 Benjamin G. Barwick
Benjamin G. Barwick Vikas A. Gupta
Vikas A. Gupta Paula M. Vertino3
Paula M. Vertino3 Lawrence H. Boise
Lawrence H. Boise
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 May 2019
Sec. B Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01121
This article is part of the Research Topic Advances in Plasma Cells in Health and Disease View all 10 articles
B cell activation and differentiation yields plasma cells with high affinity antibodies to a given antigen in a time-frame that allows for host protection. Although the end product is most commonly humoral immunity, the rapid proliferation and somatic mutation of the B cell receptor also results in oncogenic mutations that cause B cell malignancies including plasma cell neoplasms such as multiple myeloma. Myeloma is the second most common hematological malignancy and results in over 100,000 deaths per year worldwide. The genetic alterations that occur in the germinal center, however, are not sufficient to cause myeloma, but rather impart cell proliferation potential on plasma cells, which are normally non-dividing. This pre-malignant state, referred to as monoclonal gammopathy of undetermined significance or MGUS, provides the opportunity for further genetic and epigenetic alterations eventually resulting in a progressive disease that becomes symptomatic. In this review, we will provide a brief history of clonal gammopathies and detail how some of the key discoveries were interwoven with the study of plasma cells. We will also review the genetic and epigenetic alterations discovered over the past 25 years, how these are instrumental to myeloma pathogenesis, and what these events teach us about myeloma and plasma cell biology. These data will be placed in the context of normal B cell development and differentiation and we will discuss how understanding the biology of plasma cells can lead to more effective therapies targeting multiple myeloma.
The study of malignancies that would ultimately be resolved to plasma cells was intertwined with, and necessary for the discovery of plasma cells and their function. Perhaps the first report of the plasma cell malignancy multiple myeloma described as “mollities ossium” by Samuel Solly in 1844 characterized two cases of patients who presented with symptoms including fatigue, bone pain, and multiple fractures (1). The author noted, that although rare, these were certainly not the first cases. Upon autopsy it was revealed that the bone marrow of both patients was replaced with a red substance filled with distinctive looking large cells [reviewed by Kyle and Rajkumar (2)]. The second patient noted that his urine stiffened his clothes, and a sample was sent for examination by Dr. Henry Bence Jones who confirmed the semi-solid urine would liquefy upon heating but resume its viscous consistency upon cooling (3, 4). Dr. Bence Jones emphasized the importance of obtaining urine samples for diagnosis, a practice that continues today.
Contemporaneous observations in immunity would lay a foundation for understanding the cellular source of these neoplasms and the Bence Jones proteins. The seminal work of John Fewster and Edward Jenner in smallpox demonstrated acquired immunity, which Jenner would later use to successfully protect patients through inoculation with cowpox (5). This led to discoveries in 1890, where Emil von Behring and Kitasato Shibasaburo showed that the serum of animals immunized with sub-lethal doses of dipetheria and tetanus contained an antitoxin (6). This proved the existence of an adaptive humoral immune system. The following year Paul Ehrlich described this antitoxin component as an “antibody” and in his 1908 Nobel laureate speech predicted the existence of cells that recognize these toxins using a “toxin receptor” and amazingly anticipated that “the antitoxin is nothing else but discharged components of the cell, namely receptors discharged in excess” (7). Although the term “plasma cell” had already been coined (8), it would be more than 40 years before the cellular source of this immunity was discovered.
Several more cases of mollities osseum were reported and in 1873 J. von Rustizky coined the phrase multiple myeloma (9). In 1900 James H. Wright concluded that the cells prevalent in multiple myeloma “are essentially plasma cells, or immediate descendants of them” (10). However, this did not explain the presence of proteinurea or Bence Jones proteins. In 1947, plasma cell formation was correlated with antibody production implicating plasma cells as the cellular source of antibodies (11). Korngold and Lipari determined in 1956 that multiple myeloma patients often had “electrophoretically homogeneous” Bence Jones proteins (12), which would later be shown to be identical to protein in the serum of the same patients (13). These monoclonal proteins corresponded to one of the two immunoglobulin light chains that were named kappa and lambda after Korngold and Lipari. Later the delineation of T and B lymphocytes (14) [reviewed by Max Cooper (15)] would lead to the identification of B cells as the precursors to plasma cells.
Advances in electrophoresis and the invention of the immunoblot allowed for more routine testing of immunoglobulin proteins in the serum and urine. In 1961, Jan Waldenström described a monoclonal band in patients with hypergammaglobulinemia many of whom had multiple myeloma or macroglobulinemia, but other patients had no symptoms of malignancy (16). Importantly, Waldenström delineated monoclonal proteins as indicative of neoplasm or a pre-malignant disease (now known as monoclonal gammopathy of undetermined significance or MGUS). This was in contrast to polyclonal proteins that were indicative of an inflammatory response.
Today, the cellular and molecular etiology of multiple myeloma as well as the programming of normal B cell development and plasma cell differentiation have been elucidated to a great extent. Like their discoveries, we have learned much about multiple myeloma from studying the normal processes of plasma cell differentiation and vice versa. Despite the incredible progress made and knowledge gained, over 130,000 new cases of multiple myeloma occur every year worldwide (17), including over 30,000 cases in the US alone (18). It is now known that myeloma is a progressive disease preceded by an asymptomatic stage called MGUS (19, 20) often followed by an intermediate stage referred to as smoldering multiple myeloma (SMM), prior to symptomatic newly diagnosed multiple myeloma (NDMM), and finally relapsed and/or refractory multiple myeloma (RRMM). Despite the incredible progress made, it is still very difficult to identify MGUS patients who will progress from those whose condition will remain benign. This is a major problem as MGUS is present in 3% of the population over 50 years of age, and progresses to multiple myeloma at a rate of ~1% per annum (21, 22). There is now a formidable arsenal of therapies for multiple myeloma, and thus far the most successful agents are targeted at plasma cell biology, which is largely retained by multiple myeloma (23). While most patients benefit from these treatments, ultimately and unfortunately, most still succumb to disease resulting in almost 100,000 deaths per year worldwide (17).
B cell development, much like plasma cell neoplasms, progresses through a series of well-defined stages. Current data suggest that a distinguishing attribute of plasma cell malignancies is the differentiation state at which the transformation manifests. This defining characteristic can be exploited to better identify vulnerabilities of multiple myeloma through the study of non-malignant B cells and plasma cells (23). A comprehensive description of these processes has been provided for both B cell development (24, 25) and plasma cell differentiation (26–29), and is beyond the scope of this current review. However, a brief description of these processes is essential to understanding the mechanistic underpinnings and etiology of myelomagenesis.
Like all immune cells, B cells are derived from hematopoietic stem cells that primarily develop in the bone marrow (30, 31) or fetal liver (32). Hematopoietic stem cells can successively differentiate into multi-potent progenitors, common lymphoid progenitors, and eventually mature B cells through the stages pre-pro-B, pro-B, pre-B, immature B, and transitional B cells. In the mouse, this process requires the transcription factors including E2A (33), PU.1 (34), and PAX5 (35) as well as interleukin 7 (IL7) cytokine signaling (36). It is important to point out there are key differences in human B cell development (37, 38), which is not dependent upon IL7 (39). However, in both mice and humans the recombination activated genes, RAG1 and RAG2, physically recombine the variable (V), diversity (D), and joining (J) segments of the immunoglobulin genes (40, 41). Mechanistically, RAG proteins work by recognizing and excising recombination signal sequences, which are conserved heptamer and nonamer sequences separated by a spacer (42). This proceeds first at the immunoglobulin heavy chain (IgH) D → J segments (pre-pro-B), and then V → DJ segments (pro-B). If a productive (in-frame) IgH gene is recombined, it is then transcribed, translated, and expressed on the surface with a surrogate light chain (composed of VPREB and IGLL1), which triggers light chain recombination at the V → J segments (light chains contain no diversity segments) marking the pre-B stage. This occurs first at the kappa light chain and if no productive allele is made, then at the lambda light chain. Surface expression of the paired heavy and light chains—referred to as the B-cell receptor (BCR)—marks the immature B cells stage, after which B cells can transition from the bone-marrow into the periphery and secondary lymphoid tissues where they mature.
Mature naïve B cells are mitotically (43) and transcriptionally quiescent (44, 45), but surveil the environment for pathogens which are recognized by toll-like receptors (TLR) (46) and the BCR. B cell activation that occurs without cytokine help from T cells, referred to as T-cell independent activation, generally results in acute and shorter lived B cell and plasmablast responses. In contrast, antigens that invoke T cell-dependent (TD) cytokine stimulation induce a more complex B-cell activation that results in selection of B cells with higher-affinity B-cell receptors and longer lasting immunity. However, this process is prone to genomic errors that contribute to oncogenesis. Indeed, current data suggests that almost all of myeloma is initiated by mutations associated with TD responses. TD B-cell activation requires BCR-mediated endocytosis of protein antigens, which are subsequently degraded and ectopically presented by the major histocompatibility complex class II (MHC-II) (47). When an antigen peptide presented by MHC-II on a B cell is recognized by a cognate T cell receptor (TCR), this induces an immunological synapse and T cell stimulation. This causes T cell expression of CD40 ligand (CD40L) (48) that induces B cell CD40 signaling (49), as well as polar release of T cell cytokines IL4 (50, 51), IL21, and IL6 (52) resulting in potent B cell activation. In particular, IL6 not only induces B cell activation, but is a potent growth stimulant for plasma cells and myeloma (53). This stimulation induces rapid B cell proliferation, which forms a lymphoid structure called a germinal center [Figure 1A; reviewed in (54, 55)]. During the germinal center reaction, B cells continuously cycle through rounds of division and selection for high-affinity antibodies, which are made through two types of somatic alterations termed somatic hypermutation (SHM) (56) and class-switch recombination (CSR), both of which are mediated by the activation-induced cytidine deaminase (AID) (57). AID deaminates cytosines on single-stranded DNA resulting in mutations of the immunoglobulin heavy and light chains or SHM. SHM of the heavy and light chains has the potential to increase antibody-antigen affinity through mutation of the complementarity determining region. This results in more efficient antigen uptake and presentation, resulting in more T cell stimulation and selection of B cell clones with high-affinity antibodies to a given antigen. CSR occurs when IgH somatically recombines the constant region μ and its splice isoform δ with one of the alternative constant regions γ3, γ1, α1, γ2, γ4, ε, or α2. CSR occurs via AID-dependent recombination of switch regions located just 5' of each constant region resulting in recombination of a new IgH constant region (58–60). This process requires DSBs, and can result in aberrant recombination with other genomic regions causing translocations. Indeed, there is now substantial evidence that myeloma initiating alterations are a result of errors in CSR.
