 Aleksandar Ivetic
Aleksandar Ivetic Hannah Louise Hoskins Green
Hannah Louise Hoskins Green Samuel James Hart
Samuel James Hart
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 May 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01068
This article is part of the Research Topic Cell-Cell and Cell-Matrix Adhesion in Immunobiology and Cancer View all 10 articles
L-selectin (CD62L) is a type-I transmembrane glycoprotein and cell adhesion molecule that is expressed on most circulating leukocytes. Since its identification in 1983, L-selectin has been extensively characterized as a tethering/rolling receptor. There is now mounting evidence in the literature to suggest that L-selectin plays a role in regulating monocyte protrusion during transendothelial migration (TEM). The N-terminal calcium-dependent (C-type) lectin domain of L-selectin interacts with numerous glycans, including sialyl Lewis X (sLex) for tethering/rolling and proteoglycans for TEM. Although the signals downstream of L-selectin-dependent adhesion are poorly understood, they will invariably involve the short 17 amino acid cytoplasmic tail. In this review we will detail the expression of L-selectin in different immune cell subsets, and its influence on cell behavior. We will list some of the diverse glycans known to support L-selectin-dependent adhesion, within luminal and abluminal regions of the vessel wall. We will describe how each domain within L-selectin contributes to adhesion, migration and signal transduction. A significant focus on the L-selectin cytoplasmic tail and its proposed contribution to signaling via the ezrin-radixin-moesin (ERM) family of proteins will be outlined. Finally, we will discuss how ectodomain shedding of L-selectin during monocyte TEM is essential for the establishment of front-back cell polarity, bestowing emigrated cells the capacity to chemotax toward sites of damage.
The human L-selectin gene (sell) is located on the long arm of chromosome 1 (1q24.2), and is arranged in tandem with its family members (in the order: L-, P-, and E-selectin). L-selectin consists of ten exons spanning a region of ~21.0 kb. FOXO1 regulates transcription of the human sell gene (1, 2), whereas chromosome immunoprecipitation experiments identify other transcription factors (Mzf1, Klf2, Sp1, Ets1, and Irf1) in regulating the mouse sell gene (3). Splicing of the exons into mature mRNA is translated to form a protein product with a predicted molecular weight of 30 kDa. However, the actual molecular weight of L-selectin differs between cell types—ranging from 65 kDa in lymphocytes to 100 kDa in neutrophils and is due to cell type-specific glycosylation (4–10). It is highly likely that altered glycosylation patterns in L-selectin could dictate cell-specific functions, but this has not been explored in any detail. L-selectin is organized into distinct structural domains: a ligand binding calcium-dependent (C-type) lectin domain (CTLD), an EGF-like domain, two complement-like repeat sequences and an extracellular cleavage site (Figure 1A).
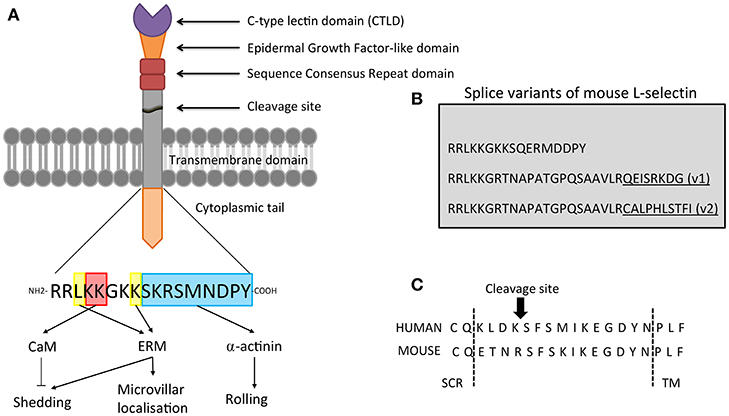
Figure 1. Domain organization of L-selectin. (A) L-selectin is a type I transmembrane glycoprotein. Going from N-terminal to C-terminal, it is broken down into: C-type lectin domain (CTLD), Epidermal Growth Factor (EGF)-like domain, two sequence consensus repeat (SCR) domains, a cleavage site, transmembrane domain and a 17 amino acid cytoplasmic tail. Amino acid sequence of the cytoplasmic tail of human L-selectin is depicted, highlighting the amino acids that support binding to calmodulin (CaM), ERM proteins, and alpha-actinin. (B) The three sequences correspond to the mouse L-selectin tail (note the mouse L-selectin tail has a single serine at position 364, whereas the tail of human L-selectin possesses an extra serine residue at position 367). Sequence conservation at the membrane-proximal region that support binding to ERM and CaM (RRLKKG) is 100% conserved. The amino acid sequences of two splice variants of mouse L-selectin (v1 and v2) are provided in the sequences below. Underlined residues represent sequences that are unique to v1 and v2. (C) Amino acid sequences surrounding the cleavage site of human and mouse L-selectin. Arrow indicates the position of cleavage. TM, transmembrane domain; SCR, sequence consensus repeat region.
Splice variants of L-selectin have been identified and characterized in both mice (11) and humans (12). The mouse sell gene is composed of 9 exons. The two splice variants, termed L-selectin-v1 and L-selectin-v2, possess an additional exon, nested between exons 7 and 8. The splice variants share the first 49bp sequence, whereas L-selectin-v2 extends for an extra 51bp that is immediately 3' to this region. Both splice variants possess longer cytoplasmic tails (WT = 17 aa; v1 = 30 aa; v2 = 32aa—see Figure 1B). The overall mRNA levels of L-selectin-v1 and -v2 constitute 2–3% of the total L-selectin mRNA, so its impact in endogenous leukocyte trafficking and signaling is not fully understood. However, over-expression of these variants in cells lacking L-selectin reveal altered capacities in adhesion to sLex under flow, ectodomain shedding in response to cellular activation, and signaling to p38 MAPK following antibody-mediated clustering (AMC) (11).
The human splice variant lacks exon 7, which codes for the transmembrane domain (12), so transcripts lacking this exon are secreted and soluble. Patients with rheumatic disease presented an increased prevalence of the splice variant transcript, which is thought to contribute to the increase in soluble L-selectin. Based on the relatively low abundance of the variant transcript, it is currently not clear what proportion of circulating soluble L-selectin is represented by either the cleaved form (e.g., through “basal shedding”—see later) or the spliced transmembrane-less forms.
The identification of human neonatal bone marrow CD10−CD62Lhi cells reveals that L-selectin is one of the earliest surface markers to be expressed on lymphoid-primed hematopoietic stem cells (13). Similar observations have been made in mice, suggesting L-selectin plays a pivotal role in stem cell trafficking and differentiation (14, 15). CD10−CD62Lhi cells can differentiate into B cells, dendritic cells, monocytes, NK cells and T cells. It is likely that the L-selectin expressed in these early progenitor cells is required for trafficking from the bone marrow toward peripheral lymphoid organs. Whether L-selectin-dependent signaling also contributes to subsequent differentiation is not known.
The average leukocyte will express ~50,000–70,000 molecules of L-selectin at the plasma membrane (16, 17). Numerous reports have shown that L-selectin is anchored on finger-like projections called microvilli, which increases tethering efficiency during recruitment (18–20). Protein expression of L-selectin is constitutive on most circulating leukocytes, and is slowly turned over at the plasma membrane through a process of ectodomain shedding (commonly referred to as “shedding”). A variety of artificial or physiological agonists of cell activation can, within minutes, promote robust L-selectin shedding in numerous leukocyte sub-types (21–24). The zinc-dependent metalloproteinase, a disintegrin and metalloproteinase (ADAM)17, is the major enzyme responsible for L-selectin shedding in leukocytes (25–28). However, ADAMs 8 and 10 have also been reported to cleave L-selectin in specific settings (29, 30). From a clinical perspective, the soluble circulating form of L-selectin (released as a consequence of ectodomain shedding) is sometimes used as a surrogate plasma/serum biomarker for leukocyte activity triggered during acute or chronic inflammation (31–35). Similarly, leukocytes expressing low levels of surface L-selectin (judged by flow cytometric analysis) are a classic indicator of cellular activation (24, 34). Paradoxically, a drop in soluble L-selectin can also be detected in certain diseases, such as sepsis (36). The drop in detectable soluble L-selectin could be due to its adsorption to luminal vascular ligands that are upregulated during sepsis. Alternatively, L-selectin could be cleaved from transmigrated neutrophils within abluminal/non-luminal regions of vessels (see later). Although soluble L-selectin can compete for cell-associated L-selectin, little is understood about how the two forms buffer leukocyte recruitment during inflammation. Soluble L-selectin is detected in the plasma of healthy humans (0.7-−1.5 μg per mL), suggesting cell-associated L-selectin is cleaved from resting circulating leukocytes at low levels. Indeed, mouse neutrophils lacking ADAM17 express higher than average surface levels of L-selectin (26, 37). This phenomenon is also observed when broad-spectrum synthetic inhibitors of ADAM17 are used, or when the cleavage site of L-selectin is mutated and rendered “sheddase-resistant” (38–40). The manner in which L-selectin expression is regulated, either at the translational or post-translational level, will be unique in different leukocyte subsets. For example, the lifespan of a central memory T-cell (TCM) far exceeds the lifespan of a neutrophil (i.e., 6 weeks compared to 8 days, respectively) (41, 42). Aging neutrophils possess waning levels of L-selectin, which contrasts with TCM (43–45). The sections below provide a few examples of how L-selectin is expressed in different leukocyte subsets and the impact that this might have on immune cell behavior.
Lymphocytes constitute ~78–88% of the total white blood cell count in mice. These values are quite different in humans (20–40%). L-selectin-deficient mice expose the absolute requirement for L-selectin in the trafficking of naïve T-cells to lymph nodes (46). Immunofluorescence imaging of frozen mouse lymph node sections reveals that L-selectin is cyclically expressed in recirculating naïve T-cells as they adhere to and extravasate across high endothelial cell (HEC) monolayers and enter the lymph node parenchyma (47). These observations would suggest ectodomain shedding is triggered during adhesion and TEM, and that re-expression of L-selectin occurs upon their entry into the lymphatics, via the thoracic duct. Increasing L-selectin expression in naïve T-cells does not enhance homing of T-cells to lymph nodes, suggesting that L-selectin densities can reach saturation within specific plasma membrane domains (e.g., on microvillar tips) (48).
Signaling downstream of the T-cell receptor triggers the activation of PI3Kδ, which leads to ectodomain shedding and concomitant inhibition of Klf2-dependent expression of L-selectin in mouse T-cells (49). The terminal differentiation of naïve T-cells into effector memory T-cells (TEM) involves both L-selectin shedding and the transcriptional shutdown of the L-selectin gene. Collectively, these events drive the trafficking of TEM away from secondary lymphoid organs toward peripheral tissues (39, 40). TCM are smaller in size and have longer telomere lengths than TEM, but it is not clear if the respective presence or absence of L-selectin expression contributes to the phenotype (45). Expression of L-selectin in TCM is essential for trafficking toward, and the immune surveillance of, peripheral lymph nodes (39, 40).
Regulatory T-cells (Tregs) play important immunosuppressive roles in diverse immune responses in health and disease [e.g., graft-vs.-host disease (50)]. Tregs are characterized by the presence of the transcription factor FoxP3, which can be further subdivided according to the expression of surface epitopes, including L-selectin. Tregs expressing L-selectin have been shown to have superior immunosuppressive roles in different disease models (51–54). Again, it is not clear if L-selectin-dependent trafficking and/or signaling contributes to improved immunosuppressive function.
The surface level of L-selectin is significantly reduced in naïve CD4 T-cells following HIV entry (55–59). The HIV accessory protein, Vpr, can increase L-selectin mRNA expression in primary human naïve T-cells and Jurkat T-cells, but its impact on the pathobiology of HIV is unclear (59). In contrast, the other accessory proteins, Nef and Vpu, can sequester L-selectin into subcellular compartments and decrease cell surface expression (57, 60). Such post-translational mechanisms of reducing L-selectin surface expression by HIV is thought to prevent trafficking back to secondary lymphoid organs, redirecting infected T-cells to the systemic circulation to increase viremia. L-selectin expression on naive CD4 T-cells has recently been defined as a novel receptor for HIV entry (58), and work from this study reveals that L-selectin shedding ensues 4–6 days post-infection. Moreover, HIV preferentially infects TCM over TEM cells – positively correlating with L-selectin expression. Blocking ectodomain shedding of L-selectin with (broad-spectrum) synthetic inhibitors to ADAM17 (BB-94, TAPI-1, TAPI-2) does not increase HIV attachment or entry, but reduces the release of viral particles (58). These findings suggest that blocking ectodomain shedding of L-selectin may alter membrane/cytoskeleton dynamics that are dependent on the release of HIV particles from infected T-cells.
One study has recently shown that increasing L-selectin expression in cytotoxic CD8 T-cells is causal to viral clearance (61). L-selectin expression is rapidly lost when antigen-presenting cells prime naïve recirculating CD8 T-cells in lymph nodes. Re-expression of L-selectin is detected when antigen-primed CD8 T-cells egress the lymph node, which is now known to be essential for trafficking toward visceral or mucosal virus-infected organs. Bestowing antigen-primed CD8 T-cells with higher levels of L-selectin, by genetically rendering the molecule non-cleavable, increases clearance of viral-infected sites without obvious signs of altering cytokine secretion profiles or clonal expansion. Interestingly, L-selectin shedding in tumor antigen-primed human CD8 TCM cells inversely correlates with the upregulation of the degranulation marker, CD107a, and enhanced tumor lytic activity (62). It is not exactly clear how L-selectin contributes to tumor or virus killing, and whether conserved mechanisms exist between mice and humans. Previous studies have shown that L-selectin clustering can augment T-cell receptor signaling, suggesting roles beyond just trafficking (63, 64).
Although L-selectin can act as a co-stimulator of specific leukocytic responses, it is much harder to model this experimentally without the prior stimulation of L-selectin—e.g., via AMC. In this regard, AMC of L-selectin on mouse splenic T-cells (CD4 and CD8) and B-cells can increase their responsiveness to the chemokine CCL21, via CCR7 (65). Interestingly, the density of surface-expressed L-selectin within different T-cell subsets (i.e. CD62LloCD44hi [TEM] or CD62LhiCD44lo [TCM] subsets within CD4 cells, or CD8 cells) positively correlated with enhanced chemotaxis toward CCL21. This model suggests that clustering of L-selectin by endothelial expressed ligands (e.g., within HEV) can facilitate the transmigration of T-cells into lymph nodes. Spleen tyrosine kinase (Syk) was shown in this study to have in vivo importance in this mechanism.
Manipulating cell surface levels of L-selectin on leukocytes can impact the pathogenesis of atherosclerosis. Mice lacking L-selectin will develop accelerated atherosclerosis (66), suggesting that L-selectin-dependent trafficking to atherosclerotic lesions is somehow protective. Flow cytometric phenotyping of immune cell infiltrates within aortae of CD62L+/+ApoE−/− and CD62L−/−ApoE−/− mice, revealed that two B-cell subsets, B1a and Bregs, provided an anti-inflammatory role in the pathogenesis of atherosclerosis (67). As atherosclerotic lesions mature, the microvascular network of the vasa vasorum can innervate into late developing lesions – acting as a portal for increased leukocyte trafficking (via “the back door”) into lesions (68). Moreover, the increased microvascular density within late developing regions is deemed a predicator for unstable plaque rupture (69, 70). Endothelial cells lining the microvessels within these late lesions were shown to upregulate P-selectin proteoglycan ligand-1 (PSGL-1), a known ligand for L-selectin (68). The data, collectively, suggests that L-selectin-dependent recruitment of leukocytes into early atherosclerotic lesions is protective, but, as the disease progresses, L-selectin-dependent recruitment of leukocytes can be pathogenic.
Human neutrophils represent 50–70% of the total white blood cell count, which contrasts with values of 10–25% in mice (71). L-selectin regulates neutrophil trafficking to sites of inflammation, but our understanding of the intracellular mechanisms underpinning this process is still incomplete. The first in vitro transmigration experiments involved the addition of primary human neutrophil suspensions onto cytokine-activated monolayers of human umbilical vein endothelial cells (HUVEC). In such assays, L-selectin is rapidly cleared from the neutrophil plasma membrane through ectodomain shedding (72). L-selectin shedding is a classic readout for neutrophil activation and/or “priming” (i.e., partial activation), which is concordant with an upregulation in CD11b expression (αMβ2 or Mac-1) (24). Blocking L-selectin shedding in human neutrophils with broad-spectrum inhibitors for ADAM17 (e.g. Ro-31- 9790, KD-IX-73-3, TAPI-0, TAPI-1, TMI005, and GM6001) does not impact rolling velocity or the kinetics of TEM (72), which is contrary to what has been observed in mice (73).
