 Felix Haussner
Felix Haussner Rebecca Halbgebauer
Rebecca Halbgebauer Markus Huber-Lang
Markus Huber-Lang- Institute of Clinical and Experimental Trauma-Immunology, University Hospital of Ulm, Ulm, Germany
Sepsis is a complex of life-threating organ dysfunction in critically ill patients, with a primary infectious cause or through secondary infection of damaged tissues. The systemic consequences of sepsis have been intensively examined and evidences of local alterations and repercussions in the intestinal mucosal compartment is gradually defining gut-associated changes during sepsis. In the present review, we focus on sepsis-induced dysfunction of the intestinal barrier, consisting of an increased permeability of the epithelial lining, which may facilitate bacterial translocation. We discuss disturbances in intestinal vascular tonus and perfusion and coagulopathies with respect to their proposed underlying molecular mechanisms. The consequences of enzymatic responses by pancreatic proteases, intestinal alkaline phosphatases, and several matrix metalloproteases are also described. We conclude our insight with a discussion on novel therapeutic interventions derived from crucial aspects of the gut mucosal dynamics during sepsis.
Introduction
The mucosa is a highly organized and compartmentalized structure, which lines our body cavities for example, the respiratory, urogenital and intestinal tracts. It provides an interface between the external environment and the host tissues (1), possessing various functions including absorption of water, nutrients and gases, secretion of molecules, clearance of waste, improvement of bio-mechanical features and maintenance of immunity. Therefore, it is not surprising that the combined surface area of the digestive and respiratory tracts by far exceeds the surface dimension of our largest organ, the skin (2). These functions also necessitate a unique immune system which is tightly regulated and this is termed as the mucosal immune system (MIS) (2). The MIS in the gut is capable of distinguishing between regular nutrient flux, self-antigens, a diverse milieu of commensal bacteria and invading pathogenic microbes (3–5). Lymphoid compartments, commonly known as the mucosa-associated lymphoid tissue (MALT), are integrated into the mucosa and perform immune-associated activities. Organized MALT has been found not only in the gut (GALT), but also in a number of other sites, like in the nasopharynx, salivary-gland and duct, larynx, bronchus and urogenital tissues (6). In the intestine, the structural organization of the coexisting symbiotic bacteria is unique, where the large intestine alone houses 1011–1012 bacteria/gram feces, the highest concentration in the entire intestinal tract (2). However, an imbalance of the co-inhabitation of the intestinal microbiome with the host can potentially threaten well-being (1, 5, 7, 8).
The homoeostatic status quo is essentially supported by the maintenance of the gut barrier integrity. In principle, any infection or severe extra intestinal trauma can cause significant alterations of the gut barrier homeostasis, which may result in a profound generation and secretion of intestinal proteolytic enzymes, alterations in mucus layer formation and composition (9, 10), increased epithelial cell permeability and damaged intestinal cells with subsequent inflammatory signaling (5, 9, 10). These distinct pathophysiological changes are frequently found in septic patients. Previously, sepsis was defined as a systemic inflammatory response (SIRS) with an underlying primary infectious cause (11). Declared as a “silent killer” in critical care units and with high global mortality rates, sepsis has been recently redefined as a “life-threatening organ dysfunction caused by a dysregulated host response to infection” (12, 13). In the clinical setting, diffused and hidden symptoms frequently make the diagnosis of sepsis difficult. To help define septic conditions, clinicians and clinical scientists can utilize the sequential (sepsis-related) organ-failure assessment (SOFA) Score, which allows more precise detection of sepsis-associated organ dysfunction compared to the SIRS-criteria (12–15). The alarming pace of sepsis with possible development of multiple organ dysfunction syndrome (MODS) frequently includes disseminated intravascular coagulopathy (DIC), making sepsis patients a colossal challenge for both clinicians and researchers.
Years of research have focused on the various intricacies, from the underlying pathology to clinical targets that could help treat sepsis patients. In the scope of our review, we consider the effects of sepsis on the intestinal mucosa regarding the main immunological mechanisms that yield a dysregulated intestinal mucosal system and the scope of associated promising therapeutic strategies.
Structure-Function Relationship of the Intestine for the Maintenance of Immune Defense
The surface of the small intestine is formed by a monolayer of highly prismatic epithelia, which are modified into structures like plications, villi (0.2–1 mm), crypts, and microvilli. Crypts contain stem cells, which generate intestinal epithelial cells (IECs). Paneth cells within the crypts secrete antimicrobial peptides (AMPs), for example, α-defensin and lysozyme, to confer intestinal protection from pathogenic insults (16, 17). The IECs in villi reabsorb nutrients and are interconnected by tight junctions (TJs) (e.g., occludins, claudins) that form apical paracellular seals thus preventing the flux of hydrophilic molecules (18). Further along the IECs lie adherens junctions (e.g., cadherins) and gap junctions (e.g., connexins), all of which determine the cellular polarity and regulate cell-cell communication and exchange of substances. The epithelium can also secrete pro-inflammatory cytokines and reactive oxygen species (ROS) in response to pathogens and metabolic stress (19). Goblet cells in the villi produce mucus, a key component of the gut barrier. A single unattached mucus layer is present superficially on the surface of the small-bowel epithelia (20, 21). Mucus contains soluble glycoproteins termed mucins, which are normally negatively charged, consisting of a core protein to which multiple polysaccharide moieties are attached, capable of binding water molecules (22). In addition to the predominant mucin-2 (MUC2), other bioactive molecules, for example, membrane-bound mucins, like MUC1, MUC3, and MUC17 and peptides, like Fc-γ binding protein and intestinal trefoil factor peptides, are secreted by goblet cells (22, 23). These play a major role in maintaining mucosal homoeostasis, mainly by limiting contact between commensals/pathogens and IECs (23). The large intestinal mucosa comprises crypts without any villi, with significantly greater numbers of goblet cells in comparison to the small bowel. The colon functions mainly as a reabsorbing organ for water and electrolytes and additionally produces mucus. One important distinction is the double layer of mucus on the colonic epithelial cell surface, where the inner layer is immediately above the epithelium, is mostly immobile and is thinner than the outer mucus layer, which is not attached to the colon wall (24). Both layers consist of gel-forming MUC2, but the glycoproteins of the inner layer form a large and dense net, whereas the outer layer consists predominantly of MUC2 cleavage products (25).
Regarding cellular immunity in the intestine, there is a well-regulated interplay between antigen-presenting dendritic cells (DCs), intestinal macrophages and adaptive immune cells. After recognition of antigens and/or pathogen-associated-molecular-patterns (PAMPs) via pattern recognition receptors (PRR), including Toll-like-receptors (TLRs) and NOD-like-receptors, intestinal DCs regulate the immune response by enhancing or suppressing T-cell activity. To achieve this, dendrites of DCs penetrate intercellular spaces through the intestinal TJs while maintaining barrier integrity (26). DCs, via these dendrites sense and bind luminal PAMPs and bacteria and present processed antigens to immune cells located in lymphoid follicles found in the connective tissue and the lamina propria. Intestinal macrophages (type CX3CR1hi) can also sense PAMPs by forming transepithelial dendrites (TEDs). Of note, this specific type of macrophage has only been observed in the murine ileum and the importance of the TEDs remains uncertain (27). Another means to reabsorb antigens is accomplished by villous microfold cells which offer antigens a channel to lymphoid tissue, where antigen presenting cells resorb the molecules and present them to CD4+T-cells via Major-Histocompatibility-Complex II (28). Moreover, DCs selectively induce a pro- or anti-inflammatory immune response by interacting with T- and B cells. IgA+-B cells colonize in the lamina propria and secrete IgA into the lumen via transcytosis (29–31) (Figure 1). This complex intestinal organization is subject to activation and dysregulation during sepsis.
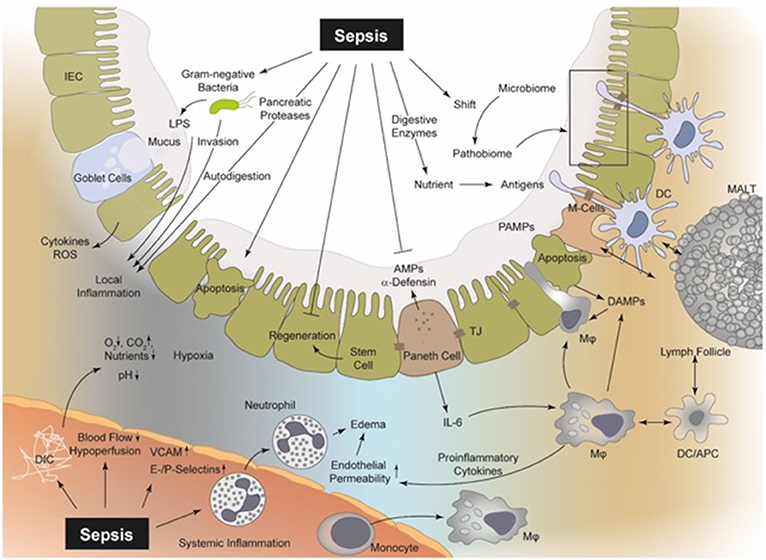
Figure 1. Sepsis is involved in several pathophysiological processes regarding the intestinal epithelial integrity, perfusion, coagulation, enzymatic response, and MIS. In sepsis, bacteria and their products (PAMPs), including LPS, PG, and bacterial DNA, can be recognized by PRRs (e.g., TLR2 and TLR4) upon the surface of macrophages, neutrophils, DCs, and even IECs (19, 32). Thereby, intestinal macrophages and DCs as part of the MIS can detect luminal PAMPs via transepithelial dendrites (TEDs) (26, 27, 33). Consequently, PAMPs induce a “cytokine storm” of pro-inflammatory mediators, which drive the local intestinal and systemic inflammation (32). The released mediators can lead to an upregulation of endothelial adhesion molecules (e.g., ICAM, VCAM, E-, and P-selectin), resulting in increased recruitment of neutrophils and monocytes and in turn to increased levels of pro-inflammatory cytokines and ROS (34, 35). These cellular responses aggravate vasodilatation and induce a high level of capillary leakage with the development of interstitial edema. Local DIC is frequently observed during sepsis with a decreased supply of oxygen and nutrients, but increased carbon dioxide concentration (36, 37). Hypoxia in turn leads to increased apoptosis and necrosis of IECs and the regeneration of these IECs is suppressed during sepsis (38–40). Furthermore, the IEC integrity is disrupted and bacterial translocation may be facilitated. Pancreatic proteases are capable of autodigestion and potentiation of MOF and self-digestion leads to an increased release of further DAMPs (10, 19, 41, 42). MIS, mucosal immune system; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; LPS, lipopolysaccharide; PG, proteoglycan; PRR, pattern-recognition-receptors; TLR, toll-like receptor; DCs, dendritic cells; IECs, intestinal epithelial cells; TEDs, transepithelial dendrites; DIC, disseminated-intravascular-coagulation; MOF, multi-organ failure; ICAM, intercellular adhesion molecule 1; VCAM, vascular cell adhesion protein 1; ROS, reactive oxygen species; M-cells, microfold cells; AMPs, antimicrobial peptides; APC, antigen presenting cells; TJ, tight junctions; MALT, mucosa-associated-molecular pattern.
