 Moriah J. Castleman1
Moriah J. Castleman1 Stephanie M. Dillon1
Stephanie M. Dillon1 Christine M. Purba1
Christine M. Purba1 Andrew C. Cogswell2
Andrew C. Cogswell2 Jon J. Kibbie1Martin D. McCarter3Mario L. Santiago1Edward Barker2
Jon J. Kibbie1Martin D. McCarter3Mario L. Santiago1Edward Barker2 Cara C. Wilson1*
Cara C. Wilson1*- 1Division of Infectious Disease, Department of Medicine, University of Colorado Anschutz Medical, Aurora, CO, United States
- 2Department of Microbial Pathogens and Immunity, Rush University Medical Center, Chicago, IL, United States
- 3Department of Surgery, University of Colorado Anschutz Medical, Aurora, CO, United States
Innate lymphoid cells (ILCs) are a diverse family of cells that play critical roles in mucosal immunity. One subset of the ILC family, Group 3 ILCs (ILC3s), has been shown to aid in gut homeostasis through the production of IL-22. IL-22 promotes gut homeostasis through its functional effect on the epithelial barrier. When gut epithelial barrier integrity is compromised, such as in Human Immunodeficiency Virus (HIV) infection and inflammatory bowel disease (IBD), microbes from the gut lumen translocate into the lamina propria, inducing a multitude of potentially pathogenic immune responses. In murine models of bacterial infection, there is evidence that bacteria can induce pro-inflammatory IFNγ production in ILC3s. However, the impact of diverse translocating bacteria, particularly commensal bacteria, in dictating IFNγ versus IL-22 production by human gut ILC3s remains unclear. Here, we utilized an in vitro human lamina propria mononuclear cell (LPMC) model to evaluate ILC3 cytokine production in response to a panel of enteric Gram-positive and Gram-negative commensal and pathogenic bacteria and determined potential mechanisms by which these cytokine responses were induced. The percentages of IL-22-producing ILC3s, but not IFNγ-producing ILC3s, were significantly increased after LPMC exposure to both Gram-positive and Gram-negative commensal or pathogenic bacterial stimuli. Stimulation of IL-22 production from ILC3s was not through direct recognition of bacterial antigen by ILC3s, but rather required the help of accessory cells within the LPMC population. CD11c+ myeloid dendritic cells generated IL-23 and IL-1β in response to enteric bacteria and contributed to ILC3 production of IL-22. Furthermore, ligation of the natural cytotoxicity receptor NKp44 on ILC3s in response to bacteria stimulation also significantly increased the percentage of IL-22-producing ILC3s. Overall, these data demonstrate that human gut microbiota, including commensal bacteria, indirectly modulate colonic ILC3 function to induce IL-22, but additional signals are likely required to induce IFNγ production by colonic ILC3s in the setting of inflammation and microbial translocation.
Introduction
Innate Lymphoid Cells (ILCs) descend from the lymphoid lineage, but unlike T cells and B cells lack rearranged antigen receptors (1). Rather than responding to antigen priming, ILCs respond in an antigen-independent manner through cytokine stimulation to produce effector cytokines (2) or through engagement of germline encoded receptor stimulation (3–5). ILCs are categorized into three groups based on the expression of specific master transcription factors and effector function (6). Group 1 ILCs (ILC1, NK cells) express T-bet and produce the pro-inflammatory cytokine IFNγ in response to IL-12 and IL-18; Group 2 ILCs (ILC2s) express GATA3 and produce IL-4, IL-13, and IL-5 in response to IL-25 and IL-33; and Group 3 ILCs (ILC3s) express RORγt and AHR and typically produce IL-22, GM-CSF, IL-17, and LT-α1β2 in response to IL-23 and IL-1β (6). ILCs are constitutively found in the gastrointestinal tract (7). ILC3s are of particular importance to gut mucosal immunity (7–9) via the secretion of effector cytokines that act directly on epithelial cells (10). Epithelial cells express the IL-22 receptor (IL-22R) (11) and IL-22 production by ILC3s promotes epithelium proliferation, survival, mucus production, upregulation of fucosylation, and in some studies increased gene expression of antimicrobial peptides (12–16). To date, the majority of studies characterizing gut ILC3 function have utilized murine models. Examination of human ILC3 function have primarily focused on tonsil tissue and demonstrated that the mechanisms contributing to IL-22 production include stimulation by the cytokines IL-23 and IL-1β (3, 17, 18) with synergistic enhancement of IL-22 production observed in the presence of the natural cytotoxicity receptor NKp44 engagement (3). Few studies have directly investigated factors driving IL-22 production by human gut ILC3s, although one study observed a requirement for IL-23, IL-1β, and IL-7, with synergy again being induced in the presence of NKp44 signaling (3).
Epithelial barrier damage and loss of function in gut-associated diseased states have correlated with alterations in ILC frequency and function. In Inflammatory Bowel Disease (IBD), patients with Crohn's disease have a loss of colonic or ileum IL-22-producing ILCs (including ILC3s) (18, 19) and an increase in IFNγ/IL-17A-producing ILCs (20–22). In Human Immunodeficiency Virus (HIV) infection, loss of colonic IL-22-producing ILCs has been reported (23). Similarly, reduced frequencies of IL-22/IL-17-producing ILCs during Simian Immunodeficiency Virus (SIV) infection (the non-human primate model of HIV) were noted (24–28). Furthermore, we and others have reported increased frequencies of IFNγ-producing ILCs both in people living with HIV (PLWH) who were not receiving anti-retroviral therapy (ART) (29) and during SIV infection (27). Since IFNγ alters epithelial tight junctions and upregulates epithelial cell expression of TNFα receptor which results in further epithelial cell damage (30, 31), increased frequencies of IFNγ-producing ILC3s may be an additional contributor to epithelial barrier breakdown. When the epithelial barrier is compromised, translocation of gut-associated bacteria into the lamina propria (LP) exposes immune cells to bacteria of different species or magnitude than what these cells typically encounter in the healthy human gut (32, 33). We previously demonstrated that increased frequencies of colonic IFNγ-producing ILCs in PLWH correlated with alterations in mucosa-associated bacterial communities (dysbiosis), specifically with increased relative abundance of Gram-negative commensal Prevotella species (29). Understanding the bacteria-specific cytokine responses of ILC3s and the mechanisms by which protective or deleterious cytokines are produced are critical to determining the effect of ILC3s on gut homeostasis, not only for their role in enteric bacterial immunity, but also for their role in influencing epithelial cell function in disease states.
Murine studies highlighted a complex role for gut microbiota in ILC subset development and functional production of IL-22 (13, 34, 35). IL-22 production by ILC3s protected against an enteric pathogen Citrobacter rodentium (34, 36, 37), and prevented systemic dissemination of the commensal Alcaligenes species in mice (15). Fucosylation of epithelial cells induced by ILC3 production of IL-22 contributed to host defense against murine S. typhimurium infection (38). Furthermore, murine ILC3s negatively regulated microbe-specific T cells in the gut to limit pathological responses to commensal bacteria (39, 40). While these studies support a homeostatic role for ILC3s in microbiota-associated gut responses in mice, gut inflammatory ILC3s in response to bacteria have been reported. ILC3s produced IFNγ in response to infection with Salmonella typhimurium (41) and IFNγ/IL-17 in response to infection with the Helicobacter hepaticus (42). Furthermore, ILC3-associated IFNγ/IL-17 production in response to H. hepaticus was linked to the development of colitis (42) highlighting a potentially deleterious role of ILC3 cytokine production. In vitro exposure of human ILC3s have also suggested a plasticity in cytokine production with the capacity to produce IFNγ or IL-22 dependent on the cytokine milieu (18, 21). These observations raise the possibility that human gut ILC3s may also have the capacity to produce IL-22 or IFNγ in response to exposure to different types of bacteria.
In this study, we hypothesized that pathogenic enteric bacteria would induce pro-inflammatory cytokine production (IFNγ) from human lamina propria ILC3s, whereas commensal bacteria would primarily elicit protective (IL-22) cytokine production. To address this, we utilized an in vitro human colonic mononuclear cell model (43, 44) to investigate ILC3 cytokine profiles induced in response to a panel of whole Gram-negative and Gram-positive, commensal and pathogenic bacteria and the mechanisms driving these responses. Overall, our observations provide insight into the ILC3 role in enteric bacteria immunity and their contribution to the inflammatory environment in disease states where microbes translocate through a compromised epithelial barrier.
Materials and Methods
Human Tissue Samples
Human colonic tissue samples were acquired from patients undergoing elective abdominal surgery at the University of Colorado Hospital and are categorized as discarded tissue from macroscopically normal sites. Samples from patients that underwent chemotherapy or radiation within 8 weeks of tissue collection were not included in the study. Other criteria for tissue exclusion include those with Inflammatory Bowel Disease, HIV infection or treatment with immunosuppressive drugs. Intraepithelial mononuclear cells (IEMC) or lamina propria mononuclear cells (LPMC) were isolated from tissue samples as previously described (43, 44) and stored in liquid nitrogen until use. Human tonsillar tissue samples were acquired from pediatric patients from Colorado Children's Hospital. Tonsillar mononuclear cells (TMCs) were isolated as previously described (45). All patients undergoing surgery signed a release to allow unrestricted use of discarded tissue and protected patient information was de-identified to the laboratory investigators. This research was reviewed by the Colorado Multiple Institutional Review Board (COMIRB) at the University of Colorado Anschutz Medical Campus and was granted exempt research status.