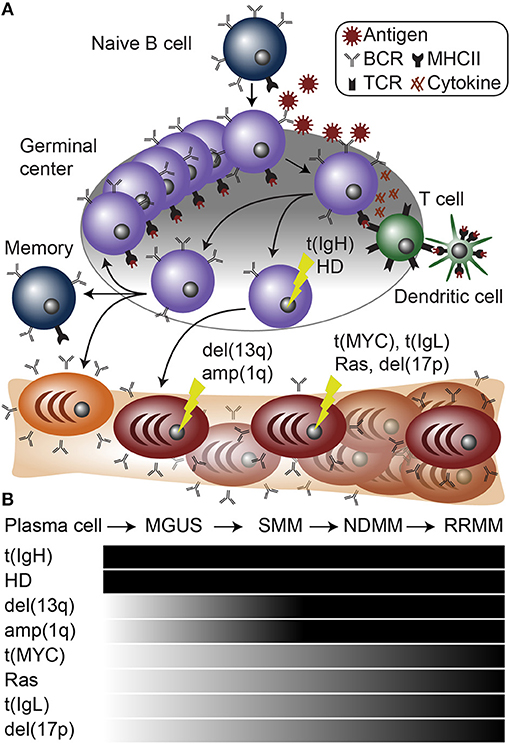
Figure 1. Plasma cell differentiation and myelomagenesis. (A) Schematic of B cell differentiation, plasma cell development, and myelomagenesis. Lightning bolts represent genetic mutations common in myeloma. (B) Diagram of stages of myeloma progressing from a normal plasma cell to monoclonal gammopathy of undetermined significance (MGUS), smoldering multiple myeloma (SMM), newly diagnosed multiple myeloma (NDMM), and relapse/refractory multiple myeloma (RRMM). Below are common mutations in myeloma and the stages at which they appear.
A dichotomy of genetic aberrations accounts for the large majority, if not all of myeloma initiating events. First, approximately half of myeloma cases contain an aneuploidy of several odd numbered chromosomes including 3, 5, 7, 9, 11, 15, 19, and 21. This is referred to as hyperdiploidy (HD), and will be further discussed below. The second type of founding genetic event is almost mutually exclusive with hyperdiploid myeloma and involves translocations of the IgH locus (61) (Figure 1B). IgH translocations juxtapose the IgH enhancers to one of a half dozen oncogenes including any of the three Cyclin D genes (CCND1-3), WHSC1 (also known as NSD2 or MMSET), MAF, or MAFB [reviewed in (62–64)] (Figure 2). When present, these translocations are clonal alterations (i.e., present in all tumor cells) in all stages of MGUS or myeloma and emanate from the IgH constant chain switch regions implicating them as errors in CSR that occurred during B cell activation in the germinal center (65, 66). Consistent with this, more than 90% of myelomas express class-switched IgH constant chains and almost all display SHM identifying them as post-germinal center cells.
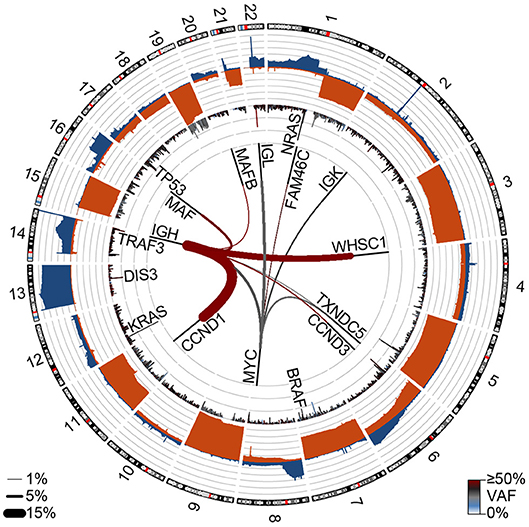
Figure 2. Genetic events in newly diagnosed multiple myeloma (NDMM). Circos plot showing copy number losses (blue) and gains (orange) in the outer ban (gray lines indicate 10% of the population). Mutations are shown on the inner ban, where the frequency of non-synonymous mutations and the variant allele frequency (VAF) are shown for 500 kb regions. Translocations are shown on the inside where the frequency is denoted by line thickness (key bottom left) and color denotes the VAF. Data are from 850 NDMM patients part of the MMRF CoMMpass study (dbGaP phs000748.v7.p4).
Cyclin D dysregulation is the most common type of IgH translocation, which involve t(11;14), t(12;14), and t(6;14) translocations that juxtapose the IgH enhancer(s) with CCND1 (15–20% of NDMM), CCND2 (~1%), and CCND3 (1–4%), respectively (65, 67, 68). All three Cyclin D genes function by activating CDK4 and CDK6 that in turn phosphorylate and inactivate RB allowing for E2F activation and cell cycle progression (63). Although these translocations result in aberrant expression of their respective Cyclin D genes, overexpression of at least one Cyclin D gene appears to be an early and unifying event in plasma cell malignancies (69). For instance, IgH translocations to MAF or MAFB result in high levels of CCND2 (70); IgH-WHSC1 translocations result in moderate levels of CCND2, and hyperdiploid disease results in overexpression of CCND1 (located on chromosome 11) or expression of both CCND1 and CCND2 (69). Conversely, CCND3 expression is less frequent and seems to be primarily a result of t(6;14) (69). Although most Cyclin D translocations occur at the switch region breakpoints, a subset of t(11;14) CCND1 translocations originate from the V(D)J region, suggesting that they may be the result of errors in V(D)J recombination during B cell development (71). Earlier work indicated myeloma-specific idiotypes reacted with some pre-B cells from the same patient, suggesting some myeloma may have origins in B cell development (72). However, these early studies were limited to two cases and it is unclear if pre-B cells with a myeloma idiotype harbor the genetic mutations that result in malignancy. Furthermore, should these errors occur during B cell development, it is not clear what causes these to manifest in myeloma rather than mantle cell lymphoma, which routinely have t(11;14) translocations originating from the V(D)J region (73). Regardless, it is clear that these translocations result in aberrant CCND1 expression, which predisposes to malignancy.
IgH-WHSC1 or t(4;14) are the second most common translocation, occurring in ~15% of NDMM, and in most cases results in the dual dysregulation of both WHSC1 and FGFR3 (74, 75). These are mostly reciprocal translocations that occur almost exclusively at the IgM switch region and split WHSC1 and FGFR3 on the telomeric side of chromosome 4p. This often results in IgH-WHSC1 fusion transcripts and/or loss of the 5′ exons of WHSC1 (76). Subsequently, the IgH Eμ enhancer drives expression of WHSC1 on the derivative of chromosome 4, while the IgH 3′ enhancers drive expression of FGFR3 on the derivative of chromosome 14 (62, 74). For some time it was unclear whether FGFR3 or WHSC1 was the definitive oncogenic factor, however, ~25% of t(4;14) myelomas do not have the reciprocal FGFR3 translocation and lack FGFR3 expression (76, 77). This suggests that WHSC1 is the essential transforming element, although FGFR3 overexpression and activating mutations likely contribute to pathogenesis. It is also possible that FGFR3 expression is required for MGUS initiation but is subsequently lost in a subset of cases. WHSC1 is now known to be a histone 3 lysine 36 (H3K36) methyltransferase that catalyzes di-methylation of histone 3 lysine 36 (H3K36me2) (78, 79). Pervasive H3K36me2 in t(4;14) myeloma is associated with accessible chromatin and gene dysregulation (79). However, how WHSC1 results in myelomagenesis or CCND2 upregulation is not well understood and difficult to trace due to the genome-wide effects H3K36me2.
IgH-MAF and IgH-MAFB translocations are the least common class of primary IgH translocation, and result from t(14;16) and t(14;20), respectively (80, 81). These are present in approximately 5–10% of NDMM cases, with MAF being more common than MAFB (66). MAF induces expression of CCND2 through a MAF binding motif in the CCND2 promoter, as well as Integrin B7 leading to increased adhesion to bone marrow stromal cells (70). In addition to upregulating MAF, t(14;16) translocation breakpoints disrupt WWOX, a tumor suppressor gene in breast and prostate cancers, where it is also commonly deleted (82, 83). The contribution of WWOX to t(14;16) myeloma is still unclear as there is little to no evidence of biallelic inactivation and most research has focused on the oncogenic effects of MAF (62).
Hyperdiploidy is the other common type of initiating genetic event in plasma cell malignancies. Hyperdiploidy is almost mutually exclusive with IgH translocations, and hyperdiploid myeloma tend to have a better prognosis than IgH-translocated multiple myeloma (61, 66, 84). Unlike IgH translocations, it is very difficult to trace the oncogenic effects of hyperdiploidy to a causative element(s) due to the aneuploidy of numerous chromosomes. Compounding the difficulty of pinpointing the pathogenic elements in hyperdiploid myeloma, model systems for hyperdiploid myeloma are lacking. For instance, of the roughly 80 multiple myeloma cell lines, more than 90% have IgH translocations and there are no commonly used hyperdiploid cell lines (62). The good prognosis of hyperdiploid myeloma and lack of cell line models suggests that hyperdiploidy rarely results in extramedullary disease or plasma cell leukemia as most cell lines are derived from patients with disease that is independent of the bone marrow microenvironment (85). Hyperdiploidy is also hypothesized to occur during rapid germinal center proliferation that results in chromosome segregation errors. However, it is not clear if this is one catastrophic event or a series of sequential errors that must occur prior to a clonal outgrowth.
Both IgH translocations and hyperdiploid myeloma are found to be clonal at all stages of gammopathy, which is consistent with them being founding genetic events (Figure 1B). Despite these large genomic changes, almost all myeloma has multiple genetic events present upon diagnosis, suggesting that primary events initiate MGUS, but are not sufficient to cause symptomatic disease.
In addition to the primary genetic events described above, presentation of myeloma is regularly accompanied by several other major chromosome abnormalities including deletion of chromosome 13q [del(13q)], amplification of chromosome 1q [amp(1q)], and deletion of chromosome 1p [del(1p)] (Figure 1). All three of these alterations involve regions tens of megabase in size and thus similar to hyperdiploidy, pinpointing the causative element(s) is difficult. However, contributing elements are speculated for all of these aberrations with varying degrees of supportive data.
Del(13q) occurs in almost 50% of NDMM (86) and is found to be clonal, but is less frequent in MGUS where it is either sub-clonal or clonal (87). Del(13q) co-occurs with t(4;14) and t(14;16) myeloma and was once considered to be marker of poor prognosis, but this outcome appears to have been overcome by the use of proteasome inhibitors (88). Generally, the entire arm of 13q is lost, and contains several loci that may contribute to myeloma pathogenesis. Notably RB1, which prevents cell cycle progression by sequestering E2F transcription factors, is located on 13q14. However, 13q loss is primarily mono-allelic and rarely are there mutations or deletions that result in biallelic RB1 inactivation (64). In contrast to RB1, the exosome endoribonuclease DIS3 is mutated in ~10% of NDMM and ~75% of these mutations occur in del(13q) myeloma suggesting biallelic loss of DIS3 occurs in most DIS3 mutated myeloma (66, 89, 90). Finally, 13q14 is also deleted in Chronic Lymphocytic Leukemia (CLL), albeit in a more punctate fashion, allowing researchers to pinpoint the DLEU2/miR-15a/16-1 locus as a minimally deleted region. Deletion of this region causes a lymphoproliferative disease in mice (91). A similar analysis independently identified the same locus as a minimally deleted region in myeloma (92), but it remains to be determined whether DLEU2, miR-15a, or miR-16-1 have tumor suppressor function in myeloma.