Gut Barrier Dysfunction and Systemic Consequences During Sepsis
A major pathophysiological mechanism of sepsis harnesses recruitment of inflammatory cells and generation of an overwhelming pro-inflammatory response. PAMPs, for example, lipopolysaccharide (LPS), peptidoglycan and bacterial DNA among others and damage-associated molecular patterns (DAMPs), including mitochondrial DNA, High-Mobility-Group-Protein-B1 and serum amyloid A, result in the upregulation of adhesion molecules on the intestinal endothelium followed by the recruitment of neutrophils and macrophages (43). Upon migration to the intestinal tissue, these cells of the first line of defense produce pro-inflammatory cytokines, clinically manifested as classical signs of local and systemic inflammation (32, 44). Cell-wall components from gram-negative and gram-positive bacteria activate PRRs like TLR4 and TLR2, respectively, resulting in a “cytokine storm” of pro-inflammatory mediators generated mainly via the mitogen-activated protein kinase and NF-κB pathways (32). Of note, pro-inflammatory responses are interspersed with anti-inflammatory responses, also termed the compensatory anti-inflammatory response syndrome (45–47), where patients with sepsis undergo a reprogramming of their defense strategies and frequently fail to eliminate primary infection, thus being unable to prevent secondary infection development (33). However, an initial hyper-inflammatory response might dominate to beneficially isolate local infectious foci and limit systemic spillover (33). Gut barrier dysfunction can be considered both a result and a cause of sepsis development, characterized by enhanced mucosal layer permeability (5, 9, 10, 23, 48–51), disturbed mucosal perfusion (38, 52–54), development of tissue edema, coagulation-associated local dysregulation (36, 37), bacterial translocation (48, 55, 56) and a shift in the gut microbiome (57, 58). Furthermore, apoptotic and necrotic mechanisms damage the mucosal epithelia, resulting in a vicious cycle of further release of DAMPs, feeding into inflammatory responses combined with the development of ulceration and hemorrhage and exacerbation of mucosal homeostatic imbalance (Figure 1) (50, 59). The causes and consequences of gut barrier dysfunction have been described in literature extensively.
Disturbances in Vascular Tonus and Perfusion
Hypoperfusion in the splanchnic region is considered one of the main reasons for mucosal gut barrier breakdown during sepsis (38). The splanchnic vasculature system normally receives about 25% of the total cardiac output, which increases up to 35% during digestion (60, 61). Perfusion is mainly controlled by local mediators, including nitric oxide and prostaglandin derivatives, but also by systemic mediators, like vasoactive substance P and by the sympathetic innervation (61). Splanchnic hypoperfusion converts the gut into a cytokine-generating organ, which releases a “toxic fluid,” containing pro-inflammatory agents and induces MODS via the circulation (48). Hypovolemia and cardiac depression during sepsis are associated with a robust inflammatory response of cytokines and other inflammatory mediators (39). Blood cells, endothelium and vascular smooth musculature are potential targets of these pro-inflammatory cytokines leading to vasodilatation, high capillary leakage, increased venous capacity and decreased venous return, all of which result in a decrease in cardiac output and tissue perfusion (39, 40). In turn, the renin-angiotensin-aldosterone-system is stimulated and increasingly generates vasoconstrictive agents, which also adds to local hypoperfusion thus developing both micro- and macro-circulatory disturbances (39, 52, 62–64). As an overall consequence, gut mucosal perfusion is reduced during sepsis, which results in further hypoxia and consequent destruction of the mucosal barrier (38). Studies using laser Doppler measurements have also revealed that CLP-induced sepsis in normotensive rats caused a decrease in the number of perfused capillaries in the small gut mucosa (65). In this context, it is also known that mucosal blood flow is dependent on inflammatory processes (52). As a result of sepsis-associated excessive inflammation, the microvasculature loses its capacity to regulate blood flow and oxygen distribution mainly based on the generation of ROS (39, 40, 66). As a consequence, increasing the blood flow by vasodilatation during hypoperfusion is not possible (53, 54), explaining why maximal O2 extraction cannot be accomplished in sepsis (Figure 1). A further process that results in an impairment of the local vasodilatory response is the pathological opening of arteriovenous shunts that alters the blood flow between hypoperfused and perfused areas (39, 67).
There exist further sepsis-induced alterations in perfusion, including increased intercapillary distances due to edema (39) and greater diffusion distances (39, 63). However, the pathophysiological details are beyond the scope of this review.
Increased Intestinal Permeability
It is well-established that sepsis results in a dysfunction of the intestinal barrier with increased permeability (5, 9, 10, 23, 48, 49, 51, 68, 69). Locally transmigrated bacteria and endotoxin exposure lead to a local activation of the MIS and in turn to the production of various pro- and anti-inflammatory cytokines by IECs and intestinal immune cells. This cellular response may also contribute to the systemic response (Figure 2). Furthermore, activation of intestinal immune cells results in a further increase in gut permeability by altering TJs (68, 69, 84, 85). Induction of experimental sepsis leads to a redistribution of the TJ proteins occludin and claudin-1, 3, 4, 5, and 8 (68, 85). In agreement with this, murine endotoxemia resulted in a disrupted ultrastructure of occludin and zonulin-1 (ZO-1) in the intestinal epithelium (86). Of note, these changes could be corrected by vagal nerve stimulation (86) or even by treatment with plant products, including berberine (87). Berberines are known to decrease downstream myosin-light chain kinase (MLCK) and NF-κB activity, representing mechanistic intermediates that may modulate TJ organization. Elaborating on this mechanism, it was found that MLCK phosphorylates myosin light chain which causes cytoskeletal contraction and junction disruption (88). Furthermore, β-catenin, another TJ organization protein, was found to be irregularly distributed in LPS-treated rats while platelet activating factor appeared to attenuate this disorganization (89). More recently, increased plasma ZO-1 levels have been found during experimental sepsis and an elevated plasma zonulin concentration, a regulator of TJs, in patients with sepsis (90). Cyclooxygenase-2 (COX-2) and particularly its product prostaglandin D2 appear to play an important role in the maintenance of epithelial TJs and barrier function, because the absence of COX-2 led to an increased permeability of the murine ileum and to a reduced expression of TJ proteins (91). Consequently, reinforced bacterial translocation and a higher mortality rate were observed in septic COX-2-knockout mice after cecal ligation and puncture (CLP) (91). With enhanced permeability, what becomes entirely imminent is the translocation of bacteria, a highly probable threat to the intestinal mucosal system.
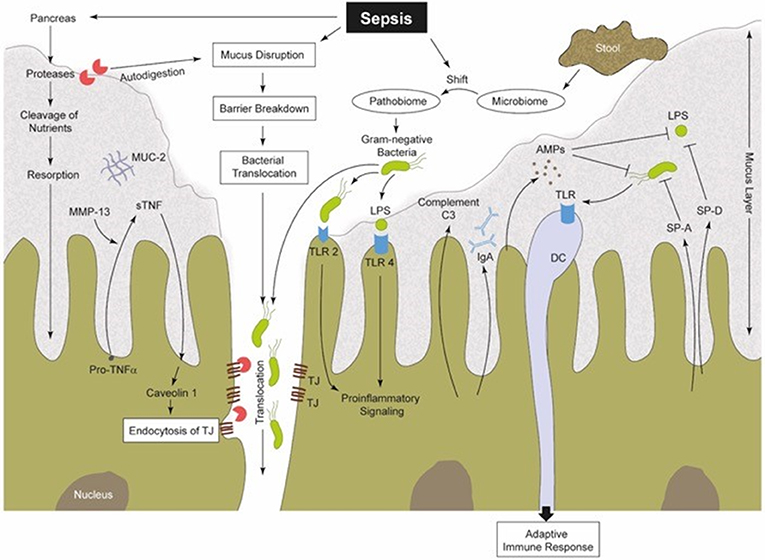
Figure 2. Sepsis-induced alterations in perfusion, vascular tonus and coagulation lead to a hypoxic microenvironment of the intestinal tissue (38). Therefore, the protective gel-forming MUC2 mucus layer becomes disrupted (22). Both, bacteria (products) and (pancreatic) proteases gain access into intestinal epithelia, inducing damage followed by increased pro-inflammatory signaling (41, 48, 70, 70, 71). Furthermore, MMPs (like MMP13) are able to cleave membrane-bound pro-TNF into sTNF, which in turn is able to stimulate caveolin-1-dependent endocytosis of TJs (72, 73). Gut barrier breakdown and dysfunction is one consequence (72). Intestinal commensal microbes regulate the maturation of the MIS and support local mucosal immunity (8, 74). During sepsis, the well-regulated interplay between the commensal microbiome, IECs and mucosal immune cells becomes imbalanced. There is a sepsis-induced shift from a physiological microbiome to a “pathobiome,” which is able to dysregulate the immune system by activating PRRs (32, 58). SP-A and SP-D can be synthetized by IECs (75, 76). These SPs are capable of increasing the permeability of bacterial membranes and in turn reduce the bacterial burden (77). IECs produce AMPs (e.g., α-defensin and lysozyme), to confer intestinal protection from pathogenic insults. Thereby, AMPs can act two ways, on the one hand directly by antimicrobial killing and on the other hand by innate immune modulation (78). Complement factors are mainly produced in the liver (79, 80), but also IECs were identified to synthesize and secrete C3 into the intestinal lumen (81), and thus may also play a role in intestinal immunity (82, 83). IECs, intestinal epithelia cells; MUC2, mucin-2; MMPs, matrix metalloproteinases; TNF, tumor necrosis factor; sTNF, soluble TNF; TJs, tight junctions; MIS, mucosal immune system; PRR, pattern recognition receptors; TLR, toll-like receptor; SP, surfactant protein; AMPs, antimicrobial peptides; C3, complement factor 3; LPS, lipopolysaccharide; DC, dendritic cell; IgA, immunoglobulin A.
Bacterial Translocation via the Mucosa
Gut permeability theoretically prompts the possibility of local bacterial translocation, supported by several pieces of evidence. For example, TLRs play a role in directing the response via MLCK activation, as shown in morphine-treated animals, which in turn facilitate bacterial translocation and even cause infection or sepsis of gut origin (92). Other factors like insulin growth factor-1 promote bacterial translocation, which induces intestinal cell apoptosis (93), or increased pneumoperitoneal pressure (during laparoscopic surgery) (94). Sepsis development has also been reported as a secondary effect of pneumonia due to mucosal and microvascular injury in the gut (95). In turn, bacterial translocation may occur as a common process, secondary to a primary infection, severe trauma, or major surgery, giving rise to sepsis and consequently supporting the “gut origin hypothesis of sepsis” (96).
There are further assumptions about the driving force of sepsis and MODS. Translocating bacteria and endotoxin from the gut lumen may not directly enter into the systemic circulation, but rather induce an immune response in the local GALT or draining lymph nodes, which results in significant systemic effects, for example, via “toxic lymph” (Figure 2) (48, 70). The mesenteric lymph contains several different proteins and lipid factors, including a modified albumin species (97), which could cause cellular damage and the activation of TLR4, resulting in priming of neutrophils and inducing remote lung injury (98). In agreement with this, a correlation between gut barrier dysfunction and secondary lung injury has been found (48, 49). Because novel techniques on the nanoscale in bacterial (product) detection have been developed over the last decade, further clinical studies may help to re-evaluate and elucidate the “bacterial translocation” paradigm and its mechanisms.