Preparation of Bacterial Stocks
Growth of anaerobic bacteria was performed using a BD GasPak EZ Anaerobe Pouch System according to manufacturer's instructions (BD Diagnostics, Franklin Lakes, NJ). Prevotella stercorea (DSM No. 18206, Braunschweig, Germany) was grown in liquid chopped meat broth (Hardy Diagnostics, Santa Maria, CA) supplemented with 1% Trace Minerals (ATCC), 1% Vitamin Supplements (ATCC), 0.05% Tween80, 29.7 mM acetic acid, 8.1 mM propionic acid and 4.4 mM butyric acid (Sigma-Aldrich) under anaerobic conditions at 37°C for 5–7 days. Ruminococcus bromii (ATCC# 27255) was grown in liquid chopped meat broth (Hardy Diagnostics) under anaerobic conditions at 37°C for 1–2 days. The long term stock of Bifidobacterium longum subsp infantis (ATCC 15697) was grown in liquid chopped meat broth (Hardy Diagnostics) under anaerobic conditions at 37°C for 2–3 days and the working stock was grown on Brucella plates (Teknova, Hollister, CA) under anaerobic conditions at 37°C for 2–3 days. Acinetobacter junii (ATCC 17908) was grown using Nutrient Agar plates (Edge Biologicals, Memphis, TN) under aerobic conditions at 26°C for 1–2 days. Salmonella typhimurium (ATCC 35986) was grown on LB agar plates (Sigma-Aldrich) under aerobic conditions at 37°C for 1–2 days. Long term stocks of all bacteria were prepared using 10% glycerol and single-use working stocks were prepared using DPBS. All stocks were stored at −80°C and bacterial cell counts were determined using the BD Cell Viability Kit (BD Bioscience).
In vitro Stimulation of LPMCs With Whole Bacteria
For the in vitro stimulations, human colonic LPMCs were thawed as previously described (43, 44) and cultured in RPMI with 10% human AB serum (Gemini Bioproducts, West Sacramento, CA), 1% Penicillin/Streptomycin/Glutamine (Life Technologies, Grand Island, NY), and 500 μg/ml Zosyn (Piperacillin and Tazobactam, Wyeth, Madison, NY) at a concentration of 1.0 × 106 million cells per mL in a 48 well plate. LPMCs were exposed to a panel of Gram-positive and Gram- negative bacteria detailed in Supplemental Table 1 including mucosa-associated colonic bacteria previously shown to be increased or decreased in relative abundance during HIV-1 infection (46, 47): Gram-negative Prevotella stercorea and Acinetobacter junii (increased) and Gram-positive Ruminococcus bromii (decreased), as well as the Gram-positive probiotic Bifidobacterium infantis and the Gram-negative pathogen Salmonella typhimurium. Broad spectrum antibiotics including Penicillin, Streptomycin, Piperacillin, and Tazobactam were present throughout the time in culture to prevent bacterial overgrowth.
For assays examining ILC3 responses, whole bacteria were added to cell cultures at a ratio of 2.5 bacteria to 1 LPMC and incubated for 16 h at 37°C + 5% CO2, followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 4 h. Cells were then collected for flow cytometry as described below.
For blocking experiments: human colonic LPMCs were first exposed to blocking antibodies targeting IL-23 p19, IL-1β (R & D Systems, Minneapolis, MN) or IL-7 (Biolegend) at 5 ug/mL or NKp44 (Biolegend) at 10 ug/mL for 30 min followed by incubation with A. junii as described above. Cells were then collected for flow cytometry as described below. The concentration of blocking antibodies was optimized for use by measurement of IL-22 + ILC3s or IL-22 + Lineage negative cells (for IL-7) in response to recombinant cytokine stimulation (50 ng/mL of IL-23, IL-1β, or IL-7) or bead ligation (for NKp44) with a dose curve of blocking antibody treatment (Supplemental Figure 1). The addition of recombinant IL-7 prevented the identification of ILC3s (defined as CD127+ which is IL-7Rα) and instead gating for IL-22 was determined on lineage- cells. Antibodies and controls used to block are listed in Supplemental Table 2.
For depletion experiments: CD11c+ cells (mDC) or CD3+ (T cells) were depleted from LPMCs using the EasySep PE Positive Selection Kit according to manufacturer's instructions (StemCell Technologies, Vancouver, Canada) and the antibody PE-CD11c (Biolegend, San Diego, CA) or PE-CD3 (Tonbo, San Diego, CA) followed by incubation with A. junii as described above. Greater than 90.25% of CD11c+ mDCs were depleted from total LPMCs. Greater than 91.92% of CD3+ T cells were depleted from total LPMCs. Cells were then collected for flow cytometry as described below.
For the measurement of secreted cytokines, LPMCS were plated at a concentration of 2.0 × 106 million cells per mL in a 96 well plate and exposed to whole bacteria R. bromii or A. junii at a ratio of 2.5 bacteria to 1 LPMC and incubated for 24 h at 37°C + 5% CO2. Supernatant was collected and saved at −20°C until use. IL-23, IL-1β, and IL-7 were measured in the supernatant using the U-PLEX Assay according to manufacturer's instructions and quantified on the QuickPlex SQ 120 Instrument (Mesoscale Discovery, Rockville, MD).
For assays examining antigen presenting cell responses (mDC, B cell, and Macrophages), whole bacteria were added to cell cultures at 2.5 bacteria to 1 LPMC and incubated for 4 h at 37°C + 5% CO2, followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 16 h. Cells were then collected for flow cytometry as described below.
In vitro Stimulation of LPMCs With Bacterial Cell Surface Components
Human colonic LPMCs were exposed to either 1 μg/mL of the TLR2 ligand, lipoteichoic acid (LTA) from B. subtilis (InvivoGen, San Diego, CA), or the TLR4 ligand, lipopolysaccharide (LPS) from E. coli (InvivoGen) for 16 h at 37°C + 5% CO2, followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 4 h. Cells were then collected for flow cytometry as described below.
In vitro Stimulation of LPMCs With Recombinant Cytokines or NKp44 Activation
Human colonic LPMCs were exposed to 50 ng/mL IL-23 or IL-1β (R & D Systems), or the combination of both, or IL-2 or IL-7 (Tonbo) for 16 h at 37°C + 5% CO2 followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 4 h. For NKp44 activation experiments; 20 ug/mL of anti-NKp44-biotin (clone P44-8, Biolegend) was combined with Anti-Biotin MACSiBead Particles (Miltenyi Biotec) according to manufacturer's instructions. The beads were then added to LPMCs at a ratio of 5 beads to 1 LPMC for 16 h at 37°C + 5% CO2 followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 4 h. Cells were then collected for flow cytometry as described below.
In vitro Stimulation of Tonsil Mononuclear Cells
Human tonsil mononuclear cells (TMCs) were cultured in RPMI with 10% human AB serum (Gemini Bioproducts), 1% penicillin/streptomycin/glutamine (Life Technologies), and 500 μg/ml Zosyn (Wyeth) at a concentration of 1.0 × 106 million cells per mL in a 48 well plate. TMCs were exposed to whole bacteria added to cell cultures at a ratio of 2.5 bacteria to 1 LPMC, or to the combination of 50 ng/mL IL-23 and IL-1β, or NKp44 activation beads at a ratio of 5 beads to 1 TMC and incubated for 16 h at 37°C + 5% CO2, followed by the addition of Golgi Plug Transport Inhibitor (BD Bioscience) for 4 h. Cells were then collected for flow cytometry as described below.
Flow Cytometry Protocol for Surface and Intracellular Staining
Using flow cytometry, viable CD45+ single cell lymphocytes were identified followed by identification of ILC3s as follows: Lineage-CD127+CD117+. The lineage negative cocktail comprised antibodies targeting CD3, CD20, CD13, CD123, CD303, CD34, FCεR1α, CD11c, and CRTH2. All data were acquired on an LSRII flow cytometer (BD Biosciences). Routine quality control using the Cytometer Setup and Tracking feature within the BD FACSDiva software version 6.1.2 (BD Biosciences) was performed daily. All antibodies and clones used for staining are listed in Supplemental Table 3.
For ex vivo phenotyping of ILC3s: IEMCs or LPMCs were thawed and surface stained to identify ILC subsets for expression of NKp44, CD56, CCR6, TLR2, TLR4, and TLR5 and intranuclear stained for the transcription factors RORγt, AHR, T-bet and EOMES using the Foxp3/Transcription Factor buffer set according to manufacturer's instructions (Thermo Fisher Scientific, Frederick, MD).
For in vitro culture examination of ILC3s; LPMCs were collected after stimulations described above and surface stained to identify ILC3s and subsets (NKp44) followed by intracellular staining for cytokines IL-22, IFNγ, and IL-17 using Fix and Perm Cell fixation and permeabilization buffer set according to manufacturer's instructions (Thermo Fisher Scientific, Frederick, MD).
