 Doumet Georges Helou
Doumet Georges Helou Stefan F. Martin
Stefan F. Martin Marc Pallardy
Marc Pallardy Sylvie Chollet-Martin
Sylvie Chollet-Martin Saadia Kerdine-Römer
Saadia Kerdine-Römer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 May 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01004
Exposure to certain chemicals disturbs skin homeostasis. In particular, protein-reactive chemical contact sensitizers trigger an inflammatory immune response resulting in eczema and allergic contact dermatitis. Chemical sensitizers activate innate immune cells which orchestrate the skin immune response. This involves oxidative and inflammatory pathways. In parallel, the Nrf2/Keap1 pathway, a major ubiquitous regulator of cellular oxidative and electrophilic stress is activated in the different skin innate immune cells including epidermal Langerhans cells and dermal dendritic cells, but also in keratinocytes. In this context, Nrf2 shows a strong protective capacity through the downregulation of both the oxidative stress and inflammatory pathways. In this review we highlight the important role of Nrf2 in the control of the innate immune response of the skin to chemical sensitizers.
Long ago, the skin was considered as a passive physical barrier protecting the internal organs from environmental assaults. In 1978, Streilein proposed that the skin has its own integrated immune function and introduced the term of skin-associated lymphoid tissues, “SALT.” He suggested that this specialized function is mediated by keratinocytes, Langerhans cells (LC) and immune-competent lymphocytes working in concert to ensure an efficient protection (1, 2). Nowadays, it is clear that the skin is an important part of the immune system. The main resident innate immune cells in the skin, such as LC in the epidermis or dendritic cell (DC) subpopulations, mast cells and macrophages in the dermis, supervise the most exposed organ, react rapidly to danger signals resulting from biological or chemical hazards and orchestrate the immune response. In addition, keratinocytes, and fibroblasts contribute to the immune response e.g., by chemokine release which leads to a rapid recruitment of circulating immune cells, mainly neutrophils and pro-inflammatory monocytes (3).
Although they form a complex and well-structured physical barrier constituting 90% of the epidermis, it is well established that keratinocytes play a major role in skin immunity, as a connector between environmental signals and the underlying immune cells (4, 5). In particular, keratinocytes can detect pathogens via their Toll-like receptors (TLRs) (6) and produce antimicrobial peptides (AMPs) such as cathelicidin and β-defensins (7). In addition, keratinocytes can produce several chemokines such as CCL1, CCL2, CCL5, CCL11, CCL13, CCL17, CCL18, CCL20, and CCL22 as well as pro-inflammatory cytokines such as IL-1β and IL-18 (8, 9). Interestingly, a recent study revealed that keratinocyte can produce IL-23 known to orchestrate chronic skin inflammation in psoriasis (10).
Discovered by Paul Langerhans in 1868, LC are antigen presenting cells deriving from the monocytic lineage and represent 2 to 4% of the epidermis (11, 12). Not long ago, LC were defined as epidermal DC that migrate to the draining lymph node and prime antigen-specific T cells. Nowadays, LC are considered to be a subset of tissue-resident macrophages that share key properties with macrophages such as embryonic origin and their capacity to self-renew within the epidermis (13, 14). LC are characterized by their expression of langerin (CD207), a C-type lectin receptor (CLR) required for antigen recognition and internalization that result in the formation of Birbeck granules (15). While LC capacity to cross-present antigens to CD8+ T cells remains a topic of debate (16–18), it is proven that LC ensure immune tolerance and protection in contact hypersensitivity (CHS) through the inhibition allergen-specific CD8+ T cells and the activation of specific regulatory T cells expressing the inducible T cell co-stimulator (ICOS) (19).
Skin DC are specialized cells that recognize and process antigens in the skin and then migrate to the draining lymph nodes to promote a T effector response. Skin DC present many subtypes such as dermal DC (dDC), conventional DC (cDC) and monocyte-derived DC (mDC) (20, 21). dDC represent the major DC population in the skin and could be divided into two populations: Langerin+ dDC that co-express the CD103 in mice or CD141high in humans and Langerin− dDC in mice or CD14−/CD1a+ in humans (22). Langerin + dDC and not LC play a key role in inducing immune responses and present a unique cross-presentation capacity among skin DC populations (18, 23, 24).
Macrophages are antigen-presenting cells better recognized for their innate functions, in particular their heterogeneity-defining phagocytic capacity during pathogen infection and tissue damage (25). Skin-resident macrophages have an embryonic origin and are self-maintained through proliferation, while infiltrating macrophages are derived from Ly-6Chi inflammatory monocytes. Nevertheless, monocytes can enter the skin in the steady state and give rise to macrophages or DC, or re-enter the circulation without any differentiation (26). Resident macrophages can be classically activated (M1) through pattern-recognition receptors (PRR) signaling pathways triggering the production of pro-inflammatory cytokines such as IL-12, tumor necrosis factor (TNF) and IL-23. Moreover, macrophages can be alternatively activated during Th2-mediated immune responses under the influence of IL-4 and IL-13 (8, 27). These alternatively activated macrophages, referred to as regulatory M2 macrophages, display distinct functions and phenotypes according to their origin, but generally promote the resolution of skin inflammation partly via IL-10 and transforming growth factor-β (TGFβ) secretion (28, 29). In CHS, M2 macrophages secrete CXCL2 to attract dDC leading to an accumulation of DC–T cell clusters in skin. Therefore, depletion of monocytes/macrophages before elicitation suppresses the inflammatory response (30, 31).
The rapid recruitment of neutrophils into the skin is a key protection mechanism against infections. In line with resident cell activation, in particular keratinocyte activation, neutrophils infiltrate the skin toward inflammatory mediators such as IL-1α, IL-1β, IL-6, TNF, CXCL1, and CXCL8 (8). Although neutrophils ensure an efficient protection against pathogens, their inappropriate activation can be responsible for severe skin inflammatory and autoimmune diseases (32). In psoriasis, neutrophils secrete IL-17 affecting thus the IL-23/IL-17 axis balance and maintaining the positive feedback loop of cutaneous inflammation (33, 34). While recent studies have evidenced that neutrophil recruitment to the skin during both phases of CHS is essential to induce a cutaneous inflammatory response, the debate is still open in relation to neutrophil involvement in antigen presentation (35–37).
Innate immune cells are the frontline of protection against invading pathogens and react immediately to infection or trauma. They are equipped with a large variety of PRR. These include TLRs, nucleotide binding oligomerization domain (NOD)-like receptors (NLRs) and RNA helicases (RIG-I (retinoid acid-inducible gene-I) that detect both microbial pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs). In addition, scavenger receptors such as CD36 and macrophage receptor with collagenous structure (MARCO) are mainly expressed on macrophages and may recognize and internalize several self and microbes-derived ligands. PRR-PAMP/DAMP interactions induce the activation of a panel of intracellular signaling pathways including TLR- and NLR-dependent signaling pathways, kinases and transcription factors (38, 39). The downstream signal cascades are therefore associated with gene transcription and expression, leading mainly to the production of inflammatory cytokines. Among PRRs, TLRs represent the major class of receptors. They are highly expressed on sentinel cells including innate immune cells and non-immune cells like fibroblasts. TLRs are non-catalytic receptors formed by an extracellular binding domain and a cytoplasmic signaling Toll/interleukin-1 (IL-1) receptor homology (TIR) domain. However, some TLRs including TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13, are exclusively intracellular. They lodge in the endosomes and recognize principally nucleic acids. In parallel, TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 represent cell surface TLRs and recognize mainly structurally conserved motifs from pathogen membranes. TLR signaling is initiated with homo- or heterodimer formation following the recruitment of TIR domain-containing adaptors including MyD88, TRIF, TIRAP/MAL, or TRAM. MyD88 and TRIF are considered as the major downstream adaptors while TIRAP and TRAM are sorting adaptors that could mediate the signal from TLR to MyD88 and TRIF, respectively. The MyD88-dependent response is very common in different TLR subfamilies except TLR3, leading to the activation of nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs) and subsequently the expression of genes coding for key inflammatory cytokines. The TRIF-dependent pathway is mainly triggered by TLR3 and TLR4 and results in the production of interferon type I as well as NF-κB and MAPKs activation (40–42).