Coagulation and Its Factors Modulating Mucosal Dysfunction
The mechanisms of sepsis-induced consumptive coagulopathy are manifold. The procoagulant tissue factor (TF), which is produced by the liver, monocytes, neutrophils and endothelial cells, is significantly increased after exposure to endotoxin or PAMPs (36, 99, 100). Synchronous inhibition of fibrinolysis occurs by an enhanced production of plasminogen activator inhibitor (PAI-1) and the downregulation of the protein-C pathway, which are important in the initiation and progression of coagulopathy with clinical manifestation of both thrombosis and DIC (36, 37). While intestinal microcirculatory disturbances are common during sepsis-induced DIC, various clotting factors may directly or indirectly affect intestinal physiology and immune-cell recruitment. For example, septic rats displayed a decrease in functional capillary density, indicating a reduction in microvascular perfusion, which could be corrected when these animals were treated with factor XIII (FXIII) (101). In addition, factor-XI deficiency could confer a survival advantage on mice with peritoneal sepsis (102). In agreement with this, microcirculatory disturbances were found in the intestinal epithelium of CLP rats associated with high intestinal TF levels, all of which could be improved by sodium tanshinone IIA sulfonate, a substance recently proposed to exhibit protective effects against coagulatory disturbances (103). Similar effects were shown for the thrombin inhibitor Argatroban (104). Dual pharmacological inhibition of factor II and factor Xa by SATI resulted in preserved activation of coagulation with no bleeding complications and protection of organ function during experimental sepsis in baboons, representing a promising tool against sepsis-induced DIC (105). Furthermore, treatment with recombinant human antithrombin has been shown to ameliorate leukocyte adhesion in mesenteric venules and to reduce intestinal injury in endotoxemic rats (106) and concomitantly improved the 28-day mortality rate in septic patients (107). Nevertheless, application of PAI-I (108) or recombinant human thrombomodulin (109) failed to reveal beneficial effects in septic patients. Linking mucosal immunity to coagulation, mucosal M2 macrophages have been recently shown to contain intracellular FXIII stores. The cell number of this subtype is decreased in inflamed mucosa in the setting of ulcerative colitis (110). Previously, macrophage procoagulant activity was found to be increased in rats with depleted intestinal microflora and orally fed with streptomycin-resistant E. coli, implying that the gut is a focal point from which systemic inflammation arises (111). Deficiency of carboxypeptidase B2, an enzyme able to cleave both fibrinogen and the central complement components C3 and C5, was shown to confer survival advantage to mice, which was mainly mediated by C3a-induced peritoneal macrophage recruitment (112). Although there is evidence of an intensive crosstalk between coagulation and the innate immune response in driving inflammation during sepsis, the exact underlying mechanisms still need to be defined.
Apoptosis as a Central Driver of Intestinal Damage
Apoptotic events play a critical role in the development of sepsis. Interestingly, in murine sepsis models and in autopsy studies of septic humans, there were barely any significant histological changes except for increased gut epithelial/lymphocyte apoptosis in comparison with non-septic deceased patients (113). Nevertheless, experimental prevention of apoptosis in sepsis models increased the survival rate (33, 113) and therefore, the hypothesis of immune cell apoptosis as a relevant pathological mechanism in sepsis could also be of special interest for mucosal immunity (114). Sepsis induced by Pseudomonas aeruginosa pneumonia was, for example, caused by apoptotic intestinal epithelia associated with reduced epithelial proliferation (45). Mechanistic investigation of intestinal cell apoptosis during sepsis identified gene overexpression of interleukin (IL)-1β-converting enzyme, which may play an important role during experimental sepsis (115). In addition, when anti-apoptotic proto-oncogene Bcl-2 was gut-specifically overexpressed, a decrease in sepsis-induced intestinal epithelial apoptosis was found in murine models (45, 116, 117). MicroRNA 195, a regulator of Bcl-2 gene expression, which assists in maintaining the pro/anti-apoptotic balance, has been shown to be upregulated in murine sepsis and its inhibition could prevent apoptosis and even the development of MODS (118). Therefore, new approaches to improve gut barrier function during sepsis could be represented by application of silencing microRNAs regulating intestinal apoptosis (117, 118).
Apart from generic apoptosis related transducers, other molecules have also been implicated to play a role in sepsis-associated apaptotic mechanisms. Cytokine IL-15 was identified to be capable of preventing apoptosis and of immune suppression as well. In sepsis, IL-15 attenuated the apoptosis rate of intestinal epithelia and increased Bcl-2 and IFN-γ expression in IECs as well as the natural killer cell population, which produced further IFN-γ (119, 120). Of note, the lung surfactant proteins SP-A and SP-D have additionally been found to be generated by epithelial cells of the small and large intestines and in gastric cells (75, 76), and the absence of SP-A and -D resulted in increased LPS-induced apoptosis of primary IECs (77). Nonetheless, to what extent surfactant molecules may therapeutically protect the gut barrier remains to be investigated.
Intestinal Microbiome as an Actor and Target
During the last decade, the commensal microbiome has been defined to play a key role in intestinal immunity because microbes regulate the maturation of the MIS (8, 74), support local mucosal immunity (7, 8) and regulate cellular growth and maintenance of epithelial barrier function (1, 5). It is likely that the human immune system not only controls bacteria, but that the microbiome also regulates the immune cell function, particularly on mucosal surfaces (8, 121). It putatively modulates neonatal immunity and determines susceptibility to infection depending on the mode of childbirth (122–125). Alterations of the lung microbiota due to colonization by gut microbes has also been shown in animal studies, which to some extent may explain the frequent simultaneous appearance of acute respiratory distress syndrome with sepsis (126). If the symbiosis between commensal bacteria and the human host becomes imbalanced, the innate and adaptive immune systems are disturbed (Figure 2) (121, 127). A decline or even a loss of protective anaerobes in fecal specimens has been observed in patients with severe sepsis (57, 58) and hypothetically this “pathobiome” is able to manipulate and dysregulate the immune system in critically septic and ill patients (58). Moreover, commensal bacteria are involved in the regulation of CD4+ T-cell immunity though the exact mechanisms remain unknown (128). Indicating the harmful effect of opioid analgesics in treating critical care patients, murine polymicrobial sepsis with opioid treatment selectively influenced gram-positive gut microbiome translocation and dissemination, inducing its pro-inflammatory effects through IL-6 and IL-17A cytokines (129). Furthermore, the function and aging of neutrophils as first cellular line of defense were also shown to be regulated by the microbiome during sepsis (130, 131). Overall, it is tempting to speculate that therapeutic interventions on the altered microbiome might improve barrier, immune and organ function as well as sepsis outcome.
Intestinal Enzymatic Response Induces Self-Destruction
The underlying mechanisms of the interplay between pancreatic enzymes, sepsis, and septic shock remain unclear, although Schmid-Schönbein and colleagues had already hypothesized in 2005 that pancreatic enzymes are capable of self-digestion and potentiation of multi-organ failure (MOF) (41). In the case of sepsis-induced hypoperfusion/ischemia of the intestine, autodigestion processes can affect the mucosal barrier (10, 42). Such self-digestion may lead to an increased release of DAMPs and enhance the systemic response due to the release of pro-inflammatory mediators by stressed IECs (Figure 2) (19, 42, 71). The inhibition of pancreatic enzymes with subsequent prevention of gut-specific autodigestion indeed improved the outcome of septic mice (132). In this regard, inhibiting pancreatic proteases with tranexamic acid reduced inflammation and could also be exploited as a future sepsis treatment beyond its application in treating traumatic coagulopathy (132). Intestinal alkaline phosphatase (IAP) is another enzyme that protects the intestinal brush borders, particularly against intestinal bacterial invasion (133). Some of the major functions of IAP are duodenal surface pH regulation (via secretion), mitigation of intestinal inflammation by PAMPs and gut microbiome control (134). IAP-mediated inactivation of bacterial products, including LPS, decreases their binding to TLR4 and reduces the resultant inflammatory responses. Interestingly, in the absence of bacteria, a lack of IAP expression results in the loss of mucosal protection (135). Mice treated with IAP after exposure to a lethal dose of Escherichia coli had an improved survival rate of 80%, compared to 20% in the control sepsis group (135, 136). In conclusion, loss of IAP expression or function increased intestinal inflammation, dysbiosis, and bacterial invasion, culminating in systemic inflammation (134).
LPS can furthermore induce matrix metalloprotease 7 (MMP7) expression and degranulation of Paneth cells, leading to increased intestinal permeability (17). MMP7 was observed as an amplifier of inflammation; MMP7-deficient mice displayed an attenuated intestinal inflammatory response (137). MMP7 is able to activate α-defensin, which in turn stimulates IL-6 release by macrophages and ileal epithelia, thereby enhancing local intestinal inflammation and damage (137). Moreover, MMP7 has also been correlated with the loss of intestinal barrier integrity, enhanced bacterial translocation and MOF development (137). Similarly, MMP13 has been described to play a role in inflammatory bowel diseases (IBD) and during sepsis (72). It is able to cleave membrane-bound pro-TNF into soluble bioactive TNF, which can affect TJs through caveolin-1-dependent endocytosis (72). The consequences are the loss of TJs, increased intestinal permeability and the creation of a new pathway for migrating bacteria, which induces further inflammation (72, 73, 138). MMPs are also present in the large intestine and play a similar role in sepsis progression through similar mechanisms. MMP-1, 2, 3, and 9 were detected in the human colon mucosa and were also increased during IBD (139, 140). However, their exact role in sepsis is yet to be investigated.
Metabolic Response Within the Intestinal Mucosa
While intestinal permeability is enhanced, amino-acid absorption by the intestine is affected as early as 24 h after sepsis onset (141). In this regard, in vivo and in realiter studies revealed that gut glutamine absorption and metabolism decreased during sepsis because of suppressed glutaminase activity (142). By contrast, glutamine supplementation improved other effects of sepsis: it reduced bacterial translocation, restored permeability and microcirculatory characteristics (143–146), and even increased the number and survival of intestinal epithelia while blocking inflammatory cytokine secretion by CD8αα(+) TCRαβ(+) IEL cells (147) and γδT-IELs (148). As a further risk factor, a high-fat diet was detrimental for sepsis outcome and worsened endotoxemia in mice by disrupting the Bifidobacterium spp. colony. Correction of the dysbiosis and its consequences by feeding prebiotic oligofructose resulted in reduced systemic inflammation in experimental (149) and clinical sepsis (150). This preliminary evidence suggests that the intricate repertoire between metabolic intermediates, gut microbiome and inflammatory responses following sepsis requires further investigation and represents a promising therapeutic potential.
Therapeutic Approaches to Improve Sepsis-Associated Mucosal Immunopathology
In the era of resurrection from the “therapeutic graveyard of sepsis,” novel pharmacological approaches address crucial aspects of gut mucosal dynamisms. For example, the dietary dipeptide gamma-l-glutamyl-l-valine (γ-EV), which leads to decreased pro-inflammatory cytokines in both plasma and the small intestine, is also effective against bacterial infections (151). γ-EV stimulates the interaction of β-arrestin-2 with toll-interleukin-1-receptor signaling proteins, including TRAF6, TAB1, and IκBα, which further suppress the inflammatory response in the small intestine (151, 152). Similar results in murine IBD models have been shown for γ-glutamyl-cysteine, which inhibits TNF signaling in intestinal epithelia (152). In other studies, the small peptide hormone ghrelin was identified to be protective by inducing autophagy in the case of tissue hypoxia. Thereby, ghrelin appears also able to protect IECs in the small intestine in the early stage of sepsis (153). Application of deacetylase sirtuin-1, a signaling intermediate that is decreased in obesity and results in enhanced microvascular inflammation within the small intestine, reduced the mortality rate in the early stage of sepsis (154, 155). Treatment with resveratrol increased the expression of sirtuin-1 in obese septic mice and the inflammatory response thereafter was diminished (154). Sirtuins also play a major role during the late onset of septic “hypo-inflammation”; SIRT-2 inhibition in obese septic mice preserved a decreased microvascular inflammation and protected against thrombotic events (155).
