 Wen-Tao Ma
Wen-Tao Ma Fei Gao1
Fei Gao1 De-Kun Chen
De-Kun Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 24 May 2019
Sec. Antigen Presenting Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01140
This article is part of the Research TopicMonocyte Heterogeneity and FunctionView all 19 articles
Monocytes (Mo) and macrophages (Mϕ) are key components of the innate immune system and are involved in regulation of the initiation, development, and resolution of many inflammatory disorders. In addition, these cells also play important immunoregulatory and tissue-repairing roles to decrease immune reactions and promote tissue regeneration. Several lines of evidence have suggested a causal link between the presence or activation of these cells and the development of autoimmune diseases. In addition, Mo or Mϕ infiltration in diseased tissues is a hallmark of several autoimmune diseases. However, the detailed contributions of these cells, whether they actually initiate disease or perpetuate disease progression, and whether their phenotype and functional alteration are merely epiphenomena are still unclear in many autoimmune diseases. Additionally, little is known about their heterogeneous populations in different autoimmune diseases. Elucidating the relevance of Mo and Mϕ in autoimmune diseases and the associated mechanisms could lead to the identification of more effective therapeutic strategies in the future.
Monocytes (Mo) and macrophages (Mϕ) possess broad immuno-modulatory, inflammatory, and tissue-repairing capabilities and actively participate in the development of many autoimmune diseases (1). These cells can secrete a wide range of cytokines and chemokines, which stimulate and recruit additional immune cells to diseased tissue (2). In many autoimmune diseases, the presence of autoantibodies and autoreactive B and T cells indicates that adaptive immune system is critical for pathogenesis, but this cannot fully account for the development of autoimmune diseases, and the innate immune response may play a necessary and irreplaceable role as well (1, 3). In fact, Mo or Mϕ infiltration is generally observed in many autoimmune diseases (4–13). Additionally, a change in the count or frequency of Mo/Mϕ is a hallmark of several autoimmune diseases, i.e., systemic sclerosis (SSc), rheumatoid arthritis (RA), primary biliary cholangitis (PBC), Sjögren's syndrome (SS), and inflammatory bowel disease (IBD) (4, 5, 10, 14–17). However, it should be noted that Mo/Mϕ frequency and count in the peripheral blood or afflicted tissues can be affected by several factors including at least bleeding regimes (for instance time of bleeding) and status of the patients (medical treatment, food intake, age, sex etc.). Thus, Mo/Mϕ frequency and count and their correlation with disease stage are usually controversial in different studies.
Although the regulatory mechanism of Mo and Mϕ in the development of autoimmune diseases has not been fully elucidated, consensus appears to suggest that their abnormal activation plays a key role. Typically, M1-polarized Mϕ are pro-inflammatory and secrete interleukin (IL)-12 and tumor necrosis factor (TNF)-α to contribute to local inflammation, while M2-polarized Mϕ produce IL-4 and IL-10 that mount immunomodulatory, wound repair and tissue remodeling functions [as reviewed by Funes et al. (18)]. However, the M1/M2 dichotomy may oversimplify a more complex activation mechanism. In fact, in certain autoimmune diseases, both M1- and M2-polarized Mϕ are detected simultaneously, and both M1- and M2-stimulating cytokines are present on a large scale (19–22). Additionally, Mϕ even exhibit an intermediate activation status by co-expressing both M1- and M2-specific markers in certain diseases (23, 24). Furthermore, in many cases, Mϕ polarization is a dynamic and reversible event that depends upon the local environment and stage of disease (25).
In the present review, we will discuss our current understanding of the properties of Mo/Mϕ in certain autoimmune diseases, highlighting the phenotypical, functional, and activation properties of these cells in disease pathogenesis and the relevant mechanisms (Summarized in Tables 1, 2). Because there are very limited reports regarding the role of Mo/Mϕ in autoimmune Addison's disease, autoimmune thyroid disease, antiphospholipid syndrome, and myasthenia gravis, these four diseases are not discussed in the present article.

Table 1. Characteristics of Mo and Mϕ in autoimmune diseases.

Table 2. Mechanisms of Mo/Mϕ activities in autoimmune diseases.
Mo percentage and count have been analyzed in SLE patients, but the findings vary among different studies. One group found that although the absolute number of the whole Mo population was similar between SLE patients and healthy controls, the rate and absolute number of CD14+CD16+ Mo was significantly higher in SLE patients, and steroid therapy could down-regulate the percentage and number of these cells in a dose-dependent manner (26). In contrast, a more recent study based on 205 SLE patients and 74 healthy controls reported decreased absolute Mo counts in SLE patients (27). However, there was no significant difference in the proportions of various Mo subpopulations. In addition, neither the absolute count nor the percentage of various Mo subsets was associated with disease activity (27). It appears that the reduction in Mo count in the latter study is supported by an independent study, which showed that Mo and Mϕ are more fragile and likely to undergo apoptosis (analyzed by flow cytometry with annexin V and propidium iodide) when induced by the sera of SLE patients (199). Although the detailed mechanism remains unclear, it appeared that C5a complement was involved in this process while serum IgG autoantibody was not involved, since Mo apoptosis profile correlated positively with C5a level, and depletion of IgG did not affect such apoptosis. In lupus mice, Mϕ depletion leads to attenuated skin and kidney disease severity, suggesting that these cells do play a critical role in SLE pathogenesis (200).
One of the contributions of Mo/Mϕ to SLE pathogenesis is modulation of the adaptive immune system. The binding of co-stimulatory molecule CD40 to its ligand CD40L is required for the activation of humoral immune responses including B cell activation, plasma cell differentiation, antibody secretion, and isotype-switching (201). In patients with SLE, a significant increase in the frequency of CD40L-expressing peripheral Mo was observed compared with healthy controls (28). Consistent with this finding, although B cells from SLE patients and normal controls showed similar CD40 expression levels, recombinant CD40L significantly stimulated the production of total IgG by SLE B cells but not normal B cells (202). In addition, data from murine studies showed that CD40L overexpression could induce lupus-like autoimmune disease, while CD40L neutralization prevented autoreactive B cell activation and autoantibody production in lupus-prone mice (203, 204). Thus, although direct evidence is still lacking, these data do suggest a potential contribution of Mo to the observed B cell hyperactivity in SLE patients through the CD40/CD40L signaling pathways. Moreover, Mo from SLE patients tend to differentiate into dendritic cells that express higher levels of CD86 when induced by IFN-α in the serum, and this potentiates them with higher abilities to present autoantigens to autoreactive T cells and B cells (29, 30).
Overexpression of adhesion molecules may lead to aberrant Mϕ migration and activation. Mϕ from active SLE patients overexpress intercellular adhesion molecule (ICAM)-1, which is associated with tissue recruitment and inflammatory cytokine production, and this is partially offset by corticosteroid therapy (31, 32). SLE Mϕ also express increased levels of sialic acid–binding Ig-like lectin 1 (Siglec-1, CD169), which could be dramatically reduced by high-dose glucocorticoid treatment (33). However, in view of the broadly anti-inflammatory effect of glucocorticoid (205), it should be noted here that this study could not rule out the possibility that the reduction in Siglec-1 expression level might result from a bystander effect of glucocorticoid treatment. Increased Mϕ Siglec-1 expression may constitute a potent inflammatory signal to promote the activation of autoimmune CD4+ or CD8+ T cells (206). In fact, it was suggested that Siglec-1 expression in Mϕ could serve as a potential biomarker for lupus activity, as the percentage of Siglec-1-expressing Mϕ was shown to positively correlate with SLE Disease Activity Index and autoantibody levels (33).
Defective phagocytosis of Mϕ has also been suggested to contribute to autoimmunity in SLE. The phagocytic capacity of Mϕ is crucial for the clearance of dead cells and debris, which otherwise can be important sources of autoantigens. Accumulating data from in vitro studies and murine models illustrate that ineffective clearance of apoptotic cells by Mϕ might be an important trigger of the autoimmune process in SLE. Two decades ago, it was observed that non-inflammatory phagocytosis of apoptotic cells by Mo-derived Mϕ (MDMs) was strikingly impaired in SLE patients (34). In addition, apoptotic cells were found to accumulate in the germinal centers of the lymph nodes in patients with SLE, and in vitro uptake of autologous apoptotic cells into Mo-derived Mϕ from SLE patients was significantly impaired (35). Interestingly, the percentage of apoptotic polymorphonuclear neutrophils (PMN) from SLE patients was significantly higher than that from healthy controls, and this percentage correlated positively with SLE Disease Activity Index and serum levels of autoantibodies (207). In addition, the phagocytosis defect may be compounded by the serum milieu of SLE patients because serum from these patients had a strong capacity to accelerate the apoptosis rate of PMN and Mϕ, which might further contribute to the high load of potential autoantigens (199, 207). Based on data from murine models, it was found that Mϕ with low expression of TLR9 and decreased TLR responsiveness to nucleic acids were largely responsible for the immunologically silent clearance of apoptotic cells (108), which was consistent with the finding that TLR9 was required in SLE pathogenesis (208). In addition, transcription factors Kruppel-like factors 2 (KLF2) and 4 (KLF4) are also important regulators of apoptotic cell clearance in SLE prone mice (108). Intriguingly, it appears that cues from the tissue microenvironment dictated these characteristics of Mϕ, as removal of these cells from specific tissues resulted in their inability to engulf apoptotic cells without generating inflammatory responses (108). A second feature of the impaired phagocytic capacity of SLE Mϕ is the delayed clearance of immune complexes (IC). Elegant work by Michael M. Frank and coworkers examined the half-time of IgG-sensitized, 51Cr-labeled erythrocytes as a measure of Fc receptor function (209). They showed that Fc-specific clearance rates were strikingly prolonged in 13 of 15 patients, and this correlated with elevated levels of IC and with disease activity. Supporting this conclusion, another study by Maria et al. has recently showed that decreased Fc receptor function correlated positively with disease activity and renal involvement (36). In addition, certain Fc receptor polymorphisms appears to determine the clearance of IC in vivo, and their heritage is associated with the course of SLE in some ethnic populations (210, 211). Abnormal Mϕ activation has also been observed in SLE patients. Labonte et al. demonstrated that higher activation profiles of Mϕ were associated with more active cases of SLE (212). In addition, Mϕ activation syndrome, a rare but usually very severe or even life-threatening complication has been reported in SLE patients (213, 214).
Accumulating findings suggest the predominance of M1 Mϕ in SLE pathogenesis. Excessive pro-inflammatory M1 Mϕ-related cytokines are produced by Mϕ from SLE patients, including IL-1β (37), interferon (IFN)-γ (19), C-X-C motif chemokine 10 (CXCL10) (38), and C-C motif chemokine ligand 2 (CCL2) (39). In addition, the pro-inflammatory serum milieu of SLE patients also favors M1 polarization, including high levels of IFN-γ, TNF-α, and granulocyte-Mϕ colony-stimulating factor (GM-CSF) (40, 112). M1 dominance may come at the expense of insufficient anti-inflammatory M2 polarization. It was shown that serum transforming growth factor (TGF)-β levels were significantly reduced in lupus patients, and TGF-β levels showed a reverse association with disease activity and organ damage in SLE patients (215). In addition, antibodies against scavenger receptors (an M2 Mϕ marker) or scavenger receptor knockout in lupus prone mice led to a compromised ability of Mϕ to engulf apoptotic cells and resulted in more aggravated SLE symptoms (109).
Considering the pro-inflammatory nature of M1 Mϕ and that M2 Mϕ are anti-inflammatory and are capable of engulfing apoptotic cells during apoptosis, it is reasonable to assume that M1 predominance and M2 insufficiency combine to worsen SLE severity. Indeed, researchers are trying to cure SLE by modulating Mϕ polarization. In a murine model of SLE, adoptive transfer of anti-inflammatory M2a Mϕ induced by IL-4 significantly decreased SLE activity (216). In patients with SLE, pioglitazone treatment enhanced M2 polarization of Mo-derived Mϕ, increasing their anti-inflammatory capacity while suppressing their production of various pro-inflammatory cytokines (217).
Available online at: Intriguingly, large amounts of IL-10, which is a hallmark of M2 Mϕ, are commonly detected in patients with SLE, and serum levels of IL-10 correlate positively with disease activity (20, 21). Contrary to its canonical anti-inflammatory functions, IL-10 in SLE acquires a pro-inflammatory capacity. This is largely dependent on high concentrations of type I IFNs, which confer a pro-inflammatory gain of function upon IL-10 and lead to a positive feedback loop of pro-inflammatory cytokine production (113). Priming of primary human Mϕ with IFN-α resulted in significantly enhanced STAT1 activation in the presence of IL-10, leading to activation of several STAT1-dependent genes such as CXCL9, CXCL10, and IFN regulatory factor 1 (113). In addition, IL-10 can directly stimulate production of platelet-activating factor (a phospholipid mediator of inflammation) of Mo of SLE patients (218). Indeed, IL-10 antagonist administration ameliorated SLE severity effectively during a 6-month therapy, even though this finding was limited by the small sample size of the study (219).
In addition to IL-10, SLE Mo or Mϕ also produce copious IL-6 and TNF-α. Elevated IL-6 levels are positively associated with disease activity or autoantibody levels (114). The underlying mechanism seems to be the stimulation of B cell hyperactivity by IL-6 (220). Indeed, in an open-label phase I dosage-escalation study, IL-6 receptor inhibition showed a significant decrease in the frequency of circulating plasma cells, reduced autoantibody levels in the serum, and significant disease improvement (221). Another cytokine, TNF-α, is generally reported to be elevated in SLE and positively associated with disease activity (112). However, TNF-α blockade therapy in SLE is controversial. Although this therapy was shown to reduce disease severity, autoantibodies to double-stranded DNA and cardiolipin increased during treatment (222). Furthermore, it seems that TNF-α blockade is safe only for short-term treatment, while long-term therapy would likely provoke severe adverse effects such as lymphoma and Legionella pneumonia (223).
Intriguingly, in lupus prone NZB/W and NZW/BXSB mice, nephritic resident CD11bhiF4/80hi Mϕ exhibit little arginase- or iNOS-producing ability even after stimulation with M1 or M2 Mϕ-inducing cytokines, irrespective of the clinical status of the mice (224). Instead, these kidney residents show a mixed pro- and anti-inflammatory phenotype during lupus nephritis (224). In contrast, Mo-derived Mϕ of the same mice were readily responsive to cytokine stimulation and can be induced to differentiate into the correspondingly M1 or M2 cells (224). In addition to these phenotypic differences, differences, functional analysis showed that nephritic resident Mϕ had higher antigen-presenting function and phagocytosis ability compared with MDMs of the kidney (224).
Several molecules and pathways have been suggested to be associated with controlling polarization and inflammatory profiles of Mϕ. For example, using MDMs of normal subjects and SLE patients, Saeed et al. found that epigenetic modification is partly responsible for the Mϕ polarization profile in SLE (225). Their data showed that sodium valproate, an histone deacetylase inhibitor, can potently induce the alternative activation of Mo-Mϕ ex vivo and inhibit the pro-inflammatory profile of these cells when stimulated by apoptotic cells in vitro (225). The same group also found that aryl hydrocarbon receptor (AhR)-mediated signaling pathway is responsible for the secretion of anti-inflammatory cytokines and expression of M2 markers from MDMs of SLE patients, as AhR agonist treatment of these cells led to a significant downregulation of M1 markers and expression of pro-inflammatory cytokines, coincided with an upregulation of M2 markers and expression of anti-inflammatory cytokines (226). In addition, decreased peroxisome proliferator-activated receptor gamma (PPAR-γ) expression has also been proposed to be involved in the defective efferocytosis and abnormal pro-inflammatory characteristics of MDMs from SLE patients (217).
More than two decades ago, Ishikawa et al. stained skin specimens from patients with SSc and found that Mϕ infiltration was generally observed around skin adnexa and vessels as well as between collagen bundles, while no close relationship with disease duration was found (6). Later, another group showed that the percentage of peripheral Mo in SSc is significantly higher than that in healthy controls. Notably, this higher percentage of Mo also correlated with worse prognosis and visceral disease involvement (14). However, in this study, Mo were not detected specifically through CD14 but instead were gated indirectly through CD3+CD4−, weakening the general application of this conclusion. Using a more specific Mϕ marker, another group showed that the number of CD68+ Mϕ was significantly higher in the skin of patients with localized SSc (41). The same group also found that the percentage of CD14+ circulating Mo was significantly greater in SSc patients than in healthy controls (43). In a more recent study, Lescoat et al. found that SSc patients had an elevated count of total peripheral Mo relative to healthy controls (42). Notably, the CD16+ subpopulation count was more significantly increased in diffuse SSc than in limited SSc. In addition, the absolute count of CD16+ Mo was significantly associated with the severity of skin fibrosis, pulmonary fibrosis, restrictive ventilatory defect, and pulmonary function impairment, suggesting a potential link between this subpopulation of Mo and the pathogenesis of fibrosis in SSc (42, 227). A potential mechanism underlying the increased Mϕ count may involve Mϕ migration inhibitory factor (MIF), which is capable of suppressing the random migration of Mϕ to concentrate them at inflammatory loci. Selvi et al. has reported the detection of high levels of MIF in the basal and suprabasal keratinocytes of SSc-affected skin (119). In addition, elevated concentrations of MIF in the peripheral blood of patients with diffuse cutaneous SSc were identified.