AMC of L-selectin on human neutrophils can result in its own ectodomain shedding (74), acting as a self-limiting mechanism by which L-selectin can behave as a cell adhesion molecule and signaling receptor. Prolonged perfusion of neutrophils over immobilized sLex can increase rolling velocity over time, which is directly due to L-selectin shedding. The activation of p38 MAPK drives L-selectin shedding during rolling, and the term “mechanical shedding” was coined to explain this phenomenon (75). Mechanical shedding has only been characterized in human neutrophils rolling on sLex, but blocking L-selectin shedding in mice with KD-IX-73-3 also reduces neutrophil rolling velocities (73, 76). These studies collectively indicate that mechanical shedding could have in vivo significance. However, it should be noted that L-selectin on human neutrophils is decorated itself with sLex, acting as a ligand for E-selectin (8, 9), so this would invariably lead to altered mechanisms of L-selectin-dependent recruitment of human neutrophils (see later).
A recent study has challenged the view that L-selectin shedding is a classic outcome of neutrophil transmigration (77). Neutrophils harvested from synovial fluid of arthritic patients registered L-selectin positive and showed little signs of priming. L-selectin shedding is therefore not a strict prerequisite for neutrophil emigration into inflamed tissue. Interestingly, neutrophils aspirated from the fluid of acutely formed skin blisters were shown to be primed (e.g., CD11b-positive) and L-selectin-negative. These observations suggest that the mechanisms of neutrophil migration toward acute or chronic inflammation could differ in the expression and turnover of adhesion molecules. Indeed, chemokine receptor expression on the neutrophils and the array of cytokines and chemokines in synovial vs. blister fluid microenvironments appear to be radically different (77).
L-selectin-null mice display significant reductions in neutrophil recruitment to a 4 h model of thioglycollate-induced peritonitis (46). Monitoring the rolling flux of neutrophils in post capillary venules of exteriorised cremasteric muscle (induced by surgical trauma) showed no differences between L-selectin-null mice and WT controls at early time points. However, a significant reduction in neutrophil rolling flux of L-selectin-deficient neutrophils was detected after 50 min of cremaster exteriorisation. Superfusing exteriorised cremasteric tissue with platelet activating factor (PAF), a potent neutrophil chemoattractant, showed only modest reductions in the rolling flux of L-selectin-null neutrophils (78). This observation supports the idea that L-selectin ligands might be absent in endothelial cells lining cremasteric post-capillary venules (79). Although no difference in neutrophil adhesion to post-capillary venules of either WT or L-selectin-null mice was observed, neutrophil emigration from post-capillary venules was significantly reduced in neutrophils lacking L-selectin. Moreover, interstitial chemotaxis toward a gradient of the chemokine CXCL1 (or Keratinocyte-derived Chemoattractant (KC)—another potent neutrophil chemoattractant) was severely impaired in the small proportion of emigrated neutrophils (78). These findings led to the hypothesis that L-selectin may regulate neutrophil chemotaxis, suggesting extended roles beyond tethering and rolling. Neither of these studies explored whether L-selectin was shed in response to PAF superfusion or chemotaxis toward KC. Interestingly, subjecting neutrophils that express non-cleavable L-selectin to a model of KC-induced interstitial chemotaxis phenocopied the behavior witnessed in neutrophils lacking L-selectin. These results seem counterintuitive, but suggest that the membrane-retained fragment (MRF - i.e., the “stump” of L-selectin retained after shedding) may be responsible for guiding neutrophil interstitial chemotaxis, as the MRF can only be processed from WT L-selectin.
The migration of circulating neutrophils into the inflamed peritoneum results in a significant reduction in L-selectin expression, matching what has been observed in vitro with human neutrophils crossing activated HUVEC monolayers (72). Knock-out studies reveal that this mechanism is through ectodomain shedding via ADAM17 (26). Mouse neutrophils lacking ADAM17 express 10 times more surface L-selectin, but will still lose a significant amount of this when entering the peritoneum (26). Whether the observed loss is through the action of a related ADAMs protease member (e.g., ADAM10 or ADAM8) (29, 30) or through the loss of membrane fragments (e.g., exosomes) has not been fully determined. The higher L-selectin expression in ADAM17-null neutrophils drives slower rolling velocities in inflamed cremasteric post-capillary venules.
Neutrophil aging in the circulation leads to the progressive increase in CXCR4 expression, with a corresponding decrease in L-selectin expression (80). An increase in CXCR4 expression is required for homing of aged neutrophils to the bone marrow for macrophage-mediated clearance (81). However, it is not clear if the reduced L-selectin expression is causal to, or an epiphenomenon of, neutrophil aging. Furthermore, it is not clear if the reduction in L-selectin expression is at the translational or post-translational level (e.g., increased basal shedding or exosome-like mediated release).
The impact of L-selectin expression on neutrophil behavior has been extensively reviewed elsewhere (82).
Monocytes constitute ~2–8% of all circulating white blood cells in the adult human, which contrasts with 0.9–1.5% in mice. Within the human population, a further three monocyte subsets are characterized according to the relative abundance of two surface markers: CD14++ CD16− or “classical” (80–95%), CD14++ CD16+ or “intermediate” (2–11%), and CD14+ CD16++ or “non-classical” (2–8%). Classical monocytes express high levels of L-selectin, which is in stark contrast to the lesser abundant intermediate and non-classical monocytes (83).
L-selectin mediates the recruitment of human monocytes to activated endothelial monolayers under flow conditions (84, 85). Subsequent experiments showed that L-selectin/PSGL-1 interactions in trans are responsible for the adhesion of monocytes through the process of secondary tethering and rolling (i.e., the interaction of bystander leukocytes with already adherent leukocytes) (86). Imaging of L-selectin on classical monocytes crossing TNF-α-activated endothelial monolayers shows that L-selectin shedding is triggered specifically during TEM and not during earlier phases of the multi-step adhesion cascade (87). These observations challenge current perceptions of L-selectin shedding occurring during the phase of “firm adhesion” in the multi-step adhesion cascade. Two recent articles suggest L-selectin plays a role in regulating monocyte pseudopod protrusion during TEM (87, 88). Interestingly, expression of L-selectin in monocyte-like THP-1 cells (which do not express endogenous L-selectin) bestows them with a more invasive phenotype (i.e., possessing a higher propensity to protrude through inflamed endothelial monolayers) (87, 88).
The secreted mucin AgC10, released from the parasite Trypanosoma cruzi, can bind to L-selectin on human monocytes, which over a 4–6 h period can induce a reduction in L-selectin expression via ectodomain shedding (89). Consequently, monocytes treated with AgC10 have a reduced capacity to interact with activated endothelial monolayers under shear stress conditions.
The equivalent of classical monocytes in mice is characterized by: CX3CR1low, CCR2+, Ly6C+, and CD26L+. On the other hand, “patrolling monocytes” are considered to be the mouse equivalent of non-classical monocytes (CX3CR1hi, CCR2−, Ly6C−, CD62L−), which will typically remain in the circulation and patrol the endothelium for infection and/or damage (90). In both human and mice, the classic monocytes share L-selectin and CCR2 expression. It would be interesting to test if the expression of L-selectin in patrolling monocytes would bestow a more pro-invasive phenotype.
Invading human cytotrophoblasts, which are tasked to bridge a gap between the placenta and the uterus, are thought to employ L-selectin to mediate interstitial migration (91, 92). These findings further support extended roles for L-selectin beyond classic leukocyte tethering and rolling—moreover within microenvironments that are devoid of hydrodynamic shear stress.
Using a mouse model of thioglycollate-induced peritonitis, researchers showed that the turnover of L-selectin is radically different between monocytes and neutrophils: L-selectin expression drops in emigrated neutrophils, but remains unchanged in monocytes (26). The differences underpinning these L-selectin dependent and independent modes of leukocyte emigration are not understood, nor are their relevance to human monocyte TEM (87, 88).
The exposure of isolated human monocytes to selenium promotes metalloproteinase-dependent shedding of L-selectin (93), leading to reduced adhesion on immobilized ligand under flow conditions. Furthermore, the observed shedding could be reversed using synthetic inhibitors of ADAM17 (i.e., GM6001). Administering selenium to mice led to a significant increase in soluble circulating L-selectin. Reducing agents such as dithiothreitol have been shown to increase ADAM17 activity (94), but whether the anti-oxidant properties of selenium could be directly/indirectly linked to reducing cysteines on ADAM17 has not been explored.
L-selectin shares a number of common extracellular domains with its family members, E-, and P-selectin (Figure 1A). In contrast, the cytoplasmic tails of the selectin family members bear no resemblance to one another and likely transduce unique intracellular signals. In this section we will discuss the roles that each domain plays with respect to adhesion, signaling, and ectodomain shedding.
L-selectin possesses an N-terminal CTLD (95), which belongs to a large superfamily of metazoan proteins with diverse functions (96, 97). The CTLD of L-selectin directly regulates leukocyte tethering and rolling by interacting with the minimal determinant tetrasaccharide, sLex (98). A unique feature of the selectins is to stabilize bond lifetimes through conformational changes in the CTLD that occurs in response to an external force, such as hydrodynamic shear stress. Mutagenesis of E-selectin showed the existence of coordinate bonds between Ca2+ and amino acid residues within the upper face of the CTLD stabilizes the interaction (99, 100). Note that these residues are also common to L-selectin. The 3- and 4-hydroxyl groups of fucose in sLex also form a coordinate bond to Ca2+, which collectively stabilize selectin/ligand binding (101). Binding of L-selectin to sLex occurs within a critical threshold of shear stress (typically between 0.3 and 1.0 dynes per cm2). L-selectin binding obeys a ‘catch' and ‘slip' bond mechanism, where optimal shear stress conditions can expose more of the ligand-binding domain and increase adhesiveness. The bond ‘slips' as the tethered cell maneuvers over the ligand-bound site, where an increase in tensile force eventually exceeds the threshold of the catch-bond (102). The catch-slip bond phenomenon is described in more detail elsewhere, elaborating on the existence of a triphasic (slip-catch-slip) behavior of binding (103–105).
X-ray crystallographic evidence of the selectins reveals that the CTLD folds onto a region of the EGF-like domain, linked by a hydrogen bond between Y37 and N138 (101, 106). This inter-domain interaction constrains L-selectin into a less adhesive conformation whilst in the circulation. The functional significance of this interaction was characterized in neutrophils, where uncoupling the Y37/N138 hydrogen bond increases the bond lifetime, which manifests in increased neutrophil priming within the circulation (107). Neutrophils bearing an N138G knock-in mutation within L-selectin displayed enhanced bacterial killing and worsened outcomes in models of sterile injury. Both of these phenotypes were directly linked to the increased priming of neutrophils, confirming causality to the knock-in mutation. It has been suggested that this mode of “mechanochemistry” exists in other immune cells that express L-selectin (107).
All three selectin family members possess a varying number of SCRs (in humans L-selectin has 2, E-selectin has 6, and P-selectin has 9), which bear homology to complement regulatory proteins. SCRs are also termed “sushi domains” and are present in a number of different cell adhesion molecules (108). In respect of the selectins, the SCR serves to distance the CTLD from the plasma membrane, placing it in a strategically advantageous position, reaching out beyond other cell adhesion molecules, to support tethering and rolling behavior. L-selectin is thought to have fewer than its family members as it is anchored to microvilli, which bestows advantageous positioning for tethering under flow.
L-selectin undergoes ectodomain shedding at a specific membrane-proximal location, eleven amino acids above the transmembrane domain—between positions K321 and S322 (109) (see Figure 1C). Alanine scanning mutations surrounding the cleavage site would suggest that there is redundancy in ADAM17 recognition of the cleavage site. Deletion of multiple amino acids suggests that the actual distance of the cleavage site from the plasma membrane is more important (110). Studies in ADAM17 knock-out mice reveal the accumulation of L-selectin on the surface of neutrophils and monocytes, strongly suggesting that both the turnover and induced ectodomain shedding is mediated by a similar enzyme. Due to the relaxed specificity of the L-selectin cleavage site, it is difficult to definitively state that both basal and activated shedding target K321/S322. Binding of calmodulin to the L-selectin cytoplasmic domain is thought to regulate the conformation of the cleavage site (see later), where binding confers protection from shedding and dissociation renders susceptibility to proteolytic attack (111).
Swapping the transmembrane domain of L-selectin with that of CD44 has been shown to alter the subcellular location away from microvilli and toward the cell body, suggesting that amino acids within this region are responsible for the anchoring L-selectin to microvilli (112, 113). It's currently not understood what the amino acids within the transmembrane domain might interact with to retain L-selectin in microvilli, but intramembrane proteases [such as presenilins (114)] could be involved. For example, CD44 is a known target of γ-secretase (115–117), the active site of which is resident within the plasma membrane. It's currently not known if γ-secretases can bind to CD44 without cleaving it and whether its interaction could influence its subcellular localization on the plasma membrane. More subtle mutations within the L-selectin tail, which have been shown to abrogate ERM binding, can phenocopy the microvillar displacement (20).
The tail of L-selectin is only 17 amino acids long, yet it has been documented to bind up to 6 intracellular proteins, which include: alpha-actinin (118), calmodulin (111), ezrin, moesin (119), protein kinase C (PKC) isozymes (120) and μ1alpha/AP-1 (121). Given the size of the L-selectin tail, not all of these proteins can bind simultaneously, but are likely to interact under tight spatio-temporal constraints—e.g., during tethering, rolling, firm adhesion, and TEM. More of this will be discussed in the section below.
Deleting the C-terminal 16 amino acids of the L-selectin tail can dramatically impact its lateral mobility along the plane of the plasma membrane, brought about by a lack of anchoring to the underlying cortical actin-based cytoskeleton (122). Such large truncations can disrupt tethering dynamics under flow conditions (123) and reduces the efficiency of L-selectin shedding (38, 110, 124).
The L-selectin tail is highly basic, with a pI of approximately 11.0, increasing its attraction toward negatively charged phospholipids within the inner leaflet of the plasma membrane. Recent evidence, both in vitro and in silico, support the view that the tail of L-selectin folds onto the inner leaflet of the plasma membrane to form an “L-shaped configuration” (125–127). Binding of ERM proteins to the tail of L-selectin is thought to desorb the tail from the inner leaflet of the plasma membrane and render it competent for calmodulin binding (126).
Numerous proteins have been shown to co-precipitate with L-selectin in biochemical experiments (mainly through standard immunoprecipitation or “pull-down” techniques). These observations do not imply direct binding, but may represent proteins belonging to higher-ordered complexes, and would require further validation by interdisciplinary means. As mentioned above, the cytosolic tail of L-selectin is highly basic and can attract false-positive binders from whole cell lysates. Experiments using the L-selectin tail peptide should undertake a “pre-clearing” step, where the exposure of the cell lysate to a scrambled form will attract false binders based on charge without sequence specificity. The sections immediately below list known and validated binding partners of the L-selectin tail.
Alpha-actinin is a classic actin filament cross-linking protein (128). There are four isoforms of alpha-actinin (1 to 4), each expressed from a different gene. Isoforms 1 and 4 are expressed in non-muscle cells and have molecular weights of approximately 100 kDa on polyacrylamide gels. In contrast, isoforms 2 and 3 are expressed in skeletal and cardiac muscle cells. Solid phase binding experiments between smooth muscle-purified alpha-actinin (specifically from chicken gizzard) and L-selectin peptide revealed that the interaction was specific, and further confirmed by immunoprecipitation (118). Deletion of the C-terminal 11 amino acids of L-selectin (called ‘LΔCyto') abrogates interaction with alpha actinin binding (118). Injection of cell lines expressing LΔCyto L-selectin into the circulation of rats revealed a significant reduction in rolling efficiency within inflamed mesenteric venules (129). These studies strongly imply the requirement for the cytoplasmic tail of L-selectin in regulating rolling interactions. However, little is known about how alpha-actinin interacts with L-selectin and whether the interaction is regulated by (serine/tyrosine) phosphorylation of the L-selectin tail or the production of secondary messengers (e.g., Ca2+). Isoforms 1 and 4 of alpha-actinin possess CaM-like EF hands, which can bind Ca2+ and inhibit actin cross-linking activity (128). Interestingly, isoforms 1 and 4 expressed in smooth muscle cells (as in chicken gizzard) exist as EF domain splice variants that are Ca2+-insensitive (128). As Ca2+-sensitive and insensitive forms of alpha-actinin were likely used by Pavalko et al. (118), it is currently unclear if Ca2+ binding to alpha-actinin might play an active role in regulating L-selectin binding. Despite its conserved identity with CaM, the EF-hand of alpha-actinin is not believed to interact with L-selectin as the amino acids that support binding of CaM and alpha-actinin are located at opposite ends of the L-selectin tail (130) (see Figure 1A). Non-muscle alpha-actinin is known to bind to other tails of cell adhesion molecules and is reviewed extensively elsewhere (131).
