 Evelien Houben
Evelien Houben Niels Hellings
Niels Hellings Bieke Broux
Bieke Broux- Department of Immunology, Biomedical Research Institute, Hasselt University, Diepenbeek, Belgium
For a long time, the central nervous system (CNS) was believed to be an immune privileged organ. In the last decades, it became apparent that the immune system interacts with the CNS not only in pathological, but also in homeostatic situations. It is now clear that immune cells infiltrate the healthy CNS as part of immune surveillance and that immune cells communicate through cytokines with CNS resident cells. In pathological conditions, an enhanced infiltration of immune cells takes place to fight the pathogen. A well-known family of cytokines is the interleukin (IL)-6 cytokine family. All members are important in cell communication and cell signaling in the immune system. One of these members is oncostatin M (OSM), for which the receptor is expressed on several cells of the CNS. However, the biological function of OSM in the CNS is not studied in detail. Here, we briefly describe the general aspects related to OSM biology, including signaling and receptor binding. Thereafter, the current understanding of OSM during CNS homeostasis and pathology is summarized.
Introduction
A well-orchestrated transmission of signals is crucial to maintain and restore homeostasis in humans. A class of messenger molecules that play an important role in interaction and communication of cells in the immune system, are cytokines. They are produced by immune cells but also by other resident cells of the human body as a response to changes in their microenvironment. The central nervous system (CNS) was long believed to be immune privileged. However, in the last decades it has become clear that communication between the CNS and the immune system is very important, even during homeostasis, and that it needs to be strictly regulated.
Many cytokines are thoroughly characterized in the CNS, yet others remain less studied, including oncostatin M (OSM). In 1991, OSM was categorized as a member of the interleukin (IL)-6 cytokine family (1, 2). Next to OSM and IL-6, this family of cytokines includes IL-11, IL-27, leukemia inhibitory factor (LIF) ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), cardiotrophin-like cytokine (CLC), neuropoietin (NP) and IL-31 (3, 4). These cytokines all signal through a multi-unit receptor complex, containing the common glycoprotein 130 (gp130) subunit, except for IL-31 which binds to the OSM receptor beta (OSMRβ)/IL-31 receptor alpha (IL-31Rα) complex (5). Because of the related receptor complexes, signaling properties are shared by the members of the IL-6 family. For more insights see (4, 6–9). Here, we briefly describe the biological effects of OSM and summarize the current understanding of OSM activity in the CNS.
OSM Biology
OSM binds to the heterodimeric OSM receptor (OSMR), consisting of the gp130/OSMRβ complex, also referred to as OSMR type II in humans. Moreover, in humans and rats, OSM signaling is also possible through the LIF receptor (LIFR), consisting of the heterodimer gp130 and LIF receptor beta (LIFRβ), also known as OSMR type I in humans (4, 10). However, in mice, murine OSM (mOSM) does not transmit signals through the LIFR (11, 12). Only extremely high concentrations of mOSM may lead to weak LIFR signaling (11). While there are reports on mOSM signaling via the LIFR in mouse osteocytes (13, 14), no literature hints to LIFR activation by mOSM in neural cells. Cross-species activities of OSM in mice, rats and humans are also possible (summarized in Table 1) (10, 15). Understanding of the cross-species activities is crucial for proper interpretation of results obtained in experimental studies. For example, experiments in which OSM only signals via the LIFR show receptor and cell signaling of LIF rather than that of OSM.
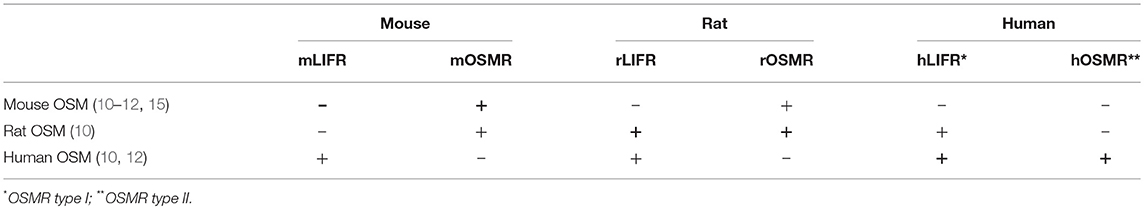
Table 1. Cross-species signaling of OSM via the LIFR and the OSMR in mice, rats and humans.
OSM contains BC loops which form a steric hindrance for OSMRβ and LIFRβ. Therefore, OSM first binds with gp130 to subsequently recruit OSMRβ or LIFRβ (16). In both cases, formation of the heterodimeric complex leads to activation of different signaling cascades [extensively reviewed in (4, 6, 7, 17)]. First, receptor binding can activate Janus kinase (JAK)s, JAK1, JAK2 and Tyk2, which recruit signal transducer and activation transcription (STAT)s, STAT1, STAT3, STAT5, and STAT6. The activated STATs translocate to the nucleus to induce transcription of target genes. Second, activation of the LIFR and OSMR can also induce the mitogen activated protein kinases (MAPK) cascade. The different MAPKs involved are extracellular signal-regulated kinases 1 and 2 (ERK1/2), p38 and c-jun N-terminal kinases (JNK) (18). Finally, activation of the phosphatidylinositol-3-kinase (PI3K)/Akt pathway and the protein kinase C delta (PKCδ) have been described after OSMR activation (19, 20). These different signaling pathways lead to the diverse nature of OSM in various cell types and environmental conditions.