For in vitro culture examination of antigen presenting cells; cells were collected after stimulation and surface stained to identify mDCs (CD45+ Viable Myeloid CD3- CD19- HLA-DR+ CD11c+), B cells (CD45+ Viable Lymphocyte CD3- CD19+) or Macrophages (CD45+ Viable Myeloid CD3- CD19- HLA-DR+ CD11c-) [as defined by Smith et al. and Bain and Mowat (48–50)] followed by intracellular staining for cytokines IL-12/IL-23 p40, IL-23p19, and IL-1β using Fix and Perm Cell fixation and permeabilization reagents according to manufacturer's instructions (Thermo Fisher Scientific). Only cells that expressed both subunits of the cytokine IL-23 (IL-12/IL-23p40 and IL-23p19) were considered to be IL-23+ cells.
In vitro Stimulation of Purified ILC3s
ILC3s were isolated from colonic LPMCs and on average purified to 91.79% purity (Supplemental Figure 2) by sorting using the MoFlo Astrios EQ (Beckman Coulter, Indianapolis, IN). ILC3s were sorted from Viable CD45+ single cell lymphocytes that were Lineage-CD127+CD117+ as described above. Purified ILC3s were then exposed to either 50 ng/mL IL-23 and IL-1β (R & D Systems) or whole bacteria at a ratio of 1 bacteria to 1 ILC3 and incubated for 24 h at 37°C + 5% CO2. Supernatant was collected and saved at −20°C until use. Secreted IL-22 was measured in the supernatant using the IL-22 U-PLEX Assay according to manufacturer's instructions and quantified on the QuickPlex SQ 120 Instrument (Mesoscale Discovery, Rockville, MD).
Data Analysis
Each patient who provided a tissue specimen for research is considered a single sample for data analysis, and figure legends indicate how many samples were examined for each assay using the following terminology: N = number of patients samples. All flow cytometer data analysis was done using FlowJo v10.0. All statistical analysis and graphing were performed using GraphPad Prism v6.00 for Windows (GraphPad Software, La Jolla California). Paired t-test was used to determine statistical differences between conditions as indicated in figure legend. Data sets without a minimum number of 25 ILC3 events captured using flow cytometry were excluded from analysis.
Results
ILC3s Are Phenotypically Similar in the Intraepithelial and Lamina Propria Layers of the Human Colon
Human ILC1 subsets were previously shown to be phenotypically different between the colonic layers (22). We therefore sought to determine if ILC3s that reside in the intraepithelial layer (IE) of the human colon (closer to the intestinal lumen where gut bacteria reside) are phenotypically different to ILC3s that reside within the lamina propria layer (LP). The frequency and phenotype of ILC3 in these locations were determined with ILC3s in colonic human tissue identified as CD45+ viable Lineage- CD127+ CD117+ (Figure 1A). ILC3s were more frequent as a fraction of CD45+ cells in the LP (1.16% ± 0.27) compared to the IE layer (0.40% ± 0.09) (Figure 1B).
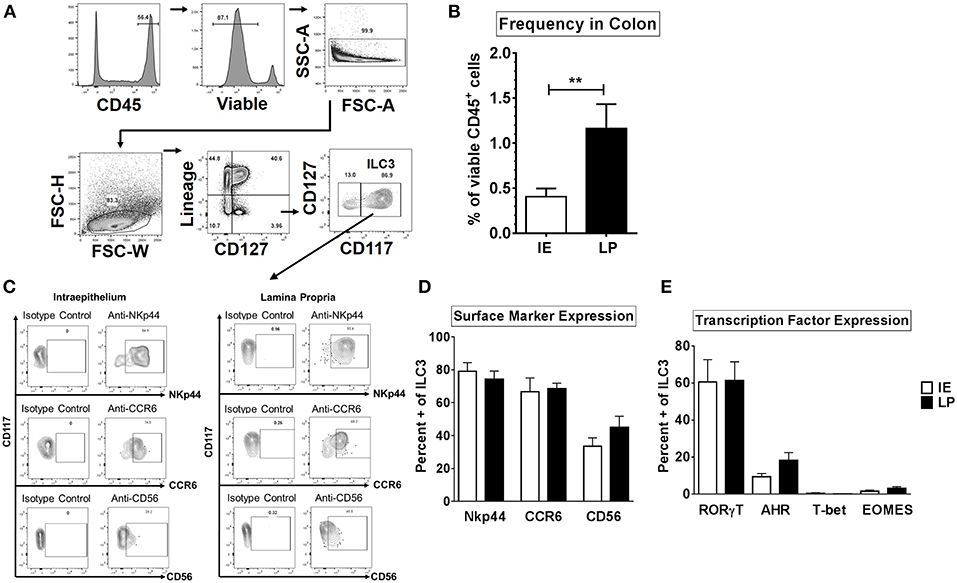
Figure 1. ILC3s are phenotypically similar between the intraepithelial and lamina propria layers of the human colon. (A) Representative flow cytometry gating strategy to identify ILC3s in human colonic tissue. (B) Frequencies of ILC3s in the colon in the intraepithelial (IE) and lamina propria (LP) layer as a percent of viable CD45+ cells. IE: N = 6, LP: N = 6. (C) Representative flow plots gated on ILC3s for the expression of surface markers NKp44, CD56, or CCR6. (D) Percentages of ILC3s expressing the surface markers NKp44, CD56, or CCR6 ex vivo. IE: N = 6, LP: N = 6. (E) Percentages of ILC3s expressing the transcription factors RORγt, AHR, T-bet, or EOMES ex vivo. IE: N = 7, LP: N = 7. Bars are mean + S.E.M. Statistical analysis performed was paired t test. **p < 0.01.
NKp44, CD56, and CCR6, have previously been utilized to identify subsets of ILC3s (17, 24, 29, 41) therefore expression of these markers was next evaluated on colonic ILC3s. The majority of ILC3s expressed NKp44 or CCR6 in both layers (NKp44: IE: 79.13% ± 5.25, LP: 74.21% ± 5.04; CCR6: IE: 66.68% ± 8.40, LP: 68.61% ± 3.27) (Figures 1C,D). On average, less than half of LP and IE ILC3s expressed CD56 (LP: 45.08% ± 6.68; IE: 33.58% ± 5.04) (Figures 1C,D).
As expected (51), the majority of ILC3s expressed the master transcription factor RORγt (IE: 60.60% ± 12.07, LP: 61.42% ± 10.10) and frequencies of RORγt + ILC3s as a percent of viable CD45 + lymphocytes were not significantly different between the IE and LP layers (Figure 1E and Supplemental Figure 3). Expression of AHR in ILC3s was lower than that of RORγt (Figure 1E and Supplemental Figure 3) but similar between tissue layers (LP: 18.23% ± 4.24, IE: 9.35% ± 1.77). Of the AHR-expressing ILC3s, the majority also co-expressed RORγt (LP: 76.77% ± 8.84, IE: 82.44% ± 9.74). Less than 1% of ILC3s expressed T-bet in both layers of the colon (LP: 0.15% ± 0.06, IE: 0.67% ± 0.49) (Figure 1E and Supplemental Figure 3). Low frequencies of EOMES expressing ILC3s were also quantified in both layers (LP: 1.56% ± 0.62, IE: 3.16% ± 0.91). Overall, of the markers examined, ILC3s were phenotypically similar between the intraepithelial and lamina propria layer in the normal human colon.
Enteric Bacteria Stimulate Production of IL-22 but Not IFNγ From ILC3s When Exposed to Total LPMCs
Mimicking the state where the epithelial barrier is damaged and LP immune cells are exposed to bacteria from the colonic lumen, we utilized an in vitro model of lamina propria mononuclear cells (LPMCs) to investigate the ILC3 response to whole enteric bacteria. LPMCs were exposed to a panel of whole bacteria representing Gram-positive and Gram-negative commensal bacteria reported to be altered in various diseases associated with epithelial barrier damage (Supplemental Table 1). This panel included commensal Gram-positive Ruminococcus bromii (Rb) which is decreased in relative abundance in colonic mucosa of people living with HIV (PLWH) and Gram-negative Acinetobacter junii (Aj) and Prevotella stercorea (Ps), which were increased in relative abundance (46, 47). The enteric gram-negative pathogen Salmonella typhimurium (St) which may contribute to the onset of IBD symptoms (52) and to which PLWH are at an increased risk of acquiring S. typhiumurium-bacteremia (53, 54) as well as the probiotic Gram-positive Bifidobacterium longum sp infantis (Bi), which is used therapeutically to reestablish a protective microbiome (55, 56) were also included. Bacterial experiments were performed in the presence of broad spectrum antibiotics in order to inhibit bacterial overgrowth. All bacteria tested, irrespective of gram-staining and commensal or pathogenic nature, induced production of IL-22 in a fraction of ILC3s relative to the no stimulation condition (Bi: 30.0% ± 7.13, Rb: 18.76% ± 6.43, Aj: 21.61% ± 3.02, Ps: 28.58% ± 3.54, St: 32.21% ± 4.65) (Figures 2A–C). Of the bacteria tested, none induced a significant increase in IL-17A+ ILC3s above the no stimuli control (data not shown: less than 0.80% IL-17+ ILC3).