The skin is frequently exposed to chemical stress by organic chemicals or metal ions that can directly or indirectly challenge its immune components and may lead to T cell-mediated delayed type hypersensitivity reactions. As for pathogens, the recognition of these chemicals depends upon PRR expression on sentinel skin cells, mainly the innate resident immune cells. Therefore, these cells could define the global immune reaction: offensive or dangerous non-self, immune tolerance or harsh hypersensitivity.
Allergic contact dermatitis (ACD) is a common inflammatory disease due to skin sensitization by chemical allergens. CHS is a well-accepted mouse model of ACD reproducing the multi-step process of skin sensitization and elicitation in human ACD. It is a manifestation of immune hypersensitivity, in contrast to irritant contact dermatitis (ICD) that results from a direct toxic effect of chemical agents on skin cells involving the innate immune system but not generating an adaptive immune response. The important prevalence of ACD, ~20%, is associated with an increased exposure to environmental and industrial products (43). Chemical sensitizers constitute an important group of low molecular weight contact allergens (MW < 500 Dalton) and behave as haptens. These include organic chemicals as usually found in fragrances, adhesives, preservatives, dyes and metal ions such as nickel, cobalt and chromium (44, 45). Complete haptens are highly reactive electrophilic molecules that bind directly to nucleophilic chains of self-proteins. Pro- and pre-haptens require enzymatic and oxidative transformation, respectively, to become reactive. Several factors drive protein haptenation within the skin proteome including the nucleophilicity, steric hindrance, competition for binding and local pH (46). While the nature of the hapten-modified proteins with functional relevance for ACD is largely unknown, it is clear that protein haptenation underlies the activation of innate immune and stress responses as well as the formation of T cell epitopes. In addition to haptenation, other types of protein modification may play an important role, e.g., oxidation as a consequence of reactive oxygen species (ROS) production induced by contact sensitizers. A disturbed redox balance may lead to thiol modification in proteins altering conformation and function (47). Thus, it has been shown that contact sensitizers induce the oxidation of cell surface thiols and that this contributes to the maturation of DC (48).
It is well established that following first skin penetration during the asymptomatic sensitization phase, haptens bind to skin proteins in order to become immunogenic. Haptens can induce oxidative stress responses in skin resident cells participating in a rapid recruitment of inflammatory cells, notably neutrophils, and an amplified activation of the cutaneous innate immunity. Subsequently, LC and dDC process haptenated proteins and migrate to the draining lymph node to prime hapten-specific T cells with a preferential Tc1/Th1 and Tc17/Th17 polarization. It is important to note here that this polarization could depend on hapten properties and the used experimental model (49). The elicitation phase of ACD results from repeated exposure to the same hapten and leads to skin infiltration by cytotoxic T cells. Local T cell activation by antigen-presenting cells is necessary for secreting interferon-gamma (IFN-γ) and IL-17, killing the haptenized cells and contributing to keratinocyte apoptosis (50). Hence, skin innate immune cell activation is maintained and amplified due to T cell cytokines and DAMPs. In this context, CD4+ and CD8+ T cells play an antagonistic role in the elicitation phase; while CD8+ T cells display cytotoxic functions, CD4+ T cells ensure a regulatory role to protect skin components from chronic inflammation. Furthermore, the resolution of inflammation relies on regulatory T cell recruitment to exert inhibitory functions in the inflamed skin. Indeed, many studies have reported a suppressive role of regulatory T cells, both during the sensitization and the elicitation phase of CHS, as their depletion results in a prolonged and exacerbated ear swelling response while their expansion reduces effector cells recruitment leading to a maintained suppression of CHS (51–53).
The sensitization potential of haptens critically depends on their irritancy, considered as necessary to promote stress responses and to supply innate immune cells with further danger signals (44, 54). The direct hapten-induced inflammation, called xenoinflammation (55), produces DAMPs that are endogenous danger signals including hyaluronic acid (HA) fragments, biglycan, heat shock proteins, uric acid and extracellular adenosine triphosphate (ATP) (56–58). Some of these DAMPs trigger TLRs which normally recognize bacterial and viral components. This is the case for TLR2 and TLR4 in the CHS response. They are triggered by HA fragments or biglycan resulting in MAPKs and NF-κB activation and production of pro-inflammatory cytokines and chemokines. Among these cytokines are pro-IL1β and pro-IL18 which are important in contact dermatitis. Their cleavage to the mature bioactive forms is achieved by contact allergen-mediated activation of the pyrin domain containing 3 (NLRP3) inflammasome (59). This involves ROS and in some cases extracellular ATP. This TLR-NLRP3 inflammasome axis seems to be a common mechanism underlying skin inflammation as shown for several contact allergens in the CHS model and in human ACD.
Mice deficient in both TLR2 and TLR4 failed to develop CHS that was normal in germ-free mice. It was shown that contact allergens trigger the production of HA fragments, endogenous host-derived DAMPs which then trigger TLR2 and TLR4 (60). Moreover, a recent study has revealed that TLR3 signaling participates in the induction of chronic CHS (61).
Upon their penetration, chemical sensitizers induce ROS production in the skin, participating in the breakdown of the extracellular matrix (ECM) (56). Among the released endogenous signals, ROS and hyaluronidase-dependent HA degradation may lead to TLR2- and TLR4-mediated DC activation (56). Direct TLR activation has been shown for nickel and cobalt ions, known for their capacity to engage two non-conserved histidine residues in human TLR4, absent in mouse TLR4 (62, 63). Palladium works in a similar manner (64). For cadmium, a toxic heavy metal that can cause sensitization, an induction of ROS production has been shown and one recent study claims a role for TLR4 in the induction of mucin 8 in human airway epithelial cells via activation of MAPKs (65).
Along with TLRs, NLRs form cytosolic multi-protein complexes called inflammasomes that activate caspase-1 leading to the cleavage of the pro-inflammatory cytokines IL-1β and IL-18 into their bioactive forms (66). Different mouse models have supported the involvement of the inflammasome in skin innate responses to chemical sensitizers, and a role for the danger signal extracellular ATP in its activation via the P2X7 receptor was demonstrated. LC migration is also impaired in caspase-1−/− mice exposed to the 1-fluoro-2,4-dinitrobenzene (DNFB) (67–70). Moreover, activation of the NLRP3 inflammasome by nickel in the human system has recently been reported (71). Many NLRs involved in the regulation of inflammation have been studied in the CHS model. It was shown that NLRP12−/− mice display a reduced CHS response to either oxazolone or fluorescein isothiocyanate (FITC). This was associated with a reduced migration of DCs from skin to draining lymph nodes. Mice lacking NLRP10 also had reduced CHS to DNFB, but not to the irritant croton oil. Interestingly, the phenotype was also seen in mice lacking NLRP10 specifically in keratinocytes. In this study the reason for the reduced CHS response was not clear. A reduction of TNF-α and CXCL1 in skin was observed indicating a reduced skin inflammation and reduced neutrophil infiltration (68, 72).
Besides direct or indirect PRR triggering, proteomic-based approaches are nowadays suggesting that chemical sensitizers may trigger intracellular signaling pathways via direct interaction with cellular proteins. For example, FITC activates p38 and c-Jun N-terminal kinase (JNK) pathways through direct haptenation of specific amino acid residues within critical intracellular proteins in THP-1 cells (human leukemic monocyte) (73). Other molecules like DNFB, induce ROS production through direct interaction and depletion of cytoplasmic glutathione (GSH) in THP-1 cells (74).Within this context, we have previously used an omic-based approach to identify proteomic alterations in DCs upon exposure to sensitizers such as dinitrochlorobenzene (DNCB), cinnamaldehyde (CinA) and Nickel II sulfate. Spot pattern analyses pointed out a metabolic reprogramming in DC (oxidative stress responses and phase II metabolism) and evidenced a positive correlation between the reactivity and the potency of the applied chemical. A total of 100 proteins regulated by the sensitizers was identified and showed a specific profile for each sensitizer (75). Such an approach based on chemical reactivity provides thereby valuable biomarker candidates for sensitizers and would be of great benefit to immuno-toxicology discipline.