The antimicrobial peptide cathelicidin-BF (C-BF) has been observed as a protective molecule, which can safeguard LPS-induced septic rodents from the development of small intestinal barrier dysfunction (156). C-BF prevented LPS-induced TJ breakdown and reduced IEC apoptosis by attenuated expression and secretion of TNF and suppression of the underlying NF-κB pathway (156). Ulinastatin is another drug able to increase the survival rate and to reduce injury of the small intestine, for example, through diminished IEC apoptosis (142). Post-treatment IL-6 and TNF plasma levels were decreased, suggesting an interesting strategy for sepsis (59, 157).
Stem-cell therapy could also represent a potential treatment approach for sepsis. In murine CLP-sepsis, human adipose-derived mesenchymal stem cells were able to modulate sepsis by downregulation of Th1-cell responses, associated with lower levels of pro-inflammatory cytokines (TNF, IL-1β, IL-6, IL-12, IFNγ) and higher levels of anti-inflammatory IL-10 derived from macrophages (158). Application of (mesenchymal) stem cells may, therefore, protect septic mice by reducing inflammatory cell infiltration and pro-inflammatory responses and enhancing anti-inflammatory signals (158). Nevertheless, to what extent stem cells or their cellular structure or secretome will modulate mucosal immunity during sepsis has to be clarified in future studies.
Defining the undisputed role of the microbiome in shaping sepsis-associated immunopathology is gradually gaining momentum, discussed in detail in several reviews (159–161). A dysregulated gut microbiome is a common causation of sepsis (162), like in late-onset sepsis development of preterm neonates (163). Conversely, burn-injury associated altered gut microbial community and leakiness of the gut have been implicated in sepsis development (164). A common approach undertaken to manage sepsis patients involving therapy with antibiotics can impair the diversity of microbes in the intestine and reduce the protective role of bacteria, which in turn leads to increased inflammation in murine models of Gram-positive as well as Gram-negative pneumosepsis (7, 165, 166). Thus, the loss of microbiome diversity was indeed identified as a predictive factor for the length of hospitalization of patients in the ICU (166, 167). Though a recent study disproved that antibiotics-mediated disrupted microbiota modulates innate immune system in endotoxemic patients (168), the exact role of how the immune system is modulated is left to be delineated. Nevertheless, to correct this ensuing dysbiosis, treatment options have included procedures like fecal microbiota transplantation (FMT) with combined usage of antibiotics in the clinical management of sepsis (169–171). In the 1950s, FMT had been developed to treat Clostridium difficile associated pseudomembranous colitis and has since subsequently proven to be an effective treatment modality in the management of C. difficile infection (172–174). Recently, the US Centers for Disease Control indicated that “death rates from sepsis following infections (e.g., C. difficile) have surged (175). Therefore, a perfect premise to facilitate new treatment approaches, FMT is a potentially effective treatment route, which could counterbalance dysbiosis, support the gut microbial barrier and improve the outcome of sepsis (171, 172, 176). Microbial dysbiosis of the gut leads to changes in the metabolism of bacteria and as a consequence to an impaired interaction between microbes, immune cells and IECs (1, 5, 7, 8). During sepsis, the exact mechanism of action for the use of FMT on the intestine is still unknown. Recolonization of the intestinal microflora has been beneficial as well, where 16 days post-FMT, improved symptoms were observed in two separate patient studies involving stroke (176) and post-surgical sepsis development (171). FMT proves to be a viable future treatment option for sepsis and further human clinical research is needed to evaluate its effectiveness in critically ill patients.
Further therapeutic approaches have diversified to include supplementing antibiotics with probiotics, prebiotics and synbiotics. Probiotics are live beneficial microorganisms, which can improve the health of hosts (170), prebiotics are non-viable and non-digestible dietary ingredients e.g., fructooligosaccharides which stimulate the growth and/or activity of a limited number of bacteria in the large intestine (170, 177) and synbiotics refer to combined usage of prebiotics with probiotics (178). Studies have shown that supplementation of Bifidobacterium breve strain Yakult and Lactobacillus casei strain Shirota as probiotics and galactooligosaccharides as prebiotics can reduce the incidence of infectious complications, e.g., enteritis, pneumonia and bacteremia in patients with severe SIRS compared to those who did not receive synbiotics (179, 180). The administration of synbiotics could maintain the gut flora and reduce septic complications in patients with severe SIRS by enhancing the levels of beneficial bacteria in the intestine. A further study suggests that the orally consumed synbiotics (Lactobacillus planatrum and fructooligosaccharide) in newborn infants improve the primary outcome (complication of sepsis or death) as well as lower respiratory tract infections compared to newborn infants with placebo treatment (181). In contradiction to these studies, other studies have suggested no difference in incidence of late-onset sepsis and mortality rate in preterm infants (182, 183). Similar results postulated that prophylactic administration of B. clausii to preterm neonates do not reduce the burden of late-onset sepsis compared to placebo (184). Further, research may be focused on dysbiosis of the gut microbiome and resultant immunosuppression as one consequence of sepsis restored by gut commensals through administration of probiotics, to reduce the incidence of late infections and the sepsis mortality rate (185, 186). Synbiotics also seem to be a potential treatment option for sepsis patients. The complications of enteritis and ventilation-associated pneumonia were significantly lowered in the patients who have been treated with synbiotics, compared to those without synbiotic administration, although the incidence of bacteremia and the mortality rates did not differ between the groups (169). The process of bacterial translocation from the intestinal lumen to systemic circulation as described in section Bacterial Translocation via the Mucosa is another interesting premise to consider as a treatment target. There are some clinical correlations showing bacterial translocation as one cause for subsequent sepsis, or in reverse, induce late onset-sepsis complications. In patients with acute pancreatitis, septic complications as a result of pancreatic necrosis is a major cause of death. Therefore, bacterial overgrowth and subsequent bacterial translocation can be prevented by administration of selected probiotics, because the usage of these bacterial supplementation have been shown to reduce infectious complications in patients with severe acute pancreatitis (187). The oral administration of Lactobacillus plantarum in combination with enteral feeding improved gut permeability and led to a significantly better clinical outcome (188). Treatment of severe acute pancreatitis could be adjusted by enteral nutrition (EN) and ecoimmunonutrition (EIN), because alone as well as in combination, both decrease the expression of plasma endotoxin, TNF, IL-6, bacterial translocation and pancreatic sepsis (189). As described in this review, synbiotics could prevent dysbiosis of the human gut, but administration of synbiotics may not affect the intestinal permeability in critically ill patients (150). A further clinical study with 72 patients have found the influence of pro- and synbiotics (termed as Synbiotic 2000FORTE) (P. pentoseceus 5-33:3, L. mesenteroides 32-77:1, L. paracasei ssp. 19, L. planatarum 2362) on the immune response in patients with multiple injuries (190). A significant decrease (p = 0.028) have been shown in the incidence of septic events as well as the occurrence of ventilation associated pneumonia by Acinetobacter baumannii. The risk of sepsis as a consequence of bacteremia was significantly decreased and even the treatment with the specific synbiotics prolonged the time of progression of primary bacteremia, compared to the placebo group (190). In the molecular level, white blood cell counts and serum C-reactive protein were significantly lower in patients treated with Synbiotic 2000FORTE compared to the placebo cohort and it could reduce the incidence of death caused by MODS (190).
Conclusion
The impact of sepsis on the gut is manifold, e.g., sepsis mediated alteration of the gut-blood barrier and increase in the intestinal permeability, which may correlate with the phenomena of bacterial translocation and lymphatic activation (“toxic-lymph”). Systemic consequences of sepsis are widespread and concern to the coagulative system, the microbiome as well as enzymes, such as pancreatic proteases, MMPs and IAPs. Nevertheless, the therapeutic approaches for modulating the mucosal immune system are still rarely effective in daily routine. Recent published studies showing that treatment with FMT, probiotics and synbiotics are new concepts for gut-specific therapeutic prevention of sepsis (Table 1). Since the past decade, several clinical trials have been completed and are underway to comprehensively actualize the currently understood putative effectiveness of targeting the gut during sepsis. This has been presented in Table 1, enlisting all completed published, completed unpublished and ongoing trials so far. One exemplary study was proven to be an effective synbiotic treatment of fructooligosaccharides and Lactoacillus plantarum to preterm neonates which prevented sepsis and mortality in the treatment group (181). However, these promising therapeutic approaches are yet to be appraised as accepted therapeutic options. More clinical investigations could help substantiate these findings and extend them into becoming alternative treatment options. This also brings into light the importance of understanding the gut mucosal immune system, where further investigation is required to evaluate unknown sepsis-induced intestinal pathophysiological processes. Scarce as of now, nonetheless, investigations to understand the MIS would prove additionally beneficial so as to identify added novel therapeutic modalities.
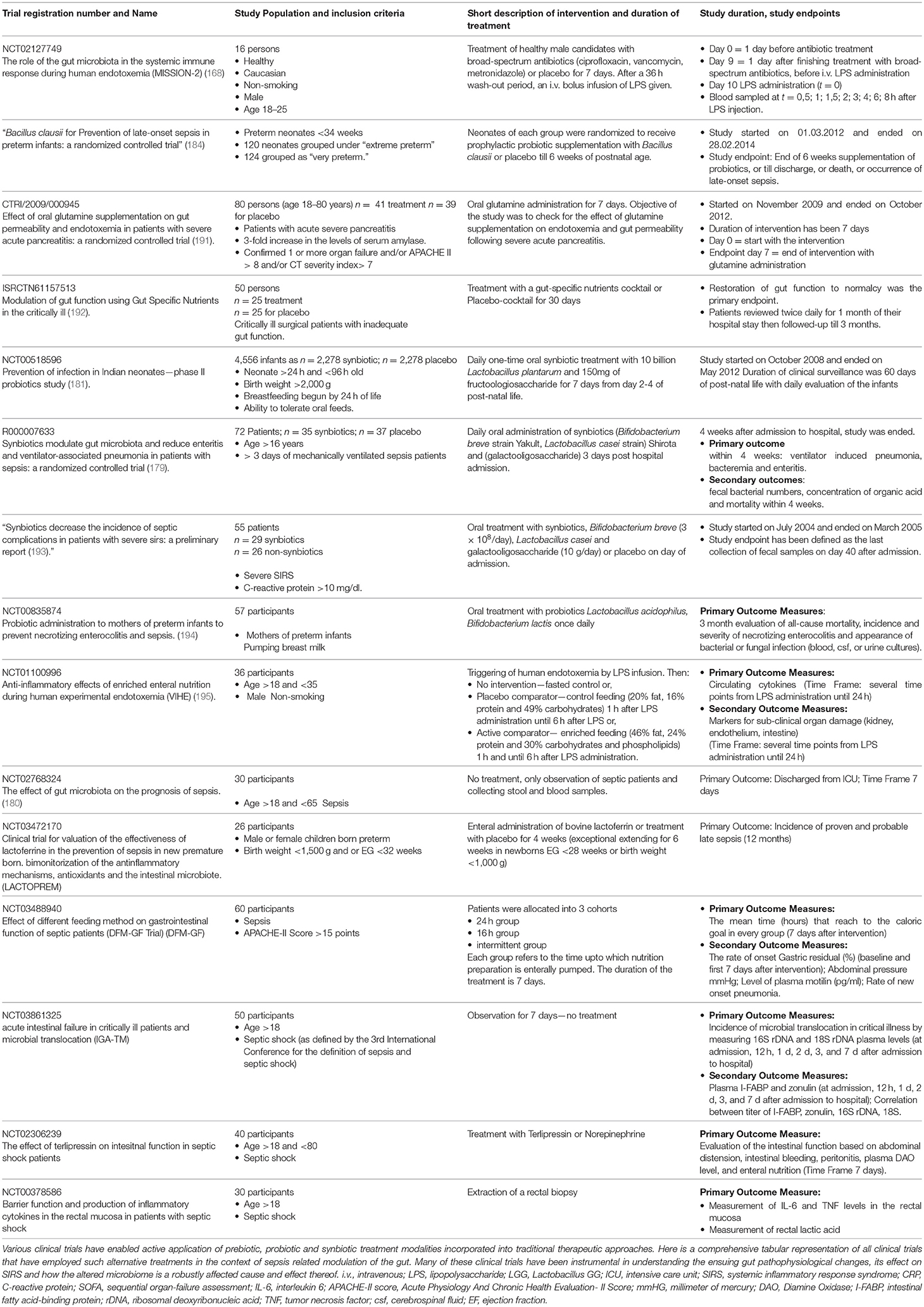
Table 1. Table with ongoing and/or completed clinical trials targeting the gut in sepsis patients.
