 Ricard Garcia-Carbonell
Ricard Garcia-Carbonell Shih-Jing Yao
Shih-Jing Yao Soumita Das1
Soumita Das1 Monica Guma
Monica Guma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 May 2019
Sec. Immunological Tolerance and Regulation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01094
This article is part of the Research Topic Regulation of Immunity by Non-Immune Cells View all 12 articles
Crohn's disease (CD) and ulcerative colitis (UC) are common intestinal bowel diseases (IBD) characterized by intestinal epithelial injury including extensive epithelial cell death, mucosal erosion, ulceration, and crypt abscess formation. Several factors including activated signaling pathways, microbial dysbiosis, and immune deregulation contribute to disease progression. Although most research efforts to date have focused on immune cells, it is becoming increasingly clear that intestinal epithelial cells (IEC) are important players in IBD pathogenesis. Aberrant or exacerbated responses to how IEC sense IBD-associated microbes, respond to TNF stimulation, and regenerate and heal the injured mucosa are critical to the integrity of the intestinal barrier. The role of several genes and pathways in which single nucleotide polymorphisms (SNP) showed strong association with IBD has recently been studied in the context of IEC. In patients with IBD, it has been shown that the expression of specific dysregulated genes in IECs plays an important role in TNF-induced cell death and microbial sensing. Among them, the NF-κB pathway and its target gene TNFAIP3 promote TNF-induced and receptor interacting protein kinase (RIPK1)-dependent intestinal epithelial cell death. On the other hand, RIPK2 functions as a key signaling protein in host defense responses induced by activation of the cytosolic microbial sensors nucleotide-binding oligomerization domain-containing proteins 1 and 2 (NOD1 and NOD2). The RIPK2-mediated signaling pathway leads to the activation of NF-κB and MAP kinases that induce autophagy following infection. This article will review these dysregulated RIPK pathways in IEC and their role in promoting chronic inflammation. It will also highlight future research directions and therapeutic approaches involving RIPKs in IBD.
Inflammatory bowel disease (IBD) is an inflammatory process with a chronic relapsing course that is characterized pathologically by intestinal inflammation and epithelial injury that affects the different gastrointestinal (GI) linings (1). IBD includes different inflammatory pathologies of the gastrointestinal track. The more prevalent IBD pathologies are Crohn's Disease (CD) and Ulcerative Colitis (UC) (1). Pathogenesis of IBD is multifactorial, involving genetic predisposition, disturbance of the commensal microbiota, epithelial barrier defects, dysregulated immune responses, and environmental factors (2). The gastrointestinal tract (in particular, the terminal ileum and colon) also contains a massive bacterial load that has the potential to initiate an acute inflammatory intestinal response if the mucosal barrier is breached and bacteria gain access to the lamina propria, as occurs in IBD (2).
The receptor interacting protein kinase (RIPK) proteins are key molecules for the maintainance of a healthy intestinal barrier (3). The RIPK family contains seven members that share a homologous serine-threonine kinase domain but has different functional domains (4). RIPK1 contains a death-domain in the C-terminal portion that allows its recruitment to different signaling complexes. RIPK2 is characterized by its caspase activation and recruitment domain (CARD). RIPK3, like RIPK1, has a RIP homotypic interaction motif (RHIM), which is necessary for RIPK1 and RIPK3 dimerization. RIPK4 (or DIK or PKK) and RIPK5 (or SgK288) contain ankyrin repeats in the C-terminal tail. Finally, RIPK6 (or LRRK1) and RIPK7 (LRRK2) have leucine-rich repeats (LRR) that could play a role in the recognition of inflammatory-associated molecular patterns. In this review, we will focus on the epithelial barrier and how an aberrant response to TNF stimulation, exarcebated, IBD-associated microbial sensing, and abnormal regeneration and healing of the injured mucosa by dysregulated RIPK pathways in IEC can critically affect the health of the intestinal barrier.
The intestinal epithelium forms the physical, protective, and host defense barrier against the harmful luminal microenvironment, while providing selective permeability for absorption of nutrients (5). The epithelium is covered by a single-cell layer composed of different subtypes of specialized intestinal epithelial cells (IECs) including enterocytes, goblet cells, enteroendocrine cells, Paneth cells, M cells, cup cells, and Tuft cells, all of which differentiate from common epithelial stem cells (5). These IECs types are functionally different and essential for maintaining intestinal homeostasis by separating the intestinal lumen from the underlying lamina propria and by controlling the crosstalk between luminal microbiota and subjacent immune cells (Figure 1). IECs not only function as a physical barrier through enteroctyes (the largest cell population in IECs), but also through other specific functions. Paneth cells, for instance, are specialized secretory epithelial cells located in the crypt of the small intestine and contribute to the host defense secreting anti-microbial peptides that are diluted in the mucus enhancing the antimicrobial barrier and shape the commensal bacterial population (6–9). Paneth cells are characterized by an extensive endoplasmatic reticulum and Golgi apparatus with big secretory granules containing a wide variety of peptides, especially those with antimicrobial activity including defensins.
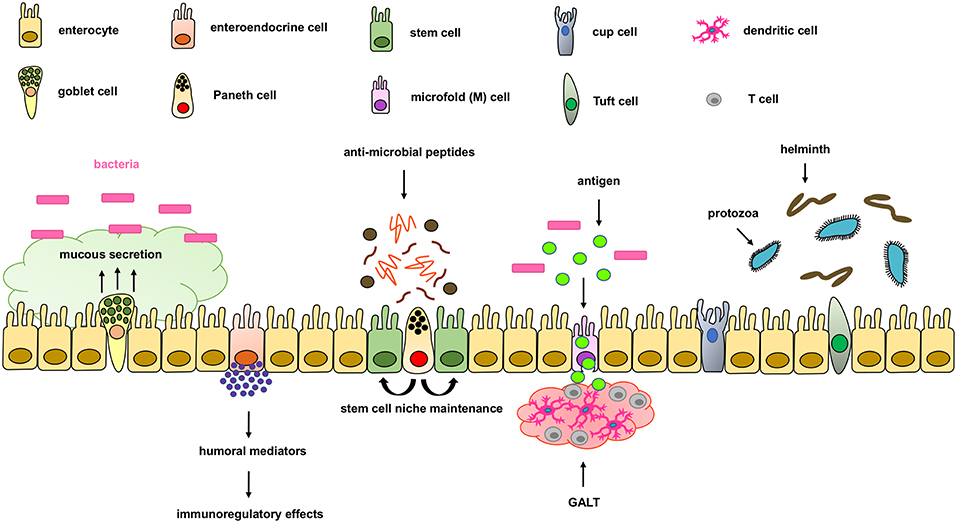
Figure 1. Components of the intestinal epithelial cell barrier. In the intestinal epithelial cell barrier we can find different specialized cell types including; enterocytes, goblet cells, enteroendocrine cells, paneth cells, stem cells, microfold (M) cells, and cup and tuft cells. Briefly, enterocytes are the most common cells with nutrient absorption functions; goblet cells secrete mucus to form an extra intestinal protective layer; enteroendocrine cells secrete humoral mediators with immunoregulatory effects; paneth cells secrete antimicrobial peptides and maintain the stem cell niche; microfold cells play an important role in transporting lumen antigens into the associated intestinal lymphoid structures; and Tuft cells play an important innate role against helminth and protozoan infections.
Goblet cells are the second most abundant cells in IECs and are specialized in mucus secretion (10). Mucins are highly O-glycosylated molecules that have gel-like properties and cover the inner walls of the gut lumen. Mucins form a bistratified mucus barrier, which becomes denser as it nears IECs, thus preventing bacteria from penetrating the barrier (11). At the same time, the mucus provides digestible glycans as a stable source of energy for the commensal microbiome (12–14). Intestinal goblet cells also sense luminal material that can be taken up delivered to lamina propria CD103+CD11c+ dendritic cells (DC) (15, 16) through goblet cell-associated antigen passages (GAPs). The DCs that interact with regulatory T cells have been suggested to induce tolerance to food antigens. Other cells, such as enteroendocrine cells, release a variety of humoral and paracrine mediators that can induce different immunoregulatory effects including cellular recruitment, activation, phagocytosis, antigen presentation and cytokine secretion (17, 18). Additionally, tuft cells, critical in the initiation of type-2 immune responses, are typically activated during intestinal protozoa or helminth parasite infections. Microfold (M) cells are epithelial cells specialized in phagocytosis and transcytosis of gut lumen antigens and pathogenic or commensal microorganisms across the intestinal epithelium toward the underlying gut-associated lymphoid tissues (GALT). M cells are also critical in maintaining a healthy intestinal barrier and control the crosstalk between luminal microbiota and subjacent immune cells.
IECs ability to act as a protective physical barrier is mediated by the formation of protein complex connections between adjacent cells, including tight junctions (TJ) and adherent junctions (AJ), which form the apical junction complex (AJC), as well as desmosomes, which are located in the basolateral membrane (19). These dynamic complexes are susceptible to endogenous and exogenous factors, such as cytokines, nutrients, and bacteria (19). TJs are the apical complexes of the AJC, connecting and sealing adjacent cells. TJ complexes are composed of junctional adhesion molecules (JAM), claudins, occludins, and zonula occludens (ZO), which seal neighboring cells together (20). AJs, composed of cadherins, form the second AJC loop, maintaining cell-to-cell connections; however, AJ are not critical for creating paracellular tightness (20). Finally, desmosomes connect intermediate filaments of neighboring cells, conferring mechanical strength to cell-to-cell junctions. They are formed by desmoplakin, plakoglobin, plakophilin, desmocollin, and desmoglein (21, 22). Tight junctions are critical for maintaining barrier function during IEC shedding, which occurs continuously from villus tips or colonic surfaces as a result of migration of the epithelial cell up the crypt–villus axis from stem cells at the base of the crypt (23). Normal cell shedding never causes a breach in the epithelial barrier because of the redistribution of tight junction proteins that facilitates the closure of the gap (24). However, in pathological conditions, when multiple neighboring cells are shed at the same time or cell death is activated, or turnover is increased a proper rearrangement of cell-to-cell contact cannot take place. Consequently, breaches appear in the intestinal epithelial barrier, which causes intestinal inflammation (23).
Autophagy is a cell stress response that causes the encapsulation of cellular contents for subsequent degradation and recycling (25). Although the first barrier against bacterial and parasitic invasion of the intestine is the mucus layer, some pathogens can penetrate this layer to reach the IECs. In this situation, autophagy plays an important role by recognizing and degrading intracellular pathogens, thus functioning as an innate barrier to infection. It has already been shown that knockdown of autophagy genes in Caenorhabditis elegans and Dictyostelium discoideum increases Salmonella typhimurium intracellular replication, decreases animal lifespan, and results in apoptotic-independent death (26).