As stated previously, redox homeostasis shapes the innate immune response to chemical sensitizers and protects against inflammation. Up to now, Nrf2 (nuclear factor erythroid-2-related factor 2) is considered as the master cytoprotective transcription factor that could dictate the outcome of oxidative and inflammatory pathways within innate immune cells. Although numerous studies have evidenced the regulatory role of Nrf2 in different inflammatory contexts, few have elucidated its capacity to manage skin primary immune responses to chemical sensitizers.
Nrf2 constitute a major mechanism in the regulation of cellular oxidative stress, particularly the detoxification and excretion of both organic xenobiotics and toxic metals (76, 77). Given its cytoprotective functions, an evolutionary conservation of Nrf2 has been defined in vertebrates, with a protein homology ranging from 49% in zebrafish to 89% in cows when compared to humans (78). Nrf2 activity is tightly regulated by its cytosolic repressor Kelch-like ECH-associated protein 1 (Keap1). At basic state, Nrf2 is sequestrated in the cytoplasm by its inhibitor the Kelch-like ECH-associated protein 1 (Keap1). Keap1 forms an E3 ubiquitin ligase complex with Cullin 3 and Ring-box 1 and triggers polyubiquitination of Nrf2 by promoting its binding to Cullin 3. Ubiquitinated Nrf2 is then degraded by the 26S proteasome with a half-life of ~20 min. Exposure to oxidants or electrophilic stress cause conformational changes in the Keap1–E3 ubiquitin ligase complex that block Nrf2 ubiquitination and favor its accumulation. Nrf2 then rapidly translocates into the nucleus, dimerizes with the small Maf protein, and binds to the antioxidant-responsive elements (AREs) in gene promoters. Consequently, Nrf2 initiates the transcription of a battery of cytoprotective genes such as NAD(P)H: quinone oxidoreductase 1 (nqo1), heme oxygenase-1 (ho-1), glutathione S-transferase (gst), catalase, superoxide dismutase (sod) and glutamate-cysteine ligase modifier subunit (gclm) (76, 79, 80) (Table 1). Among Nrf2 downstream targets, nqo1 is one of the most robustly inducible genes and its activation is strictly dependent on Nrf2 (81).
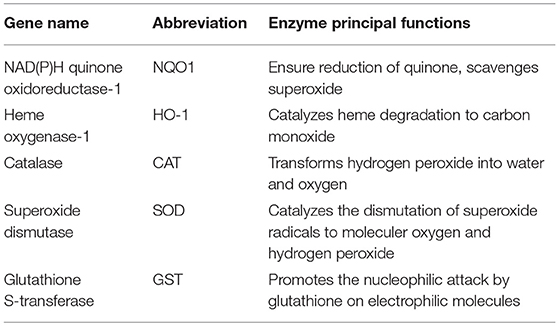
Table 1. Principal functions of five antioxidant enzymes encoded by Nrf2 target genes.
Several Nrf2 activators have been identified, with a barn capacity to modify specific cysteines in Keap1 that is known to be a cysteine-rich protein with more than 25 cysteine residues. For instance, sulforaphane (SFN) is an isothiocyanate compound derived from cruciferous vegetables (such as broccoli) used as a pharmacological activator of Nrf2 (82). SFN has a preference to modify the cysteine 151 leading to an alteration in the Keap1-Nrf2-Culllin 3 complex conformation and to further stabilization and accumulation of Nrf2 (83).
Experimental studies have evidenced that the absence of Nrf2 is always associated with an exacerbated inflammation in different animal model. The Nrf2 anti-inflammatory role is most likely attributed to a deep interaction between Nrf2 and many redox sensitive inflammatory pathways including NF-κB and inflammasomes.
IκB, the negative regulator of NF-κB transcription factor, is phosphorylated by IκB kinase (IKK) during oxidative stress, leading to the release and nuclear translocation of NF-κB. The latter induces the transcription of pro-inflammatory mediators such as IL-6, TNF-α, and cyclooxygenase-2 (COX-2) (84). Nrf2 and NF-κB signaling pathways interact via multiple mechanisms. It has been demonstrated that Keap1 mediates IKK proteasomal degradation leading to NF-κB inhibition. This could result in a competitive binding between Nrf2 and IKK with Keap1 (85). The increased HO-1 activity that depends on Nrf2 activation, also participates in NF-κB inhibition (86). In parallel, the canonical NF-κB subunit p65 can alter the Nrf2 pathway. Indeed, overexpression of p65 leads to the translocation of Keap1 into the nucleus and the disruption of Nrf2-ARE interactions (87). In addition, p65 and Nrf2 compete for the transcriptional co-activator CBP (CREB-binding protein) that acetylates non-histone proteins to improve assembly of the transcriptional machinery (88) (Figure 1).
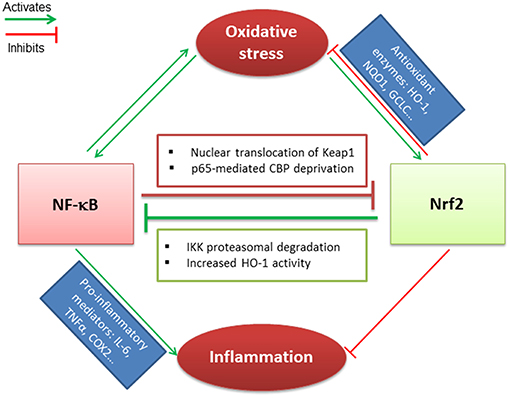
Figure 1. Crosstalk between Nrf2 and NF-κB transcription factors. Nrf2 and NF-κB related pathways are sensitive to oxidative stress that induces their activation. Nrf2 antagonizes NF-κB activation through IKK proteasomal activation and HO-1 activity end-products. In parallel, NF-κB downregulates Nrf2 via p65-mediated CBP deprivation as well as p65-induced Keap1 nuclear translocation. Nrf2 and NF-κB are regulated antagonistically: (i) if Nrf2 predominates, it decreases inflammation and oxidative stress through activation of antioxidant enzymes; (ii) when NF-κB predominates, it leads to pro-inflammatory mediators secretion and maintains the oxidative stress.
NF-κB activation is required for NLRP3 inflammasome priming, while Nrf2 activation could inhibit both pathways to limit inflammation. The activation of NLRP3 mediates IL-1β secretion followed by the process of cell death known as pyroptosis (89). Nrf2 reduces ROS levels through the activation of antioxidant target genes and inhibits NLRP3 priming. Indeed, a recent study has evidenced that Nrf2 is a key inhibitor of the NLRP3 inflammasome via the activation of the thioredoxin1 in cerebral ischemia reperfusion injury (90). Last, the multi-domain protein p62 provides a critical link between Nrf2, NF-κB and NLRP3 inflammasome pathways. Nrf2 and NF-κB contribute to p62 induction in oxidative stress, while a p62-mediated positive feedback loop supports NF-κB as well as Nrf2 activation through the autophagosomal degradation of Keap1. However, p62 inhibits the NRLP3 inflammasome (91, 92). Thus, p62 protein maintains a cellular homeostasis to ensure resistance to redox stress and inflammation depending on Nrf2.
Although literature screening suggests that Nrf2 participates in the control of inflammation through inhibitory crosstalk with redox sensitive pathways, emerging studies are suggesting a possible redox-independent regulation. Therefore, Nrf2 could directly regulate the expression of key genes to alleviate excessive immune reaction. We do not yet have enough hindsight to define all Nrf2 anti-inflammatory mechanisms; however there is a marked tendency to admit a greater role for Nrf2 in inflammation in relation with immune cell populations and pathophenotypes. In this regard, studies are continuing to report particular mechanism of regulation that is specific to chemical skin sensitization, notably to define cells- and/or chemical-specific mechanisms (86, 91).
