
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 09 April 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00678
 Bernhard Kerscher1†
Bernhard Kerscher1† Jillian L. Barlow1‡
Jillian L. Barlow1‡ Batika M. Rana1‡Helen E. Jolin1
Batika M. Rana1‡Helen E. Jolin1 Mayuri Gogoi1
Mayuri Gogoi1 Michelle A. Bartholomew2Deepali Jhamb3Ashutosh Pandey3
Michelle A. Bartholomew2Deepali Jhamb3Ashutosh Pandey3 David F. Tough4
David F. Tough4 Antoon J. M. van Oosterhout2
Antoon J. M. van Oosterhout2 Andrew N. J. McKenzie1*
Andrew N. J. McKenzie1*Group 2 innate lymphoid cells (ILC2) increase in frequency in eczema and allergic asthma patients, and thus represent a new therapeutic target cell for type-2 immune-mediated disease. The bromodomain and extra-terminal (BET) protein family of epigenetic regulators are known to support the expression of cell cycle and pro-inflammatory genes during type-1 inflammation, but have not been evaluated in type-2 immune responses. We isolated human ILC2 and examined the capacity of the BET protein inhibitor, iBET151, to modulate human ILC2 activation following IL-33 stimulation. iBET151 profoundly blocked expression of genes critical for type-2 immunity, including type-2 cytokines, cell surface receptors and transcriptional regulators of ILC2 differentiation and activation. Furthermore, in vivo administration of iBET151 during experimental mouse models of allergic lung inflammation potently inhibited lung inflammation and airways resistance in response to cytokine or allergen exposure. Thus, iBET151 effectively prevents human ILC2 activation and dampens type-2 immune responses.
Innate lymphoid cells (ILCs) have emerged as strategic players in inflammation and immunity, and represent new therapeutic targets in disease (1, 2). ILCs have been divided into five subsets based on their development and function: NK cells, ILC1s, ILC2s, ILC3s, and LTi cells (3). ILC2s were first described as playing important roles in the initiation of inflammation at mucosal barrier surfaces in response to infection or tissue damage (1, 4). However, it is now apparent that ILC2s can play more complex roles throughout the duration of immune responses, participating in the transition from innate to adaptive immunity and contributing to chronic inflammation. The proximity of ILC2s to epithelial surfaces and their constitutive strategic positioning in other tissues throughout the body ensures that, in spite of their rarity, ILC2s are able to regulate immune homeostasis and respond rapidly to damage or infection.
ILC2s are key drivers of allergic disease and upon activation express very high levels of IL-5, IL-6, and IL-13, but also secrete many other cytokines, including IL-4, GM-CSF, IL-9, and IL-10. These type-2 cytokine-expressing ILCs express receptors for epithelium-derived cytokines such as IL-25, IL-33, and TSLP required for their activation, but also depend on signals from common gamma chain cytokines including IL-2 or IL-7. It is the combination of these factors and signals transmitted by cell surface receptors such as ICOS, PD-1, ICAM-1, MHCII, OX40L that provide micro-environmental cues that lead to subtly different ILC2 subtypes in different tissues during homeostasis and in response to the variety of inflammatory stimuli encountered (5–9). Interestingly, genome-wide association studies of asthma have identified RORα (a critical transcription factor for ILC2 development), as well as TSLP, IL-33 and the IL33-receptor (IL1RL1, ST2) as asthma susceptibility genes, supporting the importance of this pathway for asthma.
In mouse models, lung-resident ILC2s contributed to airway hyperreactivity, induced by viral or allergen challenge (10). Human ILC2s were reported in lung parenchyma and bronchoalveolar lavage (BAL) fluid, and defined by Spits and colleagues as lineage negative CD45hiCD127+CD117+CRTH2+CD161+ cells that were also present in peripheral blood, fetal gut and the inflamed nasal polyps of patients with rhino-sinusitis (11). These cells responded to epithelium-derived cytokines by producing large amounts of IL-5 and IL-13. ILC2s have been shown to increase in frequency in allergic asthma patients as compared to healthy controls (12–16), and it has been reported that exposure of ILC2s to TSLP leads to their becoming steroid resistant (14). ILC2s were also found to be resident in human skin, enriched in atopic dermatitis (eczema) lesions (17), and responsive to IL-33, IL-25, TSLP, and also to the CRTH2 ligand PGD2 (18). More recently it has been reported that ILC2 also retain a degree of plasticity and can produce IFNγ or IL-17A dependent on the local cytokine environment in inflamed tissues (19, 20).
Dysregulation of ILC2 function contributes to chronic pathologies such as allergy, autoimmunity and inflammation (10). Consequently, ILC2s represent a new opportunity for therapeutic intervention in diseases including allergy and asthma. To realize this potential a better understanding of mouse and human ILC populations and biology, and their tractability to prospective therapies, is required.
BET domain-containing protein family (Brd2, Brd3, Brd4, and Brdt) are epigenetic ‘reader’ proteins that play important roles in controlling expression of cell cycle and pro-inflammatory genes (21). BET proteins bind to acetylated lysine molecules via N-terminal bromodomains, with each family member possessing two of these domains. Cell signaling-induced acetylation of histone tails and transcription factors leads to BET protein localization at gene enhancers and promoters, where they act to regulate gene expression. BET-bromodomain inhibitors (e.g., iBET151) that block the acetyl-lysine binding pocket of the BET proteins have been demonstrated to be beneficial in a number of type-1 inflammatory disease models (22–24), but little is known about their ability to modulate allergic disease.
We examined the capacity of iBET151 to block the activation of human ILC2s following IL-33 stimulation. iBET151 profoundly blocked expression of a suite of genes that are critical for type-2 immunity, including type-2 cytokines, cell surface receptors, (including ICOS, CD40 ligand (CD40L/CD154), ICAM1, and chemokine receptors), and transcriptional regulators of ILC2 differentiation and activation. Furthermore, in vivo administration of iBET151 potently inhibited lung inflammation induced by cytokine or allergen exposure. Thus, iBET151 can block human ILC2 activation and dampen type-2 responses in experimental mouse models of allergic lung inflammation.
Wildtype C57BL/6 mice and Il33-citrine (Il33cit/+) reporter mice (25) were bred inhouse. All mice were maintained in the Medical Research Council (MRC) ARES animal facility under specific pathogen-free conditions.
Lyophilized iBET151 (GSK1210151A) was dissolved in DMSO to generate a 150 mM stock solution and stored at−20°C. For in vivo experiments, iBET stock solution was diluted in PBS (pH ~5.5) containing 10% kleptose.
Lymphocytes were enriched from 150 ml human peripheral blood using lymphoprep (Axis-Shield) according to the manufacturer's instructions. The collected PBMC fraction was washed with PBS/2% FCS followed by red blood cell lysis with ACK solution for 5 min at room temperature. To pre-enrich ILCs, cells were stained with biotinylated anti-CD3, CD14 and B220 in the presence of TruStain FcX (anti-CD16/CD32) followed by depletion with M280 streptavidin beads (Invitrogen). Subsequently, the enriched fraction was stained with CD45-AF488, CD127-PE-Cy7, CD117-PE, CRTH2-BV421, a lineage cocktail (APC-streptavidin, CD8a, CD11b, CD11c, FcεRIα, CD123, CD20, CD56, CD71) and Draq7-viability dye, followed by sorting of ILCs as indicated in Supplementary Figure S1 on an Influx FACS. Sorted ILC2s were cultured in RPMI (GIBCO) supplemented with 10% FCS, 100 μM 2-mercaptoethanol, 100 μg/ml penicillin, 100 IU/ml streptomycin, 20 mM HEPES and 10 ng/ml recombinant human IL-2, IL-7 and IL-33 (as indicated).
ILC2s designated as Draq7−(live cells)CD45+Lin−(streptavidin-APC, CD8a, CD11b, CD11c, FcεRIα, CD123, CD20, CD56, CD71)CD127+CRTH2+ cells, were purified by flow cytometry from the peripheral blood of 3 individual donors and cultured separately for 24 h with IL-2, IL-7, and with or without IL-33 (all at 10 ng/ml). 250 nM iBET151 was added to some wells as indicated. Cells were pelleted and RNA prepared for RNAseq analysis.
Draq7−(live cells)CD45+Lin−(streptavidin-APC, CD8a, CD11b, CD11c, FcεRIα, CD123, CD20, CD56, CD71)CD127+CRTH2+ ILC2s were isolated from the peripheral blood of 3 individual donors and cultured separately in cycling cultures. Purified human ILC2s were grown in media containing IL-2 and IL-7 (both at 10 ng/ml) with IL-33 (10 ng/ml) added to the cultures every alternate week. ILC2s were then rested for 3 days in IL-2 and IL-7-containing media (no IL-33) before being divided and added to the following culture conditions: IL-2/IL-7, +/−IL-33 (all at 10 ng/ml), +/− 125 nM iBET151 for 24 h. Each sample group only includes genes with an expression of >5RPKM.
