
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 12 April 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00754
 Timothy R. Crother1,2
Timothy R. Crother1,2 Rebecca A. Porritt1Jargalsaikhan Dagvadorj1Gantsetseg Tumurkhuu1Anatoly V. Slepenkin3
Rebecca A. Porritt1Jargalsaikhan Dagvadorj1Gantsetseg Tumurkhuu1Anatoly V. Slepenkin3 Ellena M. Peterson4
Ellena M. Peterson4 Shuang Chen1,2
Shuang Chen1,2 Kenichi Shimada1,2
Kenichi Shimada1,2 Moshe Arditi1,2*
Moshe Arditi1,2*Autophagy can either antagonize or promote intracellular bacterial growth, depending on the pathogen. Here, we investigated the role of autophagy during a pulmonary infection with the obligate intracellular pathogen, Chlamydia pneumoniae (CP). In mouse embryonic fibroblasts (MEFs) or macrophages, deficiency of autophagy pathway components led to enhanced CP replication, suggesting that autophagy exerts a bactericidal role. However, in vivo, mice with myeloid-specific deletion of the autophagic protein ATG16L1 suffered increased mortality during CP infection, neutrophilia, and increased inflammasome activation despite no change in bacterial burden. Induction of autophagy led to reduced CP replication in vitro, but impaired survival in CP-infected mice, associated with an initial reduction in IL-1β production, followed by enhanced neutrophil recruitment, defective CP clearance, and later inflammasome activation and IL-1β production, which drove the resulting mortality. Taken together, our data suggest that a delicate interplay exists between autophagy and inflammasome activation in determining the outcome of CP infection, perturbation of which can result in inflammatory pathology or unrestricted bacterial growth.
Autophagy, the process by which cellular material is recycled (1), has been increasingly found to play a central role in diverse diseases and pathologies (2). One key role for autophagy is the removal of damaged mitochondria (3), termed mitophagy, which in turn can modulate inflammatory processes by dampening activation of the nucleotide binding domain and leucine-rich repeat (NLR) pyrin domain containing 3 (NLRP3) inflammasome (4–8). Thus, loss of autophagic function can lead to enhanced inflammation via greater NLRP3 inflammasome activation and secretion of IL-1β. Release of this pro-inflammatory cytokine is tightly regulated, because although IL-1β is critical in controlling many microbial infections, over production can lead to enhanced pathology and chronic inflammation (9, 10).
In addition to regulating cytokine production, autophagy can play a direct role in microbial clearance in a process known as xenophagy (11). This is particularly important for intracellular bacteria such as Salmonella typhimurium, group A Streptococcus, and Mycobacterium tuberculosis. In these cases, the autophagic phagophore forms around the invading bacteria, and the resulting autophagosome is targeted for fusion with the lysosome (12–14). Bacteria can escape this eradication by actively preventing lysosomal fusion (15, 16), or autophagy induction and/or elongation (17). A related process that uses many components of the autophagosome machinery, termed LC3 associated phagocytosis (LAP), targets the production of reactive oxygen species in the phagosome lumen to directly kill bacteria such as S. typhimurium and Burkholderia pseudomallei (18, 19). Finally, some bacterial agents can co-opt components of the autophagosomal machinery in order to survive and replicate (20, 21).
CP is an obligate intracellular bacterium that mainly infects macrophages and neutrophils, and exists in an inclusion inside the cell (22). While CP infection is thought to be responsible for up to 10–20% of cases of community-acquired pneumonia (23), it has also been associated with many chronic inflammatory diseases including atherosclerosis, Alzheimer's disease, and asthma (24). CP have a unique lifecycle whereby they exist as an infectious elementary body (EB), which enters the host cell and forms an inclusion (22). Once inside, it changes into the metabolically active reticulate body (RB) and replicates inside the inclusion (22). Eventually RBs revert to the EB state and burst forth from the cell (22). While CP inclusions do not fuse with the lysosome, lysosomal maturation seems to be a requirement for CP growth (25), raising the possibility that the autophagy pathway plays a role in either preventing or facilitating CP infection.
In this study we found blocking autophagy led to increased CP growth in both macrophages and mouse embryonic fibroblasts. In vivo, loss of the autophagy elongation component ATG16L1 specifically in myeloid cells led to increased mortality in response to CP infection, characterized by greater numbers of neutrophils and dendritic cells, but no change in the CP burden in the lungs. This was accompanied by an increase in inflammasome-active macrophages and IL-1β production. While induction of autophagy in macrophages led to reduced CP growth in vitro, in vivo treatment with rapamycin led to increased mortality of infected mice, likely due to initial impairment in IL-1β production resulting in increased bacterial load.
C57BL/6 (wild-type [WT]) mice were purchased from Jackson Labs (Bar Harbor, ME) and then bred in the Cedars-Sinai Medical Center Comparative Medicine facility. Atg16l1fl/fl Lysmcre mice were obtained from Dr. David Shih, Cedars-Sinai medical Center (26). Atg16l1fl/fl mice were used as littermate controls. Eight- to ten-weeks-old female mice were used in all experiments in this study. WT, Atg16l1+/−, and Atg5−/− mouse embryonic fibroblasts (MEF)s were obtained from Dr. Jae Jung (University of Southern California) (27) and grown in DMEM media containing 10% FBS. In some cases, 1 μg/ml cycloheximide (Sigma Aldrich, St. Louis MO), MEM amino acids (Sigma Aldrich, St. Louis MO), or Na-pyruvate (Sigma Aldrich, St. Louis MO) were added to the media. Atg5fl/fl Lysmcre and Atg5fl/fl bone marrow was provided by Dr. Herbert W. Virgin (Washington University, Saint Louis).
All experiments were conducted according to Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC) guidelines (IACUC# 8314).
Chlamydia pneumoniae CM-1 (American Type Culture Collection, Manassas, VA) was propagated in HEp-2 cells, as previously described (28). Both HEp-2 cells and C. pneumoniae aliquots were confirmed as being Mycoplasma- free by PCR. Mice were infected with either 1 × 106 or 2.5 × 106 inclusion forming units (IFU) C. pneumoniae by inoculating intratracheally. Bronchoalveolar lavage fluid (BALF) was collected by injecting 0.5 ml PBS containing 5 mM EDTA. BALF was separated into supernatant and cells by centrifugation. Supernatant was used for cytokine and chemokine measurements, and cells were analyzed by Flow Cytometry. In some experiments, mice were injected with rapamycin (3 mg/kg; ThermoFisher Scientific, MA, USA) i.p., every other day until sacrifice. The first injection was given 6 h prior to infection.