OSM in the CNS
The role of OSM has already been specified in joint, skin, lung, and vascular homeostasis and disease [reviewed in (4, 7, 19)]. Also in cancer, depending on the cancer type, various actions of OSM are reported (4, 7, 19). Yet, less is known about the role of OSM in the CNS. Here, we summarize the reports for which OSM is described in the healthy and pathological CNS (Figure 1).
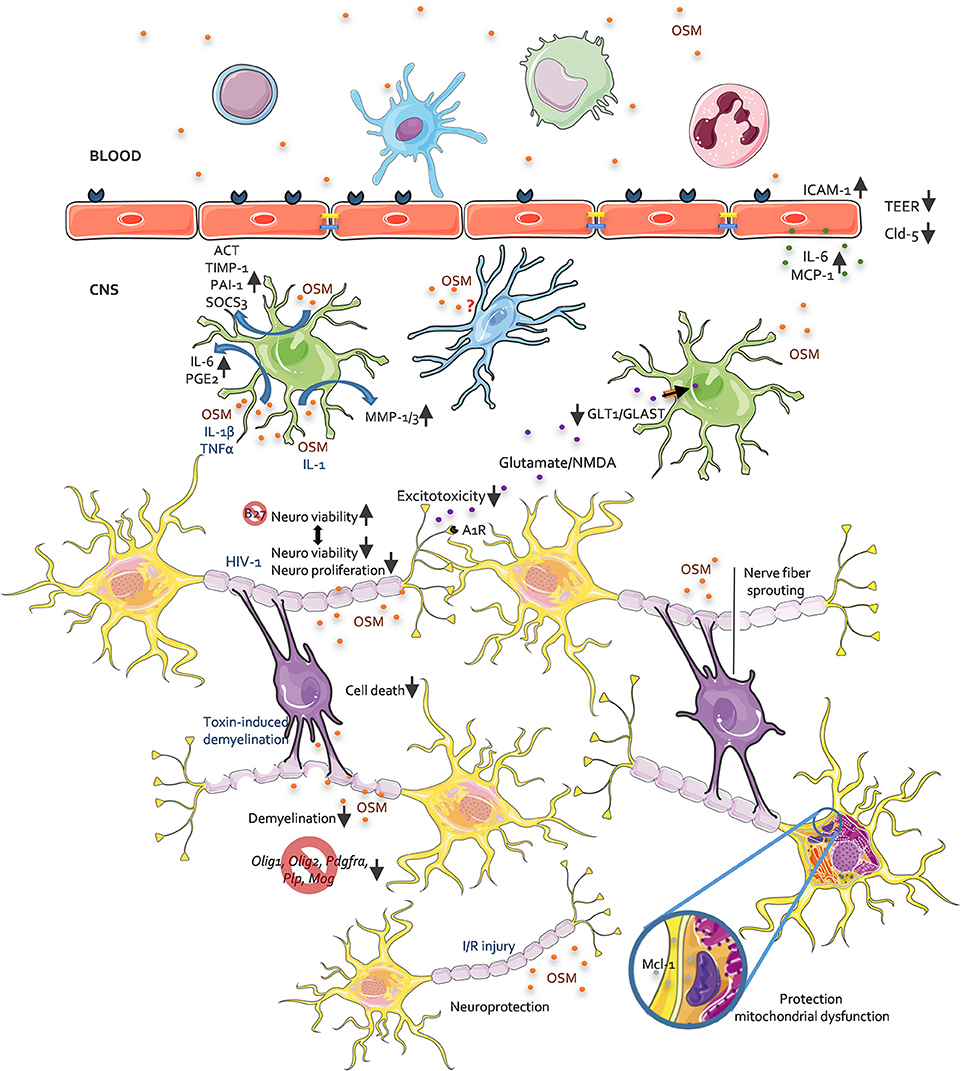
Figure 1. The role of OSM in CNS pathology, an overview. This figure depicts all reported activities of OSM on different CNS resident cells as investigated in in vitro and in vivo studies. For further details see accompanying text.
Source of OSM and OSMRβ Expression in the CNS
Many cells of the immune system, i.e. dendritic cells, neutrophils, monocytes/macrophages, and T-cells, have been identified as a source of OSM (21–23). Hematopoietic cells of the bone marrow also produce OSM, regardless of inflammation (24). In the CNS, OSM is expressed by different cell types, namely neurons, astrocytes and microglia (25–27). In pathological conditions, such as multiple sclerosis (MS), OSM expression in the CNS is increased, in part by OSM production via infiltrating leukocytes (25, 28). With regard to expression of OSMRβ in the CNS, the first reports described expression of OSMRβ RNA in most regions of the murine CNS (forebrain, cortex, midbrain, hindbrain and spinal cord) (29, 30). Later reports investigated the cellular specificity of OSMRβ expression, and indicated that the OSMRβ protein is expressed on neurons (31), astrocytes (31–34), endothelial cells (33), and oligodendrocytes (31). For microglia conflicting reports are found and discussed later. In addition, OSMRβ transcripts are also present in epithelial cells of the pia mater and the choroid plexus (29, 35). The extensive expression of the OSMRβ in the brain implies important CNS related effects of OSM in different cell types both in health and disease.