NOD2 (nucleotide-binding oligomerization domain-containing protein 2) is a critical element in regulating autophagy in IECs (27). NOD2, a cytosolic pattern recognition receptor, is activated by the peptidoglycan fragment muramyl dipeptide (MDP) to generate a proinflammatory immune response (28, 29). Over 30 cellular proteins interact with NOD2 directly and influence or regulate its functional activity (30). Among them, NOD2 recruits ATG16L1 (autophagy-related protein 16 like 1) to the plasma membrane at the bacterial entry site to induce phagophore formation. ATG16L1 then forms a complex with ATG5 and ATG12 to induce the lipidation of LC3 (microtubule-associated protein 1A/1B-light chain 3), forming an autophagosome and inducing autophagy (27). Additionally, upon activation, NOD1 and NOD2 recruit RIPK2 through CARD domains (31, 32), inducing RIPK2 k63-polyubiquitination in lysine 209 by cIAPs and the LUBAC complex (33–35). This leads to RIPK2 activation, which depends on autophosphorylation in residues Ser176 and Tyr474, an essential and enhancing site respectively (36, 37), and downstream activation of transforming growth factor beta-activated kinase 1 (TAK1) (38–40). TAK1 consecutively phosphorylates the IKK complex triggering NF-κB and MAPK pathway activation.
Travassos et al. showed that NOD1 and NOD2 can recruit ATG16L1 to the plasma membrane at the bacterial entry site in different cell types including the mouse intestinal epithelial cell line Mode-K through a RIPK2-independent mechanism (41). The role of RIPK2 as a kinase in autophagy induction downstream of NOD2 has also been investigated. In different cells, including the cell-like HCT116, RIPK2 kinase function is required for the phosphorylation of the protein kinase ULK1 at Ser555, and for the deactivation of the protein phosphatase 2A (PP2A) complex that negatively regulates autophagy induction downstream of p38 activation (42, 43). In dendritic cells, NOD2 is also able to trigger autophagy through RIPK2-mediated recruitment of ATG5, ATG7, and ATG16L1 (44). Anand et al. showed how activated RIPK2 promotes increased autophagosome formation by activating MAPK/ERK kinase 4 (MEKK4)–p38 signaling and/or extracellular signal-regulated kinase 1 (ERK1) and ERK2 signaling, which upregulates basal levels of autophagy (43).
Autophagy also plays an important role in protecting IECs from cell death. NOD2 is highly expressed in intestinal stem cells, and its activation by MDP triggers stem cell survival and strong cytoprotection against oxidative stress-mediated cell death (45). This could be due to NOD2s ability to activate the NF-κB pathway, which has protective effects in the intestinal epithelium (46). Animals lacking ATG16L1 in the epithelium were more susceptible to DSS-induced colitis, and the pathology was exacerbated when these animals were infected with murine norovirus (MNV). Further histological analysis and organoid experiments show that ATG16L1 protects cells from necroptosis by removing aberrant mitochondria and impairing downstream reactive oxygen species (ROS) accumulation (47). In another model of colitis induced by Helicobacter hepaticus, mice with a deletion of ATG16L1 in IECs had worse histopathology than their littermates. IECs in affected mice were more susceptible to TNF-induced apoptotsis, increasing inflammation and pathology of the models (48). Overall, it has been shown that autophagy adds another layer of protection from foreign organisms by preventing pathogen proliferation and dissemination to extraintestinal sites (49). It also has a protective role in IECs, and defects in the autophagy pathway increase the susceptibility of the intestine to inflammation, inducing cell death and intestinal epithelial barrier breakdown.
Two RIPK proteins have key kinase-dependent functions in deciding beneficial or deleterious effects downstream of TNF: RIPK1 and RIPK3 are two key molecules in the assembly of TNFR complexes that may trigger cell death (Figure 2).
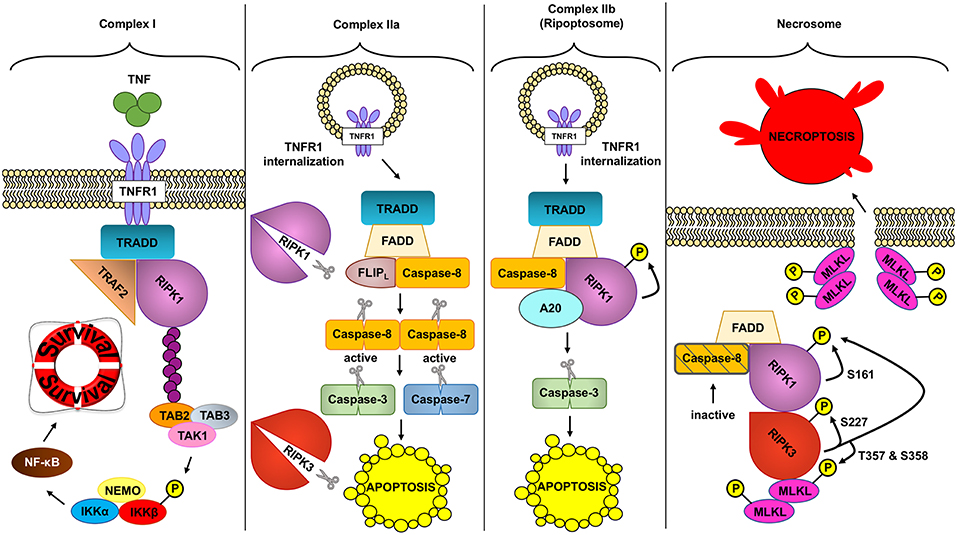
Figure 2. Different TNFR complex formed downstream of TNF. TNF (tumor necrosis factor) can induce the formation of different complexes with diverse outcomes depending on the conditions. In a homeostatic situation, TNF triggers the formation of complex I, where RIPK1 (receptor interacting protein kinase 1) acts as a scaffold protein allowing the activation of the pro-inflammatory and protective pathway NF- κB (nuclear factor kappa-light-chain-enhancer of activated B cells). In pathologic conditions, complex IIa can be formed leading to the activation of caspase-8. Active caspase-8 cleaves RIPK1 and RIPK3 (receptor interacting protein kinase 3) and downstream caspases to induce apoptosis. Unlike complex IIa, complex IIb depends on the kinase role of RIPK1 to activate caspase-8 and execute apoptosis. Inhibition of RIPK1 kinase and ROS (reactive oxygen species) prevents this type of cell death. Finally, if caspases are inhibited and the NF- κB pathway is not activated, TNF can trigger the formation of the necrosome. This complex depends on the kinase activity of RIPK1 and RIPK3 to activate MLKL (mixed lineage kinase domain like). MLKL in turn will create the necroptotic pore in the plasma membrane inducing necroptosis, a regulated type of necrosis.
- RIPK1 was the first protein of the RIPK family identified, interacting with apoptosis antigen 1 (APO-1 or FAS) through its death domain (DD), giving it its “receptor-interacting protein” name (50). Through its DD, RIPK1 can also bind other receptors such as the TNF-receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (DR4 and DR5) and death receptor 3 (DR3 or TRAMP). RIPK1 interacts with other adaptor proteins such as TRADD, Fas-associated protein with death domain (FADD), Toll/IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF), RIP-associated ICH-1 (ICE (interleukin-1β-converting enzyme)/CED-3 homolog 1) protein with a death domain (RAIDD), TNF receptor associated factor (TRAF)1, TRAF2, TRAF3, and A20 (50–60). Furthermore, it can also interact with RIPK3, through its RIP homotypic interaction motif (RHIM) domain, as well as with focal adhesion kinase, MEKK1 and MEKK3 (61–64).
- RIPK3: similar to RIPK1; it contains an N-terminal kinase domain and a RHIM domain in the C-terminal part, that allows RIPK1/RIPK3 interactions (4). However, its C-terminal domain is completely different from other RIPK proteins. This could explain its ability to interact with the liver glycogen phosphorylase (PYGL), glutamate ammonia ligase (GLUL) and glutamate dehydrogenase 1 (GLUD1), enhancing its enzymatic activity (4). These are metabolic enzymes required for ATP production, with PYGL releasing glucose-1-phosphate from liver glycogen, and GLUL and GLUD1 playing crucial roles in the use of glutamate and glutamine as substrates for ATP through oxidative phosphorylation; this suggests a link between RIPK3 and metabolism (65). In fact, RIPK3 orchestrates necroptosis, “an active cell death pathway” that requires both adenosine triphosphate (ATP) and ROS (66). Through RIPK3-enhanced aerobic respiration, mitochondria could both produce energy to execute necroptosis while increasing the amounts of ROS required for the RIPK1/RIPK3 and later MLKL (mixed lineage kinase domain-like) complex formation and activation downstream of TNF (66).
Genetic studies has helped to understand the role of these kinases in cell death. Mice lacking RIPK1 show defects in multiple tissues, triggering systemic inflammation leading to perinatal death 1-3 days after birth (67). Simultaneous deletion of TNFR1 prolonged up to 12 days post-delivery the survival in Ripk1−/− Tnfr1−/− (68). Deletion of RIPK3, mixed lineage kinase domain like pseudokinase (MLKL) or caspase-8 in Ripk1−/− did not improve the phenotype, with those mice dying soon after delivery (69, 70), suggesting that when just apoptosis or necroptosis are blocked downstream of TNF, the other pathway gets activated. In a similar manner, triple deletion of RIPK1, RIPK3, and TNFR1 allows mice survive until adulthood. Yet, shortly after birth they present intestinal apoptosis, which could contribute to the mortality associated with blood bacteremia (69). Accordingly, simultaneous deletion of RIPK1, RIPK3, and caspase-8 or FADD protect the mice and prevented any macroscopic and microscopic signs of intestinal pathology, but mice developed autoimmune lymphoproliferative syndrome (69, 70). Since double deletion of FADD and RIPK1 induces perinatal death (71) but additional deletion of RIPK3 protect the animals, these results suggest that some other mechanism, independent of RIPK1, activates RIPK3. In fact, DNA-dependent activator of interferon regulatory factors (DAI) can interact and activate RIPK3 independently of RIPK1 (72). Ripk1−/− Ripk−/− and Trif−/− or Ifnar−/− animals were generated, and although it conferred certain protection compared with Ripk1−/− Ripk−/−, those mice did not survive past weaning (70). Finally, mice lacking RIPK1 in IECs specifically (RIPK1ΔIEC), develop severe intestinal inflammation associated with IEC apoptosis leading to early post-birth death. Similarly, tamoxifen-induced deletion of Ripk1 leads to rapid weight loss and mice death. Crypt cells from RIPK1ΔIEC failed to grow into organoids (69), so in vitro deletion is required to grow RIPK1 deficient intestinal organoids (73). Unexpectedly, in those intestinal organoids the NF-κB pathway remained intact downstream of TNF, although they undergo massive cell death (73).