HEp-2 cells were infected with C. pneumoniae in lung specimens for bacterial burden quantification, as described previously (29). Briefly, HEp-2 cells were incubated with diluted lung suspensions or cell lysates in the presence of RPMI 1640 medium containing 1 μg/ml cycloheximide. Centrifugation was performed for 1 h at 800 x g, and cells were placed in an incubator (37°C, 0.5% CO2). After 72 h of culture, cells were washed with PBS, fixed with methanol, and stained with FITC-conjugated anti-Chlamydia genus-specific mAb (Pathfinder Chlamydia Culture Confirmation System; Bio-Rad, Hercules, CA), according to the manufacturer's instructions. C. pneumoniae inclusions in cells were counted by fluorescence microscopic analysis. For in vitro inclusion quantification, infected cells were directly assessed using the Pathfinder Chlamydia Culture Confirmation System as above.
BALF cellular samples were used for Flow cytometry analysis. Cells were filtered through a 70 μm cell strainer (BD Bio-sciences, San Jose, CA). Erythrocytes were obviated by RBC Lysis Buffer (eBioscience, San Diego, CA). Single cells were stained with the following antibodies (Ab)s (Tonbo Biosciences, San Diego, CA): F4/80 Ab (BM8), CD11c Ab (N418), Ly6G Ab (1A8), CD11b Ab (M1/70), and CD3 Ab (145-2C11). For BALF single-cell differentials, cell types were identified as neutrophils (CD11c− CD11b+ Ly6G+), alveolar macrophages (AM)s (CD11b+ CD11c+ F4/80+), dendritic cells (CD11c+ CD11b+ F4/80−), and T cells (CD3+). Flow cytometric analysis was performed using a CyAn flow cytometer (Beckman Coulter) and analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
The concentration of cytokine in BALF was determined using an OptEIA Mouse IL-6 ELISA Set (BD Biosciences) and Mouse IL-12p40, Mouse IFN-γ ELISA Kit (eBioscience), and DuoSet Mouse MIP-2 (CXCL2) and Mouse KC (CXCL1) (R&D Systems, Minneapolis, MN). These assays were performed according to the manufacturers' instructions. IL-1β in mouse BALF samples was measured using the U-PLEX Mouse IL-1β Assay (Meso Scale Diagnostics, Rockville, Maryland) per the manufacturer's instructions. The samples were read and analyzed by MSD QuickPlex SQ120 instrumentation and Workbench 4.0 Software (Meso Scale Diagnostics, Rockville, Maryland).
Lungs were fixed in formalin, paraffin embedded and sectioned. Lung sections were stained with hematoxylin and eosin. Inflammatory scoring was performed by a blinded pathologist, scoring 1–4 as previously described (30).
Caspase-1 activity was detected by Fluorochrome-labeled inhibitors of caspases assay (FLICA) from Immunochemistry Technologies (Bloomington, MN) per the manufacturer's instructions for either frozen sections or live tissue culture. For Immunofluorescence staining for frozen sections, fixing and antigen blocking were performed using immunoglobulin from the species of the secondary antibodies. Next, the sections were incubated with primary antibodies for 2 h RT, followed by incubation with appropriate secondary antibodies conjugated with Fluorescent dyes. For identification of FLICA+ cells, anti-F4/80 (BM8) antibody and anti CD11c (N418) (eBioscience) were used.
BMDM: Femora and tibiae of 8–10 week old C57BL/6 were flushed with RPMI 1640 medium. Bone marrow cells were cultured in RPMI 1640 medium containing 10% FBS and 15% L929 cell conditioned medium with 2 media changes. BMDM were harvested on day 7. Elicited peritoneal macrophages: 1 ml thioglycollate media was injected i.p., in mice. 3–4 days later peritoneal cells were obtained by syringe and plated. Twenty four hour later, non-adherent cells were washed away.
Data are presented as mean ± SEM. For survival studies, significance was evaluated by the log-rank test. For comparisons between two groups, statistical significance was evaluated using Student's two-tailed unpaired t-test. For non-normally distributed data, unpaired Mann–Whitney test was used. For experiments in which more than two samples were analyzed, one-way ANOVA with the Tukey post-hoc test were used to assess statistical significance between groups. p < 0.05 was considered significant.
In order to understand what role autophagy might play during CP growth, we infected MEFs with CP and observed that 48 h after infection, most of the LC3 was lipidated (LC3-II), suggestion an increase in autophagy (Supplemental Figure 1A). We next infected Atg16l1+/− MEFs and observed a significant increase in the number of inclusions present when compared with WT MEFS (Figure 1A). ATG16L1, along with ATG5 and ATG12, make up the elongation complex of autophagosome formation (31). By gross observation, the CP inclusions in Atg16l1+/− MEFs were often much larger than those in WT MEFS (Supplemental Figure 1). In addition to an increase in inclusion numbers, Atg16l1+/− cells developed a greater number of CP progeny as measured by inclusion forming units (IFU) (Figure 1B). We observed a similar increase in inclusion numbers as well as progeny in CP infected bone marrow derived macrophages from Atg16l1fl/fl LysmCre mice (Figures 1C,D). Surprisingly, we observed the opposite phenotype when cells were deficient in another autophagy component, Atg5, as CP-infected Atg5−/− MEFs with CP had significantly fewer inclusions and progeny than WT cells (Figures 1E,F). Additionally, the CP inclusions observed in Atg5−/− MEFs were significantly smaller than those in WT MEFs (Figure 1G, Supplemental Figure 1B). This discrepancy between Atg16l1+/− MEFs and Atg5−/− MEFs was surprising as both ATG16L1 and ATG5 participate in the same autophagic complex, along with ATG12. However, CP-infected Atg5−/− macrophages obtained from Atg5fl/fl LysmCre mice displayed higher inclusion numbers that WT macrophages (Figure 1H), similar to macrophages lacking ATG16L1. Thus, while it was clear that autophagic machinery played a role in CP growth in both MEFs and macrophages, the effect of loss of pathway components differed between cell types.
