. Bernardo ME, Zaffaroni N, Novara F, Cometa AM, Avanzini MA, Moretta A, et al. Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. (2007) 67:9142–9. doi: 10.1158/0008-5472.CAN-06-4690
162. Souza LEB, Almeida DC, Yaochite JNU, Covas DT, Fontes AM. Intravenous administration of bone marrow-derived multipotent mesenchymal stromal cells enhances the recruitment of CD11b(±) myeloid cells to the lungs and facilitates B16-F10 melanoma colonization. Exp Cell Res. (2016) 345:141–9. doi: 10.1016/j.yexcr.2015.05.021
163. Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H, et al. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS ONE. (2009) 4:e6278. doi: 10.1371/journal.pone.0006278
164. Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi's sarcoma. J Exp Med. (2006) 203:1235–47. doi: 10.1084/jem.20051921
165. Lalu MM, McIntyre L, Pugliese C, Fergusson D, Winston BW, Marshall JC, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS ONE. (2012) 7:e47559. doi: 10.1371/journal.pone.0047559
166. Zangi L, Margalit R, Reich-Zeliger S, Bachar-Lustig E, Beilhack A, Negrin R, et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells. (2009) 27:2865–74. doi: 10.1002/stem.217
167. Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. (2005) 106:4057–65. doi: 10.1182/blood-2005-03-1004
Keywords: mesenchymal (stromal) stem cells, transplantation, T cells, macrophage, tolerance, B cells
Citation: Podestà MA, Remuzzi G and Casiraghi F (2019) Mesenchymal Stromal Cells for Transplant Tolerance. Front. Immunol. 10:1287. doi: 10.3389/fimmu.2019.01287
Received: 21 March 2019; Accepted: 21 May 2019;
Published: 04 June 2019.
Edited by:
Martin Johannes Hoogduijn, Erasmus University Rotterdam, NetherlandsReviewed by:
Lucas Coelho Marlière Arruda, Karolinska Institute (KI), SwedenCopyright © 2019 Podestà, Remuzzi and Casiraghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Casiraghi, ZmVkZXJpY2EuY2FzaXJhZ2hpQG1hcmlvbmVncmkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.