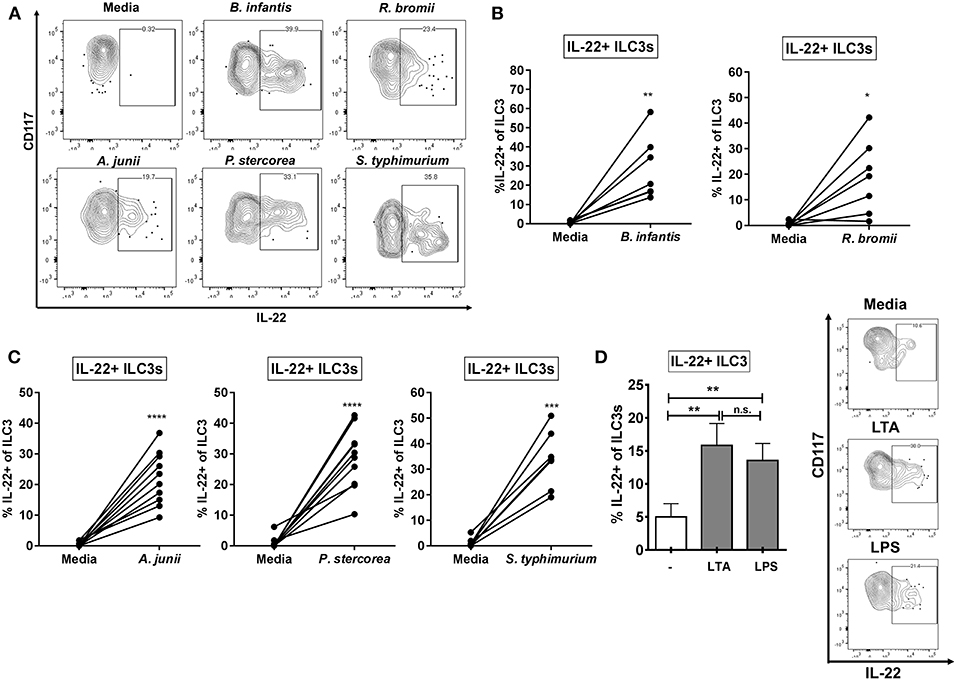
Figure 2. ILC3s produce IL-22 in response to Gram-positive and Gram-negative enteric bacteria. (A) Representative flow cytometry demonstrating cytokine staining for IL-22 gated on ILC3s after LPMC exposure to enteric bacteria in vitro. (B,C) Percentages of IL-22+ ILC3s after LPMC exposure to enteric bacteria or no bacterial control. N = 6–10. (D) Percentages of IL-22+ ILC3s after LPMC exposure to lipoteichoic acid (LTA) or lipopolysaccharide (LPS) or no stimulation control. Representative flow cytometry demonstrating cytokine staining for IL-22 gated on ILC3s after LPMC exposure to bacterial cell surface components. N = 6. Bars are mean + S.E.M. Statistical analysis performed was paired t test as indicated. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant.
To determine if bacterial surface components were important in driving the ILC3 IL-22 response to whole bacteria, LPMCs were exposed to Gram-positive and Gram-negative bacterial cell surface components (LTA and LPS, respectively). The percentage of IL-22+ ILC3s was similarly increased in response to both bacterial surface antigens (LTA: 17.98% ± 3.09, LPS: 15.18% ± 2.44) (Figure 2D).
ILC3s that produced IFNγ were associated with inflammatory responses in murine infection with bacteria (41) or bacteria-driven murine colitis (42). We next measured the frequencies of IFNγ-producing LP ILC3s in response to whole bacteria. Low frequencies of IFNγ expressing ILC3s were observed in response to exposure of LPMC to each bacteria (Bi: 0.63% ± 0.50, Rb: 0.06% ± 0.04, Aj: 1.51% ± 0.81, Ps: 0.50% ± 0.35, St: 1.77% ± 0.68), however no significant increase above the no stimuli control was detected (Supplemental Figure 4). Furthermore, IFNγ+ ILC3s were not induced by exposure to LTA and LPS (data not shown). Overall, these data indicate that the bacteria tested did not drive the production of IFNγ by ILC3s in our in vitro culture system.
Purified ILC3s Do Not Produce IL-22 in Response to Enteric Bacteria
To assess if ILC3s produced IL-22 in direct response to bacteria, ILC3s were purified from the LP (Supplemental Figure 2) and exposed in vitro to either Gram-positive (R. bromii), or Gram-negative (A. junii) bacteria or to recombinant IL-23 and IL-1β. After 24 h, the levels of secreted IL-22 were measured. Exposure of isolated ILC3s to IL-23 and IL-1β induced IL-22 production (86.74 pg/mL ± 29.60), whereas purified ILC3s did not produce IL-22 in response to either bacterial species compared to no stimulation control (C: 0.92 pg/mL ± 0.19, Rb: 1.00 pg/mL ± 0.24, Aj: 1.42 pg/mL ± 0.38) (Figure 3A). In keeping with the lack of response to direct bacterial stimulation, cell surface expression of bacterial Pattern Recognition Receptors (PRRs) TLR2, TLR4, and TLR5 on colonic ILC3s ex vivo were low or not detected (TLR2: 0.94% ± 0.36, TLR4: 0%, TLR5: 0.60% ± 0.29) compared to Lineage+ non-lymphoid cells (TLR2: 10.23% ± 1.68, TLR4: 4.28% ± 1.22, TLR5: 7.12% ± 2.96) (Figure 3B). These data suggest that human colonic ILC3s do not produce IL-22 in direct response to the enteric bacteria tested, but require additional stimulation from accessory cells in LPMCs to induce IL-22.
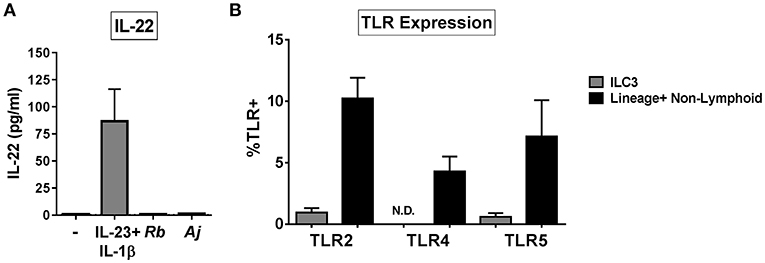
Figure 3. ILC3s do not respond directly to bacteria by producing IL-22. (A) Quantification of IL-22 (pg/mL) in the supernatant of purified ILC3s exposed to recombinant IL-23+ IL-1β (50 ng/mL) or R. bromii (Rb) or A. junii (Aj) in vitro at a ratio of 1 ILC3 to 1 bacterium or no stimulation control. N = 3. (B) Percentages of LPMCs stained ex vivo for TLR2, TLR4, or TLR5 expression gated on ILC3s or Lineage positive non-lymphoid cells. N = 6. N.D., not detected. Bars are mean + S.E.M.
IL-23 and IL-1β Contribute to the ILC3 IL-22 Response to Gut Bacteria
Given that recombinant IL-23 and IL-1β stimulated production of IL-22 from purified ILC3s, the levels of secreted IL-23 and IL-1β following exposure of LPMCs to commensal Gram-positive (R. bromii) or to Gram-negative (A. junii) bacteria were next evaluated. R. bromii and A. junii significantly induced the secretion of IL-23 (Rb: 21.57 pg/mL ± 3.54, Aj: 234.4 pg/mL ± 57.23) from LPMCs (Figure 4A) above background. Both bacteria also induced significant production of IL-1β (Rb: 34.86 pg/mL ± 7.62, Aj: 247.6 pg/mL ± 74.49) from LPMCs (Figure 4B). Although both commensal bacteria stimulated significant production of these cytokines, A. junii induced 10.8 fold more IL-23 and 7.1 fold more IL-1β than R. bromii (Figures 4A,B).
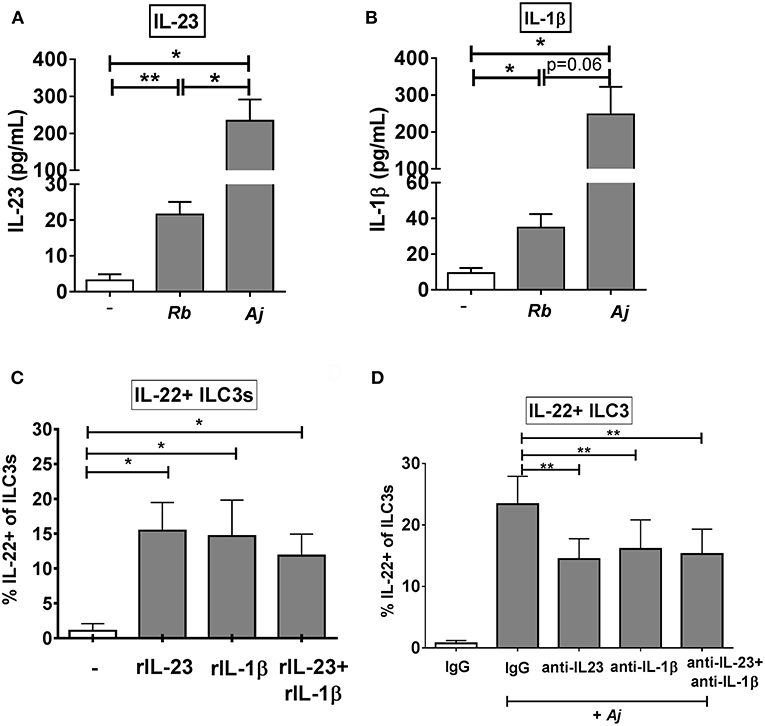
Figure 4. IL-23 and IL-1β modulate the ILC3 IL-22 response to enteric bacteria. (A) Quantification of IL-23 (pg/mL) or (B) IL-1β (pg/mL) in the supernatant of LPMCs exposed to R. bromii (Rb) or A. junii (Aj) or no bacteria control. N = 5. (C) Percentages of IL-22+ ILC3s after LPMC exposure to recombinant IL-23, IL-1β, or IL-23+ IL-1β (50 ng/mL) or no stimulation control. N = 5. (D) Percentages of IL-22+ ILC3s after LPMC exposure to no bacteria control or A. junii (Aj) in the presence of 5 ug/mL blocking antibodies targeting IL-23 and/or IL-1β or the antibody isotype control IgG. N = 8. Bars are mean + S.E.M. Statistical analysis performed was paired t-test as indicated. *p < 0.05, **p < 0.01.