Two mice models with point mutations in the kinase domain were generated to study the kinase role of RIPK1 without compromising its scaffold function. RIPK1K45A and RIPK1D138N (74, 75) mice were born at expected Mendelian ratios and showed no abnormalities, pointing out the importance of RIPK1 function as a scaffold protein. Fibroblasts and macrophages derived from these mice were stimulated with TNF and were shown to be protected from cell death demonstrating the role of the kinase domain from RIPK1 in triggering cell death (74).
Unlike RIPK1−/−, RIPK3−/− mice are indistinguible from their littermates and exhibit normal downstream pathway activation from TNFR1 and TLRs (76). However, knock-in mice harboring a kinase death form of RIPK3 (RIPK3D161N) die at embryonic day 11.5 due to high amounts of cell death in the yolk sac vasculature. The authors of this study show how this cell death was dependent on caspase-8. Kinase death RIPK3, but not wild-type RIPK3, interacted with FADD, RIPK1, and caspase-8. Similarly, expression of RIPK3D161N in the adult intestine also led to diarrhea and massive weight loss due to caspase activation, and downstream apoptosis of IECs (77). Similar results were obtained when a RIPK3 inhibitor was given to mice (78). On the contrary, another RIPK3 kinase death animal line (RIPK3K51A) did not present any embryonic abnormalities, and the mice were shown to be viable, fertile, and immunocompetent, as well as able to rescue the embryonic lethality seen in caspase-8 knock-out mice (78). However, RIPK3 inhibitors still induced apoptosis on cells expressing the RIPK3K15A. Altogether, this data suggests that RIPK3 inhibition through small molecules or the presence of the D161N mutation induces conformational changes in RIPK3 that promote apoptosis. Although those results could be secondary to a change in the RIPK3 structure due to the D161N mutation, they suggest that RIPK3 kinase inhibition leads to apoptosis.
TNF is one of numerous genes implicated in IBD pathogenesis stimulated by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). It codes for the prototypical inflammatory cytokine tumor necrosis factor (TNF), which has various functions in the intestine (79). TNF is synthesized as a transmembrane protein that forms homotrimeric structures, and is cleaved by a disintegrin and metalloprotease domain 17 (ADAM17) or by TNF-converting enzyme (or TACE), which releases its soluble form (80). TNF is able to bind two receptors: TNFR1 and TNFR2, which differ in their structure and expression pattern, as well as in the signaling pathways that they induce once they are engaged (80). TNFR1 is expressed in all cell types, whereas TNFR2 is mostly restricted to immune and endothelial cells. Both receptors are able to activate the NF-κB pathway through different signaling cascades as a result of strikingly different intracellular domains. TNFR1 contains a cytoplasmic death domain (DD), which is a conserved sequence of 80 amino acids that forms a distinctive fold (81, 82) and allows the recruitment of TNFR1-associated death domain protein (TRADD). TNFR2 lacks the death domain and recruits TNFR-associated factor 1 (TRAF1) and TRAF2, rather than TRADD (82–84). Both TNFR1 and TNFR2 can lead to NF-κB activation.
TNF has important protective functions in intestinal epithelial cells (Figure 3): (a) TNF modifies the first physical barrier of the intestine: the mucus layer. Through TNFR2, TNF sensitizes goblet cells to prostaglandin E2, a known mucus secretagoge, and protects the epithelium by increasing mucus secretion (85, 86), (b) TNF is able to induce the expression of the polymeric immunoglobulin receptor (pIgR), which is necessary for the transcytosis of secretory IgA into the mucus, and prevents bacterial translocation into the lamina propria (87), (c) TNF is critical in wound healing, which is an important step in resolving injury and preventing chronification of underlying inflammation. Two different steps occur during this process: spreading and migration of cells through the basement membrane, and redifferentiation and proliferation of cells. Through TNFR2 dependent activation of focal adhesion kinase, TNF is able to induce epithelial migration (88) and cell proliferation (89). TNF can also support wound healing and cell survival through TNF-induced TACE activation, which subsequently liberates ErbB ligands that promote cell survival (90), (d) through TNFR1, TNF is able to activate the NF-κB pathway and assemble the TNFR1 complex I, which promotes IEC survival.
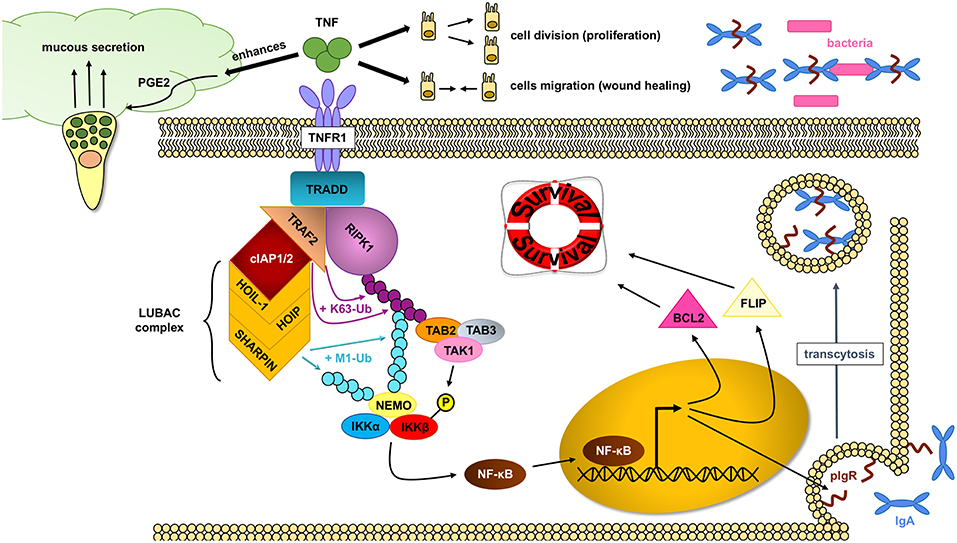
Figure 3. NF- κB pathway activation through RIPK1 protects IECs. In homeostatic conditions, TNF (tumor necrosis factor) plays an important role in maintaining an intact intestinal epithelial barrier. Upon binding to the TNFR1 (tumor necrosis factor receptor 1), the TNFR complex I is formed, where RIPK1 (receptor interacting protein kinase 1) serves as an scaffold protein upon which the key IKK (IκB kinase) complex will bind and activate. The IKK complex will in turn induce the translocation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) transcription factors into the nucleus and allow gene expression, including cell survival factors. TNF can also help in the wound healing process by inducing cell proliferation and migration as well as enhance the intestinal barrier by favoring mucus production and impairing bacterial translocation through pIgR (polymeric immunoglobulin receptor) induction.
The assembly of the TNFR1 complex I is key in intestinal barrier maintenance. In most cell types, including IEC, transient TNF signaling inhibits apoptosis due to the assembly of the TNFR complex I, and activation of IκB kinase (IKK) β-dependent NF-κB (91). Upon the binding of TNF to homotrimers of TNFR1, the adaptor molecule TRADD is recruited to the cytoplasmatic TNFR1 domain. In a step-wise process, RIPK1, TRAF2, cellular inhibitor of apoptosis protein 1 (cIAP1) or cIAP2, and linear ubiquitin chain assembly complex (LUBAC) are recruited to form signaling complex I. TRAF2 and cIAP1/2 mediate K63-linked ubiquitination of the complex. In this situation, the kinase RIPK1 acts as a scaffold protein that allows for docking of the adaptor proteins TAK1-binding protein 2 (TAB2) and (TAB3) and the kinase TAK1 through RIPK1 K63-ubiquitins (92). Meanwhile, the LUBAC complex mediates M1-ubiquitination of some components in the complex I, such as RIPK1 and NF-kappa-B essential modulator (NEMO) (93, 94). The IKK complex is also recruited to the complex, and after phosphorylation of IKKβ by TAK1, mediates the activation of the canonical NF-κB pathway and the resulting upregulation of anti-apoptotic genes such as BCL2 (B-cell lymphoma 2) and FLIP (FLICE-like inhibitory protein), to promote cell survival and cell proliferation (80).
Several previous works have highlighted the critical role of this pathway on IEC survival. Early work by Egan et al. shows that deletion of IKKβ in IECs promotes the gut damage from ionizing radiation (IR) (95). Furthermore, when LPS, a known activator of the NF-κB pathway, is administered prior to IR, IECs are also protected from massive apoptosis, suggesting the IKK complex, the main protein complex downstream of TNF, provides protective effects to IECs (95). Similarly, IKKß was shown to be protective in a model of colitis induced by C. difficile and dextran sodium sulfate (DSS) (96, 97). Although deletion of IKKß or IKKß alone does not induce spontaneous colitis, IECs lacking NEMO or TAK1 develop colon pathology, in a complete or partial TNF dependent manner, respectively, including IEC apoptosis, demonstrating that the NF-κB pathway plays key homeostatic roles (98, 99). Interestingly, unlike NEMOIEC−KO, TAK1IEC−KO develops intestinal inflammation, perhaps due to the ability of TAK1 to activate other protective pathways such as the Mitogen-activated protein kinases pathway (MAPK). Nevertheless, activated/nuclear NF-κB is present in both IECs and the lamina propria macrophages of active IBD areas (100). To determine the pathogenic function of persistent NF-κB activation, which occurs in IBD (100), we generated Ikkβ(EE)IEC mice in which a constitutively active IKKβ(EE) variant is expressed in IEC from the villin promoter (101). Surprisingly, instead of being resistant to TNF-induced mucosal erosion, Ikkβ(EE)IEC mice displayed severe TNF-dependent epithelial layer destruction when challenged with various stimuli that induce TNF production, or when given exogenous TNF (101). The mechanism by which constitutive IKKβ/NF-κB activation renders mouse IECs susceptible to TNF-induced killing rather than preventing it is unknown, but is likely to be relevant to understand the effect of NF-κB chronic activation in IECs of active IBD lesions.
As reviewed above, transient TNF signaling inhibits apoptosis due to the assembly of TNFR1 complex I and IKKβ-dependent NF-κB activation (91). However, TNFR1–TRADD signaling can result in cell death in special circumstances, when complex I shifts toward complex IIa, IIb, or the necrosome to induce different types of TNF-induced cell death (Figure 2).
