 Shivana M. Lightman
Shivana M. Lightman Adam Utley
Adam Utley Kelvin P. Lee
Kelvin P. Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 May 2019
Sec. B Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00965
This article is part of the Research TopicAdvances in Plasma Cells in Health and DiseaseView all 10 articles
Durable humoral immunity is dependent upon the generation of antigen-specific antibody titers, produced by non-proliferating bone marrow resident long-lived plasma cells (LLPC). Longevity is the hallmark of LLPC, but why and how they survive and function for years after antigen exposure is only beginning to be understood. LLPC are not intrinsically long-lived; they require continuous signals from the LLPC niche to survive. Signals unique to LLPC survival (vs. PC survival in general) most notably include those that upregulate the anti-apoptotic factor Mcl-1 and activation of the CD28 receptor expressed on LLPC. Other potential factors include expression of BCMA, upregulation of the transcription factor ZBTB20, and upregulation of the enzyme ENPP1. Metabolic fitness is another key component of LLPC longevity, facilitating the diversion of glucose to generate pyruvate during times of stress to facilitate long term survival. A third major component of LLPC survival is the microenvironment/LLPC niche itself. Cellular partners such as stromal cells, dendritic cells, and T regulatory cells establish a niche for LLPC and drive survival signaling by expressing ligands such as CD80/CD86 for CD28 and producing soluble and stromal factors that contribute to LLPC longevity. These findings have led to the current paradigm wherein both intrinsic and extrinsic mechanisms are required for the survival of LLPC. Here we outline this diverse network of signals and highlight the mechanisms thought to regulate and promote the survival of LLPC. Understanding this network of signals has direct implications in increasing our basic understanding of plasma cell biology, but also in vaccine and therapeutic drug development to address the pathologies that can arise from this subset.
Plasma cells (PC) represent an essential arm in humoral immunity as the main line of defense against infection and re-infection. As the primary producers of circulating immunoglobulin (Ig), these cells provide vital durable and protective immunity against a multitude of pathogens. Longitudinal analysis of antigen-specific antibody titers from vaccinated humans demonstrates that the predicted half-life of the measles titer is 3,014 years (1). This is a testament to the long-lived protection that PC can provide. Unlike other immune cell subsets such as T cells or B cells, the complexity of the varying PC subsets is only beginning to be understood. Plasma cell generation occurs primarily upon T cell-dependent differentiation of B lymphocytes to PC in germinal center reactions (2, 3). The current paradigm proposes that two general types of PC develop from these interactions: short-lived plasma cells (SLPC) and long-lived plasma cells (LLPC) (4, 5). The LLPC subset characteristically thought to traffic to and reside in the bone marrow (BM), is the subset that provides long-term and sustained antibody production that is maintained for decades to the lifetime of an individual (6–8). The germinal center reactions, through somatic hypermutation and class-switch recombination, allow for the selection of high-affinity antibody producing PC, which is proposed to be the major precursors of LLPC (9, 10). However, there is relatively little understanding of the driving force behind why these LLPC can become long lived.
There has been considerable research into the biology of PC as a whole, from how they are generated and the key transcriptional programs involved, to their ability to traffic to various organs (11–15). Further studies have elucidated the cellular and molecular components of various organ-specific niches occupied by PC (16–18). However, there is relatively little understanding of what distinguishes the ability of LLPC to survive in contrast to SLPC. LLPC are not intrinsically long-lived, as their survival is critically dependent on the ability to access and use a fixed number of specific pro-survival niches in the BM, secondary lymphoid organs, mucosa, and sites of inflammation (5, 7, 19–24). The BM is traditionally thought to be the primary organ of LLPC residence. It provides a dynamic infrastructure amenable to the formation of a complex microenvironment and allows for the generation of cell-type specific niches more easily than other less plastic organs (5, 6).
More recent work has illuminated the fact that LLPC do not only reside within the BM. Of human PC, about 80% are located in gut-associated lymphoid tissue (GALT) and produce primarily IgA (25). This allows for tolerance to the commensal bacteria in our gut, while also providing protection against unfavorable microbial and dietary antigens. It was originally thought that continual activation of B cells within the mucosa supplied the pool of IgA-producing PC in the gut (26, 27). However, new studies have highlighted that PC in the gut can also persist for long periods of time. Antibodies specific to Escherichia coli were detected 112 days after exposure (28). Examination of intestinal biopsies kept in culture contained non-proliferating IgA-producing PC for >4 weeks (29). Another study showed that 9 months post-immunization with both an IgA-inducing antigen (cholera toxin) and a T-dependent antigen (Ova), antigen-specific PC could be detected in the Lamina Propria (LP) but also within the BM (21). This suggests that survival niches present in the gut could be similar to those in the BM and that mucosal PC can utilize these niches in the same way as BM PC, as well as contributing to the BM LLPC pool (21).
It is traditionally thought that most BM LLPC secrete IgG antibodies, and those that reside in mucosal sites such as the gut produce IgA antibodies, however, about 40% of BM PC also produce IgA (30). It has also been reported that there are long-lived low-affinity IgM producing PC within the BM that appear to occupy different niches (20). Furthermore, allergic sensitization with ovalbumin generates IgG, IgA, and IgE secreting PC that can be found in the BM for an extended period (31). Therefore, evidence suggests that the longevity of a PC is not primarily driven by its antibody isotype—but rather by the nature of the pathogen, the characteristics of initial B cell activation elicited by the pathogen, and the niche the resultant PC homes to. This broad framework suggests that the primary role of SLPC is in protection against frequent (and less severe) endemic infections that is sustained by recurrent and antigen-dependent B cell reactivation; whereas LLPC provide sustained protective immunity against infrequent (but more severe) epidemic infections in an antigen-independent fashion. It has also been proposed that early B cell/plasmablast activation signals determine whether PC enter and respond to survival signals in the PC niche (32–34). Because the space available to LLPC is finite, LLPC longevity requires both access to the survival niche and the ability to respond to the niche's unique pro-survival signals.
It was originally proposed that sustained antibody responses resulted from constant replenishment of a SLPC pool by continuous memory B cell re-stimulation (35). However, observations that BM transplantation caused non-allergic individuals to acquire allergies through transfer of allergen-specific IgE production lent credence to the idea that these PC were long-lived, due to the absence of antigenic re-stimulation in these patients (36, 37). Further studies have shown in vivo that some PC subsets are indeed long-lived and that absence of antigen plus depletion of memory B cells through radiation did not abrogate the ability of this LLPC subset to continue to produce high-affinity antigen-specific antibodies (38, 39). Other studies have shown that prolonged therapeutic depletion of the total B cell pool did not affect antigen-specific BM PC numbers or antibody titers in vaccinated murine models (40), nor did it affect antibody titers against childhood vaccines in humans (41). In the human studies, vaccine-specific antibody titers were maintained following anti-CD20 monoclonal antibody treatment (which targets B cells but not PC), despite clear depletion of the memory B cell pool (41). Sustained B cell aplasia caused by CD19 CAR-T cells (which also target B cells but not PC) in adult and pediatric acute lymphocytic leukemia patients also had no effect on serum vaccine antigen-specific antibody titers nor PC numbers in either the BM or ileum and colon (42). A study examining PC dynamics from biopsies of transplanted duodenum found that 1-year post-transplantation, CD38+ PC from the donor could still be identified. Further characterization identified CD19− and CD19+ PC present in these biopsies, where the CD19− PC subset represented a stable population with a potentially long lifespan (24). Lastly, characterization of subsets of human PC demonstrated that CD19− PC in the BM were predominantly IgG secreting with a mature phenotype and remained settled in the BM after systemic vaccination (43). This demonstrates that LLPC are indeed distinct from other B cell subsets, however the key factors that distinguish these cells are only recently beginning to be characterized.