CaM is an 18 kDa calcium binding protein, which was first identified to bind L-selectin in immunoprecipitation experiments, and subsequently confirmed using in vitro solid-phase binding assays (109). CaM binds constitutively to L-selectin in resting cells. Its interaction is thought to render the membrane-proximal cleavage site resistant to proteolytic attack. Upon cell stimulation, CaM dissociates to promote an allosteric change in the cleavage site to drive ectodomain shedding by ADAM17. Serine 364, but not S367, on human L-selectin has been shown to be responsible for the dissociation of CaM from L-selectin (87). Indeed, mutation of S364 to alanine significantly reduces phorbol myristate acetate (PMA)-induced shedding (132). Biophysical experiments show contradictory models for how CaM interacts with L-selectin. One report suggests that CaM binds to both a membrane-proximal region of the L-selectin tail, and a region within the transmembrane domain (133). This arrangement of binding was shown to be calcium-dependent and is thought to act in a ratchet-like mechanism, bringing the cleavage site close to the plasma membrane and displacing some of the transmembrane domain into the cytosol. However, this hypothetical view is deemed thermodynamically unfavorable. Another report used lipid bilayers to address a similar question and found that CaM binds to L-selectin in an “extended conformation,” leaving one domain available for interacting with other binding partners. Biochemical and molecular modeling analyses confirmed that it is possible for the tail of L-selectin accommodate CaM and another protein (Ezrin-Radixin-Moesin—see section below). These experiments corroborated with the aforementioned biophysical data that hypothesized an extended form of CaM could accommodate ERM (134). Some suggestions of how different binding partners could be configured during different stages of the multi-step adhesion cascade is described elsewhere (135). It is still not clear whether CaM binding to the L-selectin tail occurs in a calcium-dependent or independent manner, and experiments have been able to support both views (130, 134).
The ERM proteins are a 3-member family of cytoskeletal proteins ranging from 75 to 80 kDa. They all contain very similar domains—a globular N-terminal domain, belonging to the band 4.1 ezrin-radixin-moesin (FERM) superfamily, a central alpha-helical domain and an acidic actin-binding domain (see Figure 2A). The primary role of ERM is to serve as membrane cytoskeleton linkers, however it is becoming increasingly apparent that they play essential roles in mediating signal transduction. In respect of cell adhesion molecules, such as L-selectin, ERM binding restricts their lateral mobility across the plane of the plasma membrane. Anchoring cell adhesion molecules to an underlying cortical actin framework would facilitate receptor clustering and support tethering under flow conditions. Over-expression of L-selectin in fibroblasts can drive the formation of filopodia-like extensions (134). Indeed, ERM proteins are actively involved in forming microvilli (143, 144). ERM adopt an autoinhibited folded conformation when inactive and become “open” when in contact with phosphatidylinositol 4,5-bisphosphate (88, 145–148). Phosphorylation of a conserved threonine residue stabilizes the open conformation (149), which can be targeted by a number of serine/threonine kinases (150–154). Dephosphorylation is triggered by chemokine stimulation and Rac activity in T-cells (140–142) (see Figure 2B), however the mechanism remains poorly understood. Moreover, chemokine induced stimulation of T-cells leads to microvillar collapse that is thought to increase membrane contact between T-cells and endothelial cells.
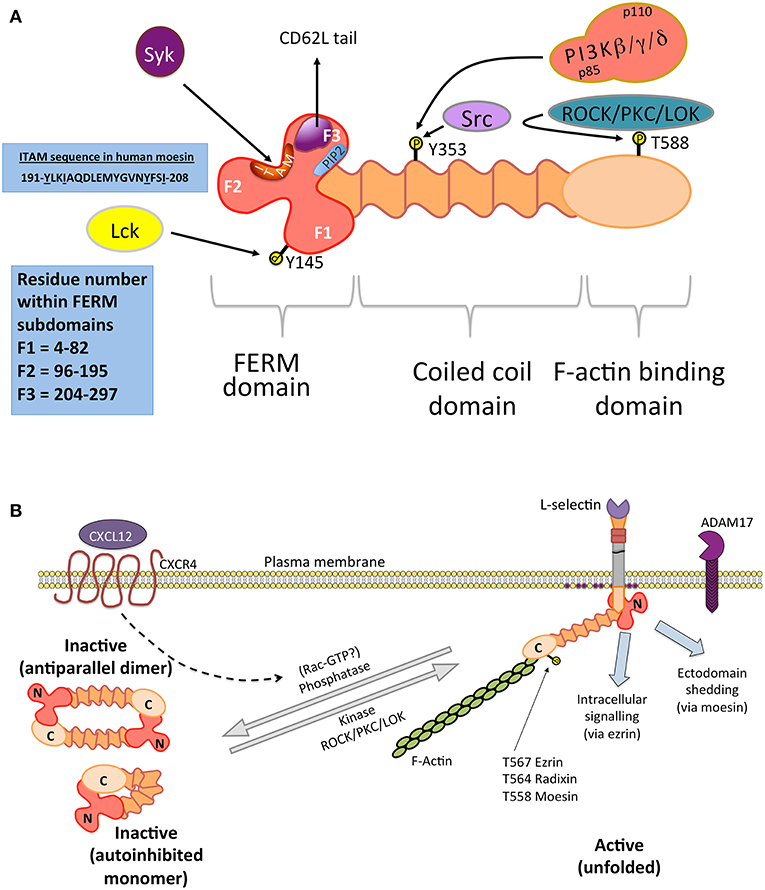
Figure 2. ERM structure and function in immune cells. (A) Domain organization of ERM: X-ray crystallographic studies reveal that the N-terminal FERM domain contains a globular clover leaf-shape (136), classified into 3 distinct subdomains (blue box indicates residue numbers that make up each F subdomain). The FERM domain contains multiple interaction sites: a cryptic ITAM (sequence shown), which can recruit Syk, a PIP2 binding site and a region responsible for binding to L-selectin. The tyrosine kinase Lck is responsible for phosphorylating Y145 of the FERM domain in T-cells (137). Src phosphorylation of Y353 in the central coiled-coil domain of ezrin can lead to the recruitment of class I PI3K, via the p85 regulatory subunit (138). Although originally identified in columnar epithelial cells, confocal microscopic colocalisation of PI3K with phospho-Y353-ezrin is witnessed in T-cell receptor-stimulated T-cells (139). These observations suggest that a PI3K/ezrin interaction is possible in leukocytes. (B) ERM proteins reside in the cytosol as inactivate parallel homodimers or in an autoinhibited conformation. Binding of ERM to PIP2 within the inner leaflet of the plasma membrane (purple circles) alleviates the inactive conformation, allowing the C-terminal domain to interact with filamentous actin (F-actin). In this manner, ERM are typically classified as membrane/cytoskeleton cross-linkers. The kinase activity of serine/threonine kinases (e.g., ROCK, PKC, or LOK) target a conserved threonine residue in all unfolded ERM (T567 in ezrin, T564 in radixin, and T558 in moesin), stabilizing the open conformation. Recent evidence suggests that ezrin might be involved in regulating intracellular signaling, whereas moesin regulates the clustering of L-selectin prior to its ectodomain shedding by ADAM17 (88). One pathway in which ERM are inactivated has been shown in T-cells, where binding of CXCR4 to CXCL12 leads to the rapid dephosphorylation and microvillar collapse of ERM (140). Although not fully understood, Rac is known to act upstream of the ERM phosphatase (141, 142).
ERM proteins can also act as adaptors of signal transduction via a number of different pathways (145, 155). In immune cells, ERM possess a cryptic immunoreceptor tyrosine-based activation motif (ITAM) nested within the FERM domain (156–158) (Figure 2A). Studies have shown that the cryptic ITAM can recruit Syk, which in turn can impact numerous downstream pathways—including the activation of integrins toward an “intermediate” state (159, 160). Ezrin is unique from moesin in that it possesses a tyrosine at position 353 (138), which, when phosphorylated by Src, can recruit the p85 regulatory subunit of PI3K. The conversion of phosphatidylinositol (4, 5)-bisphosphate (PIP2) to phosphatidylinositol (3–5)-trisphosphate (PIP3) is essential for the recruitment for guanine nucleotide exchange factors (GEFs—e.g., Vav), which serve to locally activate the Rho family of RhoGTPases, such as Rac (see later in Figure 4A).
The ERM proteins were first shown to interact with the L-selectin tail using affinity chromatography, where the 17 amino acid tail was synthesized and immobilized onto sepharose beads (119). Clarified extracts, derived from T-cells stimulated with or without PMA, were passed through these affinity columns to identify novel binders in activated and resting cells. Interestingly, moesin only bound to the column if T-cells were stimulated with PMA. In contrast, ezrin bound to the affinity column in a stimulus-independent manner. These results suggest that ezrin and moesin switch in their binding to L-selectin in a stimulus-dependent manner. Fluorescence Lifetime Imaging Microscopy (FLIM) was used to measure Fluorescence Resonance Energy Transfer (FRET) between L-selectin-GFP and ezrin/moesin-RFP, confirming that L-selectin/ezrin interaction was more readily detectable in resting cells than L-selectin/moesin interaction (88). The biological significance underlying these specific interactions is discussed later.
By tagging the tail of human L-selectin with Glutathione-S-Transferase (GST), Kilian et al. discovered that a 60 kDa PKC-like kinase activity could be precipitated in classic “pull-down” experiments from clarified extracts of Jurkat T-cells (120). Further investigations led them to show that at least 3 isozymes of PKC (α, ι, and θ) are able to phosphorylate the tail of L-selectin. PKC-α and PKC-θ bind more readily to serine phosphorylated L-selectin tail. Given that purified PKC-α can directly phosphorylate serine residues on L-selectin strongly supports the view that PKC is a direct binder of the L-selectin tail. As phosphorylation of S364 drives CaM dissociation (87), it is highly likely that serine phosphorylation of the L-selectin tail occurs during or after shedding. The turnover of the MRF (i.e., the cleavage product of L-selectin that remains in the plasma membrane) has been shown to take ~30 min to be fully degraded (161). There is a potential window of opportunity that the MRF/PKC complex may transduce unique intracellular signals. In support of the MRF/PKC complex, clustering of the T-cell receptor (TCR) has been shown to drive L-selectin shedding in a PKC-dependent manner (162). Importantly, increased retention of PKC onto the tail of L-selectin was observed only after TCR stimulation (120).
Metabolic labeling of cell lines expressing L-selectin with radiolabelled pyrophosphate has shown that S364/S367 are indeed phosphorylated following stimulation with chemoattractants (such as C5a and formyl peptides), IgE or PMA (163). Serine-to-alanine mutagenesis of both serine residues does not show any further transfer of radioactive label to L-selectin, suggesting that Y372 is not phosphorylated—at least in response to the agonists tested in this study.
The binding of L-selectin to μ1A—a 45 kDa protein of the clathrin-coated vesicle AP-1 complex—was identified in classic pull-down experiments and later verified by protein-protein interaction (121). Interestingly, binding was only witnessed in response to PMA-induced cell activation. Although the cellular mechanisms have yet to be defined, it is likely that L-selectin/μ1A interaction drives transport of de novo synthesized L-selectin from the trans golgi network toward the plasma membrane. The PMA-induced mode of interaction might suggest that the MRF is internalized by virtue of μ1A binding, once L-selectin shedding is complete. μ1A may therefore be responsible for sorting the MRF toward endosome-like vesicles for degradation. The cyclical expression of L-selectin in recirculating naïve T-cells may also suggest that, instead of L-selectin shedding, full-length L-selectin may be internalized by μ1A and taken to an endosomal recycling compartment for re-expression back to the plasma membrane at a subsequent stage during lymph node trafficking.
Son of sevenless (SOS)/Grb, p56 Lck and the Src family kinases (Fgr, Hck, Lyn, and Syk) can co-precipitate in anti-L-selectin immunoprecipitates and/or pull-down assays (164–166), but this observation does not confer direct interaction. Given that some of these proteins have already been shown to bind to ERM proteins or CaM (134, 137, 156–158, 167), it is plausible that they form indirect, higher-ordered, complexes with the tail of L-selectin. The cytoplasmic tail of L-selectin is highly basic in nature (e.g., the tail of human L-selectin possesses a predicted pI of 11.1). Based on this property, affinity chromatography experiments (such as pull-down assays) can attract false-positive binders if extracts are not pre-cleared with a scrambled peptide control. Such pre-clearing steps eliminate binders based on charge alone and selectively enrich for sequence-specific binders.
Glycans form a fundamental biological interface between adhering/migrating cells and their immediate microenvironment. Moreover, cell adhesion molecules, such as L-selectin, have the capacity to transduce intracellular signals downstream of glycan binding. There is a consensus of thought that unbound L-selectin is monomeric, which is clustered when bound to glycan (87, 134). This mode of glycan binding drives “outside-in” clustering (and signaling). Inside-out clustering of L-selectin can be driven by other input signals (168, 169). Outside-in L-selectin clustering, facilitated by monoclonal antibodies or the elegant engineering of cytoplasmic tail clustering modules, enhances bond lifetime through increased avidity, which can stabilize and/or decrease rolling speed (170–172). Both outside-in and inside-out modes of L-selectin clustering require association with the underlying actin-based cytoskeleton, which is likely to be through the ERM proteins but this has not been formally tested. How inside-out and outside-in modes of L-selectin clustering inter-relate to one another during a specific cellular process (e.g., TEM) has never been explored.
Given the diversity of L-selectin ligands, and their compartmentalisation within anatomically defined locations within vessel walls, the signals running downstream of L-selectin provides information to the leukocyte's position within the multi-step adhesion cascade (tethering, rolling, adhesion, and transmigration). Whilst L-selectin ligands on the apical aspect of the endothelium have long been characterized as tethering and rolling receptors, the glycans that are enriched in the basolateral aspect, and within the basement membrane, are likely to drive completely different signals. The bond lifetime of L-selectin with apical ligands will be within the order of subseconds [due to rapid catch-slip bond dynamics (104)]. In contrast, L-selectin-dependent adhesion within microenvironments devoid of hydrodynamic shear stress (e.g., within transmigrating pseudopods) will extend from seconds to minutes. The glycan scaffold within regions devoid of shear stress can amplify signaling downstream of L-selectin. Moreover, ectodomain shedding can also fine-tune the magnitude of input signals downstream of L-selectin—limiting signals beyond a critical threshold. In support of this view, clustering L-selectin on Jurkat T-cells can promote ectodomain shedding (173), a phenomenon that can be recapitulated in neutrophils when monoclonal antibodies are used to cluster L-selectin (74). Therefore, understanding the nature of L-selectin ligands is important for appreciating how a changing glycan scaffold can impact leukocyte behavior, for example during transmigration. This section will discuss the nature of luminal and abluminal ligands, providing some insight into how they were identified and characterized.
An excellent in-depth review on the biosynthetic pathways of glycosylation can be found in the following reference (174). Suffice to say, the majority of glycoproteins are subjected to either N- or O-linked glycosylation. N-glycosylation refers to the attachment of glycans to the amine nitrogen of asparagine (N) side chains within the N-X-S/T consensus sequence, where “X” is any amino acid except proline. In O-glycosylation, glycans are linked to the hydroxyl oxygen of serine (Ser) or Threonine (Thr) side chains. Sulfotransferases mediate the covalent attachment of sulfate groups to selected saccharide residues within specific oligosaccharide chains and therefore modulate glycan structure and function.
The vital role that L-selectin plays in the recirculation of naïve T-cells (46) led researchers to first explore the glycans presented in secondary lymphoid organs. Classic Stamper Woodruff assays (175) revealed that naïve T-cells would accumulate around vessels of thin frozen lymph node sections, which could be functionally blocked by the anti-L-selectin MEL-14 antibody (51). High endothelial venules (HEVs) are specialized microvessels that are unique to secondary lymphoid organs, allowing the entry of blood-borne naïve T-cells into the lymph node parenchyma (in search for antigen priming by resident dendritic cells). The endothelial cells lining HEVs (called high endothelial cells—HECs) can be isolated and grown in vitro. Ligands for L-selectin have also been purified and characterized from HEC culture supernatants (176).