Ubiquitin removal from RIPK1, through deubiquitination by cylindromatosis (CYLD), or ubiquitination-impairment by cIAP1/2 depletion (102–104), alters the formation of complex I, allowing its disassembly and TNFR1 internalization (105). TRADD, FADD, pro-caspase-8 (caspase-8), and FLICE-like inhibitory protein (FLIPs) are then recruited to the TNFR1. In this complex, the long isoform of FLIP (FLIPL) and the pro-caspase-8 form a heterodimeric caspase that cleaves and inactivates RIPK1 and RIPK3, as well as CYLD, to prevent necroptosis (106–108). This TRADD-dependent complex IIa also allows caspase-8 homodimerization and activation, resulting in activation of the executioners caspase-3 and caspase-7, which trigger apoptosis. This pathway, in normal conditions, would be inhibited due to previous NF-κB activation and expression of anti-apoptotic genes (108–110) but as mentioned above, ablation of IKKβ (95, 111), or its regulatory subunit NEMO (98), renders IEC susceptible to TNF-induced death.
TNFR complex IIb or the Ripoptosome has been described to occur downstream of TNF when cIAP1/2 is depleted through SMAC mimetics (SM) (112–114). SMAC (second mitochondria-derived activator of caspase) is a pro-apoptotic mitochondrial protein that inhibits IAPs. The exact mechanism that triggers the formation of complex IIb instead of IIa is unknown, although in this case the activation of NF-κB does not prevent apoptosis (115). TNF treatment together with TAK1 pharmacological inhibition also triggers RIPK1-dependent apoptosis, in a similar manner as TNF plus SM treatment, suggesting that TAK1 recruitment to cIAP1/2-ubiquitinated RIPK1 inhibits RIPK1-dependent apoptosis (115). In fact, IKKα and IKKβ, the downstream kinases of TAK1, inhibit RIPK1 in association with the Ripoptosome through direct phosphorylation of RIPK1 (116). Complex IIb, the Ripoptosome, is composed of RIPK1, FADD and caspase-8, and A20 (117). It is independent of TRAILR1/DR4, TRAILR2/DR5, and Fas/CD95 activation (112). TLR3 can also potentially induce complex IIb. TLR3 activation induces apoptotic cell death downstream of TRIF that depends on a complex formed also by RIPK1, caspase-8, and FADD, although it is unknown whether this requires RIPK1 kinase activity (118, 119). Of note, TNFR1 activation is dispensable if cell death is triggered by etoposide, a genotoxic stress inducer that also depletes cIAPs, although in this case complex IIb formation occurs 6 h after the treatment, 4 h later than when triggered downstream of TNF. Complex IIb requires the kinase activity of RIPK1 to induce cell death although the exact mechanism of its activation or its role as a kinase is unknown; in fact no targets for the kinase activity of RIPK1 have been described apart from itself (113).
Necroptosis can be triggered through different stimuli. Most studies on necroptosis have been performed after TNF, FAS, or TLRs stimulation, but it can also be triggered by intracellular events, such as viral infection through Z-DNA or Z-RNA sensing via Z-DNA binding protein 1 (ZBP1/DAI) (72). For instance, downstream of TNF, when caspases are not fully activated or their activity is blocked (ex: by viral inhibitors), the protein kinase RIPK3 is recruited and forms the necrosome, which will lead to necroptotic cell death (120, 121). Once engaged, RIPK1 and RIPK3 undergo auto and transphosphorylation leading to their activation. Interestingly, although RIPK3 can also phosphorylate RIPK1, RIPK1 does not phosphorylate RIPK3 (120). The requirement for RIPK1 and RIPK3 trans and autophosphorylation can explain the formation of RIPK1/3 amyloid structures through RIPK1 and RIPK3 RHIM domains, a required step for RIPK3 autophosphorylation (122, 123). All these signals will converge into MLKL phosphorylation and activation, and subsequent cell death (124). Phosphorylated MLKL binds to the inner leaflet of the plasma membrane and forms the necroptotic pore, executing necroptosis (125–127). Although RIPK1 can be autophosphorylated at S14/15, S20, S161, and S166, (128), only S161 has been shown to be required to induce necroptosis (129). RIPK1 phosphorylation on S89 or MK2 mediated phosphorylation of S321 impair RIPK1 mediated cell death (130, 131). Phosphorylation of S227 in RIPK3 allows the binding of RIPK3 to MLKL (124). In addition, MLKL is phosphorylated by RIPK3 at T357 and S358 residues in human, and S345, S347, and T349 residues in mouse. These phosphorylation sites are necessary for necroptosis since mutation of both sites inhibits necroptotic cell death (124, 132).
The increased areas of epithelial cell death associated with IBD are especially prevalent in UC compared to CD and controls (133). This epithelial cell death increases the chances of antigen translocation and subsequent triggering of inflammatory responses (134–137). These epithelial cell deaths present features of apoptosis and necrosis. Necrosis has long been recognized as a major trigger of inflammation; as cells die, their cellular contents activate the host immune response. Apoptotic cells will also increase the intestinal permeability by overwhelming the capacity of phagocytes to clear apoptotic cells, and by preventing proper tight junction function and intestinal barrier remodeling of patches of shedding cells. Interestingly, patients with active UC, who ultimately require surgery, had higher apoptotic indices than UC patients that were receiving medication. Also, electron microscopy on rectal biopsies of patients with CD and UC compared with normal controls showed patches of necrotic cells in four out of seven CD patients (135). Notably RIPK3 is expressed at high levels in the terminal ileums of patients with CD (138).
Several IBD pathogenic factors can promote IEC cell death in these patients. Among them, genetic predisposition, disturbance of the commensal microbiota, and dysregulated immune responses can contribute to epithelial barrier defects and promote chronic inflammation in the IBD gut.
Although family history is a risk factor for developing IBD, the concordance rate in monozygotic twins is only 10–15% in UC, and 30–35% in CD, suggesting that non-genetic factors might play a bigger role (139). Nonetheless, the first genome-wide association study (GWAS) for Crohn's disease, undertaken in 2005 in Japan, identified the susceptibility locus of the tumor necrosis factor super family 15 gene (TNFSF15) (140). Subsequently, several other studies have identified, in different ethnic cohorts, 235 genetic markers in 200 susceptibility loci (141–143). Of the 163 identified loci in the Caucasian population, 110 appear to be relevant to both CD and ulcerative colitis (TNFAIP3, IRGM, TNFSF15), 23 appear to be specifically related to CD (ATG16L1, NOD2), and 30 appear to be specifically related to UC (IRF5, NFKB1). IL-23R has also been shown to be related to CD in several studies (144–146), with rs1343151 and rs7517847 variants decreasing the risk of developing the disease. Although, most identified SNPs lack functional data, the identification of these genes elucidates the critical pathways in IBD pathogenesis.
a. The first genetic risk variant identified for CD was the NOD2 gene (147, 148). Hugo et al. found three different polymorphisms in NOD2; one is a frameshift mutation (L1007C) which causes a truncated protein transcript, and two are non-synonymous polymorphisms (R702W and G908R). Carriage of one copy of any risk allele confers a modestly increased risk of developing CD (2 to 4-fold). However, having two copies or a combination thereof is associated with a 20- to 40-fold increased risk. Another SNP in the autophagy gene ATG16L1, which is associated with CD, is responsible for a threonine to alanine substitution at amino acid 300 (T300A) that increases the odds ratio (OR) for CD to 1.62 in the Spanish population (149). Finally, another important gene related to autophagy is IRGM (immunity-related guanosine triphosphatase family M protein). It encodes a GTP-binding protein that induces autophagy and plays an important role in innate immunity against intracellular pathogens. Two flanking SNPs (rs13361189 and rs4958847) have been better associated with increased susceptibility to CD with an OR of 1.34 and 1.33; the first SNP alone also confers a small association with UC (OR: 1.16) (149).
Impaired autophagy disturbs the function of IECs and influences the inflammatory and immune responses, ROS production, and endoplasmatic reticulum (ER) stress, promoting the occurrence and development of IBD (150–153). Furthermore, it is noted that autophagy can play a role in the release and degradation of the damage-associated molecular pattern molecules (DAMPs), contributing to the alleviation of IBD (154–156). ATG16L1 deletion also increases the chance of IECs necroptosis (47), and deletion of another autophagy protein, ATG5, results in impaired intestinal permeability and protection against Toxoplasma gondii infection (157). Finally, mice deficient in Nod2 and Atg16l1 showed Paneth cell defects and susceptibility to intestinal inflammation (158, 159). These results highlight the importance of the Paneth cell, that releases antimicrobial peptides, supports stem cells, and regulates AMP production (Nod2) and granule exocytosis (Atg16l1), in the pathogenesis of the disease. Importantly, similar phenotypes have been observed in human disease, and patients with Crohn's disease carrying the ATG16L1T300A mutation showed granular abnormalities in Paneth cells (159).
Other authors looked into the role of the NOD2 L1007insC polymorphism, which results in a frameshift mutation that generates a truncated Nod2 protein. This mutation prevents peptidoglycan and MDP-dependent activation of the NF-κB pathway, and localization of NOD2 into the plasma membrane (28, 46, 160). NOD2 L1007insC did not prevent NOD2-ATG16L1 interaction, but did prevent its localization in the plasma membrane, impairing wapping of invading bacteria by autophagosomes. Furthermore, in different human epithelial cell lines, deletion of ATG16L1 or reconstitution with the common coding variant ATG16L1T300A abrogated capture and degradation of intracellular Salmonella (161, 162). Recently, Murthy et al. showed a relation between autophagy, cell death and inflammation. The authors demonstrated that caspase-3 enhances the cleavage of ATG16L1T300A, an SNP strongly associated with incidence of CD. They propose that the presence of T300A apoptotic stimuli enhances ATG16L1 cleavage, triggering cytokine production and inflammation (163). Another work has also shown how ATG16L1 prevented necroptosis in IECs (47).
b. RIP1 and RIP3: Although no SNPs in these proteins have been associated with IBD, the effect of RIPK1 deficiency in humans was studied by Cuchet-Lourenço et al. (3). In this study, they found four patients from three unrelated consanguineous families carrying homozygous loss-of-function mutations in RIPK1. The four patients had lymphopenia, suffered from recurrent viral, bacterial and fungal infections, early-onset inflammatory bowel disease, involving the upper and lower gastrointestinal tract, and developed arthritis (3). Stimulation of skin fibroblasts with TNF and poly(I:C) in vitro showed similar results to those seen in mice, with impaired activation of downstream signaling pathways from TNFR1 and TLR3 and increased cell death through necroptosis.
c. NF-κB pathway: SNPs in ubiquitously expressed genes encoding NF-κB-regulated molecules show strong association with IBD (164, 165). NF-κB stimulates transcription of numerous genes implicated in IBD pathogenesis, including TNF. TNF inhibition is one of the main therapeutic options in IBD (100), leading to reduced IEC apoptosis and enhanced mucosal repair (91). In IECs, transient TNF signaling inhibits apoptosis due to IKKβ-dependent NF-κB activation (91). On the other hand, corresponding ablation of IKKβ (95, 111), or its regulatory subunit NEMO (98), renders IEC susceptible to TNF-induced death. However, IKK or NF-κB deficiencies have never been reported in IBD.
d. A20 is a NF-κB-responsive gene that is thought to be involved in negative feedback regulation of NF-κB activation in response to many proinflammatory stimuli (166, 167). A20 contains an ovarian tumor (out) domain with deubiquitinating activity (DUB) in the amino-terminal region and seven carboxy-terminal zinc finger (ZnF) domains. A20-deficient mice have a severe inflammatory phenotype, with hypersensitivity to TNF, and die prematurely due to severe multiorgan inflammation and cachexia (168). Although several reports describe that A20 terminates the NF-κB pathway through its DUB activity by breaking down the docking sites in the TNFR1 complex I, A20 knock-in mice bearing an inactivating mutation in DUB (C103A) or ZnF4 domains do not exhibit the severe inflammatory phenotype of full A20-knockout mice (169, 170), suggesting that the function of A20 to modulate the NF-κB is not dependent on its deubiquitinase activity.