During B cell to PC differentiation, many genes are downregulated including those involved in antigen presentation and BCR function. Concurrently, there is upregulation of the PC lineage defining transcription factor PRDM1 (BLIMP-1) and genes involved in protein translation and the unfolded protein response (XBP-1) (11, 44). An avenue to distinguish this LLPC subset is through their transcriptional profile. A recent study of human PC has established a unique profile that distinguishes between early PC, circulating blood PC, and long-lived BM-resident CD138+ PC (14). The signature that distinguishes LLPC contains a significant number of genes that are downregulated and only a handful of genes that are upregulated. This latter group includes the anti-apoptotic genes Mcl-1 and ZNF667, ER stress-associated genes including EROILB and MANF, the cation transport ATPase ATP12A, and TFBS and SRF that play roles in retention of hematopoietic progenitor cells in the BM. Many of these genes are associated with the central function of PC to produce significant amounts of protein in the form of immunoglobulin—although the fact that this function is not unique to LLPC suggests that there may be additional unappreciated biology of these genes in LLPC.
Identification of a transcriptional profile that is unique to LLPC faces a significant experimental obstacle—namely the ability to identify a pure population of LLPC to analyze. Anatomic location has traditionally been the way that SLPC and LLPC were distinguished from each other, due to the lack of distinct phenotypic markers distinguishing these subsets. However, as noted above, recent sophisticated phenotypic profiling has made it increasingly clear that LLPC are not the only PC present within the BM. Recent work looking at PC precursor populations has identified that several waves of trafficking can occur post-antigen exposure and PC differentiation—resulting in other PC aside from LLPC being in the BM (34). Furthermore, surface markers that were traditionally used to identify LLPC have been recently shown to variably stain different PC subsets—for example, analysis of PC in the BM compartment have shown that there is varying expression of surface markers such as CD38 and CD19 (43, 45). These phenotypic differences are clearly associated with functional differences, such as whether these PC are producing high-affinity or low-affinity antibodies (46).
Because of the recent increase in PC subset heterogeneity, analysis of the total BM PC population compared to PC from other compartments may not pinpoint the specific factors and interactions responsible for reinforcing and sustaining the transcriptional signature necessary for the LLPC subset. Thus, one approach in defining the key transcriptional networks unique to LLPC, is to first identify the upstream receptors and signal transduction pathways that uniquely support LLPC survival and longevity.
We now know that there are intrinsic signals uniquely utilized by LLPC that regulate their long-term survival and function (Table 1). It is well-established that factors such as A Proliferation Inducing Ligand (APRIL), Interleukin-6 (IL-6), B cell Activating Factor (BAFF), CD44 and CXCL12 can support survival of both SLPC and LLPC in various organs (4, 12, 29, 47–49). One of the receptors for both BAFF and APRIL is B cell maturation antigen (BCMA), which is highly upregulated on PC (50). Genetic knockout of Tnfrsf17 (which encodes BCMA) resulted in a significant loss of antibody secreting cells (ASCs) in the BM compared to wild-type (Wt) controls, 6–8 weeks post-immunization. However, germinal center responses and early antigen-specific serum IgM and IgG titers were normal, indicating loss of BCMA primarily affected the LLPC subset (50). Additional studies have demonstrated that murine memory B cell survival is not dependent on BCMA, and BCMA is only induced in human B cells committed to PC differentiation (47, 51). This suggests that reliance on BCMA for survival is specific to the PC compartment. Furthermore, studies in multiple myeloma (MM), a BM-resident Ig-class switched LLPC neoplasm, showed that BCMA significantly promoted MM survival and proliferation (52). Therapeutically targeting BCMA in MM has shown high selectivity against the MM and PC populations (53), supporting a PC-restricted role for the receptor. Activation of BCMA by BAFF increases expression of the anti-apoptotic molecule Mcl-1, pointing to the mechanistic basis by which the BAFF/BCMA axis promotes long-term survival (50). An increase in the gene expression for Mcl-1 in BM PC was observed in Wt mice compared to BM PC from BCMA−/− mice, where both Mcl-1 mRNA and protein expression were significantly lower in the knockouts (54). Furthermore, loss of Mcl-1 causes a significant decrease in antigen-specific titers 21 days post immunization, consistent with an effect on the LLPC subset (54). As with BCMA, Mcl-1 is also a critical factor for MM survival (55, 56), which is consistent with the broader concept that much of myeloma biology is in fact the biology of normal LLPC (57).

Table 1. Summary of intrinsic factors supporting LLPC survival.
The importance of IL-6 in PC function and survival has been extensively studied (49, 58). IL-10 and IL-21 have also been implicated in human PC maturation and survival (59–61). IL-21 is predominantly produced by T follicular helper (Tfh) cells (62), which are involved in many phases of the humoral immune response from B cell activation to maturation and differentiation of B memory cells into PC (63). A study looking at human tonsil, splenic, lymph node, and BM PC indicated that BM PC had very low expression of IL-21R and that IL-21 only enhanced Ig secretion from secondary lymphoid PC but not BM PC (64). More recent work, however, has postulated that Tfh cells influence blood Ag-induced PC, which can contain precursors of the LLPC subset (65). Whether this effect is directly through IL-21 or the effect of IL-21 on B cell subsets and upregulation of receptors like IL-6R is uncertain. How these individual cytokines promote BM PC survival and function is still unclear; however, they all initiate STAT3 signaling (66–68). Activation of STAT3 plays an important role in the ability of PC to respond to these various cytokine signals (68). Impairment of this pathway inhibits cytokine-dependent increase in viability and IgG secretion. Furthermore, STAT3 activation appears to be a prerequisite for the ability of PC to respond to APRIL and BAFF. This suggests a critical role for STAT3 as a key downstream signaling node transducing stimuli in LLPC that reinforces their survival and longevity (68).
Other factors restricted to the lymphoid lineage have been studied for their possible role in supporting the LLPC subset. Aiolos, part of the Ikaros gene family of nuclear regulators that function in modulating chromatin structure, is one of these factors (69). Absence of this gene does not affect B cell development or the early SLPC response but does affect long-term PC survival (70). Immunization normally causes a first wave of low-affinity antibodies produced primarily by SLPC, and then a second wave of higher affinity antibodies from BM LLPC (10). Upon deletion of Aiolos, there is a significant decrease in the production of high-affinity antibodies in the BM 120 days after primary immunization with no effect on the low-affinity antibody response (71). Additional studies show that the deletion of this gene does not cause a homing defect. This suggests that Aiolos upregulation is required for high-affinity antigen-specific LLPC, unlike other factors like BLIMP-1, which are required for generation of all PC. The exact mechanism remains to be understood, but the ability of Aiolos to promote high-affinity antibodies is important for enhanced protective immunity long term.