Mouse ILC2s were purified from IL-33-treated lungs (see methods for IL-33-induced type 2 lung inflammation) as LiveCD45+Lineage−IL-7Rα+ICOS+ and cultured for 24 hrs with IL-7 (10 ng/ml) and with or without IL-33 (10 ng/ml). Mouse alveolar macrophages were purified from naïve lungs as LiveCD45+F4/80+ and cultured for 24 hrs with LPS (100 ng/ml). iBET151 was added to wells at 250, 125, or 62.5 nM. Golgi stop was added to the culture for the last 4 hrs and cells were then subjected to flow cytometric analysis.
Bronchoalveolar lavage (BAL) was collected by flushing the lungs with 1 ml PBS, retrieved volumes recorded, cells pelleted, and supernatants frozen at −20°C. Red blood cell lysis was performed on the pellet in ACK solution followed by staining for flow cytometry. Lungs were mechanically dissociated in RPMI-1640 (10 mM HEPES), and digested with collagenase I (500 U/ml, Life Technologies) and DNase I (0.2 mg/ml, Roche) at 37°C whilst shaking. Digested tissues were passed through a 70 μm cell strainer, connective tissue removed by centrifugation on a 25% Percoll gradient and red blood cells lysed in ACK solution for 5 min. Single cell suspensions were stained, washed and analyzed in PBS containing 2% FCS and 2 mM EDTA.
Cell suspensions were incubated with anti-mouse CD16/32 (BioXCell) to block Fc receptors and stained as indicated. Antibodies were purchased from eBioscience, Biolegend and BD and included: B220, CD3, CD4, CD5, CD8, CD11b, CD11c, CD19, CD24, CD45, CD127, Epcam, FcεR1α, GATA3, Gr1, ICOS, IL-13, IL-5, Ly6G, MHCII, NK1.1, SiglecF, and ST2. The lineage cocktail contained the antibodies CD3, CD5, CD11b, CD11c, CD19, F4/80, FcεR1α, Gr1, NK1.1, TCRβ, and Ter119, if not included separately in the panel. Live cells were identified using eFluor viability dyes (eBioscience) and cell numbers assessed with Precision Count Beads (Biolegend).
Cells were acquired on a BD Fortessa flow cytometry instrument and data analyzed using FlowJo (version 9.9). Myeloid cells were defined as eosinophils (SiglecF+, CD11c−), neutrophils (CD11b+, Ly6G+), alveolar macrophages (CD11c+, SiglecF+), interstitial macrophages (CD11b+, F4/80+), and DC (CD11c+, MHCII+). Lymphoid cells were defined as B-cells (CD3−, B220+), CD4/8-T-cells (CD3+, CD4+/CD8+), Th2-cells (CD3+, CD4+, GATA3+) and basophils (FcεR1α+). ILC2s were defined as Lin−, CD127+, and GATA3+ or ST2+ICOS+. Epithelial cells were defined with CD24 and Epcam in the CD45− fraction. Gating strategies are shown in Supplementary Figures S4 and Figure 4C.
For intracellular (IL-5, IL-13) and nuclear staining (GATA3), cells were processed following cell surface staining using the Foxp3/Transcription Factor Kit (eBioscience). For intracellular cytokine detection, cells were cultured for 4 h in the presence of 1x cell stimulation and protein transport inhibitor cocktail (eBioscience) in RPMI-1640 culture media supplemented with 10 mM HEPES, 10% FCS, 100 mM 2-mercaptoethanol, 100 mg/ml penicillin, 100 IU/ml streptomycin. Flow cytometry analysis was performed as described above.
Cells from stimulation experiments were harvested by centrifugation, transferred to Trizol (Life Technologies) and stored at −80°C until further processing. RNA was extracted from the aqueous trizol/chloroform phase using the RNeasy Mini kit (Qiagen) followed by off-column DNA digestion using Turbo DNase (Ambion). DNA-free RNA was cleaned and concentrated using an RNeasy Micro Kit (Qiagen) and assessed using a Bioanalyser (Agilent). RNA was processed for RNA sequencing using Ovation RNA-seq System V2 (Nugen), fragmented using the Covaris M220 and barcoded using Ovation Ultralow Library Systems (Nugen). Barcoded libraries were quantified using Qubit dsDNA BR assay (Invitrogen) and qPCR. Samples were sequenced using an Illumina Hiseq4000 (CRUK Cambridge Institute), and sequence data were aligned using Tophat2 with Partek Flow. RNA-seq analysis was performed using Partek® Genomics Suite® software, version 6.16 Copyright ©; 2016.
Mice were treated intraperitoneally with either vehicle (10% kleptose) or iBET151 (10, 5, or 1 mg/kg) and after 1 h challenged intranasally with LPS (10 μg in 40 μl PBS) or PBS alone. Mice were weighed regularly before being sacrificed 48 h after LPS administration.
Mice were treated intraperitoneally with either vehicle (10% kleptose) or iBET151 (10, 5, or 1 mg/kg) and challenged intranasally with IL-33 (0.2 μg in 40 μl PBS) on days 0 and 3 or PBS alone. All mice were sacrificed on day 6.
Mice were treated with two intranasal doses of papain (25 μg on day 0 and day 1) or PBS in the presence or absence of iBET151 (10 mg/kg) or kleptose vehicle given daily for six days intraperitoneally.
For the acute ragweed model, mice received either PBS alone or 100–300 μg of short ragweed (Ambrosia artemisiifolia) pollen extract (Greer Laboratories) intranasally, daily for 5 days, with or without iBET151 (10 mg/kg) treatment, and samples were taken on day 6. For the chronic lung inflammation model, mice received RWP, as above, administered intranasally three times per week over a 5-week period, and were sacrificed on day 38. iBET151 (10 mg/kg) or vehicle was given intraperitoneally for the final 10 days of the model.
Twenty-four hours after the final antigen challenge airways resistance was assessed using a restrained whole-body plethysmograph (EMMS, Hants, United Kingdom) as described previously (26). All baseline readings were found to be similar between experimental groups, and thus average pulmonary resistance after drug treatment was then divided by the baseline average for each mouse to determine fold induction. eDaq software was used to analyze airways resistance.
Supernatants were harvested 1 day after IL-33 restimulation for cytokine analysis by Magpix cytometric bead array according to the manufacturer's instructions. IL-6 in bronchoalveolar lavage fluid was determined using the mouse IL-6 Ready-SET-Go! ELISA kit according to the manufacturer's instructions.
Lung lobes were fixed in 10% formaldehyde in PBS overnight, dehydrated in 70% ethanol, and stored in PBS at 4°C until analysis. The CRUK Cambridge Institute Histology Core performed tissue embedding, sectioning and staining.
Graphpad Prism software was used for all statistical analysis. Significance levels are expressed as * <0.5, ** <0.1, *** <0.01 and **** <0.001. RNA-seq analysis was performed using Partek® Genomics Suite® software, version 6.16 Copyright ©; 2016. The threshold for statistical significance for the ex vivo stimulated ILC2 cultures was set at a differential gene expression of >2-fold (unadjusted p-value). The threshold for differential gene expression for the in vitro cultured ILC2 assays was based on Il13 expression (decreased by 1.7 fold), since IL-13 was shown by intracellular cytokine analysis and Magpix, to be decreased by iBET151 in the in vitro assays described above. Therefore, the threshold statistical significance was set at a differential gene expression of >1.7-fold (unadjusted p-value).
To evaluate the potential of iBET151 to modulate human (h)ILC2 function we purified CD45+Lin−(CD8a, CD11b, CD11c, FcεRIα, CD123, CD20, CD56, CD71)CD127+CRTH2+ ILC2s from the peripheral blood of healthy donors (Supplementary Figure S1). To replicate the initial interaction with an epithelium-derived alarmin, purified ILC2s were cultured immediately with IL-2, IL-7, and IL-33 in the presence or absence of iBET151 for 24 h and then subject to RNA sequencing (iBET ex vivo ILC2s, Figure 1A). This analysis identified 270 statistically significant differentially expressed genes (>2.0-fold, unadjusted p-value). Within this subset, iBET treatment resulted in lower expression of 236 genes and gene ontology (GO) analysis indicated a further subset of 56 genes were involved in immune system processes (Figures 1B–D and Supplementary Figure S2A). Seven of the 34 genes, which were higher in iBET treated samples were also immune-related (Supplementary Figure S2B).