Several studies have linked SNPs of TNFAIP3, which codes for the immunoregulatory protein A20, with susceptibility to multiple autoimmune human diseases. These diseases include systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), psoriasis, type 1 diabetes, coeliac disease, Crohn's disease, coronary artery disease in type 2 diabetes, and systemic sclerosis (171). Most of the SNPs related with IBD are located in non-exon areas, implying that they most likely play a role in RNA synthesis or maturation. The minor rs5029941 (alanine to valine substitution) allele is associated with increased risk for IBD with an OR of 3.75, while the rs7753394, located upstream to the coding region, has an OR of 1.21 in heterozygotes and 1.48 in homozygotes for CD. Finally the rs2327832 allele increases the OR for UC to 1.26 (172). Interestingly, the rs6927172 variant was associated with increased A20 expression, decreased TNF levels, and non-response to anti-TNF therapy in both CD and UC (173). On the other hand, the rs6927210, rs7753394, and rs7773904 variants were linked to improved response to anti-TNF drugs (174).
Given that A20 SNPs in other diseases, such as SLE, have been related to lower expression or function (175, 176), and that A20 deletion in the whole mouse or in different compartments, including the intestine, induces spontaneous inflammation (168, 174, 177, 178), it is thought (but not proven) that SNPs in the TNFAIP3 gene are associated with IBD decrease A20 expression. In IEC, deletion of A20 on those cells renders the mice more susceptible to the DSS colitis model with higher amounts of apoptotic cells in the epithelial colon (179). While the previous study did not show spontaneous intestinal inflammation, combined deletion of A20 in IEC and the myeloid compartment induces spontaneous colitis and ileitis with the presence of apoptotic cells in the crypt compartment (174). Additionally, overexpression of A20 in the IEC protects the intestinal epithelial barrier after LPS challenge and prevents colitis induced by DSS but not TNBS (180, 181).
However, A20s role in cell death seems to be more dependent on cell type than its NF-κB regulatory function. An A20 specific deletion in B and T cells actually protects them from FAS and TCR (T-cell receptor) induced cell death (178, 182). Also. two independent works have looked into the RNA expression of A20 in IBD. Although Arsenescu et al. found a decrease in the RNA levels of A20, as well as other typical proinflammatory markers of IBD in non-inflamed IBD tissue compared with control samples (183), Vereecke et al. found that A20 levels of non-responder patients to anti-TNF therapy was higher both before and after treatments compared to controls and responders. Accordingly, levels of A20 in patients that responded to anti-TNF drugs diminished to basal levels after therapy. These results could suggest that the upregulation of A20 is triggering intestinal inflammation. We have recently showed that A20 protein levels in UC and CD are increased in IECs. Using transgenic mice that overexpress A20 in the IEC, we showed that increased and prolonged recruitment of A20 to the TNFR complex I favors a shift from complex I toward complex IIb, probably through maintenance of RIPK1 linear-poly-ubiquitinated status and inducing RIPK1-dependent apoptosis in IEC (117). Of interest, we also detected A20 in the ripoptsome complex (117). Concomitant with that, pharmacological and genetic RIPK1 kinase blockade prevented apoptosis, suggesting a new therapeutical treatment for IBD.
The gut microbiome, including bacteria, fungi, virus, and other organisms, shapes host functions in both normal and disease conditions. The clinical observation that antibiotics have a modest effect in IBD (184–187) suggests that the microbiome could play a role in shaping the disease. In fact, bacterial dysbiosis has been shown to occur in IBD (188–191) with consistent reports of decreased biodiversity, both α diversity and species richness, a measure of the total number of species in a community. However, a specific role of bacterial dysbiosis in IBD is yet to be discovered. In fact, a recent paper by Halfvarson showed that inflammation was not directly correlated with increased dysbiosis (188). A similar concept was suggested by another study, which shows that there is reduced diversity in inflamed vs. non-inflamed tissues within the same patient, and a lower bacterial load in inflamed regions in CD patients (192). Also, serum reactivity against selected components of the gut microbiota is common, even in healthy individuals, and some CD associated serological markers against microbial antigens are present years before clinical manifestations in patients with CD, as well as in healthy individuals (193).
Various microorganisms that supposedly exert aggressive or protective functions relevant to Crohn's disease, such as adherent-invasive Escherichia coli and Faecalibacterium prausnitzii, respectively have been identified (194, 195). Furthermore, it is known that Helicobacter pilori has developed different mechanisms to disrupt the intracellular adhesions of the intestinal barrier (196), suggesting that other bacteria could act similarly. Yet, E. coli Nissle 1917 or ECOR63 enhance the epithelial barrier by up-regulating ZO-1 and claudin-14 and by downregulating claudin-2 (197). Also, Chelakkot et al. have demonstrated recently, that Akkermancia muciniphila, a known beneficial bacteria that reduces gut barrier disruption, upregulates occludin-2, decreasing the permeability of lipopolysaccharide-treated Caco-2 cells (198); a similar effect is seen when treating T84 monolayers with metabolites from the probiotic Bifidobacterium infantis Y1, which leads to an increase of ZO-1 while reducing claudin-2 (199). Treating Caco-2 cells with another probiotic, Lactobacillus plantarum MB452, also increased the transcription of occludins (200) and, in vivo, it increased occludin and ZO-1 (201). Some probiotics and commensals have also been shown to prevent, and even reverse, the adverse effects of pathogens on intestinal barrier function. For instance, when L. plantarum is co-cultured with enteroinvasive or enteropathogenic E. coli, it prevents the loss of permeability induced by those strains (202, 203). This data suggest that bacteria can directly regulate gut permeability by modulating cell-to-cell junctions. Thus, although it is believed that an inappropriate response against commensal gut microbiota occurs in IBD, it has been difficult to determine whether or not this process is secondary to an altered microbiota, a defective immune response, or a change in gut permeability (204), and whether these microbiome changes are primary or secondary to the disease. Of interest, some of the genes related to IBD were shown to control the bacterial microbiome and gut permeability, modulating cell-to-cell junctions. For instance, Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal bacteroides vulgatus (205).
Fungi is also a constituent of gut microbiota, however it just accounts for <0.1% of the total microbes (206). Antibiotic treatment increases fungi while decreasing bacteria populations, showing a competition between both kingdoms (207, 208). Alterations of GI bacterial populations and increased yeast can drive the development of a CD4 T-cell-mediated allergic airway response to subsequent mold spore exposure, suggesting a role for fungal microbiota in promoting immune-mediated diseases (208). In IBD patients, Basidiomycota, Ascomycota, and C. albicans are significantly elevated, whereas Saccharomyces, Candida, and Cladosporium are predominant in healthy individuals (209–211). Different components of the fungal cell wall such as chitin, β-glucans, and mannans can trigger the innate immune response, so it is not surprising that intestinal fungal invasion exacerbates colitis in mice (212).
Although virology focuses on pathogenic strains, most viruses are bacteriophages or endogenous retroviral elements. In fact, 99% of the annotated DNA viruses are bacteriophages (213). There are approximately 108-109 virus-like particles (VLP) per gram of human stool, suggesting that viruses could play an important role in the bacterial community. The human gut bacteriophage varies intensively between subjects. However, they are temporally stable within individuals with dsDNA Caudovirales and ssDNA Microviridae, the two predominant viruses in healthy humans (214, 215). In IBD, virome richness is increased with expansion from the order Caudovirales (216–218). This could be explained as a result of commensal microbes entering lytic cycles, or from new viruses introduction from new bacteria. In either case, bacteriophages can shape the gut microbiome, affecting bacterial fitness, diversity, and perhaps aiding in horizontal gene transfer (219, 220). Furthermore, viruses can translocate into the host, inducing immune responses (221–223). It is not surprising that mice with a genetic predisposition for CD (a mutation in the ATG16L1 gene) manifest the disease when infected with a gut norovirus while wild type mice controls did not (224). Another study showed that mice that were administered a cocktail of antiviral drugs had more severe colitis in the dextran sulfate sodium (DSS) model than ones treated with DSS alone. Overall, these results suggest a role for the virome in IBD, and new research will be needed to further understand its impact.
In this review we discussed the role of RIPK and autophagy in relation to IBD (Figure 4). RIPK proteins seem to be plausible candidates for new drugs to treat inflammatory flares of IBD, preventing breakdown of the intestinal epithelial barrier. Additionally, autophagy seems to be a protective pathway, mainly by regulating intestinal homeostasis and pathogen protection, especially through paneth cells. Although further research is required to completely understand the pathophysiology of IBD, great advances in the field have improved the wellbeing of patients with the disease.