Another starting point for identifying specific receptors involved in LLPC survival and longevity are those receptors that are highly upregulated on BM PC compared to mature B cells. Greenlee et al. observed that CD93, which has been largely characterized for its role in intercellular adhesion and clearance of apoptotic cells, is not present on mature B cells but is highly upregulated during PC differentiation (72). Upon immunization with either T-independent (TI) or T-dependent (TD) antigens, CD93 is further upregulated in BM PC, and the CD93+CD138+ PC have increased expression of BLIMP-1, XBP-1, IRF4, and produce higher-affinity antibodies—suggesting that this receptor identifies the LLPC subset (73). Consistent with this, loss of CD93 diminished the persistence of long-term antibody titers post immunization (73). Loss of Blimp-1 decreases CD93 expression only in PC, as CD93 is still present at low levels in Blimp-1-deficient immature B cells. This connection to Blimp-1, which is most highly expressed in LLPC, led the authors to hypothesize that CD93 is necessary to promote the mature phenotype of LLPC and production of high-affinity antibodies. This is further corroborated by a recent study looking at measles titers of vaccinated school children in which the most highly expressed genes in high responder groups were CD93, IL-6, and CXCL12- implicating CD93 involvement in the generation of long-lived antibody responses in humans (74).
Upon activation, B cells downregulate the B cell lineage-defining transcription factor Pax5 (75). Pax5 was found to directly inhibit CD28 gene expression in B cells, and the downregulation of Pax5 leads to upregulation of CD28 as an immediate/early event in B → PC differentiation. In this study loss of CD28 impaired short term induction of antigen-specific antibody titers, although long-lived antibody responses were not assessed. Previous studies in CD28 global knockouts and signaling-deficient CD28 receptor knock-in mice had demonstrated significant defects in antibody responses (76–79), but this was attributed to defects in T cell help. Subsequent work from our lab has shown that CD28 has an essential PC-intrinsic role in the survival of LLPC and the maintenance of durable antigen-specific IgG antibody titers after vaccination (80). Compared to Wt mice, unvaccinated CD28−/− mice have comparable numbers of splenic PC but have significantly fewer BM PC. In vitro studies demonstrated that CD28 activation alone (without a “signal 1” that is necessary for T cells) enhances the survival of purified BM PC, but not splenic PC, in response to serum-starvation-induced death. In vivo, the loss of CD28 expression specifically in the B cell lineage (B cells do not express CD28, only PC) resulted in the inability to sustain antigen-specific antibody titers in these murine models (80). Work done by Njau et al. also looked at the effect of CD28 on PC. Initial studies with global CD28−/− mice showed a significant increase in IgM antigen-specific responses compared to Wt mice, but found a significantly lower antigen-specific IgG response compared to Wt controls 60 days post immunization, which they attributed to a defect in T-cell help (81). However, in contrast to Rozanski et al. specific loss of CD28 in the B cell lineage resulted in a significant increase in antigen-specific antibody titers compared to Wt PC. The basis of the differences between these two studies is unclear, although the immunization strategies (both the antigen and in particular the adjuvants used) were different. Regardless both studies establish that CD28 is present on PC and has a role in affecting antibody responses.
Studies of signaling pathways emanating from the CD28 receptor in LLPC demonstrated that loss of downstream Vav/Grb2 mediated PLCγ1 signaling caused a loss of BM LLPC, while loss of downstream PI3K/AKT signaling had no effect (82). Interestingly, although murine SLPC also express CD28, direct activation of the receptor does not induce downstream activation of PLCγ1 and NFκB—indicating a signaling basis for why SLPC are not responsive to CD28 and suggesting that CD28 has different activation thresholds in SLPC vs. LLPC. Further downstream, CD28 activation augments BLIMP-1 expression, a master regulator of PC identity and antibody production (82, 83). Consistent with the role CD28 plays in normal LLPC, we have also found that activation of CD28 confers a major pro-survival/chemotherapy resistance signal in MM (84, 85).
Activation of CD28 on MM directly transduces a pro-survival signal, and also signals back to dendritic cells (DC) through the CD28 ligands CD80 and CD86 expressed on these DC (84–86). This back signaling through CD80/CD86 upregulates both IL-6 and the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) expression/activity in the DC to promote further MM survival. Thus, CD28 both directly delivers a pro-survival signal to the myeloma cell and modulates the DC in the niche to further support MM survival. Conversely, in vivo blockade of the CD28-CD80/CD86 interaction with CTLA4-Ig (which is FDA approved for the treatment of rheumatoid arthritis) significantly enhanced the sensitivity to chemotherapy in a murine model of MM (84). Given the significant shared biology between LLPC and MM, this bidirectional signaling axis between CD28 and CD80/CD86 is now being investigated as previously unrecognized pro-survival factors for LLPC.
Because LLPC are continually generating and secreting high levels of Ig protein, they require unique metabolic pathways to deal with the protein load and ER stress. Induction of the Unfolded Protein Response (UPR) has been clearly identified as essential for B cells to differentiate to PC (87). A second mechanism implicated in the adaptation to this stress is autophagy. Autophagy's primary function is to sustain cellular metabolism when nutritional starvation occurs. In yeast, to counterbalance the stress caused by ER expansion, autophagic trimming is employed to remove excess ER (88). Within the immune compartment, mature T cells rely on autophagy-mediated turnover of mitochondria (mitophagy) to control intracellular production of reactive oxygen species (89, 90). Autophagy also appears to be critical for LLPC survival. Because antibody-producing PC are highly biosynthetic, it would be predicted that autophagy plays key roles in both processing/eliminating/recycling misfolded immunoglobulins as well as supporting the fuel and metabolic substrate requirements of the PC—in particular since the BM microenvironment may face highly variable nutrient availability due to changing levels of hematopoiesis during stress (91, 92). Analysis of the secretome from mesenchymal stromal cells (known to be part of the BM niche) identified two major survival factors of BM PC, fibronectin (FN-1) and the YWHAZ protein, which were shown to downregulate mTORC1 signaling in BM PC (17). Autophagy induction normally occurs after inhibition of mTOR (target of rapamycin), an evolutionarily-conserved protein kinase. mTOR is a key regulator in cell growth and response to the nutrient status and stress signals in a cell- and is known to negatively regulate autophagy (93). Thus, these factors in the niche could further influence the ability of LLPC to upregulate autophagy as a survival mechanism. Other studies have demonstrated that in mice knocked out for Atg5, which plays a central role in initiating autophagy, there is a significant defect in high-affinity LLPC post-immunization with a TD antigen (94). While all PC are generating Ig at high levels, it is possible that LLPC are more dependent on autophagy over their lifespan for both elimination of misfolded protein as well as a recycling mechanism to supply metabolic substrates, to reinforce the metabolic fitness of these cells during periods of nutrient deprivation.
Recent studies have begun to connect the metabolic energy production pathways used by LLPC to those used by memory T cells (which need to rapidly respond to activation to become highly biosynthetic and proliferate). CD28 co-stimulation in naïve T cells switches their fuel utilization to fatty acids and mitochondrial oxidation to sustain their metabolic demands, and this has been shown to be required for the generation of memory T cells (95, 96). CD28 co-stimulation also induces changes in mitochondria in memory T cells—elongated mitochondria with increased cristae structure are associated with enhanced (spare) respiratory capacity compared with naive T cells. Similarly, work on the metabolic phenotype of LLPC by Lam et al. has interestingly shown that both mouse and human LLPC have a greater maximal respiratory capacity than SLPC, and although they have increased ability to take up glucose (via Glut1 upregulation), they constitutively use long chain fatty acids as their primary fuel source—and glucose appears to be primarily used for antibody glycosylation (97, 98). However, under possibly non-optimal conditions they are also capable of using glycolysis and mitochondrial pyruvate import. The importance of this “alternative” metabolism program was demonstrated by the finding that loss of the ability to import pyruvate into the mitochondria significantly decreased LLPC frequency and abrogated sustained long-lived antibody response 22 weeks post vaccination (97). Similar work looking at IgA-secreting PC in the LP exhibited higher expression of glycolysis-related metabolites than naïve B cells in Peyer's Patches (PP) (99). It is proposed that the switch from IgM to IgA requires a metabolic shift and preferential use of the glycolytic pathway. This group also observed that splenic SLPC did not have very many glycolytic metabolites- indicating that both the BM and gut are unique microenvironments that could contribute to this shift in the metabolic phenotype of LLPC.