Several lines of evidence have implicated the functional abnormality of SSc Mo/Mϕ. It was reported that SSc Mo were more pro-fibrotic, as they displayed increased differentiation potential toward type-1 collagen- and α-smooth muscle actin (SMA)-expressing cells after stimulation (44). In addition, the production of tissue-inhibitor of metalloproteinase-1 (TIMP-1), an important protein capable of inhibiting extracellular matrix degradation, is significantly increased in SSc Mo mediated by TLR/MyD88 signaling and the transcription factor Fos-related antigen 2 (20, 21, 126). SSc Mo/Mϕ also show abnormally expressed markers that are associated with certain functions. First, increased expression of Siglec-1 in tissue Mϕ and circulating Mo of SSc patients was identified, suggesting a potential role for type 1 IFN-mediated Mo/Mϕ activation in SSc (45). In line with this finding, levels of IFN-α mRNA were significantly increased in vascular and perivascular cells in skin samples of SSc patients (228). However, how Siglec-1 is induced and to what extent it contributes to the pathogenesis of SSc need further verification. In a recent study, Moreno-Moral et al. explored the contribution of MDMs in mediating genetic susceptibility to SSc. By conducting genome-wide genotyping and RNA-sequencing, their work shows that gene expression in Mϕ from SSc patients is altered, especially higher expression of the GSDMA and GRB10 genes (229). However, the relevance of these results at the protein level has yet to be examined in the future.
Mounting evidence suggests the predominant M2 polarization of Mϕ in SSc. The elegant work of Nobuyo et al. showed an evident increase in the number of CD14brightCD163+CD204+ Mϕ in the fibrotic areas of the SSc skin (41, 43), suggesting that this cell subset may be potential a regulator of fibrosis in SSc skin. Of note, CD204-deficient mice failed to develop silica-induced fibrosis, suggesting a critical role for this scavenger receptor in fibrosis (230). This finding was underpinned by the works of several other groups, which reported that a soluble form of CD163 (sCD163), released from the Mϕ cell surface, was increased in the sera of SSc patients relative to the general population (231–233). Intriguingly, sCD163 secretion by PBMCs ex vivo may serve as a biomarker of SSc progression, as increased production of sCD163 by PBMCs was associated with worse prognosis of SSc (233). In addition, urinary sCD163 concentrations were also higher in SSc patients, but the difference was not statistically significant (232). In line with these findings, several reports have shown elevated serum levels of M2-inducing cytokines, i.e., IL-4, IL-13, and IL-10, in patients with SSc (234–236).
A growing body of data has suggested that M2 Mϕ play crucial roles in the activation of resident fibroblasts and the progression of fibrosis, mainly through the release of TGF-β, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) (237, 238). Indeed, high levels of TGF-β and PDGF as well as their contribution to SSc have been reported by several groups (46–48). Data from skin samples of SSc patients and healthy control subjects showed that in SSc, the production of TGF-β by Mϕ was partly induced by Cadherin11, which has been implicated in both pulmonary and skin fibrosis (48). In murine studies, blockade of Cadherin11 led to fewer myofibroblasts and decreased dermal thickness in established fibrosis (48). However, whether this treatment may be therapeutically effective needs further verification.
Intriguingly, a recent study revealed that a considerable number of M2 Mo (CD204+CD163+CD206+) co-expressed M1 markers (CD80 and CD86) in the PBMCs of SSc patients, and this subset of cells constituted a significant feature that characterized SSc (23). In addition, down-regulation of the IL-6/signal transducer and activator of transcription 3 (STAT3) signaling pathway was identified in SSc Mo-derived Mϕ (239). These data suggest a more complex activation profile of SSc Mo/Mϕ, consistent with the remarkable plasticity of these cells. Further investigation into the polarization state of Mo/Mϕ in different stages of SSc is needed, and the exact role of these cells should be clarified.
Mϕ infiltration in the synovia is one of the most important hallmarks of RA. There is ample evidence that the frequency and absolute number of Mϕ are markedly increased in the synovial tissues of patients with RA (4, 5). More importantly, this phenomenon could serve as a reliable biomarker for disease activity. Mulherin et al. showed that synovial Mϕ number correlated positively with articular destruction in RA (240). In a study based on 66 patients with RA, it was found that local disease activity in particular was positively associated with the number of synovial Mϕ as well as levels of IL-6 and TNF-α, two major Mϕ-derived cytokines (241). Accordingly, it was suggested that synovial Mϕ count may also reflect the therapeutic efficacy of RA. An early study by Ghada et al. found that the number of synovial CD68+ Mϕ was significantly reduced 12 weeks after treatment with sodium aurothiomalate (242). A further study investigated synovial tissue biopsies from 88 patients with RA participating in various clinical trials, and the authors found that the number of synovial Mϕ correlated significantly with disease activity score, and that a decrease in this number was positively correlated with clinical improvement of RA, independent of the therapeutic strategies these patients received (243). In line with these findings, it was important to find that sublining Mϕ did not change in response to placebo or ineffective treatment (243, 244). These findings were corroborated by data from rodent models of arthritis. It was recently shown that experimental arthritis was accompanied by enhanced survival of synovial Mϕ and would be markedly improved in genetically modified mice in which Mϕ were more susceptible to apoptosis (131). In this study, Mϕ survival is induced by increased expression levels of nuclear factor of activated T cells 5, the expression of which is stimulated by the inflammatory tissue microenvironment of the arthritic mice. Importantly, experimental arthritis was significantly alleviated after local Mϕ depletion by knee joint clodronate liposome injection (245). Moreover, inhibition of Mϕ differentiation from Mo also ameliorated synovial inflammation in experimental arthritis (246). These findings suggest that Mϕ play a key role in RA pathogenesis.
A growing number of studies have highlighted the central role of Mϕ activation in RA pathogenesis. To be specific, unrestrained pro-inflammatory M1 polarization with incomplete M2 polarization usually leads to more severe joint pathology, and thus Mϕ polarization modulation usually alters the outcome of experimental arthritis. In a collagen II-induced arthritis mouse model, it was found that cyclophilin A, a potent pro-arthritic protein, aggravated the severity of arthritis through the induction of pro-inflammatory M1 Mϕ polarization and cytokine production in the knee joint (247). On the other hand, efficiently repressed M1 polarization or increased anti-inflammatory M2 polarization suppressed synovial inflammation and held promising potential as a targeted therapy for RA. In collagen II-induced murine arthritis and spontaneous arthritis in Hes1-GFP/TNF-transgenic mice, inhibited M1 polarization and simultaneously enhanced M2 polarization of Mϕ significantly reduced the inflammatory response in the knee joints (248, 249). Likewise, collagen-induced arthritis was efficiently ameliorated by the administration of mesenchymal stem cells, which have potent immunomodulatory capabilities (250–252). In addition, IL-10 was able to suppress the observed effects of pro-inflammatory M1 Mϕ in experimental arthritis, partly due to inhibition of the inflammation-associated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway or pro-inflammatory cytokine secretion from Mϕ (253, 254). Data from murine model of RA showed that synovial tissue-resident Mϕ and MDMs play different roles in experimental RA. Misharin et al. found that Ly6C− Mo are recruited into the synovial tissue and differentiate into pro-inflammatory M1 Mϕ during the effector phase of arthritis, thus driving initiation and progression of joint inflammation. During the resolution phase, these cells are polarized toward an alternatively activated phenotype and contribute to the resolution of arthritis (13). In comparison, synovial tissue-resident Mϕ are anti-inflammatory throughout the course of arthritis and inhibit joint inflammation during the initiation phase (13).
Activated Mϕ are a potent source of various pro-inflammatory cytokines, which are essential mediators of the effects of Mϕ during the development of RA (56, 132, 255). TNF-α is a key cytokine that is produced by synovial Mϕ and is of critical importance in the pathogenesis of RA (51, 249, 256). This cytokine is present in most arthritis biopsies, and its overexpression induces spontaneous inflammatory arthritis, whereas its inhibition suppresses various rodent arthritis models (134, 135). Accordingly, therapeutic targeting of TNF-α signaling has yielded clinical efficacy in patients with established RA, which has also been corroborated by a number of mouse model-based results (257–259). Other Mϕ-derived cytokines such as IL-1, IL-6, and IL-12 are also abundantly present in the arthritic synovium of patients with RA (134, 135). Similarly, they are indispensable for the inflammatory responses in the synovia of patients with RA, and blockade of their signaling pathways improves clinical or experimental arthritis (52–55).
In progressive MS, central nervous system (CNS) inflammation is characterized by widespread activation of mononuclear phagocytes (MPs), which include both Mo-derived Mϕ and resident microglia (58). These MPs are found in both gray and white matter lesions, are close to degenerating areas, and are associated with chronic tissue damage (11, 12). In addition, in normal-appearing white matter, MP infiltration is associated with the formation of microglial nodules that lead to disease pathology (260). It has been suggested that staging of MS lesions can be determined based on the presence of CD68-positive Mϕ and human leukocyte antigens, together with the degree of myelin loss (59). The detrimental role of MP-driven pathology in MS is also supported by evidence from murine models, which has shown that the overall burden of MPs correlates with brain atrophy (261), impaired neuronal function (262), and decreased regenerative responses (263). These findings are underpinned by evidence from clinical trials, as induction of Mϕ apoptosis by IFN-β showed a significant benefit in MS (264). In addition, in murine models, Mϕ depletion showed significantly suppressed CNS damage and clinical signs of experimental autoimmune encephalomyelitis (265, 266).
Using brain autopsy tissue from patients with MS, Tobias et al. found that the main functional changes in Mϕ and microglia are increased expression levels of molecules associated with inflammation, including CD68 (phagocytosis), human leukocyte antigen (HLA) and CD86 (antigen presentation and co-stimulation), and inducible nitric oxide synthase (iNOS) (microglia activation) (60). Another group, George et al. found that Mϕ of MS patients display deficient SHP-1 mRNA and protein expression, leading to heightened activation of STAT1, STAT6, and NF-κB signaling and a corresponding enhanced inflammatory profile (142). In addition, data from experimental autoimmune encephalomyelitis (EAE), an animal model of MS, has shown a critical role for Mϕ in triggering adaptive immune responses. For example, Mϕ NLPR3 inflammasome plays a key role in inducing migration of autoreactive T cells into the CNS in EAE (144). Mϕ also produce several key cytokines (i.e., IL-1β, IL-6, and IL-23) to promote the generation and maintenance of Th17 cells, a key cell subset mediating CNS autoimmunity in EAE (145–147). In addition, TLR7-mediated productions of IL-6 and B cell-activating factor (BAFF) are crucial cytokines for autoreactive B cell survival and differentiation (150). In consistent with these findings, Mϕ depletion or anti-GM-CSF treatment inhibits the induction of myelin antigen-specific Th17 cells and protects mice from clinical symptoms of EAE (146, 267–269).
Ample evidence indicates that inflammatory Mϕ in MS show abnormal metabolic changes. Generally, Mϕ activated by inflammatory stimuli switch their core metabolism from oxidative phosphorylation (OXPHOS) to glycolysis (61). Recent evidence shows that inflammatory Mϕ accumulate succinate, which inhibits the function of prolyl hydroxylase enzymes during this metabolic shift, thereby inducing the transcription and secretion of IL-1β as an additional pro-inflammatory signal (61). In line with this finding, Luca et al. recently showed that inhibition of succinate release from MPs can reprogram their metabolism back to OXPHOS, resulting in an anti-inflammatory phenotype of Mϕ and ameliorated experimental autoimmune encephalomyelitis (270).
Many lines of evidence indicate that Mϕ play divergent roles in the pathogenesis of MS as they exacerbate tissue injury but also show remarkable growth-promoting and neuroprotective effects (271, 272). Obviously, this dual role of Mϕ in MS can be explained by their polarization state. In fact, both M1 and M2 subsets are present in MS lesions. The pro-inflammatory M1 response is rapidly induced and then maintained at sites of CNS injury. In comparison, the immunoregulatory M2 response is comparatively weaker and more transient (271). Thus, when inflammatory signals released by type 1 MPs are suppressed by neural stem cell-derived immunoregulatory factors, significantly ameliorated CNS inflammation can be observed (270). On the contrary, sodium chloride treatment of Mϕ induced an enhanced pro-inflammatory activity of these cells and aggravated CNS autoimmunity in EAE-diseased mice (273). In addition, IL-33 treatment induced significantly ameliorated EAE, accompanied by M2 polarization of Mϕ. Of note, adoptive transfer of IL-33-treated Mϕ attenuated EAE development, suggesting the importance of IL-33-mediated Mϕ polarization in the development of EAE (274). In consistent with this finding, Miron et al. found that immunomodulatory M2 Mϕ were essential for oligodendrocyte differentiation through activin A production (275). Notably, the dichotomy of Mϕ polarization in MS is not accurate, as the majority of Mϕ in active MS lesions show an intermediate activation status, characterized by the co-expression of both M1- and M2-specific markers (24). In addition to their polarization state, the dual role of Mϕ in MS pathogenesis can also be accounted by the origins of CNS Mϕ. In fact, resident microglia and Mo can both give rise to Mϕ that exhibit distinct expression profiling in the CNS (276). Yamasaki et al. found the distinct functional capacities of these two Mϕ in EAE. They showed that resident macroglia were associated with debris clearance and demonstrated a signature of globally suppressed cellular metabolism during disease initiation, whereas Mo-derived Mϕ were highly phagocytic and inflammatory and actively participated in demyelination demyelination initiation (277).
There are scant data describing correlations between Mo and Mϕ counts and T1D development. In one study, the absolute count of circulating Mo was significantly increased in patients with T1D, while the number of CD16+ Mo decreased in patients with diabetic complications (62). Unfortunately, this study did not analyze the correlation between Mo number and T1D development. Another study found that decreased Mo counts significantly correlated with insulin resistance in T1D, although this study lacked data on healthy controls and thus could not prove a relationship between Mo number and T1D development (278).
Two independent studies showed that Mϕ from diabetes-prone non-obese diabetic (NOD) mice showed markedly compromised phagocytosis relative to those from normal mice (63, 64). Since Mϕ engulfment of apoptotic cells is an important mechanism of self-antigen clearance, it was thus suggested that deficiencies in apoptotic cell clearance by Mϕ represent a potential factor in predisposition to T1D. In addition, Mϕ from NOD mice were shown to be abnormally activated and exhibited direct cytolytic activity toward islet β-cells (65). Accordingly, in vivo depletion of Mϕ by clodronate liposomes abolished diabetes effectively.
In T1D, Mϕ play a key role in triggering the adaptive immune responses. Vomund et al. showed that islet beta cells can transfer some of their secretory granules to resident Mϕ. In autoimmune diabetes, these Mϕ present the transferred antigens to autoreactive CD4+ T cells, resulting in the activation of these cells and initiating the autoimmune diabetic process (279). Mϕ are also involved in the trafficking of autoreactive CD8+ T cells into the islets. Marro and colleagues found that depletion of Mϕ or genetic ablation of ifnar on Mϕ aborted lymphocytic choriomeningitis infection-induced T1D (280). Mechanistically, disrupted type-I IFN signaling in Mϕ restricted trafficking of CD8+ T cells into the islets, thus prohibiting the further development of murine T1D (280).
In T1D, the abnormal activation of Mϕ is exemplified by the pro-inflammatory M1 phenotype of these cells, which play a critical role in T1D pathogenesis. The pro-inflammatory serum milieu of T1D patients that favors M1 Mϕ polarization is exemplified by excessive amounts of C-reactive protein (66), IFN-γ (67), CXCL10 (68), and CCL2 (68). This M1 dominance of T1D Mo is reflected in the elevated IL-6- and IL-1β-secreting ability of these cells, regardless of whether they were in a resting state or after lipopolysaccharide stimulation (66, 69). It was suggested that a main function of these two cytokines is to induce the generation of Th17 cells, which is another key cell population in T1D pathogenesis (69). In addition to the aforementioned two cytokines, several lines of evidence have shown elevated levels of Mϕ-derived TNF-α in T1D patients (70, 71). However, the function of TNF-α in T1D pathogenesis seems controversial. Although TNF-α blockade therapy showed clinical efficacy in some cases, others showed disturbance of glycemic control after treatment, and one study even reported induction of T1D during anti-TNF-α therapy in a RA patient (281, 282).
While pro-inflammatory M1 Mϕ promote T1D development, adoptive transfer of immunosuppressive M2 Mϕ reduces the onset of T1D in NOD mice (283). In fact, more than 80% of NOD mice were protected against T1D for at least 3 months after a single transfer of M2 Mϕ, even if the treatment was conducted just prior to clinical onset. Moreover, in vitro induced M2 Mϕ can also reduce hyperglycemia, kidney injury, and insulitis in diabetic mice (284).
The pancreas contains both MDMs and resident Mϕ that exert different functional capacities. Bone marrow Mϕ have been found to prevent stem cell mobilization into peripheral blood in diabetic mice (285). In contrast, the islet resident Mϕ exhibit an activation signature with higher expression of various pro-inflammatory cytokines and mount an inflammatory immune response in NOD mice (286). Consistent with these findings, in a study conducted in C57BL/6 mice, islet Mϕ express genes and cell surface markers that categorize them as M1-like and exhibited typically pro-inflammatory characteristics. In contrast, the interacinar Mϕ expressed M2-like transcripts and exhibited anti-inflammatory and tissue-supportive functions (287). Accordingly, depletion of islet resident Mϕ through CSF-1 neutralization resulted in reduced CD4+ T cell infiltration in the pancreatic islets, impaired presentation of insulin epitopes to T cells and reduced severity of autoimmune diabetes (288).