Effects of OSM on Neural Cells
In physiological conditions, OSM has been implied in the homeostasis of neural precursor cells (NPCs). NPCs are a pool of cells for the continuous production of new neural cells, located in the subventricular zone (SVZ) (36), hippocampus (37), and olfactory bulb (38) in the adult mammalian brain. In mice, the OSMR is expressed on a subpopulation of NPC in the SVZ and in the subgranular zone of the dentate gyrus within the hippocampus (39). Functionally, OSM induces in vitro repression of neurosphere formation, indicating inhibition of NPC proliferation isolated from the SVZ and olfactory bulb, (39) while NPCs isolated from SVZ, olfactory bulb and hippocampus of OSMRβ knock-out animals lead to enhanced formation of neurospheres (39).
In pathological conditions, the vast majority of papers report neuroprotective effects of OSM. To start, OSM inhibits N-methyl-D-aspartate (NMDA)-induced excitotoxicity in a dose-dependent way. This effect is even more pronounced after pre-treatment with OSM (40). Neuroprotective effects against excitotoxicity are among others mediated by inhibitory adenosine A1 receptors (A1Rs), suppressing excitatory transmission (41). Inhibition of glutamate-induced excitotoxicity by OSM is completely abolished after A1Rs blockage and knockout, indicating the requirement of adenosine A1R function for neuroprotection (42). Also, a protective effect of OSM is observed after amyloid beta-peptide (Aβ) induced neurotoxicity (43), known to cause mitochondrial dysfunction in Alzheimer's disease (44). Furthermore, OSM protects against 3-nitropropionic acid induced mitochondrial dysfunction in rat cortical neurons through induction of myeloid cell leukemia-1 (Mcl-1). Mcl-1 enhances mitochondrial respiration and ATP production (43) and is described as an anti-apoptotic protein with neuroprotective functions (45, 46). Since rat neuronal cells and rOSM are used in these experiments, both the involvement of LIFR and OSMR signaling needs to be considered. Moreover, we reported that OSM enhances neuronal cell viability after withdrawal of B27, a vital supplement for growth and differentiation of primary neurons and enhances neurite outgrowth in vitro (47). Only one study reported a potential neurotoxic effect of OSM. In this study, neuronal cell growth was inhibited when cultured in the presence of the secretome of peripheral blood mononuclear cells (PBMC) from HIV-1-infected patients. Analysis of the secretome, identified OSM as the key molecule involved in inhibition of neuronal proliferation and viability (48). Another study reported an indirect neurotoxic activity of OSM by inducing TNF-α secretion by microglia (49). Altogether, we can conclude that OSM has been widely reported to have a direct neuroprotective activity. However, indirect neurotoxic effects are possible and need to be kept under consideration.
Astrocytes usually prevent neuronal excitotoxicity via sequestration of extracellular glutamate through the glutamate aspartate transporter (GLAST/EAAT1) and glutamate transporter-1 (GLT-1/EAAT2) (50). OSM downregulates the expression of these receptors on astrocytes, leading to reduced glutamate uptake and consequently, excitotoxic injury (26). Astrocytes also secrete different molecules in response to OSM. Plasminogen activator inhibitor-1 (PAI-1) and α1-antichymotrypsin (ACT) (51) expression is induced by OSM in astrocytes. Co-treatment of OSM and IL-1, leads to matrix metalloproteinase (MMP)-1 and MMP-3 production by astrocytes (52). Moreover, OSM works synergistically with the pro-inflammatory cytokines, IL-1β and TNF-α, to induce IL-6 (53) and prostaglandin E2 (PGE2) (54) production in human astrocytes. These OSM-induced astrocytic molecules are linked to pro-inflammatory and tissue remodeling processes. However, OSM also induces astrocytic secretion of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) (55) and SOCS3 (32), which quench inflammation. Therefore, the net outcome of OSM signaling in astrocytes depends on the microenvironment and other cytokines present herein.
For microglia/macrophages, contradictory reports are present about OSMRβ expression and therefore the effect of OSM on these cells. Different research groups do not observe OSMRβ expression in primary mouse microglia (34, 42), nor in the C8-B4 microglia cell line (34). Moreover, no phosphorylation of STAT1 or STAT3 is observed in microglia after OSM treatment (34). In contrast, tumor necrosis factor-α (TNF-α) and nitric oxide (NO) production is reported after NF-κB pathway stimulation via OSM treatment of primary microglia and the BV2 microglia cell line (49). Yet, others did not see OSM-induced activation of the NF-κB pathway nor OSM-induced NO production in microglia (34). Moreover, our group found OSMRβ expression on Iba-1+ cells, a marker for both microglia and macrophages, in naive and cuprizone treated animals (31). In naive animals, no infiltration of macrophages is expected, yet perivascular, meningeal and choroid plexus macrophages are present (56). In the cuprizone challenged mice, macrophages infiltrate the brain (57). Therefore, it is possible that the Iba-1+ cells are macrophages. However, Hsu and colleagues did not detect OSMRβ in bone marrow-derived macrophages and the RAW 264.7 macrophage cell line (34). Yet, in other tissues, OSMRβ expression is seen on macrophages, i.e. in adipose tissue and atherosclerotic lesions (58, 59). In conclusion, more research is needed to address whether OSMR signaling is active in microglia and macrophages.