Further investigation into the potentially unique metabolic characteristics of LLPC has led to the discovery of other previously unrecognized pathways. The transmembrane glycoprotein ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) is highly expressed on PC, but its function has only recently been demonstrated. This enzyme was previously characterized for its role in bone formation, glucose homeostasis and downregulation of the insulin signaling pathway (100). ENPP1−/− mice were also found to have a significant reduction of BM LLPC 12 weeks post immunization with TD dependent antigen and C. chabaudi infection, but no effect was observed on splenic PC (101). ENPP1−/− PC take up less glucose than their Wt counterparts and exhibit an impairment in glycolysis, suggesting a role for ENPP1 as a regulator of glucose and the metabolic pathway found in LLPC (101).
The relevance of some of the above factors in supporting LLPC survival were recently supported by characterization of human BM PC. These studies elegantly demonstrated the heterogeneity of BM PC in mice is also evident in humans, of which only a fraction appear to be the bonafide long-lived subset. This study defined 4 subsets of PC found in the BM: CD19+CD38hiCD138− (subset A), CD19+CD38hiCD138+ (subset B), CD19−CD38hiCD138− (subset C), and CD19−CD38hiCD138+ (subset D) (45). Subset D did not proliferate in comparison to the other three subsets and was CD20 negative, HLA-DR negative but CD28 positive (45). This subset had the highest number of IgG specific antibodies against tetanus, measles, mumps, and influenza. Interestingly, this subset also showed a distinctive RNA transcriptome signature for enhanced autophagy. This finding suggests that those components uncovered in murine studies are relevant to human PC and are fundamental to LLPC function and survival.
The PC-intrinsic signaling and downstream responses detailed above do not occur in a vacuum. LLPC are critically dependent upon signals they receive from the niche, as LLPC cultured alone in vitro rapidly die (102). Studies of the LLPC BM niche have identified several different cell types that can provide survival signals to the PC (Figure 1). The first cell type to be identified were reticular and mesenchymal stromal cells within the BM. They have high expression of CXCL12, the ligand for CXCR4 expressed on PC, which is responsible for their trafficking into the BM (18, 103). However, even though initial studies demonstrated these stromal cells supported PC survival in vitro, this survival was not sustained—and in vivo, they did not appear to have an essential role in supporting LLPC survival (48, 104). Furthermore, the PC-survival factors like APRIL, BAFF, and IL-6 are not secreted by these cells. Yet, it has recently been shown that BM mesenchymal stromal cells secrete fibronectin (FN-1) and other proteins (YWHAZ) that can sustain LLPC survival in vitro, in combination with hypoxia and factors like APRIL (17). FN-1 binds integrins, collagen and CD138 (highly expressed on LLPC), and is proposed to play an important role in tethering PC to the BM extracellular matrix. Studies in MM have also shown that integrin-mediated binding to fibronectin facilitates resistance to chemotherapy-mediated killing (105).
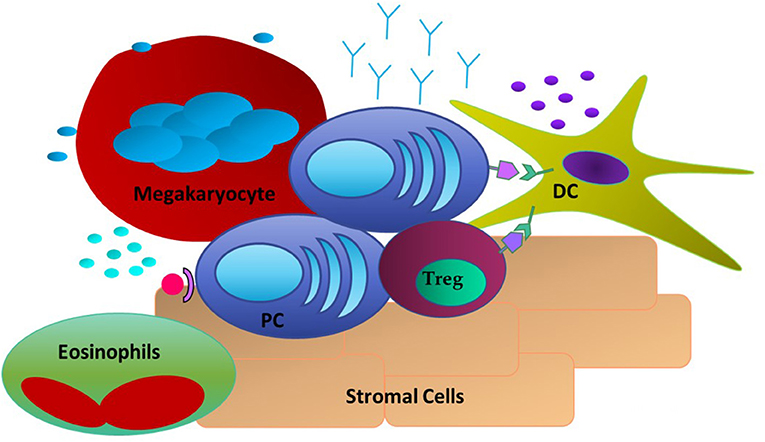
Figure 1. Extrinsic Factors in the LLPC niche. PC are associating with DC through CD28: CD80/CD86 interactions and with Tregs. Stromal cells expressing CXCL12 act as a homing signal to CXCR4+ PC. Megakaryocytes and Eosinophils are also present and have been shown to produce soluble factors such as APRIL and BAFF to promote PC survival.
Other studies have identified megakaryocytes as part of the LLPC niche. Megakaryocytes can produce APRIL and IL-6, and mice that do not have the ability to form megakaryocytes have a reduced number of BM PC when compared to Wt mice (106). Furthermore, upon immunization with ovalbumin (Ova), it was observed that Ova-specific PC were in direct contact with megakaryocytes in the BM. However, functional studies of PC post-immunization in the absence of megakaryocytes did not show any effect in the late stage of antigen-specific antibody responses. Basophils are another cell type reported to support PC survival. In in vitro cultures of basophils and BM PC, the presence of basophils supported the survival of these PC and markedly increased the Ig production of these cells (107). However, functional studies of the effect of basophils on antigen-specific BM PC responses were only assessed in vitro, therefore it is still uncharacterized how these cells might affect BM LLPC in the in vivo setting. It was determined that the survival effects seen by basophils are most likely due to their ability to secrete IL-4 and IL-6. Thus, megakaryocytes, and basophils may play an important role in establishing the LLPC niche and possibly LLPC access, however, it remains unclear how they contribute to long term survival and maintenance of LLPC.
Another cell subset thought to provide critical survival cytokines to PC, like IL-6, and APRIL, are eosinophils. It was reported that eosinophils co-localize with PC in both the BM and gut associated lymphoid tissue (GALT) and that eosinophil-deficient mouse have significantly fewer PC at steady state and post-exposure to antigen (108, 109). For IgA-secreting PC, eosinophils are necessary for promoting IgA production and maintaining immune homeostasis in the GALT (110). In a study of PC in the lamina propria one-third of the PC were found to be localized next to eosinophils (21). With respect to the BM niche, it was found that lack of eosinophils caused LLPC in the BM to undergo apoptosis. Upon reconstitution of eosinophils, the number of PC was transiently increased, suggesting a role for these cells in homing of PC to the BM as well as maintaining the PC population once there (108). Once activated an eosinophil can promote B cell differentiation into IgM secreting PC that is independent of the induction of germinal centers and generation of high-affinity IgG secreting PC (111). However, more recent studies have questioned whether eosinophils are a necessary component for LLPC survival and function, at least in murine models this has not been conclusive (112). Therefore, the current literature suggests that eosinophils may play a critical role in the generation and trafficking of PC, for both mucosal sites and the BM, but their contribution to the survival, maturity, and long term function of LLPC remains unsettled.