Sialyl Lewis X (sLex) is the minimal structural determinant for luminal ligands of L-selectin, which is composed of sialic acid, galactose, fucose, and N-acetylglucosamine (Siaα2,3Galβ1,4 (Fucα1,3)GlcNAc) (see Figure 3). sLex is typically O-linked to protein backbones, such as peripheral lymph node addressin (PNAd), CD34, glycosylation-dependent cell adhesion molecule (GlyCAM-1), mucosal vascular addressin cell adhesion molecule-1 (MAdCAM-1), podocalyxin-like protein and spg200 (95, 179–185). Monoclonal antibodies to specific glycan moieties, such as MECA-79 (see below), revealed an apical distribution of these L-selectin ligands in HECs. L-selectin ligands decorating HEV is not solely employed by recirculating naïve T-cells. NK cells are guided to lymph nodes via the same addressin “postcode” to intercept the metastatic spread of B16 melanoma cells to other sites, via local lymph nodes (186, 187). Most recently, neutrophils have been shown to access lymph nodes via the same route as recirculating naïve T-cells, during Staphylococcus aureus infection of the lymphatics. The requirement of PNAd (for L-selectin-dependent adhesion) and platelet-derived P-selectin (interacting with neutrophil P-selectin glycoprotein ligand-1—PSGL-1) were equally critical for trafficking to the infected lymph node (188). Since their identification and characterization in secondary lymphoid organs, L-selectin ligands have also been identified in tertiary lymphoid organs surrounding solid tumors (189, 190) and in microvessels within rejecting kidney (191) and heart (192) allografts.
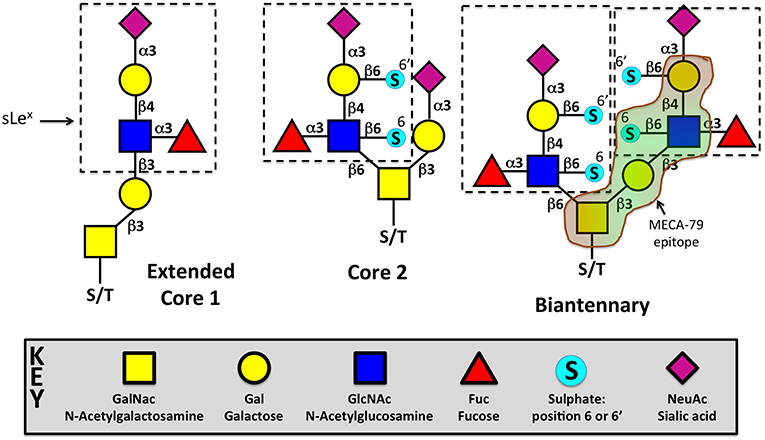
Figure 3. Different glycoforms of O-linked sialyl Lewis X (sLex). sLex can exist in multiple configurations: extended core 1, core 2, or both (biantennary). Sulfotransferases can attach sulfate esters to Gal or GlcNAc, respectively at positions 6 or 6′. Sulfates can be added to sLex one or both positions in either extended core 1, core 2, or the biantennary configuration. Recognition of the MECA-79 epitope is provided in the biantennary structure, but can exist in any of the 3 glycoforms. N-linked sLex has also been shown to act as bonafide L-selectin ligands, but are not depicted in this figure. More information can be found in a short concise commentary (177). The glycoforms are drawn in accordance with GlycanBuilder (178). S/T = serine/threonine.
The pan-selectin ligand, PSGL-1, is typically expressed on the surface of leukocytes to mediate a range of complex interactions with other leukocytes, platelets or endothelial cells (193, 194). However, endothelial-specific PSGL-1 expression has been reported in microvessels sprouting into advanced atherosclerotic lesions (68), chronic murine ileitis (195) and benign prostatic hyperplasia (196). These examples of atypical PSGL-1 expression represent unique sites for L-selectin-dependent leukocyte trafficking. PSGL-1/L-selectin interactions were first characterized to support secondary tethering and rolling between bystander leukocytes and adherent leukocytes (197). There is some debate over whether L-selectin ligands are expressed at all in the inflamed post-capillary venules of the cremasteric muscle, and whether L-selectin-dependent recruitment is purely through secondary tethering and rolling on adherent leukocytes or deposited exosome-like structures (79, 198, 199).
sLex can be linked to lipids, N- or O-linked glycans. The most comprehensively characterized biosynthetic pathway of L-selectin ligands is O-linked sLex, which will be discussed below in further detail. A concise and excellent review on N-linked sLex is provided in the following review (177).
O-linked sLex is decorated on protein backbones as single or biantennary branches that are respectively called “core-1 extended” and “core-2” glycans (Figure 3). Both branches extend from N-acetylgalactosamine (GalNac), which itself is covalently attached to a serine/threonine residue by polypeptide N-acetylgalactosamine transferase. sLex can be modified by the addition of a sulfate ester on C-6 of N-Acetylglucosamine (GlcNAc), which is specific to extended Core 1 branches, or C-6 on Galactose (Gal) of Core 2 branches—respectively termed 6-sulfo or 6'-sulfo (see Figure 3). GlcNAc-6-sulfo is recognized by the MECA-79 monoclonal antibody and is the dominant L-selectin ligand expressed in HEVs and chronically inflamed microvessels (200, 201). Unsulfated sLex is capable of supporting some lymphocyte homing, as demonstrated in mice lacking the enzymes GlcNAc 6-O-sulfotransferase (GlcNAc6ST)-1 and GlcNAc6ST-2 (201). The core 1 extension enzyme and the core 2 branching enzyme are respectively called: Core1-β3GlcNAcT and Core2GlcNAcT, which have been systematically knocked down in mice to show little impact on lymphocyte homing to lymph nodes in vivo (199, 202). Core1-β3GlcNAcT/ Core2GlcNAcT double knockout mice, which lack any detectable MECA-79 expression within HEV, reduced lymphocyte trafficking by only 50%, suggesting other ligands distinct from 6-sulfo sLex could support lymphocyte recruitment to HEV (203). Mass spectrometry discovered the presence of N-linked 6-sulfo sLex in HEV, which was responsible for the residual lymphocyte recruitment in mice lacking functional O-linked 6-sulfo sLex.
Interestingly, no recruitment of leukocytes to inflamed cremasteric venules was observed in Core2GlcNAcT knockout mice (199). As mentioned before, no detectable ligands for L-selectin are present on endothelial cells of inflamed cremasteric venules (79). It is possible that L-selectin/PSGL-1 secondary tethering and rolling at these sites is highly dependent on Core2GlcNAcT-mediated glycosylation of sLex onto PSGL-1, which has indeed been reported (204).
It has been known for some time that L-selectin is itself decorated by N-linked sLex (8, 9). This post-translational modification is exclusive to L-selectin in human neutrophils, as mouse neutrophils lack the expression of fucosyl transferase 9 (205). Clustering of L-selectin with E-selectin under flow conditions can drive the co-localization of L-selectin with ADAM17 to the uropod (the rear of the cell), where it is thought that enzyme/substrate coalesce to mediate ectodomain shedding (206). This form of ectodomain shedding is distinct from “mechanical shedding” (75), where neutrophils are perfused exclusively over immobilized sLex rather than E-selectin (discussed above). E-selectin-mediated clustering of L-selectin on human neutrophils promotes cell priming (i.e., degranulation and superoxide production), via the activation of p38MAPK (207, 208). Two more recent reports have extended our understanding of the molecular mechanisms underlying L-selectin-mediated neutrophil arrest under flow conditions, which have recently been reviewed (209). Perfusion of human neutrophils over immobilized E-selectin leads to the secretion of MRP8 in rolling cells. The secreted MRP8 then binds to TLR4 and promote integrin LFA-1 activation toward an intermediate activation state, driving the transition from rolling to slow rolling on ICAM-1 (210). A separate study showed that L-selectin-dependent adhesion to E-selectin under flow conditions could drive the clustering of L-selectin and transition LFA-1 toward an “active” state (211). Collectively, these studies suggest that signals downstream of L-selectin/E-selectin interaction and PSGL-1/E-selectin interaction converge to arrest neutrophils. Although mouse neutrophils express L-selectin that lack sLex modification, one study has shown that L-selectin and PSGL-1 co-cluster in cis in resting cells, which increases when perfused over P- or E-selectin to activate LFA-1 independently of chemokines (164). It is clear that L-selectin-dependent signaling is important in activating LFA-1 in mouse and human neutrophils, but whether any of the signaling pathways are conserved between species is yet to be determined.
It is increasingly appreciated that L-selectin ligands are present in perivascular and extravascular locations (78, 185, 212–215). This itself challenges the previously held view that the role of L-selectin is restricted to early events in the leukocyte adhesion cascade such as tethering and rolling (216), and supports more recent findings that L-selectin may have additional roles beyond luminal interactions (78, 87, 88). The nature of L-selectin/ligand interactions differs from intravascular ligands, as they occur in regions devoid of flow. Early evidence for extravascular L-selectin ligands was demonstrated by presence of the MECA-79 epitope within the abluminal aspect of HEVs in peripheral lymph nodes (185). Furthermore, in HEVs of Peyer's patches, abluminal expression appeared more abundant than luminal expression, alluding to a role for L-selectin within spatially distinct regions of the vessel wall (185).
The majority of identified extravascular ligands are basement membrane proteoglycans, including members of the heparan, dermatan, and chondroitin sulfate proteoglycan families (HSPGs, DSPGs, CSPGs) (212, 215, 217, 218). Using immunohistochemistry in kidney sections, HSPGs were identified as L-selectin ligands, using a chimeric protein consisting of the L-selectin extracellular domain fused to the Fc portion of IgM (L-selectin-IgM). L-selectin-IgM binding was abrogated by heparitinase I but not sialidase treatment (which removes sialic acid from sLex) (212, 213). However, although L-selectin co-localizes with the HSPGs perlecan, agrin, syndecan-4 and collagen type XVIII in the kidney, only collagen type XVIII was responsible for L-selectin binding, which was blocked in collagen XVIII-, but not perlecan-deficient mice (213). Interestingly, despite the presence of collagen type XVIII in some renal structures, L-selectin-IgM was unable to bind in some regions, suggesting that not all collagen XVIII molecules are permissive for L-selectin binding. This indicated that specific GAG chains may be necessary, which was further characterized as requiring O-sulphation (213). Supporting evidence was shown in heparin-mediated inhibition of L-selectin, requiring 6-O-sulphation of the glycolsaminoglycan (GAG) chains in heparin (219). GAG chain length is also an important determinant of L-selectin binding, as in vitro, L-selectin binding efficiency was reduced for lower molecular weight heparin relative to full length heparin (213).
Binding of other GAGs to L-selectin has also been demonstrated. In cartilage, L-selectin binding was removed by chondroitinase ABC but not heparitinase I, suggesting the presence of CSPG or DSPGs (213). Similarly, chondoritinase ABC reduced L-selectin binding in particular regions of the kidney, suggesting the contribution of CSPGs or DSPGs. The CSPG, versican, has been identified as an L-selectin ligand in the kidney (212, 220, 221). Like for HSPGs, these interactions depend on particular GAG chains and, similarly, require sulphation (220). Furthermore, the DSPG biglycan on microvascular endothelial cells of the endometrium binds L-selectin (218), and in vitro assays have demonstrated the ability of biglycan to induce L-selectin clustering (87). An increasing gradient from the apical to basal aspect of the endothelium has been observed for HSPGs and subsequently for biglycan (87, 222), which may influence L-selectin clustering, and its subsequent shedding, at high ligand densities (173, 223).
Interestingly, whilst binding of L-selectin to sLex modified vascular ligands occurs optimally at physiological blood pH 7.4, binding to extravascular ligands is optimal at pH 5.6 (224). This could reflect an altered conformation of L-selectin in acidic inflammatory environments that allows binding to different types of ligand, for example by protonation of histidine residues at low pH. Indeed, the CTLD of selectins contain clusters of basic amino acid residues that bind sulfate groups on GAGs via hydrogen bonds (225). At pH 5.6, the CTLD is additionally protonated, increasing the number of H-bonds between the groups (225). Perhaps, under inflammatory or hypoxic conditions in which an acidic environment is generated, the lowering of pH could act as a “switch” for L-selectin binding with greater affinity to subendothelial ligands to facilitate leukocyte tissue infiltration. In silico modeling suggests that the sulfate density of the GAG chain is also an important determinant of ligand binding. However, in contrast to Celie et al. who suggest the importance of 6-O-sulphation, here, 3-O-sulfation of HSPGs and 4,6-O-sulfation of CSPGs are reported to increase affinity (225). Whilst the binding site for sulphated proteoglycans is unknown, it seems plausible that binding may involve a cationic region of the CTLD distinct from the sLex site, as has been shown for sulphated glycolipids (226).
In a rodent model of renal ischaemia-reperfusion (I/R) injury, L-selectin binding to HSPGs was evident in the subendothelial region of interstitial capillaries, which was not detectable in healthy kidneys (214). Mice lacking functional collagen XVIII and perlecan displayed reduced monocyte influx to the kidney 24 h post-I/R injury. Consistent with this, human kidney post-transplant biopsies and acute allograft rejection biopsies showed greater HSPG dependent L-selectin binding to the basement membrane of peritubular capillaries than control kidneys (214). The mechanisms of regulation of HSPG binding of L-selectin are not fully understood, but HSulf 1 and 2 enzymes have been identified in humans to cleave 6-O-sulfate residues (227). In acute allograft rejection, HSulf 1 was downregulated (214), leading to the speculation that HSulf1 may cleave 6-O-sulphated HSPG chains constitutively under basal conditions, but, under inflammatory conditions, its downregulation causes HSPGs to retain 6-O-sulphation and bind L-selectin. The relevance of subendothelial L-selectin ligands may be of importance in inflammatory situations involving endothelial injury where basement membrane proteoglycans are exposed.
In addition to L-selectin ligands in luminal and subendothelial environments, putative ligands have been identified in extravascular locations. Using recombinant L-selectin fused to IgG as a histological probe, binding of L-selectin to ligands was identified in myelinated regions of the cerebellum and spinal cord, but not peripheral nervous system, in human, mouse or rat tissues (228, 229). L-selectin ligands were subsequently shown to localize specifically to myelin (230). Ligands were also located in the renal distal tubule, and upon obstruction of the ureter, were found to relocate to peritubular capillaries where infiltration of inflammatory cells ensued (229, 231). Binding of L-selectin-IgG to ligands in the cerebellum and renal distal tubule was abolished by organic solvents and could not be detected by SDS-PAGE, suggesting that they may be glycolipid, rather than protein ligands (229).
Whilst it seems that such ligands would seldom encounter L-selectin due to its shedding from leukocytes during extravasation, incomplete L-selectin shedding, or restoration of surface expression could allow this interaction to initiate an inflammatory response. Indeed, it has been speculated that the interaction between L-selectin on leukocytes and ligands on myelin may provoke an inflammatory response to drive demyelinating diseases (230). This is supported by the finding that L-selectin deficiency prevents macrophage-mediated destruction of myelin in the experimental allergic encephalomyelitis animal model which has clinical similarities to human multiple sclerosis (232). Thus, L-selectin appears to be involved in non-migratory functions. Shedding of L-selectin may serve, in part, to limit the interaction of leukocytes with such extravascular ligands to avoid inappropriate leukocyte activation under normal circumstances. Interestingly, L-selectin is also expressed on trophoblasts and interacts with ligands upregulated on the uterine luminal endothelium during the receptive phase for embryo implantation (233), perhaps performing a role in epithelial “capture” of the blastocyst, analogous to its role in leukocyte capture from blood.
Most textbooks will depict ectodomain shedding of L-selectin occurring on leukocytes that have adhered to the apical aspect of the endothelium, following chemokine-dependent integrin activation. Recent evidence in monocytes would suggest that this is clearly not the case. Live imaging of CD14++ CD16− classical human monocytes crossing TNF-activated HUVEC under flow conditions led to the observation that L-selectin undergoes ectodomain shedding exclusively during TEM, and not before (87). Suggestions have been proposed that L-selectin is involved in transducing signals (likely co-stimulatory signals alongside integrin- and chemokine-dependent signaling) to drive pseudopod protrusion in TEM. The window within which L-selectin can transduce signals in TEM is limited by its ectodomain shedding, which is maximal when monocytes have fully entered the subendothelial space. In support of L-selectin transducing signals prior to completion of TEM, blocking ectodomain shedding in CD14++ CD16− monocytes did not impact the kinetics of TEM per se, but monocytes entering the subendothelial space lost front-back polarity – producing unstable and excessive protrusions.
The molecular mechanism underpinning L-selectin signaling during TEM, whilst difficult to define in primary human monocytes, was determined in cell lines. Genetic engineering of THP-1 (monocyte-like) cells co-expressing L-selectin alongside a known binding partner (e.g., CaM, ezrin, or moesin), tagged to either green/red fluorescent proteins (RFP/GFP), enabled close inspection of protein-protein interactions within transmigrating pseudopods and non-transmigrated uropods. Fluorescence lifetime imaging microscopy (FLIM) was used to quantify Förster resonance energy transfer (FRET) between the GFP and RFP tags to understand the spatial organization of L-selectin with its binding partners within sub-10 nm distances. THP-1 cells captured in mid-TEM revealed that L-selectin interacts with CaM in both transmigrated pseudopods and non-transmigrated uropods (87). As TEM proceeds, CaM dissociates from the L-selectin tail, exclusively within transmigrating pseudopods, driven by phosphorylation of S364. Using similar FLIM/FRET approaches, ezrin and moesin were shown to bind sequentially with L-selectin as TEM proceeds (88). Ezrin binds initially to L-selectin, which is swapped-out by moesin, exclusively within transmigrating pseudopods. The change-over in L-selectin binding from ezrin to moesin is likely to reflect when signaling downstream of L-selectin is halted and ectodomain shedding ensues. Blocking L-selectin shedding leads to the sustained interaction of ezrin with L-selectin, where no exchange with moesin occurs. These outcomes would strongly suggest that it is the L-selectin/ezrin interaction that drives signaling for pseudopod protrusion. Interestingly, knocking out moesin, but not ezrin, in mouse T-cells can increase the surface expression of L-selectin (139, 234). These observations imply that in T-cells moesin acts as a “pro-shedding factor,” but ezrin does not.