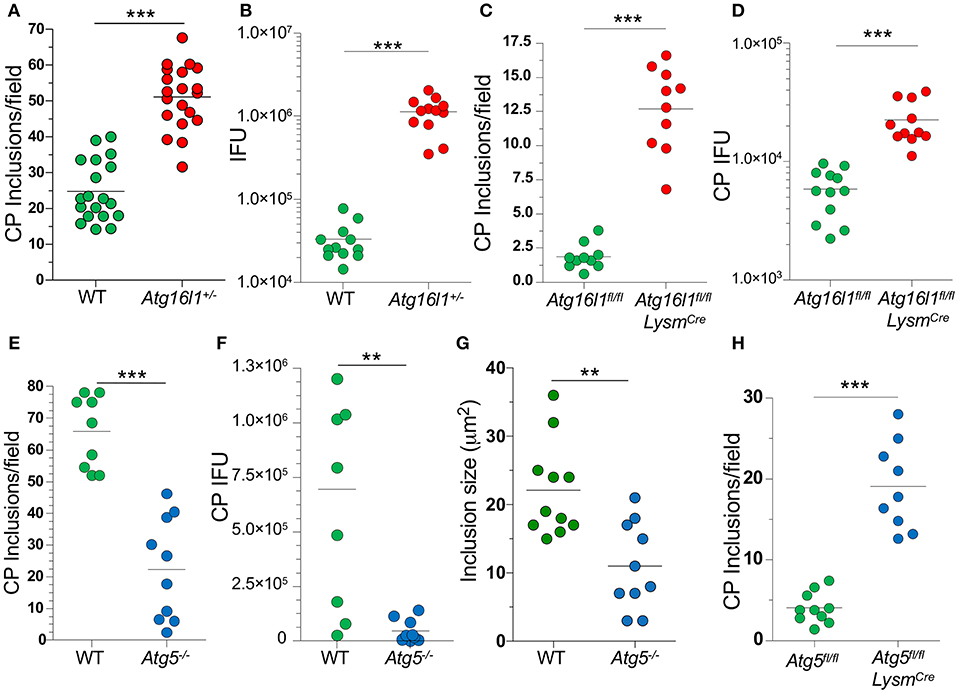
Figure 1. Autophagy plays a role during CP growth. (A,B) WT and Atg16l1+/− MEFs were infected with CP (MOI 10) and 68 h later assessed for inclusions (A) and IFUs (B). (C,D) Atg16l1fl/fl and Atg16l1fl/fl Lysmcre BMDM were infected with CP (MOI 1) and 68 h later assessed for inclusions (C) and IFUs (D). (E–G) WT and Atg5−/− MEFs were infected with CP (MOI 10) and 68 h later assessed for inclusions (E), IFUs (F), and inclusion size (G). (H) Atg5fl/fl and Atg5fl/fl Lysmcre BMDM were infected with CP (MOI 1) and 68 h later assessed for inclusions (C). All panels are representative of three experiments. Significance was assessed by students t-test. **p < 0.01, ***p < 0.001.
We next sought to understand why loss of ATG5 and ATG16L1 had differential effects on CP infection in MEFs. We observed that Atg16l1+/− MEFs divided much more slowly than WT MEFs, while Atg5−/− MEFs grew much more quickly (Figure 2A). We therefore suspected that the difference in CP inclusion formation between Atg16l1+/− MEFs and Atg5−/− MEFs may be due to cell growth rates and perhaps nutritional limits. Indeed, inhibiting cell growth with cycloheximide, which has also been reported to affect autophagy (32), resulted in a significant increase in the number and size of inclusions in Atg5−/− MEFs (Figures 2B,C). An increase was also observed in WT MEFs treated with cycloheximide. We therefore reasoned that in nutrient-replete conditions, CP inclusion formation may be affected by differential growth rates. To test this, we seeded tissue culture plates with various starting numbers of MEFs and infected them with the same CP multiplicity of infection. Regardless of the starting cell number, Atg16l1+/− MEFs were able to support high numbers of CP inclusions (Figure 2D). However, Atg5−/− MEFs could only support CP inclusion growth at the lowest cell seeding density, and no inclusions were observed at higher starting cell numbers (Figure 2D). Similarly, while WT MEFs had greater numbers of inclusions than Atg5−/− MEFs at lower seeding density, and they failed to support CP inclusion growth when seeded at high density (Figure 2D). Finally, when Atg5−/− MEFs were supplied with additional amino acid supplement and sodium pyruvate, greater numbers of inclusions were observed (Figure 2E). Taken together, these data indicate that differential growth rates and subsequent depletion of nutrients underlies the difference in CP inclusion formation in Atg16l1+/− and Atg5−/− MEFs, and in macrophages, where there is little cell growth, loss of ATG5 and ATG16L1 both lead to increased numbers of CP inclusions and progeny, suggesting that autophagy plays a role in limiting CP growth.
