 Sofia Helfrich
Sofia Helfrich Barbara C. Mindt
Barbara C. Mindt Jörg H. Fritz
Jörg H. Fritz Claudia U. Duerr
Claudia U. Duerr
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 07 June 2019
Sec. NK and Innate Lymphoid Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00930
This article is part of the Research TopicThe Role of Innate Lymphoid Cells in Mucosal ImmunityView all 17 articles
Millions of people worldwide are suffering from allergic inflammatory airway disorders. These conditions are regarded as a consequence of multiple imbalanced immune events resulting in an inadequate response with the exact underlying mechanisms still being a subject of ongoing research. Several cell populations have been proposed to be involved but it is becoming increasingly evident that group 2 innate lymphoid cells (ILC2s) play a key role in the initiation and orchestration of respiratory allergic inflammation. ILC2s are important mediators of inflammation but also tissue remodeling by secreting large amounts of signature cytokines within a short time period. Thereby, ILC2s instruct innate but also adaptive immune responses. Here, we will discuss the recent literature on allergic inflammation of the respiratory tract with a focus on ILC2 biology. Furthermore, we will highlight different therapeutic strategies to treat pulmonary allergic inflammation and their potential influence on ILC2 function as well as discuss the perspective of using human ILC2s for diagnostic purposes.
Respiratory allergic inflammatory conditions such as asthma and allergic rhinitis (hay fever) are affecting millions of people globally (1, 2). Importantly, the prevalence of these non-communicable disorders is rapidly increasing in contrast to communicable diseases of the respiratory tract, which are on the decline (1). Due to their widespread morbidity, they represent a substantial social as well as economic burden (3). The skew toward a type 2 immune response is often an important characteristic of these chronic conditions that involve both innate and adaptive branches of the immune system (4). In addition to T helper 2 (TH2) cells of the adaptive immune system, group 2 innate lymphoid cells (ILC2s) are critical in instructing a strong type 2 immune response (5). ILC2s belong to the group of innate lymphoid cells (ILCs) that provide host defense against infectious agents, participate in inflammatory responses and mediate lymphoid organogenesis and tissue repair, particularly at mucosal barriers. Taking the newest nomenclature of ILCs into account, the ILC family is comprised of five subsets including Natural Killer (NK) cells, lymphoid tissue inducer cells (LTi cells) as well as the three helper ILC members. While LTi cells are key drivers of lymphoid organogenesis and NK cells are important to fight off viral infections, helper ILCs are regarded as the innate counterpart of TH cells but lack the surface expression of common adaptive lineage markers as well as specific antigen receptors (6, 7). These helper ILCs, namely, ILC1s, ILC2s, and ILC3s are defined by their effector cytokine profile and transcription factor expression (8). The master transcription factors for the different helper subsets are T-bet for ILC1, Gata3 and Rorα for ILC2s and RORγt for ILC3s. Helper ILCs are important sources of innate effector cytokines such as ILC1-derived IFNγ, ILC2-derived IL-4, IL-5, IL-13, and amphiregulin (Areg) as well as ILC3-derived IL-17 and IL-22 (Figure 1). ILC2s are innately committed to type 2 immunity which consequently puts them in the spotlight during the onset of an allergic immune response. ILC2s are activated by local immune mediators, typically alarmins, and are able to produce large amounts of signature cytokines within a short period of time (9). Thereby, ILC2s can initiate and amplify immune responses and are able to influence innate as well as adaptive immunity by both their secreted cytokines and through cell-cell interactions. Hence, ILC2s serve as an important link between innate and adaptive effector branches of type 2 immunity. Depending on the tissue they reside in, ILC2s exhibit slightly diverse profiles shaped by their microenvironment (10). However, their specific characteristics such as their ability to produce type 2 signature cytokines in a stark and fast fashion remains unchanged. Since their detailed description in 2010 (11–13), our knowledge about this fascinating immune population has steadily increased both in mouse models but also, through applied research, in humans. In this review we will provide a brief snapshot on our current knowledge of ILC2s in mouse and human allergic respiratory inflammation. Moreover, we will summarize experimental mouse models and discuss how recent reports led to an improved understanding of therapeutic strategies in human allergic respiratory diseases with the focus on asthma.
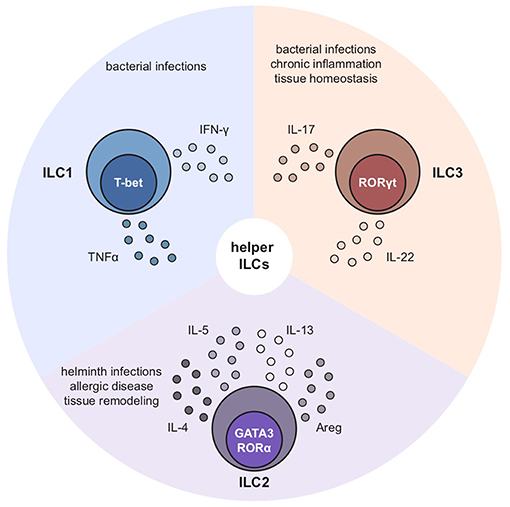
Figure 1. Characteristics of ILC1s, ILC2s, and ILC3s. ILC1s, ILC2s, and ILC3s are characterized by expression of their key transcription factors and signature cytokines: ILC1s depend on T-bet and produce IFNγ, ILC2s on Gata3 and Rorα and secrete IL-4, IL-5, IL-13 as well as amphiregulin while ILC3s depend on Rorγt and release IL-17 and IL-22 upon activation.
One of the most common human allergic diseases in the respiratory tract is asthma. Importantly, asthma is no longer considered to be one specific disease but more of an umbrella term for chronic inflammation of the lower airways with characteristics such as wheezing, bronchoconstriction and shortness of breath (4, 14). The heterogeneity of asthma is mirrored by different immune profiles. In general, asthma is subdivided into type 2 and non-type 2 with further separation of type 2 asthma in allergic or non-allergic asthma accompanied with eosinophilia (14, 15). Non-type 2 (low) asthma is defined as asthma without eosinophilia and with increased presence of neutrophils and/or IL-17 producing cells. These different asthma subtypes are termed endotypes for better classification (16). Interestingly, although there are cases of asthma onset in adulthood in a cohort of patients, most asthmatic individuals develop the disease during childhood (17). However, the exact mechanisms are still not completely understood. Different secondary diseases are correlated with asthma such as allergic rhinitis, chronic rhinosinusitis, and the development of nasal polyps. To provide optimal care to asthmatic patients of all different subgroups, a shift to a more personalized treatment approach is in the focus of discussion. In this context, mouse models of human allergic airway inflammation are an invaluable tool to understand underlying mechanisms of disease. Common allergens, including house dust mite (HDM) and papain can trigger respiratory allergic inflammation in humans as well as mice (18, 19), and mouse models can therefore be used to recapitulate these immune responses in an experimental system and thus aid in deciphering the underlying mechanism and processes of allergic respiratory diseases. To increase our knowledge of allergic lung inflammation, different experimental mouse models can be applied. Allergens such as ovalbumin (OVA), HDM, papain, fungal extracts, ILC2-eliciting cytokines and combinations thereof are used to induce and mimic allergic inflammation in the respiratory tract. The importance of ILC2s in the onset of allergic airway inflammation is highlighted by their detection in these experiments and thoroughly stratified in Table 1.