Unlike the processes of rolling, slow rolling and firm adhesion, little is known about L-selectin-dependent signaling with respect to pseudopod protrusion in TEM. One can speculate that the contribution of luminal, abluminal and interstitial ligands will play a major role in modulating signals downstream of L-selectin/ligand interaction and clustering. Luminal glycoprotein ligands must contain the sLex glycan core to participate in singular catch bonding of L-selectin, a biomechanical property that is critical for leukocyte capture under physiological shear forces. These ligands are likely to favor the binding of monomeric L-selectin on microvilli. In contrast, the subluminal ligands that bind L-selectin under shear free conditions neither contain nor depend on the sLex core, and likely share unique highly clustered arrays of sulfates [e.g., in CNS sulfolipids (232)]. These multivalent ligands are therefore specialized in clustering L-selectin and triggering different magnitudes of outside-in signals to facilitate migration or non-migratory processes [e.g., where myelin damage by L-selectin expressing effector T cells proximal to inflamed CNS (232)].
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway for human transendothelial migration shows just 4 cell adhesion molecules involved in TEM (https://www.genome.jp/kegg-bin/show_pathway?map=hsa04670&show_description=show), with little insight into the intracellular proteins that mediate this process. In contrast, nearly 20 genes within the leukocyte are required to participate in firm adhesion to the apical aspect of the endothelium. The mechanisms that drive pseudopod protrusion are likely to involve numerous integrin and non-integrin cell adhesion molecules and chemokine (or other G-protein-coupled) receptors. Many reports have documented the contribution of L-selectin in the activation of integrins (164, 211, 235–240), mobilization of chemokine receptors (241–243), and cellular responsiveness to chemokines (65)—all of which play essential roles in TEM. Blocking L-selectin shedding through the expression of ΔM-N L-selectin leads to the sustained and exclusive interaction with ezrin in transmigrated pseudopods. Engineering the R357A mutation [which blocks ERM binding (20, 88)] into ΔM-N L-selectin significantly reduces the multi-pseudopodial extension phenotype, strongly implying that L-selectin/ezrin interaction is driving pseudopod protrusion.
The events downstream of L-selectin/ERM interaction, in respect of pseudopod protrusion during TEM, remain elusive. Figure 4 proposes a mechanistic model that could link L-selectin to pseudopod protrusion. Ezrin is unique from moesin in that it can interact with the p85 regulatory subunit of PI3K (138). AMC of L-selectin in T-cells can activate the small GTPases Ras and Rac (166), which in turn can promote two inter-linked signals. Rac can target downstream effectors to remodel the actin-based cytoskeleton for pseudopod formation in leukocytes and other cell types (244–247). The catalytic activity of PI3K is significantly enhanced in the presence of activated Ras, resulting in increased local pools of PIP3 production (248, 249). Guanine nucleotide exchange factors (GEFs), such as Vav [a known Rac GEF (250, 251)], is targeted to the plasma membrane via its plextrin homology (PH) domain. Running in parallel, Syk can bind to a cryptic ITAM sequence within the FERM domain of ERM (156–158). The Syk, which is anchored to ERM, can phosphorylate GEFs within its immediate vicinity, such as Vav (252), to increase its GDP-to-GTP exchange activity. Localized Rac activation will promote actin remodeling and pseudopod protrusion during TEM. The displacement of ezrin from L-selectin by moesin would invariably shut down PI3K-mediated PIP3 production and Rac-mediated protrusion. Again, these models are purely speculative and will require further research to expose the true underlying mechanism.
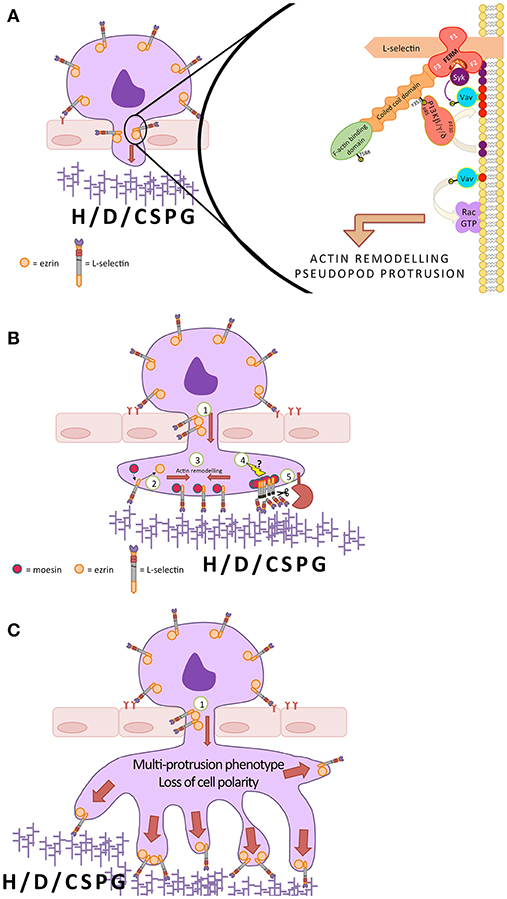
Figure 4. A possible mechanism underlying L-selectin-dependent pseudopod protrusion during monocyte TEM. (A) In early monocyte TEM, L-selectin interacts preferentially with ezrin. Moesin and ezrin are the most abundant ERM proteins in monocytes, with little to no expression of radixin. Ezrin can interact with the p85 subunit of class I PI3Ks via phospho-Y353 (a known target of Src kinase). The ezrin-anchored PI3K catalytic subunit (p110) converts PIP2 (purple spheres) into PIP3 (red spheres) within its local vicinity. The most common p110 isoforms in leukocytes are β, δ, and γ. The plekstrin homology domain of guanine nucleotide exchange factors (GEFs), such as Vav, can bind to PIP3 and mediate their effects on the small Rho family of GTPases, such as Rac. The FERM domains of ERM contain a cryptic ITAM, which, upon phosphorylation, can support the binding of spleen tyrosine kinase (Syk). Syk can phosphorylate Vav, increasing its GEF activity significantly toward GTPases like Rac. PI3K and Rac play pivotal roles in pseudopod protrusion, but the molecular mechanism in the context of TEM is currently not known. (B) As described in (A), during early TEM, the interaction between L-selectin and ezrin is thought to contribute to pseudopod protrusion (1). As TEM proceeds, the L-selectin within transmigrating pseudopods exchanges affiliation from ezrin to moesin (2). This exchange could happen when L-selectin makes contact with high-density heparan, dermatan or chondroitin sulfate proteoglycan families (H/D/CSPG) within the subendothelial space, such as the basement membrane. Phospho-cycling of serine residues within the L-selectin tail could also be contributing to the exchange mechanism, where phosphorylation drives ezrin dissociation and dephosphorylation allows binding of moesin. The moesin/L-selectin interaction leads to the remodeling of L-selectin into clusters (3), possibly by the remodeling of the cortical actin cytoskeleton. The clustered L-selectin may also trigger as yet unexplored signals (4) that lead to ectodomain shedding by ADAM17 (5). Currently, it is thought that the exchange of ezrin for moesin inhibits further protrusion and rapidly shuts down L-selectin-dependent adhesion in this region by activating shedding. (C) Blocking L-selectin shedding in monocytes during TEM sustains the L-selectin/ezrin interaction, which exacerbates the signaling mechanism proposed in (A), leading to the formation of multiple pseudopodial extensions and a loss in cell polarity of fully transmigrated cells.
Since its identification and characterization, the expression of L-selectin has been carefully interrogated in numerous leukocyte subsets. The motivation underlying this type of classification has been to unearth the ever-expanding functional diversity of individual leukocyte subsets, and their specialized roles in health and disease. A major unanswered question in many such immune cell subsets is to understand if there is a direct role for L-selectin signaling in bestowing their unique functionality.
This review also highlights how the expression of non-cleavable L-selectin in one immune cell-type may confer protection from viruses (61), whereas its expression in another cell type could block polarity necessary for migration toward inflammatory targets (40, 87). Therefore, blocking L-selectin shedding as a therapeutic target will require better understanding of the immune cells it will impact in specific disease settings.
It is highly likely that L-selectin acts as a co-receptor to many of the cellular outcomes featured in this review. Fresh insights into how L-selectin signaling synergises/antagonizes chemokine receptor signaling, integrin signaling, and signaling from non-integrin receptors during adhesion and migration is what is now required to further understand how unique signals are propagated under steady-state and pro-inflammatory conditions.
Finally, technological advances in microscopic imaging have for the first time allowed the interrogation of spatio-temporal interactions (within a sub-10 nm resolution) between L-selectin and its cytosolic binding partners during specific cellular events, such as TEM. Moreover, the recent identification of L-selectin contributing to monocyte protrusion during TEM suggests a revised view on how cell adhesion molecules contribute to the multi-step adhesion cascade. It seems that the one-molecule-to-one-step paradigm may not be as strict as once perceived.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All three authors are supported by the BHF Center of Research Excellence (RE/13/2/30182). HLHG and SJH are both supported by BHF PhD studentships (FS/18/60/34181 & RE/13/2/30182, respectively).
1. Fabre S, Carrette F, Chen J, Lang V, Semichon M, Denoyelle C, et al. FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol. (2008) 181:2980–9. doi: 10.4049/jimmunol.181.5.2980
2. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. (2009) 10:176–84. doi: 10.1038/ni.1689
3. Dang X, Raffler NA, Ley K. Transcriptional regulation of mouse L-selectin. Biochim Biophys Acta. (2009) 1789:146–52. doi: 10.1016/j.bbagrm.2008.10.004
4. Schleiffenbaum B, Spertini O, Tedder TF. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol. (1992) 119:229–38. doi: 10.1083/jcb.119.1.229
5. Griffin JD, Spertini O, Ernst TJ, Belvin MP, Levine HB, Kanakura Y, et al. Granulocyte-macrophage colony-stimulating factor and other cytokines regulate surface expression of the leukocyte adhesion molecule-1 on human neutrophils, monocytes, and their precursors. J Immunol. (1990) 145:576–84.
6. Kishimoto TK, Jutila MA, Butcher EC. Identification of a human peripheral lymph node homing receptor: a rapidly down-regulated adhesion molecule. Proc Natl Acad Sci USA. (1990) 87:2244–8. doi: 10.1073/pnas.87.6.2244
7. Tedder TF, Matsuyama T, Rothstein D, Schlossman SF, Morimoto C. Human antigen-specific memory T cells express the homing receptor (LAM-1) necessary for lymphocyte recirculation. Eur J Immunol. (1990) 20:1351–5. doi: 10.1002/eji.1830200622
8. Zollner O, Lenter MC, Blanks JE, Borges E, Steegmaier M, Zerwes HG, et al. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J Cell Biol. (1997) 136:707–16. doi: 10.1083/jcb.136.3.707
9. Picker LJ, Warnock RA, Burns AR, Doerschuk CM, Berg EL, Butcher EC. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell. (1991) 66:921–33. doi: 10.1016/0092-8674(91)90438-5
10. Wedepohl S, Kaup M, Riese SB, Berger M, Dernedde J, Tauber R, et al. N-glycan analysis of recombinant L-Selectin reveals sulfated GalNAc and GalNAc-GalNAc motifs. J Proteome Res. (2010) 9:3403–11. doi: 10.1021/pr100170c
11. Furukawa Y, Umemoto E, Jang MH, Tohya K, Miyasaka M, Hirata T. Identification of novel isoforms of mouse L-selectin with different carboxyl-terminal tails. J Biol Chem. (2008) 283:12112–9. doi: 10.1074/jbc.M801745200
12. Hirata T, Usui T, Kobayashi S, Mimori T. A novel splice variant of human L-selectin encodes a soluble molecule that is elevated in serum of patients with rheumatic diseases. Biochem Biophys Res Commun. (2015) 462:371–7. doi: 10.1016/j.bbrc.2015.05.002
13. Kohn LA, Hao QL, Sasidharan R, Parekh C, Ge S, Zhu Y, et al. Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat Immunol. (2012) 13:963–71. doi: 10.1038/ni.2405
14. Cho S, Spangrude GJ. Enrichment of functionally distinct mouse hematopoietic progenitor cell populations using CD62L. J Immunol. (2011) 187:5203–10. doi: 10.4049/jimmunol.1102119
15. Perry SS, Welner RS, Kouro T, Kincade PW, Sun XH. Primitive lymphoid progenitors in bone marrow with T lineage reconstituting potential. J Immunol. (2006) 177:2880–7. doi: 10.4049/jimmunol.177.5.2880
16. Foster GA, Gower RM, Stanhope KL, Havel PJ, Simon SI, Armstrong EJ. On-chip phenotypic analysis of inflammatory monocytes in atherogenesis and myocardial infarction. Proc Natl Acad Sci USA. (2013) 110:13944–9. doi: 10.1073/pnas.1300651110
17. Thiel M, Zourelidis C, Chambers JD, von Andrian UH, Arfors KE, Messmer K, et al. Expression of beta 2-integrins and L-selectin on polymorphonuclear leukocytes in septic patients. Eur Surg Res. (1997) 29:160–75. doi: 10.1159/000129521
18. Stein JV, Cheng G, Stockton BM, Fors BP, Butcher EC, von Andrian UH. L-selectin-mediated leukocyte adhesion in vivo: microvillous distribution determines tethering efficiency, but not rolling velocity. J Exp Med. (1999) 189:37–50. doi: 10.1084/jem.189.1.37
19. Bruehl RE, Springer TA, Bainton DF. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J Histochem Cytochem. (1996) 44:835–44. doi: 10.1177/44.8.8756756
20. Ivetic A, Florey O, Deka J, Haskard DO, Ager A, Ridley AJ. Mutagenesis of the ezrin-radixin-moesin binding domain of L-selectin tail affects shedding, microvillar positioning, and leukocyte tethering. J Biol Chem. (2004) 279:33263–72. doi: 10.1074/jbc.M312212200
21. Smalley DM, Ley K. L-selectin: mechanisms and physiological significance of ectodomain cleavage. J Cell Mol Med. (2005) 9:255–66. doi: 10.1111/j.1582-4934.2005.tb00354.x
22. Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. (1999) 18:6962–72. doi: 10.1093/emboj/18.24.6962
23. Jutila MA, Rott L, Berg EL, Butcher EC. Function and regulation of the neutrophil MEL-14 antigen in vivo: comparison with LFA-1 and MAC-1. J Immunol. (1989) 143:3318–24.
24. Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. (1989) 245:1238–41. doi: 10.1126/science.2551036
25. Ager A. ADAMs and Ectodomain Proteolytic Shedding in Leucocyte Migration: focus on L-selectin and ADAM17. Curr Immunol Rev. (2012) 8:103–17. doi: 10.2174/157339512800099657
26. Tang J, Zarbock A, Gomez I, Wilson CL, Lefort CT, Stadtmann A, et al. Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood. (2011) 118:786–94. doi: 10.1182/blood-2010-11-321406
27. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science. (1998) 282:1281–4. doi: 10.1126/science.282.5392.1281
28. Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. (2006) 108:2275–9. doi: 10.1182/blood-2006-02-005827
29. Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, et al. Expression and regulation of the metalloproteinase ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-selectin shedding. J Immunol. (2007) 178:8053–63. doi: 10.4049/jimmunol.178.12.8053
30. Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, et al. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. (2009) 20:1785–94. doi: 10.1091/mbc.e08-11-1135
31. Font J, Pizcueta P, Ramos-Casals M, Cervera R, Garcia-Carrasco M, Navarro M, et al. Increased serum levels of soluble L-selectin (CD62L) in patients with active systemic lupus erythematosus (SLE). Clin Exp Immunol. (2000) 119:169–74. doi: 10.1046/j.1365-2249.2000.01082.x
32. Kretowski A, Gillespie KM, Bingley PJ, Kinalska I. Soluble L-selectin levels in type I diabetes mellitus: a surrogate marker for disease activity? Immunology. (2000) 99:320–5. doi: 10.1046/j.1365-2567.2000.00967.x
33. Albertini JP, Valensi P, Lormeau B, Vaysse J, Attali JR, Gattegno L. Soluble L-selectin level is a marker for coronary artery disease in type 2 diabetic patients. Diabetes Care. (1999) 22:2044–8. doi: 10.2337/diacare.22.12.2044
34. Shimada Y, Sato S, Hasegawa M, Tedder TF, Takehara K. Elevated serum L-selectin levels and abnormal regulation of L-selectin expression on leukocytes in atopic dermatitis: soluble L-selectin levels indicate disease severity. J Allergy Clin Immunol. (1999) 104:163–8. doi: 10.1016/S0091-6749(99)70128-4
35. Giannitsis E, Tettenborn I, Wiegand U, Stierle U, Katus HA. Soluble L-selectin and neutrophil derived oxidative stress after pacing induced myocardial ischemia in chronic stable coronary artery disease. Pacing Clin Electrophysiol. (2000) 23:309–14. doi: 10.1111/j.1540-8159.2000.tb06754.x
36. Seidelin JB, Nielsen OH, Strom J. Soluble L-selectin levels predict survival in sepsis. Intensive Care Med. (2002) 28:1613–8. doi: 10.1007/s00134-002-1501-5
37. Long C, Hosseinkhani MR, Wang Y, Sriramarao P, Walcheck B. ADAM17 activation in circulating neutrophils following bacterial challenge impairs their recruitment. J Leukoc Biol. (2012) 92:667–72. doi: 10.1189/jlb.0312112
38. Chen A, Engel P, Tedder TF. Structural requirements regulate endoproteolytic release of the L-selectin (CD62L) adhesion receptor from the cell surface of leukocytes. J Exp Med. (1995) 182:519–30. doi: 10.1084/jem.182.2.519
39. Galkina E, Tanousis K, Preece G, Tolaini M, Kioussis D, Florey O, et al. L-selectin shedding does not regulate constitutive T cell trafficking but controls the migration pathways of antigen-activated T lymphocytes. J Exp Med. (2003) 198:1323–35. doi: 10.1084/jem.20030485
40. Venturi GM, Tu L, Kadono T, Khan AI, Fujimoto Y, Oshel P, et al. Leukocyte migration is regulated by L-selectin endoproteolytic release. Immunity. (2003) 19:713–24. doi: 10.1016/S1074-7613(03)00295-4
41. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
42. Westera L, Drylewicz J, den Braber I, Mugwagwa T, van der Maas I, Kwast L, et al. Closing the gap between T-cell life span estimates from stable isotope-labeling studies in mice and humans. Blood. (2013) 122:2205–12. doi: 10.1182/blood-2013-03-488411
43. Li XY, Matsuzaki G, Yoshikai Y, Muramori K, Nomoto K. T cells expressing both L-selectin and CD44 molecules increase in number in peritoneal exudate cells and in vitro-stimulated spleen cells from mice immunized intraperitoneally with Listeria monocytogenes. Immunology. (1993) 78:28–34.
44. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
45. Hengel RL, Thaker V, Pavlick MV, Metcalf JA, Dennis G, Jr., Yang J, et al. Cutting edge: L-selectin (CD62L) expression distinguishes small resting memory CD4+ T cells that preferentially respond to recall antigen. J Immunol. (2003) 170:28–32. doi: 10.4049/jimmunol.170.1.28
46. Arbones ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. (1994) 1:247–60. doi: 10.1016/1074-7613(94)90076-0
47. Klinger A, Gebert A, Bieber K, Kalies K, Ager A, Bell EB, et al. Cyclical expression of L-selectin (CD62L) by recirculating T cells. International immunology. (2009) 21:443–55. doi: 10.1093/intimm/dxp012
48. Galkina E, Florey O, Zarbock A, Smith BR, Preece G, Lawrence MB, et al. T lymphocyte rolling and recruitment into peripheral lymph nodes is regulated by a saturable density of L-selectin (CD62L). Eur J Immunol. (2007) 37:1243–53. doi: 10.1002/eji.200636481
49. Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. (2008) 9:513–21. doi: 10.1038/ni.1603
50. Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transpl Int. (2017) 30:745–53. doi: 10.1111/tri.12909
51. Gallatin WM, Weissman IL, Butcher EC. A cell-surface molecule involved in organ-specific homing of lymphocytes. 1983. J Immunol. (2006) 177:5–9.
52. Fu S, Yopp AC, Mao X, Chen D, Zhang N, Chen D, et al. CD4+ CD25+ CD62+ T-regulatory cell subset has optimal suppressive and proliferative potential. Am J Transl. (2004) 4:65–78. doi: 10.1046/j.1600-6143.2003.00293.x
53. Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, et al. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood. (2005) 105:2220–6. doi: 10.1182/blood-2004-05-2044
54. Lange C, Scholl M, Melms A, Bischof F. CD62L(high) Treg cells with superior immunosuppressive properties accumulate within the CNS during remissions of EAE. Brain Behav Immun. (2011) 25:120–6. doi: 10.1016/j.bbi.2010.09.004
55. Marodon G, Landau NR, Posnett DN. Altered expression of CD4, CD54, CD62L, and CCR5 in primary lymphocytes productively infected with the human immunodeficiency virus. AIDS Res Hum Retroviruses. (1999) 15:161–71. doi: 10.1089/088922299311583
56. Trinite B, Chan CN, Lee CS, Mahajan S, Luo Y, Muesing MA, et al. Suppression of Foxo1 activity and down-modulation of CD62L (L-selectin) in HIV-1 infected resting CD4 T cells. PLoS ONE. (2014) 9:e110719. doi: 10.1371/journal.pone.0110719
57. Vassena L, Giuliani E, Koppensteiner H, Bolduan S, Schindler M, Doria M. HIV-1 Nef and Vpu Interfere with L-Selectin (CD62L) Cell Surface Expression To Inhibit Adhesion and Signaling in Infected CD4+ T Lymphocytes. J Virol. (2015) 89:5687–700. doi: 10.1128/JVI.00611-15
58. Kononchik J, Ireland J, Zou Z, Segura J, Holzapfel G, Chastain A, et al. HIV-1 targets L-selectin for adhesion and induces its shedding for viral release. Nat Commun. (2018) 9:2825. doi: 10.1038/s41467-018-05197-2
59. Giuliani E, Vassena L, Galardi S, Michienzi A, Desimio MG, Doria M. Dual regulation of L-selectin (CD62L) by HIV-1: enhanced expression by Vpr in contrast with cell-surface down-modulation by Nef and Vpu. Virology. (2018) 523:121–8. doi: 10.1016/j.virol.2018.07.031
60. Stolp B, Imle A, Coelho FM, Hons M, Gorina R, Lyck R, et al. HIV-1 Nef interferes with T-lymphocyte circulation through confined environments in vivo. Proc Natl Acad Sci USA. (2012) 109:18541–6. doi: 10.1073/pnas.1204322109
61. Mohammed RN, Watson HA, Vigar M, Ohme J, Thomson A, Humphreys IR, et al. L-selectin Is Essential for Delivery of Activated CD8(+) T Cells to Virus-Infected Organs for Protective Immunity. Cell Rep. (2016) 14:760–71. doi: 10.1016/j.celrep.2015.12.090
62. Yang S, Liu F, Wang QJ, Rosenberg SA, Morgan RA. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS ONE. (2011) 6:e22560. doi: 10.1371/journal.pone.0022560
63. Nishijima K, Ando M, Sano S, Hayashi-Ozawa A, Kinoshita Y, Iijima S. Costimulation of T-cell proliferation by anti-L-selectin antibody is associated with the reduction of a cdk inhibitor p27. Immunology. (2005) 116:347–53. doi: 10.1111/j.1365-2567.2005.02234.x
64. Murakawa Y, Minami Y, Strober W, James SP. Association of human lymph node homing receptor (Leu 8) with the TCR/CD3 complex. J Immunol. (1992) 148:1771–6.
65. Subramanian H, Grailer JJ, Ohlrich KC, Rymaszewski AL, Loppnow JJ, Kodera M, et al. Signaling through L-selectin mediates enhanced chemotaxis of lymphocyte subsets to secondary lymphoid tissue chemokine. J Immunol. (2012) 188:3223–36. doi: 10.4049/jimmunol.1101032
66. Rozenberg I, Sluka SH, Mocharla P, Hallenberg A, Rotzius P, Boren J, et al. Deletion of L-selectin increases atherosclerosis development in ApoE-/- mice. PLoS ONE. (2011) 6:e21675. doi: 10.1371/journal.pone.0021675
67. Gjurich BN, Taghavie-Moghadam PL, Ley K, Galkina EV. L-selectin deficiency decreases aortic B1a and Breg subsets and promotes atherosclerosis. Thrombosis Haemosta. (2014) 112:803–11. doi: 10.1160/TH13-10-0865
68. Eriksson EE. Intravital microscopy on atherosclerosis in apolipoprotein e-deficient mice establishes microvessels as major entry pathways for leukocytes to advanced lesions. Circulation. (2011) 124:2129–38. doi: 10.1161/CIRCULATIONAHA.111.030627
69. Moreno PR, Purushothaman KR, Fuster V, Echeverri D, Truszczynska H, Sharma SK, et al. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation. (2004) 110:2032–8. doi: 10.1161/01.CIR.0000143233.87854.23
70. Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arteriosclerosis Thrombosis Vascu Biol. (2005) 25:2054–61. doi: 10.1161/01.ATV.0000178991.71605.18
71. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
72. Allport JR, Ding HT, Ager A, Steeber DA, Tedder TF, Luscinskas FW. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J Immunol. (1997) 158:4365–72.
73. Walcheck B, Kahn J, Fisher JM, Wang BB, Fisk RS, Payan DG, et al. Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature. (1996) 380:720–3. doi: 10.1038/380720a0
74. Palecanda A, Walcheck B, Bishop DK, Jutila MA. Rapid activation-independent shedding of leukocyte L-selectin induced by cross-linking of the surface antigen. Eur J Immunol. (1992) 22:1279–86. doi: 10.1002/eji.1830220524
75. Lee D, Schultz JB, Knauf PA, King MR. Mechanical shedding of L-selectin from the neutrophil surface during rolling on sialyl Lewis x under flow. J Biol Chem. (2007) 282:4812–20. doi: 10.1074/jbc.M609994200
76. Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med. (2001) 193:863–72. doi: 10.1084/jem.193.7.863
77. Bjorkman L, Christenson K, Davidsson L, Martensson J, Amirbeagi F, Welin A, et al. Neutrophil recruitment to inflamed joints can occur without cellular priming. J Leukoc Biol. (2018). doi: 10.1002/JLB.3AB0918-369R. [Epub ahead of print].
78. Hickey MJ, Forster M, Mitchell D, Kaur J, De Caigny C, Kubes P. L-selectin facilitates emigration and extravascular locomotion of leukocytes during acute inflammatory responses in vivo. J Immunol. (2000) 165:7164–70. doi: 10.4049/jimmunol.165.12.7164
79. Eriksson EE. No detectable endothelial- or leukocyte-derived L-selectin ligand activity on the endothelium in inflamed cremaster muscle venules. J Leukoc Biol. (2008) 84:93–103. doi: 10.1189/jlb.1107786
80. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, N AG, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
81. Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia. (2002) 16:1992–2003. doi: 10.1038/sj.leu.2402684
82. Ivetic A. A head-to-tail view of L-selectin and its impact on neutrophil behaviour. Cell Tissue Res. (2018) 371:437–53. doi: 10.1007/s00441-017-2774-x
83. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
84. Spertini O, Luscinskas FW, Gimbrone MA Jr., Tedder TF. Monocyte attachment to activated human vascular endothelium in vitro is mediated by leukocyte adhesion molecule-1 (L-selectin) under nonstatic conditions. J Exp Med. (1992) 175:1789–92. doi: 10.1084/jem.175.6.1789
85. Spertini O, Luscinskas FW, Kansas GS, Munro JM, Griffin JD, Gimbrone MA Jr., et al. Leukocyte adhesion molecule-1 (LAM-1, L-selectin) interacts with an inducible endothelial cell ligand to support leukocyte adhesion. J Immunol. (1991) 147:2565–73.
86. Lim YC, Snapp K, Kansas GS, Camphausen R, Ding H, Luscinskas FW. Important contributions of P-selectin glycoprotein ligand-1-mediated secondary capture to human monocyte adhesion to P-selectin, E-selectin, and TNF-alpha-activated endothelium under flow in vitro. J Immunol. (1998) 161:2501–8.
87. Rzeniewicz K, Newe A, Rey Gallardo A, Davies J, Holt MR, Patel A, et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc Natl Acad Sci USA. (2015) 112:E1461–70. doi: 10.1073/pnas.1417100112
88. Rey-Gallardo A, Tomlins H, Joachim J, Rahman I, Kitscha P, Frudd K, et al. Sequential binding of ezrin and moesin to L-selectin regulates monocyte protrusive behaviour during transendothelial migration. J Cell Sci. (2018) 131:jcs215541. doi: 10.1242/jcs.215541
89. Alcaide P, Fresno M. The Trypanosoma cruzi membrane mucin AgC10 inhibits T cell activation and IL-2 transcription through L-selectin. Int Immunol. (2004) 16:1365–75. doi: 10.1093/intimm/dxh138
90. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arteriosclerosis Thrombosis Vascular Biol. (2015) 35:1306–16. doi: 10.1161/ATVBAHA.114.304650
91. Prakobphol A, Genbacev O, Gormley M, Kapidzic M, Fisher SJ. A role for the L-selectin adhesion system in mediating cytotrophoblast emigration from the placenta. Dev Biol. (2006) 298:107–17. doi: 10.1016/j.ydbio.2006.06.020
92. Fazleabas AT, Kim JJ. Development. What makes an embryo stick? Science. (2003) 299:355–6. doi: 10.1126/science.1081277
93. Ahrens I, Ellwanger C, Smith BK, Bassler N, Chen YC, Neudorfer I, et al. Selenium supplementation induces metalloproteinase-dependent L-selectin shedding from monocytes. J Leukoc Biol. (2008) 83:1388–95. doi: 10.1189/jlb.0707497
94. Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J Immunol. (2009) 182:2449–57. doi: 10.4049/jimmunol.0802770
95. Imai Y, Singer MS, Fennie C, Lasky LA, Rosen SD. Identification of a carbohydrate-based endothelial ligand for a lymphocyte homing receptor. J Cell Biol. (1991) 113:1213–21. doi: 10.1083/jcb.113.5.1213
96. Zelensky AN, Gready JE. The C-type lectin-like domain superfamily. FEBS J. (2005) 272:6179–217. doi: 10.1111/j.1742-4658.2005.05031.x
97. Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. (2018) 18:374–89. doi: 10.1038/s41577-018-0004-8
98. Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. (2004) 22:129–56. doi: 10.1146/annurev.immunol.21.090501.080131
99. Graves BJ, Crowther RL, Chandran C, Rumberger JM, Li S, Huang KS, et al. Insight into E-selectin/ligand interaction from the crystal structure and mutagenesis of the lec/EGF domains. Nature. (1994) 367:532–8. doi: 10.1038/367532a0
100. Hession C, Osborn L, Goff D, Chi-Rosso G, Vassallo C, Pasek M, et al. Endothelial leukocyte adhesion molecule 1: direct expression cloning and functional interactions. Proc Natl Acad Sci USA. (1990) 87:1673–7. doi: 10.1073/pnas.87.5.1673
101. Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. (2000) 103:467–79. doi: 10.1016/S0092-8674(00)00138-0
102. Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol. (2004) 166:913–23. doi: 10.1083/jcb.200403144jcb.200403144[pii]
103. McEver RP, Zhu C. Rolling cell adhesion. Ann Rev Cell Dev Biol. (2010) 26:363–96. doi: 10.1146/annurev.cellbio.042308.113238
104. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res. (2015) 107:331–9. doi: 10.1093/cvr/cvv154
105. Wayman AM, Chen W, McEver RP, Zhu C. Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys J. (2010) 99:1166–74. doi: 10.1016/j.bpj.2010.05.040
106. Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI, et al. Flow-enhanced adhesion regulated by a selectin interdomain hinge. J Cell Biol. (2006) 174:1107–17. doi: 10.1083/jcb.200606056
107. Liu Z, Yago T, Zhang N, Panicker SR, Wang Y, Yao L, et al. L-selectin mechanochemistry restricts neutrophil priming in vivo. Nat Commun. (2017) 8:15196. doi: 10.1038/ncomms15196
108. Norman DG, Barlow PN, Baron M, Day AJ, Sim RB, Campbell ID. Three-dimensional structure of a complement control protein module in solution. J Mol Biol. (1991) 219:717–25. doi: 10.1016/0022-2836(91)90666-T
109. Kahn J, Ingraham RH, Shirley F, Migaki GI, Kishimoto TK. Membrane proximal cleavage of L-selectin: identification of the cleavage site and a 6-kD transmembrane peptide fragment of L-selectin. J Cell Biol. (1994) 125:461–70. doi: 10.1083/jcb.125.2.461
110. Migaki GI, Kahn J, Kishimoto TK. Mutational analysis of the membrane-proximal cleavage site of L-selectin: relaxed sequence specificity surrounding the cleavage site. J Exp Med. (1995) 182:549–57. doi: 10.1084/jem.182.2.549
111. Kahn J, Walcheck B, Migaki GI, Jutila MA, Kishimoto TK. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell. (1998) 92:809–18. doi: 10.1016/S0092-8674(00)81408-7
112. Buscher K, Riese SB, Shakibaei M, Reich C, Dernedde J, Tauber R, et al. The transmembrane domains of L-selectin and CD44 regulate receptor cell surface positioning and leukocyte adhesion under flow. J Biol Chem. (2010) 285:13490–7. doi: 10.1074/jbc.M110.102640
113. Fors BP, Goodarzi K, von Andrian UH. L-selectin shedding is independent of its subsurface structures and topographic distribution. J Immunol. (2001) 167:3642–51. doi: 10.4049/jimmunol.167.7.3642
114. Steiner H, Haass C. Intramembrane proteolysis by presenilins. Nat Rev Mol Cell Biol. (2000) 1:217–24. doi: 10.1038/35043065
115. Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, et al. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. (2001) 155:755–62. doi: 10.1083/jcb.200108159
116. Schultz K, Grieger Lindner C, Li Y, Urbanek P, Ruschel A, Minnich K, et al. Gamma secretase dependent release of the CD44 cytoplasmic tail upregulates IFI16 in cd44-/- tumor cells, MEFs and macrophages. PLoS ONE. (2018) 13:e0207358. doi: 10.1371/journal.pone.0207358
117. Pelletier L, Guillaumot P, Freche B, Luquain C, Christiansen D, Brugiere S, et al. Gamma-secretase-dependent proteolysis of CD44 promotes neoplastic transformation of rat fibroblastic cells. Cancer Res. (2006) 66:3681–7. doi: 10.1158/0008-5472.CAN-05-3870
118. Pavalko FM, Walker DM, Graham L, Goheen M, Doerschuk CM, Kansas GS. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via alpha-actinin: receptor positioning in microvilli does not require interaction with alpha-actinin. J Cell Biol. (1995) 129:1155–64. doi: 10.1083/jcb.129.4.1155
119. Ivetic A, Deka J, Ridley A, Ager A. The cytoplasmic tail of L-selectin interacts with members of the Ezrin-Radixin-Moesin (ERM) family of proteins: cell activation-dependent binding of Moesin but not Ezrin. J Biol Chem. (2002) 277:2321–9. doi: 10.1074/jbc.M109460200
120. Kilian K, Dernedde J, Mueller EC, Bahr I, Tauber R. The interaction of protein kinase C isozymes alpha, iota, and theta with the cytoplasmic domain of L-selectin is modulated by phosphorylation of the receptor. J Biol Chem. (2004) 279:34472–80. doi: 10.1074/jbc.M405916200M405916200[pii]
121. Dib K, Tikhonova IG, Ivetic A, Schu P. The cytoplasmic tail of L-selectin interacts with the adaptor-protein complex AP-1 subunit mu1A via a novel basic binding motif. J Biol Chem. (2017) 292:6703–14. doi: 10.1074/jbc.M116.768598
122. Mattila PE, Green CE, Schaff U, Simon SI, Walcheck B. Cytoskeletal interactions regulate inducible L-selectin clustering. Am J Physiol Cell Physiol. (2005) 289:C323–32. doi: 10.1152/ajpcell.00603.2004
123. Dwir O, Kansas GS, Alon R. Cytoplasmic anchorage of L-selectin controls leukocyte capture and rolling by increasing the mechanical stability of the selectin tether. J Cell Biol. (2001) 155:145–56. doi: 10.1083/jcb.200103042
124. Stoddart JH Jr., Jasuja RR, Sikorski MA, von Andrian UH, Mier JW. Protease-resistant L-selectin mutants. Down-modulation by cross-linking but not cellular activation. J Immunol. (1996) 157:5653–9.