To evaluate the relative contribution of IL-23 and IL-1β independently and in combination to ILC3 induction of IL-22, LPMC were exposed to recombinant IL-23 and/or IL-1β. Both recombinant cytokines individually significantly increased the percentage of IL-22+ ILC3s (C: 1.28% ± 1.22, IL-23: 17.59% ± 4.20, IL-1β: 17.41% ± 7.06) compared to the unstimulated control (Figure 4C). Although the combination of recombinant IL-23 and IL-1β significantly increased the percentage of IL-22+ ILC3s (11.07% ± 3.66) compared to unstimulated control, the combination response was not synergistic (Figure 4C).
To determine if production of IL-23 and/or IL-1β from LPMCs drives IL-22 induction in ILC3s in response to bacteria, blocking antibodies directed against IL-23, IL-1β, or both cytokines were added to LPMC cultures before exposure to the commensal A. junii. The frequency of IL-22 producing ILC3s in response to A. junii was significantly reduced by an average of 38.0% when blocking IL-23 and 38.5% when blocking IL-1β compared to stimulation with A. junii and the control IgG antibody (Figure 4D). The combination of blocking both IL-23 and IL-1β also significantly reduced the frequencies of IL-22 + ILC3s in response to A. junii by 40.1% (anti-IL-23: 14.46% ± 3.30, anti-IL-1β: 16.13% ± 4.71, anti-IL-23+ anti-IL1β: 15.28% ± 4.04), but did not lead to a synergistic reduction (Figure 4D).
Given the lack of complete abrogation of the ILC3 IL-22 response to bacteria when blocking IL-23 and IL-1β, the role of other cytokines (IL-7, IL-2) reported to promote ILC3 phenotype (57) were next investigated. Although secreted IL-7 was detected in unstimulated LPMC cultures, levels of IL-7 did not increase after LPMCs were exposed to commensal Gram-positive bacteria (R. bromii) or to Gram-negative bacteria (A. junii) (Supplemental Figure 5A). In keeping with this lack of production in presence of bacteria, blocking IL-7 alone did not reduce the frequency of IL-22+ ILC3s (Supplemental Figure 5B). Blocking IL-7 in combination with blocking IL-23 and IL-1β did not further reduce the frequency of IL-22+ ILC3s compared to blocking IL-23 and IL-1β (Supplemental Figure 5B). T cells are a major producer of IL-2 among other cytokines, however, depletion of CD3+ T cells from LPMCs did not alter the percentage of IL-22+ ILC3s generated in response to A. junii (Supplemental Figure 5C) and the addition of recombinant IL-2 to LPMCs did not significantly increase the percentage of IL-22+ ILC3s (data not shown). Altogether these data indicate that IL-2 and IL-7 do not have a major role in LPMCs in promoting IL-22 production by ILC3s in response to enteric bacteria in this culture system.
Myeloid Dendritic Cells Contribute to IL-22 Production of ILC3s in Response to Bacteria by Production of IL-23 and IL-1β
To identify potential cellular sources of IL-23 and IL-1β in response to A. junii and R. bromii, production of IL-23 and IL-1β by mDCs (CD3- CD19- HLA-DR+ CD11c+), macrophages (defined as CD3- CD19- HLA-DR+ CD11c-) (48–50) and CD19+ B cells in LPMCs were determined by intracellular cytokine staining and flow cytometry following exposure to bacteria (Figure 5A). A. junii, but not R. bromii significantly increased the percentage of IL-23+ mDCs (Figure 5B and Supplemental Figure 6A). Although low frequencies of IL-23+ macrophages following A. junii stimulation were detected, this was not statistically different compared to no bacteria stimulation (Supplemental Figures 7A,B). Both R. bromii and A. junii significantly increased the percentages of IL-1β+ mDCs compared to no bacteria stimulation (Figure 5C and Supplemental Figure 6B). Exposure of LPMC to R. bromii or A. junii did not significantly induce IL-1β+ macrophages above no stimuli although IL-1β+ macrophages were detected (Supplemental Figures 7D,E). No significant increases in IL-23+ or IL-1+ B cells were observed in response to either bacteria stimuli (Supplemental Figure 7). Taken together, these observations suggested that mDCs in LPMCs were a major producer of the canonical cytokines known to drive IL-22 production by ILC3s. To verify the contribution of these cells to bacteria-induced IL-22 production by ILC3s, CD11c+ mDCs were depleted from LPMCs followed by exposure to A. junii and frequencies of IL-22+ ILC3s were determined. Depletion of mDCs significantly reduced the frequency of IL-22+ ILC3s in response to A. junii by 40.9% compared to total LPMCs (Figure 5D).
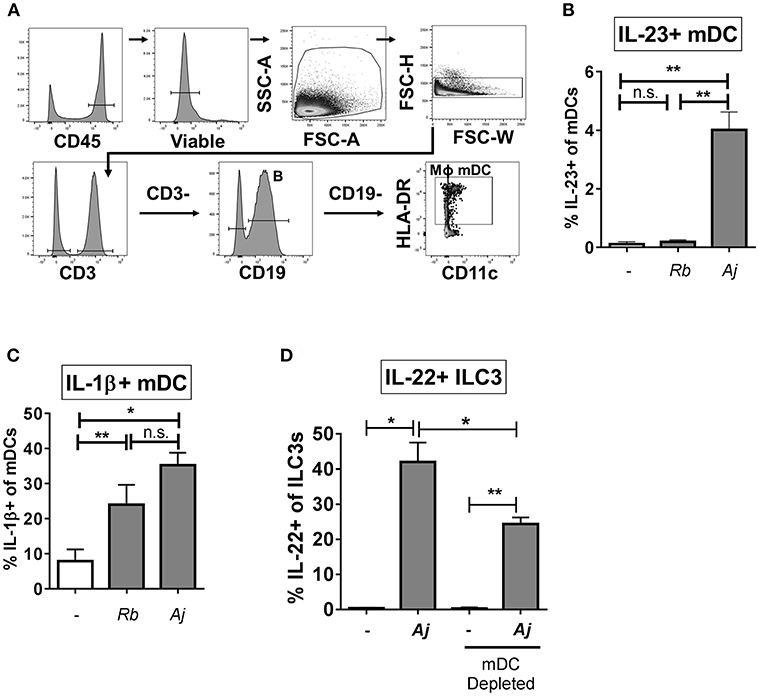
Figure 5. Myeloid dendritic cells producing IL-23 and IL-1β contribute to the ILC3 IL-22 response to bacteria. (A) Representative flow cytometry demonstrating gating strategy to identify antigen presenting cells (mDC; myeloid dendritic cells, B: B cells, MΦ; macrophages) after LPMC exposure to bacteria in vitro. (B) Percentages of IL-23+ or (C) IL-1β+ mDCs after LPMC exposure to R. bromii (Rb) or A. junii (Aj) or no bacteria control. N = 4. (D) Percentages of IL-22+ ILC3s after LPMC exposure to A. junii (Aj) or no bacteria control with CD11c mDC depletion and no depletion control. N = 3. Bars are mean + S.E.M. Statistical analysis performed was paired t-test. *p < 0.05, **p < 0.01. n.s., not significant.
NKp44 Ligation Contributes to the Gut ILC3 IL-22 Response to Enteric Bacteria
Recent work suggests a functional role for the natural cytotoxicity receptor NKp44 in driving cytokine responses of ILC3s (3), thus we sought to determine if NKp44 is critical to the IL-22 response of human colonic ILC3s to enteric bacteria. As noted previously, the majority of colon ILC3s expressed Nkp44 directly ex vivo (Figure 1C). Following exposure to enteric bacteria in vitro, a small increase in the percentage of ILC3s expressing NKp44 was noted (Figure 6A). Examination of the IL-22+ ILC3s after stimulation with bacteria revealed that the majority of IL-22-producing cells were also NKp44+ (Figure 6B). Direct ligation of NKp44 by crosslinking beads led to a significant increase in the percentage of IL-22+ ILC3s compared to the no bead control (Figure 6C). To evaluate the contribution of NKp44 to the ILC3 IL-22 response to bacteria, NKp44 was blocked in the presence of commensal A. junii. Compared to the IgG control, blocking NKp44 during A. junii stimulation resulted in a significant (IgG control: 31.08% ± 4.69, anti-NKp44: 25.52% ± 3.63) but incomplete reduction of IL-22+ ILC3s (Figure 6D). To determine if NKp44 promotion of IL-22 in ILC3s in response to bacteria was complementary or redundant with IL-23 and IL-1β, NKp44 was blocked in combination with blocking of IL-23 and IL-1β. Blocking Nkp44, IL-23, and IL-1β in combination did not further reduce the percentage of IL-22+ ILC3s generated in response to A. junii compared to only blocking IL-23 and IL-1β (Figure 6D).