Amp(1q) occurs in 40% of patients and is associated with worse prognosis (93, 94). The poor prognosis appears to be dose-dependent as patients with 4 or more copies of chromosome 1q do worse than those with three (95). These additional copies of 1q likely have a proportional effect on expression of 1q genes, as a gene signature of high-risk myeloma is composed of a large number of 1q genes (96, 97). Putative oncogenes may include CKS1B, which facilitates ubiquitinylation and degradation of the cyclin dependent kinase inhibitor CDKN1B (p27KIP1) (98). Approximately two-thirds of amp(1q) coincide with del(13q), which is a significant co-occurrence between the two events (90, 99). If CKS1B and RB1 are the myeloma-inducing genetic alterations on amp(1q) and del(13q), respectively, questions remain as to why two alterations are needed in the same pathway in addition to overexpression of a Cyclin D gene. This might be explained by the somewhat rare nature of cell cycle progression in myeloma where <1% of cells are actively synthesizing DNA (64). Alternatively, it may be a polygenic effect or other genes may be responsible for the deleterious effects of these alterations. One such gene on chromosome 1q is MCL1, a BCL2-family anti-apoptotic protein that is induced during plasma cell differentiation and essential for plasma cell and myeloma cell survival (100–102). There are now MCL1 inhibitors in early phase clinical trials, and it will be important to understand if these are more effective against myeloma with amp(1q) that overexpresses MCL1 (103–105), as discussed further below.
Del(1p) occurs in 20–25% of patients and often co-occurs with hypodiploidy (loss of chromosomes). Unlike hyperdiploidy, hypodiploidy is associated with worse prognosis (106, 107) as is del(1p) (94). The region lost on 1p often includes the cyclin dependent kinase inhibitor CDKN2C, and similar to amp(1q), two-thirds of del(1p) also coincides with del(13q) and mono-allelic loss of RB1. Another promising candidate myeloma suppressor gene located on chromosome 1p, is FAM46C, a non-canonical poly(A) polymerase (108, 109). Inactivating mutations in FAM46C result in a cell survival advantage whereas overexpression causes an unfolded protein response and cell death (110). In addition to being lost in ~25% of NDMM by del(1p), FAM46C is also mutated in ~10% and translocated in ~2.5% of NDMM, supporting its role as a bona fide tumor suppressor in multiple myeloma (66, 89, 90).
MYC structural variants are pervasive in B cell malignancies and myeloma is no exception. MYC structural variants are sometimes present in MGUS, present in ~35% of NDMM, and even more common in RRMM and myeloma cell lines (66, 111). This suggests that MYC alterations promote disease progression. This is further supported by a mouse model of myeloma, in which AID-induced MYC expression only results in myelomagenesis in mouse strains prone to MGUS (112, 113). This suggests that MYC cannot initiate MGUS, but facilitates MGUS progression to myeloma. Consistent with this, IgH-MYC [t(8;14)] translocations are distinct from other IgH translocations in that they are found at sub-clonal levels in NDMM and have extragenic IgH breakpoints (66, 112). Such MYC alterations in myeloma are distinct from other B cell malignancies such as Burkitt lymphomas, where immunoglobulin-MYC translocations are a near universal primary event and IgH-MYC translocations have breakpoints in the IgH switch regions (114, 115). In myeloma, MYC structural variants are spread across at least two broad regions and serve to amplify or transpose large enhancers to drive MYC expression (66, 112, 116). Interestingly, almost all MYC translocations are also accompanied by copy number alterations, with most showing large duplicated sequences at both translocation breakpoints (66, 117). This appears to be a common phenomenon present at other secondary translocations in myeloma and other cancers, however, it is rare at myeloma primary translocations that originate from the CSR regions (66, 117). This key insight into the mechanistic basis of secondary and complex translocations could be explained by synthesis-dependent strand annealing of DSBs with long single-stranded overhangs. Indeed, AID deaminates cytosines on single stranded DNA and is known to initiate genomic instability at heavily transcribed regions of the genome (118), such as the intragenic regions of PVT1, where MYC translocations commonly occur, as well as at the immunoglobulin enhancers.
Immunoglobulin light chain kappa (IgK) and lambda (IgL) enhancers are often co-opted in complex secondary translocations that drive oncogene expression. IgL translocations occur in ~10% of MGUS and NDMM, but up to 20% of RRMM or myeloma cell lines, whereas IgK translocations are more rare, occurring in <5% of NDMM (64, 66). This is surprising given that two-thirds of human B cells and myeloma cells express IgK and only one-third express IgL. The higher prevalence of IgL translocations can be explained by B cell ontogeny, where IgK VJ rearrangement deletes the IgK enhancer if a productive IgK product is not made (119). Thus, without an enhancer the IgK region is inert if translocated, and consequently all IgK-translocated myelomas express IgK (66). Conversely, IgL-translocated myelomas are found at the normal ratio of two-thirds IgK expressing to one-third IgL expressing, which indicates that the IgL enhancer is constitutively active, and equally prone to translocation even in IgK-expressing myeloma (66). We recently showed that translocations of the IgL locus, but not the IgK locus, were prognostic of poor outcome (66). This was even true when restricting the analysis to the same translocated oncogene. For instance, approximately 40% of both IgK and IgL translocations occur to MYC, but only patients with IgL-MYC translocations have a poor prognosis, despite similar baseline levels of MYC expression from each translocation (66). This suggests that distinct enhancers are differentially susceptible to therapeutic perturbation and myeloma is not only a disease of oncogenes but also an “enhanceropathy.”
Deletion of 17p [del(17p)] including TP53 is also a marker of poor outcome as well as of genomic instability. Unlike several other prognostic markers TP53 status as a high-risk marker has not waned in the face of modern therapies that target plasma cell biology (107). Del(17p) is rare in MGUS, present in ~10% of NDMM but present in the majority of plasma cell leukemias and associated with extramedullary disease (66, 107, 120, 121). TP53 mutations also occur in ~5% of NDMM, but are primarily restricted to samples with del(17p), suggesting a step-wise progression where del(17p) predisposes to biallelic loss of TP53 by selection for cells with TP53 mutation. The co-occurrence of TP53 mutations with 17p loss results in exceedingly poor outcomes (95), and provides strong evidence that TP53 is the functional tumor suppressor inactivated by del(17p).
Aberrant NF-κB signaling results from both inactivating mutations of genes that suppress NF-κB signaling (e.g., TRAF3) as well as aberrant upregulation of genes the promote NF-κB signaling (e.g., MAP3K14) (89). There is a broad mutational spectrum encompassing dozens of genes, mostly mutated at a low frequency, that converge on the non-canonical NF-κB pathway in ~20% of NDMM (122, 123). Non-canonical NF-κB signaling provides a pro-survival signal and growth advantage to myeloma cells (122, 123), but it is possible that it occurs by a variety of mechanisms. For instance, NF-κB was discovered as a transcription factor that regulates kappa light chain expression (124), but is now known to also regulate IgH and human IgL expression (125). This suggests that NF-κB signaling may enhance expression of oncogenes translocated to immunoglobulin enhancers in myeloma.
This non-canonical NF-κB signaling in myeloma is in contrast to other B cell malignancies such as Waldenstrom's macroglobulinemia (lymphoplasmacytic lymphoma), where over 80% of cases harbor activating mutations in MYD88 that result in canonical NF-κB signaling (126, 127). This may impart a more “innate-like” B cell response and explain why Waldenstrom's macroglobulinemia is almost exclusively IgM expressing whereas IgM expressing myeloma is very rare. Indeed, long-term follow-up of MGUS cases, 15% of which are of the IgM isotype, indicate that IgM MGUS patients progress almost exclusively to Waldenstrom's macroglobulinemia or non-Hodgkin's lymphoma whereas class-switched MGUS cases progress to multiple myeloma (128).
Ras signaling is a common alteration in myeloma but rare in MGUS (129). KRAS and NRAS are the two most commonly mutated genes in myeloma, each present in ~20% of NDMM (66, 89, 95). Counterintuitively, 15% of KRAS-mutated patients also have NRAS mutations. Given the subclonal nature of most Ras mutations, it is conceivable that in cases where both KRAS and NRAS are mutated, these occur in distinct and non-overlapping clonal populations. Alternatively, it may suggest that not all Ras mutations uniformly activate MAPK signaling. Indeed, this has been recently confirmed by phosphoproteomics in myeloma cell lines (130). In contrast, FGFR3 mutations appear to be a more potent inducer of MAPK signaling and are mutually exclusive with NRAS and KRAS mutations (130, 131).
As noted above, translocations and chromosomal aberrations serve to dysregulate oncogenes and tumor suppressor genes and given the broad array of mutations in myeloma, it is not surprising that these result in distinct gene expression subtypes. Over a decade ago Zhan et al. used cDNA microarrays to classify myeloma into 7 gene expression subtypes, which mostly reflected the founding genetic mutations (132). These expression subtypes include two CCND1 subtypes, CD-1 and CD-2, both driven by t(11;14) translocations, but CD-2 tended to express more B-cell like markers such as CD20; the HY subtype corresponded with genetic hyperdiploidy; the MF subtype corresponded with MAF and MAFB translocations; the MS corresponded with WHSC1 translocations (WHSC1 was commonly referred to as MMSET at the time); LB or low bone disease was not well defined by gene expression or discernable baseline genetics; PR represented a proliferative disease with poor outcome (132). These subtypes of myeloma are well conserved, as segregation of myeloma based on translocations and Cyclin D expression (TC subtype) resulted in groups with similar characteristics (69). Another independent study from Europe identified 10 groups based on gene expression, which corresponded with those from Zhan et al. but provided slightly more granularity (133). Furthermore, we recently found that gene expression subtypes from Zhan et al. were largely conserved in yet another independent data set using a newer technology (RNA-seq) (66). Thus, it appears the initiating genetic alterations of myeloma appear to imprint a gene expression program such that myeloma is really several diseases.