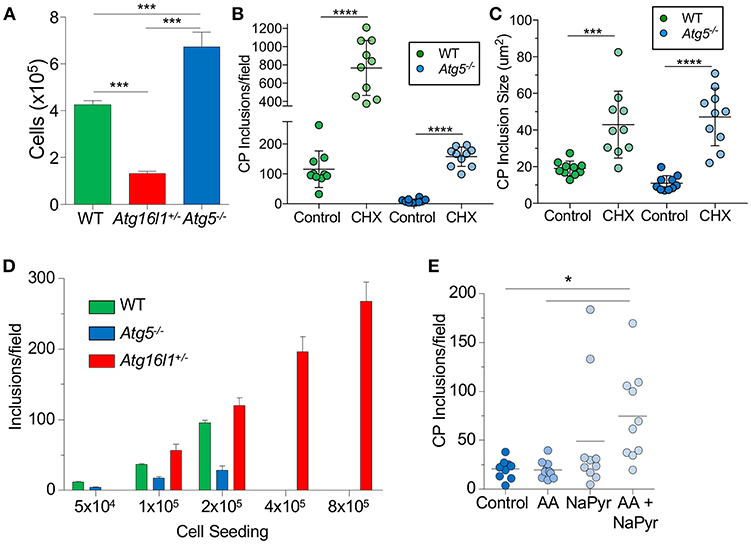
Figure 2. CP growth requires adequate nutrition available to the host. (A) WT, Atg16l1+/−, and Atg5−/− MEFs were seeded at 5 × 104 in a 24-well plate. Total cell numbers were counted after 3 days. (B,C) Atg5−/− MEFs were infected with CP (MOI 10) and treated with 1 μg/ml cycloheximide. Inclusions were counted 68 h post infection (B), as was inclusion size (C). (D) WT, Atg16l1+/−, and Atg5−/− MEFs were seeded at the indicated amounts in a 24-well plate and infected with CP (MOI 10). Inclusions were counted 68 h post infection. (E) Atg5−/− MEFs were infected with CP (MOI 10) and the media was supplemented with either MEM amino acids (AA), Na-pyruvate (NaPyr), or both. Inclusions were counted 68 h post infection. All panels are representative of three experiments. Data are mean ± SEM. Significance was assessed by students t-test (B,C) or one-way ANOVA (A,E). *p < 0.05, ***p < 0.001, **** p < 0.0001.
We next investigated how the loss of autophagy in phagocytic cells [macrophages (Supplemental Figure 2A), neutrophils, dendritic cells (DC)s etc.] would affect CP infection and clearance in mice. We found that Atg16l1fl/fl LysmCre mice were significantly more susceptible to CP infection than littermate Atg16l1fl/fl mice, displaying 100% lethality within 8 days of infection (Figure 3A). In order to investigate the course of infection and immune responses, we infected Atg16l1fl/fl LysmCre and littermate Atg16l1fl/fl controls with a lower inclusion forming unit (IFU) dose of CP. To our surprise, despite the differences observed in mortality, the bacterial burden was equivalent in the lungs of mice with myeloid autophagy deficiency and controls (Figure 3B). Bronchoalveolar lavage fluid (BALF) analysis revealed an increase in the number of cells in Atg16l1fl/fl LysmCre lungs 5 days after infection that resolved by day 9 (Figure 3C). While there were no differences in the numbers of AMs and T-cells between genotypes (Figures 3D,E), flow cytometry revealed a significant increase in both neutrophils and dendritic cells in the BALF of Atg16l1fl/fl LysmCre mice 5 and 9 days after infection (Figures 3F,G, Supplemental Figure 2B). Autophagy deficiency did not alter the levels of the critical pro-inflammatory cytokines required for normal immune responses to CP: IFN-γ, IL-6, or IL-12p40 (Figures 3H–J). However, in support of our finding of increased neutrophils, neutrophilic chemokines CXCL1 and CXCL2 were elevated in BALF from Atg16l1fl/fl LysmCre mice compared with controls (Figures 3K, L). Despite the increase in neutrophils and DCs, we did not observe a significant difference in gross pathology in the lungs by H&E between the conditional knockout and control mice (Supplemental Figure 2C). Thus, loss of autophagy in myeloid cells led to increased lung infiltration by neutrophils and DCs in the setting of CP infection and impaired survival but does not affect CP burden.
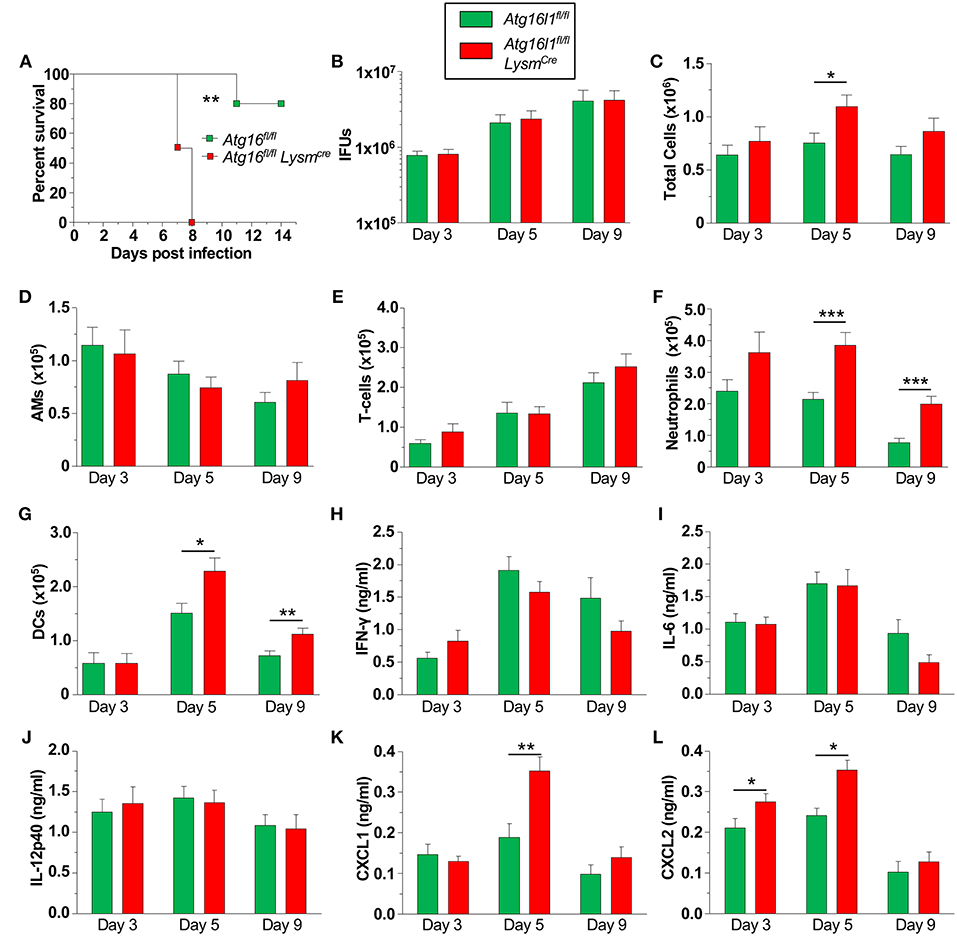
Figure 3. Loss of autophagy in myeloid cells leads to neutrophilia and mortality during CP infection. (A) Atg16l1fl/fl and Atg16l1fl/fl Lysmcre mice were infected with 2.5 × 106 IFU CP i.t. and mortality was assessed (n = 5–6). (B–L) Atg16l1fl/fl and Atg16l1fl/fl Lysmcre mice were infected with 1.0 × 106 IFU CP i.t. and mice were sacrificed 3, 5, and 9 days after infection (n = 9–13, pooled from two experiments). (B) Bacterial burden in the lungs. (C) Total cells in the BALF. (D) BALF Alveolar macrophages. (E) BALF T-cells. (F) BALF neutrophils. (G) BALF DCs. (H) BALF IFN-γ. (I) BALF IL-6. (J) BALF IL-12p40. (K) BALF CXCL1. (L) BALF CXCL2. Data are mean ± SEM. Significance was assessed by students t-test. Survival curve was compared using the Log-Rank (Mantel-Cox) Test. *p < 0.05, **p < 0.01, ***p < 0.001.