125. Sun F, Schroer CFE, Xu L, Yin H, Marrink SJ, Luo SZ. Molecular Dynamics of the Association of L-Selectin and FERM Regulated by PIP2. Biophys J. (2018) 114:1858–68. doi: 10.1016/j.bpj.2018.02.034
126. Deng W, Cho S, Li R. FERM domain of moesin desorbs the basic-rich cytoplasmic domain of l-selectin from the anionic membrane surface. J Mol Biol. (2013) 425:3549–62. doi: 10.1016/j.jmb.2013.06.008
127. Deng W, Putkey JA, Li R. Calmodulin adopts an extended conformation when interacting with L-selectin in membranes. PLoS ONE. (2013) 8:e62861. doi: 10.1371/journal.pone.0062861
128. Foley KS, Young PW. An analysis of splicing, actin-binding properties, heterodimerization and molecular interactions of the non-muscle alpha-actinins. Biochem J. (2013) 452:477–88. doi: 10.1042/BJ20121824
129. Kansas GS, Ley K, Munro JM, Tedder TF. Regulation of leukocyte rolling and adhesion to high endothelial venules through the cytoplasmic domain of L-selectin. J Exp Med. (1993) 177:833–8. doi: 10.1084/jem.177.3.833
130. Matala E, Alexander SR, Kishimoto TK, Walcheck B. The cytoplasmic domain of L-selectin participates in regulating L-selectin endoproteolysis. J Immunol. (2001) 167:1617–23. doi: 10.4049/jimmunol.167.3.1617
131. Foley KS, Young PW. The non-muscle functions of actinins: an update. Biochem J. (2014) 459:1–13. doi: 10.1042/BJ20131511
132. Killock DJ, Ivetic A. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem J. (2010) 428:293–304. doi: 10.1042/BJ20091611
133. Gifford JL, Ishida H, Vogel HJ. Structural insights into calmodulin-regulated L-selectin ectodomain shedding. J Biol Chem. (2012) 287:26513–27. doi: 10.1074/jbc.M112.373373
134. Killock DJ, Parsons M, Zarrouk M, Ameer-Beg SM, Ridley AJ, Haskard DO, et al. In Vitro and in vivo Characterization of Molecular Interactions between Calmodulin, Ezrin/Radixin/Moesin, and L-selectin. J Biol Chem. (2009) 284:8833–45. doi: 10.1074/jbc.M806983200
135. Ivetic A. Signals regulating L-selectin-dependent leucocyte adhesion and transmigration. Int J Biochem Cell Biol. (2013) 45:550–5. doi: 10.1016/j.biocel.2012.12.023
136. Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. (2000) 101:259–70. doi: 10.1016/S0092-8674(00)80836-3
137. Autero M, Heiska L, Ronnstrand L, Vaheri A, Gahmberg CG, Carpen O. Ezrin is a substrate for Lck in T cells. FEBS Lett. (2003) 535:82–6. doi: 10.1016/S0014-5793(02)03861-9
138. Gautreau A, Poullet P, Louvard D, Arpin M. Ezrin, a plasma membrane-microfilament linker, signals cell survival through the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA. (1999) 96:7300–5. doi: 10.1073/pnas.96.13.7300
139. Shaffer MH, Dupree RS, Zhu P, Saotome I, Schmidt RF, McClatchey AI, et al. Ezrin and moesin function together to promote T cell activation. J Immunol. (2009) 182:1021–32. doi: 10.4049/jimmunol.182.2.1021
140. Brown MJ, Nijhara R, Hallam JA, Gignac M, Yamada KM, Erlandsen SL, et al. Chemokine stimulation of human peripheral blood T lymphocytes induces rapid dephosphorylation of ERM proteins, which facilitates loss of microvilli and polarization. Blood. (2003) 102:3890–9. doi: 10.1182/blood-2002-12-3807
141. Nijhara R, van Hennik PB, Gignac ML, Kruhlak MJ, Hordijk PL, Delon J, et al. Rac1 mediates collapse of microvilli on chemokine-activated T lymphocytes. J Immunol. (2004) 173:4985–93. doi: 10.4049/jimmunol.173.8.4985
142. Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VL, Bismuth G, Trautmann A, et al. ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation. Nat Immunol. (2004) 5:272–9. doi: 10.1038/ni1039
143. Bretscher A. Purification of an 80,000-dalton protein that is a component of the isolated microvillus cytoskeleton, and its localization in nonmuscle cells. J Cell Biol. (1983) 97:425–32. doi: 10.1083/jcb.97.2.425
144. Bretscher A, Reczek D, Berryman M. Ezrin: a protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J Cell Sci. (1997) 110:3011–8.
145. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. (2010) 11:276–87. doi: 10.1038/nrm2866
146. Niggli V, Andreoli C, Roy C, Mangeat P. Identification of a phosphatidylinositol-4,5-bisphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett. (1995) 376:172–6. doi: 10.1016/0014-5793(95)01270-1
147. Barret C, Roy C, Montcourrier P, Mangeat P, Niggli V. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate [PIP(2)] binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. J Cell Biol. (2000) 151:1067–80. doi: 10.1083/jcb.151.5.1067
148. Bretscher A, Gary R, Berryman M. Soluble ezrin purified from placenta exists as stable monomers and elongated dimers with masked C-terminal ezrin-radixin-moesin association domains. Biochemistry. (1995) 34:16830–7. doi: 10.1021/bi00051a034
149. Nakamura F, Amieva MR, Furthmayr H. Phosphorylation of threonine 558 in the carboxyl-terminal actin-binding domain of moesin by thrombin activation of human platelets. J Biol Chem. (1995) 270:31377–85. doi: 10.1074/jbc.270.52.31377
150. Lee JH, Katakai T, Hara T, Gonda H, Sugai M, Shimizu A. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J Cell Biol. (2004) 167:327–37. doi: 10.1083/jcb.200403091
151. Pelaseyed T, Viswanatha R, Sauvanet C, Filter JJ, Goldberg ML, Bretscher A. Ezrin activation by LOK phosphorylation involves a PIP2-dependent wedge mechanism. Elife. (2017) 6:22759. doi: 10.7554/eLife.22759
152. Yoshinaga-Ohara N, Takahashi A, Uchiyama T, Sasada M. Spatiotemporal regulation of moesin phosphorylation and rear release by Rho and serine/threonine phosphatase during neutrophil migration. Exp Cell Res. (2002) 278:112–22. doi: 10.1006/excr.2002.5571
153. Belkina NV, Liu Y, Hao JJ, Karasuyama H, Shaw S. LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc Natl Acad Sci USA. (2009) 106:4707–12. doi: 10.1073/pnas.0805963106
154. Pietromonaco SF, Simons PC, Altman A, Elias L. Protein kinase C-theta phosphorylation of moesin in the actin-binding sequence. J Biol Chem. (1998) 273:7594–603. doi: 10.1074/jbc.273.13.7594
155. Ivetic A, Ridley AJ. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology. (2004) 112:165–76. doi: 10.1111/j.1365-2567.2004.01882.x
156. Mu L, Tu Z, Miao L, Ruan H, Kang N, Hei Y, et al. A phosphatidylinositol 4,5-bisphosphate redistribution-based sensing mechanism initiates a phagocytosis programing. Nat Commun. (2018) 9:4259. doi: 10.1038/s41467-018-06744-7
157. Urzainqui A, Serrador JM, Viedma F, Yanez-Mo M, Rodriguez A, Corbi AL, et al. ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity. (2002) 17:401–12. doi: 10.1016/S1074-7613(02)00420-X
158. Rozsnyay Z, Sarmay G, Zoller M, Gergely J. Membrane-bound ezrin is involved in B-cell receptor-mediated signaling: potential role of an ITAM-like ezrin motif. Immunol Lett. (1996) 54:163–9. doi: 10.1016/S0165-2478(96)02667-3
159. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood. (2010) 116:485–94. doi: 10.1182/blood-2009-12-259556
160. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med. (2008) 205:2339–47. doi: 10.1084/jem.20072660
161. Zhao L, Shey M, Farnsworth M, Dailey MO. Regulation of membrane metalloproteolytic cleavage of L-selectin (CD62l) by the epidermal growth factor domain. J Biol Chem. (2001) 276:30631–40. doi: 10.1074/jbc.M103748200
162. Gharbi SI, Avila-Flores A, Soutar D, Orive A, Koretzky GA, Albar JP, et al. Transient PKCalpha shuttling to the immunological synapse is governed by DGKzeta and regulates L-selectin shedding. J Cell Sci. (2013) 126:2176–86. doi: 10.1242/jcs.118513
163. Haribabu B, Steeber DA, Ali H, Richardson RM, Snyderman R, Tedder TF. Chemoattractant receptor-induced phosphorylation of L-selectin. J Biol Chem. (1997) 272:13961–5. doi: 10.1074/jbc.272.21.13961
164. Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med. (2013) 210:2171–80. doi: 10.1084/jem.20130664
165. Xu T, Chen L, Shang X, Cui L, Luo J, Chen C, et al. Critical role of Lck in L-selectin signaling induced by sulfatides engagement. J Leukoc Biol. (2008) 84:1192–201. doi: 10.1189/jlb.0208084
166. Brenner B, Gulbins E, Schlottmann K, Koppenhoefer U, Busch GL, Walzog B, et al. L-selectin activates the Ras pathway via the tyrosine kinase p56lck. Proc Natl Acad Sci USA. (1996) 93:15376–81. doi: 10.1073/pnas.93.26.15376
167. Corti C, Leclerc L'Hostis E, Quadroni M, Schmid H, Durussel I, Cox J, et al. Tyrosine phosphorylation modulates the interaction of calmodulin with its target proteins. Eur J Biochem. (1999) 262:790–802. doi: 10.1046/j.1432-1327.1999.00441.x
168. Evans SS, Schleider DM, Bowman LA, Francis ML, Kansas GS, Black JD. Dynamic association of L-selectin with the lymphocyte cytoskeletal matrix. J Immunol. (1999) 162:3615–24.
169. Chen Q, Wang WC, Bruce R, Li H, Schleider DM, Mulbury MJ, et al. Central role of IL-6 receptor signal-transducing chain gp130 in activation of L-selectin adhesion by fever-range thermal stress. Immunity. (2004) 20:59–70. doi: 10.1016/S1074-7613(03)00358-3
170. Dwir O, Steeber DA, Schwarz US, Camphausen RT, Kansas GS, Tedder TF, et al. L-selectin dimerization enhances tether formation to properly spaced ligand. J Biol Chem. (2002) 277:21130–9. doi: 10.1074/jbc.M201999200
171. Li X, Steeber DA, Tang ML, Farrar MA, Perlmutter RM, Tedder TF. Regulation of L-selectin-mediated rolling through receptor dimerization. J Exp Med. (1998) 188:1385–90. doi: 10.1084/jem.188.7.1385
172. Riha P, Dumas D, Latger V, Muller S, Stoltz JF. The cooperative effect of L-selectin clusters and velocity-dependent bond formation that stabilizes leukocyte rolling. Biorheology. (2003) 40:161–6.