Parallel studies in the niche have focused on cellular components that express the ligands that can engage receptors on LLPC that support survival and function. Our work has demonstrated a key role for CD80, and CD86, the natural ligands for CD28. An absence of these ligands in global knockout mice significantly diminished BM LLPC numbers and durable antigen-specific titers (80). The cells that have the highest expression of CD80 and CD86 are myeloid (conventional) DC. Other myeloid cells (monocytes, eosinophils) can also have significant expression. Our lab and others have demonstrated co-localization of DC with PC occurs within the BM. Additionally this supports LLPC survival in vitro (in a contact and CD28-dependent fashion) and in vivo (80, 113). This interaction has further significance as DC can produce significant amounts of the pro-PC survival cytokine IL-6, and that CD80/CD86 activation is one pathway that induces DC IL-6 production (86). As noted above, DC also significantly enhances the survival of MM in a CD28-CD80/CD86 dependent manner. This suggests a bidirectional signaling axis in the niche to support LLPC both directly and indirectly through modulation of cellular partners to produce essential survival factors.
Another significant partner recently identified within the LLPC BM niche are T regulatory (Tregs) cells in close physical association with CD11c+ DC. High number of Tregs have been shown to co-localize with PC in the BM in vivo, and this is necessary to homeostatically support BM PC populations (113). Ablation of Tregs during infection caused a significant reduction in the number of BM PC. The phenotype of these BM Tregs was consistent with an activated subset with increased suppressive capabilities, and most closely matched the profile observed in injured muscle. Interestingly, unlike splenic Treg cells that have little CTLA-4 expression, these BM Tregs express CTLA-4—which is a member of the CD28 superfamily that also binds CD80 and CD86. In the absence of Treg expression of CTLA-4, there was a 3-fold increase in the number of BM PC. The authors proposed that CTLA-4+ Tregs contribute to the maintenance of an immune-privileged niche for LLPC in part by buffering this niche against systemic inflammatory changes, as well as maintaining LLPC homeostasis by limiting the PC pool (113). However, what role CTLA-4 itself is playing within the PC niche is unclear. It is possible that through expression on Tregs it acts in a homeostatic way to limit the amount of PC able to traffic into and reside within the BM. However, it could also be binding to the CD80/CD86 receptors on niche DC, causing upregulation of cytokines like IL-6 to help support PC survival. Alternatively, Treg CTLA-4 may compete for and limit LLPC CD28 binding to these ligands on the DC, as part of homeostatic regulation of the LLPC population. It is interesting to speculate that this cellular triumvirate of LLPC, DC, and Tregs is modulated by dynamic molecular interactions between CD28, CD80/CD86, and CTLA-4, facilitating a stable niche that critically supports LLPC survival and continued function.
This review has identified some of the components that form the networks that allow for survival and maintenance of LLPC. Long-term generation of protective antibodies by this subset is crucial for durable protective immunity. Understanding these key molecular and cellular components that support LLPC survival and longevity gives us tools for utilization of new vaccination strategies as well as therapeutic strategies against diseases that arise from this cell, such as MM and autoimmune syndromes.
It has become clear that there are distinct subsets of PC defined by anatomic location, phenotype, function, and longevity (Table 2). Evidence suggests that PC making high affinity neutralizing Ig (which would be particularly important in rapidly lethal pathogens that characteristically cause epidemics) are key members of the LLPC subset and are generated upon germinal center reactions and then traffic to the BM; where they have unique characteristics that allow them to unlock and occupy/utilize the LLPC niche. It is now clear that this is a significant oversimplification given the heterogeneity of the BM PC populations and niches present in other organs like the gut. Nonetheless, a growing body of evidence demonstrates that the “traditional” LLPC that produce lifelong protective antibody titers, requires both PC intrinsic and niche components to be long-lived. These interactions can then lead to downstream PC-intrinsic signaling that buttresses the ability of the LLPC subset to survive over decades in their microenvironment; which in the BM is hypoxic and crowded with rapidly proliferating cells (especially reactive BM responding to infection) competing for space and nutrients, and in mucosal sites where there is constant exposure to both helpful and harmful microbes. Thus, the key aspects that underlie the longevity of LLPC likely reinforce the mechanisms that allow for effective adaptation to these stresses over a lifetime.
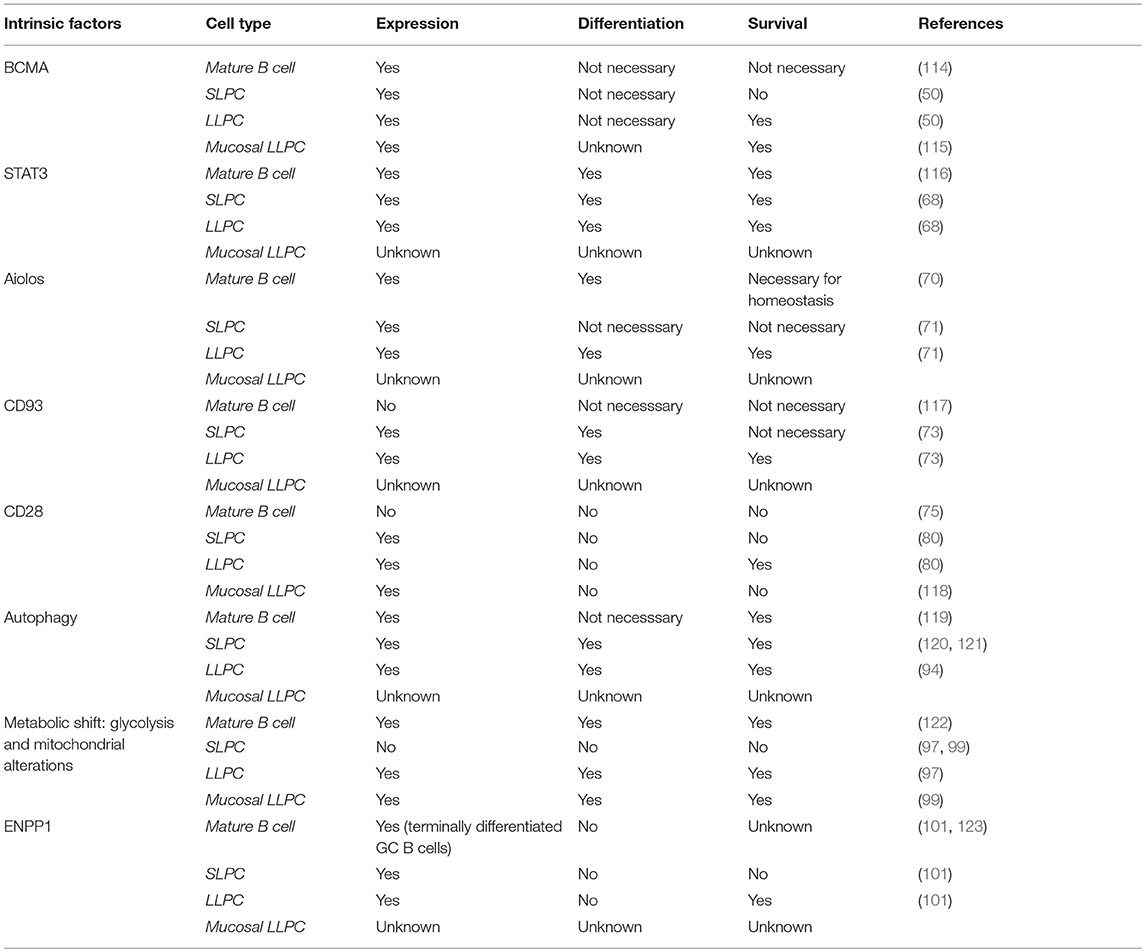
Table 2. Importance of intrinsic factors in B cell and PC subsets.