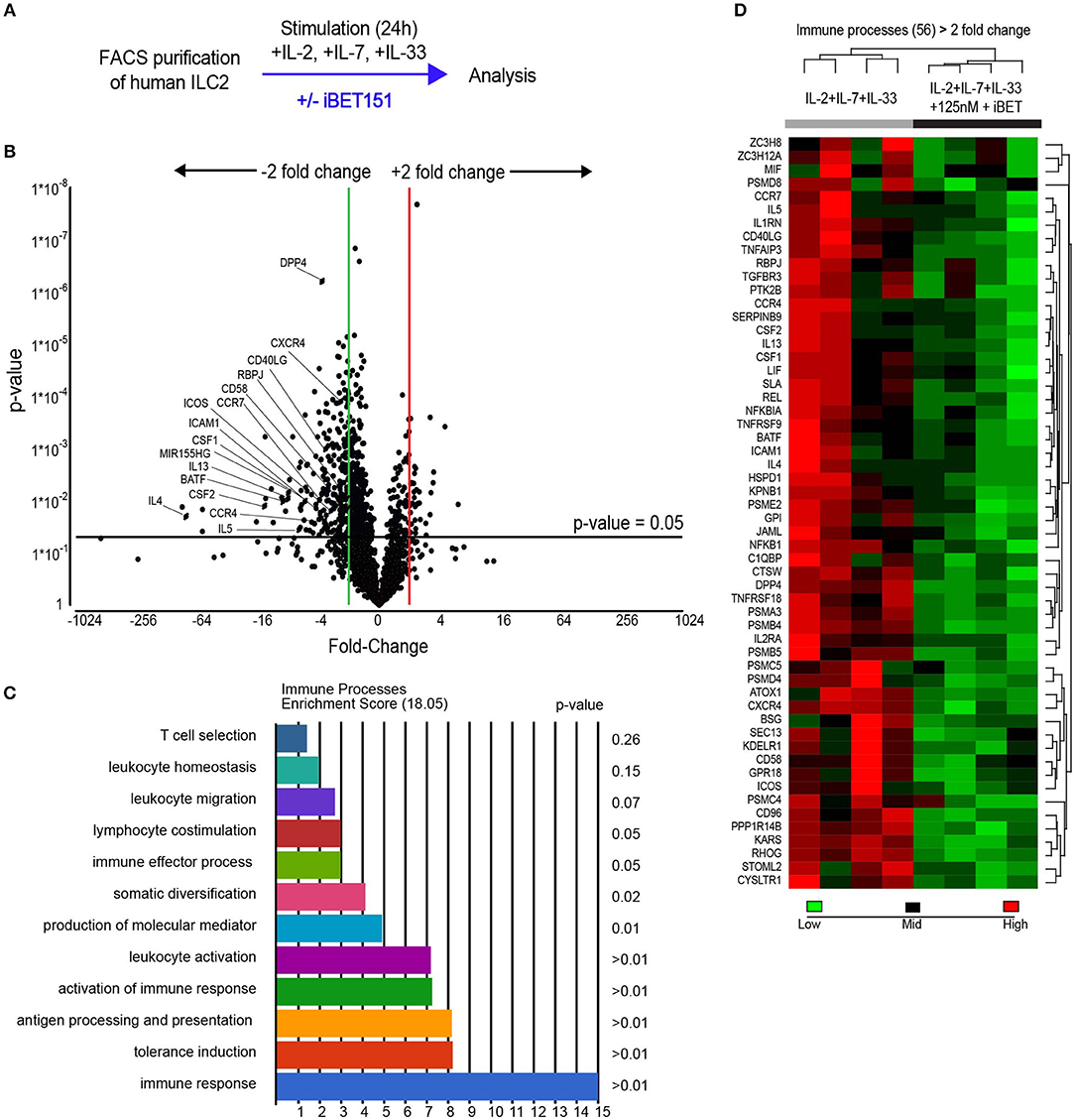
Figure 1. RNAseq analysis of iBET151-modulated genes expressed by ex vivo human ILC2s. (A) Method for stimulating hILC2s ex vivo with IL-33 and iBET151. (B) Volcano plot of genes differentially expressed between hILC2s stimulated with IL-33 alone or with IL-33 and 125 nM iBET151 (>2.0 fold change). (C) Gene ontology summary of immune system process genes lowered by iBET151. (D) Heat map of immune system process genes from gene ontology analysis in (C). Data from four individual donors processed independently before library preparation.
A characteristic feature of ILC2s is their copious expression of type-2 effector cytokines. Notably, treatment of ILC2s with iBET151 potently inhibited the transcription of the genes encoding IL-4, IL-5, and IL-13, as well as GM-CSF and CSF1 (Figures 1B,D). In addition, a number of cell surface receptors that are known to regulate ILC2 function were also decreased by iBET151. These included ICOS, CD40LG (CD154), ICAM1, and also CD58, a ligand for CD2, CD26 (DPP4), and receptors for lipid mediators from the eicosanoid pathway, for example CYSLTR1 (Figures 1B,D). ILC2 expression of the chemokine receptors CCR4, CXCR4, CCR7 that are known regulators of lymphocyte migration were also lower following iBET treatment (Figures 1B,D). The screen also identified the inhibition of RBPJ, a critical transcription factor in Notch signaling and ILC2 differentiation; BATF, a component of the BATF–AP1–IRF4 complex that regulates the type-2 cytokine locus in Th2 cells; and microRNA-155 host gene (HG) that is processed to give miR-155, which has been reported to support mouse ILC2 function (Figures 1B,D).
To mimic chronic and highly activated type-2 inflammation, purified hILC2s were cultured in media containing IL-2 and IL-7 with IL-33 added to the cultures in cycles every alternate week (Figure 2A). After a period of expansion, the ILC2s were rested for 3 days in IL-2 and IL-7-containing media before being stimulated with IL-33 in the presence or absence of 125 nM iBET151 for 24 h. After 24 h, intracellular cytokine analysis was performed to detect the type-2 cytokines IL-13 and IL-5 in ILC2s. Inclusion of iBET151 in the cultures inhibited the production of IL-13 (Figure 2B), and to a lesser extent IL-5, following IL-33 stimulation (Figure 2C). Furthermore, analysis of culture supernatants indicated that type-2 cytokines (IL-4, IL-5, and IL-13) were inhibited by the presence of iBET151, as was GM-CSF (Figure 2D). Gene expression analysis using RNAseq was performed on the cultured ILC2s. In order to focus our analysis on functionally relevant genes we used the change in Il13 expression (1.7 fold decreased) to set our threshold for genes that were lower following iBET treatment. This was based on intracellular cytokine analysis and Magpix, showing IL-13 to be decreased by iBET151 in the in vitro assays described above. Therefore, the threshold statistical significance was set at a differential gene expression of >1.7-fold (unadjusted p-value). We identified 204 genes with statistically significant differential expression between ILC2s stimulated in the presence or absence of iBET (Figure 2E and Supplementary Figure S3A). Within this subset, iBET treatment was found to lower the expression of 188 genes and GO analysis indicated 58 genes were involved in immune system processes (Figures 2F,G and Supplementary Figures S3A,B). A further subset of four of the 16 genes that showed iBET-dependent increases were immune system-associated (Supplementary Figures S3A,C).

Figure 2. Analysis of iBET151-modulated proteins and genes expressed by in vitro-cultured human ILC2s. (A) Method for stimulating hILC2s in vitro with IL-33 and iBET151. Intracellular flow cytometry detection of (B), IL-13 and (C), IL-5 protein expression in hILC2 cultures in the absence or presence of iBET151. (D) Cytokine analysis of culture supernatant taken from in vitro expanded hILC2s following IL-33 restimulation for 24 h. (E) Volcano plot of genes differentially expressed between hILC2s stimulated with IL-33 alone or with IL-33 and 125 nM iBET151 (>1.7 fold change). (F) Gene ontology summary of immune system process genes lowered by iBET151. (G) Heat map of immune system process genes from gene ontology analysis in (F). Data include three individual donors, processed independently before restimulation and library preparation. (H) Venn diagram of common and distinct genes from Figure 1C and Figure 2F.
Although several of the genes that were reduced by iBET151 in the long-term cultured ILC2s were in common with those from the 24-h ex vivo ILC2 cultures, there were a number of interesting additional targets (Figure 2H). These included Neuromedin U receptor (NMUR1, though at relatively low amounts), IL-9R and CRTH2 (prostaglandin D2 receptor); antigen processing and presentation genes (proteasome complex genes, CD74); chemokines and chemokine receptors (CCL3 (MIP1α), CCL4 (MIP1β), CCL5, CCR3, CCR5, CXCR3) (Figure 2E and Supplementary Figure S3). Interestingly, and mirroring recent reports on ILC plasticity, we observed increases in the transcription of NK/ILC3-like molecules in the long-term cultures. For example, Cathepsin (CTSW), Granzymes A and B (GZMA/B), and IL-22 (though at relatively low amounts) were increased relative to the ex vivo ILC2s. These molecules were inhibited by iBET151 treatment.
Taken together these results demonstrate that iBET151 is a potent inhibitor of type-2 responses in ILC2s, capable of repressing the expression of the signature type-2 cytokines, receptor-mediated ILC2 activation, and chemokine-regulated cell migration pathways.
BET inhibitors have been reported previously to be beneficial in type-1 inflammatory disease models (22, 24). To assess the ability of iBET151 to inhibit type-1 responses in the lung we administered LPS intranasally to mice to induce airways inflammation 1 h after intraperitoneal administration of iBET151 (Figure 3A). As expected, LPS treatment resulted in pronounced (~15%) weight loss within 2 days as compared to controls (Figure 3B). However, mice pre-treated with 10 mg/kg iBET151 were protected from LPS-induced weight loss (Figure 3B). This protective effect was maintained when iBET151 was given at 5 mg/kg, but the effect was lost at a dose of 1 mg/kg. iBET treatment at all doses (10, 5, and 1 mg/kg) was also associated with reduced infiltration of CD45+ cells into the bronchoalveolar lavage, primarily attributable to a fall in neutrophil (Ly6G+CD11b+) numbers (Figures 3C,D). Treatment of mice with 10 mg/kg iBET151 also reduced production of the pro-inflammatory cytokine IL-6 (Figure 3E). However, ILC2s (Figure 3F), CD103+ DCs, eosinophils (SiglecF+CD11c−) and alveolar macrophages (SiglecF+CD11c+) were not altered (data not shown). Furthermore, iBET151 treatment of in vitro cultured LPS-stimulated alveolar macrophages resulted in a significant decrease in IL-6 production at 250 nM of iBET151, but not at 125 or 62.5 nM (Figure 3G). Taken together these results indicate that iBET151 protected mice from LPS-induced weight loss, inflammatory cell infiltration and inflammatory cytokine production.