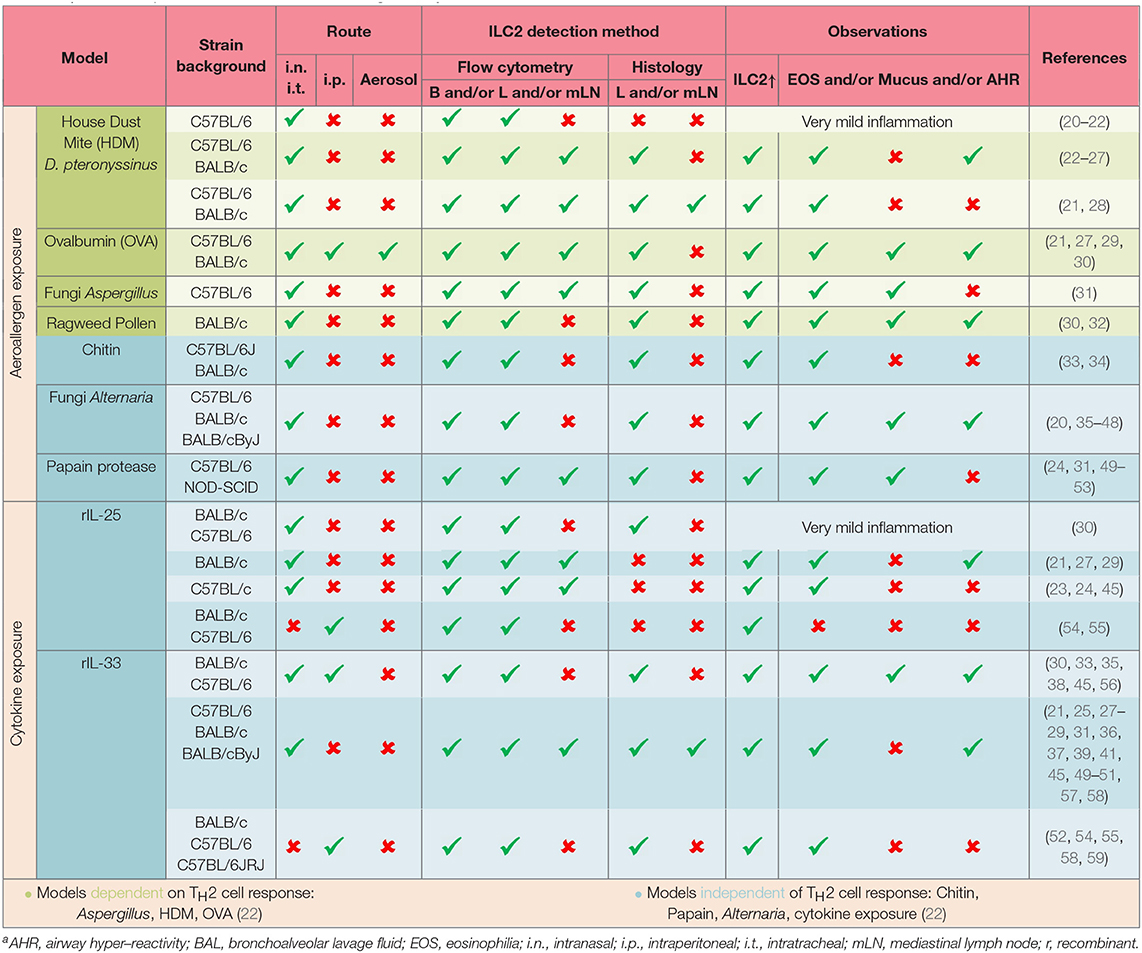
Table 1. ILC2s in experimental mouse models of allergic airway inflammationa.
Allergic respiratory diseases are characterized in general by a dysregulated immune response targeting a harmless and non-pathogenic allergen. Several immune populations participate in an allergic respiratory disease including populations of the innate as well as the adaptive immune system. Different subgroups of T and B cells as well as eosinophils, basophils, NK cells and, last but not least, ILC2s are major players. Although a direct mechanistical link of ILC2s to asthma pathogenesis still needs to be established in humans, an increasing body of evidence supports an association of ILC2s with disease in asthmatic patients. These include GWAS studies revealing several genes essential in ILC2 biology as susceptibility markers for human asthma such as the ILC2-activating alarmin IL-33 and its cognate receptor ST2 (IL1RL1) as well as the transcription factors GATA3 and RORα (57, 60, 61).
ILC2s are the predominant ILC population in the lung, in contrast to other mucosal surfaces such as small intestine and colon. However, the reason for this skewed distribution is still not clear. ILC2s are activated mainly by the cytokines interleukin (IL)-25, IL-33, and thymic stromal lymphopoietin (TSLP) in the lungs, however, ILC2s accumulate in tissues independent of these signals and pulmonary ILC2s can also be detected in triple knockout mice deficient in the TSLP receptor, ST2, and IL-25 (10, 62). However, ILC2s show reduced IL-5 reporter expression in lung, fat, and gut tissue, but not in the skin indicating that TSLP, ST2, and IL-25 signaling is indeed needed for the functional maintenance of ILC2s in the local microenvironment of the lung (10). In addition to IL-5, ILC2s commonly secrete IL-4, IL-9, IL-13, and amphiregulin. IL-4 triggers the differentiation of TH2 cells and induces class switching of B cells to IgE. IL-5 activates B cells and plays a role in eosinophil homeostasis (63). IL-9 is needed by ILC2s for their survival and maintenance (64, 65), and IL-13 induces goblet cell hyperplasia and mucus secretion but can also act on alveolar macrophages (13, 66) and initiate the migration of dendritic cells to the mediastinal lymph nodes (49). Moreover, ILC2s are able to serve as antigen presenting cells for T cells by expressing MHCII (67), although this appears to be less pronounced in the lungs compared to the gut (67). Additional functional characteristics of pulmonary ILC2s include their expression of Il5 as well as ST2 (IL-33R) at steady state in contrast to intestinal ILC2s which express both IL-5 and IL-13 mRNA and mainly the IL-25 receptor chain (IL-25RB) further demonstrating that ILC2s are imprinted by their microenvironment (10, 63).
Furthermore, ILC2s get support from basophil-derived IL-4 (68), and T cell-derived IL-2 (56), thus, establishing a quick but robust allergic reaction (Figure 2). In addition to the typical type 2 cytokines, IL-17 secreting ILC2s have been described, a cytokine known to be regulated by RORγt that has been correlated to severe asthma phenotypes (69). However, while one group reported IL-17 expression by KLRG1hiST2− inflammatory ILC2s (iILC2s) in the lungs, which correlated with their expression of RORγt (70) a more recent report showed increased IL-17+ST2+ ILC2s (ILC217s) upon IL-33 or allergen challenge, independently of RORγt expression (52, 70). Finally, in line with their immunomodulatory potential, ILC2s can also acquire a regulatory phenotype and memory-like properties upon IL-33 and IL-2 stimulation in vivo, decreasing the expression of their pro-inflammatory repertoire as well as eosinophilic recruitment and accumulation (71). Further studies on this subset could certainly open up new venues in allergic disease therapies. Overall, ILC2s are key in the initiation, amplification, and modulation of type 2 immune responses in the respiratory tract by exhibiting these fundamental characteristics.