The blood brain-barrier (BBB) is very important to protect the brain from unwanted intruders. On human cerebral endothelial cells (HCECs), expression of OSMRβ, but not LIFRβ, is seen, despite low RNA levels of LIFRβ (25). The latter implies that OSM only signals through the OSMR type II in HCECs. OSM treatment increases the percentage of HCECs expressing intracellular adhesion molecule-1 (ICAM-1), yet no effect on vascular cell adhesion molecule-1 (VCAM-1) expression is detectable (25). Next to adhesion molecules, OSM augments the secretion of IL-6 and monocyte chemotactic protein-1 (MCP-1) in HCECs (25). This effect is further enhanced after co-treatment of HCECs with OSM and TNF-α (25). Also, a decreased BBB permeability is attributed to persistently high activation of the JAK/STAT3 signaling pathway (60). Here, rat brain capillary endothelial cells (RBECs) were treated with mOSM, implying OSMR and not LIFR signaling as indicated in Table 1 (10). Both increased permeability for sodium fluorescein and decreased transendothelial electrical resistance (TEER) are seen in mOSM treated RBECs (60, 61). Moreover, delocalization of the tight junction molecules, claudin-5 and zonula occludens-1 (ZO-1) is apparent after OSM treatment (60). Together, these studies imply a pro-inflammatory state of BBB-ECs when treated with OSM.
Finally, for oligodendrocytes and myelination, protective effects of LIF have been described [reviewed in (62, 63)] and tested preclinically via therapeutic delivery through nanoparticles or lentiviral vectors (64, 65). However, only a few in vivo studies reported on the role of OSM on oligodendrocytes and its repair mechanisms after myelin damage. These results are described in the next section. Overall, both protective and detrimental effects on cells of the CNS are described for OSM. Even though, the described in vitro experiments investigated the effect of OSM on distinctive cell types, the interplay between different cells is more complex and needs to be studied using in vivo models.
OSM in Murine Models of CNS Pathology
Murine neurological disease models are used to investigate the biological role of OSM in a more complex in vivo setting and to further allow development of OSM based treatment strategies based on these insights. Inducing disease in these models can influence the level of OSM and OSMRβ expression. Indeed, upregulation of OSM and OSMRβ in whole spinal cord is observed in the mouse spinal cord hemisection model (47). During cuprizone-induced demyelination increased OSMRβ expression is seen mainly on astrocytes and Iba-1+ cells (microglia/macrophages) (31). In contrast, middle cerebral artery occlusion (MCAO)/reperfusion reduces expression of OSMRβ on neurons in brain areas of disturbed perfusion, i.e. ipsilateral cortex and striatum (66).
To test the effect of OSM signaling, both OSM treatment and transgenic mice [OSMRβ overexpressing animals or OSMRβ knock-out (KO) mice] can be used. While OSM KO and OSMRβ KO mice are healthy and fertile, phenotypical changes observed are a disturbed hematopoiesis in both KO strains (67, 68) and severe obesity upon a high-fat diet in OSMRβ KO animals (69). It is unknown whether there is a phenotype in the CNS, since no neurological deficits are reported to date. For the OSMRβ KO mice, the IL-31 receptor consisting of OSMRβ/IL-31Rα is also affected. However, to date there are no reports of IL-31 signaling in the CNS, only the presence of the IL-31 receptor is described in dorsal root ganglia (70–73).Therefore, it is difficult to evaluate the effect of disturbed IL-31 signaling in CNS studies using OSMRβ deficient mice.
OSM was studied in the experimental autoimmune encephalomyelitis (EAE) mouse model, a MOG autoreactive T-cell mediated model for MS. Mice receiving intraperitoneal injections with OSM did not develop any sign of paralysis, the clinical outcome of EAE induction (74). The absence of symptoms was in agreement with limited immune cell infiltration in the brain of these animals (74). However, it needs to be mentioned that human OSM was used to treat the animals. Therefore, LIFR signaling and not OSMR signaling is studied here as indicated in Table 1. Indeed, systemic LIF treatment of EAE mice reduces the disease symptoms (75). We have shown that local overexpression of OSM through lentiviral vectors reduces cell-death of oligodendrocytes in the cuprizone model and therefore limits subsequent demyelination. The latter is linked to a reduced microglial response, increased IL-4 expression and M2 polarization (31). OSM-induced M2 polarization is also seen in other organ/tissue models, i.e. lung, adipose tissue (58, 76–78) and cancer models (79, 80). In line with the cuprizone model, OSM treatment in the ethidium bromide (EtBr) toxin-induced demyelination model counteracts a reduction in mRNA expression of oligodendrocyte precursor marker (Pdgfra), oligodendrocyte lineage transcription factors (Olig1 and Olig2) and myelin genes (Plp and Mog) indicative for oligodendrocyte and myelin sparing (33). Moreover, neuroprotective effects are reported in vivo. Local OSM treatment in a spinal cord injury (SCI) mouse model improves functional recovery, and histological analysis reveals a reduced lesion size with less astrogliosis, less CD4+ T cell infiltration and an increased nerve fiber sprouting (47). Also, stereotaxic injection of NMDA together with OSM reduces the NMDA-induced lesion volume in a model of neurotoxic injury. This could be attributed to a reduced expression of the NR2C subunit of the NMDA-receptor and attenuated increase of intracellular calcium, preceding NMDA-induced cell death (40). Correspondingly, OSMRβ overexpression in neurons improves stroke outcome following ischemia/reperfusion (I/R)-induced cerebral injury (66). The latter is due to a protective role of OSMRβ against neuronal apoptosis via JAK2/STAT3 signal activation, leading to transcription of genes involved in neuronal survival (66). When mice lack OSMRβ, opposite effects on neurons and oligodendrocytes are described. OSMRβ deficiency leads to an increased infarct volume and more severe neurological deficits after I/R damage (66) and an aggravation of demyelination in the cuprizone model (31). Taken together, these studies indicate protective effects of OSM signaling in different mouse models for neurodegenerative diseases, with both oligodendrocytes and neurons being directly or indirectly protected by OSM. The most important intermediate players in these processes are astrocytes and microglia/macrophages.