In 1994, Mathew et al. found that while Kupffer cell counts were not altered significantly in stage 1 and 2 PBC, increased Kupffer cell numbers were clearly identified in periportal and periseptal zones of stage 3 PBC and in the parenchymal areas of stage 3 and stage 4 cases (10). This finding was supported by another independent study (72). In contrast to these findings, the work of Leicester and colleagues showed that the total number of CD68+ Mϕ in the liver remained constant at different stages of fibrosis and did not differ significantly from that of controls (73). This discrepancy may result from distinct disease staging strategies or different hepatic Mϕ immune-staining and quantification methods. In addition to Mϕ, several lines of evidence also showed increased Mo counts in PBC patients. Leicester et al. revealed that while few CD14+ Mo could be observed in control livers, these cells were increased markedly in PBC livers, especially in patients with advanced stage of fibrosis (73). The work of Peng et al. showed that the frequencies of peripheral blood CD14highCD16+ and CD14lowCD16+ subpopulations of Mo were elevated in patients with PBC (74). Intriguingly, the frequency of CD14lowCD16+ cells was positively associated with disease progress. Consistent with these findings, increased levels of Mo chemotactic proteins were also identified in PBC livers (289). These findings are supported by data from murine models of PBC. In dominant-negative TGF-β receptor type II transgenic mice, clusters of Mϕ are observed in the parenchyma and portal tracts of the liver (290). In another PBC mouse model, the 2-octynoic acid-conjugated bovine serum albumin immunization-induced autoimmune cholangitis, interestingly, it was found that while MDMs (CD11bhiF4/80intCX3CR1hi) were enriched around the portal triads, liver resident Kupffer cells (CD11bintF4/80hiCX3CR1neg) were significantly reduced (161). In this study, MDMs play a key role in the development of experimental PBC, as inhibition of their recruitment either by genetic deletion of CCR2 or by pharmacological antagonization of CCR2 resulted in ameliorated autoimmune cholangitis (161).
The dysfunction of Mϕ in PBC is reflected in several findings. In 2005, Mao et al. showed that Mo isolated from the peripheral blood of patients with PBC were more sensitive to toll-like receptor (TLR) ligation and thus produced higher levels of pro-inflammatory cytokines (75). This finding was supported by another independent study, which demonstrated that the expression of TLR4 and its negative regulator RP105 were altered on PBC Mo, making them hyperreactive to LPS and leading to increased production of various pro-inflammatory cytokines (78). In an in vitro co-culture model using human peripheral blood Mo and T cells, it was shown that circulating CD14lowCD16+ Mo could promote Th1 cell proliferation by IL-12 production and direct contact of CD4+ T cells (presumably through HLA-DR-, CD80-, and CD86-mediated mechanisms). In line with these findings, circulating CD14lowCD16+ Mo were positively associated with Th1 cell frequency in PBC patients (74). Other molecules, such as Siglec-1, were also found to be abnormally overexpressed by PBC Mo (76). A great breakthrough in the abnormally altered functions of Mo and Mϕ in PBC may be achieved in studies illustrating their ability to recognize anti-mitochondrial antibody (AMA)-apotope complexes (77, 164). Apoptotic biliary epithelial cell-derived autoantigens might remain immunologically intact and can be recognized by circulating AMAs in apoptotic bodies (164). Of note, these AMA-apotope complexes are capable of activating Mo-derived Mϕ of the liver, thus stimulating the secretion of various pro-inflammatory cytokines from these cells. This effect leads to further biliary epithelial cell apoptosis, thus perpetuating local inflammation and eventually causing bile duct damage (77).
Many lines of evidence indicate a pro-inflammatory M1 polarization of Mϕ in PBC. These Mϕ express high levels of TLR4 and are highly sensitive to endotoxin stimulation, leading to markedly increased secretion of several pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8, IL-12, and TNF-α (75, 78). Interestingly, endotoxin, which is a strong stimulator of M1 Mϕ activation, is increased in biliary epithelial cells of patients with PBC (79). In addition, levels of CD40L, which interacts with its corresponding receptor CD40 and mediates potent inflammatory signals, are significantly elevated in PBC Mϕ (72). The same study also found that this increase in CD40L expression was mainly stimulated by LPS and IFN-mediated signals.
Increased levels of peripheral mature (CD14lowCD16+) Mo were described in patients with SS (15), even though their direct aetiopathogenic role remains undefined. Another Mo subset, pro-inflammatory CD14brightCD16+ Mo, is also increased in the salivary glands of SS patients, accompanied by overexpression of IL-34, a cytokine that specifically stimulates the growth and differentiation of Mo (80). In addition, the salivary profile of CCL2, a potent Mo chemoattractant, is highly expressed in patients with SS (85). Until now, there has been no direct evidence concerning the association of Mϕ or Mo numbers with human SS disease activity, even though elevated expression of Mϕ-derived molecules (i.e., molecules of the chitinase family) indeed corresponded to more severe SS (291). In addition, a study analyzing saliva proteomics showed that proteins associated with Mϕ differentiation represented one of the biomarker signatures of SS (292). In mouse models, it has been shown that Mϕ are critical mediators of SS pathogenesis and have intimate crosstalks with autoreactive T cells. Using autoimmune regulator-deficient mice as an animal model of SS, Zhou et al. demonstrated that Mϕ infiltration the limbus, corneal stroma, and lacrimal glands were mediated by autoreactive CD4+ T cells (293). Importantly, local infiltration of Mϕ correlates with ocular surface damage, and Mϕ depletion by clodronate liposomes led to significant improvements in lacrimal gland pathology (293), indicating the immunopathologic involvement of these cells in SS. In another mouse model of SS wherein NFS/sld mice are thymectomized on day 3 after birth, Ushio and colleagues found that tissue resident Mϕ of the salivary gland mediated CD4+ T cell recruitment by effective production of CCL22 (171). Moreover, CCL22 was found to enhance IFN-γ production from T cells in these mice (171). Of note, numerous CCL22-producing Mϕ can be observed in the salivary gland tissue specimens of SS patients (171).
Functional abnormalities of SS Mϕ are exemplified by impaired phagocytosis ability of them. Mϕ isolated from an SS mouse model showed defective phagocytosis of apoptotic cells (294). This finding is in line with previous reports in SS patients, as Mo from these patients showed reduced engulfment of apoptotic epithelial cells and were unable to promote an immunosuppressant cytokine profile (81). In addition, elevated levels of MIF have been shown to be associated with hypergammaglobulinemia in patients with SS (295).
There is a paucity of data on the polarization of Mϕ in patients with SS. Although Baban et al. reported the presence of M1 and M2 Mϕ along with T and B cells in the salivary glands of SS mouse model, the balance of M1 and M2 Mϕ has not been characterized (296). However, accumulating data indicate that pro-inflammatory M1 polarization is the predominant phenotype of SS Mϕ. It has been reported that systemic and local concentrations of IL-6 are significantly increased in SS patients (82). In addition, serum IL-12 levels are associated with more active disease, while an immunosuppressant cytokine, IL-35, is associated with lower disease activity (83). It has also been shown that peripheral IFN-γ levels are increased in patients with SS (84), which is suggested to be stimulated by the synergistic functions of IL-33, IL-12, and IL-23 (297). Additionally, salivary levels of the pro-inflammatory cytokines and chemokines TNF-α, IL-1β, IL-18, CXCL8, and CXCL10 are also significantly higher in SS patients than in non-SS controls (80, 85–87). Notably, levels of pro-inflammatory cytokines or chemokines that are directly secreted by Mo and Mϕ, i.e., IL-6, IL-18, type I IFN and BAFF, are significantly higher in SS patients (87, 172, 173). In accordance with the increased pro-inflammatory cytokine levels of SS Mo, these cells express reduced levels of NF-κB inhibitor (IκBα), indicating the abnormal activation of the NFκB signaling pathway (88). In addition, Adrienne et al. used freshly isolated peripheral blood Mo and found that SS-associated microRNAs collectively suppressed immunoregulatory TGF-β signaling as opposed to the pro-inflammatory IL-12 and NF-κB signaling pathways (170). Interestingly, in thymectomized NFS/sld mice, an animal model of SS, tissue resident Mϕ of the salivary gland contain two main subsets (CD11blowF4/80+ and CD11bhighF4/80+) (171). These two subsets of Mϕ display different phenotypes and functions. For example, CD11blowF4/80+ Mϕ express higher levels of pro-inflammatory M1 markers including MHC-II, CD11c, and CD86, while CD11bhighF4/80+ Mϕ express higher levels of M2 markers such as CD206 and CD204 (171). In addition, CD11bhighF4/80+ Mϕ showed significantly higher phagocytic activity compared with CD11blowF4/80+ ones (171).
Numerous CD68+ tissue Mϕ were present in duodenal biopsies from patients with celiac disease (7). Of note, these Mϕ showed strikingly impaired phagocytosis ability, as reduced expression levels of Mϕ-associated scavenger receptors, i.e., CD36, thrombospondin-1 and CD61, were identified in the duodenal mucosae of patients with the active phase of celiac disease, accompanied by the accumulation of apoptotic bodies in these areas (89). However, direct evidence for the phagocytosis ability of Mϕ is lacking. In addition, Mϕ from patients with celiac disease exhibit greater antigen-presenting ability, which is exemplified by the upregulated expression of the co-stimulatory molecules CD80, CD86, and CD40, in concert with higher CD40L expression and a more highly activated state of T cells (90, 91). However, more direct evidence is warranted to support this conclusion.
The cytokine milieu of patients with celiac disease implicates a simultaneous M1- and M2-related profiles. For one thing, significantly higher levels of M1-associated pro-inflammatory cytokines, i.e., IFN-γ, IL-1β, TNF-α, and IL-8 have been identified in celiac disease sera (22). More specifically, gliadin peptides could induce significantly higher levels of IL-8 and TNF-α production by Mo from patients with celiac disease relative to those from healthy donors. This pro-inflammatory cytokine secretion is accompanied by a more pro-inflammatory activation state of Mo expressing higher levels of M1 markers, i.e., CD80, CD86, and CD40, as well as higher activation of the NF-κB signaling (90). In addition, it was shown that gliadin fragments could induce RAW264.7 cells and mouse peritoneal Mϕ to secrete TNF-α and CCL5, and to produce increased levels of nitric oxide in the presence of IFN-γ, which is also associated with the activation of NF-κB signaling (298–300). The interaction of gliadin with Mϕ involved a myeloid differentiation factor 88 (MyD88)-dependent pro-inflammatory cascade, while this was neither TLR2- nor TLR4-dependent (176). Intriguingly, even in patients with celiac disease on a gluten-free diet whose duodenal biopsy specimens are histologically normal, intraepithelial lymphocytes and intestinal epithelial cells exhibit increased expression of TNF-α and MIF (301). This may help explain the rapidity with which the celiac mucosa responds to gliadin challenge.
Additionally, M2-associated immunosuppressive cytokines are also frequently detected in celiac disease. For example, IL-10 concentration is significantly higher in celiac disease sera (22). Importantly, serum levels of IL-10 is significantly correlated with levels of autoantibody titers (22). In addition, IL-10 polymorphisms are correlated with more severe mucosal damage and early-onset of celiac disease (302), even though IL-10 secretion abnormalities are suggested to be more a cause than a consequence of this disease (303). Using Mo from patients with celiac disease or healthy subjects, Amelia et al. found that gluten peptides induced the expression of arginase 1 and arginase 2, both of which are typical markers of M2 Mϕ (92). This finding was supported by data from the same group showing that gliadin stimulation significantly activated the arginase pathway in human Mo as well as in RAW264.7 cells (93).
In IBD, the intestinal mucosa is characterized by extensive Mϕ infiltration (8, 9). Elevated CD68+ Mϕ count in the colonic and ileal mucosae were observed in both Crohn's disease (CD) and ulcerative colitis (UC), while a CD163-positive subset in the colon mucosa was increased only in CD but not UC patients (16). In patients with CD, the mesenteric fat tissue also exhibits considerable Mϕ infiltration (9, 304). Regarding circulating Mo, it was found that Mo with a CD14+CD16+ phenotype are increased significantly and are the main contributor to the inflammatory infiltrate in the CD mucosa, while classical Mo (CD14hiCD16−) are decreased (94, 95). A dramatic increase in peripheral CD14+CD16+ Mo was observed in patients with active CD, particularly in those with colonic involvement and a high Disease Activity Index (95). Intriguingly, a significant correlation between the percentage of CD14+CD16+ Mo and clinical activity index has been shown in both CD and UC patients, suggesting the potential involvement of this cell subset in the inflammatory drive of IBD (305). Of note, computational simulations conducted by Wendelsdorf et al. identified that Mϕ and their mechanisms of plasticity are key reasons for mucosal inflammation (188).
The expression level of aldehyde dehydrogenase (ALDH), which is necessary for the synthesis of retinoic acid, is significantly reduced in Mϕ populations of the UC colon, both in active disease and remission (8). Given that retinoic acid has important immunoregulatory properties and is critical for the generation of regulatory T cells (Tregs), local suppressive failure due to a lack of retinoic acid may be involved in driving UC. In line with this finding, Treg numbers in UC patients were lower than that of healthy controls, and Treg number was negatively associated with the clinical activity index of UC (306). In comparison, the percent change in ALDH+ Mϕ in CD is controversial, as one study showed that this fraction is similar to that in controls, while another study identified up-regulated ALDH activity in CD14+ Mϕ from CD patients (8, 307). CD Mϕ also showed an abnormally accelerated breakdown of pro-inflammatory cytokines due to faster lysosomal degradation, while cytokine messenger RNA showed normal stability and levels (96). This was shown to lead to impaired neutrophil attraction, causing defective bacterial clearance and thereby boosting the formation of granulomas. However, this case differs strikingly from UC Mϕ, which showed similar or even significantly higher secretion of various cytokines relative to healthy controls in the same study. In addition, there is proof that IBD patients showed defective Mo GM-CSF receptor (CD116) expression and function, which was more prominent in UC than in CD patients, indicating a causal link between the innate immune defect in IBD patients and Mo CD116 expression (97). Intriguingly, CD116 expression in IBD patients was independent of current medications and was not influenced by disease activity.
Several studies have reported the potential interactions between colonic Mϕ and lymphocytes in IBD. Abnormally activated intestinal Mϕ in CD patients produce various cytokines (i.e., IL-1β, IL-6, IL-23, TNF-α, and TNF-like protein 1A) necessary for T cell differentiation, specifically promoting the generation of Th1 and Th17 cells (191–194). A subset of CD14 and CD209 dual positive Mϕ in the lamina propria also possess potent antigen-presenting ability and can strongly evoke the differentiation of Th1 and Th17 cells (194). In addition, these Mϕ can induce the proliferation of naive CD4+ T cells (194). Similarly, in UC patients, IL-23 from CD68+ Mϕ promotes the differentiation of Th17 cells, which are important contributors to the pathogenesis of UC (195–197). In addition, Mϕ-derived IL-23 can strongly promote the activation and cytolytic activities of intestinal NK cells crucially contributing to tissue pathology of UC patients (195). Data from murine model-based studies showed that adoptive transfer of M2a Mϕ to IBD mice increased Th17 and Treg generation, while M1 Mϕ contributed to the disruption of the intestinal epithelial barrier during IBD development (308, 309).
The polarization profile of IBD Mϕ is a complex issue. In CD, Mϕ are more polarized to an M2 profile, which is reflected by several findings. First, CD163 is expressed on a substantial percent of Mϕ in the colonic mucosa as well as in the peripheral blood of CD patients (16). In addition, sCD163 levels are significantly increased in CD patients (310). Upon successful treatment, serum sCD163 levels are dramatically decreased (310). Second, large numbers of Mϕ are found in fibrotic lesions of CD patients, consistent with the potent tissue-repairing and pro-fibrotic capacity of M2 Mϕ (311, 312). Third, defective bacterial clearance by Mϕ is frequently observed in CD patients, which is presumably due to the impaired pro-inflammatory cytokine secretion of these cells (96). Fourth, IL-13, which is a potent M2 Mϕ inducer, was dramatically upregulated in CD patients (100). In comparison, the Mϕ polarization profile seems much more complex in UC patients. The fact that CD163+ Mϕ numbers and serum sCD163 levels are increased in UC patients, coupled with the finding that CD206+ Mϕ are enriched in the injured mucosa of these patients, indicates an M2 polarization profile for these Mϕ (16, 101). However, the continuous excessive inflammation in the gut mucosa of UC patients, as well as the significant increase in pro-inflammatory M1 while decrease in M2 Mϕ accompanied by suppressed IL-10 production in mouse models of UC also points to the evident M1 polarization of these Mϕ (98, 99). In various mouse models of IBD, inhibition of the pro-inflammatory activities of M1 Mϕ or induction of tissue-repairing/immunomodulatory M2 Mϕ usually results in attenuated experimental IBD (185, 187, 313, 314).