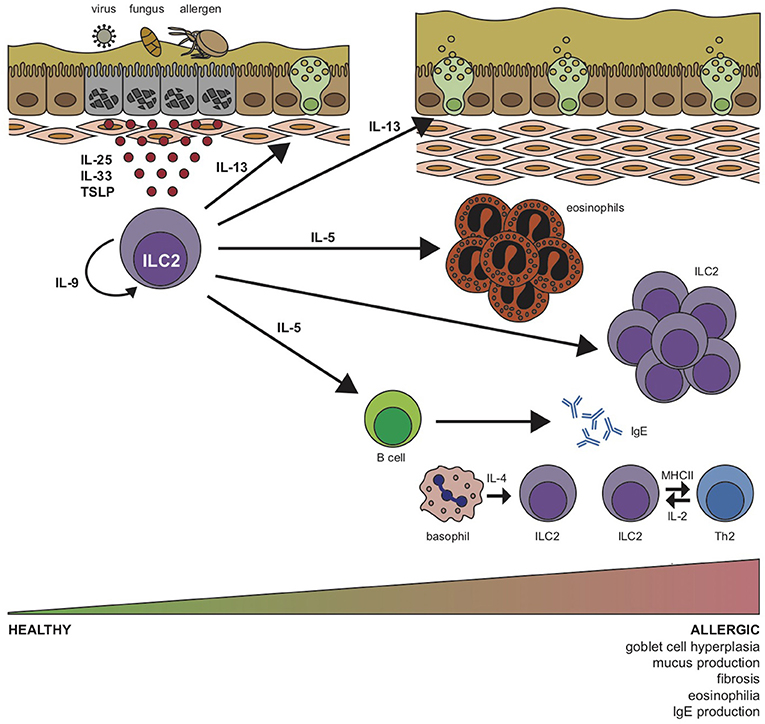
Figure 2. Group 2 innate lymphoid cell (ILC2) stimulation by airway pathogens drives allergic respiratory inflammation. In response to allergens, fungi or viruses, lung epithelial cells release alarmins IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) that induce expansion and cytokine production by ILC2s. Activated ILC2s initiate an innate type 2 inflammatory response through production of IL-5, which induces eosinophilia, and IL-13, which promotes airway hyper-reactivity, goblet cell hyperplasia, mucus production and fibrosis. B cells and basophils are stimulated to release IgE and IL-4, respectively, and ILC2-derived IL-9 acts in an autocrine manner to prolong survival of ILC2s in the lung. Expression of MHCII enables ILC2 interaction with TH2 cells, which subsequently promote ILC2 function via T cell-derived IL-2.
Due to their scarcity and phenotypical heterogeneity in mice and humans, the detection of ILC2s by flow cytometry requires some considerations. ILC2s are characterized by the absence of known lineage markers and antigen receptors as well as the expression of surface markers CD45, CD25 (IL-2Rα), CD127 (IL-7Rα), and ST2 (IL-33R) in human asthma as well as in rodent models of respiratory allergic inflammation. Interestingly, CD127 low/negative ILC2s have also been reported in asthmatic patients as well as in an allergic experimental mouse model (72, 73). Of note, iILC2s show reduced CD127 expression upon systemic challenge with IL-25 (70). Whether these ILC2s have just downregulated the expression or endocytosed CD127 due to strong activity still needs to be investigated. In addition, mouse ILC2s are further identified by the surface expression of Thy1, a marker that is not present on human ILC2s. Another difference is the expression of CD161 by human ILC2s. The functions of these two glycoproteins are still not completely understood. Both mouse and human ILC2s can express the prostaglandin receptor family member CRTH2. CRTH2 is important in the migration of ILC2s and thereby expressed to a different extent depending on location (74). However, the detection of mouse CRTH2 is limited by the availability of a specific detection antibody (74, 75). Our current strategies to detect ILC2s are largely based on surface receptors, which are actively used in an immune response by the cell to sense its environment and act accordingly. In addition, surface receptors and Gata3 are often used in combination to detect human and murine ILC2s (76, 77). The caveat with Gata3 is that ILC2s in the lungs are able to downregulate Gata3 expression under specific conditions such as viral (78), or helminth infections (70), however, under steady state conditions Gata3 is a reliable marker for pulmonary ILC2s.
Interestingly, ILC2s are not a homogenous population and several different subtypes of ILC2s have been reported at mucosal surfaces (79), including the lungs (31, 70). Memory ILC2s can be elicited upon repeated challenge of mice with IL-33, or when using papain as an allergen (31), thereby intensifying the immune response. Thus, ILC2s are specifically shaped by their local microenvironment and are reported to exhibit a sedentary lifestyle (45, 80). However, a subgroup of murine ILC2s, intestinally-derived inflammatory ILC2s (iILC2s) are able to traffic through the lymphatics and blood toward the lung to aid in the local response to helminth infection or systemic challenge with IL-25 (81). Moreover, the identification of circulating multipotent ILC progenitors in peripheral blood in humans indicates that ILC2 progenitors may be able to traffic to local pools of tissue-resident cells and replenish them (82). Since iILC2s are able to further differentiate into natural ILC2s (nILCs), the ability to traffic might be limited to specific circumstances and confined to not yet fully matured or differentiated ILC2 populations. However, additional studies are needed to pinpoint the potential of ILC2s to migrate within the body. nILC2s, which are present at steady state in the lungs and characterized by their ST2 expression, are typically increased upon stimulation with IL-33 (70). Both populations, iILC2s and memory ILC2s are characterized by elevated expression of the IL-25 receptor in the lungs (31, 70), however, it is still not completely understood how similar the biology and function of both populations is. So far, IL-25 receptor-expressing ILC2s have only been described in the human skin (83), and it still needs to be determined whether IL-25 receptor expression is present and regulated in human ILC2s during respiratory inflammation. KLRG1 is another commonly used marker to identify mature ILC2s in both human and mouse lungs. However, its expression is greatly influenced by androgens with ILC2s from male mice exhibiting higher expression of KLRG1 while female lungs harbor significantly higher numbers of KLRG1− ILC2s promoting lung inflammation (59, 84). The ligand for KLRG1 is E-cadherin and ILC2s isolated from human skin were shown to be restrained by this interaction (83). Testosterone can additionally regulate ILC2s by controlling their response to IL-2 as well as by the restraint of IL-5 and IL-13 production and thus resulting in decreased pulmonary pathology upon Alternaria challenge (42). In the same study, elevated levels of ILC2s in peripheral blood of female asthma patients in comparison to male patients have been reported which is especially interesting in the light of the increased prevalence of asthma in women. The exact role of androgens such as testosterone in asthma still needs further investigation since testosterone is able to induce IL-33 mRNA in mast cells (85), however, lower levels of IL-33 and TSLP have been detected in the bronchoalveolar lavage of Alternaria challenged mice.
Insights into the function of ILC2s in allergic inflammation has mainly been generated using experimental mouse models. However, a substantial amount of reports provide evidence that ILC2s are also key in human allergic respiratory inflammation. Of note, the first reports on human ILC2s provided a detailed description of ILC2s in polyp tissue of chronic rhinosinusitis patients (75, 77). Moreover, increased levels and activity of ILC2s have also been reported in asthmatic patients. ILC2s could be detected in bronchoalveolar lavage, lung tissue, sputum and blood of patients with respiratory inflammation. Although the gating strategies of ILC2s slightly vary between the distinct reports, all studies demonstrate expression of CD127 in combination with CRTH2 and/or CD44 and ST2 on ILC2s. A positive correlation of eosinophilia and ILC2s levels has been further reported in human patients similar to the observation in mouse respiratory inflammation (86, 87). Recent work opened the discussion of functional redundancy of TH2 cells and ILC2s in humans (88). However, even if this is the case, pulmonary ILC2s have a critical function in the development of allergic diseases being innately committed to type 2 immunity and strong and immediate amplifiers of initial responses.