Conclusion
Within the CNS, the major cellular sources of OSM are astrocytes, neurons, microglia and infiltrating immune cells. OSM can signal though both the LIFR (OSMR type I in humans) and OSMR (OSMR type II in humans) in humans and rats, while in mice, only signaling via the OSMR is possible. Since OSM can signal via two receptors, caution should be taken when interpreting research findings, because OSM-mediated effects can be attributed to LIFR and/or OSMR signaling, depending on the model and species used. With regard to the effect of OSM on neurons, the majority of papers report protective effects of OSM. For oligodendrocytes, astrocytes, and the BBB a limited amount of studies is available. OSM has protective effects at the level of myelination, which is very important for good signal transduction and protection of axons. For astrocytes, there is a role in excitotoxicity and secretion of inflammatory molecules. Finally, at the level of the BBB a pro-inflammatory readout is observed. To conclude, OSM can exhibit different functions depending on the variety of cell types that express the receptor and the cellular and molecular microenvironment. While in vivo models demonstrated that OSM has beneficial effects in the diseased CNS, more research is warranted to reveal the true role of OSM in the CNS.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by the Research Foundation of Flanders (FWO Vlaanderen, 1106817N). Figure 1 was adapted from smart servier medical art (https://smart.servier.com/).
References
1. Rose TM, Bruce AG. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc Natl Acad Sci USA. (1991) 88:8641–5. doi: 10.1073/pnas.88.19.8641
2. Liu J, Modrell B, Aruffo A, Marken JS, Taga T, Yasukawa K, et al. Interleukin-6 signal transducer gp 130 mediates Oncostatin M signaling. J Biol Chem. (1992) 267:16763–6.
3. Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci USA. (2004) 101:4827–32. doi: 10.1073/pnas.0306178101
4. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol. (2018) 18:773–89. doi: 10.1038/s41577-018-0066-7
5. Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. (2005) 6:114. doi: 10.1038/ni0105-114a
6. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J. (2003) 374(Pt 1):1–20. doi: 10.1042/bj20030407
7. Hermanns HM. Oncostatin M and interleukin- 31: cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. (2015) 26:545–58. doi: 10.1016/j.cytogfr.2015.07.006
8. Janssens K, Slaets H, Hellings N. Immunomodulatory properties of the IL-6 cytokine family in multiple sclerosis. Ann NY Acad Sci. (2015) 1351:52–60. doi: 10.1111/nyas.12821
9. Garbers C, Hermanns HM, Schaper F, Muller-Newen G, Grotzinger J, Rose-John S, et al. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. (2012) 23:85–97. doi: 10.1016/j.cytogfr.2012.04.001
10. Drechsler J, Grotzinger J, Hermanns HM. Characterization of the rat Oncostatin M receptor complex which resembles the human, but differs from the murine cytokine receptor. PLoS ONE. (2012) 7:e43155. doi: 10.1371/journal.pone.0043155
11. Ichihara M, Hara T, Kim H, Murate T, Miyajima A. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood. (1997) 90:165–73.
12. Lindberg RA, Juan TS, Welcher AA, Sun Y, Cupples R, Guthrie B, et al. Cloning and characterization of a specific receptor for mouse Oncostatin M. Mol Cell Biol. (1998) 18:3357–67. doi: 10.1128/MCB.18.6.3357
13. Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. (2010) 120:582–92. doi: 10.1172/JCI40568
14. Walker EC, Johnson RW, Hu Y, Brennan HJ, Poulton IJ, Zhang JG, et al. Murine Oncostatin M acts via leukemia inhibitory factor receptor to phosphorylate signal transducer and activator of transcription 3 (STAT3) but not STAT1, an effect that protects bone mass. J Biol Chem. (2016) 291:21703–16. doi: 10.1074/jbc.M116.748483
15. Adrian-Segarra JM, Sreenivasan K, Gajawada P, Lorchner H, Braun T, Poling J. The AB loop of Oncostatin M (OSM) determines species-specific signaling in humans and mice. J Biol Chem. 293:20181–99 (2018). doi: 10.1074/jbc.RA118.004375
16. Chollangi S, Mather T, Rodgers KK, Ash JD. A unique loop structure in Oncostatin M determines binding affinity toward Oncostatin M receptor and leukemia inhibitory factor receptor. J Biol Chem. (2012) 287:32848–59. doi: 10.1074/jbc.M112.387324
17. Chen SH, Benveniste EN. Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev. (2004) 15:379–91. doi: 10.1016/j.cytogfr.2004.06.002
18. Thoma B, Bird TA, Friend D, Gearing D, Dower S. Oncostatin-M and leukemia inhibitory factor trigger overlapping and different signals through partially shared receptor complexes. J Biol Chem. (1994) 269:6215–22.