Nrf2 is a ubiquitously expressed transcription factor required for skin homeostasis mostly through modulation of the redox balance. Indeed, the electrophilic property of chemical sensitizers promotes the activation of Nrf2 in the skin. However, Nrf2 can modulate innate immune cell activation and function in different ways. Using a model of normal human epidermal keratinocytes (NHEK), it has been demonstrated that Nrf2 improves keratinocyte differentiation through the upregulation of specific differentiation markers such as loricrin and keratin 10 (93). Interestingly, a recent study has revealed that Nrf2 expression in keratinocytes ensures a rescue effect on UV-induced DNA damage and apoptosis in melanocytes (94). Furthermore, Nrf2 activation increases IL-36γ expression in keratinocytes leading to an increased proliferation (95). In psoriasis, Nrf2 is activated in response to inflammatory cytokines and enhances the expression of hyperproliferation-related keratins (96). Few studies have focused on the role of Nrf2 in the immune response of keratinocytes to chemical sensitizers, in particular its effect on cytokine production. Nevertheless, given its importance in sensing chemical stress, evaluation of Nrf2 activation in keratinocyte in vitro models becomes a useful tool participating in the prediction of chemical skin sensitization potential (97, 98). OECD guideline test 442D is based on a luciferase reporter of Nrf2 activation in the human keratinocyte cell line HaCaT (99).
Numerous studies have specifically investigated the role of Nrf2 in the DC response to chemical sensitizers in different cell models. We have demonstrated that chemical sensitizers such as nickel, DNCB and CinA were able to increase Nrf2 protein level in CD34-DC and THP-1 cells associated with an up-regulation of the target genes ho-1 and nqo1, while irritants such as sodium dodecyl sulfate (SDS) and benzalkonium chloride (BZK) failed to induce Nrf2 accumulation. A positive correlation between Nrf2 target gene induction and chemical reactivity to cysteine has been established; however lysine-reactive chemicals were unable to activate Nrf2. These results proved that chemical reactivity of contact sensitizers provides necessary danger signals for Nrf2-dependent DC activation (100, 101). The use of Nrf2 knock-out (KO) mice allowed pinpointing the specific effects of Nrf2 in the DC response to chemical sensitizers. Indeed, using a proteomic approach, we identified a set of Nrf2-dependent and Nrf2-independent promising biomarkers that are regulated in hapten-induced DC activation. Thus, 7 ARE-containing Nrf2 targets were described relevant in response to chemical sensitizers: HSPA9, voltage-dependent anion-selective channel protein 1 (VDAC1), glutathione S-transferase omega-1 (GSTO1), ferritin light chain 1 (FTL1), peroxiredoxin 1 (PRDX1), SOD2 and transketolase TKT (75). Then using a transcriptional approach, we showed that the lack of Nrf2 in BMDC prevents the up-regulation of antioxidant genes in response to DNCB and CinA leading notably to a higher production of ROS and reduced levels of intracellular GSH. Furthermore, Nrf2 was able to control the chemically-induced apoptotic pathways in DC through the up-regulation of bcl-2 gene expression (102). Apart from chemical sensitizers, the majority of studies in different inflammatory contexts revealed that Nrf2 controls DC activation, notably the expression of co-stimulatory molecules such as CD86 and CD80 and affects the subsequent activation of T cells (103–106). Although Nrf2 may play a pivotal role in all skin immune cell populations, only its involvement in DC responses to chemical sensitizers was widely investigated, while its implication in other population such as LC, macrophages and neutrophils remains underappreciated. One question raises in all cases: is the anti-inflammatory role of Nrf2 independent from its capacity to regulate oxidative stress?
The best answer could be found in the study of Nrf2 implication in macrophage biology. A breakthrough study revealed that Nrf2 directly counteracts the transcription of pro-inflammatory cytokine genes revising a long-held view that Nrf2 regulates inflammation by redox control. This study provides a chromatin immunoprecipitation (Chip-seq)-based molecular approach suggesting that Nrf2 binds to the proximity of the IL-6 and IL-1β gene transcription start site, interfering thus with the recruitment of RNA polymerase. In contrast to the classically adopted hypothesis, this mechanism is totally independent from ROS levels and ARE, and is not a simple consequence to anti-oxidant upstream event (107). In line with these data, Nrf2 has the capacity to up-regulate the expression of MARCO and CD36, two scavenger receptors that are highly required for bacteria phagocytosis and elimination of apoptotic neutrophils, respectively, promoting thus the resolution of inflammation (108–111).
Although clear evidence about the role of Nrf2 in macrophage functions has emerged, its involvement in neutrophil biology remains a topic of debate. Many studies have argued a correlation between Nrf2 activation in neutrophils and the modulation of their hyper-reactive phenotype in inflammatory contexts such as sepsis and chronic periodontitis (112–114). More recently, a transgenic mouse model displaying Nrf2-specific deletion in myeloid cells was created. Interestingly, high Nrf2 mRNA levels were quantified in neutrophils of wound tissue and blood, 10-fold higher than in monocytes and lymphocytes. However, Nrf2 deletion in myeloid cells did not exacerbate the inflammatory response or alter the wound healing capability in this study (115). These striking results may put into question the role of Nrf2 in neutrophils suggesting the existence of redundant and alternative antioxidant mechanisms. Nevertheless, neutrophils are among the first cells to be recruited to chemically-treated skin in response to chemokines secreted by resident cells (36). Thus, their recruitment and their state of activation may indirectly rely on Nrf2 expression in skin resident cells.
Altogether, Nrf2 is a key regulator of innate immune cells, in particular the most common pathway induced by chemical sensitizers (Figures 2, 3). Consistently, Nrf2 is actually considered as a key biomarker able to differentiate contact sensitizers from non-sensitizers. Furthermore, the important need for alternatives to animal testing, led to the development of a keratinocyte-based in vitro assay, relying on Nrf2 activation as a key event to identify sensitizers (97, 99, 116). Although numerous toxicological studies have characterized at the molecular level the underlying mechanisms in the principal skin cells (keratinocytes and DC), few immunological studies took into consideration the complexity of interactions between the different actors of the immune system that could drive the response to chemical sensitizers in an unpredictable way. To overcome the lack, our group addressed the question in vivo, using a Nrf2 KO mouse model. In response to moderate and strong chemical sensitizers, CinA and DNCB respectively, Nrf2 decreases skin inflammation during the elicitation phase of CHS in a concentration-dependent manner. Indeed, the mouse ear swelling test showed an approximatively 2-fold higher percentage of ear thickness increase, 48 h after challenge. Moreover, using the local lymph node assay (LLNA), we detected an enhanced proliferation of lymph node cells during the sensitization phase of CHS, suggesting that Nrf2 controls activation and migration of skin DC. This enhanced proliferation was not observed exclusively in response to DNCB and CinA that react specifically with cysteine residues, but also in response to trimethylaluminium (TMA) that reacts with lysine residues and to mixed reactive compound like isophorone diisocyanate (IPDI). Interestingly, Nrf2 is also able to regulate skin irritation in response to croton oil which fails to induce Nrf2 in vitro (117). This last result is in accordance with a recent study showing the Nrf2 target gene nqo1 controls skin irritation in response to croton oil through the protection and maintenance of dendritic epidermal T cells (DETC) (118).
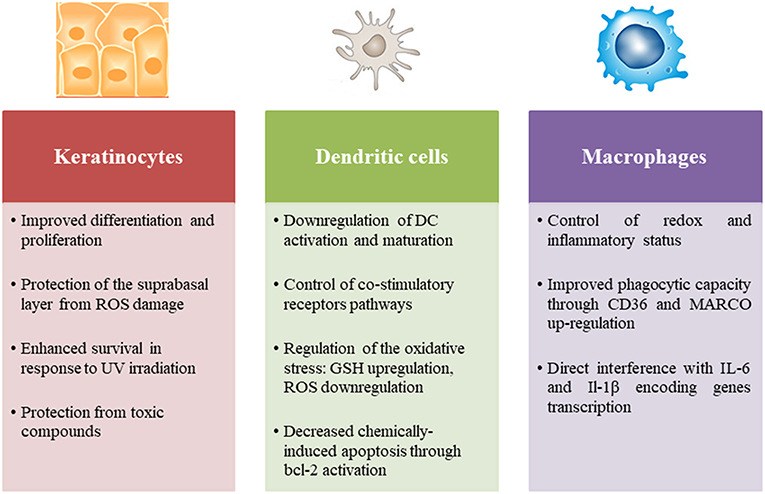
Figure 2. Role of Nrf2 in the principal skin innate immune cells including keratinocytes, dendritic cells, and macrophages.