SL primary author and drafted the bulk of the review, substantial contributions to conception and design. AU provided revisions to content of manuscript. KL contributed to conception and design, provided critical revisions and final approval, and provided funding.
This work has been supported in part by three National Institutes of Health (NIH) grants: awards R01CA121044 and T32CA085183, from the National Cancer Institute (NCI), and R01AI100157, from the National Institute of Allergy and Infectious Diseases (NIAID).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. (2007) 357:1903–15. doi: 10.1056/NEJMoa066092
2. Smith KG, Light A, OReilly LA, Ang SM, Strasser A, Tarlinton D. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J Exp Med. (2000) 191:475–84. doi: 10.1084/jem.191.3.475
3. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Brink R. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med. (2006) 203:2419–24. doi: 10.1084/jem.20061254
4. Moser K, Tokoyoda K, Radbruch A, MacLennan I, Manz RA. Stromal niches, plasma cell differentiation and survival. Curr Opin Immunol. (2006) 18:265–70. doi: 10.1016/j.coi.2006.03.004
5. Sze DM, Toellner KM, de Vinuesa CG, Taylor DR, Ian MacLennan CM. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med. (2000) 192:813–22. doi: 10.1084/jem.192.6.813
6. Hofer T, Muehlinghaus G, Moser K, Yoshida TE, Mei H, Hebel K, et al. Adaptation of humoral memory. Immunol Rev. (2006) 211:295–302. doi: 10.1111/j.0105-2896.2006.00380.x
7. Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol. (1995) 69:1895–902.
8. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. (1997) 388:133–4. doi: 10.1038/40540
9. Kometani K, Kurosaki T. Differentiation and maintenance of long-lived plasma cells. Curr Opin Immunol. (2015) 33:64–9. doi: 10.1016/j.coi.2015.01.017
10. Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. (1997) 16:2996–3006. doi: 10.1093/emboj/16.11.2996
11. Tarte K, Zhan F, De Vos J, Klein B, Shaughnessy J. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood. (2003) 102:592–600. doi: 10.1182/blood-2002-10-3161
12. Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Manz RA. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. (2003) 171:1684–90. doi: 10.4049/jimmunol.171.4.1684
13. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. (2003) 19:607–20. doi: 10.1016/S1074-7613(03)00267-X
14. Valor LM, Rodriguez-Bayona B, Ramos-Amaya AB, Brieva JA, Campos-Caro A. The transcriptional profiling of human in vivo-generated plasma cells identifies selective imbalances in monoclonal gammopathies. PLoS ONE. (2017) 12:e0183264. doi: 10.1371/journal.pone.0183264
15. Kassambara A, Rème T, Michel J, Thierry F, Dirk H, Karin T, et al. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput Biol. (2015) 11:e1004077–e77. doi: 10.1371/journal.pcbi.1004077
16. O'Connor BP, Gleeson MW, Noelle RJ, Erickson LD. The rise and fall of long-lived humoral immunity: terminal differentiation of plasma cells in health and disease. Immunol Rev. (2003) 194:61–76. doi: 10.1034/j.1600-065X.2003.00055.x
17. Nguyen DC, Garimalla S, Xiao H, Kyu S, Albizua I, Galipeau J, et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat Commun. (2018) 9:3698. doi: 10.1038/s41467-018-05853-7
18. Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol. (2003) 3:822–9. doi: 10.1038/nri1203
19. Ellyard JI, Avery DT, Phan TG, Hare NJ, Hodgkin PD, Tangye SG. Antigen-selected, immunoglobulin-secreting cells persist in human spleen and bone marrow. Blood. (2004) 103:3805–12. doi: 10.1182/blood-2003-09-3109
20. Bohannon C, Powers R, Satyabhama L, Cui A, Tipton C, Michaeli M, et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat Commun. (2016) 7:12687. doi: 10.1038/ncomms12687
21. Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, Hauser AE. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. (2016) 9:83–97. doi: 10.1038/mi.2015.38
22. Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten GA. Inflamed kidneys of NZB / W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. (2001) 31:2726–32. doi: 10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H
23. Manz RA, Arce S, Cassese G, Hauser AE, Hiepe F, Radbruch A. Humoral immunity and long-lived plasma cells. Curr Opin Immunol. (2002) 14:517–21. doi: 10.1016/S0952-7915(02)00356-4
24. Landsverk OJ, Snir O, Casado RB, Richter L, Mold JE, Jahnsen FL. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med. (2017) 214:309–17. doi: 10.1084/jem.20161590
25. Brandtzaeg P, Johansen FE. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev. (2005) 206:32–63. doi: 10.1111/j.0105-2896.2005.00283.x
26. Kawanishi H, Saltzman LE, Strober W. Mechanisms regulating IgA class-specific immunoglobulin production in murine gut-associated lymphoid tissues. I. T cells derived from Peyers patches that switch sIgM B cells to sIgA B cells in vitro. J Exp Med. (1983) 157:433–50. doi: 10.1084/jem.157.2.433
27. Dullaers M, Li D, Xue Y, Ni L, Gayet I, Morita R, Oh S. A T cell-dependent mechanism for the induction of human mucosal homing immunoglobulin A-secreting plasmablasts. Immunity. (2009) 30:120–9. doi: 10.1016/j.immuni.2008.11.008
28. Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Macpherson AJ. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. (2010) 328:1705–9. doi: 10.1126/science.1188454
29. Mesin L, Di Niro R, Thompson KM, Lundin KE, Sollid LM. Long-lived plasma cells from human small intestine biopsies secrete immunoglobulins for many weeks in vitro. J Immunol. (2011) 187:2867–74. doi: 10.4049/jimmunol.1003181
30. Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz R, Dorner T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood. (2009) 113:2461–69. doi: 10.1182/blood-2008-04-153544
31. Luger EO, Fokuhl V, Wegmann M, Abram M, Tillack K, Achatz G, Renz H. Induction of long-lived allergen-specific plasma cells by mucosal allergen challenge. J Allergy Clin Immunol. (2009) 124:819–26.e4. doi: 10.1016/j.jaci.2009.06.047
32. Tarlinton D, Radbruch A, Hiepe F, Dorner T. Plasma cell differentiation and survival. Curr Opin Immunol. (2008) 20:162–9. doi: 10.1016/j.coi.2008.03.016
33. Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. (2002) 99:2154–61. doi: 10.1182/blood.V99.6.2154
34. O'Connor BP, Cascalho M, Noelle RJ. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J Exp Med. (2002) 195:737–45. doi: 10.1084/jem.20011626
35. Zinkernagel RM, Bachmann MF, Kundig TM, Oehen S, Pirchet H, Hengartner H. On immunological memory. Annu Rev Immunol. (1996) 14:333–67. doi: 10.1146/annurev.immunol.14.1.333
36. Schuurman HJ, Verdonck LF, Geertzema JG, van der Linden JA, de Gast GC. Monotypic immunoglobulin E plasma cells in an allogeneic bone marrow transplant recipient. Histopathology. (1986) 10:963–9. doi: 10.1111/j.1365-2559.1986.tb02594.x
37. Abedi MR, Backman L, Persson U, Ringden O. Serum IgE levels after bone marrow transplantation. Bone Marrow Transplant. (1989) 4:255–60.