The prevalence of both obesity and asthma has increased drastically in recent years. Although asthma in obese patients is characterized mainly as non-allergic with an increase in neutrophils, eosinophils have been reported to be present in elevated numbers in the lung tissue of obese asthma patients as well (89). Of note, in addition to their mucosal location, ILC2s were originally identified as fat-associated lymphoid cluster (FALC) Lineage−ckit+Sca-1+ cells in the mesentery (12). Here, adipocytes and endothelial cells within the adipose tissue are sources of ILC2-activating IL-25 and IL-33 (90, 91). ILC2s are able to maintain the metabolic status of healthy adipose tissue by secreting IL-5 for eosinophil homeostasis, IL-13 to trigger alternative macrophage differentiation and methionine-enkephalin which directly acts on adipocytes and induces beiging of fat (92). However, in obesity ILC2s are decreased in adipose tissue and in ILC2-deficient mice, a high-fat diet accelerates obesity and insulin resistance indicating that ILC2s in adipose tissue are important for homeostasis. It thus seems contrary at first to link obesity and asthma. However, although obese mice have lower ILC2s and eosinophils in their adipose tissue, the levels of these populations are increased in the lungs in obese mice at steady state and upon allergen challenge such as with HDM. Not only ILC2s but also ILC3s are increased in the lungs of obese mice which can be enhanced by ozone triggered IL-33 or Nlrp3 inflammasome induced IL-1β, respectively (93, 94). It has been suggested that ILC2s and eosinophils might migrate from adipose tissue into lung tissue during obesity and thereby influence pulmonary immunity and may trigger asthma. This represents an interesting potential mechanism but further research will be needed to fully support this idea.
Interestingly, in humans, an allergic response is provoked in the skin as a first assessment of an allergic response in general but also as a first evaluation of asthma. Different tests can be used depending on the way of application using subcutaneous injections or exposure to allergen by topical application with the most common test in clinical practice still being the prick test. It still needs to be determined to what extent and how ILC2s directly contribute to the assessment of allergy via the prick test. Upon external stimuli or cellular damage, IL-33 is released from cells and engages ST2 on ILC2s but also on TH2 cells, eosinophils, mast cells, and basophils, contributing to cutaneous allergic inflammation with increased levels of local and peripheral blood ILC2s, eosinophils, IgE, and histamine (95). IL-33 can be released locally and systemically after mechanical skin injury (i.e., scratching), promoting IgE-mediated degranulation (96). Of note, ILC2s present in the skin can control mast cell (MC) activity by direct interaction (97). Conversely, MC are an important source of IL-33 in vivo, contributing to ILC2 activation and type 2 immune response in disease models of multiple sclerosis (85), and helminth infection (98), a MC-ILC2 crosstalk also occurs in the lung (99). It is not yet fully understood how cutaneous antigen exposure could activate ILC2s but it is conceivable that dysregulation between MC and ILC2s could exacerbate the immune response during allergic airway inflammation and anaphylactic reaction. Moreover, the evaluation of the eosinophil count, as well as the level of IgE, including allergen-specific IgE in blood, or less common in sputum, is used to assess the grade of the allergic response. Type 2 signature cytokines (IL-4, IL-5, and IL-13) are used in addition as biomarkers. However, to assess the severity of asthma, lung function tests are routinely carried out in humans. Sequential examinations are performed on the patient such as allergy tests to pinpoint the responsible allergen(s), bronchial provocation or exhaled nitric oxide tests; before formulating any therapeutic recommendation (100). Since asthma and allergic inflammation of the respiratory tract can have multiple underlying causes, the aim is to personalize the treatment as much as possible depending on the results of the examinations. Thus, achieving control is the main objective currently proposed in asthma management where pharmacological and non-pharmacological treatment is adjusted in a continuous cycle that involves assessment, treatment and review. In mouse models, experimentally-induced airway hyper-reactivity is usually analyzed upon challenge with increasing doses of inhaled methacholine. The concept of using ILC2 prevalence as a biomarker for diagnostic purposes in lung disease is appealing. Screening of ILC2s as an early hierarchical population might already indicate asthma susceptibility before the start of symptoms or pathology in the lungs. However, due to their high phenotypic diversity, ILC2 characterization in different asthmatic subgroups and their comparison is necessary, and even then, ILC2 level and functionality should be carefully evaluated for each subgroup. Nevertheless, biomarkers and the assessment of allergic respiratory diseases show overall important similarities between rodent models of disease and clinical practice.
In humans, as a first and often immediate treatment inhaled corticosteroids are commonly used in both allergic and non-allergic respiratory inflammation. Corticosteroids reduce the general inflammation of the lung and provide relief of symptoms for the patient (101), but can also cause adverse side effects especially when given systemically in high doses and during long-term treatment. Importantly, corticosteroids dampen the activity of both mouse and human ILC2s (102). However, under specific conditions, corticosteroids are less able to act on ILC2 activity. This has been reported to be the case in situations of enhanced STAT5 activation upon TSLP stimulation. This increased activation of STAT5 by TSLP has been identified as a regulatory mechanism in mice (102), was further confirmed in human ILC2s (103) and helps to explain why not all asthma patients respond to corticosteroid therapy. Corticosteroid resistance can also occur in neutrophilic asthma. IL-17 contributes to neutrophil accumulation in the lungs but also increases the expression of the glucocorticoid receptor beta (GRβ) (104). GRβ inhibits the activity of GRα by direct competition for glucocorticoids. Increased expression of GRβ has been reported on cell populations in glucocorticoid-resistant patients (105, 106), and is discussed to contribute to steroid resistance in neutrophilic asthma (104). IL-17 can also be derived from pulmonary iILC2s (70) as well as ILC217s, which were described to be the main source of IL-17 in the lung after IL-33-induced lung inflammation (52). However, if and which role ILC2s may have in neutrophilic asthma still needs to be investigated.
Like corticosteroids, β2-agonists that act on β2-adrenergic receptors are frequently used to treat asthmatic patients. A recent report showed that both human and mouse ILC2s express the β2AR (β2-adrenergic receptor) for epinephrine and norepinephrine and that the use of an agonist during lung inflammation in mice impaired ILC2 proliferation, cytokine production and effector function (107). These findings highlight the importance of ILC2s in integrating neuroimmune signals. In humans, short acting β-agonists (SABA, e.g., Salbutamol) and long acting β-agonists (LABA, e.g., Salmeterol) are routinely used for asthma treatment. LABA is used in combination with inhaled corticosteroids whereas SABA is also approved as a monotherapy in mild asthma (108). Adverse effects of β2-agonists have been reported and the discussion was stirred up upon the report of detrimental effects upon excessive Fenoterol (SABA) treatment (long time and high dose) (109, 110). The underlying mechanisms of these severe consequences are only incompletely understood. However, the use of β2-agonists for asthmatic patients under treatment is generally regarded as safe and successful (101).