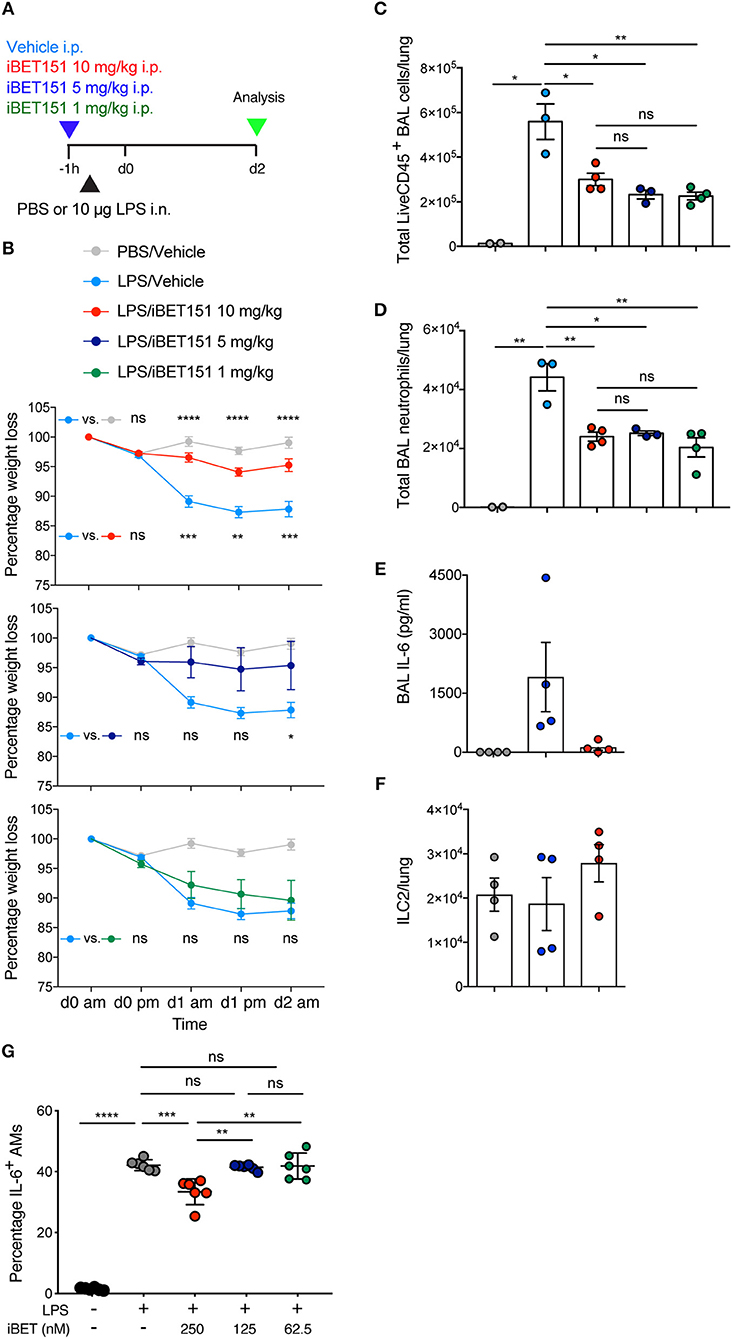
Figure 3. iBET-151 inhibits LPS-induced lung inflammation. (A) Model of LPS intranasal administration used to investigate the effects of iBET151. (B) Percentage weight loss, (C) total live CD45+ BAL cells, (D) total BAL neutrophils (liveCD45+SiglecF−Ly6G+F4/80−) of mice following treatment with LPS and vehicle (light blue circles), or LPS and iBET151 at 10 mg/kg (red circles), 5 mg/kg (dark blue circles), or 1 mg/kg (green circles). (E) IL-6 production in BAL fluid and (F), total numbers of lung ILC2s following LPS administration and iBET151 treatment at 10 mg/kg. iBET151 10 mg/kg data are pooled from three repeat experiments, with iBET151 titration data being representative of one additional experiment. (G) Percentage of IL-6+ alveolar macrophages (AMs) following stimulation with LPS (100 ng/ml) and either 250, 125, or 62.5 nM iBET151 for 24 h.
Papain is a cysteine protease that when administered intranasally to mice leads to lung damage and results in type-2 inflammation. This has been shown to be highly dependent on IL-33 (27). Mice were treated with intranasal papain in the presence or absence of iBET151 and lung inflammation was assessed 5 days after the final administration of papain (Figure 4A). At this timepoint iBET151 inhibited eosinophil accumulation in the lungs (Figure 4B), but we could not detect changes in ILC2 or T cell numbers (data not shown). We speculated whether this result may be due to iBET151 blocking the expression of Il33 by the epithelial cells, thereby curtailing the response at initiation, or by directly altering the cell-intrinsic properties of the inflammatory cells. To address this question, we used Il33-citrine (Il33cit/+) reporter mice in which the citrine fluorescent protein is expressed from the start site of the Il33 gene providing a surrogate marker for Il33 transcription. Il33-citrine mice were challenged intranasally with papain in the presence or absence of iBET151 and the proportions of Il33-citrine-positive cells determined after 6 days. Although papain induced Il33-citrine expression in CD45−CD24−Epcam+ epithelial cells this was unaltered by co-treatment of the mice with iBET151 (Figures 4C–E). These data indicate that whilst iBET151 can inhibit type-2 lung inflammation in a papain-induced model this is not due to altered transcription of the Il33 locus in epithelial cells.
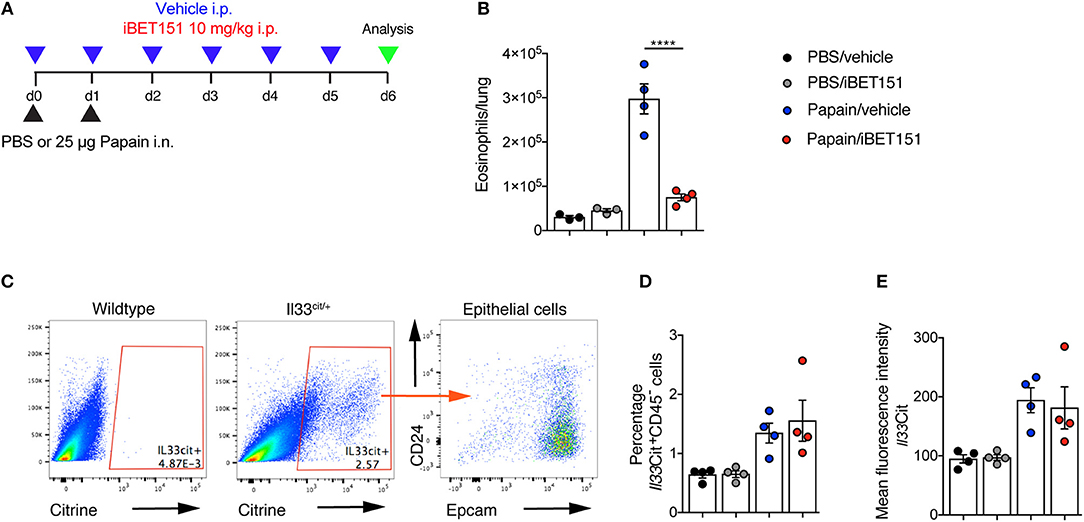
Figure 4. iBET inhibits papain-induced eosinophilia in the lung in an IL-33-independent manner. (A) Model of papain-induced lung inflammation used to investigate the effects of iBET151. (B) Total number of eosinophils in the lung following treatment with papain and vehicle (blue circles) or papain and iBET151 at 10 mg/kg (red circles). (C) Gating strategy for the identification of Il33-citrine+ lung epithelial cells, (D) percentage, and (E) mean fluorescence intensity of Il33-citrine+ cells in the lungs following intranasal papain challenge in the presence or absence of iBET-151. Data are representative of two repeat experiments.