38. Hammarlund E, Thomas A, Amanna IJ, Holden LA, Slayden OD, Slifka MK. Plasma cell survival in the absence of B cell memory. Nat Commun. (2017) 8:1781. doi: 10.1038/s41467-017-01901-w
39. Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. (1998) 8:363–72. doi: 10.1016/S1074-7613(00)80541-5
40. Ahuja A, Anderson MS, Khalil A, Shlomchik JM. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci USA. (2008) 105:4802–7. doi: 10.1073/pnas.0800555105
41. DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas K, Tedder TF. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. (2007) 180:361–71. doi: 10.4049/jimmunol.180.1.361
42. Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Milone M. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. (2016) 128:360–70. doi: 10.1182/blood-2016-01-694356
43. Mei HE, Wirries I, Frolich D, Brisslert M, Giesecke C, Grun J, Dorner T. A unique population of IgG-expressing plasma cells lacking CD19 is enriched in human bone marrow. Blood. (2015) 125:1739–48. doi: 10.1182/blood-2014-02-555169
44. Underhill GH, George D, Bremer EG, Kansas GS. Gene expression profiling reveals a highly specialized genetic program of plasma cells. Blood. (2003) 101:4013–21. doi: 10.1182/blood-2002-08-2673
45. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Lee FE. Long-lived plasma cells are contained within the CD19(-)CD38(hi)CD138(+) subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
46. Reynolds AE, Kuraoka M, Kelsoe G. Natural IgM is produced by CD5- plasma cells that occupy a distinct survival niche in bone marrow. J Immunol. (2015) 194:231–42. doi: 10.4049/jimmunol.1401203
47. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. (2008) 180:3655–9. doi: 10.4049/jimmunol.180.6.3655
48. Minges Wols HA, Ippolito JA, Yu Z, Palmer JL, White FA, Witte PL. The effects of microenvironment and internal programming on plasma cell survival. Int Immunol. (2007) 19:837–46. doi: 10.1093/intimm/dxm051
49. Jourdan M, Cren M, Robert N, Bollore K, Fest T, Duperray C. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia. (2014) 28:1647–56. doi: 10.1038/leu.2014.61
50. O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. (2004) 199:91–8. doi: 10.1084/jem.20031330
51. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. (2003) 21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152
52. Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Anderson KC. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. (2016) 127:3225–36. doi: 10.1182/blood-2016-01-691162
53. Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. (2018) 9:1821. doi: 10.3389/fimmu.2018.01821
54. Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Tarlinton DM. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. (2013) 14:290–7. doi: 10.1038/ni.2527
55. Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. (2002) 100:194–9. doi: 10.1182/blood.V100.1.194
56. Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. (2002) 99:1885–93. doi: 10.1182/blood.V99.6.1885
57. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. (2014) 124:1873–9. doi: 10.1182/blood-2014-05-578732
58. Kawano MM, Mihara K, Huang N, Tsujimoto T, Kuramoto A. Differentiation of early plasma cells on bone marrow stromal cells requires interleukin-6 for escaping from apoptosis. Blood. (1995) 85:487–94.
59. Choe J, Choi Y S. IL-10 interrupts memory B cell expansion in the germinal center by inducing differentiation into plasma cells. Eur J Immunol. (1998) 28:508–15. doi: 10.1002/(SICI)1521-4141(199802)28:02<508::AID-IMMU508>3.0.CO;2-I
60. Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Lipsky PE. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. (2005) 175:7867–79. doi: 10.4049/jimmunol.175.12.7867
61. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J Immunol. (2013) 190:1827–36. doi: 10.4049/jimmunol.1201678
62. Bryant VL, Ma CS, Avery DT, Li Y, Good KLG. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. (2007) 179:8180–90. doi: 10.4049/jimmunol.179.12.8180
63. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly - TFH cells in human health and disease. Nat Rev Immunol. (2013) 13:412–26. doi: 10.1038/nri3447
64. Rodriguez-Bayona B, Ramos-Amaya A, Bernal J, Campos-Caro A, Brieva JA. Cutting edge: IL-21 derived from human follicular helper T cells acts as a survival factor for secondary lymphoid organ, but not for bone marrow, plasma cells. J Immunol. (2012) 188:1578–81. doi: 10.4049/jimmunol.1102786
65. Ramos-Amaya A, Rodriguez-Bayona B, Lopez-Blanco R, Andujar E, Perez-Alegre M, Campos-Caro A. Survival of human circulating antigen-induced plasma cells is supported by plasma cell-niche cytokines and T follicular helper lymphocytes. J Immunol. (2015) 194:1031–8. doi: 10.4049/jimmunol.1402231
66. Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Leonard WJ. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. (2009) 31:941–52. doi: 10.1016/j.immuni.2009.10.008
67. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Tangye SG. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. (2010) 207:155–71. doi: 10.1084/jem.20091706
68. Rodriguez-Bayona B, Ramos-Amaya A, Lopez-Blanco R, Campos-Caro A, Brieva J A. STAT-3 activation by differential cytokines is critical for human in vivo-generated plasma cell survival and Ig secretion. J Immunol. (2013) 191:4996–5004. doi: 10.4049/jimmunol.1301559
69. Rebollo A, Schmitt C. Ikaros, Aiolos and Helios: transcription regulators and lymphoid malignancies. Immunol Cell Biol. (2003) 81:171–5. doi: 10.1046/j.1440-1711.2003.01159.x
70. Wang JH, Avitahl N, Cariappa A, Friedrich C, Ikeda T, Renold A, et al. Aiolos regulates B cell activation and maturation to effector state. Immunity. (1998) 9:543–53. doi: 10.1016/S1074-7613(00)80637-8
71. Cortes M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med. (2004) 199:209–19. doi: 10.1084/jem.20031571
72. Greenlee MC, Sullivan SA, Bohlson SS. CD93 and related family members: their role in innate immunity. Curr Drug Targets. (2008) 9:130–8. doi: 10.2174/138945008783502421
73. Chevrier S, Genton C, Kallies A, Karnowski A, Otten L, Orbea A. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc Natl Acad Sci USA. (2009) 106:3895–900. doi: 10.1073/pnas.0809736106
74. Haralambieva IH, Zimmermann MT, Ovsyannikova IG, Grill DE, Oberg AL, Poland GA. Whole transcriptome profiling identifies CD93 and other plasma cell survival factor genes associated with measles-specific antibody response after vaccination. PLoS ONE. (2016) 11:e0160970. doi: 10.1371/journal.pone.0160970
75. Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. (2006) 24:269–81. doi: 10.1016/j.immuni.2006.01.012
76. Ferguson SE, Han S, Kelsoe G, Thompson CB. CD28 is required for germinal center formation. J Immunol. (1996) 156:4576–81.
77. Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. (1993) 261:609–12. doi: 10.1126/science.7688139
78. Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Green JM. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol. (2009) 29:3710–21. doi: 10.1128/MCB.01869-08
79. Friend LD, Shah DD, Deppong C, Lin J, Bricker TL, Green JM. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J Exp Med. (2006) 203:2121–33. doi: 10.1084/jem.20052230
80. Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Lee KP. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. (2011) 208:1435–46. doi: 10.1084/jem.20110040
81. Njau MN, Kim JH, Chappell CP, Ravindran R, Thomas L, Jacob J. CD28-B7 interaction modulates short- and long-lived plasma cell function. J Immunol. (2012) 189:2758–67. doi: 10.4049/jimmunol.1102728
82. Rozanski CH, Utley A, Carlson LM, Farren MR, Murray M, Lee KP. CD28 promotes plasma cell survival, sustained antibody responses, and BLIMP-1 upregulation through its distal PYAP proline motif. J Immunol. (2015) 194:4717–28. doi: 10.4049/jimmunol.1402260
83. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth G, Nutt SL. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. (2016) 17:323–30. doi: 10.1038/ni.3348
84. Murray ME, Gavile CM, Nair JR, Koorella C, Carlson LM, Lee KP. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood. (2014) 123:3770–9. doi: 10.1182/blood-2013-10-530964
85. Nair JR, Carlson LM, Koorella C, Rozanski CH, Byrne GE, Lee KP. CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J Immunol. (2011) 187:1243–53. doi: 10.4049/jimmunol.1100016
86. Koorella C, Nair JR, Murray ME, Carlson LM, Watkins SK, Lee KP. Novel regulation of CD80/CD86-induced phosphatidylinositol 3-kinase signaling by NOTCH1 protein in interleukin-6 and indoleamine 2,3-dioxygenase production by dendritic cells. J Biol Chem. (2014) 289:7747–62. doi: 10.1074/jbc.M113.519686
87. Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. (2002) 277:49047–54. doi: 10.1074/jbc.M205011200
88. Yorimitsu T, Usha N, Zhifen Y, Daniel Klionsky J. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. (2006) 281:30299–304. doi: 10.1074/jbc.M607007200
89. Pua HH, Ivan D, Mariana C, Noboru M, You-Wen HE. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. (2007) 204:25. doi: 10.1084/jem.20061303
90. McLeod IX, Jia W, He YW. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev. (2012) 249:195–204. doi: 10.1111/j.1600-065X.2012.01143.x
91. Cenci S. The proteasome in terminal plasma cell differentiation. Semin Hematol. (2012) 49:215–22. doi: 10.1053/j.seminhematol.2012.04.005
92. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. (2007) 447:859–63. doi: 10.1038/nature05853
93. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. (2010) 584:1287–95. doi: 10.1016/j.febslet.2010.01.017
94. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A.Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. (2013) 14:298–305. doi: 10.1038/ni.2524
95. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Pearce EL. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
96. Buck MD, OSullivan D, Klein Geltink RI, Curtis JD, Chang CH, Pearce EL. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
97. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Bhattacharya D. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. (2016) 45:60–73. doi: 10.1016/j.immuni.2016.06.011
98. Lam WY, Jash A, Yao CH, DSouza L, Wong R, Bhattacharya D. Metabolic and transcriptional modules independently diversify plasma cell lifespan and function. Cell Rep. (2018) 24:2479–92.e6. doi: 10.1016/j.celrep.2018.07.084
99. Kunisawa J, Sugiura Y, Wake T, Nagatake T, Suzuki H, Nagasawa R. Mode of bioenergetic metabolism during B cell differentiation in the intestine determines the distinct requirement for vitamin B1. Cell Rep. (2015)13:122–31. doi: 10.1016/j.celrep.2015.08.063
100. Kato K, Nishimasu H, Okudaira S, Mihara E, Ishitani R, Takagi J. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc Natl Acad Sci USA. (2012) 109:16876–81. doi: 10.1073/pnas.1208017109
101. Wang H, Gonzalez-Garcia I, Traba J, Jain S, Conteh S, Shin D, et al. ATP-degrading ENPP1 is required for survival (or persistence) of long-lived plasma cells. Sci Rep. (2017) 7:17867. doi: 10.1038/s41598-017-18028-z
102. Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. (2002) 169:4213–21. doi: 10.4049/jimmunol.169.8.4213
103. Wilmore JR, Allman D. Here, there, and anywhere? Arguments for and against the physical plasma cell survival Niche. J Immunol. (2017) 199:839–45. doi: 10.4049/jimmunol.1700461
104. Moore N, Moreno Gonzales M, Bonner K, Smith B, Park W, Stegall M. Impact of CXCR4/CXCL12 blockade on normal plasma cells in vivo. Am J Transplant. (2017) 17:1663–9. doi: 10.1111/ajt.14236
105. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. (1999) 93:1658–67.
106. Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, Manz R A. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. (2010) 116:1867–75. doi: 10.1182/blood-2009-12-259457
107. Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. J Immunol. (2010) 185:7180–5. doi: 10.4049/jimmunol.1002319
108. Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. (2011) 12:151–9. doi: 10.1038/ni.1981
109. Brynjolfsson SF, Persson Berg L, Olsen Ekerhult T, Rimkute I, Wick MJ, Grimsholm O. Long-lived plasma cells in mice and men. Front Immunol. (2018) 9:2673. doi: 10.3389/fimmu.2018.02673
110. Chu VT, Beller A, Rausch S, Strandmark J, Zanker M, Arbach O. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity. (2014) 40:582–93. doi: 10.1016/j.immuni.2014.02.014
111. Berek C. Eosinophils: important players in humoral immunity. Clin Exp Immunol. (2016) 183:57–64. doi: 10.1111/cei.12695
112. Bortnick A, Chernova I, Spencer SP, Allman D. No strict requirement for eosinophils for bone marrow plasma cell survival. Eur J Immunol. (2018) 48:815–21. doi: 10.1002/eji.201747229
113. Glatman Zaretsky A, Konradt C, Depis F, Wing JB, Goenka R, Hunter CA. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. (2017) 18:1906–16. doi: 10.1016/j.celrep.2017.01.067
114. Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol. (2001) 21:4067–74. doi: 10.1128/MCB.21.12.4067-4074.2001
115. Gustafson CE, Higbee D, Yeckes AR, Wilson CC, De Zoeten EF, Jedlicka P. Limited expression of APRIL and its receptors prior to intestinal IgA plasma cell development during human infancy. Mucosal Immunol. (2013) 7:467. doi: 10.1038/mi.2013.64
116. Chou WC, Levy DE, Lee CK. STAT3 positively regulates an early step in B-cell development. Blood. (2006) 108:3005–11. doi: 10.1182/blood-2006-05-024430
117. McKearn JP, Baum C, Davie JM. Cell surface antigens expressed by subsets of pre-B cells and B cells. J Immunol. (1984) 132:332–9.
118. Gärdby E, Wrammert J, Schön K, Ekman L, Leanderson T, Lycke N. Strong differential regulation of serum and mucosal IgA responses as revealed in CD28-deficient mice using cholera toxin adjuvant. J Immunol. (2003) 170:55–63. doi: 10.4049/jimmunol.170.1.55
119. Arnold J, Murera D, Arbogast F, Fauny JD, Muller S, Gros F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. (2016) 23:853–64. doi: 10.1038/cdd.2015.149
120. Milan E, Fabbri M, Cenci S. Autophagy in plasma cell ontogeny and malignancy. J Clin Immunol. (2016) 36 (Suppl. 1):18–24. doi: 10.1007/s10875-016-0254-9
121. Oliva L, Cenci S. Autophagy in plasma cell pathophysiology. Front Immunol. (2014) 5:103. doi: 10.3389/fimmu.2014.00103
122. Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience. (2018) 5:99–109. doi: 10.1016/j.isci.2018.07.005
Keywords: long-lived plasma cells (LLPC), plasma cell survival, plasma cell niche, plasma cell function, humoral responses
Citation: Lightman SM, Utley A and Lee KP (2019) Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 10:965. doi: 10.3389/fimmu.2019.00965
Received: 16 November 2018; Accepted: 15 April 2019;
Published: 03 May 2019.
Edited by:
Simone Cenci, San Raffaele Hospital (IRCCS), ItalyReviewed by:
F. Eun-Hyung Lee, Emory University, United StatesCopyright © 2019 Lightman, Utley and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shivana M. Lightman, U2hpdmFuYS5saWdodG1hbkByb3N3ZWxscGFyay5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.