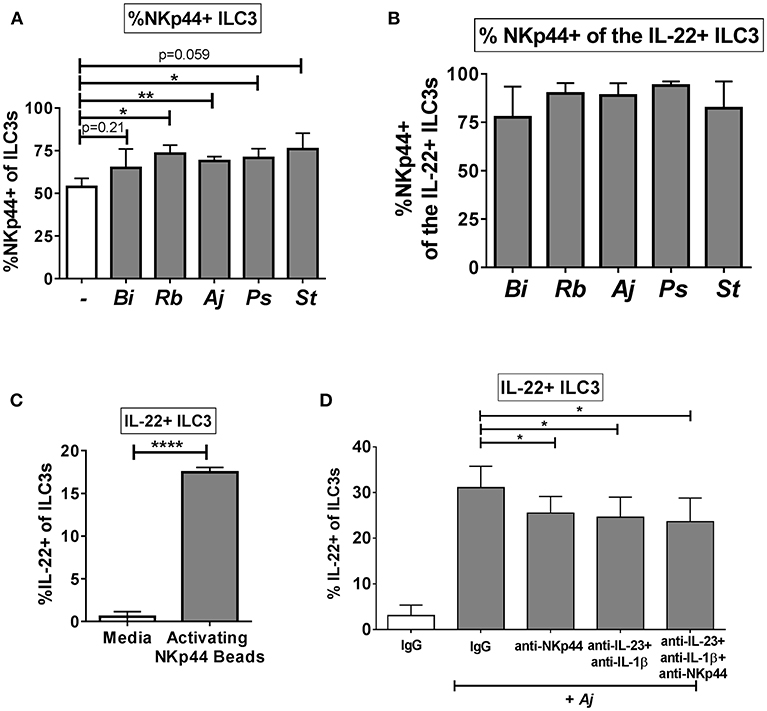
Figure 6. NKp44 contributes to the gut ILC3 IL-22 response to bacteria. (A) Percentages of NKp44 + ILC3s after LPMC exposure to enteric bacteria or no bacteria control. N = 5. (B) Percentages of NKp44+ when gating on IL-22 + ILC3s after LPMC exposure to enteric bacteria. N = 5. (C) Percentages of IL-22+ ILC3s after LPMC stimulation with NKp44 cross-linking beads. N = 3. (D) Percentages of IL-22+ ILC3s after LPMC exposure to no bacteria control or A. junii (Aj) in the presence of blocking antibodies targeting IL-23 and IL-1β (5 ug/mL) or NKp44 (10 ug/mL) or the antibody isotype controls IgG. N = 5. Bars are mean + S.E.M. Statistical analysis performed was paired t-test. *p < 0.05, **p < 0.01, ****p < 0.0001.
Bacteria-Induced IL-22 Production by ILC3s Is Enhanced in LPMCs Relative to TMCs
To evaluate if the ILC3 IL-22 response to bacteria is unique to the colonic environment, tonsil mononuclear cells (TMCs) were stimulated with commensal bacteria R.bromii and A.junii as well as the combination of recombinant IL-23 + IL-1β or NKp44 activating beads and induction of IL-22 by ILC3s determined by flow cytometry. The combination of IL-23+IL-1β significantly induced IL-22 production by tonsillar ILC3s as expected (Figure 7A). A small but significant increase in the percentage of IL-22+ ILC3s was observed following exposure to both commensal bacteria tested, whereas a similarly small increase in IL-22+ ILC3s following ligation of NKp44 was not statistically significant (Figure 7A). NKp44 expression on tonsillar ILC3s was characterized ex vivo and was not found to differ significantly from NKp44 expression on colon ILC3s (T: 60.17% ± 4.19, L: 74.21% ± 5.05) (Figure 7B). Despite the majority of the tonsillar ILC3s expressing the receptor NKp44, stimulation with NKp44 activation beads resulted in significantly lower percentages of IL-22+ ILC3s (T: 1.11% ± 1.11, L: 16.89% ± 0.12) compared to the colon (Figure 7C). Furthermore, when stimulated with bacteria, the percentage of tonsillar IL-22+ ILC3s was significantly lower in response to R. bromii (T: 1.11% ± 0.37, L: 18.67% ± 5.41) and A. junii (T: 0.93% ± 0.30, L: 21.38% ± 2.81) compared to the colon (Figures 7D,E). Although there was minimal induction of IL-22+ ILC3s in response to NKp44 ligation in tonsil cultures, the canonical cytokines, IL-23+IL-1β similarly induced ILC3 IL-22 responses (T: 8.15% ± 1.61, L: 10.79% ± 3.11) between the colon and the tonsil (Figure 7F).
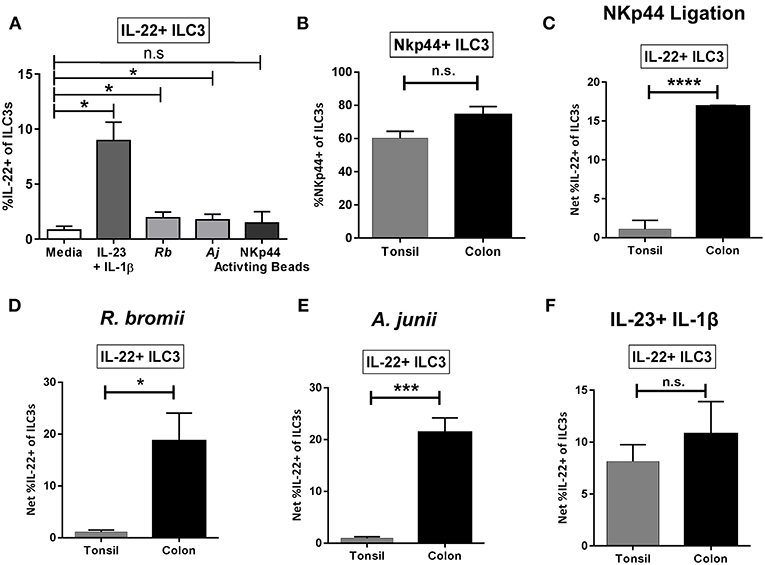
Figure 7. Tonsillar and gut ILC3 IL-22 responses to stimulation. (A) Percentages of IL-22+ ILC3s after TMC exposure in vitro to R. bromii, A. junii, NKp44 cross-linking beads, recombinant IL-23+ IL-1β (50 ng/mL) or no stimulation control. N = 5. Bars are mean + S.E.M. Statistical analysis performed was paired t-test. *p < 0.05, n.s., not significant. (B) Percentages of ILC3s expressing NKp44 ex vivo. TMC: N = 3, LPMC: N = 9. (C) Percentages of IL-22+ ILC3s after TMC or LPMC stimulation in vitro with NKp44 cross-linking beads. TMC: N = 5, LPMC: N = 3. (D) Percentages of IL-22+ ILC3s after TMC or LPMC stimulation with R. bromii. TMC: N = 5, LPMC: N = 7. (E) Percentages of IL-22+ ILC3s after TMC or LPMC stimulation with A. junii. TMC: N = 5, LPMC: N = 11. (F) Percentages of IL-22+ ILC3s after TMC or LPMC exposure to recombinant IL-23 + IL-1β (50 ng/mL). TMC: N = 5, LPMC: N = 6. Bars are mean + S.E.M. Statistical analysis performed was unpaired t-test. *p < 0.05, ***p < 0.001, ****p < 0.0001, n.s., not significant.
Discussion
Numerous murine studies have highlighted the importance of gut ILC3s in immunity to bacteria, but few studies have directly investigated how human lamina propria ILC3s respond to enteric bacteria, particularly commensal bacteria, and the mechanisms driving these responses. The major findings of the present study demonstrate that: (1) both Gram-positive and Gram-negative commensal and pathogenic bacteria induced similar frequencies of IL-22-producing ILC3s, (2) ILC3 production of IL-22 was not mediated through direct ILC3 recognition of bacteria, but rather mediated indirectly by mDCs, (3) IL-22 production was partly dependent on IL-23 and IL-1β, (4) and ligation of the NKp44 receptor stimulated IL-22 production. Overall, this work expands on the basic biology of human gut ILC3s and provides insight into their contribution to the innate immune response to enteric bacteria.