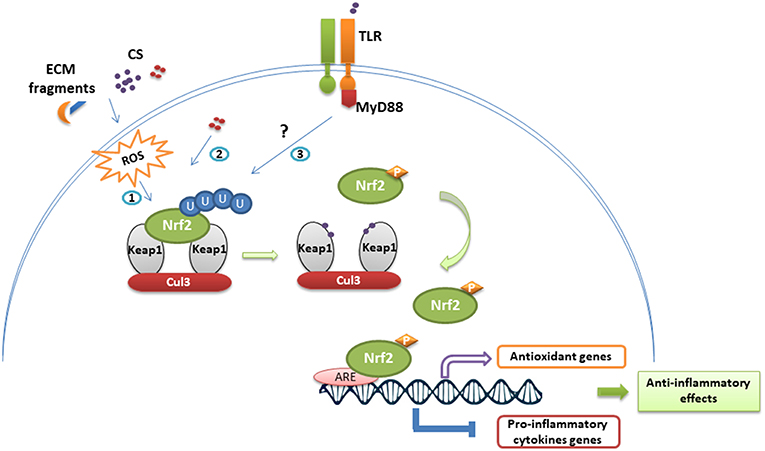
Figure 3. Nrf2 stabilization and anti-inflammatory effects following immune cell exposure to chemical sensitizers. In homeostatic conditions, Nrf2 is continuously repressed by the Keap1-Cul3 complex and ubiquitinylated. Exposure to chemical sensitizers (CS) activates the Nrf2/Keap1 pathway through different ways: (1) ROS-mediated activation resulting from CS irritancy and degradation of extracellular matrix (ECM), (2) direct chemical modification of Keap1, (3) TLR-dependent activation. Thus, modifications on Keap1 lead to Nrf2 stabilization and phosphorylation. Nrf2 translocates then to the nucleus where it binds to ARE sequences promoting the transcription of antioxidant genes and inhibiting the transcriptional activity of several genes encoding pro-inflammatory cytokines.
These in vivo studies in the CHS model clearly show that the threshold for sensitization to contact allergens is significantly lowered in Nrf2 KO mice. This is most likely due to the increased inflammatory response in absence of Nrf2 due to loss of redox homeostasis. A single contact sensitizer application, but also croton oil application produces a significantly increased ear swelling compared to wildtype mice. The fact that Nrf2 KO mice can even mount CHS responses to weak contact sensitizers which fail to do so in wildtype mice underlines the crucial role of the Keap1/Nrf2 system in balancing inflammation and immuno-regulation which are inversely correlated. A recent in vivo study conducted by our group has dissected the mechanisms allowing Nrf2 to regulate CHS, more specifically the early immune events following skin sensitization. Interestingly, Nrf2 inhibits neutrophil recruitment to the skin that was dependent on ROS elimination and is able to shorten the persistence of neutrophils in the inflamed skin. Indeed, Nrf2 enhances neutrophil clearance by resident macrophages through a direct activation of the macrophage-specific gene encoding CD36, which is essential for efferocytosis. In this regard, the mentioned study reports a novel Nrf2-regulated mechanism of CHS development independently from the redox balance (119). Of note, the sum of different mechanisms allows Nrf2 to play a remarkable role in inhibiting the sensitization process.
Interestingly, we have evidence now that Nrf2 downregulates the adaptive immune response in CHS indirectly via the modulation of the early innate immune response to chemical sensitizers in xenoinflammation. Thus, Nrf2-dependent immuno-regulation started very early after chemical sensitizer diffusion into the skin, highlighting the necessity of an optimal activation of Nrf2 during the first contact between the chemical and the skin in order to avoid the set-up of the sensitization process. In particular, variability in the level of Nrf2 activation in response to chemical sensitizers could bring important elements explaining the particular susceptibility of certain individuals to develop cutaneous allergies. The same concept applies to define a polymorphism in allergic patients based on the level of Nrf2 activation in the skin. Indeed, we can suggest that patients with ACD associated with low expression or reduced inducibility of Nrf2 in skin cells may benefit from specific treatment based on skin Nrf2 activation. However, a therapeutic strategy dependent on Nrf2 in the ACD should take into consideration the benefit/risk ratio. In particular, prolonged activation of Nrf2 can be dangerous and lead to alteration of the epidermal barrier, uncontrolled hyper-proliferation of keratinocytes and pro-tumorigenic effects related to immunosuppression.
Taken together, Nrf2 is a major regulator in skin immune responses to chemical sensitizers. These latter trigger inflammatory responses in skin innate immune cells associated with Nrf2 activation. Consequently, a battery of cytoprotective enzymes counterbalances the cellular oxidative stress mainly through ROS elimination. Although Nrf2 regulates pro-inflammatory cytokine pathways and efferocytosis in macrophages via direct gene regulation, further studies are required to better understand the mechanisms of Nrf2 contribution in other skin innate immune cells. Nowadays, there is a growing interest in new pertinent tests to evaluate the sensitization potential of frequently used chemicals that are flooding the market every day, with new potential sensitizers. On the other hand, several Nrf2 activators, such as modified triterpenoids, are proving anti-inflammatory protective effects in clinical trials (120, 121). Thus, the importance of Nrf2 emerges not only in risk assessment but also as a potential therapeutic target in chemically-induced skin inflammation and other diseases such as cancer.
DH wrote the manuscript and designed figures. MP critically reviewed the manuscript. SM, SC-M and SK-R critically reviewed the manuscript and applied important modifications. All authors have approved the last version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACD, Allergic contact dermatitis; ARE, Antioxidant response element; ATP, Adenosine triphosphate; CHS, Contact hypersensitivity; CinA, Cinnamaldehyde; DAMP, Damage-associated molecular patterns; DC, Dendritic cells; DNCB, dinitrochlorobenzene; GSH, Glutathione; HO-1, Heme oxygenase-1; IKK, IκB kinase; Keap-1, Kelch-like ECH-associated protein 1; KO, Knock-out; LC, Langerhans cells; NF-κB, nuclear factor-kappa B; NLR, (NOD)-like receptors; NQO1, NAD(P)H, quinone oxidoreductase 1; Nrf2, Nuclear factor erythroid-2-related factor 2; PAMP, Pathogen-associated molecular patterns; PRR, Pattern-recognition receptors; ROS, Reactive oxygen species; Tc, Cytotoxic T cells; Th, Helper T cells; TLR, Toll-like receptors; TNF, Tumor necrosis factor.
1. Streilein JW. Skin-associated lymphoid tissues (SALT): origins and functions. J Invest Dermatol. (1983) 80 (Suppl.):12s−6s.