Bioactive lipid mediators are important regulators of ILC2s (111). Indeed, leukotrienes (LTs) including cysteinyl LTs (CysLTs) are generated by arachidonic acid metabolism and CysLTs have been linked to the initiation of asthma and bronchoconstriction since a long time (112). Leukotriene receptor antagonists such as Montelukast, a CysLT1 receptor antagonist, are commonly prescribed to improve asthma symptoms in humans (113). Both mouse and human ILC2s have been reported to express CysLT receptors and it was shown that CysLTs positively regulate ILC2 activation (47, 114–116).
In addition, Leukotriene B4 (LTB4) has also been linked to asthma. LTB4 is a neutrophil chemoattractant in pulmonary inflammation (117), and neutrophils can be increased during the exacerbation phase of asthma (118). However, sputum samples of asthmatic patients showed an increased level of LTB4 which strikingly did not correlate with neutrophil levels in the samples of the analyzed patients (119). The high affinity receptor for LTB4, LTB4R1, has been reported on mouse ILC2s but its presence and role in human ILC2s still needs to be elucidated. Interestingly, LTB4 also plays an important role in the development of insulin resistance in obese mice (120). However, how and if ILC2s are involved in this context still needs to be addressed.
Similar to LTs, prostaglandins are products of arachidonic acid metabolism. As mentioned previously, human and mouse ILC2s express the receptor for prostaglandin D2, CRTH2 (74, 75). CRTH2 plays a potent role in activation, migration and cytokine release of TH2 cells and eosinophils. Curiously, accumulation of pulmonary ILC2s is regulated via CRTH2 and differences in its expression on ILC2s have been reported in inflamed pulmonary tissue (121). Use of monoclonal antibodies against CRTH2 resulted in a reduction of CRTH2 expressing cells including ILC2s in mice (122). In addition to its role in migration of ILC2s, PGD2 has been reported to potentiate the action of ILC2s eliciting cytokines leading to an increase in effector cytokine expression. Consequently, small-molecules antagonists of the CRTH2 receptor, have been promising in human trials for asthma patients (123–126). One example is OC000459 which was reported to be a safe and effective alternative treatment of eosinophilic asthma improving lung function and asthma symptoms (127, 128).
Monoclonal antibodies are used to treat allergic respiratory diseases such as asthma in humans. These antibodies are directed against either type 2 signature cytokines IL-4, IL-5, and IL-13, their respective surface receptors or against IgE. Both TH2 cells and ILC2s are potential sources of type 2 signature cytokines. Moreover, ILC2s have been reported to enhance the adaptive immune response and thereby influence IgE production. The following monoclonal antibodies to treat asthma are currently used in clinical practice: Dupilumab (moderate to severe asthma and severe asthma) targets IL-4 and IL-13 by binding to the IL-4Rα subunit thereby acting as a blocking antibody for these signaling pathways (129, 130). Mepolizumab and Reslizumab (both eosinophilic asthma) target IL-5 directly and thereby neutralize this signature type 2 cytokine, reducing the rate of exacerbations (131, 132). Benralizumab (severe eosinophilic asthma) is directed against the IL-5Rα subunit (133). Lebrikizumab (severe asthma) and Tralokinumab (moderate to severe asthma) are directed against IL-13 (134, 135), and Omalizumab (severe allergic asthma in adults and children) targets and neutralizes IgE (136, 137). The ILC2-eliciting cytokine TSLP plays a critical role in human asthma and antibodies to neutralize TSLP (Tezepelumab) have been tested to treat allergen-induced asthma (138, 139). Overall, these antibodies block and thereby neutralize important immune mediators secreted by ILC2s as well as TH2 cells.
ILC2s in mouse and human can sense and respond to neuropeptides such as neuromedin U (25, 140, 141). Although pulmonary ILC2s exhibit a more moderate response to neuromedin U when compared to intestinal ILC2s under the tested conditions (140), several different neuropeptides are present in the lungs (142), including vasoactive intestinal peptide (VIP). VIP can induce cytokine stimulation of intestinal but also pulmonary ILC2s (63). Inhalation of a VIP agonist (Ro 25-1553) resulted in a short but significant bronchodilatory effect (143), however the exact mechanism and a possible link to pulmonary ILC2s is unclear. In contrast to VIP, calcitonin gene-related peptide (CGRP) is elevated in some asthmatic patients (144), and neuroendocrine cells, which co-localize in the airways with ILC2s are an important source thereof (145). ILC2s respond to CGRP by secreting more IL-5, however, targeting CGRP and thereby ILC2s still needs to be evaluated in asthma patients. Targeting neuropeptides and their receptors may be a promising concept for future therapy but still requires further investigation.
GATA3 is the master transcription factor of ILC2s and critical in regulating asthmatic responses in patients with a predominant TH2 phenotype. Targeting GATA3 is complex due to its intranuclear location but would enable to already intervene at a very early stage in the disease formation process. Novel approaches to antagonize GATA3 using antisense molecules (DNAzymes) overcome this challenge by cleaving and inactivating GATA3 messenger RNA (mRNA). GATA3-specific DNAzyme SB010 has shown to significantly attenuate both early and late-phase asthmatic responses after allergen exposure in a phase IIa proof-of-concept trial (146).
Since allergic respiratory diseases are rapidly increasing worldwide, there is a critical need to optimize current and develop novel therapeutic strategies. Reports in recent years have shown that ILC2s are important players in experimental mouse models of allergic airway inflammation and their role in asthmatic patients is just starting to unveil itself. Although neutralizing ILC2-elicited immune mediators or blocking respective signaling pathways are currently used to treat asthma, it is of great interest to develop alternative strategies to target not only the consequence but the cause of respiratory inflammation. This may be achieved by blocking ILC2s fairly early in disease and re-directing their activity. Moreover, the merge of immunology with other fields such as neurobiology opens new concepts and will reveal novel targets of translational interest. The unique microenvironment of the respiratory tract with its diversity of non-hematopoietic cells and their close proximity to ILC2s will as well unfold thrilling answers of ILC2 maintenance and activation in the future. We are excited to see further research on ILC2 biology in respiratory allergic inflammation which will surely provide essential knowledge to develop novel concepts and strategies for asthma treatment and improving overall respiratory health.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
CD and SH acknowledge support by a Rahel Hirsch Habilitationsstipendium (CD) and by a Boehringer Ingelheim Fonds travel grant (SH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
1. Yach D, Hawkes C, Gould CL, Hofman KJ. The global burden of chronic diseases: overcoming impediments to prevention and control. JAMA. (2004) 291:2616–22. doi: 10.1001/jama.291.21.2616
2. Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. (2008) 454:445–54. doi: 10.1038/nature07204
3. Nunes C, Pereira AM, Morais-Almeida M. Asthma costs and social impact. Asthma Res Pract. (2017) 3:1. doi: 10.1186/s40733-016-0029-3
4. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
5. Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. (2013) 14:536–42. doi: 10.1038/ni.2617
6. Artis D, Spits H. The biology of innate lymphoid cells. Nature. (2015) 517:293–301. doi: 10.1038/nature14189
7. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
8. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
9. Duerr CU, Fritz JH. Regulation of group 2 innate lymphoid cells. Cytokine. (2016) 87:1–8. doi: 10.1016/j.cyto.2016.01.018
10. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol. (2018) 19:1093–99. doi: 10.1038/s41590-018-0201-4
11. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
12. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature. (2010) 463:540–4. doi: 10.1038/nature08636
13. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
14. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol. (2015) 15:57–65. doi: 10.1038/nri3786
15. Koczulla AR, Vogelmeier CF, Garn H, Renz H. New concepts in asthma: clinical phenotypes and pathophysiological mechanisms. Drug Discov Today. (2017) 22:388–96. doi: 10.1016/j.drudis.2016.11.008
16. Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. (2011) 127:355–60. doi: 10.1016/j.jaci.2010.11.037
17. van Aalderen WM. Childhood asthma: diagnosis and treatment. Scientifica. (2012) 2012:674204. doi: 10.6064/2012/674204
18. Novey HS, Marchioli LE, Sokol WN, Wells ID. Papain-induced asthma–physiological and immunological features. J Allergy Clin Immunol. (1979) 63:98–103.