Similar frequencies of IL-22-producing ILC3s were induced irrespective of bacterial cell surface structure by Gram stain (i.e., Gram-positive or Gram-negative) or characterization as a human commensal or pathogenic bacteria suggesting a commonality between bacteria which drive ILC3 cytokine responses. In the context of gut ILC3 biology, this suggests that different enteric bacteria induce immune responses by LPMCs that are then “sensed” by ILC3s as the same. Using purified ILC3s, we demonstrated that induction of IL-22 was not due to direct recognition of bacteria or bacterial antigens. Presumably, direct recognition of bacteria by purified ILC3s would occur through external expression of TLRs specific to bacterial ligands. In keeping with this concept and a lack of direct induction of IL-22 by bacteria, we detected minimal expression of bacteria-associated TLRs on colonic ILC3s. This contrasts to a previous report demonstrating TLR2 expression by ILC3s in human duodenum (58) suggesting possible tissue site differences in PRR expression. Importantly, production of IL-22 by tonsillar ILC3s in response to TLR2 ligand required co-stimulation with IL-2, IL-15, or IL-23 (59).
We determined that IL-23 and IL-1β were important for bacteria-driven IL-22 production by human gut ILC3s similar to multiple murine studies highlighting IL-23-mediated regulation of IL-22 production in the context of pathogenic C. rodentium infection (12, 36) and responses to bacterial flagella (60, 61). IL-1β signaling through the IL-1R1 and MyD88 pathway was also shown to be critical for murine ILC3 production of IL-22 (62) and in human secondary lymphoid tissue, continuous IL-1β signaling was required to preserve the ILC capacity to produce IL-22 (63). Interestingly, in our in vitro model, frequencies of IL-22-producing ILC3s were not synergistically increased by the combined addition of IL-23 and IL-1β or decreased with antibody-mediated blocking of both cytokines in the context of bacteria exposure, suggesting that IL-23 and IL-1β may stimulate colonic ILC3 production of IL-22 in a redundant manner. Alternatively, ILC3s that express the IL-23R could also be the same subset that express the IL-1R indicating that only a fraction of ILC3s have to capacity to be stimulated in this manner. Taken together, these observations highlight a role for accessory cell mediated production of IL-23 and IL1β in regulating IL-22 from human gut ILC3s in response to enteric bacteria.
The importance of crosstalk between accessory cells such as mDCs and ILCs for induction of IL-22 has been implicated in a number of studies including the observation that a loss of a subset of mDCs correlated with a loss of IL-22-producing ILCs during SIV infection (25). Additionally, mouse intestinal mDC production of IL-23 in response to flagella derived from Salmonella was important in the stimulation of IL-22 from ILC3s (60, 61) and a need for physical contact between DCs and ILCs was necessary for the protective IL-22 response to infection with C. rodentium (64). We provide evidence that depletion of mDC resulted in a decrease in IL-22 production by ILC3s; however, purified colonic ILC3s responded directly to IL-23 and IL-1β, suggesting that contact dependence between human colonic ILC3s and mDCs may not be a requirement for induction of IL-22. Importantly, despite removing mDCs, complete abrogation of the bacteria-induced IL-22 response in ILC3s was not achieved, highlighting that other accessory cell types may contribute to the regulation of bacteria-specific ILC3 responses in the colon. Indeed, we detected low percentages of IL-23 and IL-1β-expressing macrophages in response to representative commensal bacteria in a subset of donors, suggesting that macrophages have the potential to contribute to the ILC3 IL-22 response to bacteria. A number of studies have implicated monocytes in driving IL-22 production. For example, soluble factors from LPS-activated human monocytes stimulated IL-22 from tonsillar ILC3s (17). In murine studies CX3CR1+ phagocytes (including mDCs and macrophages) stimulated a protective IL-22 ILC3 response during C. rodentium infection (65, 66) and mice deficient in CX3CR1+ phagocytes had impaired IL-22 production by ILC3s leading to increased microbial translocation and bacterial dissemination (65). Thus, exposure of multiple types of human gut antigen presenting cells to bacteria and the subsequent induction of IL-23 and IL-1β likely contributes to the production of IL-22 by ILC3s.
In Crohn's disease patients, loss of NKp44+ ILC3s from the ileum correlated with an increase in pro-inflammatory T cell subsets implying a role for NKp44+ ILC3s in mucosal regulation (67). In our study, we show that the majority of IL-22-producing ILC3s expressed the NKp44 receptor and direct ligation of the NKp44 receptor induced IL-22 from colonic ILC3s. Interestingly, the role of NKp44 in directly driving IL-22 production was unique to the gut and not observed with tonsillar ILC3s in keeping with a previous report on NKp44 ligation of tonsillar ILC3s (3). This is intriguing since we show that there are similar percentages of NKp44-expressing ILC3s ex vivo, as well as, similar percentages of IL-22-expressing ILC3s after in vitro exposure to exogenous IL-23 and IL-1β in both the tonsil and colon. In the context of bacterial stimulation, blocking NKp44 signaling partially reduced the percentage of IL-22-producing ILC3s demonstrating that NKp44 also plays a contributory role in cytokine stimulation of ILC3s. Ligands for NKp44 have been identified during pathological conditions such as tumor development or immortalized cell lines of cancerous origin, and include an isoform of the protein MLL5, proliferating cell nuclear antigen (PCNA), and platelet derived growth factor (PDGF)-DD (5, 68, 69). Further studies will be needed to determine if these, or yet to be identified ligands, can stimulate IL-22 production by ILC3s in our in vitro model. Interestingly, NKp44 has been shown to directly bind to bacteria (70) as well as viral hemagglutinin of influenza (71), thus, investigations will need to be undertaken to determine if enteric bacteria are an additional source of NKp44L. It is possible that other factors not identified here may contribute to the IL-22 response by ILC3s by bacteria. A recent study determined that IL-18 production by mDCs, in conjunction with IL-15, was able to induce IL-22 in tonsillar ILC3s after longer term exposure (14 days) to these cytokines (72). It is therefore possible, that similar combinations of signals could induce IL-22 from colonic ILC3s, although if there is a role for enteric bacteria in initiating this signaling cascade would need to be determined.
Reports have indicated that IFNγ production by ILC3s is possible in the context of murine models of GI bacterial infection (41, 42). Furthermore, in vitro human tonsillar ILC3s can be functionally plastic in terms of cytokine production including a switch from IL-22 to IFNγ production dependent on the cytokine milieu (18, 21). In this current study, exposure of LPMC to enteric bacteria did not induce a significant increase in IFNγ+ ILC3s suggesting that while our in vitro model has the appropriate microenvironment to drive IL-22 production from ILC3s, additional signals would be required for robust IFNγ production. It is possible that cellular movement as is seen in vivo could promote interactions that may be needed for IFNγ induction in response to bacteria, and the in vitro model used in this study may not recapitulate those interactions. It is also important to note that in our model system, broad spectrum antibiotics were present to prevent bacterial overgrowth. Thus, functional responses are likely driven by static whole bacteria and/or processed bacterial antigens. Perhaps when bacteria are able to be metabolically active and/or replicate, additional components (such as virulence factors in regards to S. typhimurium) would be produced leading to the subsequent production of IFNγ from ILC3s.
To the best of our knowledge, this study is the first to undertake an extensive evaluation of the mechanisms by which in vitro exposure to whole enteric commensal and pathogenic bacteria drive human colonic ILC3 cytokine production. Here we demonstrate that IL-22 production was driven indirectly in LPMCs, mediated in part by mDCs, and driven by multiple mechanisms including IL-23, IL-1β, and NKp44 signaling. The complexity of the gut environment was emphasized by the observation that the combination of these processes did not fully account for all of the bacteria-driven IL-22 responses. Remarkably, IL-22 production from ILC3s were induced in response to all bacteria tested, potentially highlighting an evolutionarily conserved response by ILC3s to both commensal and pathogenic bacterial antigens. IL-22 has important gut homeostatic functions and thus the production of IL-22 by ILC3s would be a critical component of the innate response to enteric pathogenic challenge. This function of ILC3s may serve as a means to repair the epithelial barrier during disease states when there is a breach in epithelium integrity to prevent the induction of further inflammation and damage by translocating microbes. Our in vitro observations highlight that production of IL-22 by ILC3s in response to commensal bacteria is also likely a significant component of GI tract bacterial immunity.
Author Contributions
MC, SD, AC, EB, and CW designed the study and interpreted the work. MC, SD, and CW wrote the manuscript. MC and CP performed experiments. MM and MS provided tissue specimens. JK provided technical assistance and interpretation of the work. All authors contributed to manuscript revision, and read and approved the submitted version.