IL-33 is an epithelium-derived “alarmin” cytokine that potently induces type-2 immune reactions. To determine if the iBET151 suppression of allergen-induced type 2 lung inflammation was up- or down-stream of IL-33, we took the approach of treating mice with IL-33 in the presence or absence of iBET151 at 10 mg/kg (Figure 5A). Intranasal administration of IL-33, with intraperitoneal treatment of vehicle, resulted in an increase of IL-13-producing ILC2s (Figures 5B–D), GATA3+CD4+ Th2 cells (Figure 5E), and eosinophils (Figure 5F) by day 6 post-treatment. Notably, daily administration of iBET151 completely prevented all facets of IL-33-induced type-2 immunity measured, including inhibiting ILC2 numbers and their expression of IL-13, blocking Th2 cells and preventing lung eosinophilia (Figures 5B–F). Titration of iBET151 to 5 mg/kg, but not 1 mg/kg, also caused significant decreases in type 2 inflammation (Supplementary Figure S5). However, titration of iBET151 to a dose of 1 mg/kg still resulted in a statistically significant reduction in lung ILC2 percentage (Supplementary Figure S5A). Furthermore, in vitro culture of IL-33-induced ILC2s with decreasing doses of iBET151 (250, 125, and 62.5 nM) also showed a dose dependent effect on IL-13 production (Supplementary Figure S5E). However, iBET151 showed no ability to effect changes in ILC2 viability (live/dead staining) or proliferation (Ki67 expression, data not shown). Thus, iBET151 is a potent inhibitor of IL-33-induced inflammation in vivo.
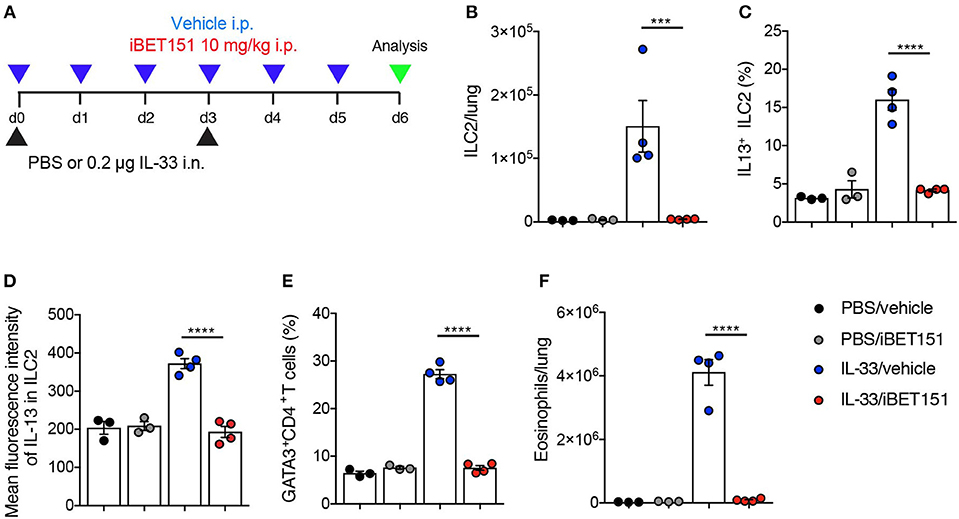
Figure 5. iBET inhibits IL-33-induced type-2 inflammation in the lungs. (A) Model of IL-33-induced lung inflammation used to investigate the effects of iBET151. (B) Total number of lung ILC2s, (C) percentage of IL-13+ ILC2s, (D) mean fluorescence intensity of IL-13 expression by ILC2s, (E) percentage of lung Th2 cell number (GATA3+CD4+) and (F) total number of eosinophils in the lung following IL-33 intranasal challenge and vehicle (blue circles), or IL-33 and iBET151 at 10 mg/kg (red circles). Data are representative of three repeat experiments.
Next, we wished to assess the ability of iBET151 to modulate type-2 immunity induced in response to a clinically relevant allergen (Figure 6A). Ragweed pollen (RWP) is one of the most common causes of allergies, especially in the United States. As expected, RWP challenge over 6 days resulted in increased numbers of ILC2s (Figure 6B), CD4+ T cells (Figure 6C) and eosinophils (Figure 6D) in the lungs, and IL-13 (Figure 6E) in the BAL. Notably, co-administration of iBET151 completely inhibited this inflammatory response with lung ILC2s, CD4+ T cells, eosinophils and BAL IL-13 remaining at baseline (Figures 6B–E). Thus, iBET151 has a potent inhibitory effect on acute type-2 inflammation in the lungs.
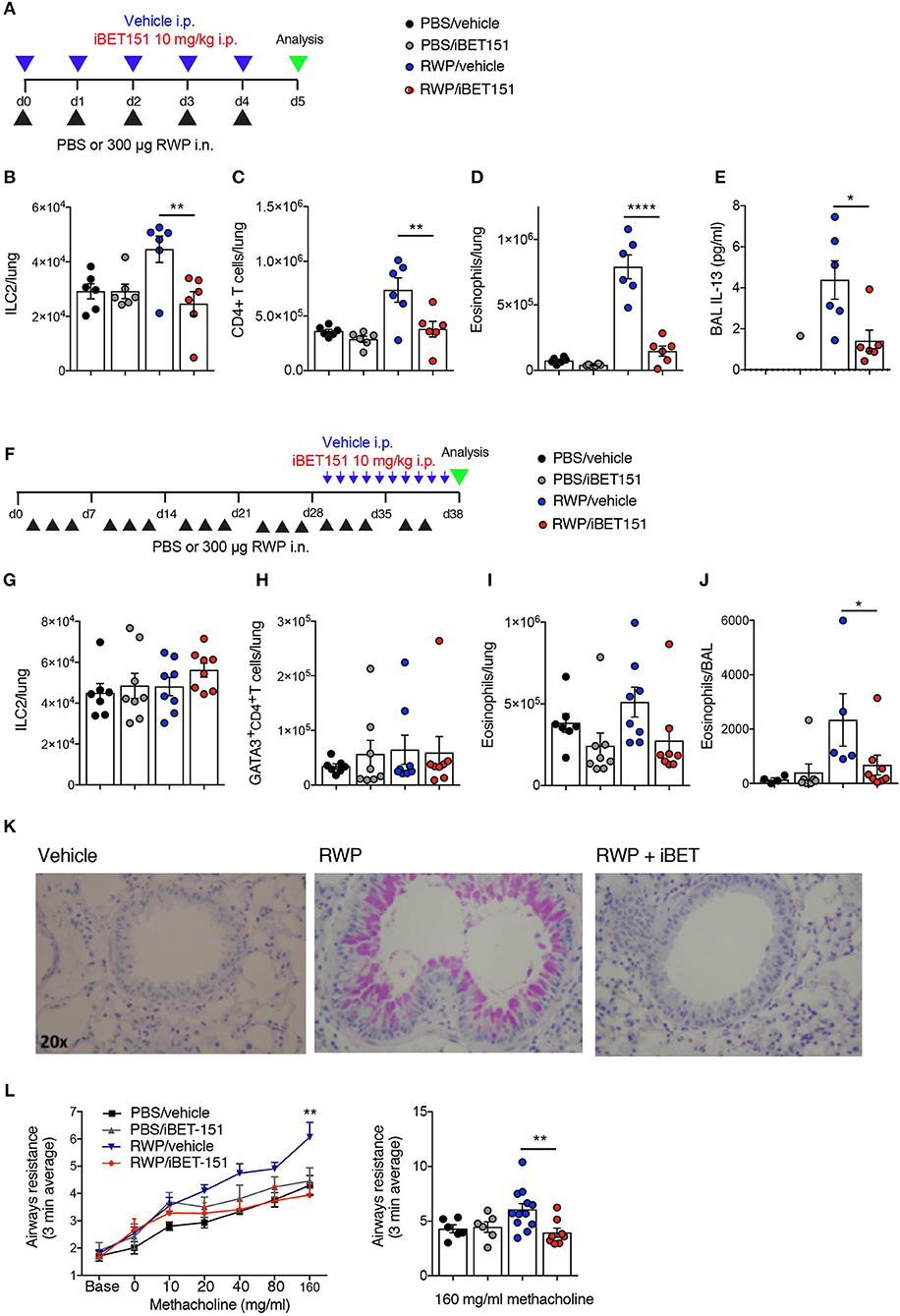
Figure 6. iBET reduces type-2 allergic inflammation in acute and chronic ragweed protein (RWP) challenge models. (A) Model of RWP-induced acute lung inflammation used to investigate the effects of iBET151. (B–D) Total number of immune cells in the lung, and (E) IL-13 in the BAL fluid of mice challenged with intranasal RWP for 5 consecutive days in the presence or absence of iBET-151. (F) Model of RWP-induced chronic lung inflammation used to investigate the effects of iBET151. Total number of immune cells (G–I) in the lung, and (J) in the BAL fluid of mice challenged with intranasal RWP three times per week over a 5-week period in the presence or absence of iBET-151 given intraperitoneally for the final 10 days of the model. (K) Lung histology and (L) airways resistance measurements from mice described in (G–J). Data are representative of two repeat experiments.
We have previously established a prolonged lung inflammation model in which RWP is administered intranasally over a 5-week period (Figure 6F). Following RWP challenge, flow cytometry and lung histology confirmed the existence of lung pathology, characterized by eosinophilia and goblet cell hyperplasia (Figures 6I–K). When iBET151 was given intraperitoneally for the final 10 days of the model it inhibited allergen-induced eosinophilia and mucus production (Figures 6I–K). We were unable to detect changes in ILC2s or Th2 cell numbers in this model in the presence or absence of RWP or iBET151 (Figures 6G,H). However, increases in methacholine-induced airways hyperreactivity in RWP challenged mice were blocked by daily administration of iBET151 during the final 10 days of allergen challenge (Figures 6L).