We previously showed that IL-1β production via the NLRP3 inflammasome is critically required to control CP growth to successfully resolve the infection in mice (33). Furthermore, autophagy and mitophagy are known to negatively regulate the NLRP3 inflammasome and subsequent IL-1β secretion (5–7). Indeed, Atg16l1fl/fl LysmCre macrophages produced more IL-1β than controls in response to CP infection (Figure 4A), which correlated with increased Caspase-1 activity (Figure 4B, Supplemental Figure 3A).
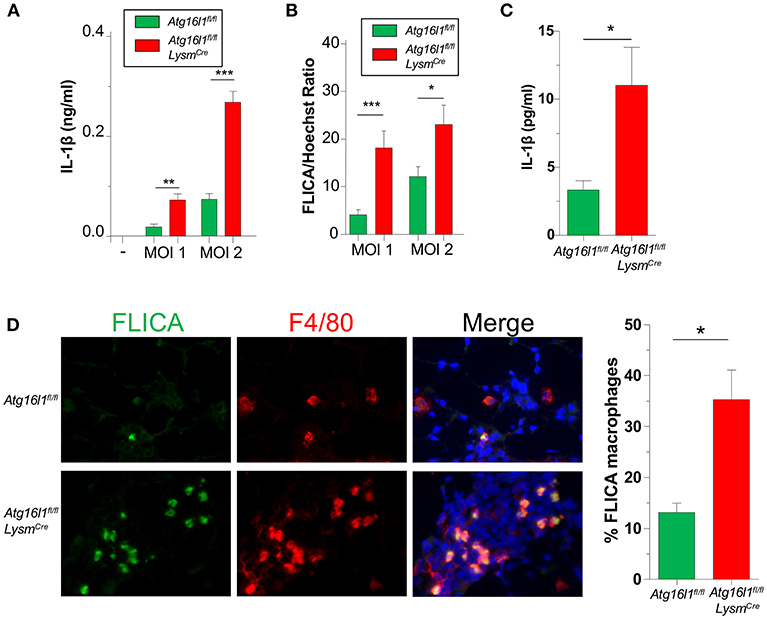
Figure 4. Loss of autophagy in myeloid cells leads to an increase in inflammasome activation and IL-1β during CP infection. (A,B) Atg16l1fl/fl and Atg16l1fl/fl Lysmcre peritoneal macrophages were infected with CP at indicated MOI and (A) IL-1β was measured in the supernatants by ELISA 20 h later. (B) % FLICA positive cells. (C) IL-1β in BALF 1 day after CP infection in Atg16l1fl/fl and Atg16l1fl/fl Lysmcre mice (n = 9–11 pooled from two separate experiments). (D) Caspase-1 activity measured by FLICA in frozen lung sections 1 day after CP infection in Atg16l1fl/fl and Atg16l1fl/fl Lysmcre mice (n = 7). Data are mean ± SEM. Significance was assessed by students t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
We next assessed the effect of autophagy deficiency on IL-1 β production in vivo. Atg16l1fl/fl LysmCre mice had significantly more IL-1β in BALF 1 day after infection compared with control animals (Figure 4C), corroborating out in vitro data. Finally, we also assessed Caspase-1 activity in the lungs of CP-infected mice 1 day post infection. Confirming our in vitro results, lung sections from Atg16l1fl/fl LysmCre showed significantly more FLICA positive macrophages compared with littermate controls (Figure 4D, Supplemental Figure 3B). Interestingly, the FLICA positive macrophages (F4/80+) that we observed were not CD11c+ (Supplemental Figure 3C), suggesting that these were not alveolar macrophages (AMs), but instead of a different origin, such as interstitial macrophages or newly recruited monocytes. Additionally, while it does not completely rule out other cell types secreting IL-1β in this model, we did not observe any non F4/80+ FLICA+ cells.
Since our data indicated that loss of autophagy in macrophages led to increased CP growth due to reduced bacterial killing, we asked whether induction of autophagy by chemical means would lead to reduced CP inclusion formation. We therefore treated WT MEFs with tamoxifen, a potent autophagy inducer (34), for the first 2 h of CP infection, and found that autophagy induction led to a significant reduction in CP inclusion formation, as well as progeny (Figures 5A, B). We next assessed whether induction of autophagy in vivo could rescue mice infected with a high dose of CP. Mice were i.p., injected with the autophagy inducer rapamycin (35), 6 h prior to CP infection, and every other day through the duration of the study. Surprisingly, the mice receiving rapamycin perished significantly earlier than control mice (Figure 5C). In order to determine why the rapamycin-treated mice succumbed to infection more quickly, we repeated the study with a lower infective dose and sacrificed mice 3 and 6 days after infection. Three days after infection we observed a reduction in T-cells in the BALF of rapamycin treated mice, but no changes in DCs, AMs, or neutrophils (Figures 5D–H). In contrast, while there was a decrease in the number of AMs 6 days after infection, there was a significant overall increase in cell counts in the airways primarily due to a large increase in the number of neutrophils (Figures 5D–H) in the rapamycin-treated animals. There was no difference in the bacterial burden at day 3 between control and rapamycin-treated mice, but there was a significantly greater CP load in the lungs of rapamycin-treated mice 6 days after infection (Figure 5I). Since autophagy induction can limit inflammasome activation, we measured IL-1β in the BALF and found a significant decrease in rapamycin-treated mice compared with controls 3 days after infection (Figure 5J). However, at 6 days after infection we found a significant increase in IL-1β production. We also found a clear increase in inflammation in the lungs 6 days after infection (Figure 5K, Supplemental Figure 4), correlating with the increase in neutrophils and IL-1β in the BALF, as well as the greater bacterial burden. We did not observe a difference in lung pathology 3 days after infection (data not shown). Taken together, these data indicate that the rapamycin-treated mice had greater mortality due to increased inflammation as a consequence of the greater bacterial load, likely as a result of the initial reduction in IL-1β production.