Author Contributions
FH and MH-L conceptualized the review. FH and SC performed the literature search for the review. FH wrote the review as a first author, with written contribution by SC as the second, RH as the third and MH-L as the senior author. The table was compiled and prepared by FH and SC. Figures were prepared with the help of Dr. Stephanie Denk as mentioned in Acknowledgments.
Funding
This work is supported by grants from the German Research Foundation (DFG) CRC1149 to MH-L (INST 40/479-1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Stephanie Denk for illustrating the figures for this review.
Abbreviations
AMP, antimicrobial peptide; APC, antigen presenting cell; APACHE-II, Acute Physiology and Chronic Health Evaluation- II Score; Bcl-2, B-cell lymphoma gene-2; C-BF, cathelicidin-BF; C3, complement factor 3; C3a, activated complement factor 3; C5, complement factor 5; CD4, cluster of differentiation 4; CLP, cecal ligation and puncture; COX-2, cyclooxygenase-2; CRP, C-reactive protein; CSF, cerebrospinal fluid; DAMP, danger-associated molecular pattern; DAO, Diamine Oxidase; DC, dendritic cell; DIC, disseminated intravascular coagulopathy; DNA, deoxyribonucleic acid; EF, ejection fraction; FXIII, clotting factor XIII; γ-EV, dietary dipeptide gamma-l-glutamyl-l-valine; GALT, gut-associated lymphoid tissue; HMGB1, high-mobility-group-protein-B1; IAP, intestinal alkaline phosphatase; IBD, inflammatory bowel disease; ICAM, intercellular adhesion molecule; ICU, intensive care unit; IEC, intestinal epithelial cell; IEL, intraepithelial lymphocyte; I-FABP, intestinal fatty acid-binding protein; IFN, interferon; IgA, immunoglobulin A; IL, interleukin; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; i. v., intravenous; LPS, lipopolysaccharide; LGG, Lactobacillus GG; M-cells, microfold cells; MALT, mucosa-associated lymphoid tissue; MIS, mucosal immune system; MLCK, myosin-light chain kinase; mmHG, millimeter of mercury; MMP, matrix metalloprotease; MODS, multiple organ dysfunction syndrome; MOF, multi-organ failure; MUC, mucin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B-cells; NOD, nucleotide-binding oligomerization domain; PAI-1, plaminogen activator inhibitor-1; PAMP, pathogen-associated molecular pattern; PG, proteoglycan; PRR, pattern recognition receptor; rDNA, ribosomal deoxyribonucleic acid; RNA, ribonucleic acid; ROS, reactive oxygen species; SATI, short acting thrombin (factor II) and factor Xa (FXa) inhibitor; SIRS, systemic inflammatory response syndrome; SIRT2, NAD-dependent deacetylase sirtuin 2; SOFA, sequential organ-failure assessment; SP, surfactant protein; sTNF, soluble TNF; TAB, TAK1-binding protein; TCR, T-cell receptor; TED, transepithelial dendrites; TF, tissue factor; Th, T helper lymphocyte; TJ, tight junction; TLR, Toll-like-receptor; TNF, tumor necrosis factor; TRAF, TNF receptor associated factor; VCAM, vascular cell adhesion protein; ZO-1, zonulin-1.
References
1. Neish AS. Mucosal immunity and the microbiome. Ann Am Thorac Soc. (2014) 11(Suppl. 1):S28–32. doi: 10.1513/AnnalsATS.201306-161MG
2. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. (2014) 14:667–85. doi: 10.1038/nri3738
3. Ahluwalia B, Magnusson MK, Ohman L. Mucosal immune system of the gastrointestinal tract: maintaining balance between the good and the bad. Scand J Gastroenterol. (2017) 52:1185–93. doi: 10.1080/00365521.2017.1349173
4. Hase K, Ohno H. Epithelial cells as sentinels in mucosal immune barrier. Nihon Rinsho Meneki Gakkai Kaishi. (2006) 29:16–26. doi: 10.2177/jsci.29.16
5. Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, et al. Homeostasis of the gut barrier and potential biomarkers. Am J Physiol Gastrointest Liver Physiol. (2017) 312:G171–93. doi: 10.1152/ajpgi.00048.2015
6. Brandtzaeg P, Kiyono H, Pabst R, Russell MW. Terminology: nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunol. (2008) 1:31–7. doi: 10.1038/mi.2007.9
7. Lankelma JM, Birnie E, Weehuizen TAF, Scicluna BP, Belzer C, Houtkooper RH, et al. The gut microbiota as a modulator of innate immunity during melioidosis. PLoS Negl Trop Dis. (2017) 11:e0005548. doi: 10.1371/journal.pntd.0005548
8. O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. (2006) 7:688–93. doi: 10.1038/sj.embor.7400731
9. Altshuler AE, Lamadrid I, Li D, Ma SR, Kurre L, Schmid-Schonbein GW, et al. Transmural intestinal wall permeability in severe ischemia after enteral protease inhibition. PLoS ONE. (2014) 9:e96655. doi: 10.1371/journal.pone.0096655
10. Chang M, Alsaigh T, Kistler EB, Schmid-Schonbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS ONE. (2012) 7:e40087. doi: 10.1371/journal.pone.0040087
11. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. (1992) 20:864–74. doi: 10.1097/00003246-199206000-00025
12. Gul F, Arslantas MK, Cinel I, Kumar A. Changing Definitions of Sepsis. Turk J Anaesthesiol Reanim. (2017) 45:129–38. doi: 10.5152/TJAR.2017.93753
13. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
14. Raith EP, Udy AA, Bailey M, McGloughlin S, MacIsaac C, Bellomo R, et al. Prognostic accuracy of the SOFA score, SIRS criteria, and qSOFA score for in-hospital mortality among adults with suspected infection admitted to the intensive care unit. JAMA. (2017) 317:290–300. doi: 10.1001/jama.2016.20328
15. Carneiro AH, Povoa P, Gomes JA. Dear Sepsis-3, we are sorry to say that we don't like you. Rev Bras Ter Intensiva. (2017) 29:4–8. doi: 10.5935/0103-507X.20170002
16. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. (2013) 75:289–311. doi: 10.1146/annurev-physiol-030212-183744
17. Elphick DA, Mahida YR. Paneth cells: their role in innate immunity and inflammatory disease. Gut. (2005) 54:1802–9. doi: 10.1136/gut.2005.068601
18. Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol (Oxf). (2018) 222:e12860. doi: 10.1111/apha.12860
19. Stadnyk AW. Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can J Gastroenterol. (2002) 16:241–6. doi: 10.1155/2002/941087
20. Ermund A, Schutte A, Johansson ME, Gustafsson JK, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer's patches. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G341–7. doi: 10.1152/ajpgi.00046.2013
21. Rodriguez-Pineiro AM, Bergstrom JH, Ermund A, Gustafsson JK, Schutte A, Johansson ME, et al. Studies of mucus in mouse stomach, small intestine, and colon. II. Gastrointestinal mucus proteome reveals Muc2 and Muc5ac accompanied by a set of core proteins. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G348–56. doi: 10.1152/ajpgi.00047.2013
22. Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. (2010) 12:319–30. doi: 10.1007/s11894-010-0131-2
23. Dharmani P, Srivastava V, Kissoon-Singh V, Chadee K. Role of intestinal mucins in innate host defense mechanisms against pathogens. J Innate Immun. (2009) 1:123–35. doi: 10.1159/000163037
24. Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA. (2011) 108(Suppl. 1):4659–65. doi: 10.1073/pnas.1006451107
25. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. (2008) 105:15064–9. doi: 10.1073/pnas.0803124105
26. Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. (2001) 2:361–7. doi: 10.1038/86373
27. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol. (2014) 35:270–7. doi: 10.1016/j.it.2014.04.003
28. Mach J, Hshieh T, Hsieh D, Grubbs N, Chervonsky A. Development of intestinal M cells. Immunol Rev. (2005) 206:177–89. doi: 10.1111/j.0105-2896.2005.00281.x
29. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. (2004) 303:1662–5. doi: 10.1126/science.1091334
30. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
31. Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. (2000) 288:2222–6. doi: 10.1126/science.288.5474.2222
32. Oliveira-Nascimento L, Massari P, Wetzler LM. The Role of TLR2 in Infection and Immunity. Front Immunol. (2012) 3:79. doi: 10.3389/fimmu.2012.00079
33. Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. (2009) 15:496–7. doi: 10.1038/nm0509-496
34. Amalakuhan B, Habib SA, Mangat M, Reyes LF, Rodriguez AH, Hinojosa CA, et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine. (2016) 88:267–73. doi: 10.1016/j.cyto.2016.08.028
35. Silswal N, Reddy NS, Qureshi AA, Qureshi N. Resveratrol downregulates biomarkers of sepsis via inhibition of proteasome's proteases. Shock. (2017) 50:579–588. doi: 10.1097/SHK.0000000000001080
36. Wang L, Bastarache JA, Ware LB. The coagulation cascade in sepsis. Curr Pharm Des. (2008) 14:1860–9. doi: 10.2174/138161208784980581
38. De BD, Creteur J, Preiser JC, Dubois MJ, Vincent JL. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. (2002) 166:98–104. doi: 10.1164/rccm.200109-016OC
39. Morelli A, Passariello M. Hemodynamic coherence in sepsis. Best Pract Res Clin Anaesthesiol. (2016) 30:453–63. doi: 10.1016/j.bpa.2016.10.009
40. Cepinskas G, Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. J Clin Biochem Nutr. (2008) 42:175–84. doi: 10.3164/jcbn.2008026
41. Schmid-Schonbein GW, Hugli TE. A new hypothesis for microvascular inflammation in shock and multiorgan failure: self-digestion by pancreatic enzymes. Microcirculation. (2005) 12:71–82. doi: 10.1080/10739680590896009
42. Schmid-Schonbein GW, Penn A, Kistler E. The autodigestion hypothesis in shock and multi-organ failure: degrading protease activity. Bol Soc Port Hemorreol Microcirc. (2011) 26:6–15.
43. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. (2007) 81:1–5. doi: 10.1189/jlb.0306164
44. Schnoor M, Garcia PA, Vadillo E, Pelayo R, Rossaint J, Zarbock A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell Mol Life Sci. (2017) 74:1985–97. doi: 10.1007/s00018-016-2449-x
45. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. (2012) 72:1491–501. doi: 10.1097/TA.0b013e318256e000
46. Keel M, Trentz O. Pathophysiology of polytrauma. Injury. (2005) 36:691–709. doi: 10.1016/j.injury.2004.12.037
47. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. (2011) 208:2581–90. doi: 10.1084/jem.20111354
48. Deitch EA. Bacterial translocation or lymphatic drainage of toxic products from the gut: what is important in human beings? Surgery. (2002) 131:241–4. doi: 10.1067/msy.2002.116408
49. Lemaire LC, van Lanschot JB, Stoutenbeek CP, van Deventer SJ, Dankert J, Oosting H, et al. Thoracic duct in patients with multiple organ failure: no major route of bacterial translocation. Ann Surg. (1999) 229:128–36. doi: 10.1097/00000658-199901000-00017
50. Jiang LY, Zhang M, Zhou TE, Yang ZF, Wen LQ, Chang JX. Changes of the immunological barrier of intestinal mucosa in rats with sepsis. World J Emerg Med. (2010) 1:138–43.
51. Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. (1996) 20:411–7. doi: 10.1007/s002689900065
52. Theuer CJ, Wilson MA, Steeb GD, Garrison RN. Microvascular vasoconstriction and mucosal hypoperfusion of the rat small intestine during bacteremia. Circ Shock. (1993) 40:61–8.
53. Spronk PE, Zandstra DF, Ince C. Bench-to-bedside review: sepsis is a disease of the microcirculation. Crit Care. (2004) 8:462–8. doi: 10.1186/cc2894
54. Ellis CG, Jagger J, Sharpe M. The microcirculation as a functional system. Crit Care. (2005) 9(Suppl. 4):S3–8. doi: 10.1186/cc3751
55. Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. (2011) 17:153–9. doi: 10.1097/MCC.0b013e328344b446
56. Moore FA, Moore EE, Poggetti R, McAnena OJ, Peterson VM, Abernathy CM, et al. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. (1991) 31:629–36. doi: 10.1097/00005373-199105000-00006
57. Defazio J, Fleming ID, Shakhsheer B, Zaborina O, Alverdy JC. The opposing forces of the intestinal microbiome and the emerging pathobiome. Surg Clin North Am. (2014) 94:1151–61. doi: 10.1016/j.suc.2014.08.002
58. Shimizu K, Ogura H, Hamasaki T, Goto M, Tasaki O, Asahara T, et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci. (2011) 56:1171–7. doi: 10.1007/s10620-010-1418-8
59. Li KY, Jiang LY, Zhang M, Zhong M, Xie WZ. Effects of ulinastatin on gut mucosal apoptosis and bacterial translocation in rats with sepsis. Nan Fang Yi Ke Da Xue Xue Bao. (2008) 28:1244–6.
60. Prin M, Bakker J, Wagener G. Hepatosplanchnic circulation in cirrhosis and sepsis. World J Gastroenterol. (2015) 21:2582–92. doi: 10.3748/wjg.v21.i9.2582
61. Hoyt DB. Looking forward. The trauma, critical care and acute care surgery program. Bull Am Coll Surg. (2012) 97:4–5.
62. Ince C. The microcirculation is the motor of sepsis. Crit Care. (2005) 9(Suppl. 4):S13–9. doi: 10.1186/cc3753
63. Miranda M, Balarini M, Caixeta D, Bouskela E. Microcirculatory dysfunction in sepsis: pathophysiology, clinical monitoring, and potential therapies. Am J Physiol Heart Circ Physiol. (2016) 311:H24–35. doi: 10.1152/ajpheart.00034.2016
64. Lipinska-Gediga M. Sepsis and septic shock-is a microcirculation a main player? Anaesthesiol Intensive Ther. (2016) 48:261–5. doi: 10.5603/AIT.a2016.0037
65. Farquhar I, Martin CM, Lam C, Potter R, Ellis CG, Sibbald WJ. Decreased capillary density in vivo in bowel mucosa of rats with normotensive sepsis. J Surg Res. (1996) 61:190–6. doi: 10.1006/jsre.1996.0103
66. Trzeciak S, Cinel I, Phillip DR, Shapiro NI, Arnold RC, Parrillo JE, et al. Resuscitating the microcirculation in sepsis: the central role of nitric oxide, emerging concepts for novel therapies, and challenges for clinical trials. Acad Emerg Med. (2008) 15:399–413. doi: 10.1111/j.1553-2712.2008.00109.x
67. Sielenkamper AW, Meyer J, Kloppenburg H, Eicker K, Van AH. The effects of sepsis on gut mucosal blood flow in rats. Eur J Anaesthesiol. (2001) 18:673–8. doi: 10.1097/00003643-200110000-00006
68. Yoseph BP, Klingensmith NJ, Liang Z, Breed ER, Burd EM, Mittal R, et al. Mechanisms of intestinal barrier dysfunction in sepsis. Shock. (2016) 46:52–9. doi: 10.1097/SHK.0000000000000565
69. Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. (2003) 9:143–51. doi: 10.1097/00075198-200304000-00011
70. Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. (2012) 10:350–6. doi: 10.1016/j.surge.2012.03.003
71. Penn AH, Hugli TE, Schmid-Schonbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. (2007) 27:296–304. doi: 10.1097/01.shk.0000235139.20775.7f
72. Vandenbroucke RE, Dejonckheere E, Van HF, Lodens S, De RR, Van WE, et al. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. (2013) 5:1000–16. doi: 10.1002/emmm.201202100
73. Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. (2010) 189:111–26. doi: 10.1083/jcb.200902153
74. Shi N, Li N, Duan X, Niu H. (2017). Interaction between the gut microbiome and mucosal immune system. Mil Med Res. (2017) 4:14. doi: 10.1186/s40779-017-0122-9
75. Rubio S, Lacaze-Masmonteil T, Chailley-Heu B, Kahn A, Bourbon JR, Ducroc R. Pulmonary surfactant protein A (SP-A) is expressed by epithelial cells of small and large intestine. J Biol Chem. (1995) 270:12162–9. doi: 10.1074/jbc.270.20.12162
76. Fisher JH, Mason R. Expression of pulmonary surfactant protein D in rat gastric mucosa. Am J Respir Cell Mol Biol. (1995) 12:13–8. doi: 10.1165/ajrcmb.12.1.7811466
77. Zhang L, Meng Q, Yepuri N, Wang G, Xi X, Cooney RN. Surfactant Proteins-A and -D attenuate LPS-induced Apoptosis in Primary Intestinal Epithelial Cells (IECs). Shock.
78. Ganz T. (2004). Defensins: antimicrobial peptides of vertebrates. C R Biol. (2017) 327:539–49. doi: 10.1016/j.crvi.2003.12.007
79. Alper CA, Johnson AM, Birtch AG, Moore FD. Human C'3: evidence for the liver as the primary site of synthesis. Science. (1969) 163:286–8. doi: 10.1126/science.163.3864.286
80. Torisu M, Yokoyama T, Kohler PF, Durst AL, Martineau G, Schroter G, et al. Serum complement after orthotopic transplantation of the human liver. Clin Exp Immunol. (1972) 12:21–30.
81. Jain U, Otley AR, Van LJ, Stadnyk AW. The complement system in inflammatory bowel disease. Inflamm Bowel Dis. (2014) 20:1628–37. doi: 10.1097/MIB.0000000000000056
82. Moon R, Parikh AA, Szabo C, Fischer JE, Salzman AL, Hasselgren PO. Complement C3 production in human intestinal epithelial cells is regulated by interleukin 1beta and tumor necrosis factor alpha. Arch Surg. (1997) 132:1289–93. doi: 10.1001/archsurg.1997.01430360035007
83. Sunderhauf A, Skibbe K, Preisker S, Ebbert K, Verschoor A, Karsten CM, et al. Regulation of epithelial cell expressed C3 in the intestine - Relevance for the pathophysiology of inflammatory bowel disease? Mol Immunol. (2017) 90:227–38. doi: 10.1016/j.molimm.2017.08.003
84. Sipola S, Ala-Kokko TI, Laurila JJ, Saarnio J, Ohtonen P, Syrjala H, et al. Colon epithelial injury in critically ill colectomized patients: aberration of tight junction proteins and Toll-like receptors. Minerva Anestesiol. (2017) 83:1017–25. doi: 10.23736/S0375-9393.17.11715-3
85. Li Q, Zhang Q, Wang C, Liu X, Li N, Li J. Disruption of tight junctions during polymicrobial sepsis in vivo. J Pathol. (2009) 218:210–21. doi: 10.1002/path.2525
86. Zhou H, Liang H, Li ZF, Xiang H, Liu W, Li JG. Vagus nerve stimulation attenuates intestinal epithelial tight junctions disruption in endotoxemic mice through alpha7 nicotinic acetylcholine receptors. Shock. (2013) 40:144–51. doi: 10.1097/SHK.0b013e318299e9c0
87. Gu L, Li N, Gong J, Li Q, Zhu W, Li J. Berberine ameliorates intestinal epithelial tight-junction damage and down-regulates myosin light chain kinase pathways in a mouse model of endotoxinemia. J Infect Dis. (2011) 203:1602–12. doi: 10.1093/infdis/jir147
88. Moriez R, Salvador-Cartier C, Theodorou V, Fioramonti J, Eutamene H, Bueno L. Myosin light chain kinase is involved in lipopolysaccharide-induced disruption of colonic epithelial barrier and bacterial translocation in rats. Am J Pathol. (2005) 167:1071–9. doi: 10.1016/S0002-9440(10)61196-0
89. Wang LJ, Sun M. Effect of platelet activating factor receptor antagonist on the tight junction associated protein between the epithelial cells of intestinal mucosa during endotoxemia in young rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. (2007) 19:477–480.
90. Klaus DA, Motal MC, Burger-Klepp U, Marschalek C, Schmidt EM, Lebherz-Eichinger D, et al. Increased plasma zonulin in patients with sepsis. Biochem Med. (2013) 23:107–111. doi: 10.11613/BM.2013.013
91. Fredenburgh LE, Velandia MM, Ma J, Olszak T, Cernadas M, Englert JA, et al. Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis. J Immunol. (2011) 187:5255–5267. doi: 10.4049/jimmunol.1101186
92. Meng J, Yu H, Ma J, Wang J, Banerjee S, Charboneau R, et al. Morphine induces bacterial translocation in mice by compromising intestinal barrier function in a TLR-dependent manner. PLoS ONE. (2013) 8:e54040. doi: 10.1371/journal.pone.0054040
93. Hunninghake GW, Doerschug KC, Nymon AB, Schmidt GA, Meyerholz DK, Ashare A. Insulin-like growth factor-1 levels contribute to the development of bacterial translocation in sepsis. Am J Respir Crit Care Med. (2010) 182:517–25. doi: 10.1164/rccm.200911-1757OC
94. Kesici U, Kesici S, Polat E, Agca B, Turkmen UA, Ozcan D, et al. Effects of intra-abdominal pressure increase on intestinal ischemia and bacterial translocation in experimental sepsis model. Saudi Med J. (2011) 32:813–7.