The gene expression subtypes described above correlate with primary genetic events in myeloma, but the impact of secondary and tertiary genetic mutations on gene expression are harder to discern. This may be due to the often sub-clonal nature of these alterations, which likely result in their effects on gene expression being diluted out by cells without the alteration when profiled en masse. Emerging single-cell technologies may eventually be able to address this difficult problem, which would ideally require simultaneous profiling of DNA and RNA. Early experiments of single-cell RNA-seq have provided intriguing data indicating the inter-sample heterogeneity of myeloma cells was segmented into distinct gene expression programs, whereas those from SMM, MGUS, and plasma cells from healthy donors where more homogenous (134). This suggests that sub-clonal genetic differences may underlie these variations in the gene expression program, and has important implications for myeloma cell plasticity and the ability of current therapies to effectively eradicate all clones.
Although the gene expression program appears to be driven by primary genetic alterations, there is clearly a cascade of molecular events that result from these abnormalities. Furthermore, genetic events alone cannot fully explain the gene expression program or phenotype. For instance, the most common translocation in myeloma, t(11;14), is also a defining characteristic of mantle cell lymphoma (73), and t(8;14) translocations that occur as secondary events in myeloma are present as primary events in the majority of Burkitt lymphomas (114, 115). Unlike myeloma, both these lymphomas are believed to originate from pre-germinal center B cell, suggesting that the combination of cancer genetics and cell differentiation state determine the cellular phenotype. Indeed, plasma cell differentiation involves dramatic changes in gene expression, epigenetic reprogramming, and cell morphology (45, 135–140). Thus, genetic alterations may manifest in different phenotypes given their timing in the context of the epigenetic landscape of the cell of origin. These epigenetic changes involve DNA methylation, which primarily occurs on cytosines in CpG dinucleotides (Figure 3). DNA methylation at promoters or enhancers usually functions to repress gene expression by occluding transcription factor binding (141), whereas intragenic DNA methylation corresponds with high levels of gene expression and serves to help prevent transcription from cryptic promoters (142). During germinal center formation and plasma cell differentiation, the histone 3 lysine 27 (H3K27) methyltransferase EZH2 represses plasma cell differentiation genes (e.g., PRDM1, IRF4) by depositing the repressive H3K27me3 histone modification, thereby prolonging the germinal center response (143, 144). The rapid cellular proliferation during the germinal center results in a genome-wide DNA hypomethylation, thereby facilitating activation of plasma cell enhancers, which have the ability to induce plasma cell differentiation (45, 137–139, 145). This explains why activating mutations in EZH2 give rise to germinal center B cell lymphomas (143). These and other epigenetic processes serve to activate plasma cell enhancers and super-enhancers, which are often co-opted to drive oncogene expression (112, 116, 146). Determining the unique trans-acting factors in plasma cell and myeloma cell enhancers may provide an effective way to therapeutically target multiple myeloma (147).
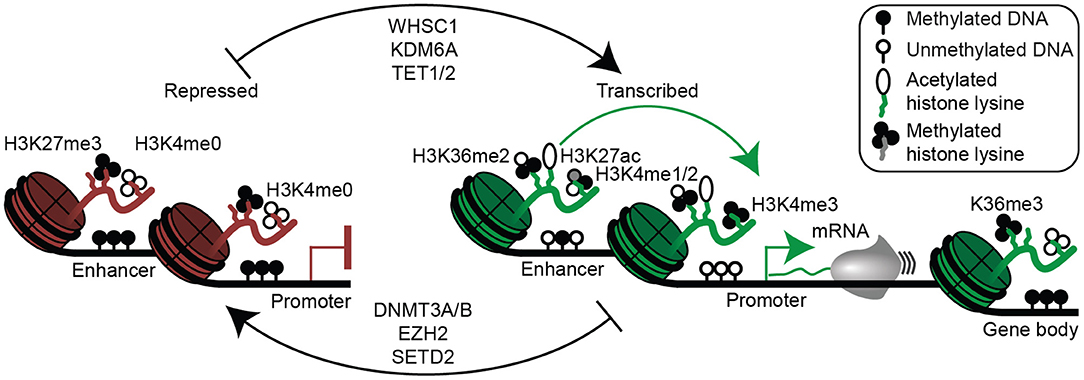
Figure 3. Epigenetic mechanisms of gene regulation in plasma cells and multiple myeloma. Epigenetic modifications associated with gene repression are shown (left) and include histone 3 lysine 27 trimethylation (H3K27me3), lack of histone 3 lysine 4 methylation (H3K4me0), as well as enhancer and/or promoter DNA methylation. Epigenetic modifications permissive to gene transcription are shown (right) and include H3K36me2 (mediated in part by WHSC1), histone 3 lysine 27 acetylation (H3K27ac), H3K4me1/2 at enhancers, H3K4me3 at promoters, and absence of enhancer and promoter DNA methylation. Gene bodies (far right) of actively transcribed genes are often demarcated with H3K36me3 and DNA methylation. Certain enzymes that mediate activating (top) and repressive (bottom) epigenetic modifications relevant to multiple myeloma are shown (middle).
As noted above, one of the most common translocated genes in myeloma is the H3K36 methyltransferase, WHSC1, resulting in a genetic alteration with widespread epigenetic effects. This results in a global increase of H3K36me2 and increased chromatin accessibility as well as a commensurate inhibition of the repressive mark H3K27me3 (78, 79). How this may specifically promote oncogenesis, is still being determined.
In addition to aberrant WHSC1 expression due to t(4;14) translocations, there are a number of mutations in epigenetic enzymes that confer survival advantages to myeloma cells. These include mutations in the H3K27me3 demethylase KDM6A (also known as UTX), where loss of KDM6A function results in increased proliferation, adhesion, and clonogenicity of myeloma cells (148). Unlike the genome-wide effects seen as a result of WHSC1 overexpression, only focal changes on H3K27me3 were observed with KDM6A ablation (148).
Recently, mutations in the isocitrate dehydrogenases, IDH1 and IDH2, have been reported (90). Isocitrate dehydrogenases normally produce α-ketoglutarate, but when mutated result in the accumulation of D-2-hydroxyglutarate, which inhibits Jumonji-C histone demethylases and TET family methylcytosine hydroxylases that require α-ketoglutarate as a co-factor. A consequence of IDH mutations includes altered histone modifications and DNA hypermethylation (149). This may alter the function of transcription factors, such as MYC and MAX, which bind CpG containing E-box elements and are sensitive to DNA methylation state (146, 150). Indeed, we recently showed that loss of function mutations in MAX occur in ~3% of myelomas and alter its binding affinity to methylated and hydroxymethylated E-box transcription factor binding sites (150).
Other modifiers of the DNA methylation pathway mutated in myeloma include TET2 (90), which oxidizes DNA methylation (151) allowing for its removal by base excision repair, as well as the de novo DNA methyltransferase DNMT3A (90), which catalyzes DNA methylation at unmethylated CpGs (152). We recently showed that conditional deletion of both de novo DNA methyltransferases in B cells results in a loss of DNA methylation at B cell enhancers as well as increased B cell activation and plasma cell differentiation in response to immunization (137). However, the functional impact of these enzymes in myeloma has yet to be elucidated.
In addition to the recent discoveries of mutations in enzymes that regulate DNA methylation, early observations in multiple myeloma showed promoter DNA hypermethylation and gene silencing of the cyclin-dependent kinases inhibitors CDKN2B (p15) and CDKN2A (p16), suggesting they were incapable of preventing cell cycle progression (153). SOCS1, a suppressor of cytokine signaling including the key myeloma cytokine IL6 (53), is also aberrantly silenced by DNA hypermethylation (154, 155). Despite these punctate hypermethylation events, recent genome-wide analyses have found myeloma is mostly characterized by widespread hypomethylation as compared to plasma cells from healthy individuals (156–159). This DNA hypomethylation appears to be progressive as it is more severe in NDMM and RRMM than in MGUS and SMM (156, 159). Indeed, as part of the Multiple Myeloma Research Foundation's CoMMpass project we are performing whole genome bisulfite sequencing on a large cohort of multiple myeloma and have found pervasive hypomethylation organized into megabase domains that are devoid of gene expression. In contrast, DNA methylation was retained in the gene bodies of highly expressed genes. Given the pre-clinical data showing that myeloma cells are sensitive to the DNA methylation inhibitors, such as 5-azacytidine and decitabine (160, 161), the selective sensitivity of multiple myeloma to demethylating agents has yet to be shown in vivo.
As our understanding of plasma cell and myeloma biology has improved, so too has our ability to treat myeloma effectively. Like most malignancies diagnosed in the mid twentieth century, myeloma was initially treated with cytotoxic chemotherapy that derived its benefit from attacking any rapidly dividing cell in the body. The alkylating agent melphalan was the first effective treatment for myeloma and in combination with the corticosteroid prednisone formed the backbone of myeloma therapy for 40 years (162, 163). The next major advance came in the 1980's with the introduction of high dose chemotherapy and autologous stem cell rescue, a procedure that is still routinely performed on the majority of eligible myeloma patients today (164, 165). However, it is the addition of novel plasma cell targeted therapy that has had the greatest impact on the improvement in overall outcomes for myeloma patient over the last two decades.
Proteasome inhibitors target the ability of both normal and malignant plasma cells to produce thousands of antibodies per second. In order to sustain such rapid levels of protein production, the cells rely heavily upon a number of quality control pathways for survival, and it is these pathways that have proven to be an Achilles heel for myeloma. In all cells, protein synthesis occasionally results in the production of misfolded and non-functional peptides that must be quickly disposed of to prevent their accumulation. These peptides are tagged with ubiquitin, which targets them for degradation by the proteasome system. Given the marked protein synthesis activity in myeloma cells, the amount of misfolded protein is similarly amplified, making myeloma even more dependent on the proteasome (166). Proteasome inhibitors block the degradation of misfolded proteins, allowing them to accumulate and ultimately induce cell death through the unfolded protein response (Figure 4). Although the proteasome plays an outsized role in myeloma cells by controlling the unfolded protein response, it has a number of other functions including regulation of signaling pathways, cell-cycle, and DNA repair. Proteasome inhibitors may therefore contribute to cell death through multiple mechanisms. There are currently three proteasome inhibitors approved for myeloma, bortezomib (167), carfilzomib (168), and ixazomib (169). These agents are often combined with dexamethasone, a corticosteroid with anti-lymphocyte activity, and an immunomodulatory drug, particularly during induction therapy (170), but also during maintenance (171) and relapse (172, 173).