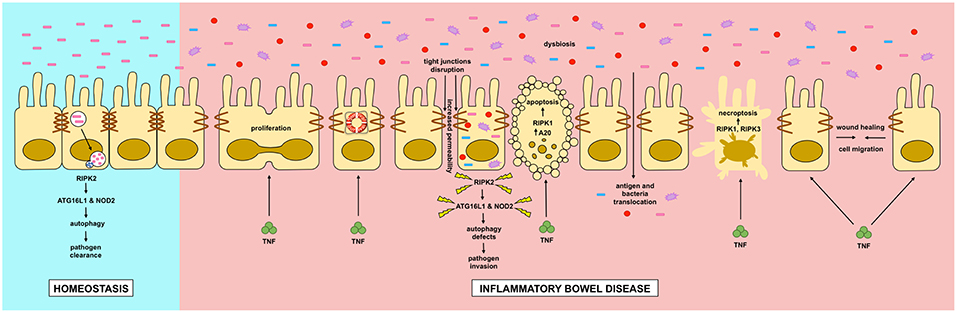
Figure 4. Dysregulation of intestinal epithelial cell RIPK pathways promotes cell death in IBD. Dysregulation of RIPK (receptor interacting protein kinase) pathways play a key role in the inflammatory processes occurring in IBD (inflammatory bowel disease). TNF (tumor necrosis factor) has pleyotropic roles in intestinal epithelial cells. In homeostasis, TNF through the activation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway, where RIPK1 has a scaffold function, it promotes cell proliferation, migration and survival, helping to a proper intestinal barrier regeneration. In IBD, TNF can induce apoptosis or necroptosis in a kinase dependent function of RIPK1 and RIPK1/3 respectively. Cell-to-cell adhesions are also loosened in IBD, allowing the translocation of food antigens and bacteria from the gut lumen. Another feature of IBD is microbial dysbiosis. Genetic mutations in RIPK2, NOD2 (nucleotide-binding oligomerization domain-containing protein 2) or ATG16L1 (autophagy-related protein 16 like 1) can impair a proper autophagy response allowing the proliferation and invasion of the host by pathogenic bacteria.
RG-C, S-JY, SD, and MG contributed to the literature review. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
Supported by grants from the National Institutes of Health: AR073324 to MG, and DK107585 and DK099275 to SD. MG also acknowledges funding from GlaxoSmithKline. GlaxoSmithKline had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. (2016) 13:13–27. doi: 10.1038/nrgastro.2015.186
2. Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol. (2018) 15:39–49. doi: 10.1038/nrgastro.2017.136
3. Cuchet-Lourenco D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science. (2018) 361:810–3. doi: 10.1126/science.aar2641
4. Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. (2010) 7:243–9. doi: 10.1038/cmi.2010.10
5. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. (2014) 14:141–53. doi: 10.1038/nri3608
6. Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. (2003) 422:522–6. doi: 10.1038/nature01520
7. Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. (2007) 204:1891–900. doi: 10.1084/jem.20070563
8. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. (2011) 9:356–68. doi: 10.1038/nrmicro2546
9. Lueschow SR, Stumphy J, Gong H, Kern SL, Elgin TG, Underwood MA, et al. Loss of murine Paneth cell function alters the immature intestinal microbiome and mimics changes seen in neonatal necrotizing enterocolitis. PLoS ONE. (2018) 13:e0204967. doi: 10.1371/journal.pone.0204967
10. Johansson ME, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. (2016) 16:639–49. doi: 10.1038/nri.2016.88
11. van der Waaij LA, Harmsen HJ, Madjipour M, Kroese FG, Zwiers M, van Dullemen HM, et al. Bacterial population analysis of human colon and terminal ileum biopsies with 16S rRNA-based fluorescent probes: commensal bacteria live in suspension and have no direct contact with epithelial cells. Inflamm Bowel Dis. (2005) 11:865–71. doi: 10.1097/01.mib.0000179212.80778.d3
12. Corfield AP. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim Biophys Acta. (2015) 1850:236–52. doi: 10.1016/j.bbagen.2014.05.003
13. Marcobal A, Southwick AM, Earle KA, Sonnenburg JL. A refined palate: bacterial consumption of host glycans in the gut. Glycobiology. (2013) 23:1038–46. doi: 10.1093/glycob/cwt040
14. Pudlo NA, Urs K, Kumar SS, German JB, Mills DA, Martens EC. Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. MBio. (2015) 6:e01282–15. doi: 10.1128/mBio.01282-15
15. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. (2012) 483:345–9. doi: 10.1038/nature10863
16. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. (2013) 342:447–53. doi: 10.1126/science.1237910
17. Shajib MS, Khan WI. The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol. (2015) 213:561–74. doi: 10.1111/apha.12430
18. Ponomarev ED. Fresh evidence for platelets as neuronal and innate immune cells: their role in the activation, differentiation, and deactivation of Th1, Th17, and tregs during tissue inflammation. Front Immunol. (2018) 9:406. doi: 10.3389/fimmu.2018.00406
19. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. (2009) 124:3–20; quiz 1–2. doi: 10.1016/j.jaci.2009.05.038
20. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. (2016) 17:564–80. doi: 10.1038/nrm.2016.80
21. Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. (2008) 1778:572–87. doi: 10.1016/j.bbamem.2007.07.014
22. Schmidt A, Koch PJ. Desmosomes: just cell adhesion or is there more? Cell Adh Migr. (2007) 1:28–32. doi: 10.4161/cam.1.1.4204
23. Blander JM. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J. (2016) 283:2720–30. doi: 10.1111/febs.13771
24. Patterson AM, Watson AJM. Deciphering the complex signaling systems that regulate intestinal epithelial cell death processes and shedding. Front Immunol. (2017) 8:841. doi: 10.3389/fimmu.2017.00841
25. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
26. Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, et al. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci USA. (2009) 106:14564–9. doi: 10.1073/pnas.0813319106
27. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. (2014) 41:898–908. doi: 10.1016/j.immuni.2014.12.010
28. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
29. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. (2003) 278:5509–12. doi: 10.1074/jbc.C200673200
30. Boyle JP, Parkhouse R, Monie TP. Insights into the molecular basis of the NOD2 signalling pathway. Open Biol. (2014) 4:140178. doi: 10.1098/rsob.140178
31. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. (2001) 276:4812–8. doi: 10.1074/jbc.M008072200
32. Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. (2000) 275:27823–31. doi: 10.1074/jbc.M003415200
33. Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. (2007) 282:36223–9. doi: 10.1074/jbc.M703079200
34. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. (2008) 27:373–83. doi: 10.1038/sj.emboj.7601962
35. Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. (2009) 106:14524–9. doi: 10.1073/pnas.0907131106
36. Tigno-Aranjuez JT, Asara JM, Abbott DW. Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. (2010) 24:2666–77. doi: 10.1101/gad.1964410
37. Dorsch M, Wang A, Cheng H, Lu C, Bielecki A, Charron K, et al. Identification of a regulatory autophosphorylation site in the serine-threonine kinase RIP2. Cell Signal. (2006) 18:2223–9. doi: 10.1016/j.cellsig.2006.05.005
38. Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. (2009) 30:789–801. doi: 10.1016/j.immuni.2009.04.011
39. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. (2012) 46:746–58. doi: 10.1016/j.molcel.2012.04.014
40. Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker-Jensen S, et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol Cell. (2013) 50:818–30. doi: 10.1016/j.molcel.2013.06.004
41. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. (2010) 11:55–62. doi: 10.1038/ni.1823
42. Homer CR, Kabi A, Marina-Garcia N, Sreekumar A, Nesvizhskii AI, Nickerson KP, et al. A dual role for receptor-interacting protein kinase 2 (RIP2) kinase activity in nucleotide-binding oligomerization domain 2 (NOD2)-dependent autophagy. J Biol Chem. (2012) 287:25565–76. doi: 10.1074/jbc.M111.326835
43. Anand PK, Tait SW, Lamkanfi M, Amer AO, Nunez G, Pages G, et al. TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J Biol Chem. (2011) 286:42981–91. doi: 10.1074/jbc.M111.310599
44. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. (2010) 16:90–7. doi: 10.1038/nm.2069
45. Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe. (2014) 15:792–8. doi: 10.1016/j.chom.2014.05.003
46. Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol. (2005) 170:21–6. doi: 10.1083/jcb.200502153
47. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. (2017) 214:3687–705. doi: 10.1084/jem.20170558
48. Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe. (2018) 23:191–202.e4. doi: 10.1016/j.chom.2017.12.017
49. Benjamin JL, Sumpter R Jr, Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. (2013) 13:723–34. doi: 10.1016/j.chom.2013.05.004
50. Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. (1995) 81:513–23. doi: 10.1016/0092-8674(95)90072-1
51. Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. (1997) 7:821–30. doi: 10.1016/S1074-7613(00)80400-8
52. Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J Biol Chem. (2003) 278:39251–8. doi: 10.1074/jbc.M305833200
53. Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. (1996) 4:387–96. doi: 10.1016/S1074-7613(00)80252-6
54. Varfolomeev EE, Boldin MP, Goncharov TM, Wallach D. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: proteins binding to the death domains of the two receptors also bind to each other. J Exp Med. (1996) 183:1271–5. doi: 10.1084/jem.183.3.1271
55. Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, et al. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. (1997) 57:615–9.