3. Silvestre MC, Sato MN, dos Reis VMS. Innate immunity and effector and regulatory mechanisms involved in allergic contact dermatitis. An Bras Dermatol. (2018) 93:242–50. doi: 10.1590/abd1806-4841.20186340
4. Pivarcsi A, Kemény L, Dobozy A. Innate immune functions of the keratinocytes. A review. Acta Microbiol Immunol Hung. (2004) 51:303–10. doi: 10.1556/AMicr.51.2004.3.8
5. Sugita K, Kabashima K, Atarashi K, Shimauchi T, Kobayashi M, Tokura Y. Innate immunity mediated by epidermal keratinocytes promotes acquired immunity involving Langerhans cells and T cells in the skin. Clin Exp Immunol. (2007) 147:176–83. doi: 10.1111/j.1365-2249.2006.03258.x
6. Lebre MC, van der Aar AMG, van Baarsen L, van Capel TMM, Schuitemaker JHN, Kapsenberg ML, et al. Human keratinocytes express functional toll-like receptor 3, 4, 5, and 9. J Invest Dermatol. (2007) 127:331–41. doi: 10.1038/sj.jid.5700530
7. Kisich KO, Howell MD, Boguniewicz M, Heizer HR, Watson NU, Leung DYM. The Constitutive capacity of human keratinocytes to kill staphylococcus aureus is dependent on β-Defensin 3. J Invest Dermatol. (2007) 127:2368–80. doi: 10.1038/sj.jid.5700861
8. Mann ER, Smith KM, Bernardo D, Al-Hassi HO, Knight SC, Hart AL. Review: skin and the immune system. J Clin Exp Dermatol Res. (2012) S2:003. doi: 10.4172/2155-9554.S2-003
9. Nedoszytko B, Sokołowska-Wojdyło M, Ruckemann-Dziurdzinska K, Roszkiewicz J, Nowicki RJ. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: atopic dermatitis, psoriasis and skin mastocytosis. Adv Dermatol Allergol Dermatol Alergol. (2014) 31:84–91. doi: 10.5114/pdia.2014.40920
10. Li H, Yao Q, Mariscal AG, Wu X, Hülse J, Pedersen E, et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun. (2018) 9:1420. doi: 10.1038/s41467-018-03704-z
11. Langerhans P. Ueber die Nerven der menschlichen Haut. Arch Für Pathol Anat Physiol Für Klin Med. (1868) 44:325–37. doi: 10.1007/BF01959006
12. Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai XM, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. (2006) 7:265–73. doi: 10.1038/ni1307
13. Doebel T, Voisin B, Nagao K. Langerhans cells – the macrophage in dendritic cell clothing. Trends Immunol. (2017) 38:817–28. doi: 10.1016/j.it.2017.06.008
14. Deckers J, Hammad H, Hoste E. Langerhans cells: sensing the environment in health and disease. Front Immunol. (2018) 9:93. doi: 10.3389/fimmu.2018.00093
15. Ota F, Hirayama T, Kizuka Y, Yamaguchi Y, Fujinawa R, Nagata M, et al. High affinity sugar ligands of C-type lectin receptor langerin. Biochim Biophys Acta. (2018) 1862:1592–601. doi: 10.1016/j.bbagen.2018.04.004
16. Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, et al. Functional specializations of human epidermal langerhans cells and CD14+ dermal dendritic cells. Immunity. (2008) 29:497–510. doi: 10.1016/j.immuni.2008.07.013
17. Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med. (2010) 207:189–206. doi: 10.1084/jem.20091964
18. Kaplan DH. In vivo function of langerhans cells and dermal DC. Trends Immunol. (2010) 31:446–51. doi: 10.1016/j.it.2010.08.006
19. Gomez de Agüero M, Vocanson M, Hacini-Rachinel F, Taillardet M, Sparwasser T, Kissenpfennig A, et al. Langerhans cells protect from allergic contact dermatitis in mice by tolerizing CD8(+) T cells and activating Foxp3(+) regulatory T cells. J Clin Invest. (2012) 122:1700–11. doi: 10.1172/JCI59725
20. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J. (2014) 33:1104–16. doi: 10.1002/embj.201488027
21. Yazdi AS, Röcken M, Ghoreschi K. Cutaneous immunology: basics and new concepts. Semin Immunopathol. (2016) 38:3–10. doi: 10.1007/s00281-015-0545-x
22. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
23. Fukunaga A, Khaskhely NM, Sreevidya CS, Byrne SN, Ullrich SE. Dermal dendritic cells, and not Langerhans cells, play an essential role in inducing an immune response. J Immunol Baltim Md. (2008) 180:3057–64.
24. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. (2009) 10:488–95. doi: 10.1038/ni.1724
25. A-Gonzalez N, Quintana JA, García-Silva S, Mazariegos M, de la Aleja AG, Nicolás-Ávila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. (2017) 214:1281–96. doi: 10.1084/jem.20161375
26. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
27. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
28. Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. (2014) 123:e110–22. doi: 10.1182/blood-2013-08-520619
29. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. (2014) 14:289–301. doi: 10.1038/nri3646
30. Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T, et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat Immunol. (2014) 15:1064–9. doi: 10.1038/ni.2992
31. Suwanpradid J, Shih M, Pontius L, Yang B, Birukova A, Guttman-Yassky E, et al. Arginase1 deficiency in monocytes/macrophages upregulates inducible nitric oxide synthase to promote cutaneous contact hypersensitivity. J Immunol. (2017) 199:1827–34. doi: 10.4049/jimmunol.1700739
32. Sadik CD, Miyabe Y, Sezin T, Luster AD. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol. (2018) 37:21–9. doi: 10.1016/j.smim.2018.03.002
33. Keijsers RRMC, Hendriks AGM, van Erp PEJ, van Cranenbroek B, van de Kerkhof PCM, Koenen HJPM, et al. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap-forming neutrophils expressing RORγt and IL-17. J Invest Dermatol. (2014) 134:1276–84. doi: 10.1038/jid.2013.526
34. Schön MP, Broekaert SMC, Erpenbeck L. Sexy again: the renaissance of neutrophils in psoriasis. Exp Dermatol. (2017) 26:305–11. doi: 10.1111/exd.13067
35. Christensen AD, Skov S, Haase C. The role of neutrophils and G-CSF in DNFB-induced contact hypersensitivity in mice. Immun Inflamm Dis. (2014) 2:21–34. doi: 10.1002/iid3.16
36. Weber FC, Németh T, Csepregi JZ, Dudeck A, Roers A, Ozsvári B, et al. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med. (2015) 212:15–22. doi: 10.1084/jem.20130062
37. Moniaga CS, Watanabe S, Honda T, Nielsen S, Hara-Chikuma M. Aquaporin-9-expressing neutrophils are required for the establishment of contact hypersensitivity. Sci Rep. (2015) 5:15319. doi: 10.1038/srep15319
38. Fukata M, Arditi M. The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol. (2013) 6:451–63. doi: 10.1038/mi.2013.13
39. Krishnaswamy JK, Chu T, Eisenbarth SC. Beyond pattern recognition: NOD-like receptors in dendritic cells. Trends Immunol. (2013) 34:224–33. doi: 10.1016/j.it.2012.12.003
40. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
41. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
42. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. (2011) 3:920–40. doi: 10.3390/v3060920
43. Alinaghi F, Bennike NH, Egeberg A, Thyssen JP, Johansen JD. Prevalence of contact allergy in the general population: a systematic review and meta-analysis. Contact Dermatitis. (2019) 80:77–85. doi: 10.1111/cod.13119
44. Kaplan DH, Igyártó BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. (2012) 12:114–24. doi: 10.1038/nri3150
45. Yim E, Baquerizo Nole KL, Tosti A. Contact dermatitis caused by preservatives. Dermat Contact Atopic Occup Drug. (2014) 25:215–31. doi: 10.1097/DER.0000000000000061
46. Parkinson E, Aleksic M, Cubberley R, Kaur-Atwal G, Vissers JPC, Skipp P. Determination of protein haptenation by chemical sensitizers within the complexity of the human skin proteome. Toxicol Sci. (2018) 162:429–38. doi: 10.1093/toxsci/kfx265
47. Baba SP, Bhatnagar A. Role of thiols in oxidative stress. Curr Opin Toxicol. (2018) 7:133–9. doi: 10.1016/j.cotox.2018.03.005
48. Kagatani S, Sasaki Y, Hirota M, Mizuashi M, Suzuki M, Ohtani T, et al. Oxidation of cell surface thiol groups by contact sensitizers triggers the maturation of dendritic cells. J Invest Dermatol. (2010) 130:175–83. doi: 10.1038/jid.2009.229
49. Honda T, Egawa G, Grabbe S, Kabashima K. Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J Invest Dermatol. (2013) 133:303–15. doi: 10.1038/jid.2012.284
50. Akiba H, Kehren J, Ducluzeau M-T, Krasteva M, Horand F, Kaiserlian D, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. (2002) 168:3079–87. doi: 10.4049/jimmunol.168.6.3079
51. Beidaq AE, Link CWM, Hofmann K, Frehse B, Hartmann K, Bieber K, et al. In vivo expansion of endogenous regulatory t cell populations induces long-term suppression of contact hypersensitivity. J Immunol. (2016) 197:1567–76. doi: 10.4049/jimmunol.1600508
52. Honda T, Otsuka A, Tanizaki H, Minegaki Y, Nagao K, Waldmann H, et al. Enhanced murine contact hypersensitivity by depletion of endogenous regulatory T cells in the sensitization phase. J Dermatol Sci. (2011) 61:144–7. doi: 10.1016/j.jdermsci.2010.11.001
53. Tomura M, Honda T, Tanizaki H, Otsuka A, Egawa G, Tokura Y, et al. Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest. (2010) 120:883–93. doi: 10.1172/JCI40926