19. Calderon MA, Linneberg A, Kleine-Tebbe J, De Blay F, Hernandez Fernandez de Rojas D, Virchow JC, et al. Respiratory allergy caused by house dust mites: what do we really know? J Allergy Clin Immunol. (2015) 136:38–48. doi: 10.1016/j.jaci.2014.10.012
20. Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L577–88. doi: 10.1152/ajplung.00174.2012
21. Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. (2012) 42:1106–16. doi: 10.1002/eji.201142018
22. Li BWS, de Bruijn MJW, Tindemans I, Lukkes M, KleinJan A, Hoogsteden HC, et al. T cells are necessary for ILC2 activation in house dust mite-induced allergic airway inflammation in mice. Eur J Immunol. (2016) 46:1392–403. doi: 10.1002/eji.201546119
23. Coquet JM, Schuijs MJ, Smyth MJ, Deswarte K, Beyaert R, Braun H, et al. Interleukin-21-producing CD4+ T cells promote type 2 immunity to house dust mites. Immunity. (2015) 43:318–30. doi: 10.1016/j.immuni.2015.07.015
24. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
25. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour R-EE., Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. (2017) 549:351–56. doi: 10.1038/nature24029
26. Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol. (2014) 133:1142–8. doi: 10.1016/j.jaci.2014.02.033
27. Li Y, Chen S, Chi Y, Yang Y, Chen X, Wang H, et al. Kinetics of the accumulation of group 2 innate lymphoid cells in IL-33-induced and IL-25-induced murine models of asthma: a potential role for the chemokine CXCL16. Cell Mol Immunol. (2019) 16:75–86. doi: 10.1038/s41423-018-0182-0
28. Li BWS, Stadhouders R, de Bruijn MJW, Lukkes M, Beerens D, Brem MD, et al. Group 2 innate lymphoid cells exhibit a dynamic phenotype in allergic airway inflammation. Front Immunol. (2017) 8:1684. doi: 10.3389/fimmu.2017.01684
29. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. (2012) 129:191–8.e1–4. doi: 10.1016/j.jaci.2011.09.041
30. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. (2013) 132:933–41. doi: 10.1016/j.jaci.2013.05.012
31. Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. (2016) 45:198–208. doi: 10.1016/j.immuni.2016.06.017
32. Kato Y, Akasaki S, Muto-Haenuki Y, Fujieda S, Matsushita K, Yoshimoto T. Nasal sensitization with ragweed pollen induces local-allergic-rhinitis-like symptoms in mice. PLoS One. (2014) 9:e103540. doi: 10.1371/journal.pone.0103540
33. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. (2016) 9:275–86. doi: 10.1038/mi.2015.59
34. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity. (2014) 40:414–24. doi: 10.1016/j.immuni.2014.02.003
35. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. (2012) 188:1503–13. doi: 10.4049/jimmunol.1102832
36. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity. (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
37. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol. (2017) 139:1468–77.e2. doi: 10.1016/j.jaci.2016.08.034
38. Shen X, Pasha MA, Hidde K, Khan A, Liang M, Guan W, et al. Group 2 innate lymphoid cells promote airway hyperresponsiveness through production of VEGFA. J Allergy Clin Immunol. (2018) 141:1929–31.e4. doi: 10.1016/j.jaci.2018.01.005
39. Toki S, Goleniewska K, Reiss S, Zhou W, Newcomb DC, Bloodworth MH, et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax. (2016) 71:633–45. doi: 10.1136/thoraxjnl-2015-207728
40. Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. (2016) 193:31–42. doi: 10.1164/rccm.201410-1793OC
41. Zhou Y, Wang W, Zhao C, Wang Y, Wu H, Sun X, et al. Prostaglandin E2 inhibits group 2 innate lymphoid cell activation and allergic airway inflammation through E-prostanoid 4-cyclic adenosine monophosphate signaling. Front Immunol. (2018) 9:501. doi: 10.3389/fimmu.2018.00501
42. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep. (2017) 21:2487–99. doi: 10.1016/j.celrep.2017.10.110
43. Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, et al. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol. (2012) 188:2622–9. doi: 10.4049/jimmunol.1101632
44. Kim HK, Lund S, Baum R, Rosenthal P, Khorram N, Doherty TA. Innate type 2 response to Alternaria extract enhances ryegrass-induced lung inflammation. Int Arch Allergy Immunol. (2014) 163:92–105. doi: 10.1159/000356341
45. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. (2016) 17:76–86. doi: 10.1038/ni.3309
46. Karta MR, Rosenthal PS, Beppu A, Vuong CY, Miller M, Das S, et al. beta2 integrins rather than beta1 integrins mediate Alternaria-induced group 2 innate lymphoid cell trafficking to the lung. J Allergy Clin Immunol. (2018) 141:329–38.e12. doi: 10.1016/j.jaci.2017.03.010
47. Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. (2013) 132:205–13. doi: 10.1016/j.jaci.2013.03.048
48. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med. (2018) 215:263–81. doi: 10.1084/jem.20170449
49. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
50. Cao Y, He Y, Wang X, Liu Y, Shi K, Zheng Z, et al. Polymorphonuclear myeloid-derived suppressor cells attenuate allergic airway inflammation by negatively regulating group 2 innate lymphoid cells. Immunology. (2019) 156:402–12. doi: 10.1111/imm.13040
51. Xiong Y, Cui X, Li W, Lv J, Du L, Mi W, et al. BLT1 signaling in epithelial cells mediates allergic sensitization via promotion of IL-33 production. Allergy. (2019) 74:495–506. doi: 10.1111/all.13656
52. Cai T, Qiu J, Ji Y, Li W, Ding Z, Suo C, et al. IL-17-producing ST2+ group 2 innate lymphoid cells play a pathogenic role in lung inflammation. J Allergy Clin Immunol. (2019) 143:229–44.e9. doi: 10.1016/j.jaci.2018.03.007
53. Kamachi F, Isshiki T, Harada N, Akiba H, Miyake S. ICOS promotes group 2 innate lymphoid cell activation in lungs. Biochem Biophys Res Commun. (2015) 463:739–45. doi: 10.1016/j.bbrc.2015.06.005
54. Drake LY, Iijima K, Bartemes K, Kita H. Group 2 innate lymphoid cells promote an early antibody response to a respiratory antigen in mice. J Immunol. (2016) 197:1335–42. doi: 10.4049/jimmunol.1502669
55. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. (2014) 69:1300–7. doi: 10.1111/all.12446
56. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol. (2015) 136:1653–63.e7. doi: 10.1016/j.jaci.2015.03.043
57. Stadhouders R, Li BWS, de Bruijn MJW, Gomez A, Rao TN, Fehling HJ, et al. Epigenome analysis links gene regulatory elements in group 2 innate lymphocytes to asthma susceptibility. J Allergy Clin Immunol. (2018) 142:1793–807. doi: 10.1016/j.jaci.2017.12.1006
58. Paclik D, Stehle C, Lahmann A, Hutloff A, Romagnani C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur J Immunol. (2015) 45:2766–72. doi: 10.1002/eji.201545635
59. Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med. (2017) 214:1581–92. doi: 10.1084/jem.20161807
60. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
61. Vicente CT, Revez JA, Ferreira MAR. Lessons from ten years of genome-wide association studies of asthma. Clin Transl Immunol. (2017) 6:e165. doi: 10.1038/cti.2017.54
62. Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, et al. A tissue checkpoint regulates type 2 immunity. Nat Immunol. (2016) 17:1381–87. doi: 10.1038/ni.3582
63. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. (2013) 502:245–8. doi: 10.1038/nature12526
64. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951–65. doi: 10.1084/jem.20130071
65. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol. (2011) 12:1071–7. doi: 10.1038/ni.2133
66. Saluzzo S, Gorki AD, Rana BM, Martins R, Scanlon S, Starkl P, et al. First-breath-induced type 2 pathways shape the lung immune environment. Cell Rep. (2017) 18:1893–905. doi: 10.1016/j.celrep.2017.01.071
67. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
68. Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. (2014) 40:758–71. doi: 10.1016/j.immuni.2014.04.013
69. Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med. (2014) 190:1094–101. doi: 10.1164/rccm.201405-0859PP
70. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat Immunol. (2015) 16:161–9. doi: 10.1038/ni.3078
71. Seehus CR, Kadavallore A, Torre BDl, Yeckes AR, Wang Y, Tang J, et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun. (2017) 8:1900. doi: 10.1038/s41467-017-02023-z
72. Saglani S, Gregory LG, Manghera AK, Branchett WJ, Uwadiae F, Entwistle LJ, et al. Inception of early-life allergen-induced airway hyperresponsiveness is reliant on IL-13+CD4+ T cells. Sci Immunol. (2018) 3:eaan4128. doi: 10.1126/sciimmunol.aan4128
73. Nagakumar P, Denney L, Fleming L, Bush A, Lloyd CM, Saglani S. Type 2 innate lymphoid cells in induced sputum from children with severe asthma. J Allergy Clin Immunol. (2016) 137:624–26.e6. doi: 10.1016/j.jaci.2015.06.038
74. Tait Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. (2015) 8:1313–23. doi: 10.1038/mi.2015.21
75. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. (2011) 12:1055–62. doi: 10.1038/ni.2104
76. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
77. Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. (2012) 37:649–59. doi: 10.1016/j.immuni.2012.08.015
78. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. (2016) 17:626–35. doi: 10.1038/ni.3443
79. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell. (2016) 166:1231–46.e13. doi: 10.1016/j.cell.2016.07.043
80. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. (2015) 350:981–5. doi: 10.1126/science.aac9593
81. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science. (2018) 359:114–19. doi: 10.1126/science.aam5809
82. Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. (2017) 168:1086–100.e10. doi: 10.1016/j.cell.2017.02.021
83. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. (2013) 210:2939–50. doi: 10.1084/jem.20130351
84. Kadel S, Ainsua-Enrich E, Hatipoglu I, Turner S, Singh S, Khan S, et al. A major population of functional KLRG1− ILC2s in female lungs contributes to a sex bias in ILC2 numbers. Immunohorizons. (2018) 2:74–86. doi: 10.4049/immunohorizons.1800008
85. Russi AE, Ebel ME, Yang Y, Brown MA. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc Natl Acad Sci USA. (2018) 115:E1520–E29. doi: 10.1073/pnas.1710401115
86. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. (2016) 137:75–86.e8. doi: 10.1016/j.jaci.2015.05.037
87. Liu T, Wu J, Zhao J, Wang J, Zhang Y, Liu L, et al. Type 2 innate lymphoid cells: a novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respir Med. (2015) 109:1391–6. doi: 10.1016/j.rmed.2015.09.016
88. Vely F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. (2016) 17:1291–9. doi: 10.1038/ni.3553
89. Desai D, Newby C, Symon FA, Haldar P, Shah S, Gupta S, et al. Elevated sputum interleukin-5 and submucosal eosinophilia in obese individuals with severe asthma. Am J Respir Crit Care Med. (2013) 188:657–63. doi: 10.1164/rccm.201208-1470OC
90. Feng J, Li L, Ou Z, Li Q, Gong B, Zhao Z, et al. IL-25 stimulates M2 macrophage polarization and thereby promotes mitochondrial respiratory capacity and lipolysis in adipose tissues against obesity. Cell Mol Immunol. (2018) 15:493–505. doi: 10.1038/cmi.2016.71
91. Zeyda M, Wernly B, Demyanets S, Kaun C, Hammerle M, Hantusch B, et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int J Obes. (2013) 37:658–65. doi: 10.1038/ijo.2012.118
92. Cautivo KM, Molofsky AB. Regulation of metabolic health and adipose tissue function by group 2 innate lymphoid cells. Eur J Immunol. (2016) 46:1315–25. doi: 10.1002/eji.201545562
93. Mathews JA, Krishnamoorthy N, Kasahara DI, Cho Y, Wurmbrand AP, Ribeiro L, et al. IL-33 drives augmented responses to ozone in obese mice. Environ Health Perspect. (2017) 125:246–53. doi: 10.1289/EHP272
94. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. (2014) 20:54–61. doi: 10.1038/nm.3423
95. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA. (2013) 110:13921–6. doi: 10.1073/pnas.1307321110
96. Galand C, Leyva-Castillo JM, Yoon J, Han A, Lee MS, McKenzie ANJ, et al. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J Allergy Clin Immunol. (2016) 138:1356–66. doi: 10.1016/j.jaci.2016.03.056
97. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. (2013) 14:564–73. doi: 10.1038/ni.2584
98. Shimokawa C, Kanaya T, Hachisuka M, Ishiwata K, Hisaeda H, Kurashima Y, et al. Mast cells are crucial for induction of group 2 innate lymphoid cells and clearance of helminth infections. Immunity. (2017) 46:863–74.e4. doi: 10.1016/j.immuni.2017.04.017
99. Mortaz E, Amani S, Mumby S, Adcock IM, Movassaghi M, Folkerts J, et al. Role of mast cells and type 2 innate lymphoid (ILC2) cells in lung transplantation. J Immunol Res. (2018) 2018:2785971. doi: 10.1155/2018/2785971
100. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. Vancouver, WA: NIH Publication (2017). Available online at: www.ginasthma.org
101. Crompton G. A brief history of inhaled asthma therapy over the last fifty years. Prim Care Respir J. (2006) 15:326–31. doi: 10.1016/j.pcrj.2006.09.002
102. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun. (2013) 4:2675. doi: 10.1038/ncomms3675
103. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: the role of thymic stromal lymphopoietin. J Allergy Clin Immunol. (2018) 141:257–68.e6. doi: 10.1016/j.jaci.2017.03.032
104. Vazquez-Tello A, Halwani R, Hamid Q, Al-Muhsen S. Glucocorticoid receptor-beta up-regulation and steroid resistance induction by IL-17 and IL-23 cytokine stimulation in peripheral mononuclear cells. J Clin Immunol. (2013) 33:466–78. doi: 10.1007/s10875-012-9828-3
105. Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung D.Y. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. (2006) 173:607–16. doi: 10.1164/rccm.200507-1046OC
106. Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, et al. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. (1999) 159 (5 Pt 1):1600–4. doi: 10.1164/ajrccm.159.5.9804131
107. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. beta2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science. (2018) 359:1056–61. doi: 10.1126/science.aan4829
108. Johnston SL, Edwards MR. Mechanisms of adverse effects of {beta}-agonists in asthma. Thorax. (2009) 64:739–41. doi: 10.1136/thx.2009.119230
109. Crane J, Pearce N, Flatt A, Burgess C, Jackson R, Kwong T, et al. Prescribed fenoterol and death from asthma in New Zealand, 1981–83: case-control study. Lancet. (1989) 1:917–22.