Together, these data demonstrate that iBET151 is a potent inhibitor of both acute and established type-2 immune mediated lung inflammation.
We have assessed the effects of the bromodomain inhibitor iBET151 on human ILC2 gene expression in response to activation in the presence of IL-33. Gene expression analysis using RNA-sequencing identified that iBET151 was a potent inhibitor of several immune regulatory molecules that have been shown to be important for ILC2-mediated immune activation. Critically, this included the type-2 effector cytokines IL-4, IL-5, and IL-13, as well as GM-CSF and CSF1. The roles for IL-4, IL-5, IL-13, and GM-CSF in allergic diseases are well established (28), and it is clear that iBET151 has the potential to suppress these responses in ILC2. Although there is currently no report of a role for CSF1 relating to human or mouse ILC2s, airway epithelial cell-derived CSF1 was reported recently to activate cDC2 and promote allergen sensitization (29).
To complement the in vitro analysis of human ILC2 we performed additional studies to assess the efficacy of iBET151 in inhibiting type-2 immune-mediated inflammation in mouse lungs. Strikingly, iBET151 completely blocked all IL-33-induced responses in the lungs following intranasal challenge. iBET151 treatment also successfully prevented type-2 responses induced by more complex allergens, including papain and RWP extract. To determine if iBET151 was acting directly on the epithelial cells to preclude the transcriptional activation of the Il33 gene we examined the expression of citrine in epithelial cells derived from Il33-citrine reporter mice. Interestingly, citrine levels were not altered by iBET151 treatment, suggesting that iBET151 inhibition of the type-2 response acts downstream of Il33 gene expression by altering gene expression in IL-33-responsive cells such as ILC2. As shown previously, and herein, ILC2 are key IL-33-responsive cells that help drive IL-5 and IL-13 levels in acute type-2 responses. Indeed, in the case of the type-2 response to RWP, administration of iBET151 correlated with fewer ILC2 and fewer CD4+ T cells. Furthermore, in a long-term model of lung inflammation induced by RWP, iBET151 treatment administered over only the last 10 days of the allergen challenge model inhibited mucus production, eosinophil infiltration and airway hyperreactivity, though we could not detect changes in ILC2s or T cells. Taken together these data demonstrate that iBET151 has potent inhibitory effects on ILC2s in type-2 immunity and successfully counteracts lung inflammation. Also, the results may suggest alternative ILC2- and T cell-proliferation independent pathways for iBET151 inhibition of chronic eosinophilia and mucus production. This could perhaps be explained by a direct effect of iBET151 on the epithelium (30), or by changes in type-2 cytokine production by ILC2s and T cells, though this was not measured in our studies.
In addition to effector cytokines, iBET151 inhibited the expression of numerous other genes in ILC2s that may contribute to the efficacy observed in in vivo models. This includes a number of cell surface receptors with known roles as ILC2 or lymphocyte activators, including ICOS, CD40L and ICAM1. ICOS expression was first reported on mouse ILC2 (4, 31), and ICOS:ICOS ligand (ICOSL) signaling has since been shown to be important for ILC2 function, including their role in the induction of airway hyperreactivity (7). Additionally, recent evidence indicates that induced Tregs (iTregs) can suppress the production of ILC2-derived IL-5 and IL-13 via the ICOS-ICOSL pathway (32, 33). CD40 ligand was reported recently to be induced on ILC2s in response to stimulation with phorbol ester and ionomycin, IL-25 or IL-33, or a number of TLR agonists. Notably, following the induction of CD40L, these ILC2s induced immunoglobulin production, including IgE, by B cells (34). Furthermore, ICAM1-deficient ILC2s were reported to produce diminished levels of type-2 cytokines in response to IL-33 stimulation, and ICAM1-deficient mice developed less severe airway inflammation following lung challenge with Alternaria alternata fungal extract or papain (6).
Although there are currently no reports of roles for CD58, CD26/DPP4, or CD96 in human or mouse ILC2-mediated responses, these molecules are expressed on ILC2 and inhibited by iBET151. CD58 is a ligand for CD2 and antibodies binding to CD2 resulted in increased T cell production of IL-4 (35), suggesting that modulating CD58 expression on ILC2s may have consequences for interactions between ILC2 and Th2 cell and subsequently for cytokine production. Interestingly, CD26 (DPP4) is a dipeptidyl peptidase-4 known to play roles in T cell homeostasis and activation, including the interaction of APCs with T cells. Many neuropeptides, hormones, growth factors, and chemokines have the X-Pro or -Ala motif at their N-terminus and have been shown to be cleaved by DPP4, leading to their enhanced degradation by further peptidases. In addition, DPP4 has also been reported to bind several cell surface receptors (e.g., Caveolin on APCs) to regulate various physiological and immune responses. Although soluble CD26/DPP4 has been reported to increase in asthmatics, its contribution is still contentious (36).
In contrast to the extensive number of activation molecules that were inhibited by iBET151 we also noted that CD96, an inhibitory receptor for CD155 was also lower (37). CD96 is expressed by CD4+ and CD8+ T cells, and NK cells in humans and mice and is a member of the nectin-like protein family. The absence of CD96-signaling in mice resulted in improved resistance to melanoma metastasis (38), and it will be interesting to discover how expression of this molecule specifically on ILC2s may regulate immunity.
We also identified that in ILC2s iBET151 repressed the expression of pathways that are known to regulate lymphocyte migration, including the chemokine receptors CCR4, CXCR4, and CCR7. CCR4 has been associated with asthma (39), and CCR4-deficient mice display reduced airway hyperresponsiveness following chronic lung allergen challenge (40). Furthermore, a CCR4 antagonist attenuated allergic lung inflammation in an experimental mouse model (41). Although impaired migration of ILC2s may be related to these observations, an effect on CCR4+ Th2 migration cannot be excluded. CXCR4 binds stromal-derived-factor-1 (SDF-1 also called CXCL12), a potent chemotactic molecule for lymphocytes, and a CXCR4 inhibitor has been reported to attenuate allergen-induced lung inflammation (42). CCR7 is also involved in homing of T cells to secondary lymphoid tissues including the lymph nodes and spleen. It is expressed on mouse ILC1 and ILC3, but only at low levels on ILC2 (43). The potential roles of these chemokines in human ILC2 function requires further investigation.
The expression of bioactive lipid products by ILC2 is also inhibited by iBET151. Arachidonic acid metabolism leads to the production of either prostaglandins via the cyclooxygenase (COX) pathway, or leukotrienes through the 5-lipooxygenase (5LO) pathway (44). Our data indicate that ILC2 expression of the receptor for cysteinyl leukotrienes, CYSLTR1, was lowered by iBET151. CysLTs have been reported to potentiate type-2 cytokine production from ILC2s stimulated with IL-33 and CYSLTR-deficient mice display reduced ILC2 responses following exposure to A. alternata allergen (45).
A number of notable transcription factors were also reduced by treatment of ILC2 with iBET151, including RBPJ, BATF, and EGR2. RBPJ is a transcriptional regulator critical for Notch signaling and has been shown to be essential for mouse and human ILC2 development (31, 46) and plasticity (47). BATF–AP1–IRF4 complexes regulate the type-2 cytokine locus in Th2 cells to induce IL-4 that feeds back to induce Th2 cell differentiation and amplify the subsequent Th2 cell immune response (48–52). Recently, BACH2 has been shown to antagonize this feedback loop by disrupting BATF–AP1–IRF4 complex binding to AICE motifs in the type 2 cytokine locus (50). It has been reported that BATF transcription is significantly higher in the PBMC of children with recurrent asthma who have not been treated with corticosteroid (53). In addition, Batf-deficient mice were less susceptible to experimental allergic lung inflammation. Though there is currently no report of a role for BATF in human or mouse ILC2s it is highly likely that this pathway will be important for ILC2-derived type-2 cytokine production. The transcription factor EGR2 (early growth response 2) suppresses excessive immune activation and Egr2-deficient T cells are hyperproliferative (54). There is currently no report of a role for EGR2 in human or mouse ILC2s. Furthermore, a recent report demonstrates that in cat dander extract-induced lung inflammation, bromodomain-containing protein 4 (Brd4) and transcription factor NF-κB/RELA interaction is crucial. Inhibition of Brd4 confers protection by inhibiting IKK-NF-κB/RELA mediated epithelial to mesenchymal transition and mucosal inflammation (30). In our study, iBET151 treatment shows downregulation of NFKB1 gene in human ILCs indicating the involvement of common pathways in immune and non-immune cells.
Interestingly, we also found that miR-155HG expression was lowered in ILC2 in response to iBET151. MiR-155HG is processed to give miR-155, and inhibitors of miR-155 have been reported to decrease IL-13 and IL-5 expression by Th2 cells, and miR-155-deficient mice exhibited reduced airway inflammation and eosinophilia in response to house dust mite challenge or a model of ovalbumin-induced allergic airway inflammation (55, 56). Recently, miR-155 has also been reported to regulate ILC2 and it was observed that miR-155-deficient mice have reduced numbers of ILC2 in response to ovalbumin-induced lung inflammation (57, 58).