173. Liu S, Kiick K. Architecture effects on L-selectin shedding induced by polypeptide-based multivalent ligands. Polymer Chem. (2011) 2:1513–22. doi: 10.1039/c1py00063b
174. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. (2006) 126:855–67. doi: 10.1016/j.cell.2006.08.019
175. Stamper HB Jr., Woodruff JJ. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. (1976) 144:828–33. doi: 10.1084/jem.144.3.828
176. Derry CJ, Mordsley KR, Preece G, Ager A. Purification of L-selectin ligands synthesised by rat peripheral lymph nodes and cultured high endothelial cells. Biochem Soc Trans. (1997) 25:260S. doi: 10.1042/bst025260s
177. Alon R, Rosen S. Rolling on N-linked glycans: a new way to present L-selectin binding sites. Nat Immunol. (2007) 8:339–41. doi: 10.1038/ni0407-339
178. Ceroni A, Dell A, Haslam SM. The GlycanBuilder: a fast, intuitive and flexible software tool for building and displaying glycan structures. Source Code Biol Med. (2007) 2:3. doi: 10.1186/1751-0473-2-3
179. Puri KD, Finger EB, Gaudernack G, Springer TA. Sialomucin CD34 is the major L-selectin ligand in human tonsil high endothelial venules. J Cell Biol. (1995) 131:261–70. doi: 10.1083/jcb.131.1.261
180. Sassetti C, Tangemann K, Singer MS, Kershaw DB, Rosen SD. Identification of podocalyxin-like protein as a high endothelial venule ligand for L-selectin: parallels to CD34. J Exp Med. (1998) 187:1965–75. doi: 10.1084/jem.187.12.1965
181. Samulowitz U, Kuhn A, Brachtendorf G, Nawroth R, Braun A, Bankfalvi A, et al. Human endomucin: distribution pattern, expression on high endothelial venules, and decoration with the MECA-79 epitope. Am J Pathol. (2002) 160:1669–81. doi: 10.1016/S0002-9440(10)61114-5
182. Baumheter S, Singer MS, Henzel W, Hemmerich S, Renz M, Rosen SD, et al. Binding of L-selectin to the vascular sialomucin CD34. Science. (1993) 262:436–8. doi: 10.1126/science.7692600
183. Lasky LA, Singer MS, Dowbenko D, Imai Y, Henzel WJ, Grimley C, et al. An endothelial ligand for L-selectin is a novel mucin-like molecule. Cell. (1992) 69:927–38. doi: 10.1016/0092-8674(92)90612-G
184. Berg EL, McEvoy LM, Berlin C, Bargatze RF, Butcher EC. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature. (1993) 366:695–8. doi: 10.1038/366695a0
185. Streeter PR, Rouse BT, Butcher EC. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J Cell Biol. (1988) 107:1853–62. doi: 10.1083/jcb.107.5.1853
186. Sobolev O, Stern P, Lacy-Hulbert A, Hynes RO. Natural killer cells require selectins for suppression of subcutaneous tumors. Cancer Res. (2009) 69:2531–9. doi: 10.1158/0008-5472.CAN-08-3126
187. Chen S, Kawashima H, Lowe JB, Lanier LL, Fukuda M. Suppression of tumor formation in lymph nodes by L-selectin-mediated natural killer cell recruitment. J Exp Med. (2005) 202:1679–89. doi: 10.1084/jem.20051473
188. Bogoslowski A, Butcher EC, Kubes P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus. Proc Natl Acad Sci USA. (2018) 115:2449–54. doi: 10.1073/pnas.1715756115
189. Ager A. High endothelial venules and other blood vessels: critical regulators of lymphoid organ development and function. Front Immunol. (2017) 8:45. doi: 10.3389/fimmu.2017.00045
190. Colbeck EJ, Ager A, Gallimore A, Jones GW. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or bystander sentinels in disease? Front Immunol. (2017) 8:1830. doi: 10.3389/fimmu.2017.01830
191. Kirveskari J, Paavonen T, Hayry P, Renkonen R. De novo induction of endothelial L-selectin ligands during kidney allograft rejection. J Am Soc Nephrol. (2000) 11:2358–65.
192. Toppila S, Paavonen T, Nieminen MS, Hayry P, Renkonen R. Endothelial L-selectin ligands are likely to recruit lymphocytes into rejecting human heart transplants. Am J Pathol. (1999) 155:1303–10. doi: 10.1016/S0002-9440(10)65232-7
193. Tinoco R, Otero DC, Takahashi AA, Bradley LM. PSGL-1: a new player in the immune checkpoint landscape. Trends Immunol. (2017) 38:323–35. doi: 10.1016/j.it.2017.02.002
194. Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ. PSGL-1 function in immunity and steady state homeostasis. Immunol Rev. (2009) 230:75–96. doi: 10.1111/j.1600-065X.2009.00797.x
195. Rivera-Nieves J, Burcin TL, Olson TS, Morris MA, McDuffie M, Cominelli F, et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med. (2006) 203:907–17. doi: 10.1084/jem.20052530
196. Laszik Z, Jansen PJ, Cummings RD, Tedder TF, McEver RP, Moore KL. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood. (1996) 88:3010–21.
197. Alon R, Fuhlbrigge RC, Finger EB, Springer TA. Interactions through L-selectin between leukocytes and adherent leukocytes nucleate rolling adhesions on selectins and VCAM-1 in shear flow. J Cell Biol. (1996) 135:849–65. doi: 10.1083/jcb.135.3.849
198. Shigeta A, Matsumoto M, Tedder TF, Lowe JB, Miyasaka M, Hirata T. An L-selectin ligand distinct from P-selectin glycoprotein ligand-1 is expressed on endothelial cells and promotes neutrophil rolling in inflammation. Blood. (2008) 112:4915–23. doi: 10.1182/blood-2008-04-153866
199. Sperandio M, Forlow SB, Thatte J, Ellies LG, Marth JD, Ley K. Differential requirements for core2 glucosaminyltransferase for endothelial L-selectin ligand function in vivo. J Immunol. (2001) 167:2268–74. doi: 10.4049/jimmunol.167.4.2268
200. Uchimura K, Kadomatsu K, El-Fasakhany FM, Singer MS, Izawa M, Kannagi R, et al. N-acetylglucosamine 6-O-sulfotransferase-1 regulates expression of L-selectin ligands and lymphocyte homing. J Biol Chem. (2004) 279:35001–8. doi: 10.1074/jbc.M404456200
201. Kawashima H, Petryniak B, Hiraoka N, Mitoma J, Huckaby V, Nakayama J, et al. N-acetylglucosamine-6-O-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nat Immunol. (2005) 6:1096–104. doi: 10.1038/ni1259
202. Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. (1998) 9:881–90. doi: 10.1016/S1074-7613(00)80653-6
203. Mitoma J, Bao X, Petryanik B, Schaerli P, Gauguet JM, Yu SY, et al. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol. (2007) 8:409–18. doi: 10.1038/ni1442
204. Leppanen A, Yago T, Otto VI, McEver RP, Cummings RD. Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J Biol Chem. (2003) 278:26391–400. doi: 10.1074/jbc.M303551200
205. Buffone A, Jr., Mondal N, Gupta R, McHugh KP, Lau JT, Neelamegham S. Silencing alpha1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J Biol Chem. (2013) 288:1620–33. doi: 10.1074/jbc.M112.400929
206. Schaff U, Mattila PE, Simon SI, Walcheck B. Neutrophil adhesion to E-selectin under shear promotes the redistribution and co-clustering of ADAM17 and its proteolytic substrate L-selectin. J Leukoc Biol. (2008) 83:99–105. doi: 10.1189/jlb.0507304
207. Green CE, Pearson DN, Christensen NB, Simon SI. Topographic requirements and dynamics of signaling via L-selectin on neutrophils. Am J Physiol Cell Physiol. (2003) 284:C705–17. doi: 10.1152/ajpcell.00331.2002
208. Smolen JE, Petersen TK, Koch C, O'Keefe SJ, Hanlon WA, Seo S, et al. L-selectin signaling of neutrophil adhesion and degranulation involves p38 mitogen-activated protein kinase. J Biol Chem. (2000) 275:15876–84. doi: 10.1074/jbc.M906232199
209. Morikis VA, Simon SI. Neutrophil mechanosignaling promotes integrin engagement with endothelial cells and motility within inflamed vessels. Front Immunol. (2018) 9:2774. doi: 10.3389/fimmu.2018.02774
210. Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker X, Bieber S, Nussbaum CF, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun. (2015) 6:6915. doi: 10.1038/ncomms7915
211. Morikis VA, Chase S, Wun T, Chaikof EL, Magnani JL, Simon SI. Selectin catch-bonds mechanotransduce integrin activation and neutrophil arrest on inflamed endothelium under shear flow. Blood. (2017) 130:2101–10. doi: 10.1182/blood-2017-05-783027
212. Kawashima H, Li YF, Watanabe N, Hirose J, Hirose M, Miyasaka M. Identification and characterization of ligands for L-selectin in the kidney. I. Versican, a large chondroitin sulfate proteoglycan, is a ligand for L-selectin. Int Immunol. (1999) 11:393–405. doi: 10.1093/intimm/11.3.393
213. Celie JW, Keuning ED, Beelen RH, Drager AM, Zweegman S, Kessler FL, et al. Identification of L-selectin binding heparan sulfates attached to collagen type XVIII. J Biol Chem. (2005) 280:26965–73. doi: 10.1074/jbc.M502188200
214. Celie JW, Rutjes NW, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. (2007) 170:1865–78. doi: 10.2353/ajpath.2007.070061
215. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. (2006) 6:633–43. doi: 10.1038/nri1918
216. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
217. Kawashima H, Watanabe N, Hirose M, Sun X, Atarashi K, Kimura T, et al. Collagen XVIII, a basement membrane heparan sulfate proteoglycan, interacts with L-selectin and monocyte chemoattractant protein-1. J Biol Chem. (2003) 278:13069–76. doi: 10.1074/jbc.M212244200
218. Kitaya K, Yasuo T. Dermatan sulfate proteoglycan biglycan as a potential selectin L/CD44 ligand involved in selective recruitment of peripheral blood CD16(-) natural killer cells into human endometrium. J Leukoc Biol. (2009) 85:391–400. doi: 10.1189/jlb.0908535
219. Wang L, Brown JR, Varki A, Esko JD. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. (2002) 110:127–36. doi: 10.1172/JCI0214996
220. Kawashima H, Hirose M, Hirose J, Nagakubo D, Plaas AH, Miyasaka M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J Biol Chem. (2000) 275:35448–56. doi: 10.1074/jbc.M003387200
221. Li YF, Kawashima H, Watanabe N, Miyasaka M. Identification and characterization of ligands for L-selectin in the kidney. II. Expression of chondroitin sulfate and heparan sulfate proteoglycans reactive with L-selectin. FEBS Lett. (1999) 444:201–5. doi: 10.1016/S0014-5793(99)00046-0
222. Stoler-Barak L, Moussion C, Shezen E, Hatzav M, Sixt M, Alon R. Blood vessels pattern heparan sulfate gradients between their apical and basolateral aspects. PLoS ONE. (2014) 9:e85699. doi: 10.1371/journal.pone.0085699
223. Mowery P, Yang ZQ, Gordon EJ, Dwir O, Spencer AG, Alon R, et al. Synthetic glycoprotein mimics inhibit L-selectin-mediated rolling and promote L-selectin shedding. Chem Biol. (2004) 11:725–32. doi: 10.1016/j.chembiol.2004.03.027
224. Hirose M, Matsumura R, Sato K, Murai T, Kawashima H. Binding of L-selectin to its vascular and extravascular ligands is differentially regulated by pH. Biochem Biophys Res Commun. (2011) 414:437–42. doi: 10.1016/j.bbrc.2011.09.123
225. Martinez P, Vergoten G, Colomb F, Bobowski M, Steenackers A, Carpentier M, et al. Over-sulfated glycosaminoglycans are alternative selectin ligands: insights into molecular interactions and possible role in breast cancer metastasis. Clin Exp Metasta. (2013) 30:919–31. doi: 10.1007/s10585-013-9592-7
226. Malhotra R, Taylor NR, Bird MI. Anionic phospholipids bind to L-selectin (but not E-selectin) at a site distinct from the carbohydrate-binding site. Biochem J. (1996) 314:297–303. doi: 10.1042/bj3140297
227. Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. (2002) 277:49175–85. doi: 10.1074/jbc.M205131200
228. Huang K, Geoffroy JS, Singer MS, Rosen SD. A lymphocyte homing receptor (L-selectin) mediates the in vitro attachment of lymphocytes to myelinated tracts of the central nervous system. J Clin Invest. (1991) 88:1778–83. doi: 10.1172/JCI115498
229. Tamatani T, Kuida K, Watanabe T, Koike S, Miyasaka M. Molecular mechanisms underlying lymphocyte recirculation. III. Characterization of the LECAM-1 (L-selectin)-dependent adhesion pathway in rats. J Immunol. (1993) 150:1735–45.
230. Huang K, Kikuta A, Rosen SD. Myelin localization of a central nervous system ligand for L-selectin. J Neuroimmunol. (1994) 53:133–41. doi: 10.1016/0165-5728(94)90023-X
231. Shikata K, Suzuki Y, Wada J, Hirata K, Matsuda M, Kawashima H, et al. L-selectin and its ligands mediate infiltration of mononuclear cells into kidney interstitium after ureteric obstruction. J Pathol. (1999) 188:93–9. doi: 10.1002/(SICI)1096-9896(199905)188:1<93::AID-PATH305>3.0.CO;2-#
232. Grewal IS, Foellmer HG, Grewal KD, Wang H, Lee WP, Tumas D, et al. CD62L is required on effector cells for local interactions in the CNS to cause myelin damage in experimental allergic encephalomyelitis. Immunity. (2001) 14:291–302. doi: 10.1016/S1074-7613(01)00110-8
233. Genbacev OD, Prakobphol A, Foulk RA, Krtolica AR, Ilic D, Singer MS, et al. Trophoblast L-selectin-mediated adhesion at the maternal-fetal interface. Science. (2003) 299:405–8. doi: 10.1126/science.1079546
234. Hirata T, Nomachi A, Tohya K, Miyasaka M, Tsukita S, Watanabe T, et al. Moesin-deficient mice reveal a non-redundant role for moesin in lymphocyte homeostasis. Int Immunol. (2012) 24:705–17. doi: 10.1093/intimm/dxs077
235. Giblin PA, Hwang ST, Katsumoto TR, Rosen SD. Ligation of L-selectin on T lymphocytes activates beta1 integrins and promotes adhesion to fibronectin. J Immunol. (1997) 159:3498–507.
236. Hwang ST, Singer MS, Giblin PA, Yednock TA, Bacon KB, Simon SI, et al. GlyCAM-1, a physiologic ligand for L-selectin, activates beta 2 integrins on naive peripheral lymphocytes. J Exp Med. (1996) 184:1343–8. doi: 10.1084/jem.184.4.1343
237. Simon SI, Burns AR, Taylor AD, Gopalan PK, Lynam EB, Sklar LA, et al. L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J Immunol. (1995) 155:1502–14.
238. Steeber DA, Engel P, Miller AS, Sheetz MP, Tedder TF. Ligation of L-selectin through conserved regions within the lectin domain activates signal transduction pathways and integrin function in human, mouse, and rat leukocytes. J Immunol. (1997) 159:952–63.
239. Tsang YT, Neelamegham S, Hu Y, Berg EL, Burns AR, Smith CW, et al. Synergy between L-selectin signaling and chemotactic activation during neutrophil adhesion and transmigration. J Immunol. (1997) 159:4566–77.
240. Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol. (2004) 172:7780–90. doi: 10.4049/jimmunol.172.12.7780
241. Duchesneau P, Gallagher E, Walcheck B, Waddell TK. Up-regulation of leukocyte CXCR4 expression by sulfatide: an L-selectin-dependent pathway on CD4+ T cells. Eur J Immunol. (2007) 37:2949–60. doi: 10.1002/eji.200737118
242. Perfilyeva YV, Kustova EA, Urazalieva NT, Baisheva SA, Aubakirova AT, Tleulieva RT, et al. Effects of L-selectin stimulation of the expression of chemokine receptor CXCR4 on NK cells of healthy donors and tumor patients. Bull Exp Biol Med. (2012) 153:86–8. doi: 10.1007/s10517-012-1650-7
243. Ding Z, Issekutz TB, Downey GP, Waddell TK. L-selectin stimulation enhances functional expression of surface CXCR4 in lymphocytes: implications for cellular activation during adhesion and migration. Blood. (2003) 101:4245–52. doi: 10.1182/blood-2002-06-1782
244. Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. (2000) 287:1040–6. doi: 10.1126/science.287.5455.1040
245. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. (2000) 287:1049–53. doi: 10.1126/science.287.5455.1049
246. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. (1999) 144:1235–44. doi: 10.1083/jcb.144.6.1235
247. Cho SY, Klemke RL. Purification of pseudopodia from polarized cells reveals redistribution and activation of Rac through assembly of a CAS/Crk scaffold. J Cell Biol. (2002) 156:725–36. doi: 10.1083/jcb.200111032
248. Castellano E, Downward J. RAS Interaction with PI3K: More than just another effector pathway. Genes Cancer. (2011) 2:261–74. doi: 10.1177/1947601911408079
249. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. (1994) 370:527–32. doi: 10.1038/370527a0
250. Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. (2000) 20:1461–77. doi: 10.1128/MCB.20.5.1461-1477.2000
251. Vedham V, Phee H, Coggeshall KM. Vav activation and function as a rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol Cell Biol. (2005) 25:4211–20. doi: 10.1128/MCB.25.10.4211-4220.2005
Keywords: L-selectin (CD62, SELL), migration, inflammation, signaling, adhesion, monocyte, lymphocyte, Neutrophil (PMN)
Citation: Ivetic A, Hoskins Green HL and Hart SJ (2019) L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 10:1068. doi: 10.3389/fimmu.2019.01068
Received: 15 February 2019; Accepted: 26 April 2019;
Published: 14 May 2019.
Edited by:
Toshiyuki Murai, Osaka University, JapanReviewed by:
Francis William Luscinskas, Harvard Medical School, United StatesCopyright © 2019 Ivetic, Hoskins Green and Hart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandar Ivetic, YWxla3NhbmRhci5pdmV0aWNAa2NsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.