56. Duan H, Dixit VM. RAIDD is a new 'death' adaptor molecule. Nature. (1997) 385:86–9. doi: 10.1038/385086a0
57. Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. (2000) 254:14–24. doi: 10.1006/excr.1999.4733
58. Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene. (2001) 20:6482–91. doi: 10.1038/sj.onc.1204788
59. Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. (2004) 5:503–7. doi: 10.1038/ni1061
60. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. (2004) 430:694–9. doi: 10.1038/nature02794
61. Kim JW, Joe CO, Choi EJ. Role of receptor-interacting protein in tumor necrosis factor-alpha -dependent MEKK1 activation. J Biol Chem. (2001) 276:27064–70. doi: 10.1074/jbc.M009364200
62. Kurenova E, Xu LH, Yang X, Baldwin AS Jr., Craven RJ, Hanks SK, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. (2004) 24:4361–71. doi: 10.1128/MCB.24.10.4361-4371.2004
63. Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. (1999) 274:16871–5. doi: 10.1074/jbc.274.24.16871
64. Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. (2001) 2:620–4. doi: 10.1038/89769
65. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. (2009) 325:332–6. doi: 10.1126/science.1172308
66. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
67. Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. (1998) 8:297–303. doi: 10.1016/S1074-7613(00)80535-X
68. Cusson N, Oikemus S, Kilpatrick ED, Cunningham L, Kelliher M. The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2-induced cell death. J Exp Med. (2002) 196:15–26. doi: 10.1084/jem.20011470
69. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. (2014) 157:1189–202. doi: 10.1016/j.cell.2014.04.018
70. Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. (2014) 157:1175–88. doi: 10.1016/j.cell.2014.04.019
71. Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. (2011) 471:373–6. doi: 10.1038/nature09878
72. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. (2012) 11:290–7. doi: 10.1016/j.chom.2012.01.016
73. Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. (2014) 513:95–9. doi: 10.1038/nature13706
74. Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. (2014) 192:5476–80. doi: 10.4049/jimmunol.1400499
75. Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. (2014) 193:1539–43. doi: 10.4049/jimmunol.1400590
76. Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. (2004) 24:1464–9. doi: 10.1128/MCB.24.4.1464-1469.2004
77. Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. (2014) 343:1357–60. doi: 10.1126/science.1249361
78. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. (2014) 56:481–95. doi: 10.1016/j.molcel.2014.10.021
79. Leppkes M, Roulis M, Neurath MF, Kollias G, Becker C. Pleiotropic functions of TNF-alpha in the regulation of the intestinal epithelial response to inflammation. Int Immunol. (2014) 26:509–15. doi: 10.1093/intimm/dxu051
80. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
81. Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. (2005) 118(Pt 2):265–7. doi: 10.1242/jcs.01610
82. Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. (1993) 74:845–53. doi: 10.1016/0092-8674(93)90464-2
83. Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. (1995) 81:495–504. doi: 10.1016/0092-8674(95)90070-5
84. Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. (1995) 269:1424–7. doi: 10.1126/science.7544915
85. McElroy SJ, Prince LS, Weitkamp JH, Reese J, Slaughter JC, Polk DB. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: a potential role in neonatal necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. (2011) 301:G656–66. doi: 10.1152/ajpgi.00550.2010
86. Iwashita J, Sato Y, Sugaya H, Takahashi N, Sasaki H, Abe T. mRNA of MUC2 is stimulated by IL-4, IL-13 or TNF-alpha through a mitogen-activated protein kinase pathway in human colon cancer cells. Immunol Cell Biol. (2003) 81:275–82. doi: 10.1046/j.1440-1711.2003.t01-1-01163.x
87. Bruno ME, Frantz AL, Rogier EW, Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor by the classical and alternative NF-kappaB pathways in intestinal epithelial cells. Mucosal Immunol. (2011) 4:468–78. doi: 10.1038/mi.2011.8
88. Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, et al. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. (2003) 284:C953–61. doi: 10.1152/ajpcell.00309.2002
89. Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology. (2002) 122:134–44. doi: 10.1053/gast.2002.30347
90. Hilliard VC, Frey MR, Dempsey PJ, Peek RM Jr., Polk DB. TNF-alpha converting enzyme-mediated ErbB4 transactivation by TNF promotes colonic epithelial cell survival. Am J Physiol Gastrointest Liver Physiol. (2011) 301:G338–46. doi: 10.1152/ajpgi.00057.2011
91. Lin A, Karin M. NF-kappaB in cancer: a marked target. Semin Cancer Biol. (2003) 13:107–14. doi: 10.1016/S1044-579X(02)00128-1
92. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
93. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
94. Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature. (2011) 471:637–41. doi: 10.1038/nature09814
95. Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA. (2004) 101:2452–7. doi: 10.1073/pnas.0306734101
96. Chae S, Eckmann L, Miyamoto Y, Pothoulakis C, Karin M, Kagnoff MF. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J Immunol. (2006) 177:1214–20. doi: 10.4049/jimmunol.177.2.1214
97. Eckmann L, Nebelsiek T, Fingerle AA, Dann SM, Mages J, Lang R, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. (2008) 105:15058–63. doi: 10.1073/pnas.0808216105
98. Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. (2007) 446:557–61. doi: 10.1038/nature05698
99. Kajino-Sakamoto R, Inagaki M, Lippert E, Akira S, Robine S, Matsumoto K, et al. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J Immunol. (2008) 181:1143–52. doi: 10.4049/jimmunol.181.2.1143
100. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. (2010) 28:573–621. doi: 10.1146/annurev-immunol-030409-101225
101. Guma M, Stepniak D, Shaked H, Spehlmann ME, Shenouda S, Cheroutre H, et al. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med. (2011) 208:1889–900. doi: 10.1084/jem.20110242
102. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. (2008) 30:689–700. doi: 10.1016/j.molcel.2008.05.014
103. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. (2008) 105:11778–83. doi: 10.1073/pnas.0711122105
104. Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. (2008) 283:24295–9. doi: 10.1074/jbc.C800128200
105. Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. (2004) 21:415–28. doi: 10.1016/j.immuni.2004.08.017
106. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. (1999) 13:2514–26. doi: 10.1101/gad.13.19.2514
107. Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. (2007) 19:2056–67. doi: 10.1016/j.cellsig.2007.05.016
108. Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. (2002) 277:45162–71. doi: 10.1074/jbc.M206882200
109. O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. (2011) 13:1437–42. doi: 10.1038/ncb2362
110. Feng P, Liang C, Shin YC, Xiaofei E, Zhang W, Gravel R, et al. A novel inhibitory mechanism of mitochondrion-dependent apoptosis by a herpesviral protein. PLoS Pathog. (2007) 3:e174. doi: 10.1371/journal.ppat.0030174
111. Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. (2003) 9:575–81. doi: 10.1038/nm849
112. Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. (2007) 12:445–56. doi: 10.1016/j.ccr.2007.08.029
113. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. (2011) 43:432–48. doi: 10.1016/j.molcel.2011.06.006
114. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. (2008) 133:693–703. doi: 10.1016/j.cell.2008.03.036
115. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. (2013) 20:1381–92. doi: 10.1038/cdd.2013.94
116. Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. (2009) 16:1279–88. doi: 10.1038/cdd.2009.41
117. Garcia-Carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL, et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc Natl Acad Sci USA. (2018) 115:E9192–E200. doi: 10.1073/pnas.1810584115
118. Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J Biol Chem. (2004) 279:15652–61. doi: 10.1074/jbc.M311629200
119. Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. (2005) 174:4942–52. doi: 10.4049/jimmunol.174.8.4942
120. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
121. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. (2009) 137:1100–11. doi: 10.1016/j.cell.2009.05.021
122. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. (2012) 150:339–50. doi: 10.1016/j.cell.2012.06.019
123. Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. (2014) 21:1511–21. doi: 10.1038/cdd.2014.76
124. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
125. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. (2014) 16:55–65. doi: 10.1038/ncb2883
126. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
127. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
128. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. (2008) 4:313–21. doi: 10.1038/nchembio.83
129. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. (2017) 8:14329. doi: 10.1038/ncomms14329
130. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell. (2017) 66:698–710 e5. doi: 10.1016/j.molcel.2017.05.003
131. McQuade T, Cho Y, Chan FK. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem J. (2013) 456:409–15. doi: 10.1042/BJ20130860
132. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. (2013) 39:443–53. doi: 10.1016/j.immuni.2013.06.018
133. Souza HS, Tortori CJ, Castelo-Branco MT, Carvalho AT, Margallo VS, Delgado CF, et al. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: evidence of altered expression of FasL and perforin cytotoxic pathways. Int J Colorectal Dis. (2005) 20:277–86. doi: 10.1007/s00384-004-0639-8
134. Di Sabatino A, Calarota SA, Vidali F, Macdonald TT, Corazza GR. Role of IL-15 in immune-mediated and infectious diseases. Cytokine Growth Factor Rev. (2011) 22:19–33. doi: 10.1016/j.cytogfr.2010.09.003
135. Dourmashkin RR, Davies H, Wells C, Shah D, Price A, O'Morain C, et al. Epithelial patchy necrosis in Crohn's disease. Hum Pathol. (1983) 14:643–8. doi: 10.1016/S0046-8177(83)80207-X
136. Hagiwara C, Tanaka M, Kudo H. Increase in colorectal epithelial apoptotic cells in patients with ulcerative colitis ultimately requiring surgery. J Gastroenterol Hepatol. (2002) 17:758–64. doi: 10.1046/j.1440-1746.2002.02791.x
137. Iwamoto M, Koji T, Makiyama K, Kobayashi N, Nakane PK. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. (1996) 180:152–9.
138. Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. (2011) 477:335–9. doi: 10.1038/nature10400
139. Spehlmann ME, Begun AZ, Burghardt J, Lepage P, Raedler A, Schreiber S. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflamm Bowel Dis. (2008) 14:968–76. doi: 10.1002/ibd.20380
140. Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. (2005) 14:3499–506. doi: 10.1093/hmg/ddi379
141. Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. (2011) 43:246–52. doi: 10.1038/ng.764
142. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. (2010) 42:1118–25. doi: 10.1038/ng.717
143. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
144. Ferguson LR, Han DY, Fraser AG, Huebner C, Lam WJ, Morgan AR. IL23R and IL12B SNPs and haplotypes strongly associate with Crohn's disease risk in a New Zealand population. Gastroenterol Res Pract. (2010) 2010:539461. doi: 10.1155/2010/539461
145. McGovern DP, Jones MR, Taylor KD, Marciante K, Yan X, Dubinsky M, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn's disease. Hum Mol Genet. (2010) 19:3468–76. doi: 10.1093/hmg/ddq248
146. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
147. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. (2001) 411:599–603. doi: 10.1038/35079107
148. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. (2001) 411:603–6. doi: 10.1038/35079114
149. Palomino-Morales RJ, Oliver J, Gomez-Garcia M, Lopez-Nevot MA, Rodrigo L, Nieto A, et al. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes Immun. (2009) 10:356–64. doi: 10.1038/gene.2009.25
150. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. (2007) 39:207–11. doi: 10.1038/ng1954
151. Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. (2001) 2:211–6. doi: 10.1038/35056522
152. Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol. (2013) 4:301. doi: 10.3389/fimmu.2013.00301
153. Di Rocco C, Rende M. Neural tube defects: considerations on the pathogenesis and correlation with myelodysplasia in man. II. An experimental model. Fetal Ther. (1989) 4 (Suppl. 1):99–103. doi: 10.1159/000263473
154. Zhang Q, Kang R, Zeh HJ III, Lotze MT, Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. (2013) 9:451–8. doi: 10.4161/auto.23691
155. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
156. Li W, Zhu S, Li J, Assa A, Jundoria A, Xu J, et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol. (2011) 81:1152–63. doi: 10.1016/j.bcp.2011.02.015
157. Burger E, Araujo A, Lopez-Yglesias A, Rajala MW, Geng L, Levine B, et al. Loss of paneth cell autophagy causes acute susceptibility to toxoplasma gondii-mediated inflammation. Cell Host Microbe. (2018) 23:177–90 e4. doi: 10.1016/j.chom.2018.01.001
158. Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci USA. (2010) 107:14739–44. doi: 10.1073/pnas.1003363107
159. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. (2008) 456:259–63. doi: 10.1038/nature07416
160. Lecine P, Esmiol S, Metais JY, Nicoletti C, Nourry C, McDonald C, et al. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem. (2007) 282:15197–207. doi: 10.1074/jbc.M606242200
161. Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS ONE. (2008) 3:e3391. doi: 10.1371/journal.pone.0003391
162. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. (2007) 39:596–604. doi: 10.1038/ng2032
163. Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, et al. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. (2014) 506:456–62. doi: 10.1038/nature13044
164. Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. (2009) 30:383–91. doi: 10.1016/j.it.2009.05.007
165. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. (2011) 474:307–17. doi: 10.1038/nature10209