110. Beasley R, Nishima S, Pearce N, Crane J. Beta-agonist therapy and asthma mortality in Japan. Lancet. (1998) 351:1406–7. doi: 10.1016/S0140-6736(05)79446-0
111. Kabata H, Moro K, Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. (2018) 286:37–52. doi: 10.1111/imr.12706
112. Hallstrand TS, Henderson WR Jr. An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol. (2010) 10:60–6. doi: 10.1097/ACI.0b013e32833489c3
113. Kittana N, Hattab S, Ziyadeh-Isleem A, Jaradat N, Zaid AN. Montelukast, current indications and prospective future applications. Expert Rev Respir Med. (2016) 10:943–56. doi: 10.1080/17476348.2016.1207533
114. Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. (2014) 133:1184–94. doi: 10.1016/j.jaci.2013.10.056
115. von Moltke J, O'Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. (2017) 214:27–37. doi: 10.1084/jem.20161274
116. Salimi M, Stoger L, Liu W, Go S, Pavord I, Klenerman P, et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol. (2017) 140:1090–100.e11. doi: 10.1016/j.jaci.2016.12.958
117. Corhay JL, Henket M, Nguyen D, Duysinx B, Sele J, Louis R. Leukotriene B4 contributes to exhaled breath condensate and sputum neutrophil chemotaxis in COPD. Chest. (2009) 136:1047–54. doi: 10.1378/chest.08-2782
118. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. (1995) 95:843–52.
119. Higham A, Cadden P, Southworth T, Rossall M, Kolsum U, Lea S, et al. Leukotriene B4 levels in sputum from asthma patients. ERJ Open Res. (2016) 2:00088–2015. doi: 10.1183/23120541.00088-2015
120. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med. (2015) 21:239–47. doi: 10.1038/nm.3800
121. Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol. (2016) 17:656–65. doi: 10.1038/ni.3421
122. Huang T, Hazen M, Shang Y, Zhou M, Wu X, Yan D, et al. Depletion of major pathogenic cells in asthma by targeting CRTh2. JCI Insight. (2016) 1:e86689. doi: 10.1172/jci.insight.86689
123. Singh D, Ravi A, Southworth T. CRTH2 antagonists in asthma: current perspectives. Clin Pharmacol. (2017) 9:165–73. doi: 10.2147/CPAA.S119295
124. Hall IP, Fowler AV, Gupta A, Tetzlaff K, Nivens MC, Sarno M, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther. (2015) 32:37–44. doi: 10.1016/j.pupt.2015.03.003
125. Kuna P, Bjermer L, Tornling G. Two Phase II randomized trials on the CRTh2 antagonist AZD1981 in adults with asthma. Drug Des Devel Ther. (2016) 10:2759–70. doi: 10.2147/DDDT.S105142
126. Gonem S, Berair R, Singapuri A, Hartley R, Laurencin MFM, Bacher G, et al. Fevipiprant, a prostaglandin D 2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. (2016) 4:699–707. doi: 10.1016/s2213-2600(16)30179-5
127. Pettipher R, Hunter MG, Perkins CM, Collins LP, Lewis T, Baillet M, et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy. (2014) 69:1223–32. doi: 10.1111/all.12451
128. Barnes N, Pavord I, Chuchalin A, Bell J, Hunter M, Lewis T, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. (2012) 42:38–48. doi: 10.1111/j.1365-2222.2011.03813.x
129. Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, et al. Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med. (2018) 378:2475–85. doi: 10.1056/NEJMoa1804093
130. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. (2018) 378:2486–96. doi: 10.1056/NEJMoa1804092
131. Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. (2016) 388:2115–27. doi: 10.1016/S0140-6736(16)31324-1
132. Castro M, Bacharier LB. Treatment for severe eosinophilic asthma-consistent effect of anti-interleukin-5 antibodies? Lancet. (2016) 388:2059–60. doi: 10.1016/S0140-6736(16)31537-9
133. FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. (2016) 388:2128–41. doi: 10.1016/S0140-6736(16)31322-8
134. Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med. (2016) 4:781–96. doi: 10.1016/S2213-2600(16)30265-X
135. Piper E, Brightling C, Niven R, Oh C, Faggioni R, Poon K, et al. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. (2013) 41:330–8. doi: 10.1183/09031936.00223411
136. Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF. Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol. (2009) 124:1210–6. doi: 10.1016/j.jaci.2009.09.021
137. Berger W, Gupta N, McAlary M, Fowler-Taylor A. Evaluation of long-term safety of the anti-IgE antibody, omalizumab, in children with allergic asthma. Ann Allergy Asthma Immunol. (2003) 91:182–8. doi: 10.1016/S1081-1206(10)62175-8
138. Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. (2017) 377:936–46. doi: 10.1056/NEJMoa1704064
139. Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. (2014) 370:2102–10. doi: 10.1056/NEJMoa1402895
140. Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. (2017) 549:277–81. doi: 10.1038/nature23469
141. Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. (2017) 549:282–86. doi: 10.1038/nature23676
142. Barnes PJ. Neuropeptides and asthma. Am Rev Respir Dis. (1991) 143 (3 Pt 2):S28–32. doi: 10.1164/ajrccm/143.3_Pt_2.S28
143. Linden A, Hansson L, Andersson A, Palmqvist M, Arvidsson P, Lofdahl CG, et al. Bronchodilation by an inhaled VPAC(2) receptor agonist in patients with stable asthma. Thorax. (2003) 58:217–21. doi: 10.1136/thorax.58.3.217
144. Springer J, Geppetti P, Fischer A, Groneberg DA. Calcitonin gene-related peptide as inflammatory mediator. Pulmonary Pharmacol Therapeut. (2003) 16:121–30. doi: 10.1016/s1094-5539(03)00049-x
145. Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. (2018) 360:eaan8546. doi: 10.1126/science.aan8546
Keywords: group 2 innate lymphoid cells (ILC2s), mucosal immunity, respiratory tract, allergic inflammation, therapeutic strategies
Citation: Helfrich S, Mindt BC, Fritz JH and Duerr CU (2019) Group 2 Innate Lymphoid Cells in Respiratory Allergic Inflammation. Front. Immunol. 10:930. doi: 10.3389/fimmu.2019.00930
Received: 07 January 2019; Accepted: 11 April 2019;
Published: 07 June 2019.
Edited by:
Jessica Borger, Monash University, AustraliaReviewed by:
Rudi W. Hendriks, Erasmus University Rotterdam, NetherlandsCopyright © 2019 Helfrich, Mindt, Fritz and Duerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia U. Duerr, Y2xhdWRpYS5kdWVyckBjaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.