19. Richards CD. The enigmatic cytokine Oncostatin m and roles in disease. ISRN Inflamm. (2013) 2013:512103. doi: 10.1155/2013/512103
20. Chipoy C, Berreur M, Couillaud S, Pradal G, Vallette F, Colombeix C, et al. Downregulation of osteoblast markers and induction of the glial fibrillary acidic protein by Oncostatin M in osteosarcoma cells require PKCdelta and STAT3. J Bone Miner Res. (2004) 19:1850–61. doi: 10.1359/JBMR.040817
21. Alain G, Danièle C, Jean C, Marie Anne G-P, Claude G, Monique D, et al. Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab Investig. (2001) 81:133–41. doi: 10.1038/labinvest.3780220
22. Suda T, Chida K, Todate A, Ide K, Asada K, Nakamura Y, et al. Oncostatin m production by human dendritic cells in response to bacterial products. Cytokine. (2002) 17:335–40. doi: 10.1006/cyto.2002.1023
23. Brown TJ, Lioubin MN, Marquardt H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and Oncostatin M for human melanoma cells. J Immunol. (1987) 139:2977–83.
24. Sato F, Miyaoka Y, Miyajima A, Tanaka M. Oncostatin M maintains the hematopoietic microenvironment in the bone marrow by modulating adipogenesis and osteogenesis. PLoS ONE. (2014) 9:e116209. doi: 10.1371/journal.pone.0116209
25. Ruprecht K, Kuhlmann T, Seif F, Hummel V, Kruse N, Bruck W, et al. Effects of Oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J Neuropathol Exp Neurol. (2001) 60:1087–98. doi: 10.1093/jnen/60.11.1087
26. Moidunny S, Matos M, Wesseling E, Banerjee S, Volsky DJ, Cunha RA, et al. Oncostatin M promotes excitotoxicity by inhibiting glutamate uptake in astrocytes: implications in HIV-associated neurotoxicity. J Neuroinflamm. (2016) 13:144. doi: 10.1186/s12974-016-0613-8
27. Znoyko I, Sohara N, Spicer SS, Trojanowska M, Reuben A. Comparative studies of Oncostatin M expression in the tissues of adult rodents. Anat Rec Part A Discov Mol Cell Evol Biol. (2005) 283:182–6. doi: 10.1002/ar.a.20159
28. Kastl PS, Speidl SW, Kaun MC, Katsaros NK, Rega WG, Afonyushkin WT, et al. In human macrophages the complement component C5a induces the expression of Oncostatin M via AP-1 activation. Arterioscler Thromb Vasc Biol. (2008) 28:498–503. doi: 10.1161/ATVBAHA.107.160580
29. Tamura S, Morikawa Y, Senba E. Localization of Oncostatin M receptor beta in adult and developing CNS. Neuroscience. (2003) 119:991–7. doi: 10.1016/S0306-4522(03)00240-9
30. Tamura S, Morikawa Y, Miyajima A, Senba E. Expression of Oncostatin M receptor beta in a specific subset of nociceptive sensory neurons. Eur J Neurosci. (2003) 17:2287–98. doi: 10.1046/j.1460-9568.2003.02681.x
31. Janssens K, Maheshwari A, Van den Haute C, Baekelandt V, Stinissen P, Hendriks JJ, et al. Oncostatin M protects against demyelination by inducing a protective microglial phenotype. Glia. (2015) 63:1729–37. doi: 10.1002/glia.22840
32. Baker BJ, Qin H, Benveniste EN. Molecular basis of Oncostatin M- induced SOCS- 3 expression in astrocytes. Glia. (2008) 56:1250–62. doi: 10.1002/glia.20694
33. Glezer I, Rivest S. Oncostatin M is a novel glucocorticoid-dependent neuroinflammatory factor that enhances oligodendrocyte precursor cell activity in demyelinated sites. Brain Behav Immun. (2010) 24:695–704. doi: 10.1016/j.bbi.2010.01.005
34. Hsu MP, Frausto R, Rose-John S, Campbell IL. Analysis of IL-6/gp130 family receptor expression reveals that in contrast to astroglia, microglia lack the Oncostatin M receptor and functional responses to Oncostatin M. Glia. (2015) 63:132–41. doi: 10.1002/glia.22739
35. Tanaka M, Hara T, Copeland NG, Gilbert DJ, Jenkins NA, Miyajima A. Reconstitution of the functional mouse Oncostatin M (OSM) receptor: molecular cloning of the mouse OSM receptor beta subunit. Blood. (1999) 93:804–15.
36. Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, et al. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. (1994) 13:1071–82. doi: 10.1016/0896-6273(94)90046-9
37. Bull ND, Bartlett PF. The adult mouse hippocampal progenitor is neurogenic but not a stem cell. J Neurosci. (2005) 25:10815–21. doi: 10.1523/JNEUROSCI.3249-05.2005
38. Liu Z, Martin LJ. Olfactory bulb core is a rich source of neural progenitor and stem cells in adult rodent and human. J Comp Neurol. (2003) 459:368–91. doi: 10.1002/cne.10664
39. Beatus P, Jhaveri DJ, Walker TL, Lucas PG, Rietze RL, Cooper HM, et al. Oncostatin M regulates neural precursor activity in the adult brain. Dev Neurobiol. (2011) 71:619–33. doi: 10.1002/dneu.20871
40. Weiss TW, Samson AL, Niego B, Daniel PB, Medcalf RL. Oncostatin M is a neuroprotective cytokine that inhibits excitotoxic injury in vitro and in vivo. FASEB J. (2006) 20:2369–71. doi: 10.1096/fj.06-5850fje
41. Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. BBA Biomembr. (2011) 1808:1380–99. doi: 10.1016/j.bbamem.2010.12.001
42. Moidunny S, Dias RB, Wesseling E, Sekino Y, Boddeke HW, Sebastiao AM, et al. Interleukin-6-type cytokines in neuroprotection and neuromodulation: Oncostatin M, but not leukemia inhibitory factor, requires neuronal adenosine A1 receptor function. J Neurochem. (2010) 114:1667–77. doi: 10.1111/j.1471-4159.2010.06881.x
43. Chang S-H, Hwang C-S, Yin J-H, Chen S-D, Yang D-I. Oncostatin M- dependent Mcl- 1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. BBA Mol Cell Res. (2015) 1853:2306–25. doi: 10.1016/j.bbamcr.2015.05.014
44. Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta- amyloid fragment 25- 35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. (2002) 10:258–67. doi: 10.1006/nbdi.2002.0516
45. Ekholm-Reed S, Goldberg M, Schlossmacher M, Reed S. Parkin- dependent degradation of the F-box protein Fbw7β promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Mol Cell Biol. (2013) 33:3627–43. doi: 10.1128/MCB.00535-13
46. Magiera MM, Mora S, Mojsa B, Robbins I, Lassot I, Desagher S. Trim17- mediated ubiquitination and degradation of Mcl- 1 initiate apoptosis in neurons. Cell Death Differ. (2012) 20:28–921. doi: 10.1038/cdd.2012.124
47. Slaets H, Nelissen S, Janssens K, Vidal PM, Lemmens E, Stinissen P, et al. Oncostatin M reduces lesion size and promotes functional recovery and neurite outgrowth after spinal cord injury. Mol Neurobiol. (2014) 50:1142–51. doi: 10.1007/s12035-014-8795-5
48. Ensoli F, Fiorelli V, Decristofaro M, Santini Muratori D, Novi A, Vannelli B, et al. Inflammatory cytokines and HIV- 1- associated neurodegeneration: Oncostatin-M produced by mononuclear cells from HIV- 1-infected individuals induces apoptosis of primary neurons. J Immunol. (1999) 162:6268–77.
49. Baker BJ, Park KW, Qin H, Ma X, Benveniste EN. IL-27 inhibits OSM-mediated TNF-alpha and iNOS gene expression in microglia. Glia. (2010) 58:1082–93. doi: 10.1002/glia.20989
50. Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. (1996) 16:675–86. doi: 10.1016/S0896-6273(00)80086-0
51. Kordula T, Rydel RE, Brigham EF, Horn F, Heinrich PC, Travis J. Oncostatin M and the interleukin- 6 and soluble interleukin- 6 receptor complex regulate α 1 -antichymotrypsin expression in human cortical astrocytes. J Biol Chem. (1998) 273:4112–8. doi: 10.1074/jbc.273.7.4112
52. Korzus E, Nagase H, Rydell R, Travis J. The mitogen- activated protein kinase and JAK- STAT signaling pathways are required for an Oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J Biol Chem. (1997) 272:1188–96. doi: 10.1074/jbc.272.2.1188
53. Van Wagoner NJ, Choi C, Repovic P, Benveniste EN. Oncostatin M regulation of interleukin- 6 expression in astrocytes. J Neurochem. (2000) 75:563–75. doi: 10.1046/j.1471-4159.2000.0750563.x
54. Repovic P, Mi K, Benveniste EN. Oncostatin M enhances the expression of prostaglandin E2 and cyclooxygenase-2 in astrocytes: synergy with interleukin-1beta, tumor necrosis factor-alpha, and bacterial lipopolysaccharide. Glia. (2003) 42:433–46. doi: 10.1002/glia.10182
55. Kasza A, Kiss DL, Gopalan S, Xu W, Rydel RE, Koj A, et al. Mechanism of plasminogen activator inhibitor- 1 regulation by Oncostatin M and interleukin- 1 in human astrocytes. J Neurochem. (2002) 83:696–703. doi: 10.1046/j.1471-4159.2002.01163.x
56. Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia. (2005) 49:375–84. doi: 10.1002/glia.20124
57. McMahon EJ, Suzuki K, Matsushima GK. Peripheral macrophage recruitment in cuprizone- induced CNS demyelination despite an intact blood– brain barrier. J Neuroimmunol. (2002) 130:32–45. doi: 10.1016/S0165-5728(02)00205-9
58. Komori T, Tanaka M, Senba E, Miyajima A, Morikawa Y. Lack of Oncostatin M receptor beta leads to adipose tissue inflammation and insulin resistance by switching macrophage phenotype. J Biol Chem. (2013) 288:21861–75. doi: 10.1074/jbc.M113.461905