In the present review, we mainly discussed the association of Mo/Mϕ with the development of certain autoimmune diseases. It has been quite well elucidated that Mo/Mϕ are key component of the innate immune system and are involved in both amplifying and suppressing inflammation (2). Mounting evidence suggests that these cells participate in the pathogenesis of autoimmune diseases, mainly through their remarkably pro-inflammatory or fibrogenic properties (1, 2). As discussed above, in different autoimmune diseases, the heterogeneity of Mo/Mϕ subpopulations varies dramatically, and their polarization profile usually plays a key role in disease progression (Figure 1). However, in many autoimmune diseases, the phenotypic and functional characteristics of Mo/Mϕ have not been classified unambiguously, as many pro-inflammatory M1-polarized Mo/Mϕ simultaneously express M2-related markers or exhibit immunomodulatory functions (19–22). In addition, in several cases, Mϕ activation is a dynamic and reversible event in which pro-inflammatory Mϕ can be re-programmed into Mϕ with immunosuppressive or tissue-repairing cells by local microenvironment (13, 25). Thus, future investigation into explaining the seemingly opposing phenotypic and functional programs of Mo/Mϕ and identifying the dynamic changes is clearly needed.
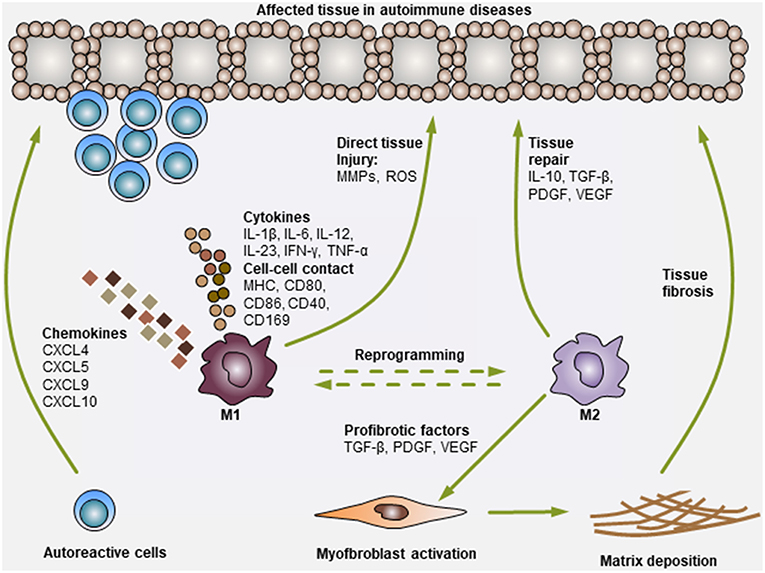
Figure 1. Modulation of autoimmune diseases by Mo and Mϕ. Mo and Mϕ are key players in autoimmune diseases. During the development of autoimmune diseases, pro-inflammatory M1 Mo or Mϕ can secrete various chemokines to recruit additional immune cells (i.e., T cells, B cells, neutrophils, NK cells, and NKT cells) to the affected tissues. Then, Mo or Mϕ can activate these cells via the secretion of various pro-inflammatory cytokines (i.e., IL-1β, IL-6, IL-12, IL-23, IFN-γ, and TNF-α) or through direct cell-cell contact (antigen presentation: MHC, co-stimulation: CD80, CD86 and CD40, and adhesion molecules: CD169). In addition, Mo or Mϕ can also exert direct tissue injury functions by producing matrix metalloproteinases (MMPs) and reactive oxygen species (ROS). Consequently, the activation of Mo or Mϕ and other immune cells synergistically leads to tissue damage. On the other hand, M2 Mo or Mϕ mediate immunosuppressive or tissue-repairing effects during this process, mainly by producing cytokines (i.e., IL-10 and TGF-β) and growth factors (i.e., PDGF and VEGF). M2 Mo or Mϕ can also secrete various pro-fibrotic factors, such as TGF-β, PDGF and VEGF, to activate myofibroblasts in certain tissues, leading to extracellular matrix deposition and fibrosis generation (i.e., cases in PBC and SSc).
Several possible mechanisms responsible for Mϕ phenotype in autoimmune diseases in general have been suggested by recent findings. For example, genome-wide association studies have identified several candidate genes responsible for the pathogenesis of autoimmune diseases. Among the susceptibility genes, HLA, which is closely with the antigen-presenting ability of Mϕ, has been suggested to be involved in the development of SLE (315), SSc (316), RA (317), MS (318), T1D (319–322), SS (323), Celiac disease (324), and IBD (325). In addition, protein tyrosine phosphatase, non-receptor type 22 (PTPN22), which can be expressed in Mϕ and controls Mϕ activation and polarization, has been identified as a risk gene for RA (317) and IBD (17). Interferon regulatory factor 5 (IRF5), which is mainly expressed by myeloid cells and is a key regulator of Mϕ activation and polarization, has been identified as an important predisposed factor in patients with SLE (326), SS (323), RA (327), PBC (328), and IBD (329, 330). However, functional studies investigating the actual function of these genes in Mϕ should be done to confirm whether they really play a critical role in controlling Mϕ activation in autoimmune diseases.
In recent years, mounting reports have overturned the long-held knowledge that Mϕ in the adult are merely replenished by circulating Mo from bone marrow progenitors (331–333). The new paradigm supports that some Mϕ are embryo-derived and are maintained by self-renewal independent of hematopoietic contribution (332). Intriguingly, this heterogeneity of Mϕ results in distinct phenotypes and, more importantly, totally different biologic functions (334, 335). Thus, it is necessary for future studies to elucidate the roles of tissue-resident Mϕ and bone marrow-derived Mϕ in the initiation, progression and termination of different autoimmune diseases.
Although Mo and Mϕ play a key role in the pathogenesis of certain autoimmune diseases, the development of these diseases is not solely Mo/Mϕ-dependent, and this process involves the interplay of these cells with other immune cells, i.e., autoreactive T and B cells (2). However, most studies fail to explore the interactions of Mo/Mϕ with other immune cells in the local microenvironment. Thus, future work is needed to better determine the synergistic effects and related mechanisms of the interactions between Mo/Mϕ and other immune cells in the development of autoimmune diseases.
To date, although the functions of Mo/Mϕ in several autoimmune diseases have been determined, the clinical translation of this knowledge is still challenging. Certain Mo- or Mϕ-targeted therapies have been developed (see Table 3), but whether they are more effective and safer than traditional treatment remains to be verified, and some of them have already proven disappointing (52, 54, 281, 282). However, this does not rule out a potential effective role for Mo/Mϕ as an attractive therapeutic strategy for autoimmune diseases. Thus, further studies are needed to elucidate a more detailed and comprehensive mechanism of Mo/Mϕ regulation in autoimmune diseases; such work, coupled with a wider understanding of the determinant factors of autoimmune diseases (i.e., sex, age, genetics, and environmental factors), which act together but differ between patients, will probably lead to the development of more specific and effective therapies in the future.

Table 3. Pathogenic functions of Mo and Mϕ in autoimmune diseases and the relevant treatment strategies.
W-TM and D-KC designed the structure of this article. W-TM wrote the manuscript. FG and KG revised the manuscript. All authors have reviewed the final version of this article.
This research was supported by the Qinghai Province Major R&D and Transformation Project (2018-NK-125), Xianyang Science and Technology Major Project (2017K01-34), Key Industrial Innovation Chains of Shaanxi Province (2018ZDCXL-NY-01-06), the Youth Innovation Team of Shaanxi Universities, and PhD Research Startup Fund of Northwest Agriculture and Forestry University (00500/Z109021716).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Laria A, Lurati A, Marrazza M, Mazzocchi D, Re KA, Scarpellini M. The macrophages in rheumatic diseases. J Inflamm Res. (2016) 9:1–11. doi: 10.2147/JIR.S82320
2. Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med. (2017) 15:36. doi: 10.1186/s12967-017-1141-8
3. Ma WT, Chang C, Gershwin ME, Lian ZX. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: a comprehensive review. J Autoimmun. (2017) 83:95–112. doi: 10.1016/j.jaut.2017.07.003
4. Sack U, Stiehl P, Geiler G. Distribution of macrophages in rheumatoid synovial membrane and its association with basic activity. Rheumatol Int. (1994) 13:181–6. doi: 10.1007/BF00390265
5. Janossy G, Panayi G, Duke O, Bofill M, Poulter LW, Goldstein G. Rheumatoid arthritis: a disease of T-lymphocyte/macrophage immunoregulation. Lancet. (1981) 2:839–42. doi: 10.1016/S0140-6736(81)91107-7
6. Ishikawa O, Ishikawa H. Macrophage infiltration in the skin of patients with systemic sclerosis. J Rheumatol. (1992) 19:1202–6.
7. Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood. (2004) 104:3021–7. doi: 10.1182/blood-2004-02-0701
8. Magnusson MK, Brynjolfsson SF, Dige A, Uronen-Hansson H, Borjesson LG, Bengtsson JL, et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. (2016) 9:171–82. doi: 10.1038/mi.2015.48
9. Kuhl AA, Erben U, Kredel LI, Siegmund B. Diversity of intestinal macrophages in inflammatory bowel diseases. Front Immunol. (2015) 6:613. doi: 10.3389/fimmu.2015.00613
10. Mathew J, Hines JE, Toole K, Johnson SJ, James OF, Burt AD. Quantitative analysis of macrophages and perisinusoidal cells in primary biliary cirrhosis. Histopathology. (1994) 25:65–70. doi: 10.1111/j.1365-2559.1994.tb00599.x
11. Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. (2001) 50:389–400. doi: 10.1002/ana.1123
12. Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sorensen PS, et al. Demyelination versus remyelination in progressive multiple sclerosis. Brain. (2010) 133:2983–98. doi: 10.1093/brain/awq250
13. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK III, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. (2014) 9:591–604. doi: 10.1016/j.celrep.2014.09.032
14. Lopez-Cacho JM, Gallardo S, Posada M, Aguerri M, Calzada D, Mayayo T, et al. Association of immunological cell profiles with specific clinical phenotypes of scleroderma disease. Biomed Res Int. (2014) 2014:148293. doi: 10.1155/2014/148293
15. Wildenberg ME, Welzen-Coppens JM, van Helden-Meeuwsen CG, Bootsma H, Vissink A, van Rooijen N, et al. Increased frequency of CD16+ monocytes and the presence of activated dendritic cells in salivary glands in primary Sjogren syndrome. Ann Rheum Dis. (2009) 68:420–6. doi: 10.1136/ard.2008.087874
16. Demetter P, De Vos M, Van Huysse JA, Baeten D, Ferdinande L, Peeters H, et al. Colon mucosa of patients both with spondyloarthritis and Crohn's disease is enriched with macrophages expressing the scavenger receptor CD163. Ann Rheum Dis. (2005) 64:321–4. doi: 10.1136/ard.2003.018382
17. Yilmaz B, Spalinger MR, Biedermann L, Franc Y, Fournier N, Rossel JB, et al. The presence of genetic risk variants within PTPN2 and PTPN22 is associated with intestinal microbiota alterations in Swiss IBD cohort patients. PLoS ONE. (2018) 13:e0199664. doi: 10.1371/journal.pone.0199664
18. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
19. Jin O, Sun LY, Zhou KX, Zhang XS, Feng XB, Mok MY, et al. Lymphocyte apoptosis and macrophage function: correlation with disease activity in systemic lupus erythematosus. Clin Rheumatol. (2005) 24:107–10. doi: 10.1007/s10067-004-0972-x
20. Godsell J, Rudloff I, Kandane-Rathnayake R, Hoi A, Nold MF, Morand EF, et al. Clinical associations of IL-10 and IL-37 in systemic lupus erythematosus. Sci Rep. (2016) 6:34604. doi: 10.1038/srep34604
21. Mellor-Pita S, Citores MJ, Castejon R, Yebra-Bango M, Tutor-Ureta P, Rosado S, et al. Monocytes and T lymphocytes contribute to a predominance of interleukin 6 and interleukin 10 in systemic lupus erythematosus. Cytometry B Clin Cytom. (2009) 76:261–70. doi: 10.1002/cyto.b.20468
22. Manavalan JS, Hernandez L, Shah JG, Konikkara J, Naiyer AJ, Lee AR, et al. Serum cytokine elevations in celiac disease: association with disease presentation. Hum Immunol. (2010) 71:50–7. doi: 10.1016/j.humimm.2009.09.351
23. Soldano SP, Trombetta CP, Contini PP, Tomatis VM, Ruaro BM, Brizzolara RP, et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann Rheum Dis. (2018) 2018:213648. doi: 10.1136/annrheumdis-2018-213648
24. Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflamm. (2013) 10:35. doi: 10.1186/1742-2094-10-35
25. Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabo A, et al. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat Immunol. (2017) 18:530–540. doi: 10.1038/ni.3710
26. Sumegi A, Antal-Szalmas P, Aleksza M, Kovacs I, Sipka S, Zeher M, et al. Glucocorticosteroid therapy decreases CD14-expression and CD14-mediated LPS-binding and activation of monocytes in patients suffering from systemic lupus erythematosus. Clin Immunol. (2005) 117:271–9. doi: 10.1016/j.clim.2005.09.002
27. Li Y, Lee PY, Sobel ES, Narain S, Satoh M, Segal MS, et al. Increased expression of FcgammaRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res Ther. (2009) 11:R6. doi: 10.1186/ar2591
28. Katsiari CG, Liossis SN, Souliotis VL, Dimopoulos AM, Manoussakis MN, Sfikakis PP. Aberrant expression of the costimulatory molecule CD40 ligand on monocytes from patients with systemic lupus erythematosus. Clin Immunol. (2002) 103:54–62. doi: 10.1006/clim.2001.5172
29. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. (2001) 294:1540–3. doi: 10.1126/science.1064890
30. Decker P, Kotter I, Klein R, Berner B, Rammensee HG. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology. (2006) 45:1087–95. doi: 10.1093/rheumatology/kel061
31. Orme J, Mohan C. Macrophages and neutrophils in SLE-An online molecular catalog. Autoimmun Rev. (2012) 11:365–72. doi: 10.1016/j.autrev.2011.10.010
32. Hepburn AL, Mason JC, Davies KA. Expression of Fcgamma and complement receptors on peripheral blood monocytes in systemic lupus erythematosus and rheumatoid arthritis. Rheumatology. (2004) 43:547–54. doi: 10.1093/rheumatology/keh112
33. Biesen R, Demir C, Barkhudarova F, Grun JR, Steinbrich-Zollner M, Backhaus M, et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis Rheum. (2008) 58:1136–45. doi: 10.1002/art.23404
34. Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. (1998) 41:1241–50. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H
35. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
36. Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus. Autoimmun Rev. (2007) 6:497–502. doi: 10.1016/j.autrev.2007.01.017
37. Umare V, Pradhan V, Nadkar M, Rajadhyaksha A, Patwardhan M, Ghosh KK, et al. Effect of proinflammatory cytokines (IL-6, TNF-alpha, and IL-1beta) on clinical manifestations in Indian SLE patients. Mediators Inflamm. (2014) 2014:385297. doi: 10.1155/2014/385297
38. Santer DM, Yoshio T, Minota S, Moller T, Elkon KB. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol. (2009) 182:1192–201. doi: 10.4049/jimmunol.182.2.1192
39. Wagrowska-Danilewicz M, Stasikowska O, Danilewicz M. Correlative insights into immunoexpression of monocyte chemoattractant protein-1, transforming growth factor beta-1 and CD68+ cells in lupus nephritis. Pol J Pathol. (2005) 56:115–20.
40. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
41. Higashi-Kuwata N, Makino T, Inoue Y, Takeya M, Ihn H. Alternatively activated macrophages (M2 macrophages) in the skin of patient with localized scleroderma. Exp Dermatol. (2009) 18:727–9. doi: 10.1111/j.1600-0625.2008.00828.x
42. Lescoat A, Lecureur V, Roussel M, Sunnaram BL, Ballerie A, Coiffier G, et al. CD16-positive circulating monocytes and fibrotic manifestations of systemic sclerosis. Clin Rheumatol. (2017) 36:1649–54. doi: 10.1007/s10067-017-3597-6
43. Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther. (2010) 12:R128. doi: 10.1186/ar3066
44. Binai N, O'Reilly S, Griffiths B, van Laar JM, Hugle T. Differentiation potential of CD14+ monocytes into myofibroblasts in patients with systemic sclerosis. PLoS ONE. (2012) 7:e33508. doi: 10.1371/journal.pone.0033508
45. York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. (2007) 56:1010–20. doi: 10.1002/art.22382
46. Christmann RB, Lafyatis R. The cytokine language of monocytes and macrophages in systemic sclerosis. Arthritis Res Ther. (2010) 12:146. doi: 10.1186/ar3167
47. Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, de Carvalho CR, Kairalla R, et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. (2014) 66:714–25. doi: 10.1002/art.38288
48. Wu M, Pedroza M, Lafyatis R, George AT, Mayes MD, Assassi S, et al. Identification of cadherin 11 as a mediator of dermal fibrosis and possible role in systemic sclerosis. Arthritis Rheumatol. (2014) 66:1010–21. doi: 10.1002/art.38275
49. Yoon BR, Yoo SJ, Choi Y, Chung YH, Kim J, Yoo IS, et al. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PLoS ONE. (2014) 9:e109775. doi: 10.1371/journal.pone.0109775
50. Xiong YS, Cheng Y, Lin QS, Wu AL, Yu J, Li C, et al. Increased expression of Siglec-1 on peripheral blood monocytes and its role in mononuclear cell reactivity to autoantigen in rheumatoid arthritis. Rheumatology. (2014) 53:250–9. doi: 10.1093/rheumatology/ket342
51. Yamanaka H. TNF as a target of inflammation in rheumatoid arthritis. Endocr Metab Immune. (2015) 15:129–34. doi: 10.2174/1871530315666150316121808
52. Dayer JM, Bresnihan B. Targeting interleukin-1 in the treatment of rheumatoid arthritis. Arthritis Rheum. (2002) 46:574–8. doi: 10.1002/art.10168
53. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. (2000) 191:313–20. doi: 10.1084/jem.191.2.313
54. Semerano L, Thiolat A, Minichiello E, Clavel G, Bessis N, Boissier MC. Targeting IL-6 for the treatment of rheumatoid arthritis: phase II investigational drugs. Expert Opin Investig Drugs. (2014) 23:979–99. doi: 10.1517/13543784.2014.912276
55. Pope RM, Shahrara S. Possible roles of IL-12-family cytokines in rheumatoid arthritis. Nat Rev Rheumatol. (2013) 9:252–6. doi: 10.1038/nrrheum.2012.170
56. Palacios BS, Estrada-Capetillo L, Izquierdo E, Criado G, Nieto C, Municio C, et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J Pathol. (2015) 235:515–26. doi: 10.1002/path.4466
57. Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, et al. M1 and M2 monocytes in rheumatoid arthritis: a contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol. (2017) 8:1958. doi: 10.3389/fimmu.2017.01958
58. Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol. (2015) 127–128:1–22. doi: 10.1016/j.pneurobio.2015.02.003
59. van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. (2000) 26:2–10. doi: 10.1046/j.1365-2990.2000.00217.x
60. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
61. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
62. Min D, Brooks B, Wong J, Salomon R, Bao W, Harrisberg B, et al. Alterations in monocyte CD16 in association with diabetes complications. Mediat Inflamm. (2012) 2012:649083. doi: 10.1155/2012/649083
63. Maree FM, Komba M, Dyck C, Labecki M, Finegood DT, Edelstein-Keshet L. Quantifying macrophage defects in type 1 diabetes. J Theor Biol. (2005) 233:533–51. doi: 10.1016/j.jtbi.2004.10.030
64. O'Brien BA, Geng X, Orteu CH, Huang YQ, Ghoreishi M, Zhang YQ, et al. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun. (2006) 26:104–15. doi: 10.1016/j.jaut.2005.11.006
65. Calderon B, Suri A, Unanue ER. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am J Pathol. (2006) 169:2137–47. doi: 10.2353/ajpath.2006.060539
66. Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. (2006) 55:774–9. doi: 10.2337/diabetes.55.03.06.db05-1417
67. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. (2004) 113:451–63. doi: 10.1172/JCI200419585
68. Antonelli A, Fallahi P, Ferrari SM, Pupilli C, d'Annunzio G, Lorini R, et al. Serum Th1 (CXCL10) and Th2 (CCL2) chemokine levels in children with newly diagnosed Type 1 diabetes: a longitudinal study. Diabet Med. (2008) 25:1349–53. doi: 10.1111/j.1464-5491.2008.02577.x
69. Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol. (2009) 183:4432–9. doi: 10.4049/jimmunol.0900576
70. Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia. (2007) 50:596–601. doi: 10.1007/s00125-006-0569-9
71. Lechleitner M, Koch T, Herold M, Dzien A, Hoppichler F. Tumour necrosis factor-alpha plasma level in patients with type 1 diabetes mellitus and its association with glycaemic control and cardiovascular risk factors. J Intern Med. (2000) 248:67–76. doi: 10.1046/j.1365-2796.2000.00705.x
72. Alabraba EB, Lai V, Boon L, Wigmore SJ, Adams DH, Afford SC. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology. (2008) 47:552–62. doi: 10.1002/hep.22011
73. Leicester KL, Olynyk JK, Brunt EM, Britton RS, Bacon BR. Differential findings for CD14-positive hepatic monocytes/macrophages in primary biliary cirrhosis, chronic hepatitis C and nonalcoholic steatohepatitis. Liver Int. (2006) 26:559–65. doi: 10.1111/j.1478-3231.2006.01255.x
74. Peng A, Ke P, Zhao R, Lu X, Zhang C, Huang X, et al. Elevated circulating CD14(low)CD16(+) monocyte subset in primary biliary cirrhosis correlates with liver injury and promotes Th1 polarization. Clin Exp Med. (2016) 16:511–21. doi: 10.1007/s10238-015-0381-2
75. Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. (2005) 42:802–8. doi: 10.1002/hep.20859
76. Bao G, Han Z, Yan Z, Wang Q, Zhou Y, Yao D, et al. Increased Siglec-1 expression in monocytes of patients with primary biliary cirrhosis. Immunol Invest. (2010) 39:645–60. doi: 10.3109/08820139.2010.485625
77. Lleo A, Bowlus CL, Yang GX, Invernizzi P, Podda M, Van de Water J, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. (2010) 52:987–98. doi: 10.1002/hep.23783
78. Honda Y, Yamagiwa S, Matsuda Y, Takamura M, Ichida T, Aoyagi Y. Altered expression of TLR homolog RP105 on monocytes hypersensitive to LPS in patients with primary biliary cirrhosis. J Hepatol. (2007) 47:404–11. doi: 10.1016/j.jhep.2007.03.012
79. Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol. (1998) 29:409–16. doi: 10.1016/S0168-8278(98)80058-5
80. Ciccia F, Alessandro R, Rodolico V, Guggino G, Raimondo S, Guarnotta C, et al. IL-34 is overexpressed in the inflamed salivary glands of patients with Sjogren's syndrome and is associated with the local expansion of pro-inflammatory CD14(bright)CD16(+) monocytes. Rheumatology. (2013) 52:1009–17. doi: 10.1093/rheumatology/kes435
81. Hauk V, Fraccaroli L, Grasso E, Eimon A, Ramhorst R, Hubscher O, et al. Monocytes from Sjogren's syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin Exp Immunol. (2014) 177:662–70. doi: 10.1111/cei.12378
82. Tishler M, Yaron I, Shirazi I, Yossipov Y, Yaron M. Increased salivary interleukin-6 levels in patients with primary Sjogren's syndrome. Rheumatol Int. (1999) 18:125–7. doi: 10.1007/s002960050070
83. Fogel O, Riviere E, Seror R, Nocturne G, Boudaoud S, Ly B, et al. Role of the IL-12/IL-35 balance in patients with Sjogren syndrome. J Allergy Clin Immunol. (2017) 2017:3586. doi: 10.1136/annrheumdis-2017-eular.3586
84. Willeke P, Schluter B, Schotte H, Domschke W, Gaubitz M, Becker H. Interferon-gamma is increased in patients with primary sjogren's syndrome and raynaud's phenomenon. Semin Arthritis Rheu. (2009) 39:197–202. doi: 10.1016/j.semarthrit.2008.04.002
85. Hernandez-Molina G, Michel-Peregrina M, Hernandez-Ramirez DF, Sanchez-Guerrero J, Llorente L. Chemokine saliva levels in patients with primary Sjogren's syndrome, associated Sjogren's syndrome, pre-clinical Sjogren's syndrome and systemic autoimmune diseases. Rheumatology. (2011) 50:1288–92. doi: 10.1093/rheumatology/ker019
86. Lee YJ, Scofield RH, Hyon JY, Yun PY, Lee HJ, Lee EY, et al. Salivary chemokine levels in patients with primary Sjogren's syndrome. Rheumatology. (2010) 49:1747–52. doi: 10.1093/rheumatology/keq121
87. Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren's syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol. (2008) 181:2898–906. doi: 10.4049/jimmunol.181.4.2898
88. Lisi S, Sisto M, Lofrumento DD, D'Amore M. Altered IkappaBalpha expression promotes NF-kappaB activation in monocytes from primary Sjogren's syndrome patients. Pathology. (2012) 44:557–61. doi: 10.1097/PAT.0b013e3283580388
89. Cupi ML, Sarra M, De Nitto D, Franze E, Marafini I, Monteleone I, et al. Defective expression of scavenger receptors in celiac disease mucosa. PLoS ONE. (2014) 9:e100980. doi: 10.1371/journal.pone.0100980
90. Cinova J, Palova-Jelinkova L, Smythies LE, Cerna M, Pecharova B, Dvorak M, et al. Gliadin peptides activate blood monocytes from patients with celiac disease. J Clin Immunol. (2007) 27:201–9. doi: 10.1007/s10875-006-9061-z
91. Di Sabatino A, Rovedatti L, Vetrano S, Vidali F, Biancheri P, Rescigno M, et al. Involvement of CD40-CD40 ligand in uncomplicated and refractory celiac disease. Am J Gastroenterol. (2011) 106:519–27. doi: 10.1038/ajg.2010.450
92. Barilli A, Gaiani F, Prandi B, Cirlini M, Ingoglia F, Visigalli R, et al. Gluten peptides drive healthy and celiac monocytes toward an M2-like polarization. J Nutr Biochem. (2018) 54:11–17. doi: 10.1016/j.jnutbio.2017.10.017
93. Barilli A, Rotoli BM, Visigalli R, Dall'Asta V. Gliadin activates arginase pathway in RAW264.7 cells and in human monocytes. Biochim Biophys Acta. (2014) 1842:1364–71. doi: 10.1016/j.bbadis.2014.04.021
94. Thiesen S, Janciauskiene S, Uronen-Hansson H, Agace W, Hogerkorp CM, Spee P, et al. CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn's disease. J Leukoc Biol. (2014) 95:531–41. doi: 10.1189/jlb.0113021
95. Koch S, Kucharzik T, Heidemann J, Nusrat A, Luegering A. Investigating the role of proinflammatory CD16+ monocytes in the pathogenesis of inflammatory bowel disease. Clin Exp Immunol. (2010) 161:332–41. doi: 10.1111/j.1365-2249.2010.04177.x
96. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. (2009) 206:1883–97. doi: 10.1084/jem.20091233
97. Goldstein JI, Kominsky DJ, Jacobson N, Bowers B, Regalia K, Austin GL, et al. Defective leukocyte GM-CSF receptor (CD116) expression and function in inflammatory bowel disease. Gastroenterology. (2011) 141:208–16. doi: 10.1053/j.gastro.2011.03.060
98. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. (2003) 3:521–33. doi: 10.1038/nri1132
99. Zhu W, Yu J, Nie Y, Shi X, Liu Y, Li F, et al. Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol Invest. (2014) 43:638–52. doi: 10.3109/08820139.2014.909456
100. Bailey JR, Bland PW, Tarlton JF, Peters I, Moorghen M, Sylvester PA, et al. IL-13 promotes collagen accumulation in Crohn's disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS ONE. (2012) 7:e52332. doi: 10.1371/journal.pone.0052332
101. Cosin-Roger J, Ortiz-Masia D, Calatayud S, Hernandez C, Alvarez A, Hinojosa J, et al. M2 macrophages activate WNT signaling pathway in epithelial cells: relevance in ulcerative colitis. PLoS ONE. (2013) 8:e78128. doi: 10.1371/journal.pone.0078128
102. Shi X, Qian T, Li M, Chen F, Chen Y, Hao F. Aberrant low expression of A20 in tumor necrosis factor-alpha-stimulated SLE monocytes mediates sustained NF-kappaB inflammatory response. Immunol Invest. (2015) 44:497–508. doi: 10.3109/08820139.2015.1037957
103. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med. (2016) 14:156. doi: 10.1186/s12967-016-0911-z
104. Burbano C, Villar-Vesga J, Orejuela J, Munoz C, Vanegas A, Vasquez G, et al. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:322. doi: 10.3389/fimmu.2018.00322
105. Lopez P, Scheel-Toellner D, Rodriguez-Carrio J, Caminal-Montero L, Gordon C, Suarez A. Interferon-alpha-induced B-lymphocyte stimulator expression and mobilization in healthy and systemic lupus erthymatosus monocytes. Rheumatology. (2014) 53:2249–58. doi: 10.1093/rheumatology/keu249
106. Thanei S, Trendelenburg M. Anti-C1q autoantibodies from systemic lupus erythematosus patients induce a proinflammatory phenotype in macrophages. J Immunol. (2016) 196:2063–74. doi: 10.4049/jimmunol.1501659
107. Lu M, Yu S, Xu W, Gao B, Xiong S. HMGB1 promotes systemic lupus erythematosus by enhancing macrophage inflammatory response. J Immunol Res. (2015) 2015:946748. doi: 10.1155/2015/946748
108. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. (2017) 47:913–27 e6. doi: 10.1016/j.immuni.2017.10.006
109. Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. (2007) 204:2259–65. doi: 10.1084/jem.20070600
110. Oxer DS, Godoy LC, Borba E, Lima-Salgado T, Passos LA, Laurindo I, et al. PPAR gamma expression is increased in systemic lupus erythematosus patients and represses CD40/CD40L signaling pathway. Lupus. (2011) 20:575–87. doi: 10.1177/0961203310392419
111. Liu J, Berthier CC, Kahlenberg JM. Enhanced inflammasome activity in systemic lupus erythematosus is mediated via type I interferon-induced up-regulation of interferon regulatory factor 1. Arthritis Rheumatol. (2017) 69:1840–9. doi: 10.1002/art.40166
112. Aringer M, Feierl E, Steiner G, Stummvoll GH, Hofler E, Steiner CW, et al. Increased bioactive TNF in human systemic lupus erythematosus: associations with cell death. Lupus. (2002) 11:102–8. doi: 10.1191/0961203302lu160oa
113. Sharif MN, Tassiulas I, Hu Y, Mecklenbrauker I, Tarakhovsky A, Ivashkiv LB. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. (2004) 172:6476–81. doi: 10.4049/jimmunol.172.10.6476
114. Grondal G, Gunnarsson I, Ronnelid J, Rogberg S, Klareskog L, Lundberg I. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin Exp Rheumatol. (2000) 18:565–70.
115. Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology. (2015) 54:324–31. doi: 10.1093/rheumatology/keu318
116. Yamamoto T, Eckes B, Hartmann K, Krieg T. Expression of monocyte chemoattractant protein-1 in the lesional skin of systemic sclerosis. J Dermatol Sci. (2001) 26:133–9. doi: 10.1016/S0923-1811(00)00169-9
117. Cho JS, Fang TC, Reynolds TL, Sofia DJ, Hamann S, Burkly LC. PDGF-BB promotes type I IFN-dependent vascular alterations and monocyte recruitment in a model of dermal fibrosis. PLoS ONE. (2016) 11:e0162758. doi: 10.1371/journal.pone.0162758
118. Hasegawa M, Sato S, Echigo T, Hamaguchi Y, Yasui M, Takehara K. Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann Rheum Dis. (2005) 64:21–8. doi: 10.1136/ard.2003.018705
119. Selvi E, Tripodi SA, Catenaccio M, Lorenzini S, Chindamo D, Manganelli S, et al. Expression of macrophage migration inhibitory factor in diffuse systemic sclerosis. Ann Rheum Dis. (2003) 62:460–4. doi: 10.1136/ard.62.5.460
120. Ciechomska M, O'Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone demethylation and toll-like receptor 8-dependent cross-talk in monocytes promotes transdifferentiation of fibroblasts in systemic sclerosis via Fra-2. Arthritis Rheumatol. (2016) 68:1493–504. doi: 10.1002/art.39602
121. Reich N, Maurer B, Akhmetshina A, Venalis P, Dees C, Zerr P, et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. (2010) 62:280–90. doi: 10.1002/art.25056
122. Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology. (2008) 47(Suppl 5):v2–4. doi: 10.1093/rheumatology/ken265
123. Carvalheiro T, Horta S, van Roon JAG, Santiago M, Salvador MJ, Trindade H, et al. Increased frequencies of circulating CXCL10-, CXCL8- and CCL4-producing monocytes and Siglec-3-expressing myeloid dendritic cells in systemic sclerosis patients. Inflamm Res. (2018) 67:169–77. doi: 10.1007/s00011-017-1106-7
124. Taniguchi T, Miyagawa T, Toyama S, Yamashita T, Nakamura K, Saigusa R, et al. CXCL13 produced by macrophages due to Fli1 deficiency may contribute to the development of tissue fibrosis, vasculopathy and immune activation in systemic sclerosis. Exp Dermatol. (2018) 27:1030–7. doi: 10.1111/exd.13724
125. Masuda A, Yasuoka H, Satoh T, Okazaki Y, Yamaguchi Y, Kuwana M. Versican is upregulated in circulating monocytes in patients with systemic sclerosis and amplifies a CCL2-mediated pathogenic loop. Arthritis Res Ther. (2013) 15:R74. doi: 10.1186/ar4251
126. Ciechomska M, Huigens CA, Hugle T, Stanly T, Gessner A, Griffiths B, et al. Toll-like receptor-mediated, enhanced production of profibrotic TIMP-1 in monocytes from patients with systemic sclerosis: role of serum factors. Ann Rheum Dis. (2013) 72:1382–9. doi: 10.1136/annrheumdis-2012-201958
127. Rhys HI, Dell'Accio F, Pitzalis C, Moore A, Norling LV, Perretti M. Neutrophil microvesicles from healthy control and rheumatoid arthritis patients prevent the inflammatory activation of macrophages. EBioMedicine. (2018) 29:60–9. doi: 10.1016/j.ebiom.2018.02.003
128. Tsai CH, Liu SC, Wang YH, Su CM, Huang CC, Hsu CJ, Tang CH. Osteopontin inhibition of miR-129-3p enhances IL-17 expression and monocyte migration in rheumatoid arthritis. Biochim Biophys Acta. (2017) 1861:15–22. doi: 10.1016/j.bbagen.2016.11.015
129. Rajasekhar M, Olsson AM, Steel KJ, Georgouli M, Ranasinghe U, Brender Read C, et al. MicroRNA-155 contributes to enhanced resistance to apoptosis in monocytes from patients with rheumatoid arthritis. J Autoimmun. (2017) 79:53–62. doi: 10.1016/j.jaut.2017.01.002
130. Darrieutort-Laffite C, Boutet MA, Chatelais M, Brion R, Blanchard F, Heymann D, et al. IL-1beta and TNFalpha promote monocyte viability through the induction of GM-CSF expression by rheumatoid arthritis synovial fibroblasts. Mediat Inflamm. (2014) 2014:241840. doi: 10.1155/2014/241840
131. Choi S, You S, Kim D, Choi SY, Kwon HM, Kim HS, et al. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J Clin Invest. (2017) 127:954–69. doi: 10.1172/JCI87880
132. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. (2016) 213:1655–62. doi: 10.1084/jem.20160061
133. Asquith DL, Ballantine LE, Nijjar JS, Makdasy MK, Patel S, Wright PB, et al. The liver X receptor pathway is highly upregulated in rheumatoid arthritis synovial macrophages and potentiates TLR-driven cytokine release. Ann Rheum Dis. (2013) 72:2024–31. doi: 10.1136/annrheumdis-2012-202872
134. Lin NY, Beyer C, Giessl A, Kireva T, Scholtysek C, Uderhardt S, et al. Autophagy regulates TNF alpha-mediated joint destruction in experimental arthritis. Ann Rheumat Dis. (2013) 72:761–8. doi: 10.1136/annrheumdis-2012-201671
135. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. (1991) 10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x
136. Scott BB, Weisbrot LM, Greenwood JD, Bogoch ER, Paige CJ, Keystone EC. Rheumatoid arthritis synovial fibroblast and U937 macrophage/monocyte cell line interaction in cartilage degradation. Arthritis Rheum. (1997) 40:490–8. doi: 10.1002/art.1780400315
137. Moreno M, Bannerman P, Ma J, Guo F, Miers L, Soulika AM, et al. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J Neurosci. (2014) 34:8175–85. doi: 10.1523/JNEUROSCI.1137-14.2014
138. Vogel DY, Kooij G, Heijnen PD, Breur M, Peferoen LA, van der Valk P, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol. (2015) 45:1808–19. doi: 10.1002/eji.201444960
139. Imitola J, Rasouli J, Watanabe F, Mahajan K, Sharan AD, Ciric B, et al. Elevated expression of granulocyte-macrophage colony-stimulating factor receptor in multiple sclerosis lesions. J Neuroimmunol. (2018) 317:45–54. doi: 10.1016/j.jneuroim.2017.12.017
140. Bsibsi M, Peferoen LAN, Holtman IR, Nacken PJ, Gerritsen WH, Witte ME, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-gamma and alpha B-crystallin. Acta Neuropathol. (2014) 128:215–29. doi: 10.1007/s00401-014-1317-8
141. Jiang W, Li D, Han R, Zhang C, Jin WN, Wood K, et al. Acetylcholine-producing NK cells attenuate CNS inflammation via modulation of infiltrating monocytes/macrophages. Proc Natl Acad Sci USA. (2017) 114:E6202–11. doi: 10.1073/pnas.1705491114
142. Christophi GP, Panos M, Hudson CA, Christophi RL, Gruber RC, Mersich AT, et al. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab Invest. (2009) 89:742–59. doi: 10.1038/labinvest.2009.32
143. Shi H, Sheng B, Zhang C, Nayak L, Lin Z, Jain MK, et al. Myeloid Kruppel-like factor 2 deficiency exacerbates neurological dysfunction and neuroinflammation in a murine model of multiple sclerosis. J Neuroimmunol. (2014) 274:234–9. doi: 10.1016/j.jneuroim.2014.06.023
144. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2012) 109:10480–5. doi: 10.1073/pnas.1201836109
145. Zhang H, Bi J, Yi H, Fan T, Ruan Q, Cai L, et al. Silencing c-Rel in macrophages dampens Th1 and Th17 immune responses and alleviates experimental autoimmune encephalomyelitis in mice. Immunol Cell Biol. (2017) 95:593–600. doi: 10.1038/icb.2017.11
146. Holz K, Prinz M, Brendecke SM, Holscher A, Deng F, Mitrucker HW, et al. Differing outcome of experimental autoimmune encephalitis in macrophage/neutrophil- and T cell-specific gp130-deficient mice. Front Immunol. (2018) 9:836. doi: 10.3389/fimmu.2018.00836
147. Wang LM, Zhang Y, Li X, Zhang ML, Zhu L, Zhang GX, et al. Nr4a1 plays a crucial modulatory role in Th1/Th17 cell responses and CNS autoimmunity. Brain Behav Immun. (2018) 68:44–55. doi: 10.1016/j.bbi.2017.09.015
148. Oleszak EL, Zaczynska E, Bhattacharjee M, Butunoi C, Legido A, Katsetos CD. Inducible nitric oxide synthase and nitrotyrosine are found in monocytes/macrophages and/or astrocytes in acute, but not in chronic, multiple sclerosis. Clin Diagn Lab Immunol. (1998) 5:438–45.
149. Hill KE, Zollinger LV, Watt HE, Carlson NG, Rose JW. Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: distribution, cellular expression and association with myelin damage. J Neuroimmunol. (2004) 151:171–9. doi: 10.1016/j.jneuroim.2004.02.005
150. Giacomini E, Severa M, Rizzo F, Mechelli R, Annibali V, Ristori G, et al. IFN-beta therapy modulates B-cell and monocyte crosstalk via TLR7 in multiple sclerosis patients. Eur J Immunol. (2013) 43:1963–72. doi: 10.1002/eji.201243212
151. Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes. (2008) 57:3025–33. doi: 10.2337/db08-0625
152. Sanchez-Zamora YI, Juarez-Avelar I, Vazquez-Mendoza A, Hiriart M, Rodriguez-Sosa M. Altered macrophage and dendritic cell response in Mif-/- mice reveals a role of Mif for inflammatory-Th1 response in type 1 diabetes. J Diabetes Res. (2016) 2016:7053963. doi: 10.1155/2016/7053963
153. Bojunga J, Kusterer K, Bacher M, Kurek R, Usadel KH, Renneberg H. Macrophage migration inhibitory factor and development of type-1 diabetes in non-obese diabetic mice. Cytokine. (2003) 21:179–86. doi: 10.1016/S1043-4666(03)00076-0
154. Stosic-Grujicic S, Stojanovic I, Maksimovic-Ivanic D, Momcilovic M, Popadic D, Harhaji L, et al. Macrophage migration inhibitory factor (MIF) is necessary for progression of autoimmune diabetes mellitus. J Cell Physiol. (2008) 215:665–75. doi: 10.1002/jcp.21346
155. Garrigan E, Belkin NS, Alexander JJ, Han Z, Seydel F, Carter J, et al. Persistent STAT5 phosphorylation and epigenetic dysregulation of GM-CSF and PGS2/COX2 expression in Type 1 diabetic human monocytes. PLoS ONE. (2013) 8:e76919. doi: 10.1371/journal.pone.0076919
156. Jain SK, Kannan K, Lim G, Matthews-Greer J, McVie R, Bocchini JA Jr. Elevated blood interleukin-6 levels in hyperketonemic type 1 diabetic patients and secretion by acetoacetate-treated cultured U937 monocytes. Diabetes Care. (2003) 26:2139–43. doi: 10.2337/diacare.26.7.2139
157. Rains JL, Jain SK. Hyperketonemia increases monocyte adhesion to endothelial cells and is mediated by LFA-1 expression in monocytes and ICAM-1 expression in endothelial cells. Am J Physiol. (2011) 301:E298–306. doi: 10.1152/ajpendo.00038.2011
158. Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA. Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes. (2004) 53:1979–86. doi: 10.2337/diabetes.53.8.1979
159. Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci USA. (2012) 109:E715–24. doi: 10.1073/pnas.1111600109
160. Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. (2005) 41:506–16. doi: 10.1002/hep.20582
161. Reuveni D, Gore Y, Leung SC, Lichter Y, Moshkovits I, Kaminitz A, et al. The critical role of chemokine (C-C Motif) receptor 2-positive monocytes in autoimmune cholangitis. Front Immunol. (2018) 9:1852. doi: 10.3389/fimmu.2018.01852
162. Harada K, Shimoda S, Ikeda H, Chiba M, Hsu M, Sato Y, et al. Significance of periductal Langerhans cells and biliary epithelial cell-derived macrophage inflammatory protein-3alpha in the pathogenesis of primary biliary cirrhosis. Liver Int. (2011) 31:245–53. doi: 10.1111/j.1478-3231.2010.02367.x
163. Harada K, Ozaki S, Sudo Y, Tsuneyama K, Ohta H, Nakanuma Y. Osteopontin is involved in the formation of epithelioid granuloma and bile duct injury in primary biliary cirrhosis. Pathol Int. (2003) 53:8–17. doi: 10.1046/j.1440-1827.2003.01426.x
164. Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. (2009) 49:871–9. doi: 10.1002/hep.22736
165. Qin B, Wei T, Wang L, Ma N, Tang Q, Liang Y, et al. Decreased expression of TIPE2 contributes to the hyperreactivity of monocyte to Toll-like receptor ligands in primary biliary cirrhosis. J Gastroenterol Hepatol. (2016) 31:1177–83. doi: 10.1111/jgh.13251
166. Tomiyama T, Yang GX, Zhao M, Zhang W, Tanaka H, Wang J, et al. The modulation of co-stimulatory molecules by circulating exosomes in primary biliary cirrhosis. Cell Mol Immunol. (2017) 14:276–84. doi: 10.1038/cmi.2015.86
167. Tian J, Yang G, Chen HY, Hsu DK, Tomilov A, Olson KA, et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB J. (2016) 30:4202–13. doi: 10.1096/fj.201600392RR
168. Aota K, Yamanoi T, Kani K, Nakashiro KI, Ishimaru N, Azuma M. Inverse correlation between the number of CXCR3(+) macrophages and the severity of inflammatory lesions in Sjogren's syndrome salivary glands: a pilot study. J Oral Pathol Med. (2018) 47:710–8. doi: 10.1111/jop.12756
169. Vakrakou AG, Boiu S, Ziakas PD, Xingi E, Boleti H, Manoussakis MN. Systemic activation of NLRP3 inflammasome in patients with severe primary Sjogren's syndrome fueled by inflammagenic DNA accumulations. J Autoimmun. (2018) 91:23–33. doi: 10.1016/j.jaut.2018.02.010
170. Williams AE, Choi K, Chan AL, Lee YJ, Reeves WH, Bubb MR, et al. Sjogren's syndrome-associated microRNAs in CD14(+) monocytes unveils targeted TGFbeta signaling. Arthritis Res Ther. (2016) 18:95. doi: 10.1186/s13075-016-0987-0
171. Ushio A, Arakaki R, Otsuka K, Yamada A, Tsunematsu T, Kudo Y, et al. CCL22-producing resident macrophages enhance T cell response in sjogren's syndrome. Front Immunol. (2018) 9:2594. doi: 10.3389/fimmu.2018.02594
172. Yoshimoto K, Tanaka M, Kojima M, Setoyama Y, Kameda H, Suzuki K, et al. Regulatory mechanisms for the production of BAFF and IL-6 are impaired in monocytes of patients of primary Sjogren's syndrome. Arthritis Res Ther. (2011) 13:R170. doi: 10.1186/ar3493
173. Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren's syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis. (2013) 72:728–35. doi: 10.1136/annrheumdis-2012-201381
174. Jelinkova L, Tuckova L, Cinova J, Flegelova Z, Tlaskalova-Hogenova H. Gliadin stimulates human monocytes to production of IL-8 and TNF-alpha through a mechanism involving NF-kappaB. FEBS Lett. (2004) 571:81–5. doi: 10.1016/j.febslet.2004.06.057
175. Harris KM, Fasano A, Mann DL. Monocytes differentiated with IL-15 support Th17 and Th1 responses to wheat gliadin: implications for celiac disease. Clin Immunol. (2010) 135:430–9. doi: 10.1016/j.clim.2010.01.003
176. Thomas KE, Sapone A, Fasano A, Vogel SN. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. (2006) 176:2512–21. doi: 10.4049/jimmunol.176.4.2512
177. Junker Y, Zeissig S, Kim SJ, Barisani D, Wieser H, Leffler DA, et al. Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J Exp Med. (2012) 209:2395–408. doi: 10.1084/jem.20102660
178. Zanoni G, Navone R, Lunardi C, Tridente G, Bason C, Sivori S, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. (2006) 3:1637–53. doi: 10.1371/journal.pmed.0030358
179. Palova-Jelinkova L, Danova K, Drasarova H, Dvorak M, Funda DP, Fundova P, et al. Pepsin digest of wheat gliadin fraction increases production of IL-1beta via TLR4/MyD88/TRIF/MAPK/NF-kappaB signaling pathway and an NLRP3 inflammasome activation. PLoS ONE. (2013) 8:e62426. doi: 10.1371/journal.pone.0062426
180. Castellanos-Rubio A, Kratchmarov R, Sebastian M, Garcia-Etxebarria K, Garcia L, Irastorza I, et al. Cytoplasmic form of Carlr lncRNA facilitates inflammatory gene expression upon NF-kappa B activation. J Immunol. (2017) 199:581–8. doi: 10.4049/jimmunol.1700023
181. Frisullo G, Nociti V, Iorio R, Patanella AK, Plantone D, Bianco A, et al. T-bet and pSTAT-1 expression in PBMC from coeliac disease patients: new markers of disease activity. Clin Exp Immunol. (2009) 158:106–14. doi: 10.1111/j.1365-2249.2009.03999.x
182. Chrobok NL, Sestito C, Wilhelmus MM, Drukarch B, van Dam AM. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids. (2017) 49:441–52. doi: 10.1007/s00726-016-2334-9
183. Brynychova V, Vaclavikova R, Hlavac V, Trnkova M, Kozevnikovova R, Mrhalova M, et al. Downregulation of miR-195 and miR-497 in luminal breast carcinoma is associated with high grade of tumors, expression of HER2, and luminal B subtype but not with presence of LN metastasis in patients. Febs Open Bio. (2018) 8:303.