95. Yu P, Martin CM. Increased gut permeability and bacterial translocation in Pseudomonas pneumonia-induced sepsis. Crit Care Med. (2000) 28:2573–7. doi: 10.1097/00003246-200007000-00065
96. O'Boyle CJ, MacFie J, Mitchell CJ, Johnstone D, Sagar PM, Sedman PC. Microbiology of bacterial translocation in humans. Gut. (1998) 42:29–35. doi: 10.1136/gut.42.1.29
97. Kaiser VL, Sifri ZC, Dikdan GS, Berezina T, Zaets S, Lu Q, et al. Trauma-hemorrhagic shock mesenteric lymph from rat contains a modified form of albumin that is implicated in endothelial cell toxicity. Shock. (2005) 23:417–25. doi: 10.1097/01.shk.0000160524.14235.6c
98. Reino DC, Pisarenko V, Palange D, Doucet D, Bonitz RP, Lu Q, et al. Trauma hemorrhagic shock-induced lung injury involves a gut-lymph-induced TLR4 pathway in mice. PLoS ONE. (2011) 6:e14829. doi: 10.1371/journal.pone.0014829
99. Osterud B. Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul Fibrinol. (1998) 9(Suppl. 1):S9–14.
100. Todoroki H, Nakamura S, Higure A, Okamoto K, Takeda S, Nagata N, et al. Neutrophils express tissue factor in a monkey model of sepsis. Surgery. (2000) 127:209–16. doi: 10.1067/msy.2000.103027
101. Birnbaum J, Hein OV, Luhrs C, Ruckbeil O, Spies C, Ziemer S, et al. Effects of coagulation factor XIII on intestinal functional capillary density, leukocyte adherence and mesenteric plasma extravasation in experimental endotoxemia. Crit Care. (2006) 10:R29. doi: 10.1186/cc3994
102. Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, Gruber A. Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis. (2008) 198:271–4. doi: 10.1086/589514
103. Zhu W, Lv Q, Chen H, Wang Z, Zhong Q. Protective effect and mechanism of sodium tanshinone II A sulfonate on microcirculatory disturbance of small intestine in rats with sepsis. J Huazhong Univ Sci Technolog Med Sci. (2011) 31:441–5. doi: 10.1007/s11596-011-0470-8
104. Fuchs C, Ladwig E, Zhou J, Pavlovic D, Behrend K, Whynot S, et al. Argatroban administration reduces leukocyte adhesion and improves capillary perfusion within the intestinal microcirculation in experimental sepsis. Thromb Haemost. (2010) 104:1022–8. doi: 10.1160/TH10-04-0241
105. Schochl H, van GM, Heitmeier S, Laux V, Kipman U, Roodt J, et al. Dual inhibition of thrombin and activated factor X attenuates disseminated intravascular coagulation and protects organ function in a baboon model of severe Gram-negative sepsis. Crit Care. (2017) 21:51. doi: 10.1186/s13054-017-1636-y
106. Neviere R, Tournoys A, Mordon S, Marechal X, Song FL, Jourdain M, et al. Antithrombin reduces mesenteric venular leukocyte interactions and small intestine injury in endotoxemic rats. Shock. (2001) 15:220–5. doi: 10.1097/00024382-200115030-00010
107. Tagami T, Matsui H, Fushimi K, Yasunaga H. Supplemental dose of antithrombin use in disseminated intravascular coagulation patients after abdominal sepsis. Thromb Haemost. (2015) 114:537–45. doi: 10.1160/TH15-01-0053
108. Raeven P, Drechsler S, Weixelbaumer KM, Bastelica D, Peiretti F, Klotz A, et al. Systemic inhibition and liver-specific over-expression of PAI-1 failed to improve survival in all-inclusive populations or homogenous cohorts of CLP mice. J Thromb Haemost. (2014) 12:958–69. doi: 10.1111/jth.12565
109. Tagami T, Matsui H, Fushimi K, Yasunaga H. Use of recombinant human soluble thrombomodulin in patients with sepsis-induced disseminated intravascular coagulation after intestinal perforation. Front Med. (2015) 2:7. doi: 10.3389/fmed.2015.00007
110. Soendergaard C, Kvist PH, Seidelin JB, Pelzer H, Nielsen OH. Systemic and intestinal levels of factor XIII-A: the impact of inflammation on expression in macrophage subtypes. J Gastroenterol. (2016) 51:796–807. doi: 10.1007/s00535-015-1152-2
111. Sullivan BJ, Swallow CJ, Girotti MJ, Rotstein OD. Bacterial translocation induces procoagulant activity in tissue macrophages. A potential mechanism for end-organ dysfunction. Arch Surg. (1991) 126:586–590. doi: 10.1001/archsurg.1991.01410290062013
112. Shao Z, Nishimura T, Leung LL, Morser J. Carboxypeptidase B2 deficiency reveals opposite effects of complement C3a and C5a in a murine polymicrobial sepsis model. J Thromb Haemost. (2015) 13:1090–102. doi: 10.1111/jth.12956
113. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. (1999) 27:1230–51. doi: 10.1097/00003246-199907000-00002
114. Tiesi G, Reino D, Mason L, Palange D, Tomaio JN, Deitch EA. Early trauma-hemorrhage-induced splenic and thymic apoptosis is gut-mediated and toll-like receptor 4-dependent. Shock. (2013) 39:507–13. doi: 10.1097/SHK.0b013e318293d020
115. Chen L, Song X, Meng X. Correlation of interleukin 1 beta-converting enzyme(ICE) gene expression with gut epithelial cell apoptosis in septic mice. Zhonghua Yi Xue Za Zhi. (1998) 78:544–6.
116. Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, et al. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. (2002) 30:195–201. doi: 10.1097/00003246-200201000-00028
117. Iwata A, Stevenson VM, Minard A, Tasch M, Tupper J, Lagasse E, et al. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J Immunol. (2003) 171:3136–41. doi: 10.4049/jimmunol.171.6.3136
118. Zheng D, Yu Y, Li M, Wang G, Chen R, Fan GC, et al. Inhibition of MicroRNA 195 prevents apoptosis and multiple-organ injury in mouse models of sepsis. J Infect Dis. (2016) 213:1661–70. doi: 10.1093/infdis/jiv760
119. Wesche-Soldato DE, Swan RZ, Chung CS, Ayala A. The apoptotic pathway as a therapeutic target in sepsis. Curr Drug Targets. (2007) 8:493–500. doi: 10.2174/138945007780362764
120. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. (2010) 184:1401–9. doi: 10.4049/jimmunol.0902307
121. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. (2009) 9:313–23. doi: 10.1038/nri2515
122. Berrington JE, Stewart CJ, Cummings SP, Embleton ND. The neonatal bowel microbiome in health and infection. Curr Opin Infect Dis. (2014) 27:236–43. doi: 10.1097/QCO.0000000000000061
123. Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis. (2014) 58:1211–8. doi: 10.1093/cid/ciu084
124. Stewart CJ, Marrs EC, Nelson A, Lanyon C, Perry JD, Embleton ND, et al. Development of the preterm gut microbiome in twins at risk of necrotising enterocolitis and sepsis. PLoS ONE. (2013) 8:e73465. doi: 10.1371/journal.pone.0073465
125. Madan JC, Salari RC, Saxena D, Davidson L, O'Toole GA, Moore JH, et al. Gut microbial colonisation in premature neonates predicts neonatal sepsis. Arch Dis Child Fetal Neonatal Ed. (2012) 97:F456–62. doi: 10.1136/fetalneonatal-2011-301373
126. Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. (2016) 1:16113. doi: 10.1038/nmicrobiol.2016.113
127. Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. (2011) 10:311–23. doi: 10.1016/j.chom.2011.10.004
128. Cabrera-Perez J, Badovinac VP, Griffith TS. Enteric immunity, the gut microbiome, and sepsis: rethinking the germ theory of disease. Exp Biol Med. (2017) 242:127–39. doi: 10.1177/1535370216669610
129. Meng J, Banerjee S, Li D, Sindberg GM, Wang F, Ma J, et al. Opioid exacerbation of gram-positive sepsis, induced by gut microbial modulation, is rescued by IL-17A neutralization. Sci Rep. (2015) 5:10918. doi: 10.1038/srep10918
130. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
131. Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. (2014) 20:524–30. doi: 10.1038/nm.3542
132. DeLano FA, Hoyt DB, Schmid-Schonbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. (2013) 5:169ra11. doi: 10.1126/scitranslmed.3005046
133. Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut. (2010) 59:1476–84. doi: 10.1136/gut.2010.211706
134. Fawley J, Gourlay DM. Intestinal alkaline phosphatase: a summary of its role in clinical disease. J Surg Res. (2016) 202:225–34. doi: 10.1016/j.jss.2015.12.008
135. Verweij WR, Bentala H, Huizinga-van der Vlag A, Miek van Loenen-Weemaes A, Kooi K, Meijer DK Protection against an Escherichia coli-induced sepsis by alkaline phosphatase in mice. Shock. (2004) 22:174–9. doi: 10.1097/01.shk.0000132485.05049.8a
136. Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock. (2002) 18:561–66. doi: 10.1097/00024382-200212000-00013
137. Vandenbroucke RE, Vanlaere I, Van HF, Van WE, Wilson C, Libert C. Pro-inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunol. (2014) 7:579–88. doi: 10.1038/mi.2013.76
138. He F, Peng J, Deng XL, Yang LF, Camara AD, Omran A, et al. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. (2012) 59:264–72. doi: 10.1016/j.cyto.2012.04.008
139. Baugh MD, Perry MJ, Hollander AP, Davies DR, Cross SS, Lobo AJ, et al. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology. (1999) 117:814–22. doi: 10.1016/S0016-5085(99)70339-2
140. Gao Q, Meijer MJ, Kubben FJ, Sier CF, Kruidenier L, van DW, et al. Expression of matrix metalloproteinases-2 and−9 in intestinal tissue of patients with inflammatory bowel diseases. Dig Liver Dis. (2005) 37:584–92. doi: 10.1016/j.dld.2005.02.011
141. Sodeyama M, Gardiner KR, Regan MC, Kirk SJ, Efron G, Barbul A. Sepsis impairs gut amino acid absorption. Am J Surg. (1993) 165:150–54. doi: 10.1016/S0002-9610(05)80419-2
142. Souba WW, Herskowitz K, Klimberg VS, Salloum RM, Plumley DA, Flynn TC, et al. The effects of sepsis and endotoxemia on gut glutamine metabolism. Ann Surg. (1990) 211:543–49. doi: 10.1097/00000658-199005000-00004
143. Foitzik T, Kruschewski M, Kroesen AJ, Hotz HG, Eibl G, Buhr HJ. Does glutamine reduce bacterial translocation? A study in two animal models with impaired gut barrier. Int J Colorectal Dis. (1999) 14:143–9. doi: 10.1007/s003840050200
144. Chang X, Wang LL, Lian SJ, Tang Q, Chen P, Wang H. Effect of oral glutamine on intestinal barrier function in young rats with endotoxemia. Zhongguo Dang Dai Er Ke Za Zhi. (2010) 12:809–11.