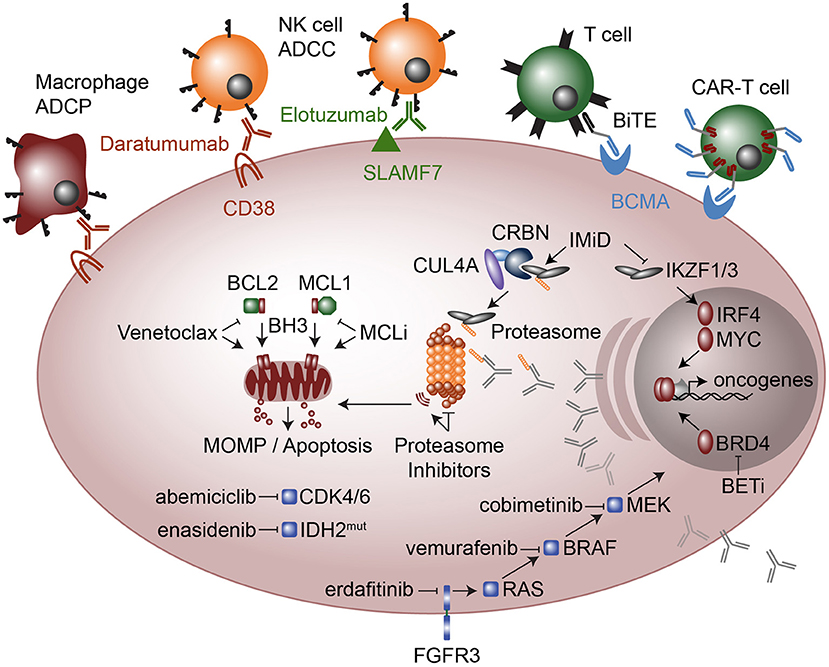
Figure 4. Therapeutic modalities in multiple myeloma. Cellular targeted therapies (top) include chimeric antigen receptor T-cells (CAR-T cells) that target B cell maturation antigen (BCMA) and Bispecific T cell engagers (BiTE), which are two conjugated antibodies, one that recognizes the CD3 receptor on T cells while the other antibody recognizes BCMA. Monoclonal antibodies elotuzumab and daratumumab target SLAMF7 and CD38, respectively and result in myeloma cell killing by Natural Killer (NK) cell mediated antibody-dependent cellular cytotoxicity (ADCC) and in the case of daratumumab also by Macrophage antibody-dependent cellular phagocytosis (ADCP). Molecular modalities include immunomodulatory imide drugs (IMiD; top right) that bind Ikaros (IKZF1) and Aiolos (IKZF3) to Cereblon (CRBN) as part of an E3 ubiquitin ligase complex, which subsequently ubiquitinates IKZF1 and IKZF3 marking them for proteasomal degradation. Proteasome inhibitors (center) result in proteotoxic stress and the unfolded protein response, which plasma cells are particularly sensitive due to their high levels of antibody production. Anti-apoptosis inhibitors (middle left) include MCL1 inhibitors (MCLi) and BCL2 inhibitors such as venetoclax which induce mitochondrial outer membrane permeabilization (MOMP) and apoptosis. Therapeutics targeted at intracellular signaling include the cyclin dependent kinase 4 and 6 (CDK4/6) inhibitor abemiciclib and the mutant IDH2 inhibitor enasidenib. FGFR3 which is highly expressed in most t(4;14) myeloma and sometimes has activating mutations, is targeted with erdafitinib. FGFR3 feeds into RAS / MEK / MAPK signaling, which is targeted with drugs against BRAF (vemurafenib) and MEK (cobimetinib). Finally, a new class of drugs that target transcriptional activators such as bromodomain and extra-terminal (BET) inhibitors the block or degrade BRD4 are being used to target the enhancer machinery present at large enhancers that are often translocated in myeloma such as those found at the immunoglobulin loci.
Immunomodulatory imide drugs (IMiDs) include thalidomide, the notorious anti-nausea medicine developed in Europe during the 1950s. Despite extensive testing in animals with no side-effects, thalidomide resulted in severe birth defects and in most cases death. However, discovery of the anti-angiogenic properties of thalidomide (174) led to clinical trials which showed it to be an effective agent in treating myeloma (175). This spurred the development of more potent and less toxic analogs of thalidomide, including lenalidomide (176, 177) and pomalidomide (178), now approved for the treatment of myeloma. Despite their efficacy, the mechanism by which IMiDs exert their effect was only recently discovered. IMiDs alter the target specificity of the CUL4A-DDB1-Cereblon E3 ubiquitin ligase (179), and in myeloma, this leads to the degradation of two key plasma cell transcription factors, Ikaros (IKZF1) and Aiolos (IKZF3) (180, 181) (Figure 4). Importantly, IMiDs bind Cereblon through an interaction at residue 391, which is not conserved in mice (182), explaining why thalidomide had no effect on animal studies originally conducted in the 1950s. IMiD-mediated degradation of IKZF1 and IKZF3 results in myeloma cell growth arrest as well as activation of T cells (180, 181), both of which may contribute to anti-myeloma effects of IMiDs. Through loss of IKZF1 and IKZF3, IMiDs also lead to down regulation of IRF4, another essential plasma cell transcription factor (183). IRF4 in turn regulates the expression of MYC (184), a potent oncogene in numerous lymphoid malignancies.
MYC aberrant expression occurs in the majority of myeloma cases through amplification, translocation, or transcriptional dysregulation (66, 112). Many MYC translocations result in its juxtaposition to immunoglobulin enhancers where the BET bromodomain protein BRD4 is highly associated. As a result, BRD4 inhibitors and degraders are currently being investigated as a method of downregulating MYC expression and inhibiting myeloma cell proliferation (185–187). IMiDs may also target MYC expression through inhibition of IKZF1- and/or IKZF3-regulated enhancers translocated to MYC (66) (Figure 4).
Monoclonal antibodies against cell surface antigens highly expressed on malignant cells have been an important part of cancer therapy since the introduction of rituximab two decades ago. Like other cells of the immune system, plasma cells express cell surface markers that distinguish them from other cells, many of which continue to be expressed on myeloma. The transmembrane glycoprotein CD38 and the immunoreceptor SLAMF7 are the targets of the two monoclonal antibodies currently approved for the treatment of multiple myeloma, daratumamab (188, 189) and eloztuzumab (190), respectively. Daratumumab is capable of inducing complement dependent cytotoxicity, antibody dependent cellular cytotoxicity (ADCC) by NK cells, and antibody dependent cell phagocytosis (ADCP) by macrophages (191, 192), while elotuzumab acts primarily through ADCC and enhancement of anti-myeloma NK cell activity by crosslinking SLAMF7 on the two cell types (193–195) (Figure 4). Development of biologics that target plasma cells has been limited by the number of plasma cell specific markers, and thus a number of other potential targets on myeloma cells are being studied, including GPRC5D (196) and sulfated HLA-I epitopes (197). BCMA is an important cell survival receptor on plasma cells and is the target of the first generation of myeloma directed chimeric antigen receptor (CAR)-T cells and bi-specific T cell engaging (BiTE) antibodies, which are conjugated antibodies binding both myeloma cells and T cells. A neutralizing antibody against the BCMA ligand APRIL is also being developed, as are monoclonal antibodies that deliver cytotoxic drugs more specifically to the antigen expressing cell, so called antibody drug conjugates (ADCs). ADCs targeting the plasma cell markers CD138, CD74, and CD48 are currently undergoing clinical trials.
Targeting tumor specific biology has been successfully used to treat CML and a number of solid tumors with common driver mutations, but given the degree of genetic heterogeneity in myeloma, this has been less successful than plasma cell directed therapy. Nonetheless, treatment guided by specific oncogenic events in an individual patient's tumor remains an active area of investigation. As described above, alterations in the Ras-MAPK pathway occur in approximately 40% of patients. Although no direct Ras inhibitors exist, treatment with inhibitors of downstream kinases such as BRAF (vemurafenib) and MEK (trametinib, cobimetinib) have been reported in a small number of cases (198–201). Cobimetinib for Ras and Raf mutated patients is also being incorporated into a larger precision medicine trial known as MyDrug (202). Additional arms of this study include inhibitors of IDH2 (enasidenib), FGFR3 (erdafitinib), and CDK (abemiciclib) (Figure 4).
BCL2 family inhibitors represent a new class of drugs that may have applications in a broad range of malignancies. Pro- and anti-apoptotic members of the BCL2 family exist in a delicate state of balance that regulates the survival of both normal and malignant cells (203, 204). The anti-apoptotic proteins BCL2, BCL2L1 (also known as BCL-xL), and MCL1 bind to and sequester pro-apoptotic proteins BIM, BAX, and BAK, preventing them from activating the apoptotic pathway. As normal cells transform into malignant cells they become even more dependent on the anti-apoptotic proteins for survival, leaving them sensitive to inhibitors of the BCL2 family and providing a potential therapeutic window (205–207). Venetoclax, navitoclax (ABT-263), AZD5991, AMG176, and S63845 induce tumor apoptosis by disrupting the function of BCL2, BCL2L1, and/or MCL1 (103, 104, 208). Venetoclax, a BCL2 specific inhibitor, has been approved for the treatment of chronic lymphocytic leukemia (CLL), which originates from BCL2-dependent B cells (209, 210). In contrast, plasma cells upregulate and become dependent upon MCL1, reducing their dependence upon BCL2 (100, 211). As a consequence, myeloma is primarily dependent on MCL1 and inhibitors of MCL1 have shown promising pre-clinical activity (103, 104). Surprisingly, a subset of myeloma characterized by the t(11;14) translocation is co-dependent on BCL2 and responds to BCL2 inhibition with venetoclax (212–218). Dexamethasone further enhances sensitivity to venetoclax by increasing expression of BIM and its binding to BCL2 (219). The biological basis for the BCL2 co-dependence in t(11;14) myeloma remains a mystery, however gene expression profiling of myeloma patient samples did reveal that t(11;14) were composed of two gene expression groups, CD1 and CD2, where CD2 had increased expression of B cell markers such as CD20, PAX5, and VPREB3, suggesting a possible connection with B cells and BCL2 dependence. The bone marrow microenvironment may also play a role in maintaining plasma cell MCL1 dependence through stromal cell mediated secretion of the plasma cell survival cytokine IL6 (220, 221).
Throughout the history of multiple myeloma, we have learned a great deal about normal plasma cells from studying the malignant form and vice versa. While tremendous progress in the treatment of myeloma has been made over the past 25 years, due in a large part to therapies targeting plasma cell biology, myeloma remains an incurable disease. This necessitates not only the continued study of plasma cell and myeloma biology, but also the germinal center B cell origins of the disease. Clues of these origins are provided by epidemiological correlations. For instance, patients with Gaucher's disease accumulate lysolipids due to a deficiency in glucocerebrosidase, and are more prone to monoclonal gammopathies (222). This was recently leveraged to identify lysolipids as an antigen driving the gammopathy (223). Similarly, it is realized that MGUS incidence increases with obesity and has a higher prevalence in African Americans and in males (64, 223, 224). It will be important to sort out the genetic vs. environmental factors in each of these cases in hopes of minimizing risk of MGUS development. Likewise, it will be very important to identify factors that influence progression of MGUS to myeloma. Clinical trials testing therapeutic intervention to minimize risk of disease progression in SMM are already underway. However, given that 3% of adults over the age of 50 have MGUS (64), less toxic approaches at early stages of clonal gammopathy are needed to minimize chances of disease. Here, even interventions with small effects may have a large impact on cumulative disease burden. It would also be very valuable to accurately identify cases of MGUS that will develop into myeloma. Given the likelihood that myeloma may never be completely eliminated by preventative approaches, better models of disease will be needed to effectively develop the next generation of therapies. For instance, although we have learned a lot from patient derived cell lines models, these do not provide tractable comparisons of different genetic alterations without confounding background genetics. Although CRISPR has revolutionized gene editing, it has yet to be co-opted to induce myeloma translocations and it is unclear if this is possible. However, it is encouraging that other genetic approaches have been able to induce such translocations in murine B cells (225). Finally, given the dependence of most myeloma on the microenvironment, better in vivo models will also be needed. Significant efforts have yielded a mouse model of myeloma driven by AID-dependent MYC expression (113), and a humanized mouse capable of sustaining the human immune system including myeloma (226). These systems will need to be further exploited and expanded to better understand how the different genetic subtypes of myeloma respond to therapy and to delineate microenvironmental interactions and dependencies that can be leveraged to better treat multiple myeloma.