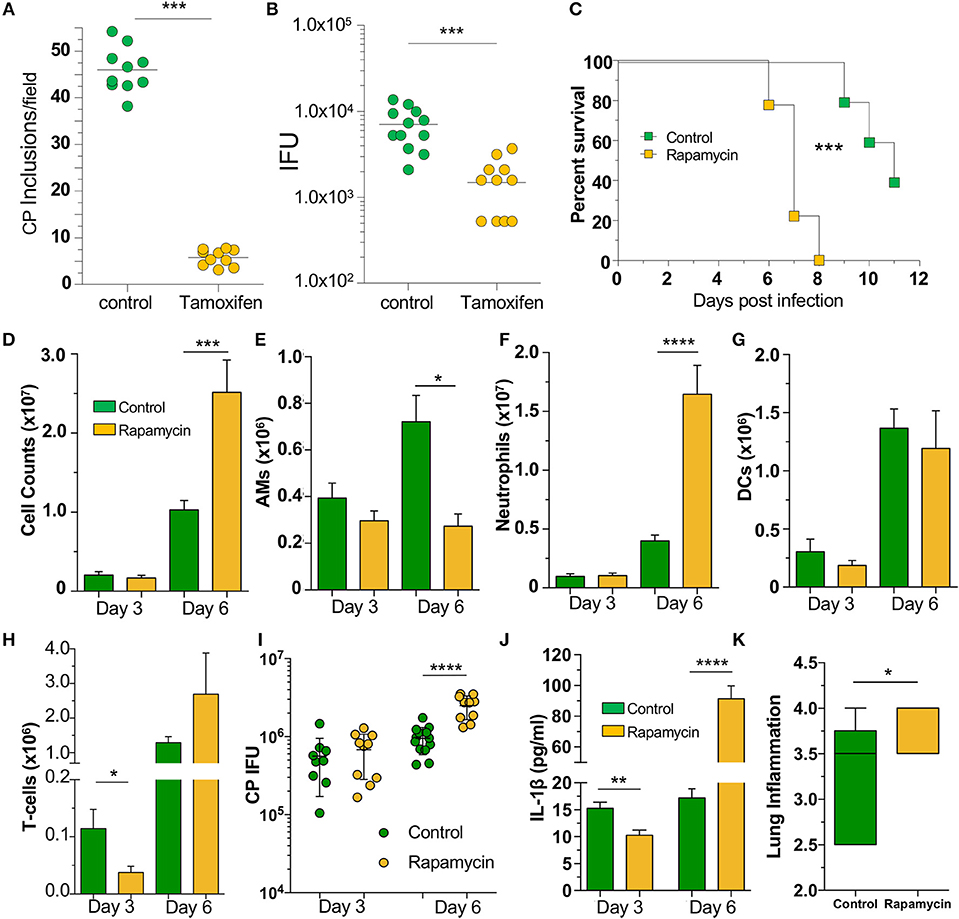
Figure 5. Autophagy induction fails to rescue mice from CP infection. WT MEFs were infected with CP (MOI 10) and treated with 10 μM Tamoxifen during the first 2 h of infection. (A) CP inclusions. (B) CP progeny. (C) C57Bl/6 mice were infected with 2 × 106 IFU CP. Six hour prior to infection, mice received either sham or 3 mg/kg rapamycin i.p., and again every other day. Mortality was assessed (n = 5–9). (D–J) C57Bl/6 mice were infected with 1 × 106 IFU CP. Six hour prior to infection, mice received either sham or 3 mg/kg rapamycin i.p., and again every other day (n = 8–12, pooled from two separate experiments). Mice were sacrificed on days 3, 6. (D) Total cells in the BALF. (E) BALF Alveolar macrophages. (F) BALF neutrophils. (G) BALF DCs. (H) BALF T-cells. (I) Bacterial burden in the lungs. (J) IL-1β in the BALF after CP infection and rapamycin treatment. (K) Lung inflammation score on day 6. Significance was assessed by students t-test. Survival curves was compared using the Log-Rank (Mantel-Cox) Test. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Our data investigating the role of autophagy during CP infection highlights the delicate balance in biological systems. This is not surprising considering the pathways investigated here and their relative importance toward immune responses. While our in vitro experiments demonstrate the key role that autophagy plays in controlling CP inclusion formation and bacterial growth, the overall balance in vivo is much more complex. Loss of autophagy in myeloid cells led to increased mortality during CP infection, but not due to increased bacterial burden. Instead, we observed a sustained increase in neutrophil recruitment to the lungs, with a corresponding increase in neutrophilic chemokines. Importantly, this was accompanied by an increase in inflammasome positive macrophages in the lung and greater IL-1β production. This sustained excess inflammation was the likely driver of excess mortality. While induction of autophagy in vitro limited CP growth, rapamycin treatment in vivo during CP infection increased mortality of WT mice, likely due to the reduction in early IL-1β production leading to an increase in bacterial burden, followed by increased IL-1β and neutrophil recruitment. IL-1β is a critical cytokine in controlling CP infection (33), yet can also be a powerful driver of pathology (36). Thus, since autophagy antagonizes inflammasome activation, any perturbation of these pathways during CP infection seems to result in a worsened outcome, creating a Goldilocks situation in which the level of autophagy and inflammasome activation must be perfectly balanced to eliminate the infection.