Following extended culture in the presence of IL-33 the ILC2s retained their propensity for type-2 cytokine expression, but also showed a higher expression of additional markers of allergic-type activation including iBET-sensitive CRTH2, IL-9 receptor and the receptor for the neuropeptide, Neuromedin U. CRTH2, encoded by the PTGDR2 gene, is the receptor for prostaglandin D2 and is expressed by a spectrum of type-2 leukocytes, including Th2 cells, eosinophils and ILC2s. CRTH2 is a key marker for the identification of ILC2s (11) and its ligation regulates ILC2 migration and activation (18). Notably, fewer ILC2 accumulate in the lungs of CRTH2-deficient mice in response to pulmonary challenge (59). Inhibitors of the PGD2 pathway are in clinical trials for allergic diseases and it is likely that CRTH2 and PGD2 inhibitors will also inhibit ILC2 biology. IL-9R expression was increased in the long-term ILC2 cultures and was decreased by iBET151. IL-9 has been shown to act as an autocrine growth factor for ILC2 (60). Notably, we also identified that the gene encoding Neuromedin U receptor 1 (NMUR1) is induced following long-term ILC2 culture and is reduced by iBET151. Several groups have recently reported that Neuromedin U receptor expression on mouse ILC2s is critical for their activation by Neuromedin U produced by mucosal neurons. Without this signal helminth worms are expelled less efficiently (61–63). Our results suggest that human ILC2 also upregulate the Neuromedin U receptor and therefore may acquire responsiveness to neuron-derived signals.
Extended culture also led to the expression of additional chemokines and chemokine receptors that could be inhibited by co-culture with iBET151. The chemokines CCL3, CCL4, and CCL5 are all associated with the migration of macrophages and other leukocytes through their binding to CCR1, CCR4, and CCR5, and have been linked with asthma. Notably, CCR3 is the receptor for eotaxins that contribute to the accumulation and activation of eosinophils and T cells in the airways (64). Eotaxins are increased in asthma and correlate with disease severity. CCR3 also binds MCP-3 (CCL7), MCP-4 (CCL13), and RANTES (CCL5). There is currently no report of a role for CCR3 in human ILC2s, but it would be predicted that the expression of this receptor would facilitate ILC2 migration to sites of type-2 inflammation. Inhibitors of CCR3 are in clinical trials for asthma (65).
We have shown previously that mouse and human ILC2s are capable of processing and presenting antigens to T cells (8). RNAseq analysis identified that iBET151 treatment efficiently inhibited the transcription of ILC2-expressed genes encoding proteins involved in antigen processing and presentation. These included subunits of the proteasome complex that is required for enzymatically processing proteins into oligopeptides in the cytoplasm. In addition, CD74, the invariant chain of the MHCII complex, and HLA-DR and DQ were also inhibited. These results suggest that iBET151-treated ILC2 would be less efficient at processing and presenting antigens to T cells. Taken together, our results indicate iBET151 could be acting as an inhibitor of multiple transcriptional pathways responsible for mediating ILC2 migration, proliferation, and activation. Further studies would be required to delineate the epigenetic changes involved in these processes.
Our data suggest that iBET inhibitors could have therapeutic potential in allergic diseases and asthma. Based on their anti-tumor effects in vitro and in animal models, several BET bromodomain inhibitors have entered clinical trials in oncology. In addition to evaluating therapeutic efficacy in cancer, these trials are important for identifying potential side effects of BET inhibition in humans, which have included thrombocytopenia, diarrhea, and vomiting. We found that iBET151 was well-tolerated in mouse models of allergic lung inflammation and it is possible that treatment of inflammation in humans may require lower doses than have been administered in cancer studies and led to unwanted side-effects. At present this type of pan-iBET inhibitor would downmodulate multiple inflammatory networks, which could promote an increased susceptibility to microbial infections. However, such effects also arise when giving high dose oral steroids to severe asthmatics, and indeed perhaps pan-iBET inhibitors would offer an alternative to steroid use in such patients, and in cases of non-allergic or steroid resistant asthma. As inhibitors of individual BET bromodomain family members become available it will be important to determine if these may be used to preferentially modulate type-2 immunity. Their potential efficacy also raises the possibility of using BET inhibitors in combination therapies for allergic disease, possibly to synergise with anti-cytokine treatments.
All mouse experiments undertaken in this study were performed with the approval of the UK Home Office. All experiments involving human subjects were performed according to the research ethics committee of NHS health and research authority guidelines, reference no. 15/LO/0321.
BK designed and performed experiments and wrote the paper. JB and HJ performed experiments, provided advice on experimental design. BR, MG, DJ, and AP analyzed the gene expression data. MB, DT, and AvO provided expertise, advice on experimental design and interpretation, and reagents. AM supervised the project, designed the experiments, and wrote the paper. All authors commented on the manuscript.
This study was supported by grants from GSK and the UK Medical Research Council (U105178805).
AM has grant funding from GSK and AstraZeneca/MedImmune. MB, DJ, AP, DT, and AvO are employees of GSK.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the Ares staff, genotyping facility, and flow cytometry core for their technical assistance. We thank Jen Walker for advice on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00678/full#supplementary-material
Supplementary Figure S1. Human blood ILC subsets analyzed by flow cytometry.
Supplementary Figure S2. RNAseq analysis of iBET151-modulated genes expressed by ex vivo human ILC2s. (A) List of the 270 differentially expressed genes between IL-33-stimulated hILC2 and IL-33-stimulated iBET (125 nM)-treated hILC2 from heatmap in Figure 1. (B) List of immune system process-associated genes increased by iBET151 following ex vivo culture of ILC2s.
Supplementary Figure S3. Gene ontology analysis of iBET151-modulated genes expressed by in vitro-cultured human ILC2s. (A) List of the 204 differentially expressed genes between IL-33-stimulated hILC2 and IL-33-stimulated iBET (125 nM)-treated hILC2 from heatmap in Figure 2. (B) List of immune system process-associated genes reduced by iBET151 following in vitro culture of ILC2s. (C) List of immune system process genes from gene ontology analysis increased by iBET151 following in vitro culture of ILC2s.
Supplementary Figure S4. Flow cytometry gating strategy for (A) mouse lymphoid cell lineages, and (B) myeloid cell lineage.
Supplementary Figure S5. Titration of iBET151 in IL-33-induced type-2 inflammation in the lungs. (A) Percentage of ILC2s, (B) percentage of IL-13+ ILC2s, (C) mean fluorescence intensity of IL-13 expression by ILC2s, and (D) percentage of eosinophils in the lung following IL-33 intranasal challenge and vehicle (light blue circles), or IL-33 and iBET151 at 10 mg/kg (red circles), 5 mg/kg (dark blue circles), or 1 mg/kg (green circles) (as per Figure 5A). (E) Percentage of IL-13+ ILC2s following stimulation in vitro with IL-7 and IL-33 and either 250, 125, or 62.5 nM iBET151 for 24 h. iBET151 titration data are from one experiment.
1. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nat Rev Immunol. (2013) 13:75–87. doi: 10.1038/nri3349
2. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
3. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
4. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
5. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity. (2018) 48:1195–207.e6. doi: 10.1016/j.immuni.2018.05.003
6. Lei AH, Xiao Q, Liu GY, Shi K, Yang Q, Li X, et al. ICAM-1 controls development and function of ILC2. J Exp Med. (2018) 215:2157–74. doi: 10.1084/jem.20172359
7. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity. (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
8. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
9. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature. (2016) 539:102–6. doi: 10.1038/nature20105
10. McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. (2014) 41:366–74. doi: 10.1016/j.immuni.2014.09.006
11. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. (2011) 12:1055–62. doi: 10.1038/ni.2104
12. Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. (2014) 134:671–8.e4. doi: 10.1016/j.jaci.2014.06.024
13. Jia Y, Fang X, Zhu X, Bai C, Zhu L, Jin M, et al. IL-13(+) type 2 innate lymphoid cells correlate with asthma control status and treatment response. Am J Respir Cell Mol Biol. (2016) 55:675–83. doi: 10.1165/rcmb.2016-0099OC
14. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: the role of thymic stromal lymphopoietin. J Allergy Clin Immunol. (2018) 141:257–68.e6. doi: 10.1016/j.jaci.2017.03.032
15. Nagakumar P, Denney L, Fleming L, Bush A, Lloyd CM, Saglani S. Type 2 innate lymphoid cells in induced sputum from children with severe asthma. J Allergy Clin Immunol. (2016) 137:624–6.e6. doi: 10.1016/j.jaci.2015.06.038
16. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. (2016) 137:75–86.e8. doi: 10.1016/j.jaci.2015.05.037
17. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. (2013) 210:2939–50. doi: 10.1084/jem.20130351
18. Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. (2014) 133:1184–94. doi: 10.1016/j.jaci.2013.10.056
19. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol. (2015) 16:161–9. doi: 10.1038/ni.3078
20. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. (2016) 17:626–35. doi: 10.1038/ni.3443
21. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. (2017) 28:1776–87. doi: 10.1093/annonc/mdx157
22. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. (2010) 468:1119–23. doi: 10.1038/nature09589
23. Nadeem A, Al-Harbi NO, Al-Harbi MM, El-Sherbeeny AM, Ahmad SF, Siddiqui N, et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharmacol Res. (2015) 99:248–57. doi: 10.1016/j.phrs.2015.06.001
24. Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci USA. (2012) 109:14532–7. doi: 10.1073/pnas.1212264109
25. Hardman CS, Panova V, McKenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. (2013) 43:488–98. doi: 10.1002/eji.201242863
26. Barlow JL, Flynn RJ, Ballantyne SJ, McKenzie AN. Reciprocal expression of IL-25 and IL-17A is important for allergic airways hyperreactivity. Clin Exp Allergy. (2011) 41:1447–55. doi: 10.1111/j.1365-2222.2011.03806.x
27. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
28. Lloyd CM, Snelgrove RJ. Type 2 immunity: expanding our view. Sci Immunol. (2018) 3:eaat1604. doi: 10.1126/sciimmunol.aat1604
29. Moon HG, Kim SJ, Jeong JJ, Han SS, Jarjour NN, Lee H, et al. Airway epithelial cell-derived colony stimulating factor-1 promotes allergen sensitization. Immunity. (2018) 49:275–87.e5. doi: 10.1016/j.immuni.2018.06.009
30. Tian B, Hosoki K, Liu Z, Yang J, Zhao Y, Sun H, et al. Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J Allergy Clin Immunol. (2018). doi: 10.1016/j.jaci.2018.09.029. [Epub ahead of print].
31. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol. (2012) 13:229–36. doi: 10.1038/ni.2208
32. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol. (2017) 139:1468–77.e2. doi: 10.1016/j.jaci.2016.08.034
33. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity. (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
34. Maggi L, Montaini G, Mazzoni A, Rossettini B, Capone M, Rossi MC, et al. Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J Allergy Clin Immunol. (2017) 139:964–76.e4. doi: 10.1016/j.jaci.2016.06.032
35. Biancone L, Andres G, Ahn H, Lim A, Dai C, Noelle R, et al. Distinct regulatory roles of lymphocyte costimulatory pathways on T helper type-2 mediated autoimmune disease. J Exp Med. (1996) 183:1473–81. doi: 10.1084/jem.183.4.1473
36. Klemann C, Wagner L, Stephan M, von Horsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase-4's (DPP4) entanglement in the immune system. Clin Exp Immunol. (2016) 185:1–21. doi: 10.1111/cei.12781
37. Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. (2018) 9:1072. doi: 10.3389/fimmu.2018.01072
38. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. (2014) 15:431–8. doi: 10.1038/ni.2850
39. Garcia G, Godot V, Humbert M. New chemokine targets for asthma therapy. Curr Allergy Asthma Rep. (2005) 5:155–60. doi: 10.1007/s11882-005-0090-0
40. Schuh JM, Power C, Proudfoot AE, Kunkel SL, Lukacs NW, Hogaboam CM. Airway hyperresponsiveness, but not airway remodeling, is attenuated during chronic pulmonary allergic responses to Aspergillus in CCR4-/- mice. FASEB J. (2002) 16:1313–5. doi: 10.1096/fj.02-0193fje
41. Zhang Y, Wu Y, Qi H, Xiao J, Gong H, Zhang Y, et al. A new antagonist for CCR4 attenuates allergic lung inflammation in a mouse model of asthma. Sci Rep. (2017) 7:15038. doi: 10.1038/s41598-017-11868-9
42. Chen H, Xu X, Teng J, Cheng S, Bunjhoo H, Cao Y, et al. CXCR4 inhibitor attenuates allergen-induced lung inflammation by down-regulating MMP-9 and ERK1/2. Int J Clin Exp Pathol. (2015) 8:6700–7.
43. Kim MH, Taparowsky EJ, Kim CH. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity. (2015) 43:107–19. doi: 10.1016/j.immuni.2015.06.009
44. Cavagnero K, Doherty TA. Cytokine and lipid mediator regulation of group 2 Innate Lymphoid Cells (ILC2s) in human allergic airway disease. J Cytokine Biol. (2017) 2:116. doi: 10.4172/2576-3881.1000116
45. Lund SJ, Portillo A, Cavagnero K, Baum RE, Naji LH, Badrani JH, et al. Leukotriene C4 potentiates IL-33-induced group 2 innate lymphoid cell activation and lung inflammation. J Immunol. (2017) 199:1096–104. doi: 10.4049/jimmunol.1601569
46. Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting edge: notch signaling promotes the plasticity of group-2 innate lymphoid cells. J Immunol. (2017) 198:1798–803. doi: 10.4049/jimmunol.1601421
47. Gentek R, Munneke JM, Helbig C, Blom B, Hazenberg MD, Spits H, et al. Modulation of signal strength switches notch from an inducer of T cells to an inducer of ILC2. Front Immunol. (2013) 4:334. doi: 10.3389/fimmu.2013.00334
48. Bao K, Carr T, Wu J, Barclay W, Jin J, Ciofani M, et al. BATF Modulates the Th2 locus control region and regulates CD4+ T cell fate during antihelminth immunity. J Immunol. (2016) 197:4371–81. doi: 10.4049/jimmunol.1601371
49. Iwata A, Durai V, Tussiwand R, Briseno CG, Wu X, Grajales-Reyes GE, et al. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat Immunol. (2017) 18:563–72. doi: 10.1038/ni.3714
50. Kuwahara M, Ise W, Ochi M, Suzuki J, Kometani K, Maruyama S, et al. Bach2-Batf interactions control Th2-type immune response by regulating the IL-4 amplification loop. Nat Commun. (2016) 7:12596. doi: 10.1038/ncomms12596
51. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. (2012) 490:543–6. doi: 10.1038/nature11530
52. Sahoo A, Alekseev A, Tanaka K, Obertas L, Lerman B, Haymaker C, et al. Batf is important for IL-4 expression in T follicular helper cells. Nat Commun. (2015) 6:7997. doi: 10.1038/ncomms8997
53. Ubel C, Sopel N, Graser A, Hildner K, Reinhardt C, Zimmermann T, et al. The activating protein 1 transcription factor basic leucine zipper transcription factor, ATF-like (BATF), regulates lymphocyte- and mast cell-driven immune responses in the setting of allergic asthma. J Allergy Clin Immunol. (2014) 133:198–206.e1–9. doi: 10.1016/j.jaci.2013.09.049
54. Singh R, Miao T, Symonds ALJ, Omodho B, Li S, Wang P. Egr2 and 3 inhibit T-bet-mediated IFN-gamma production in T cells. J Immunol. (2017) 198:4394–402. doi: 10.4049/jimmunol.1602010
55. Malmhall C, Alawieh S, Lu Y, Sjostrand M, Bossios A, Eldh M, et al. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophilic inflammation in the lung. J Allergy Clin Immunol. (2014) 133:1429–38, 38.e1–7. doi: 10.1016/j.jaci.2013.11.008
56. Okoye IS, Czieso S, Ktistaki E, Roderick K, Coomes SM, Pelly VS, et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc Natl Acad Sci USA. (2014) 111:E3081–90. doi: 10.1073/pnas.1406322111
57. Johansson K, Malmhall C, Ramos-Ramirez P, Radinger M. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J Allergy Clin Immunol. (2017) 139:1007–16.e9. doi: 10.1016/j.jaci.2016.06.035
58. Knolle MD, Chin SB, Rana BMJ, Englezakis A, Nakagawa R, Fallon PG, et al. MicroRNA-155 protects group 2 innate lymphoid cells from apoptosis to promote type-2 immunity. Front Immunol. (2018) 9:2232. doi: 10.3389/fimmu.2018.02232
59. Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. (2015) 8:1313–23. doi: 10.1038/mi.2015.21
60. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951–65. doi: 10.1084/jem.20130071
61. Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. (2017) 549:277–81. doi: 10.1038/nature23469
62. Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. (2017) 549:282–6. doi: 10.1038/nature23676
63. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. (2017) 549:351–6. doi: 10.1038/nature24029
64. Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. (1997) 277:2005–7. doi: 10.1126/science.277.5334.2005
Keywords: ILC2, Th2, asthma, allergy, bromodomain, extra-terminal motif protein
Citation: Kerscher B, Barlow JL, Rana BM, Jolin HE, Gogoi M, Bartholomew MA, Jhamb D, Pandey A, Tough DF, van Oosterhout AJM and McKenzie ANJ (2019) BET Bromodomain Inhibitor iBET151 Impedes Human ILC2 Activation and Prevents Experimental Allergic Lung Inflammation. Front. Immunol. 10:678. doi: 10.3389/fimmu.2019.00678
Received: 08 January 2019; Accepted: 12 March 2019;
Published: 09 April 2019.
Edited by:
Masato Kubo, Tokyo University of Science, JapanReviewed by:
Koichiro Asano, Tokai University, JapanCopyright © 2019 Kerscher, Barlow, Rana, Jolin, Gogoi, Bartholomew, Jhamb, Pandey, Tough, van Oosterhout and McKenzie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew N. J. McKenzie, YW5tQG1yYy1sbWIuY2FtLmFjLnVr
†Present Address: Bernhard Kerscher, Paul-Ehrlich-Institute, Federal Institute for Vaccines and Biomedicines, Langen, Germany
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.