Figure 2 was generated by analysis from 850 newly diagnosed patients from the MMRF CoMMpass study (dbGAP phs000748.v7.p4).
BB and VG wrote the manuscript. LB and PV provided editorial input.
This study was supported in part by Winship Cancer Institute of Emory University and NIH/NCI award number P30CA138292. LB is supported by NIH/NCI R01 CA192844 and an Answer Fund award from the MMRF. BB is supported by Postdoctoral Fellowship PF-17-109-1-TBG from the American Cancer Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the Emory Myeloma Team for insightful discussions that helped in the development of this review as well as the Multiple Myeloma Research Foundation for their support and public disclosure of data from the CoMMpass trial (NCT01454297).
1. Solly S. Remarks on the pathology of mollities ossium; with cases. Med Chir Trans. (1844) 27:435–98.
2. Kyle RA, Rajkumar SV. Multiple myeloma ASH 50th anniversary review Multiple myeloma. Blood. (2008) 111:2962–72. doi: 10.1182/blood-2007-10-078022
4. Jones HB. On a New Substance Occurring in the Urine of a Patient with Mollities Ossium. In: Jones HB, editor. Philosophical Transactions of the Royal Society of London. London: Royal Society Stable (1848) 138:55–62.
5. Jenner EI. An inquiry into the causes and effects of the variolae vaccinae, or cow-pox. 1798. In: Edward J editor. The Three Original Publications on Vaccination Against Smallpox. The Harvard Classics (1798). Available online at: http://www.bartleby.com/38/4/1.html (accessed March 22, 2016).
6. van Behring E, Kitasato S. The mechanism of immunity in animals to diphtheria and tetanus. Deutsche Medizinsche Wochenschrift. (1890) 16:1113–4.
7. Ehrlich P. Paul ehrlich - nobel lecture: partial cell functions. Scand J Immunol. (1990) 31:4–13. doi: 10.1111/j.1365-3083.1990.tb02737.x
8. Waldeyer W. Ueber Bindegewebszellen. Arch für Mikroskopische Anat. (1875) 11:176–94. doi: 10.1007/BF02933794
9. Rustizky J. Multiples myelom. Dtsch Zeitschrift für Chir. (1873) 3:162–72. doi: 10.1007/BF02911073
10. Wright J. A case of multiple myeloma. Br Soc Med Sci. (1900) 15:137–47. doi: 10.1002/bjs.1800249517
11. Fagraeus A. Plasma cellular reaction and its relation to the formation of antibodies in vitro. Nature. (1947) 159:499. doi: 10.1038/159499a0
12. Korngold L, Lipari R. Multiple-myeloma proteins.III. The antigenic relationship of Bence Jones proteins to normal gamma-globulin and multiple-myeloma serum proteins. Cancer. (1956) 9:262–72. doi: 10.1002/1097-0142(195603/04)9:2<262::AID-CNCR2820090210>3.0.CO;2-B
13. Edelman GM, Gally JA. The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins. J Exp Med. (1962) 116:207–27. doi: 10.1084/JEM.116.2.207
14. Cooper MD, Peterson RDA, Good RA. Delineation of the thymic and bursal lymphoid systems in the chicken. Nature. (1965) 205:143–6. doi: 10.1038/205143a0
16. Waldenstrom J. Studies on conditions associated with disturbed gamma globulin formation (gammopathies). Harvey Lect. (1960–1961) 56:211–31.
17. Cowan AJ, Allen C, Barac A, Basaleem H, Bensenor I, Curado MP, et al. Global burden of multiple myeloma: a systematic analysis for the global burden of disease study 2016. JAMA Oncol. (2018) 4:1221–7. doi: 10.1001/jamaoncol.2018.2128
18. Cancer Statistics Review, 1975-2013 - SEER Statistics. Available online at: https://seer.cancer.gov/csr/1975_2013/ (accessed August 10, 2016).
19. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. (2009) 113:5418–22. doi: 10.1182/blood-2008-12-195008
20. Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. (2009) 113:5412–7. doi: 10.1182/blood-2008-12-194241
21. Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. (2002) 346:564–9. doi: 10.1056/NEJMoa01133202
22. Kyle RA, Buadi F, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncology. (2011) 25:578–86.
23. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. (2014) 124:1873–79. doi: 10.1182/blood-2014-05-578732
24. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. (2001) 19:595–621. doi: 10.1146/annurev.immunol.19.1.595
25. Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. (2007) 26:715–25. doi: 10.1016/j.immuni.2007.05.010
26. Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. (2005) 5:230–42. doi: 10.1038/nri1572
27. Oracki SA, Walker JA, Hibbs ML, Corcoran LM, Tarlinton DM. Plasma cell development and survival. Immunol Rev. (2010) 237:140–59. doi: 10.1111/j.1600-065X.2010.00940.x
28. Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. (2008) 20:49–58. doi: 10.1016/j.smim.2007.12.002
29. Boothby MR, Hodges E, Thomas JW. Molecular regulation of peripheral B cells and their progeny in immunity. Genes Dev. (2019) 33:26–48. doi: 10.1101/gad.320192.118
30. Osmond DG, Nossal GJV. Differentiation of lymphocytes in mouse bone marrow. Cell Immunol. (1974) 131:117–31.
31. Ryser J-E, Vassalli P. Mouse bone marrow lymphocytes and their differentiation. J Immunol. (1974) 113:719–28.
32. Owen JJT, Cooper MD, Raff MC. In vitro generation of B lymphocytes in mouse foetal liver, a mammalian ‘bursa equivalent.' Nature. (1974) 249:361–3. doi: 10.1038/249361a0
33. Bain G, Maandag ECR, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. (1994) 79:885–92. doi: 10.1016/0092-8674(94)90077-9
34. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU .1 in the development of multiple hematopoietic lineages. Science. (1994) 265:1573–7. doi: 10.1126/science.8079170
35. Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. (1999) 401:556–62. doi: 10.1038/44076
36. Sudo T, Nishikawa S, Ohno N, Akiyama N, Tamakoshi M, Yoshida H, et al. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc Natl Acad Sci USA. (1993) 90:9125–9.
38. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. (2008) 112:1570–80. doi: 10.1182/blood-2008-02-078071
39. Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. (1993) 73:147–57. doi: 10.1016/0092-8674(93)90167-O
40. Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. (1989) 59:1035–48. doi: 10.1016/0092-8674(89)90760-5
41. Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. (1990) 248:1517–23. doi: 10.1126/science.2360047
42. Sakano H, Hüppi K, Heinrich G, Tonegawa S. Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature. (1979) 280:288–94. doi: 10.1038/280288a0
43. Macallan DC, Wallace DL, Zhang Y, Ghattas H, Asquith B, de Lara C, et al. B-cell kinetics in humans: rapid turnover of peripheral blood memory cells. Blood. (2005) 105:3633–40. doi: 10.1182/blood-2004-09-3740
44. Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer-Kwon K-R, Bandle R, et al. Global regulation of promoter melting in naive lymphocytes. Cell. (2013) 153:988–99. doi: 10.1016/j.cell.2013.04.033
45. Barwick BG, Scharer CD, Bally APR, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol. (2016) 17:1216–25. doi: 10.1038/ni.3519
46. Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. (1997) 388:394–7. doi: 10.1038/41131
47. Kindred B, Shreffler DC. H-2 Dependence of co-operation between T and B cells in vivo. J Immunol. (1972) 109:940–3.
48. Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, et al. Molecular and biological characterization of a murine ligand for CD40. Nature. (1992) 357:80–2. doi: 10.1038/357080a0
49. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA. (1992) 89:6550–4.
50. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57.
51. Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. (1987) 236:944–7. doi: 10.1126/science.3107127
52. Muraguchi A, Hirano T, Tang B, Matsuda T, Horii Y, Nakajima K, et al. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. J Exp Med. (1988) 167:332–44. doi: 10.1084/JEM.167.2.332
53. Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. (1988) 332:83–5. doi: 10.1038/332083a0
54. MacLennan ICM. Germinal centers. Annu Rev Immunol. (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
55. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
56. Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. (1991) 354:389–92. doi: 10.1038/354389a0
57. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. (2000) 102:553–63. doi: 10.1016/S0092-8674(00)00078-7
58. Iwasato T, Shimizu A, Honjo T, Yamagishi H. Circular DNA is excised by immunoglobulin class switch recombination. Cell. (1990) 62:143–9. doi: 10.1016/0092-8674(90)90248-D
59. Matsuoka M, Yoshida K, Maeda T, Usuda S, Sakano H. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. (1990) 62:135–42. doi: 10.1016/0092-8674(90)90247-C
60. Von Schwedler U, Jäck HM, Wabl M. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. (1990) 345:452–6. doi: 10.1038/345452a0
61. Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. (2003) 102:2562–7. doi: 10.1182/blood-2003-02-0493
62. Bergsagel PL, Kuehl WM. Chromosomal translocations in multiple myeloma. Oncogene. (2001) 20:5611. doi: 10.1038/sj.onc.1204641
63. Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. (2005) 23:6333–8. doi: 10.1200/JCO.2005.05.021
64. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. (2002) 2:175–87. doi: 10.1038/nrc746
65. Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci USA. (1996) 93:13931–6. doi: 10.1073/pnas.93.24.13931
66. Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun. (2019) 10:1911. doi: 10.1038/s41467-019-09555-6
67. Shaughnessy J, Gabrea A, Qi Y, Brents L, Zhan F, Tian E, et al. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood. (2001) 98:217–23. doi: 10.1182/blood.V98.1.217
68. Avet-Loiseau H, Li JY, Facon T, Brigaudeau C, Morineau N, Maloisel F, et al. High incidence of translocations t(11;14)(q13;q32) and t(4;14)(p16;q32) in patients with plasma cell malignancies. Cancer Res. (1998) 58:5640–5.
69. Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood. (2005) 106:296–303. doi: 10.1182/blood-2005-01-0034
70. Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. (2004) 5:191–9. doi: 10.1016/S1535-6108(04)00019-4
71. Walker BA, Wardell CP, Johnson DC, Kaiser MF, Begum DB, Dahir NB, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. (2013) 121:3413–9. doi: 10.1182/blood-2012-12-471888
72. Kubagawa H, Vogler LB, Capra JD, Conrad ME, Lawton AR, Cooper MD. Studies on the clonal origin of multiple myeloma. Use of individually specific (idiotype) antibodies to trace the oncogenic event to its earliest point of expression in B-cell differentiation. J Exp Med. (1979) 150:792–807. doi: 10.1084/JEM.150.4.792
73. Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. (2011) 117:26 LP−38. doi: 10.1182/blood-2010-04-189977
74. Chesi M, Nardini E, Lim RSC, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. (1998) 92:3025–34.
75. Chesi M, Nardini E, Brents LA, Schröck E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. (1997) 16:260–4. doi: 10.1038/ng0797-260
76. Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. (2003) 101:1520–9. doi: 10.1182/blood-2002-06-1675
77. Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. (2005) 105:4060–9. doi: 10.1182/blood-2004-09-3704
78. Martinez-Garcia E, Popovic R, Min D-J, Sweet SMM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. (2011) 117:211–20. doi: 10.1182/blood-2010-07-298349
79. Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. (2014) 10:e1004566. doi: 10.1371/journal.pgen.1004566
80. Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood. (1998) 91:4457–63.
81. Hanamura I, Iida S, Akano Y, Hayami Y, Kato M, Miura K, et al. Ectopic expression of MAFB gene in human myeloma cells carrying (14;20)(q32;q11) chromosomal translocations. Japanese J Cancer Res. (2001) 92:638–44. doi: 10.1111/j.1349-7006.2001.tb01142.x
82. Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX , a novel WW domain-containing protein mapping to affected in breast cancer advances in brief. Cancer Res. (2000) 60:2140–5.
83. Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, et al. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res. (2001) 61:8068–73.
84. Avet-Loiseau H, Li C, Magrangeas F, Gouraud W, Charbonnel C, Harousseau JL, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. (2009) 27:4585–90. doi: 10.1200/JCO.2008.20.6136
85. Besse L, Sedlarikova L, Greslikova H, Kupska R, Almasi M, Penka M, et al. Cytogenetics in multiple myeloma patients progressing into extramedullary disease. Eur J Haematol. (2016) 97:93–100. doi: 10.1111/ejh.12688
86. Attal M, Moreau P, Charbonnel C, Hulin C, Leyvraz S, Michallet M, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. (2007) 109:3489–95. doi: 10.1182/blood-2006-08-040410
87. Avet-Loiseau H, Facon T, Daviet A, Godon C, Rapp MJ, Harousseau JL, et al. 14Q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Cancer Res. (1999) 59:4546–50.
88. Anderson KC, Sonneveld P, Stadtmauer EA, Harousseau J-L, Schuster MW, Irwin D, et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia. (2006) 21:151–7. doi: 10.1038/sj.leu.2404442
89. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. (2011) 471:467–72. doi: 10.1038/nature09837
90. Walker BA, Mavrommatis K, Wardell CP, Cody Ashby T, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. (2018) 132:587–97. doi: 10.1182/blood-2018-03-840132
91. Ambesi-Impiombato A, Mo T, Califano A, Lia M, Dalla-Favera R, Bhagat G, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. (2010) 17:28–40. doi: 10.1016/j.ccr.2009.11.019
92. Elnenaei MO, Hamoudi RA, Swansbury J, Gruszka-Westwood AM, Brito-Babapulle V, Matutes E, et al. Delineation of the minimal region of loss at 13q14 in multiple myeloma. Genes, Chromosom Cancer. (2003) 36:99–106. doi: 10.1002/gcc.10140
93. Fonseca R, Van Wier SA, Chng WJ, Ketterling R, Lacy MQ, Dispenzieri A, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. (2006) 20:2034–40. doi: 10.1038/sj.leu.2404403
94. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma associated chromosomal copy number abnormalities and their prognostic value. Blood. (2010) 116:56–65. doi: 10.1182/blood-2010-04-279596
95. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. (2019) 33:159–70. doi: 10.1038/s41375-018-0196-8
96. Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27 Kip1 and an aggressive clinical course in multiple myeloma. Hematology. (2005) 10:117–26. doi: 10.1080/10245330512331390140
97. Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. (2007) 109:2276–84. doi: 10.1182/blood-2006-07-038430
98. Tyers M, Ko TK, Pagano M, Ganoth D, Bornstein G, Hershko A, et al. The cell-cycle regulatory protein Cks1 is required for SCFSkp2-mediated ubiquitinylation of p27. Nat Cell Biol. (2002) 3:321–4. doi: 10.1038/35060126
99. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. (2015) 33:3911–20. doi: 10.1200/JCO.2014.59.1503
100. Peperzak V, Vikström I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. (2013) 14:290–7. doi: 10.1038/ni.2527
101. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. (1993) 90:3516–20. doi: 10.1073/PNAS.90.8.3516
102. Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. (2002) 99:1885–93. doi: 10.1182/blood.V99.6.1885
103. Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. (2018) 9:5341. doi: 10.1038/s41467-018-07551-w
104. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. (2016) 538:477–82. doi: 10.1038/nature19830
105. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. (2018) 8:1582–97. doi: 10.1158/2159-8290.CD-18-0387
106. Vé Ronique Smadja N, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. (2001) 98:2229–38.
107. Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International myeloma working group molecular classification of multiple myeloma: spotlight review. Leukemia. (2009) 23:2210–21. doi: 10.1038/leu.2009.174
108. Pawlowski K, Knizewski L, Kuchta K, Wyrwicz LS, Ginalski K, Rychlewski L, et al. FAM46 proteins are novel eukaryotic non-canonical poly(A) polymerases. Nucleic Acids Res. (2016) 44:3534–48. doi: 10.1093/nar/gkw222
109. Mroczek S, Chlebowska J, Kulinski TM, Gewartowska O, Gruchota J, Cysewski D, et al. The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat Commun. (2017) 8:619. doi: 10.1038/s41467-017-00578-5
110. Zhu YX, Shi CX, Bruins LA, Jedlowski P, Wang X, Kortüm KM, et al. Loss of FAM46C promotes cell survival in myeloma. Cancer Res. (2017) 77:4317–27. doi: 10.1158/0008-5472.CAN-16-3011
111. Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA. (2000) 97:228–33. doi: 10.1073/pnas.97.1.228
112. Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. (2014) 28:1725–35. doi: 10.1038/leu.2014.70
113. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. (2008) 13:167–80. doi: 10.1016/j.ccr.2008.01.007
114. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. (1982) 79:7824–7. doi: 10.1073/pnas.79.24.7824
115. Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA. (1982) 79:7837–41. doi: 10.1073/pnas.79.24.7837
116. Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. (2014) 4:e191. doi: 10.1038/bcj.2014.13
117. Demchenko Y, Roschke A, Chen W-D, Asmann Y, Bergsagel PL, Kuehl WM. Frequent occurrence of large duplications at reciprocal genomic rearrangement breakpoints in multiple myeloma and other tumors. Nucleic Acids Res. (2016) 44:8189–98. doi: 10.1093/nar/gkw527
118. Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. (2014) 159:1538–48. doi: 10.1016/j.cell.2014.11.014
119. Siminovitch KA, Bakhshi A, Goldman P, Korsmeyer SJ. A uniform deleting element mediates the loss of κ genes in human B cells. Nature. (1985) 316:260–2. doi: 10.1038/316260a0
120. Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. (2008) 22:1044–52. doi: 10.1038/leu.2008.4
121. Manier S, Chauvet P, Willaume A, Farhat M, Escure G, Demonchy J, et al. Deregulation and targeting of TP53 pathway in multiple myeloma. Front Oncol. (2019) 8:665. doi: 10.3389/fonc.2018.00665
122. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. (2007) 12:131–44. doi: 10.1016/j.ccr.2007.07.003
123. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. (2007) 12:115–30. doi: 10.1016/j.ccr.2007.07.004
124. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. (1986) 46:705–16. doi: 10.1016/0092-8674(86)90346-6
125. Combriato G, Klobeck H-G. Regulation of human ig light chain gene expression by NF- B. J Immunol. (2014) 168:1259–66. doi: 10.4049/jimmunol.168.3.1259
126. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
127. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P Somatic Mutation in Waldenström's Macroglobulinemia. N Engl J Med. (2012) 367:826–33. doi: 10.1056/NEJMoa1200710
128. Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. (2018) 378:241–9. doi: 10.1056/NEJMoa1709974
129. Liu P, Leong T, Quam L, Billadeau D, Kay N, Greipp P, et al. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: analysis of the Eastern Cooperative Oncology Group Phase III Trial. Blood. (1996) 88:2699–706.
130. Lin Y-HT, Way GP, Barwick BG, Mariano MC, Marcoulis M, Ferguson ID, et al. Integrated phosphoproteomics and transcriptional classifiers reveal hidden RAS signaling dynamics in multiple myeloma. bioRxiv. (2019) 563312. doi: 10.1101/563312
131. Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. (2001) 97:729–36. doi: 10.1182/blood.V97.3.729
132. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. (2006) 108:2020–8. doi: 10.1182/blood-2005-11-013458
133. Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. (2010) 116:2543–53. doi: 10.1182/blood-2009-12-261032
134. Ledergor G, Weiner A, Zada M, Wang S-Y, Cohen YC, Gatt ME, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. (2018) 24:1867–76. doi: 10.1038/s41591-018-0269-2
135. Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. (2015) 16:663–73. doi: 10.1038/ni.3154
136. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. (2011) 23:341–9. doi: 10.1016/j.smim.2011.08.010
137. Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun. (2018) 9:1900. doi: 10.1038/s41467-018-04234-4
138. Kulis M, Merkel A, Heath S, Queirós AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. (2015) 47:746–56. doi: 10.1038/ng.3291
139. Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. (2013) 23:2030–41. doi: 10.1101/gr.155473.113
140. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long-lived plasma cells are contained within the CD19–CD38hiCD138+ subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
141. Schübeler D. Function and information content of DNA methylation. Nature. (2015) 517:321–6. doi: 10.1038/nature14192
142. Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. (2017) 543:72–7. doi: 10.1038/nature21373
143. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. (2013) 23:677–92. doi: 10.1016/j.ccr.2013.04.011
144. Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, et al. EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol. (2017) 200:ji1701470. doi: 10.4049/jimmunol.1701470
145. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood. (2011) 118:3559–69. doi: 10.1182/blood-2011-06-357996
146. Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. (2012) 151:56–67. doi: 10.1016/j.cell.2012.08.026
147. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
148. Ezponda T, Dupéré-Richer D, Will CM, Small EC, Varghese N, Patel T, et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. (2017) 21:628–40. doi: 10.1016/j.celrep.2017.09.078
149. Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. (2012) 22:2339–55. doi: 10.1101/gr.132738.111
150. Wang D, Hashimoto H, Zhang X, Barwick BG, Lonial S, Boise LH, et al. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res. (2017) 45:2396–407. doi: 10.1093/nar/gkw1184
151. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in mammalian DNA by MLL Partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
152. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
153. Ng MHL, Chung YF, Lo KW, Wickham NWR, Lee JCK, Huang DP. Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood. (1997) 89:2500–06.
154. Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood. (2003) 101:2784–88. doi: 10.1182/blood-2002-06-1735
155. Chim CS, Fung TK, Cheung WC, Liang R, Kwong YL. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood. (2004) 103:4630–5. doi: 10.1182/blood-2003-06-2007
156. Agirre X, Castellano G, Pascual M, Heath S, Kulis M, Segura V, et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. (2015) 25:478–87. doi: 10.1101/gr.180240.114
157. Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. (2011) 117:553–62. doi: 10.1182/blood-2010-04-279539
158. Kaiser MF, Johnson DC, Wu P, Walker BA, Brioli A, Mirabella F, et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood. (2013) 122:219–26. doi: 10.1182/blood-2013-03-487884
159. Heuck CJ, Mehta J, Bhagat T, Gundabolu K, Yu Y, Khan S, et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J Immunol. (2013) 190:2966–75. doi: 10.4049/jimmunol.1202493
160. Kiziltepe T, Hideshima T, Catley L, Raje N, Yasui H, Shiraishi N, et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol Cancer Ther. (2007) 6:1718–27. doi: 10.1158/1535-7163.MCT-07-0010
161. Maes K, De Smedt E, Kassambara A, Hose D, Seckinger A, Van Valckenborgh E, et al. In vivo treatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget. (2015) 6:3319–34. doi: 10.18632/oncotarget.3207
162. Hoogstraten B, Sheehe PR, Cuttner J, Cooper T, Kyle RA, Oberfield RA, et al. Melphalan in multiple myeloma. Blood. (1967) 30:74–83.
163. Alexanian R, Haut A, Khan AU, Lane M, McKelvey EM, Migliore PJ, et al. Treatment for multiple myeloma. JAMA. (1969) 208:1680. doi: 10.1001/jama.1969.03160090040009
164. Barlogie B, Hall R, Zander A, Dicke K, Alexanian R. High-dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood. (1986) 67:1298–301.
165. Attal M, Harousseau J-L, Stoppa A-M, Sotto J-J, Fuzibet J-G, Rossi J-F, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. N Engl J Med. (1996) 335:91–7. doi: 10.1056/NEJM199607113350204
166. Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. (2006) 107:4907–16. doi: 10.1182/blood-2005-08-3531
167. Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. (2005) 352:2487–98. doi: 10.1056/NEJMoa043445
168. Hájek R, Masszi T, Petrucci MT, Palumbo A, Rosiñol L, Nagler A, et al. A randomized phase III study of carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia. (2017) 31:107–14. doi: 10.1038/leu.2016.176
169. Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. (2016) 374:1621–34. doi: 10.1056/NEJMoa1516282
170. Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. (2017) 376:1311–20. doi: 10.1056/NEJMoa1611750
171. Nooka AKA, Kaufman JLJ, Muppidi S, Langston A, Heffner LT, Gleason C, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib and dexamethasone (RVD) in high-risk myeloma patients. Leukemia. (2014) 28:690–3. doi: 10.1038/leu.2013.335
172. Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. (2015) 372:142–52. doi: 10.1056/NEJMoa1411321
173. Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood. (2015) 126:2284–90. doi: 10.1182/blood-2015-05-643320
174. D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. (1994) 91:4082–5. doi: 10.1073/PNAS.91.9.4082
175. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. (1999) 341:1565–71. doi: 10.1056/NEJM199911183412102
176. Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in north america. N Engl J Med. (2007) 357:2133–42. doi: 10.1056/NEJMoa070596
177. Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau J-L, Dmoszynska A, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. (2007) 357:2123–32. doi: 10.1056/NEJMoa070594
178. Miguel JS, Weisel K, Moreau P, Lacy M, Song K, Delforge M, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. (2013) 14:1055–66. doi: 10.1016/S1470-2045(13)70380-2
179. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. (2010) 327:1345–50. doi: 10.1126/science.1177319
180. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. (2014) 343:301–5. doi: 10.1126/science.1244851
181. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science. (2014) 343:305–9. doi: 10.1126/science.1244917
182. Fink EC, McConkey M, Adams DN, Haldar SD, Kennedy JA, Guirguis AA, et al. Crbn I391V is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood. (2018) 132:blood-2018-05-852798. doi: 10.1182/blood-2018-05-852798
183. Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH, et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. (2018) 132:2166–78. doi: 10.1182/blood-2018-05-850727
184. Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. (2008) 454:226–31. doi: 10.1038/nature07064
185. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. (2010) 468:1067–73. doi: 10.1038/nature09504
186. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. (2011) 146:904–17. doi: 10.1016/j.cell.2011.08.017
187. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. (2015) 348:1376–81. doi: 10.1126/science.aab1433
188. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet. (2016) 387:1551–60. doi: 10.1016/S0140-6736(15)01120-4
189. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. (2016) 375:754–66. doi: 10.1056/NEJMoa1606038
190. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. (2015) 373:621–31. doi: 10.1056/NEJMoa1505654
191. Overdijk MB, Verploegen S, Bögels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. (2015) 7:311–21. doi: 10.1080/19420862.2015.1007813
192. de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
193. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. (2008) 14:2775–84. doi: 10.1158/1078-0432.CCR-07-4246
194. Tai Y-T, Dillon M, Song W, Leiba M, Li X-F, Burger P, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. (2008) 112:1329–37. doi: 10.1182/blood-2007-08-107292
195. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. (2013) 62:1841–9. doi: 10.1007/s00262-013-1493-8
196. Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. (2019) 11:eaau7746. doi: 10.1126/scitranslmed.aau7746
197. Chan JTH, Liu Y, Khan S, St-Germain JR, Zou C, Leung LYT, et al. A tyrosine sulfation–dependent HLA-I modification identifies memory B cells and plasma cells. Sci Adv. (2018) 4:eaar7653. doi: 10.1126/sciadv.aar7653
198. Heuck CJ, Jethava Y, Khan R, van Rhee F, Zangari M, Chavan S, et al. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia. (2016) 30:976–80. doi: 10.1038/leu.2015.208
199. Sharman JP, Chmielecki J, Morosini D, Palmer GA, Ross JS, Stephens PJ, et al. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clin Lymphoma Myeloma Leuk. (2014) 14:e161–3. doi: 10.1016/j.clml.2014.06.004
200. Mey UJM, Renner C, von Moos R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol Oncol. (2017) 35:890–93. doi: 10.1002/hon.2353
201. Andrulis M, Lehners N, Capper D, Penzel R, Heining C, Huellein J, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. (2013) 3:862–9. doi: 10.1158/2159-8290.CD-13-0014
202. Kumar SK. Targeted management strategies in multiple myeloma. Cancer J. (2019) 25:59–64. doi: 10.1097/PPO.0000000000000353
203. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. (2010) 37:299–310. doi: 10.1016/j.molcel.2010.01.025
204. Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. (2011) 30:3667–83. doi: 10.1038/emboj.2011.307
205. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. (2012) 151:344–55. doi: 10.1016/j.cell.2012.08.038
206. Ni Chonghaile T, Sarosiek KA, Vo T-T, Ryan JA, Tammareddi A, Moore VDG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. (2011) 334:1129–33. doi: 10.1126/science.1206727
207. Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. (2016) 16:99–109. doi: 10.1038/nrc.2015.17
208. Scarfò L, Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett. (2013) 155:36–9. doi: 10.1016/j.imlet.2013.09.015
209. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
210. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. (2016) 374:311–22. doi: 10.1056/NEJMoa1513257
211. Vikstrom I, Carotta S, Lüthje K, Peperzak V, Jost PJ, Glaser S, et al. Mcl-1 is essential for germinal center formation and B cell memory. Science. (2010) 330:1095–9. doi: 10.1126/science.1191793
212. Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maïga S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. (2011) 118:3901–10. doi: 10.1182/blood-2010-11-317438
213. Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. (2011) 118:1329–39. doi: 10.1182/blood-2011-01-327197
214. Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. (2014) 28:210–2. doi: 10.1038/leu.2013.216
215. Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. (2016) 128:1834–44. doi: 10.1182/blood-2016-03-704908
216. Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. (2017) 130:2401–9. doi: 10.1182/blood-2017-06-788786
217. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood. (2018) 132:2656–69. doi: 10.1182/blood-2018-03-836718
218. Matulis SM, Gupta VA, Neri P, Bahlis NJ, Maciag P, Leverson JD, et al. Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia. (2019) 33:1291–96. doi: 10.1038/s41375-018-0374-8
219. Matulis SM, Gupta VA, Nooka AK, Hollen HV, Kaufman JL, Lonial S, et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia. (2016) 30:1086–93. doi: 10.1038/leu.2015.350
220. Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment–derived signals induce Mcl-1 dependence in multiple myeloma. Blood. (2017) 129:1969–79. doi: 10.1182/blood-2016-10-745059
221. Puthier D, Bataille R, Amiot M. IL-6 up-regulates Mcl-1 in human myeloma cells through JAK/STAT rather than Ras/MAP kinase pathway. Eur J Immunol. (1999) 29:3945–50. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O
222. Mistry PK, Taddei T, vom Dahl S, Rosenbloom BE. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog. (2013) 18:235–46. doi: 10.1615/CritRevOncog.2013006145
223. Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N Engl J Med. (2016) 374:555–61. doi: 10.1056/NEJMoa1508808
224. Landgren O, Gridley G, Turesson I, Caporaso NE, Goldin LR, Baris D, et al. Risk of monoclonal gammopathy of undetermined significance (MGUS) and subsequent multiple myeloma among African American and white veterans in the United States. Blood. (2006) 107:904–6. doi: 10.1182/blood-2005-08-3449
225. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. (2011) 147:107–19. doi: 10.1016/j.cell.2011.07.049
Keywords: multiple myeloma, MGUS, plasma cell, B cell, genetics, epigenetics, IgH translocations, MYC
Citation: Barwick BG, Gupta VA, Vertino PM and Boise LH (2019) Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 10:1121. doi: 10.3389/fimmu.2019.01121
Received: 06 March 2019; Accepted: 02 May 2019;
Published: 21 May 2019.
Edited by:
Simone Cenci, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Peter Daniel Burrows, University of Alabama at Birmingham, United StatesCopyright © 2019 Barwick, Gupta, Vertino and Boise. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lawrence H. Boise, bGJvaXNlQGVtb3J5LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.