Common to both scenarios in which CP infected mice had reduced survival was an increase in neutrophil recruitment. CXCL1 and CXCL2 were both elevated in infected Atg16l1fl/fl Lysmcre mice, likely as a result of increased IL-1β signaling (37). Pulmonary neutrophilia is often associated with pathology and more recent studies have found that neutrophil extracellular traps can also lead to enhanced tissue damage (38). While we did not assess this in our model, this could be a mechanism by which pulmonary damage leads to decreased survival. We also observed an increase in DC recruitment to the lungs in CP infected Atg16l1fl/fl Lysmcre mice, but this did not lead to an increased T-cell presence in the lung.
The process of recycling cellular components and removal of damaged organelles has been found to play critical roles in a wide variety of diseases and cellular responses. Importantly, in the setting of bacterial infections, autophagy can be either beneficial and detrimental to the host. In multiple infections, such as S. typhimurium, Pseudomonas aeruginosa, or M. tuberculosis, loss of autophagy led to reduced bacterial clearance and subsequent enhanced pathology (13, 39, 40). In contrast, we showed that despite a clear role for autophagy in limiting CP growth in vitro, loss of autophagy in vivo did not impair bacterial clearance. This difference is likely due to the enhanced IL-1β production observed in CP-infected autophagy impaired mice, as CP is exquisitely sensitive to IL-1β (33).
While the development of novel antibiotics is one approach to combating the ever increasing number of resistant strains of bacteria, other mechanisms of enhancing bacterial clearance have also been investigated. Given that, in many cases, autophagy induction can help cells eliminate intracellular infections, several studies have investigated the antibacterial potential of autophagy induction. Supporting this effect, it has been shown that rapamycin given prophylactically in the drinking water before P. aeruginosa lung infection significantly reduced the bacterial burden (41), and that rapamycin treatment given i.p., reduced Leishmania parasite load (42). However, we found here that rapamycin treatment during CP infection actually increased CP burden and mortality. One possibility for these differing results may be the particular sensitivity of CP to IL-1β, such that the rapamycin-induced initial reduction in IL-1β resulted in enhanced CP overgrowth followed by subsequent immune-derived pathology. Perhaps a selective autophagy inducer that did not affect mitophagy and inflammasome activation might have a more beneficial outcome. Additionally, rapamycin is known to suppress T-cell responses and neutrophil extracellular traps (43, 44), which could also impair bacterial clearance in this model. Regardless, these investigations make clear that the antibacterial effects of autophagy induction are context- and pathogen-dependent.
Activation of the NLRP3 inflammasome involves damage to the mitochondria in the presence of NF-κB signaling (6, 45). Damaged mitochondria are sensed by various cellular pathways and mitophagy is induced to clear them. This, in effect, leads to a reduction in inflammasome activity, and thus the manipulation of mitophagy with inducers and inhibitors directly affects inflammasome activity and IL-1β production (7, 46). We observed that disrupted autophagy in macrophages led to enhanced IL-1β secretion by CP infected macrophages in vitro and more IL-1β found in BALF of CP-infected mice in vivo. ATG7 deficiency in mice has similarly been shown to lead to increased IL-1β production during P. aeruginosa infection (47). However, in contrast to CP infection, autophagy disruption resulted in increased bacterial CFU in that study, corroborating earlier work showing that loss of IL-1β leads to a reduction in bacterial burden and that enhanced inflammation is beneficial to P. aeruginosa growth in the lung (48). In contrast, the loss of autophagy may lead to excessive IL-1β production and enhanced pathology (36), as in our CP study.
In many instances, intracellular bacterial infections manipulate or require components of the autophagy machinery to successfully infect host cells. After invading host cells, Staphylococcus aureus grows in double membraned autophagosomes which do not fuse with the lysosomal machinery (20). Similarly, Yersinia pestis can prevent acidification of the autophagosome, thereby preventing xenophagy (16). In other instances, bacteria can utilize specific components of the autophagy machinery to form the intracellular niche in which they can replicate. For example, Brucella abortus requires ATG14L, Beclin 1, and ULK1 to form the vacuole where it replicates (49). However, while CP grows inside a protected inclusion, loss of either ATG16L1 or ATG5 did not affect CP growth and instead led to enhanced inclusion formation and CP replication. However, as ATG16L1 and ATG5 are part of the same elongation complex in autophagosome development, it is possible that other parts of the autophagy pathway are required for CP inclusion formation.
In this study we investigated the role of autophagy during a pulmonary CP infection. While impairing autophagy limited intracellular CP growth in vitro, in vivo studies demonstrated that loss of autophagy in myeloid cells increased mortality, likely due to the complex antagonistic interplay between the inflammasome and autophagy. All these data suggest that, at least for some infections such as CP, any perturbation to the delicate balance between inflammasome activation and IL-1β secretion can have negative results. However, given the highly distinctive nature of bacterial pathogens, understanding each infection separately is required to fully appreciate how modulating the system would affect the end result.
All experiments were conducted according to Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC) guidelines (IACUC# 8314).
TC conceived and led the project and wrote the manuscript. TC, RP, JD, and GT performed all of the experiments. AS and EP provided critical materials to this project. SC, KS, and MA provided critical editing and content to the manuscript as well as experimental design. MA supervised the study. All of the authors read and approved the final manuscript.