145. De-Souza DA, Greene LJ. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit Care Med. (2005) 33:1125–35. doi: 10.1097/01.CCM.0000162680.52397.97
146. Ding LA, Li JS. Effects of glutamine on intestinal permeability and bacterial translocation in TPN-rats with endotoxemia. World J Gastroenterol. (2003) 9:1327–32. doi: 10.3748/wjg.v9.i6.1327
147. Tung JN, Lee WY, Pai MH, Chen WJ, Yeh CL, Yeh SL. Glutamine modulates CD8alphaalpha(+) TCRalphabeta(+) intestinal intraepithelial lymphocyte expression in mice with polymicrobial sepsis. Nutrition. (2013) 29:911–7. doi: 10.1016/j.nut.2013.01.001
148. Lee WY, Hu YM, Ko TL, Yeh SL, Yeh CL. Glutamine modulates sepsis-induced changes to intestinal intraepithelial gammadeltaT lymphocyte expression in mice. Shock. (2012) 38:288–93. doi: 10.1097/SHK.0b013e3182655932
149. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. (2007) 50:2374–83. doi: 10.1007/s00125-007-0791-0
150. Jain PK, McNaught CE, Anderson AD, MacFie J, Mitchell CJ. Influence of synbiotic containing Lactobacillus acidophilus La5, Bifidobacterium lactis Bb 12, Streptococcus thermophilus, Lactobacillus bulgaricus and oligofructose on gut barrier function and sepsis in critically ill patients: a randomised controlled trial. Clin Nutr. (2004) 23:467–75. doi: 10.1016/j.clnu.2003.12.002
151. Chee ME, Majumder K, Mine Y. Intervention of dietary dipeptide gamma-l-glutamyl-l-valine (gamma-EV) ameliorates inflammatory response in a mouse model of LPS-induced sepsis. J Agric Food Chem. (2017) 65:5953–60. doi: 10.1021/acs.jafc.7b02109
152. Zhang H, Kovacs-Nolan J, Kodera T, Eto Y, Mine Y. γ-Glutamyl cysteine and γ-glutamyl valine inhibit TNF-α signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim Biophys Acta. (2015) 1852:792–804. doi: 10.1016/j.bbadis.2014.12.023
153. Wan SX, Shi B, Lou XL, Liu JQ, Ma GG, Liang DY, et al. Ghrelin protects small intestinal epithelium against sepsis-induced injury by enhancing the autophagy of intestinal epithelial cells. Biomed Pharmacother. (2016) 83:1315–20. doi: 10.1016/j.biopha.2016.08.048
154. Wang X, Buechler NL, Yoza BK, McCall CE, Vachharajani VT. Resveratrol attenuates microvascular inflammation in sepsis via SIRT-1-Induced modulation of adhesion molecules in ob/ob mice. Obesity. (2015) 23:1209–17. doi: 10.1002/oby.21086
155. Wang X, Buechler NL, Martin A, Wells J, Yoza B, McCall CE, et al. Sirtuin-2 Regulates Sepsis Inflammation in ob/ob Mice. PLoS ONE. (2016) 11:e0160431. doi: 10.1371/journal.pone.0160431
156. Song D, Zong X, Zhang H, Wang T, Yi H, Luan C, et al. Antimicrobial peptide Cathelicidin-BF prevents intestinal barrier dysfunction in a mouse model of endotoxemia. Int Immunopharmacol. (2015) 25:141–7. doi: 10.1016/j.intimp.2015.01.017
157. Jiang L, Yang L, Zhang M, Fang X, Huang Z, Yang Z, et al. Beneficial effects of ulinastatin on gut barrier function in sepsis. Indian J Med Res. (2013) 138:904–11.
158. Gonzalez-Rey E, Anderson P, Gonzalez MA, Rico L, Buscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. (2009) 58:929–39. doi: 10.1136/gut.2008.168534
159. Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol. (2017) 2:135–43. doi: 10.1016/S2468-1253(16)30119-4
160. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. (2017) 17:407–20. doi: 10.1038/nri.2017.36
161. Alverdy JC, Krezalek MA. Collapse of the microbiome, emergence of the pathobiome, the immunopathology of sepsis. Crit Care Med. (2017) 45:337–47. doi: 10.1097/CCM.0000000000002172
162. Krezalek MA, Defazio J, Zaborina O, Zaborin A, Alverdy JC. The shift of an intestinal “microbiome” to a “pathobiome” governs the course and outcome of sepsis following surgical injury. Shock. (2016) 45:475–82. doi: 10.1097/SHK.0000000000000534
163. Stewart CJ, Embleton ND, Marrs ECL, Smith DP, Fofanova T, Nelson A, et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome. (2017) 5:75. doi: 10.1186/s40168-017-0295-1
164. Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, et al. Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS ONE. (2015) 10:e0129996. doi: 10.1371/journal.pone.0129996
165. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. (2016) 65:575–83. doi: 10.1136/gutjnl-2015-309728
166. Baggs J, Jernigan JA, Halpin AL, Epstein L, Hatfield KM, McDonald LC. Risk of subsequent sepsis within 90 days after a hospital stay by type of antibiotic exposure. Clin Infect Dis. (2018) 66:1004–12. doi: 10.1093/cid/cix947
167. McDonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, et al. Extreme dysbiosis of the microbiome in critical illness. mSphere. (2016) 1:e00199–16. doi: 10.1128/mSphere.00199-16
168. Lankelma JM, Cranendonk DR, Belzer C, de Vos AF, de Vos WM, van der Poll T, et al. Antibiotic-induced gut microbiota disruption during human endotoxemia: a randomised controlled study. Gut. (2017) 66:1623–30. doi: 10.1136/gutjnl-2016-312132
169. Haak BW, Prescott HC, Wiersinga WJ. Therapeutic Potential of the Gut Microbiota in the Prevention and Treatment of Sepsis. Front Immunol. (2018) 9:2042. doi: 10.3389/fimmu.2018.02042
170. Akrami K, Sweeney DA. The microbiome of the critically ill patient. Curr Opin Crit Care. (2018) 24:49–54. doi: 10.1097/MCC.0000000000000469
171. Li Q, Wang C, Tang C, He Q, Zhao X, Li N, et al. Successful treatment of severe sepsis and diarrhea after vagotomy utilizing fecal microbiota transplantation: a case report. Crit Care. (2015) 19:37. doi: 10.1186/s13054-015-0738-7
172. Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. (1958) 44:854–9.
173. Kelly CR, Ihunnah C, Fischer M, Khoruts A, Surawicz C, Afzali A, et al. Fecal microbiota transplant for treatment of Clostridium difficile infection in immunocompromised patients. Am J Gastroenterol. (2014) 109:1065–71. doi: 10.1038/ajg.2014.133
174. Hvas CL, Dahl Jørgensen SM, Jørgensen SP, Storgaard M, Lemming L, Hansen MM, et al. Fecal microbiota transplantation is superior to fidaxomicin for treatment of recurrent clostridium difficile infection. Gastroenterology. (2019) 156:1324–32. doi: 10.1053/j.gastro.2018.12.019
175. Milbrandt EB, Kersten A, Rahim MT, Dremsizov TT, Clermont G, Cooper LM, et al. Growth of intensive care unit resource use and its estimated cost in Medicare. Crit Care Med. (2008) 36:2504–10. doi: 10.1097/CCM.0b013e318183ef84
176. Wei Y, Yang J, Wang J, Yang Y, Huang J, Gong H, et al. Successful treatment with fecal microbiota transplantation in patients with multiple organ dysfunction syndrome and diarrhea following severe sepsis. Crit Care. (2016) 20:332. doi: 10.1186/s13054-016-1491-2
178. Pandey KR, Naik SR, Vakil BV. Probiotics, prebiotics and synbiotics- a review. J Food Sci Technol. (2015) 52:7577–87. doi: 10.1007/s13197-015-1921-1
179. Shimizu K, Ogura H, Goto M, Asahara T, Nomoto K, Morotomi M, et al. Synbiotics decrease the incidence of septic complications in patients with severe SIRS: a preliminary report. Dig Dis Sci. (2009) 54:1071–8. doi: 10.1007/s10620-008-0460-2
180. Shimizu K, Yamada T, Ogura H, Mohri T, Kiguchi T, Fujimi S, et al. Synbiotics modulate gut microbiota and reduce enteritis and ventilator-associated pneumonia in patients with sepsis: a randomized controlled trial. Crit Care. (2018) 22:239. doi: 10.1186/s13054-018-2167-x
181. Panigrahi P, Parida S, Nanda NC, Satpathy R, Pradhan L, Chandel DS, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature. (2017) 548:407–12. doi: 10.1038/nature23480
182. Jacobs SE, Tobin JM, Opie GF, Donath S, Tabrizi SN, Pirotta M, et al. Probiotic effects on late-onset sepsis in very preterm infants: a randomized controlled trial. Pediatrics. (2013) 132:1055–62. doi: 10.1542/peds.2013-1339
183. Costeloe K, Hardy P, Juszczak E, Wilks M, Millar MR. Bifidobacterium breve BBG-001 in very preterm infants: a randomised controlled phase 3 trial. Lancet. (2016) 387:649–60. doi: 10.1016/S0140-6736(15)01027-2
184. Tewari VV, Dubey SK, Gupta G. Bacillus clausii for prevention of late-onset sepsis in preterm infants: a randomized controlled trial. J Trop Pediatr. (2015) 61:377–85. doi: 10.1093/tropej/fmv050
185. Nascimento DC, Melo PH, Pineros AR, Ferreira RG, Colon DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. (2017) 8:14919. doi: 10.1038/ncomms14919
186. Nascimento DC, Alves-Filho JC, Sonego F, Fukada SY, Pereira MS, Benjamim C, et al. Role of regulatory T cells in long-term immune dysfunction associated with severe sepsis. Crit Care Med. (2010) 38:1718–25. doi: 10.1097/CCM.0b013e3181e78ad0
187. Besselink MG, Timmerman HM, Buskens E, Nieuwenhuijs VB, Akkermans LM, Gooszen HG. Probiotic prophylaxis in patients with predicted severe acute pancreatitis (PROPATRIA): design and rationale of a double-blind, placebo-controlled randomised multicenter trial [ISRCTN38327949]. BMC Surg. (2004) 4:12. doi: 10.1186/1471-2482-4-12
188. Qin HL, Zheng JJ, Tong DN, Chen WX, Fan XB, Hang XM, et al. Effect of Lactobacillus plantarum enteral feeding on the gut permeability and septic complications in the patients with acute pancreatitis. Eur J Clin Nutr. (2008) 62:923–30. doi: 10.1038/sj.ejcn.1602792
189. Wang G, Wen J, Xu L, Zhou S, Gong M, Wen P, et al. Effect of enteral nutrition and ecoimmunonutrition on bacterial translocation and cytokine production in patients with severe acute pancreatitis. J Surg Res. (2013) 183:592–7. doi: 10.1016/j.jss.2012.12.010
190. Giamarellos-Bourboulis EJ, Bengmark S, Kanellakopoulou K, Kotzampassi K. Pro- and synbiotics to control inflammation and infection in patients with multiple injuries. J Trauma. (2009) 67:815–21. doi: 10.1097/TA.0b013e31819d979e
191. Singh N, Mishra SK, Sachdev V, Sharma H, Upadhyay AD, Arora I, et al. Effect of oral glutamine supplementation on gut permeability and endotoxemia in patients with severe acute pancreatitis: a randomized controlled trial. Pancreas. (2014) 43:867–73. doi: 10.1097/MPA.0000000000000124
192. Gatt M, MacFie J. Randomized clinical trial of gut-specific nutrients in critically ill surgical patients. Br J Surg. (2010) 97:1629–36. doi: 10.1002/bjs.7155
193. Stadlbauer V, Horvath A, Komarova I, Schmerboeck B, Feldbacher N, Klymiuk I, et al. Dysbiosis in early sepsis can be modulated by a multispecies probiotic: a randomised controlled pilot trial. Benef Microbes. (2019) 29:1–14. doi: 10.3920/BM2018.0067
194. Benor S, Marom R, Ben TA, Armoni DK, Zaidenberg-Israeli G, Dollberg S. Probiotic supplementation in mothers of very low birth weight infants. Am J Perinatol. (2014) 31:497–504. doi: 10.1055/s-0033-1353490
Keywords: sepsis, innate immunity, gut-barrier dysfunction, perfusion disturbances, enzymatic response, microbiome
Citation: Haussner F, Chakraborty S, Halbgebauer R and Huber-Lang M (2019) Challenge to the Intestinal Mucosa During Sepsis. Front. Immunol. 10:891. doi: 10.3389/fimmu.2019.00891
Received: 14 November 2018; Accepted: 08 April 2019;
Published: 30 April 2019.
Edited by:
Johannes Trück, University Children's Hospital Zurich, SwitzerlandReviewed by:
Chenyang Wang, Nanjing University, ChinaEvangelos Giamarellos-Bourboulis, National and Kapodistrian University of Athens, Greece
Copyright © 2019 Haussner, Chakraborty, Halbgebauer and Huber-Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Huber-Lang, bWFya3VzLmh1YmVyLWxhbmdAdW5pa2xpbmlrLXVsbS5kZQ==