54. Martin SF, Esser PR, Weber FC, Jakob T, Freudenberg MA, Schmidt M, et al. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. (2011) 66:1152–63. doi: 10.1111/j.1398-9995.2011.02652.x
55. Martin SF. Allergic contact dermatitis: xenoinflammation of the skin. Curr Opin Immunol. (2012) 24:720–9. doi: 10.1016/j.coi.2012.08.003
56. Esser PR, Wölfle U, Dürr C, von Loewenich FD, Schempp CM, Freudenberg MA, et al. Contact sensitizers induce skin inflammation via ROS production and hyaluronic acid degradation. PLoS ONE. (2012) 7:e41340. doi: 10.1371/journal.pone.0041340
57. Weber FC, Esser PR, Müller T, Ganesan J, Pellegatti P, Simon MM, et al. Lack of the purinergic receptor P2X7 results in resistance to contact hypersensitivity. J Exp Med. (2010) 207:2609–19. doi: 10.1084/jem.20092489
58. Esser PR, Zech A, Idzko M, Martin SF. Lack of biglycan reduces contact hypersensitivity in mice. Contact Dermatitis. (2018) 79:326–8. doi: 10.1111/cod.13082
59. Martin SF, Rustemeyer T, Thyssen JP. Recent advances in understanding and managing contact dermatitis. F1000Res. (2018) 7:F1000 Faculty Rev-810. doi: 10.12688/f1000research.13499.1
60. Martin SF, Dudda JC, Bachtanian E, Lembo A, Liller S, Dürr C, et al. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J Exp Med. (2008) 205:2151–62. doi: 10.1084/jem.20070509
61. Yasuike R, Tamagawa-Mineoka R, Ueta M, Nakamura N, Kinoshita S, Katoh N. The role of toll-like receptor 3 in chronic contact hypersensitivity induced by repeated elicitation. J Dermatol Sci. (2017) 88:184–91. doi: 10.1016/j.jdermsci.2017.07.017
62. Schmidt M, Raghavan B, Müller V, Vogl T, Fejer G, Tchaptchet S, et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. (2010) 11:814–9. doi: 10.1038/ni.1919
63. Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M. Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep. (2012) 13:1109–15. doi: 10.1038/embor.2012.155
64. Rachmawati D, Bontkes HJ, Verstege MI, Muris J, von Blomberg BME, Scheper RJ, et al. Transition metal sensing by Toll-like receptor-4: next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermatitis. (2013) 68:331–8. doi: 10.1111/cod.12042
65. Song S-Y, Bae CH, Choi YS, Kim Y-D. Cadmium induces mucin 8 expression via Toll-like receptor 4–mediated extracellular signal related kinase 1/2 and p38 mitogen-activated protein kinase in human airway epithelial cells. Int Forum Allergy Rhinol. (2016) 6:638–45. doi: 10.1002/alr.21705
66. Liu D, Rhebergen AM, Eisenbarth SC. Licensing adaptive immunity by NOD-like receptors. Front Immunol. (2013) 4:486. doi: 10.3389/fimmu.2013.00486
67. Antonopoulos C, Cumberbatch M, Dearman RJ, Daniel RJ, Kimber I, Groves RW. Functional caspase-1 is required for langerhans cell migration and optimal contact sensitization in mice. J Immunol. (2001) 166:3672–7. doi: 10.4049/jimmunol.166.6.3672
68. Arthur JC, Lich JD, Wilson JE, Ye Z, Allen IC, Gris D, et al. NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol Baltim Md. (2010) 185:4515–9. doi: 10.4049/jimmunol.1002227
69. Watanabe H, Gaide O, Pétrilli V, Martinon F, Contassot E, Roques S, et al. Activation of the IL-1β-Processing Inflammasome Is Involved in Contact Hypersensitivity. J Invest Dermatol. (2007) 127:1956–63. doi: 10.1038/sj.jid.5700819
70. Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. (2006) 24:317–27. doi: 10.1016/j.immuni.2006.02.004
71. Li X, Zhong F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation. (2014) 37:457–66. doi: 10.1007/s10753-013-9759-z
72. Damm A, Giebeler N, Zamek J, Zigrino P, Kufer TA. Epidermal NLRP10 contributes to contact hypersensitivity responses in mice. Eur J Immunol. (2016) 46:1959–69. doi: 10.1002/eji.201646401
73. Guedes S, Neves B, Vitorino R, Domingues R, Cruz MT, Domingues P. Contact dermatitis: in pursuit of sensitizer's molecular targets through proteomics. Arch Toxicol. (2017) 91:811–25. doi: 10.1007/s00204-016-1714-y
74. Ferreira I, Silva A, Martins JD, Neves BM, Cruz MT. Nature and kinetics of redox imbalance triggered by respiratory and skin chemical sensitizers on the human monocytic cell line THP-1. Redox Biol. (2018) 16:75–86. doi: 10.1016/j.redox.2018.02.002
75. Mussotter F, Tomm JM, El Ali Z, Pallardy M, Kerdine-Römer S, Götz M, et al. Proteomics analysis of dendritic cell activation by contact allergens reveals possible biomarkers regulated by Nrf2. Toxicol Appl Pharmacol. (2016) 313:170–9. doi: 10.1016/j.taap.2016.11.001
76. Huang Y, Li W, Su Z, Kong A-NT. The complexity of the Nrf2 pathway: beyond the antioxidant response. J Nutr Biochem. (2015) 26:1401–13. doi: 10.1016/j.jnutbio.2015.08.001
77. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta BBA Mol Cell Res. (2018) 1865:721–33. doi: 10.1016/j.bbamcr.2018.02.010
78. Maher J, Yamamoto M. The rise of antioxidant signaling—The evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. (2010) 244:4–15. doi: 10.1016/j.taap.2010.01.011
79. Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
80. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. (2002) 62:5196–203.
81. Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. (2010) 501:116–23. doi: 10.1016/j.abb.2010.03.019
82. Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen J-G, et al. Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Top Curr Chem. (2013) 329:163–77. doi: 10.1007/128_2012_339
83. Hu C, Eggler AL, Mesecar AD, van Breemen RB. Modification of keap1 cysteine residues by sulforaphane. Chem Res Toxicol. (2011) 24:515–21. doi: 10.1021/tx100389r
84. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
85. Tian H, Zhang B, Di J, Jiang G, Chen F, Li H, et al. Keap1: one stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett. (2012) 325:26–34. doi: 10.1016/j.canlet.2012.06.007
86. Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans. (2015) 43:621–6. doi: 10.1042/BST20150014
87. Yu M, Li H, Liu Q, Liu F, Tang L, Li C, et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal. (2011) 23:883–92. doi: 10.1016/j.cellsig.2011.01.014
88. Liu G-H, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. (2008) 1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002
89. Rabolli V, Lison D, Huaux F. The complex cascade of cellular events governing inflammasome activation and IL-1β processing in response to inhaled particles. Part Fibre Toxicol. (2016) 13:40. doi: 10.1186/s12989-016-0150-8
90. Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, et al. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res. (2018) 336:32–9. doi: 10.1016/j.bbr.2017.06.027
91. Hennig P, Garstkiewicz M, Grossi S, Di Filippo M, French LE, Beer H-D. The crosstalk between Nrf2 and Inflammasomes. Int J Mol Sci. (2018) 19:E562. doi: 10.3390/ijms19020562
92. Ichimura Y, Waguri S, Sou Y-S, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. (2013) 51:618–31. doi: 10.1016/j.molcel.2013.08.003
93. Lee Y, Shin J-M, Jang S, Choi D-K, Seo M-S, Kim H-R, et al. Role of nuclear factor E2-related factor 2 (Nrf2) in epidermal differentiation. Arch Dermatol Res. (2014) 306:677–82. doi: 10.1007/s00403-014-1470-x
94. Jeayeng S, Wongkajornsilp A, Slominski AT, Jirawatnotai S, Sampattavanich S, Panich U. Nrf2 in keratinocytes modulates UVB-induced DNA damage and apoptosis in melanocytes through MAPK signaling. Free Radic Biol Med. (2017) 108:918–28. doi: 10.1016/j.freeradbiomed.2017.05.009
95. Kurinna S, Muzumdar S, Köhler UA, Kockmann T, Auf dem Keller U, Schäfer M, et al. Autocrine and paracrine regulation of keratinocyte proliferation through a novel Nrf2-IL-36γ pathway. J Immunol Baltim Md. (2016) 196:4663–70. doi: 10.4049/jimmunol.1501447
96. Yang L, Fan X, Cui T, Dang E, Wang G. Nrf2 promotes keratinocyte proliferation in psoriasis through up-regulation of keratin 6, Keratin 16, and Keratin 17. J Invest Dermatol. (2017) 137:2168–76. doi: 10.1016/j.jid.2017.05.015
97. Emter R, van der Veen JW, Adamson G, Ezendam J, van Loveren H, Natsch A. Gene expression changes induced by skin sensitizers in the KeratinoSensTM cell line: Discriminating Nrf2-dependent and Nrf2-independent events. Toxicol In Vitro. (2013) 27:2225–32. doi: 10.1016/j.tiv.2013.09.009
98. Alloul-Ramdhani M, Tensen CP, El Ghalbzouri A. Performance of the N/TERT epidermal model for skin sensitizer identification via Nrf2-Keap1-ARE pathway activation. Toxicol Vitro Int J Publ Assoc BIBRA. (2014) 28:982–9. doi: 10.1016/j.tiv.2014.04.007
99. OECD Guidelines for the Testing of Chemicals Section 4. Available online at: https://www.oecd-ilibrary.org/environment/oecd-guidelines-for-the-testing-of-chemicals-section-4-health-effects_20745788 (accessed November 1, 2018).