This work was supported by National Institutes of Health (NIH) grants 2R56AI067995-06 and AI117968 to MA and AI112826 to TC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Wenxuan Zhang, Ganghua Huang, Amanda Abholhsen, and Debbie Moreira for their expert technical skills.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00754/full#supplementary-material
1. Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. (2012) 13:7–12. doi: 10.1038/nrm3249
2. Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. (2013) 368:651–62. doi: 10.1056/NEJMra1205406
3. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. (2012) 441:523–40. doi: 10.1042/BJ20111451
4. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. (2016) 138:28–36. doi: 10.1016/j.jaci.2016.05.009
5. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. (2008) 456:264–8. doi: 10.1038/nature07383
6. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
7. Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
8. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
9. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
10. Storek KM, Monack DM. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev. (2015) 265:112–29. doi: 10.1111/imr.12289
11. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. (2014) 12:101–14. doi: 10.1038/nrmicro3160
12. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. (2006) 281:11374–83. doi: 10.1074/jbc.M509157200
13. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
14. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. (2004) 306:1037–40. doi: 10.1126/science.1103966
15. Lapaquette P, Bringer M-A, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. (2012) 14:791–807. doi: 10.1111/j.1462-5822.2012.01768.x
16. Pujol C, Klein KA, Romanov GA, Palmer LE, Cirota C, Zhao Z, et al. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun. (2009) 77:2251–61. doi: 10.1128/IAI.00068-09
17. Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. (2005) 307:727–31. doi: 10.1126/science.1106036
18. Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MAO, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA. (2009) 106:6226–31. doi: 10.1073/pnas.0811045106
19. Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE. (2011) 6:e17852. doi: 10.1371/journal.pone.0017852
20. Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. (2007) 282:2695–706. doi: 10.1074/jbc.M609784200
21. Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci USA. (2012) 109:20800–7. doi: 10.1073/pnas.1218674109
22. Kuo C-C, Jackson LA, Campbell LA, Grayston JT. Chlamydia pneumoniae (TWAR). Clin Microbiol Rev. (1995) 8:451–61. doi: 10.1128/CMR.8.4.451
23. Teh B, Grayson ML, Johnson PDR, Charles PGP. Doxycycline vs. macrolides in combination therapy for treatment of community-acquired pneumonia. Clin Microbiol Infect. (2012) 18:E71–3. doi: 10.1111/j.1469-0691.2011.03759.x
24. Porritt RA, Crother TR. Chlamydia pneumoniae infection and inflammatory diseases. For Immunopathol Dis Therap. (2016) 7:237–54. doi: 10.1615/ForumImmunDisTher.2017020161
25. Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL, Carabeo RA. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS ONE. (2011) 6:e16783. doi: 10.1371/journal.pone.0016783
26. Zhang H, Zheng L, McGovern DPB, Hamill AM, Ichikawa R, Kanazawa Y, et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. J Immunol. (2017) 198:2133–46. doi: 10.4049/jimmunol.1601293
27. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. (2004) 432:1032–6. doi: 10.1038/nature03029
28. Naiki Y, Michelsen KS, Schröder NWJ, Alsabeh R, Slepenkin A, Zhang W, et al. MyD88 is pivotal for the early inflammatory response and subsequent bacterial clearance and survival in a mouse model of Chlamydia pneumoniae pneumonia. J Biol Chem. (2005) 280:29242–9. doi: 10.1074/jbc.M503225200
29. Peterson EM, la Maza de LM, Brade L, Brade H. Characterization of a neutralizing monoclonal antibody directed at the lipopolysaccharide of Chlamydia pneumoniae. Infect Immun. (1998) 66:3848–55.
30. Jupelli M, Shimada K, Chiba N, Slepenkin A, Alsabeh R, Jones HD, et al. Chlamydia pneumoniae infection in mice induces chronic lung inflammation, iBALT formation, and fibrosis. PLoS ONE. (2013) 8:e77447. doi: 10.1371/journal.pone.0077447
31. Bah A, Vergne I. Macrophage autophagy and bacterial infections. Front Immunol. (2017) 8:1483. doi: 10.3389/fimmu.2017.01483
32. Watanabe-Asano T, Kuma A, Mizushima N. Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem Biophys Res Commun. (2014) 445:334–9. doi: 10.1016/j.bbrc.2014.01.180
33. Shimada K, Crother TR, Karlin J, Chen S, Chiba N, Ramanujan VK, et al. Caspase-1 dependent IL-1β secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PLoS ONE. (2011) 6:e21477. doi: 10.1371/journal.pone.0021477
34. Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. (2008) 7:2977–87. doi: 10.1158/1535-7163.MCT-08-0447
35. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DHA, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. (2011) 7:1359–70. doi: 10.4161/auto.7.11.17660
36. Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. (2012) 11:633–52. doi: 10.1038/nrd3800
37. Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, et al. The interleukin-1β/CXCL1/2/neutrophil axis mediates host protection against group B streptococcal infection. Infect Immun. (2014) 82:4508–17. doi: 10.1128/IAI.02104-14
38. Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. (2016) 7:311. doi: 10.3389/fimmu.2016.00311
39. Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. (2013) 145:1347–57. doi: 10.1053/j.gastro.2013.08.035
40. Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. (2012) 125:507–15. doi: 10.1242/jcs.094573
41. Junkins RD, Shen A, Rosen K, McCormick C, Lin T-J. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS ONE. (2013) 8:e72263. doi: 10.1371/journal.pone.0072263
42. Khadir F, Shaler CR, Oryan A, Rudak PT, Mazzuca DM, Taheri T, et al. Therapeutic control of leishmaniasis by inhibitors of the mammalian target of rapamycin. PLoS Negl Trop Dis. (2018) 12:e0006701. doi: 10.1371/journal.pntd.0006701
43. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood. (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
44. Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol. (1999) 162:2775–84.
45. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-J, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
46. Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. (2012) 13:255–63. doi: 10.1038/ni.2215
47. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol. (2017) 198:3205–13. doi: 10.4049/jimmunol.1601196
48. Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol. (2002) 282:L285–90. doi: 10.1152/ajplung.00461.2000
Keywords: autophagy, Chlamydia pneumoniae, inflammasome, macrophages, IL-1β
Citation: Crother TR, Porritt RA, Dagvadorj J, Tumurkhuu G, Slepenkin AV, Peterson EM, Chen S, Shimada K and Arditi M (2019) Autophagy Limits Inflammasome During Chlamydia pneumoniae Infection. Front. Immunol. 10:754. doi: 10.3389/fimmu.2019.00754
Received: 15 January 2019; Accepted: 21 March 2019;
Published: 12 April 2019.
Edited by:
Ken J. Ishii, University of Tokyo, JapanReviewed by:
Roberta Olmo Pinheiro, Fundação Oswaldo Cruz (Fiocruz), BrazilCopyright © 2019 Crother, Porritt, Dagvadorj, Tumurkhuu, Slepenkin, Peterson, Chen, Shimada and Arditi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moshe Arditi, bW9zaGUuYXJkaXRpQGNzaHMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.