166. Opipari AW Jr., Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. (1990) 265:14705–8.
167. Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci USA. (1989) 86:2336–40. doi: 10.1073/pnas.86.7.2336
168. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. (2000) 289:2350–4. doi: 10.1126/science.289.5488.2350
169. De A, Dainichi T, Rathinam CV, Ghosh S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep. (2014) 15:775–83. doi: 10.15252/embr.201338305
170. Lu TT, Onizawa M, Hammer GE, Turer EE, Yin Q, Damko E, et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity. (2013) 38:896–905. doi: 10.1016/j.immuni.2013.03.008
171. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. (2012) 12:774–85. doi: 10.1038/nri3313
172. Wang K, Baldassano R, Zhang H, Qu HQ, Imielinski M, Kugathasan S, et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet. (2010) 19:2059–67. doi: 10.1093/hmg/ddq078
173. Bank S, Andersen PS, Burisch J, Pedersen N, Roug S, Galsgaard J, et al. Associations between functional polymorphisms in the NFkappaB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenomics J. (2014) 14:526–34. doi: 10.1038/tpj.2014.19
174. Vereecke L, Vieira-Silva S, Billiet T, van Es JH, Mc Guire C, Slowicka K, et al. A20 controls intestinal homeostasis through cell-specific activities. Nat Commun. (2014) 5:5103. doi: 10.1038/ncomms6103
175. Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. (2011) 43:253–8. doi: 10.1038/ng.766
176. Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. (2008) 40:1062–4. doi: 10.1038/ng.202
177. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. (2011) 117:2227–36. doi: 10.1182/blood-2010-09-306019
178. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. (2010) 33:181–91. doi: 10.1016/j.immuni.2010.07.017
179. Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. (2010) 207:1513–23. doi: 10.1084/jem.20092474
180. Kolodziej LE, Lodolce JP, Chang JE, Schneider JR, Grimm WA, Bartulis SJ, et al. TNFAIP3 maintains intestinal barrier function and supports epithelial cell tight junctions. PLoS ONE. (2011) 6:e26352. doi: 10.1371/journal.pone.0026352
181. Rhee L, Murphy SF, Kolodziej LE, Grimm WA, Weber CR, Lodolce JP, et al. Expression of TNFAIP3 in intestinal epithelial cells protects from DSS- but not TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G220–7. doi: 10.1152/ajpgi.00077.2012
182. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. (2015) 16:618–27. doi: 10.1038/ni.3172
183. Arsenescu R, Bruno ME, Rogier EW, Stefka AT, McMahan AE, Wright TB, et al. Signature biomarkers in Crohn's disease: toward a molecular classification. Mucosal Immunol. (2008) 1:399–411. doi: 10.1038/mi.2008.32
184. Ohkusa T, Kato K, Terao S, Chiba T, Mabe K, Murakami K, et al. Newly developed antibiotic combination therapy for ulcerative colitis: a double-blind placebo-controlled multicenter trial. Am J Gastroenterol. (2010) 105:1820–9. doi: 10.1038/ajg.2010.84
185. Turner D, Levine A, Kolho KL, Shaoul R, Ledder O. Combination of oral antibiotics may be effective in severe pediatric ulcerative colitis: a preliminary report. J Crohns Colitis. (2014) 8:1464–70. doi: 10.1016/j.crohns.2014.05.010
186. Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, et al. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. (2011) 106:661–73. doi: 10.1038/ajg.2011.72
187. Wang SL, Wang ZR, Yang CQ. Meta-analysis of broad-spectrum antibiotic therapy in patients with active inflammatory bowel disease. Exp Ther Med. (2012) 4:1051–6. doi: 10.3892/etm.2012.718
188. Halfvarson J, Brislawn CJ, Lamendella R, Vazquez-Baeza Y, Walters WA, Bramer LM, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. (2017) 2:17004. doi: 10.1038/nmicrobiol.2017.4
189. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
190. Casen C, Vebo HC, Sekelja M, Hegge FT, Karlsson MK, Ciemniejewska E, et al. Deviations in human gut microbiota: a novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment Pharmacol Ther. (2015) 42:71–83. doi: 10.1111/apt.13236
191. Putignani L, Del Chierico F, Vernocchi P, Cicala M, Cucchiara S, Dallapiccola B, et al. Gut microbiota dysbiosis as risk and premorbid factors of IBD and IBS along the childhood-adulthood transition. Inflamm Bowel Dis. (2016) 22:487–504. doi: 10.1097/MIB.0000000000000602
192. Sepehri S, Kotlowski R, Bernstein CN, Krause DO. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm Bowel Dis. (2007) 13:675–83. doi: 10.1002/ibd.20101
193. Choung RS, Princen F, Stockfisch TP, Torres J, Maue AC, Porter CK, et al. Serologic microbial associated markers can predict Crohn's disease behaviour years before disease diagnosis. Aliment Pharmacol Ther. (2016) 43:1300–10. doi: 10.1111/apt.13641
194. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. (2004) 127:412–21. doi: 10.1053/j.gastro.2004.04.061
195. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
196. Backert S, Schmidt TP, Harrer A, Wessler S. Exploiting the Gastric Epithelial Barrier: Helicobacter pylori's Attack on Tight and Adherens Junctions. Curr Top Microbiol Immunol. (2017) 400:195–226. doi: 10.1007/978-3-319-50520-6_9
197. Alvarez CS, Badia J, Bosch M, Gimenez R, Baldoma L. Outer Membrane vesicles and soluble factors released by probiotic Escherichia coli Nissle 1917 and Commensal ECOR63 enhance barrier function by regulating expression of tight junction proteins in intestinal epithelial cells. Front Microbiol. (2016) 7:1981. doi: 10.3389/fmicb.2016.01981
198. Chelakkot C, Choi Y, Kim DK, Park HT, Ghim J, Kwon Y, et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med. (2018) 50:e450. doi: 10.1038/emm.2017.282
199. Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, Backer J, et al. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol. (2008) 295:G1025–34. doi: 10.1152/ajpgi.90227.2008
200. Anderson RC, Cookson AL, McNabb WC, Park Z, McCann MJ, Kelly WJ, et al. Lactobacillus plantarum MB452 enhances the function of the intestinal barrier by increasing the expression levels of genes involved in tight junction formation. BMC Microbiol. (2010) 10:316. doi: 10.1186/1471-2180-10-316
201. Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, et al. Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G851–9. doi: 10.1152/ajpgi.00327.2009
202. Qin H, Zhang Z, Hang X, Jiang Y. L. plantarum prevents enteroinvasive Escherichia coli-induced tight junction proteins changes in intestinal epithelial cells. BMC Microbiol. (2009) 9:63. doi: 10.1186/1471-2180-9-63
203. Anderson RC, Cookson AL, McNabb WC, Kelly WJ, Roy NC. Lactobacillus plantarum DSM 2648 is a potential probiotic that enhances intestinal barrier function. FEMS Microbiol Lett. (2010) 309:184–92. doi: 10.1111/j.1574-6968.2010.02038.x
204. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. (2012) 9:599–608. doi: 10.1038/nrgastro.2012.152
205. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. (2014) 41:311–24. doi: 10.1016/j.immuni.2014.06.015
206. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464:59–65. doi: 10.1038/nature08821
207. Dollive S, Chen YY, Grunberg S, Bittinger K, Hoffmann C, Vandivier L, et al. Fungi of the murine gut: episodic variation and proliferation during antibiotic treatment. PLoS ONE. (2013) 8:e71806. doi: 10.1371/journal.pone.0071806
208. Noverr MC, Noggle RM, Toews GB, Huffnagle GB. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect Immun. (2004) 72:4996–5003. doi: 10.1128/IAI.72.9.4996-5003.2004
209. Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD. Gut. (2017) 66:1039–48. doi: 10.1136/gutjnl-2015-310746
210. Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE. (2013) 8:e66019. doi: 10.1371/journal.pone.0066019
211. Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol. (2008) 43:831–41. doi: 10.1080/00365520801935434
212. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. (2012) 336:1314–7. doi: 10.1126/science.1221789
213. Kim MS, Park EJ, Roh SW, Bae JW. Diversity and abundance of single-stranded DNA viruses in human feces. Appl Environ Microbiol. (2011) 77:8062–70. doi: 10.1128/AEM.06331-11
214. Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. (2010) 466:334–8. doi: 10.1038/nature09199
215. Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci USA. (2013) 110:12450–5. doi: 10.1073/pnas.1300833110
216. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. (2015) 160:447–60. doi: 10.1016/j.cell.2015.01.002
217. Wagner J, Maksimovic J, Farries G, Sim WH, Bishop RF, Cameron DJ, et al. Bacteriophages in gut samples from pediatric Crohn's disease patients: metagenomic analysis using 454 pyrosequencing. Inflamm Bowel Dis. (2013) 19:1598–608. doi: 10.1097/MIB.0b013e318292477c
218. Lepage P, Colombet J, Marteau P, Sime-Ngando T, Dore J, Leclerc M. Dysbiosis in inflammatory bowel disease: a role for bacteriophages? Gut. (2008) 57:424–5. doi: 10.1136/gut.2007.134668
219. Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. (2004) 68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004
220. Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci USA. (2013) 110:20236–41. doi: 10.1073/pnas.1319470110
221. Uhr JW, Dancis J, Franklin EC, Finkelstein MS, Lewis EW. The antibody response to bacteriophage phi-X 174 in newborn premature infants. J Clin Invest. (1962) 41:1509–13. doi: 10.1172/JCI104606
222. Gorski A, Wazna E, Dabrowska BW, Dabrowska K, Switala-Jelen K, Miedzybrodzki R. Bacteriophage translocation. FEMS Immunol Med Microbiol. (2006) 46:313–9. doi: 10.1111/j.1574-695X.2006.00044.x
223. Parent K, Wilson ID. Mycobacteriophage in Crohn's disease. Gut. (1971) 12:1019–20. doi: 10.1136/gut.12.12.1019
Keywords: IBD, apoptosis, RIPK, A20, RIPK1, NOD2, autophagy
Citation: Garcia-Carbonell R, Yao S-J, Das S and Guma M (2019) Dysregulation of Intestinal Epithelial Cell RIPK Pathways Promotes Chronic Inflammation in the IBD Gut. Front. Immunol. 10:1094. doi: 10.3389/fimmu.2019.01094
Received: 15 January 2019; Accepted: 29 April 2019;
Published: 20 May 2019.
Edited by:
Teruki Dainichi, Kyoto University, JapanReviewed by:
Marie-Claude Gaudreau, Bristol Myers Squibb, United StatesCopyright © 2019 Garcia-Carbonell, Yao, Das and Guma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Guma, bWd1bWFAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.