59. Zhang X, Li J, Qin JJ, Cheng WL, Zhu X, Gong FH, et al. Oncostatin M receptor beta deficiency attenuates atherogenesis by inhibiting JAK2/STAT3 signaling in macrophages. J Lipid Res. (2017) 58:895–906. doi: 10.1194/jlr.M074112
60. Takata F, Dohgu S, Matsumoto J, Machida T, Sakaguchi S, Kimura I, et al. Oncostatin M–induced blood-brain barrier impairment is due to prolonged activation of STAT3 signaling in vitro. J Cell Biochem. (2018) 119:9055–63. doi: 10.1002/jcb.27162
61. Takata F, Sumi N, Nishioku T, Harada E, Wakigawa T, Shuto H, et al. Oncostatin M induces functional and structural impairment of blood-brain barriers comprised of rat brain capillary endothelial cells. Neurosci Lett. (2008) 441:163–6. doi: 10.1016/j.neulet.2008.06.030
62. Ostasov P, Houdek Z, Cendelin J, Kralickova M. Role of leukemia inhibitory factor in the nervous system and its pathology. Rev Neurosci. (2015) 26:443–59. doi: 10.1515/revneuro-2014-0086
63. Slaets H, Hendriks JJ, Stinissen P, Kilpatrick TJ, Hellings N. Therapeutic potential of LIF in multiple sclerosis. Trends Mol Med. (2010) 16:493–500. doi: 10.1016/j.molmed.2010.08.007
64. Metcalfe SM. LIF and multiple sclerosis: one protein with two healing properties. Mult Scler Relat Disord. (2018) 20:223–7. doi: 10.1016/j.msard.2018.01.018
65. Slaets H, Dumont D, Vanderlocht J, Noben JP, Leprince P, Robben J, et al. Leukemia inhibitory factor induces an antiapoptotic response in oligodendrocytes through Akt-phosphorylation and up-regulation of 14-3-3. Proteomics. (2008) 8:1237–47. doi: 10.1002/pmic.200700641
66. Guo S, Li Z-Z, Gong J, Xiang M, Zhang P, Zhao G-N, et al. Oncostatin M confers neuroprotection against ischemic stroke. J Neurosci. (2015) 35:12047–62. doi: 10.1523/JNEUROSCI.1800-15.2015
67. Tanaka M, Hirabayashi Y, Sekiguchi T, Inoue T, Katsuki M, Miyajima A. Targeted disruption of Oncostatin M receptor results in altered hematopoiesis. Blood. (2003) 102:3154–62. doi: 10.1182/blood-2003-02-0367
68. Minehata K, Takeuchi M, Hirabayashi Y, Inoue T, Donovan PJ, Tanaka M, et al. Oncostatin m maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int J Hematol. (2006) 84:319–27. doi: 10.1532/IJH97.06090
69. Komori T, Tanaka M, Senba E, Miyajima A, Morikawa Y. Deficiency of Oncostatin M receptor beta (OSMRbeta) exacerbates high-fat diet-induced obesity and related metabolic disorders in mice. J Biol Chem. (2014) 289:13821–37. doi: 10.1074/jbc.M113.542399
70. Arai I, Tsuji M, Miyagawa K, Takeda H, Akiyama N, Saito S. Repeated administration of IL-31 upregulates IL-31 receptor A (IL-31RA) in dorsal root ganglia and causes severe itch-associated scratching behaviour in mice. Exp Dermatol. (2015) 24:75–8. doi: 10.1111/exd.12587
71. Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. (2006) 117:411–7. doi: 10.1016/j.jaci.2005.10.033
72. Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. (2014) 133:448–60. doi: 10.1016/j.jaci.2013.10.048
73. Feld M, Garcia R, Buddenkotte J, Katayama S, Lewis K, Muirhead G, et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol. (2016) 138:500–8 e24. doi: 10.1016/j.jaci.2016.02.020
74. Wallace PM, MacMaster JF, Rouleau KA, Brown TJ, Loy JK, Donaldson KL, et al. Regulation of inflammatory responses by Oncostatin M. J Immunol. (1999) 162:5547–55.
75. Butzkueven H, Zhang J-G, Soilu-Hanninen M, Hochrein H, Chionh F, Shipham KA, et al. LIF receptor signaling limits immune- mediated demyelination by enhancing oligodendrocyte survival. Nat Med. (2002) 8:613–9. doi: 10.1038/nm0602-613
76. Dubey A, Izakelian L, Ayaub EA, Ho L, Stephenson K, Wong S, et al. Separate roles of IL-6 and Oncostatin M in mouse macrophage polarization in vitro and in vivo. Immunol Cell Biol. (2018) 96:257–72. doi: 10.1111/imcb.1035
77. Ayaub EA, Dubey A, Imani J, Botelho F, Kolb MRJ, Richards CD, et al. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci Rep. (2017) 7:13281. doi: 10.1038/s41598-017-13511-z
78. Lauber S, Wong S, Cutz JC, Tanaka M, Barra N, Lhotak S, et al. Novel function of Oncostatin M as a potent tumour-promoting agent in lung. Int J Cancer. (2015) 136:831–43. doi: 10.1002/ijc.29055
79. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
80. Shrivastava R, Asif M, Singh V, Dubey P, Ahmad Malik S, Lone MU, et al. M2 polarization of macrophages by Oncostatin M in hypoxic tumor microenvironment is mediated by mTORC2 and promotes tumor growth and metastasis. Cytokine. (2018) S1043-4666(18)30118-2. doi: 10.1016/j.cyto.2018.03.032. [Epub ahead of print].
Keywords: Oncostatin M (OSM), central nervous system, cell biology, homeostasis, pathology
Citation: Houben E, Hellings N and Broux B (2019) Oncostatin M, an Underestimated Player in the Central Nervous System. Front. Immunol. 10:1165. doi: 10.3389/fimmu.2019.01165
Received: 21 February 2019; Accepted: 08 May 2019;
Published: 29 May 2019.
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Carl D. Richards, McMaster University, CanadaYoshiro Ohara, Kanazawa Medical University, Japan
Copyright © 2019 Houben, Hellings and Broux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bieke Broux, Ymlla2UuYnJvdXhAdWhhc3NlbHQuYmU=