184. Harris KM, Fasano A, Mann DL. Cutting edge: IL-1 controls the IL-23 response induced by gliadin, the etiologic agent in celiac disease. J Immunol. (2008) 181:4457–60. doi: 10.4049/jimmunol.181.7.4457
185. Seo DH, Che X, Kwak MS, Kim S, Kim JH, Ma HW, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci Rep. (2017) 7:851. doi: 10.1038/s41598-017-00840-2
186. Mitsuhashi S, Feldbrugge L, Csizmadia E, Mitsuhashi M, Robson SC, Moss AC. Luminal extracellular vesicles (EVs) in inflammatory bowel disease (IBD) exhibit proinflammatory effects on epithelial cells and macrophages. Inflamm Bowel Dis. (2016) 22:1587–95. doi: 10.1097/MIB.0000000000000840
187. Kanai T, Mikami Y, Hayashi A. A breakthrough in probiotics: Clostridium butyricum regulates gut homeostasis and anti-inflammatory response in inflammatory bowel disease. J Gastroenterol. (2015) 50:928–39. doi: 10.1007/s00535-015-1084-x
188. Wendelsdorf K, Bassaganya-Riera J, Hontecillas R, Eubank S. Model of colonic inflammation: immune modulatory mechanisms in inflammatory bowel disease. J Theor Biol. (2010) 264:1225–39. doi: 10.1016/j.jtbi.2010.03.027
189. Hontecillas R, Horne WT, Climent M, Guri AJ, Evans C, Zhang Y, et al. Immunoregulatory mechanisms of macrophage PPAR-gamma in mice with experimental inflammatory bowel disease. Mucosal Immunol. (2011) 4:304–13. doi: 10.1038/mi.2010.75
190. Liu R, Tang A, Wang X, Chen X, Zhao L, Xiao Z, et al. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int J Mol Med. (2018) 42:2903–13. doi: 10.3892/ijmm.2018.3829
191. Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Takayama T, et al. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn's disease. Inflamm Bowel Dis. (2010) 16:568–75. doi: 10.1002/ibd.21124
192. Ogino T, Nishimura J, Barman S, Kayama H, Uematsu S, Okuzaki D, et al. Increased Th17-inducing activity of CD14+ CD163 low myeloid cells in intestinal lamina propria of patients with Crohn's disease. Gastroenterology. (2013) 145:1380–91 e1. doi: 10.1053/j.gastro.2013.08.049
193. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. (2008) 118:2269–80. doi: 10.1172/JCI34610
194. Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Kitazume MT, et al. Human CD14+ macrophages in intestinal lamina propria exhibit potent antigen-presenting ability. J Immunol. (2009) 183:1724–31. doi: 10.4049/jimmunol.0804369
195. Liu Z, Yadav PK, Xu X, Su J, Chen C, Tang M, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. (2011) 89:597–606. doi: 10.1189/jlb.0810456
196. Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. (2008) 57:1682–9. doi: 10.1136/gut.2007.135053
197. Globig AM, Hennecke N, Martin B, Seidl M, Ruf G, Hasselblatt P, et al. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-gamma+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm Bowel Dis. (2014) 20:2321–9. doi: 10.1097/MIB.0000000000000210
198. Liu L, Dong Y, Ye M, Jin S, Yang J, Joosse ME, et al. The pathogenic role of NLRP3 inflammasome activation in inflammatory bowel diseases of both mice and humans. J Crohns Colitis. (2017) 11:737–50. doi: 10.1093/ecco-jcc/jjw219
199. Bengtsson AA, Sturfelt G, Gullstrand B, Truedsson L. Induction of apoptosis in monocytes and lymphocytes by serum from patients with systemic lupus erythematosus - an additional mechanism to increased autoantigen load? Clin Exp Immunol. (2004) 135:535–43. doi: 10.1111/j.1365-2249.2003.02386.x
200. Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun. (2015) 57:42–52. doi: 10.1016/j.jaut.2014.11.007
201. Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front Immunol. (2018) 9:427. doi: 10.3389/fimmu.2018.00427
202. Harigai M, Hara M, Fukasawa C, Nakazawa S, Kawaguchi Y, Kamatani N, et al. Responsiveness of peripheral blood B cells to recombinant CD40 ligand in patients with systemic lupus erythematosus. Lupus. (1999) 8:227–33. doi: 10.1191/096120399678847678
203. Higuchi T, Aiba Y, Nomura T, Matsuda J, Mochida K, Suzuki M, et al. Cutting edge: ectopic expression of CD40 ligand on B cells induces lupus-like autoimmune disease. J Immunol. (2002) 168:9–12. doi: 10.4049/jimmunol.168.1.9
204. Wang X, Huang W, Schiffer LE, Mihara M, Akkerman A, Hiromatsu K, et al. Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis Rheum. (2003) 48:495–506. doi: 10.1002/art.10929
205. Ronchetti S, Migliorati G, Bruscoli S, Riccardi C. Defining the role of glucocorticoids in inflammation. Clin Sci. (2018) 132:1529–43. doi: 10.1042/CS20171505
206. Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. (2014) 14:653–66. doi: 10.1038/nri3737
207. Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. (2003) 48:2888–97. doi: 10.1002/art.11237
208. Summers SA, Hoi A, Steinmetz OM, O'Sullivan KM, Ooi JD, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. (2010) 35:291–8. doi: 10.1016/j.jaut.2010.05.004
209. Frank MM, Hamburger MI, Lawley TJ, Kimberly RP, Plotz PH. Defective reticuloendothelial system Fc-receptor function in systemic lupus erythematosus. N Engl J Med. (1979) 300:518–23. doi: 10.1056/NEJM197903083001002
210. Dijstelbloem HM, Bijl M, Fijnheer R, Scheepers RH, Oost WW, Jansen MD, et al. Fcgamma receptor polymorphisms in systemic lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis Rheum. (2000) 43:2793–800. doi: 10.1002/1529-0131(200012)43:12<2793::AID-ANR20>3.0.CO;2-6
211. Salmon JE, Millard S, Schachter LA, Arnett FC, Ginzler EM, Gourley MF, et al. Fc gamma RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J Clin Invest. (1996) 97:1348–54. doi: 10.1172/JCI118552
212. Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS ONE. (2018) 13:e0208132. doi: 10.1371/journal.pone.0208132
213. Liu AC, Yang Y, Li MT, Jia Y, Chen S, Ye S, et al. Macrophage activation syndrome in systemic lupus erythematosus: a multicenter, case-control study in China. Clin Rheumatol. (2018) 37:93–100. doi: 10.1007/s10067-017-3625-6
214. Bracaglia C, Prencipe G, De Benedetti F. Macrophage activation syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J. (2017) 15:5. doi: 10.1186/s12969-016-0130-4
215. Becker-Merok A, Eilertsen GO, Nossent JC. Levels of transforming growth factor-beta are low in systemic lupus erythematosus patients with active disease. J Rheumatol. (2010) 37:2039–45. doi: 10.3899/jrheum.100180
216. Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage polarization modulates development of systemic lupus erythematosus. Cell Physiol Biochem. (2015) 37:1279–88. doi: 10.1159/000430251
217. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. (2017) 26:1318–27. doi: 10.1177/0961203317701842
218. Bussolati B, Rollino C, Mariano F, Quarello F, Camussi G. IL-10 stimulates production of platelet-activating factor by monocytes of patients with active systemic lupus erythematosus (SLE). Clin Exp Immunol. (2000) 122:471–6. doi: 10.1046/j.1365-2249.2000.01392.x
219. Llorente L, Richaud-Patin Y, Garcia-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, et al. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. (2000) 43:1790–800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2
220. Tackey E, Lipsky PE, Illei GG. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus. (2004) 13:339–43. doi: 10.1191/0961203304lu1023oa
221. Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. (2010) 62:542–52. doi: 10.1002/art.27221
222. Aringer M, Graninger WB, Steiner G, Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum. (2004) 50:3161–9. doi: 10.1002/art.20576
223. Aringer M, Houssiau F, Gordon C, Graninger WB, Voll RE, Rath E, et al. Adverse events and efficacy of TNF-alpha blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatology. (2009) 48:1451–4. doi: 10.1093/rheumatology/kep270
224. Sahu R, Bethunaickan R, Singh S, Davidson A. Structure and function of renal macrophages and dendritic cells from lupus-prone mice. Arthritis Rheumatol. (2014) 66:1596–607. doi: 10.1002/art.38410
225. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Sodium valproate modulates immune response by alternative activation of monocyte-derived macrophages in systemic lupus erythematosus. Clin Rheumatol. (2018) 37:719–27. doi: 10.1007/s10067-017-3922-0
226. Mohammadi S, Memarian A, Sedighi S, Behnampour N, Yazdani Y. Immunoregulatory effects of indole-3-carbinol on monocyte-derived macrophages in systemic lupus erythematosus: a crucial role for aryl hydrocarbon receptor. Autoimmunity. (2018) 51:199–209. doi: 10.1080/08916934.2018.1494161
227. Laurent P, Sisirak V, Lazaro E, Richez C, Duffau P, Blanco P, et al. Innate immunity in systemic sclerosis fibrosis: recent advances. Front Immunol. (2018) 9:1702. doi: 10.3389/fimmu.2018.01702
228. Duan H, Fleming J, Pritchard DK, Amon LM, Xue J, Arnett HA, et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum. (2008) 58:1465–74. doi: 10.1002/art.23451
229. Moreno-Moral A, Bagnati M, Koturan S, Ko JH, Fonseca C, Harmston N, et al. Changes in macrophage transcriptome associate with systemic sclerosis and mediate GSDMA contribution to disease risk. Ann Rheum Dis. (2018) 77:596–601. doi: 10.1136/annrheumdis-2017-212454
230. Beamer CA, Holian A. Scavenger receptor class A type I/II (CD204) null mice fail to develop fibrosis following silica exposure. Am J Physiol. (2005) 289:L186–95. doi: 10.1152/ajplung.00474.2004
231. Nakayama W, Jinnin M, Makino K, Kajihara I, Makino T, Fukushima S, et al. Serum levels of soluble CD163 in patients with systemic sclerosis. Rheumatol Int. (2012) 32:403–7. doi: 10.1007/s00296-010-1691-z
232. Frantz C, Pezet S, Avouac J, Allanore Y. Soluble CD163 as a potential biomarker in systemic sclerosis. Dis Markers. (2018) 2018:8509583. doi: 10.1136/annrheumdis-2018-eular.2631
233. Bielecki M, Kowal K, Lapinska A, Chyczewski L, Kowal-Bielecka O. Increased release of soluble CD163 by the peripheral blood mononuclear cells is associated with worse prognosis in patients with systemic sclerosis. Adv Med Sci. (2013) 58:126–33. doi: 10.2478/v10039-012-0076-9
234. Sato S, Hasegawa M, Takehara K. Serum levels of interleukin-6 and interleukin-10 correlate with total skin thickness score in patients with systemic sclerosis. J Dermatol Sci. (2001) 27:140–6. doi: 10.1016/S0923-1811(01)00128-1
235. Huang XL, Wang YJ, Yan JW, Wan YN, Chen B, Li BZ, et al. Role of anti-inflammatory cytokines IL-4 and IL-13 in systemic sclerosis. Inflamm Res. (2015) 64:151–9. doi: 10.1007/s00011-015-0806-0
236. Fuschiotti P. Role of IL-13 in systemic sclerosis. Cytokine. (2011) 56:544–9. doi: 10.1016/j.cyto.2011.08.030
237. Chia JJ, Lu TT. Update on macrophages and innate immunity in scleroderma. Curr Opin Rheumatol. (2015) 27:530–6. doi: 10.1097/BOR.0000000000000218
238. Braga TT, Agudelo JS, Camara NO. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol. (2015) 6:602. doi: 10.3389/fimmu.2015.00602
239. Lescoat A, Jego P, Lecureur V. M-CSF and GM-CSF monocyte-derived macrophages in systemic sclerosis: the two sides of the same coin? Ann Rheum Dis. (2018) 2018:213112. doi: 10.1136/annrheumdis-2018-213112
240. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. (1996) 39:115–24. doi: 10.1002/art.1780390116
241. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. (1997) 40:217–25. doi: 10.1002/art.1780400206
242. Yanni G, Nabil M, Farahat MR, Poston RN, Panayi GS. Intramuscular gold decreases cytokine expression and macrophage numbers in the rheumatoid synovial membrane. Ann Rheum Dis. (1994) 53:315–22. doi: 10.1136/ard.53.5.315
243. Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. (2005) 64:834–8. doi: 10.1136/ard.2004.029751
244. Bresnihan B, Gerlag DM, Rooney T, Smeets JM, Wijbrandts CA, Boyle D, et al. Synovial macrophages as a biomarker of response to therapeutic intervention in rheumatoid arthritis: standardization and consistency across centers. J Rheumatol. (2007) 34:620–2.
245. Van Lent PL, Holthuysen AE, Van Rooijen N, Van De Putte LB, Van Den Berg WB. Local removal of phagocytic synovial lining cells by clodronate-liposomes decreases cartilage destruction during collagen type II arthritis. Ann Rheum Dis. (1998) 57:408–13. doi: 10.1136/ard.57.7.408
246. Park SY, Lee SW, Kim HY, Lee SY, Lee WS, Hong KW, et al. SIRT1 inhibits differentiation of monocytes to macrophages: amelioration of synovial inflammation in rheumatoid arthritis. J Mol Med. (2016) 94:921–31. doi: 10.1007/s00109-016-1402-7
247. Dongsheng Z, Zhiguang F, Junfeng J, Zifan L, Li W. Cyclophilin A aggravates collagen-induced arthritis via promoting classically activated macrophages. Inflammation. (2017) 2017:619. doi: 10.1007/s10753-017-0619-0
248. Park SY, Lee SW, Lee SY, Hong KW, Bae SS, Kim K, et al. SIRT1/adenosine monophosphate-activated protein kinase alpha signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front Immunol. (2017) 8:1135. doi: 10.3389/fimmu.2017.01135
249. Sun W, Zhang HW, Wang H, Chiu YG, Wang MM, Ritchlin CT, et al. Targeting notch-activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res. (2017) 32:1469–80. doi: 10.1002/jbmr.3117
250. Shin TH, Kim HS, Kang TW, Lee BC, Lee HY, Kim YJ, et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. (2016) 7:e2524. doi: 10.1038/cddis.2016.442
251. Donlin LT, Jayatilleke A, Giannopoulou EG, Kalliolias GD, Ivashkiv LB. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol. (2014) 193:2373–83. doi: 10.4049/jimmunol.1400486
252. Li N, Hua JL. Interactions between mesenchymal stem cells and the immune system. Cell Mol Life Sci. (2017) 74:2345–60. doi: 10.1007/s00018-017-2473-5
253. Chen S, Chen BN, Wen ZY, Huang Z, Ye L. IL-33/ST2-mediated inflammation in macrophages is directly abrogated by IL-10 during rheumatoid arthritis. Oncotarget. (2017) 8:32407–18. doi: 10.18632/oncotarget.16299
254. Ye L, Wen ZY, Li YQ, Chen BN, Yu T, Liu LY, et al. Interleukin-10 attenuation of collagen-induced arthritis is associated with suppression of interleukin-17 and retinoid-related orphan receptor gamma t production in macrophages and repression of classically activated macrophages. Arthritis Res Ther. (2014) 16:4544. doi: 10.1186/ar4544
255. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nat Rev Rheumatol. (2016) 12:63–8. doi: 10.1038/nrrheum.2015.171
256. van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. (2013) 9:164–72. doi: 10.1038/nrrheum.2013.4
257. Quinn MA, Conaghan PG, O'Connor PJ, Karim Z, Greenstein A, Brown A, et al. Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2005) 52:27–35. doi: 10.1002/art.20712
258. Yarilina A, Xu K, Chan C, Ivashkiv LB. Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum. (2012) 64:3856–66. doi: 10.1002/art.37691
259. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. (2007) 7:429–42. doi: 10.1038/nri2094
260. Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, et al. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. (2011) 70:764–73. doi: 10.1002/ana.22521
261. Tambalo S, Peruzzotti-Jametti L, Rigolio R, Fiorini S, Bontempi P, Mallucci G, et al. Functional magnetic resonance imaging of rats with experimental autoimmune encephalomyelitis reveals brain cortex remodeling. J Neurosci. (2015) 35:10088–100. doi: 10.1523/JNEUROSCI.0540-15.2015
262. Planche V, Panatier A, Hiba B, Ducourneau EG, Raffard G, Dubourdieu N, et al. Selective dentate gyrus disruption causes memory impairment at the early stage of experimental multiple sclerosis. Brain Behav Immun. (2017) 60:240–54. doi: 10.1016/j.bbi.2016.11.010
263. Jiang Z, Jiang JX, Zhang GX. Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol Lett. (2014) 160:17–22. doi: 10.1016/j.imlet.2014.03.006
264. Van Weyenbergh J, Wietzerbin J, Rouillard D, Barral-Netto M, Liblau R. Treatment of multiple sclerosis patients with interferon-beta primes monocyte-derived macrophages for apoptotic cell death. J Leukoc Biol. (2001) 70:745–8. doi: 10.1189/jlb.70.5.745
265. Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. (1990) 172:1025–33. doi: 10.1084/jem.172.4.1025
266. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. (2005) 11:146–52. doi: 10.1038/nm1177
267. Moreno MA, Burns T, Yao P, Miers L, Pleasure D, Soulika AM. Therapeutic depletion of monocyte-derived cells protects from long-term axonal loss in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2016) 290:36–46. doi: 10.1016/j.jneuroim.2015.11.004
268. Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2008) 105:9041–6. doi: 10.1073/pnas.0802218105
269. Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. (1998) 161:3767–75.
270. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa SH, Kwok CK, Leonardi T, et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell. (2018) 22:355–68 e13. doi: 10.1016/j.stem.2018.01.020
271. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. (2009) 29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009
272. Abdul-Majid KB, Stefferl A, Bourquin C, Lassmann H, Linington C, Olsson T, et al. Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scand J Immunol. (2002) 55:70–81. doi: 10.1046/j.1365-3083.2002.01024.x
273. Hucke S, Eschborn M, Liebmann M, Herold M, Freise N, Engbers A, et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J Autoimmun. (2016) 67:90–101. doi: 10.1016/j.jaut.2015.11.001
274. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol. (2012) 42:1804–14. doi: 10.1002/eji.201141947
275. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
276. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol. (2018) 9:697. doi: 10.3389/fimmu.2018.00697
277. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. (2014) 211:1533–49. doi: 10.1084/jem.20132477
278. Bulum T, Kolaric B, Duvnjak L. Decreased serum monocytes and elevated neutrophils as additional markers of insulin resistance in type 1 diabetes. Int J Diabetes Dev C. (2014) 34:150–5. doi: 10.1007/s13410-013-0176-5
279. Vomund AN, Zinselmeyer BH, Hughes J, Calderon B, Valderrama C, Ferris ST, et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci USA. (2015) 112:E5496–502. doi: 10.1073/pnas.1515954112
280. Marro BS, Legrain S, Ware BC, Oldstone BA. Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight. (2019) 4:125067. doi: 10.1172/jci.insight.125067
281. Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S, et al. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care. (2009) 32:1244–9. doi: 10.2337/dc09-0054
282. Tack CJ, Kleijwegt FS, Van Riel PL, Roep BO. Development of type 1 diabetes in a patient treated with anti-TNF-alpha therapy for active rheumatoid arthritis. Diabetologia. (2009) 52:1442–4. doi: 10.1007/s00125-009-1381-0
283. Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, et al. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes. (2012) 61:2881–92. doi: 10.2337/db11-1635
284. Zheng D, Wang Y, Cao Q, Lee VW, Zheng G, Sun Y, et al. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp Nephrol. (2011) 118:e87-99. doi: 10.1159/000321034
285. Albiero M, Poncina N, Ciciliot S, Cappellari R, Menegazzo L, Ferraro F, et al. Bone marrow macrophages contribute to diabetic stem cell mobilopathy by producing oncostatin M. Diabetes. (2015) 64:2957–68. doi: 10.2337/db14-1473
286. Ferris ST, Zakharov PN, Wan XX, Calderon B, Artyomov MN, Unanue ER, et al. The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J Exp Med. (2017) 214:2369–85. doi: 10.1084/jem.20170074
287. Carrero JA, Ferris ST, Unanue ER. Macrophages and dendritic cells in islets of Langerhans in diabetic autoimmunity: a lesson on cell interactions in a mini-organ. Curr Opin Immunol. (2016) 43:54–59. doi: 10.1016/j.coi.2016.09.004
288. Carrero JA, McCarthy DP, Ferris ST, Wan XX, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci USA. (2017) 114:E10418–27. doi: 10.1073/pnas.1713543114
289. Tsuneyama K, Harada K, Yasoshima M, Hiramatsu K, Mackay CR, Mackay IR, et al. Monocyte chemotactic protein-1,−2, and−3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: implications for pathogenesis. J Pathol. (2001) 193:102–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH725>3.0.CO;2-P
290. Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. (2006) 177:1655–60. doi: 10.4049/jimmunol.177.3.1655
291. Greenwell-Wild T, Moutsopoulos NM, Gliozzi M, Kapsogeorgou E, Rangel Z, Munson PJ, et al. Chitinases in the salivary glands and circulation of patients with sjogren's syndrome macrophage harbingers of disease severity. Arthritis Rheum. (2011) 63:3103–15. doi: 10.1002/art.30465
292. Delaleu N, Mydel P, Kwee I, Brun JG, Jonsson MV, Jonsson R. High fidelity between saliva proteomics and the biologic state of salivary glands defines biomarker signatures for primary sjogren's syndrome. Arthritis Rheumatol. (2015) 67:1084–95. doi: 10.1002/art.39015
293. Zhou DL, Chen YT, Chen FL, Gallup M, Vijmasi T, Bahrami AF, et al. Critical involvement of macrophage infiltration in the development of sjogren's syndrome-associated dry eye. Am J Pathol. (2012) 181:753–60. doi: 10.1016/j.ajpath.2012.05.014
294. Hauk V, Calafat M, Larocca L, Fraccaroli L, Grasso E, Ramhorst R, et al. Vasoactive intestinal peptide/vasoactive intestinal peptide receptor relative expression in salivary glands as one endogenous modulator of acinar cell apoptosis in a murine model of Sjogren's syndrome. Clin Exp Immunol. (2011) 166:309–16. doi: 10.1111/j.1365-2249.2011.04478.x
295. Willeke P, Gaubitz M, Schotte H, Maaser C, Domschke W, Schluter B, et al. Increased serum levels of macrophage migration inhibitory factor in patients with primary Sjogren's syndrome. Arthritis Res Ther. (2007) 9:R43. doi: 10.1186/ar2182
296. Baban B, Liu JY, Abdelsayed R, Mozaffari MS. Reciprocal relation between GADD153 and Del-1 in regulation of salivary gland inflammation in Sjogren syndrome. Exp Mol Pathol. (2013) 95:288–97. doi: 10.1016/j.yexmp.2013.09.002
297. Awada A, Nicaise C, Ena S, Schandene L, Rasschaert J, Popescu I, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary Sjogren's syndrome. Ann Rheumat Dis. (2014) 73:1259–63. doi: 10.1136/annrheumdis-2012-203187
298. Tuckova L, Flegelova Z, Tlaskalova-Hogenova H, Zidek Z. Activation of macrophages by food antigens: enhancing effect of gluten on nitric oxide and cytokine production. J Leukoc Biol. (2000) 67:312–8. doi: 10.1002/jlb.67.3.312
299. De Stefano D, Maiuri MC, Iovine B, Ialenti A, Bevilacqua MA, Carnuccio R. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J Mol Med. (2006) 84:65–74. doi: 10.1007/s00109-005-0713-x
300. Maiuri MC, De Stefano D, Mele G, Iovine B, Bevilacqua MA, Greco L, et al. Gliadin increases iNOS gene expression in interferon-gamma-stimulated RAW 264.7 cells through a mechanism involving NF-kappa B. Naunyn Schmiedebergs Arch Pharmacol. (2003) 368:63–71. doi: 10.1007/s00210-003-0771-y
301. O'Keeffe J, Lynch S, Whelan A, Jackson J, Kennedy NP, Weir DG, et al. Flow cytometric measurement of intracellular migration inhibition factor and tumour necrosis factor alpha in the mucosa of patients with coeliac disease. Clin Exp Immunol. (2001) 125:376–82. doi: 10.1046/j.1365-2249.2001.01594.x
302. Barisani D, Ceroni S, Meneveri R, Cesana BM, Bardella MT. IL-10 polymorphisms are associated with early-onset celiac disease and severe mucosal damage in patients of Caucasian origin. Genet Med. (2006) 8:169–74. doi: 10.1097/01.gim.0000204464.87540.39
303. Nunez C, Alecsandru D, Varade J, Polanco I, Maluenda C, Fernandez-Arquero M, et al. Interleukin-10 haplotypes in Celiac Disease in the Spanish population. BMC Med Genet. (2006) 7:32. doi: 10.1186/1471-2350-7-32
304. Kredel LI, Batra A, Stroh T, Kuhl AA, Zeitz M, Erben U, et al. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn's disease. Gut. (2013) 62:852–62. doi: 10.1136/gutjnl-2011-301424
305. Hanai H, Iida T, Takeuchi K, Watanabe F, Yamada M, Kikuyama M, et al. Adsorptive depletion of elevated proinflammatory CD14+CD16+DR++ monocytes in patients with inflammatory bowel disease. Am J Gastroenterol. (2008) 103:1210–6. doi: 10.1111/j.1572-0241.2007.01714.x
306. Gong Y, Lin Y, Zhao N, He X, Lu A, Wei W, et al. The Th17/treg immune imbalance in ulcerative colitis disease in a chinese han population. Mediat Inflamm. (2016) 2016:7089137. doi: 10.1155/2016/7089137
307. Sanders TJ, McCarthy NE, Giles EM, Davidson KL, Haltalli ML, Hazell S, et al. Increased production of retinoic acid by intestinal macrophages contributes to their inflammatory phenotype in patients with Crohn's disease. Gastroenterology. (2014) 146:1278–88 e1-2. doi: 10.1053/j.gastro.2014.01.057
308. Lissner D, Schumann M, Batra A, Kredel LI, Kuhl AA, Erben U, et al. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm Bowel Dis. (2015) 21:1297–305. doi: 10.1097/MIB.0000000000000384
309. Haribhai D, Ziegelbauer J, Jia S, Upchurch K, Yan K, Schmitt EG, et al. Alternatively activated macrophages boost induced regulatory T and Th17 cell responses during immunotherapy for colitis. J Immunol. (2016) 196:3305–17. doi: 10.4049/jimmunol.1501956
310. Dige A, Stoy S, Thomsen KL, Hvas CL, Agnholt J, Dahlerup JF, et al. Soluble CD163, a specific macrophage activation marker, is decreased by anti-TNF-alpha antibody treatment in active inflammatory bowel disease. Scand J Immunol. (2014) 80:417–23. doi: 10.1111/sji.12222
311. Scharl M, Huber N, Lang S, Furst A, Jehle E, Rogler G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn's disease associated intestinal fibrosis. Clin Transl Med. (2015) 4:1. doi: 10.1186/s40169-015-0046-5
312. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
313. Zhu Y, Li X, Chen J, Chen T, Shi Z, Lei M, et al. The pentacyclic triterpene Lupeol switches M1 macrophages to M2 and ameliorates experimental inflammatory bowel disease. Int Immunopharmacol. (2016) 30:74–84. doi: 10.1016/j.intimp.2015.11.031
314. Song WJ, Li Q, Ryu MO, Ahn JO, Ha Bhang D, Chan Jung Y, et al. TSG-6 secreted by human adipose tissue-derived mesenchymal stem cells ameliorates DSS-induced colitis by inducing M2 macrophage polarization in mice. Sci Rep. (2017) 7:5187. doi: 10.1038/s41598-017-04766-7
315. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. (2006) 15:308–18. doi: 10.1191/0961203306lu2305xx
316. Zhou X, Lee JE, Arnett FC, Xiong M, Park MY, Yoo YK, et al. HLA-DPB1 and DPB2 are genetic loci for systemic sclerosis: a genome-wide association study in Koreans with replication in North Americans. Arthritis Rheum. (2009) 60:3807–14. doi: 10.1002/art.24982
317. C. Wellcome Trust Case Control. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
318. Bashinskaya VV, Kulakova OG, Boyko AN, Favorov AV, Favorova OO. A review of genome-wide association studies for multiple sclerosis: classical and hypothesis-driven approaches. Hum Genet. (2015) 134:1143–62. doi: 10.1007/s00439-015-1601-2
319. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. (2009) 41:703–7. doi: 10.1038/ng.381
320. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. (2009) 360:1646–54. doi: 10.1056/NEJMra0808284
321. Qu HQ, Bradfield JP, Li QA, Kim C, Frackelton E, Grant FA, et al. In silico replication of the genome-wide association results of the Type 1 Diabetes Genetics Consortium. Hum Mol Genet. (2010) 19:2534–8. doi: 10.1093/hmg/ddq133
322. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what's next? Diabetes. (2010) 59:1561–71. doi: 10.2337/db10-0076
323. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren's syndrome. Nat Genet. (2013) 45:1284. doi: 10.1038/ng.2792
324. Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. (2008) 40:395–402. doi: 10.1038/ng.102
325. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
326. Armstrong DL, Zidovetzki R, Alarcon-Riquelme ME, Tsao BP, Criswell LA, Kimberly RP, et al. GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immunity. (2014) 15:347–54. doi: 10.1038/gene.2014.23
327. Kottyan LC, Zoller EE, Bene J, Lu XM, Kelly JA, Rupert AM, et al. The IRF5-TNPO3 association with systemic lupus erythematosus has two components that other autoimmune disorders variably share. Hum Mol Genet. (2015) 24:582–96. doi: 10.1093/hmg/ddu455
328. Hirschfield GM, Liu XD, Han YH, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5-TNPO3:17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. (2010) 42:655–7. doi: 10.1038/ng.631
329. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–124. doi: 10.1038/nature11582
330. McGovern DPB, Kugathasan S, Cho JH. Genetics of inflammatory bowel diseases. Gastroenterology. (2015) 149:1163. doi: 10.1053/j.gastro.2015.08.001
331. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
332. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
333. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
334. Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. (2007) 110:4077–85. doi: 10.1182/blood-2007-02-073841
335. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
336. Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. (1996) 97:2063–73. doi: 10.1172/JCI118643
337. Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. (1996) 98:826–37. doi: 10.1172/JCI118855
338. Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. (2016) 387:2630–40. doi: 10.1016/S0140-6736(16)00232-4
339. Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. (2015) 125:2795–807. doi: 10.1172/JCI77958
340. Davignon JL, Hayder M, Baron M, Boyer JF, Constantin A, Apparailly F, et al. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology. (2013) 52:590–8. doi: 10.1093/rheumatology/kes304
341. Inui T, Katsuura G, Kubo K, Kuchiike D, Chenery L, Uto Y, et al. Case report: GcMAF treatment in a patient with multiple sclerosis. Anticancer Res. (2016) 36:3771–4.
342. Liu C, Li Y, Yu J, Feng L, Hou S, Liu Y, et al. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS ONE. (2013) 8:e54841. doi: 10.1371/journal.pone.0054841
343. Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes. (2011) 60:2144–51. doi: 10.2337/db10-1222
344. Daneshmandi S, Karimi MH, Pourfathollah AA. TGF-beta engineered mesenchymal stem cells (TGF-beta/MSCs) for treatment of Type 1 diabetes (T1D) mice model. Int Immunopharmacol. (2017) 44:191–6. doi: 10.1016/j.intimp.2017.01.019
345. Wang L, Li J, Liu H, Li Y, Fu J, Sun Y, et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol. (2013) 28(Suppl 1):85–92. doi: 10.1111/jgh.12029
346. Wang L, Han Q, Chen H, Wang K, Shan GL, Kong F, et al. Allogeneic bone marrow mesenchymal stem cell transplantation in patients with UDCA-resistant primary biliary cirrhosis. Stem Cells Dev. (2014) 23:2482–9. doi: 10.1089/scd.2013.0500
347. Dimopoulou D, Dimitroulas T, Akriviadis E, Garyfallos A. Infliximab as a treatment option for patients with rheumatoid arthritis and primary biliary cirrhosis. Rheumatol Int. (2015) 35:1913–6. doi: 10.1007/s00296-015-3366-2
348. Kovacs A, Siminischi AG, Baksay B, Gall A, Takacs M, Szekanecz Z. Successful etanercept treatment for primary biliary cirrhosis associated with rheumatoid arthritis. Isr Med Assoc J. (2015) 17:114–6.
349. Hirschfield GM, Gershwin ME, Strauss R, Mayo MJ, Levy C, Zou B, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: A proof-of-concept study. Hepatology. (2016) 64:189–99. doi: 10.1002/hep.28359
350. Mariette X, Ravaud P, Steinfeld S, Baron G, Goetz J, Hachulla E, et al. Inefficacy of infliximab in primary Sjogren's syndrome: results of the randomized, controlled Trial of Remicade in Primary Sjogren's Syndrome (TRIPSS). Arthritis Rheum. (2004) 50:1270–6. doi: 10.1002/art.20146
351. Moutsopoulos NM, Katsifis GE, Angelov N, Leakan RA, Sankar V, Pillemer S, et al. Lack of efficacy of etanercept in Sjogren syndrome correlates with failed suppression of tumour necrosis factor alpha and systemic immune activation. Ann Rheum Dis. (2008) 67:1437–43. doi: 10.1136/ard.2007.077891
352. Croese J, Giacomin P, Navarro S, Clouston A, McCann L, Dougall A, et al. Experimental hookworm infection and gluten microchallenge promote tolerance in celiac disease. J Allergy Clin Immunol. (2015) 135:508–16. doi: 10.1016/j.jaci.2014.07.022
353. Gillett HR, Arnott ID, McIntyre M, Campbell S, Dahele A, Priest M, et al. Successful infliximab treatment for steroid-refractory celiac disease: a case report. Gastroenterology. (2002) 122:800–5. doi: 10.1053/gast.2002.31874
354. Costantino G, della Torre A, Lo Presti MA, Caruso R, Mazzon E, Fries W. Treatment of life-threatening type I refractory coeliac disease with long-term infliximab. Dig Liver Dis. (2008) 40:74–7. doi: 10.1016/j.dld.2006.10.017
355. Rugtveit J, Nilsen EM, Bakka A, Carlsen H, Brandtzaeg P, Scott H. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology. (1997) 112:1493–505. doi: 10.1016/S0016-5085(97)70030-1
356. Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology. (2004) 126:989–96; discussion 947. doi: 10.1053/j.gastro.2004.01.012
357. Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. (2004) 351:2069–79. doi: 10.1056/NEJMoa033402
358. Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, et al. Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. (2010) 16:233–42. doi: 10.1002/ibd.21038
359. Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, et al. Infliximab, azathioprine, or combination therapy for Crohn's disease. N Engl J Med. (2010) 362:1383–95. doi: 10.1056/NEJMoa0904492
360. Marshall DC, Lyman SK, McCauley S, Kovalenko M, Spangler R, Liu C, et al. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS ONE. (2015) 10:e0127063. doi: 10.1371/journal.pone.0127063
361. Panes J, Garcia-Olmo D, Van Assche G, Colombel JF, Reinisch W, Baumgart DC, et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. Lancet. (2016) 388:1281–90. doi: 10.1016/S0140-6736(16)31203-X
362. Garcia-Arranz M, Herreros MD, Gonzalez-Gomez C, de la Quintana P, Guadalajara H, Georgiev-Hristov T, et al. Treatment of crohn's-related rectovaginal fistula with allogeneic expanded-adipose derived stem cells: a phase I-IIa clinical trial. Stem Cells Transl Med. (2016) 5:1441–6. doi: 10.5966/sctm.2015-0356
Keywords: monocyte, macrophage, autoimmune disease, count, function, polarization
Citation: Ma W-T, Gao F, Gu K and Chen D-K (2019) The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 10:1140. doi: 10.3389/fimmu.2019.01140
Received: 01 February 2019; Accepted: 07 May 2019;
Published: 24 May 2019.
Edited by:
Chen Varol, Tel Aviv Sourasky Medical Center, IsraelReviewed by:
Felix Heymann, University Hospital RWTH Aachen, GermanyCopyright © 2019 Ma, Gao, Gu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Tao Ma, bWF3ZW50YW9AbndhZnUuZWR1LmNu; De-Kun Chen, Y2RrQG53c3VhZi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.