100. Ade N, Leon F, Pallardy M, Peiffer J-L, Kerdine-Romer S, Tissier M-H, et al. HMOX1 and NQO1 genes are upregulated in response to contact sensitizers in dendritic cells and THP-1 cell line: role of the Keap1/Nrf2 pathway. Toxicol Sci Off J Soc Toxicol. (2009) 107:451–60. doi: 10.1093/toxsci/kfn243
101. Migdal C, Botton J, El Ali Z, Azoury M-E, Guldemann J, Giménez-Arnau E, et al. Reactivity of chemical sensitizers toward amino acids in cellulo plays a role in the activation of the Nrf2-ARE pathway in human monocyte dendritic cells and the THP-1 cell line. Toxicol Sci. (2013) 133:259–74. doi: 10.1093/toxsci/kft075
102. El Ali Z, Deloménie C, Botton J, Pallardy M, Kerdine-Römer S. Dendritic cells' death induced by contact sensitizers is controlled by Nrf2 and depends on glutathione levels. Toxicol Appl Pharmacol. (2017) 322:41–50. doi: 10.1016/j.taap.2017.02.014
103. Al-Huseini LMA, Yeang HXA, Sethu S, Alhumeed N, Hamdam JM, Tingle Y, et al. Nuclear factor-erythroid 2 (NF-E2) p45-related Factor-2 (Nrf2) modulates dendritic cell immune function through regulation of p38 MAPK-cAMP-responsive element binding protein/activating transcription factor 1 signaling. J Biol Chem. (2013) 288:22281–8. doi: 10.1074/jbc.M113.483420
104. Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, et al. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol Baltim Md. (2008) 181:4545–59.
105. Wang J, Liu P, Xin S, Wang Z, Li J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp Cell Res. (2017) 360:66–73. doi: 10.1016/j.yexcr.2017.07.031
106. Yeang HXA, Hamdam JM, Al-Huseini LMA, Sethu S, Djouhri L, Walsh J, et al. Loss of Transcription Factor Nuclear Factor-Erythroid 2 (NF-E2) p45-related Factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem. (2012) 287:10556–64. doi: 10.1074/jbc.M111.322420
107. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:ncomms11624. doi: 10.1038/ncomms11624
108. Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K, et al. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys. (2008) 477:139–45. doi: 10.1016/j.abb.2008.06.004
109. Kim W, Lee H-N, Jang J-H, Kim SH, Lee Y-H, Hahn Y-I, et al. 15-Deoxy-Δ12,14-prostaglandin J2 exerts proresolving effects through nuclear factor E2-related factor 2-induced expression of CD36 and heme oxygenase-1. Antioxid Redox Signal. (2017) 27:1412–31. doi: 10.1089/ars.2016.6754
110. Olagnier D, Lavergne R-A, Meunier E, Lefèvre L, Dardenne C, Aubouy A, et al. Nrf2, a PPARγ alternative pathway to promote CD36 expression on inflammatory macrophages: implication for Malaria. PLoS Pathog. (2011) 7:e1002254. doi: 10.1371/journal.ppat.1002254
111. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. (2011) 3:78ra32. doi: 10.1126/scitranslmed.3002042
112. Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, et al. Nrf2 dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. (2006) 351:883–9. doi: 10.1016/j.bbrc.2006.10.102
113. Dias IHK, Chapple ILC, Milward M, Grant MM, Hill E, Brown J, et al. Sulforaphane restores cellular glutathione levels and reduces chronic periodontitis neutrophil hyperactivity in vitro. PLoS ONE. (2013) 8: 66407. doi: 10.1371/journal.pone.0066407
114. Araújo VC, Demasi APD, Soares AB, Passador-Santos F, Napimoga MH, Martinez EF, et al. Neutrophils in oral paracoccidioidomycosis and the involvement of Nrf2. PLoS ONE. (2013) 8:e76976. doi: 10.1371/journal.pone.0076976
115. Joshi N, Werner S. Nrf2 is highly expressed in neutrophils, but myeloid cell-derived Nrf2 is dispensable for wound healing in mice. PLoS ONE. (2017) 12:e0187162. doi: 10.1371/journal.pone.0187162
116. Natsch A. The Nrf2-Keap1-ARE toxicity pathway as a cellular sensor for skin sensitizers—functional relevance and a hypothesis on innate reactions to skin sensitizers. Toxicol Sci. (2010) 113:284–92. doi: 10.1093/toxsci/kfp228
117. El Ali Z, Gerbeix C, Hemon P, Esser PR, Martin SF, Pallardy M, et al. Allergic skin inflammation induced by chemical sensitizers is controlled by the transcription factor Nrf2. Toxicol Sci. (2013) 134:39–48. doi: 10.1093/toxsci/kft084
118. Kitajima M, Kimura A, Suzuki H. Cutting edge: Nqo1 regulates irritant contact hypersensitivity against croton oil through maintenance of dendritic epidermal T cells. J Immunol Baltim Md. (2018) 200:1555–9. doi: 10.4049/jimmunol.1701389
119. Helou DG, Noël B, Gaudin F, Groux H, Ali ZE, Pallardy M, et al. Cutting edge: Nrf2 regulates neutrophil recruitment and accumulation in skin during contact hypersensitivity. J Immunol. (2019) 202:2189–94. doi: 10.4049/jimmunol.1801065
120. Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. (2018) 70:348–83. doi: 10.1124/pr.117.014753
Keywords: Nrf2, skin cells, innate immunity, contact sensitizers, xenoinflammation
Citation: Helou DG, Martin SF, Pallardy M, Chollet-Martin S and Kerdine-Römer S (2019) Nrf2 Involvement in Chemical-Induced Skin Innate Immunity. Front. Immunol. 10:1004. doi: 10.3389/fimmu.2019.01004
Received: 23 November 2018; Accepted: 18 April 2019;
Published: 07 May 2019.
Edited by:
Miriam Wittmann, University of Leeds, United KingdomReviewed by:
Caroline Jefferies, Cedars-Sinai Medical Center, United StatesCopyright © 2019 Helou, Martin, Pallardy, Chollet-Martin and Kerdine-Römer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saadia Kerdine-Römer, c2FhZGlhLmtlcmRpbmUtcm9tZXJAdS1wc3VkLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.