Funding
This work was supported by National Institute of Health Grants R01 AI118983 (EB, CW) and T32 AI007405 (MC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Flow Cytometry Shared Resource at the University of Colorado Cancer Center (P30CA046934) for technical assistance with sorting ILC3s. We also acknowledge the help of Sabrina Nesladek, Allison Christians, and other members of the Wilson lab for technical assistance with processing of the tissue specimens.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00649/full#supplementary-material
References
1. Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity. (2012) 37:601–10. doi: 10.1016/j.immuni.2012.10.003
2. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells: a new paradigm in immunology. Science. (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
3. Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, et al. RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity. (2013) 38:1223–35. doi: 10.1016/j.immuni.2013.05.013
4. Killig M, Glatzer T, Romagnani C. Recognition strategies of group 3 innate lymphoid cells. Front Immunol. (2014) 5:142. doi: 10.3389/fimmu.2014.00142
5. Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell. (2018) 172:534–48 e519. doi: 10.1016/j.cell.2017.11.037
6. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
7. Sonnenberg GF. Regulation of intestinal health and disease by innate lymphoid cells. Int Immunol. (2014) 26:501–7. doi: 10.1093/intimm/dxu052
8. Bostick JW, Zhou L. Innate lymphoid cells in intestinal immunity and inflammation. Cell Mol Life Sci. (2016) 73:237–52. doi: 10.1007/s00018-015-2055-3
9. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/ni.3489
10. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. (2011a) 12:383–90. doi: 10.1038/ni.2025
11. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
12. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. (2008) 14:282–9. doi: 10.1038/nm1720
13. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. (2009) 10:83–91. doi: 10.1038/ni.1684
14. Sanos SL, Vonarbourg C, Mortha A, Diefenbach A. Control of epithelial cell function by interleukin-22-producing RORgammat+ innate lymphoid cells. Immunology. (2011) 132:453–65. doi: 10.1111/j.1365-2567.2011.03410.x
15. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. (2012) 336:1321–5. doi: 10.1126/science.1222551
16. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
17. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. (2009) 457:722–5. doi: 10.1038/nature07537
18. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and−23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
19. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn's disease. Gastroenterology. (2010) 139:882–92, 892 e881–3. doi: 10.1053/j.gastro.2010.05.040
20. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
21. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. (2013) 14:221–9. doi: 10.1038/ni.2534
22. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
23. Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, et al. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J Clin Invest. (2015) 125:3692–703. doi: 10.1172/JCI82124
24. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, et al. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood. (2011) 118:3321–30. doi: 10.1182/blood-2011-04-347260
25. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. (2012) 5:646–57. doi: 10.1038/mi.2012.38
26. Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. (2012) 5:658–69. doi: 10.1038/mi.2012.39
27. Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, et al. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog. (2014) 10:e1004551. doi: 10.1371/journal.ppat.1004551
28. Xu H, Wang X, Lackner AA, Veazey RS. Type 3 innate lymphoid cell depletion is mediated by TLRs in lymphoid tissues of simian immunodeficiency virus-infected macaques. FASEB J. (2015) 29:5072–80. doi: 10.1096/fj.15-276477
29. Dillon SM, Castleman MJ, Frank DN, Austin GL, Gianella S, Cogswell AC, et al. Inflammatory colonic innate lymphoid cells are increased during untreated HIV-1 infection and associated with markers of gut dysbiosis and mucosal immune activation. J Acquir Immune Defic Syndr. (2017) 76:431–7. doi: 10.1097/QAI.0000000000001523
30. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-γ and tumor necrosis factor-α synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. (2005) 166:409–19. doi: 10.1016/S0002-9440(10)62264-X
31. Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res. (2009) 29:133–44. doi: 10.1089/jir.2008.0057
32. Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev. (2013) 26:2–18. doi: 10.1128/CMR.00050-12
33. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. (2013) 4:280. doi: 10.3389/fimmu.2013.00280
34. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
35. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. (2015) 163:381–93. doi: 10.1016/j.cell.2015.08.061
36. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. (2011b) 34:122–34. doi: 10.1016/j.immuni.2010.12.009
37. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. (2014) 40:25–39. doi: 10.1016/j.immuni.2013.10.021
38. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. (2014) 345:1254009. doi: 10.1126/science.1254009
39. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
40. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science. (2015) 348:1031–5. doi: 10.1126/science.aaa4812
41. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. (2013) 494:261–5. doi: 10.1038/nature11813
42. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
43. Howe R, Dillon S, Rogers L, McCarter M, Kelly C, Gonzalez R, et al. Evidence for dendritic cell-dependent CD4(+) T helper-1 type responses to commensal bacteria in normal human intestinal lamina propria. Clin Immunol. (2009) 131:317–32. doi: 10.1016/j.clim.2008.12.003
44. Dillon SM, Rogers LM, Howe R, Hostetler LA, Buhrman J, McCarter MD, et al. Human intestinal lamina propria CD1c+ dendritic cells display an activated phenotype at steady state and produce IL-23 in response to TLR7/8 stimulation. J Immunol. (2010) 184:6612–21. doi: 10.4049/jimmunol.1000041
45. Miller SM, Miles B, Guo K, Folkvord J, Meditz AL, McCarter MD, et al. Follicular regulatory T cells are highly permissive to R5-tropic HIV-1. J Virol. (2017) 91:e00430–17. doi: 10.1128/JVI.00430-17
46. Dillon SM, Lee EJ, Kotter CV, Austin GL, Dong Z, Hecht DK, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. (2014) 7:983–94. doi: 10.1038/mi.2013.116
47. Dillon SM, Lee EJ, Donovan AM, Guo K, Harper MS, Frank DN, et al. Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology. (2016) 13:5. doi: 10.1186/s12977-016-0237-1
48. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. (2011) 4:31–42. doi: 10.1038/mi.2010.66
49. Bain CC, Mowat AM. The monocyte-macrophage axis in the intestine. Cell Immunol. (2014b) 291:41–8. doi: 10.1016/j.cellimm.2014.03.012
50. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev. (2014a) 260:102–17. doi: 10.1111/imr.12192
51. Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol. (2016) 17:451–60. doi: 10.1038/ni.3368
52. Schultz BM, Paduro CA, Salazar GA, Salazar-Echegarai FJ, Sebastian VP, Riedel CA, et al. A potential role of salmonella infection in the onset of inflammatory bowel diseases. Front Immunol. (2017) 8:191. doi: 10.3389/fimmu.2017.00191
53. Hung CC, Hung MN, Hsueh PR, Chang SY, Chen MY, Hsieh SM, et al. Risk of recurrent nontyphoid Salmonella bacteremia in HIV-infected patients in the era of highly active antiretroviral therapy and an increasing trend of fluoroquinolone resistance. Clin Infect Dis. (2007) 45:e60–67. doi: 10.1086/520681
54. Taramasso L, Tatarelli P, Di Biagio A. Bloodstream infections in HIV-infected patients. Virulence. (2016) 7:320–8. doi: 10.1080/21505594.2016.1158359
55. d'Ettorre G, Ceccarelli G, Giustini N, Serafino S, Calantone N, De Girolamo G, et al. Probiotics reduce inflammation in antiretroviral treated, HIV-infected individuals: results of the “Probio-HIV” clinical trial. PLoS ONE. (2015) 10:e0137200. doi: 10.1371/journal.pone.0137200
56. Eom T, Kim YS, Choi CH, Sadowsky MJ, Unno T. Current understanding of microbiota- and dietary-therapies for treating inflammatory bowel disease. J Microbiol. (2018) 56:189–98. doi: 10.1007/s12275-018-8049-8
57. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA. (2010) 107:10961–6. doi: 10.1073/pnas.1005641107
58. Marafini I, Monteleone I, Di Fusco D, Cupi ML, Paoluzi OA, Colantoni A, et al. TNF-alpha producing innate lymphoid cells (ILCs) are increased in active celiac disease and contribute to promote intestinal atrophy in mice. PLoS ONE. (2015) 10:e0126291. doi: 10.1371/journal.pone.0126291
59. Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. (2010) 33:752–64. doi: 10.1016/j.immuni.2010.10.012
60. Van Maele L, Carnoy C, Cayet D, Songhet P, Dumoutier L, Ferrero I, et al. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127+ immune cells in spleen and mucosa. J Immunol. (2010) 185:1177–85. doi: 10.4049/jimmunol.1000115
61. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
62. Reynders A, Yessaad N, Vu Manh TP, Dalod M, Fenis A, Aubry C, et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J. (2011) 30:2934–47. doi: 10.1038/emboj.2011.201
63. Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity. (2010) 32:803–14. doi: 10.1016/j.immuni.2010.06.007
64. Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. (2011) 10:44–53. doi: 10.1016/j.chom.2011.06.002
65. Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, et al. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. (2013) 6:177–88. doi: 10.1038/mi.2012.61
66. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. (2014) 211:1571–83. doi: 10.1084/jem.20140678
67. Li J, Doty AL, Tang Y, Berrie D, Iqbal A, Tan SA, et al. Enrichment of IL-17A(+) IFN-gamma(+) and IL-22(+) IFN-gamma(+) T cell subsets is associated with reduction of NKp44(+) ILC3s in the terminal ileum of Crohn's disease patients. Clin Exp Immunol. (2017) 190:143–53. doi: 10.1111/cei.12996
68. Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F, et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J Immunol. (2011) 187:5693–702. doi: 10.4049/jimmunol.1102267
69. Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debre P, Vieillard V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood. (2013) 122:2935–42. doi: 10.1182/blood-2013-03-489054
70. Esin S, Batoni G, Counoupas C, Stringaro A, Brancatisano FL, Colone M, et al. Direct binding of human NK cell natural cytotoxicity receptor NKp44 to the surfaces of mycobacteria and other bacteria. Infect Immun. (2008) 76:1719–27. doi: 10.1128/IAI.00870-07
71. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. (2001) 31:2680–9. doi: 10.1002/1521-4141(200109)31:9<2680::AID-IMMU2680>3.0.CO;2-A
Keywords: human, innate lymphoid cells, commensal bacteria, colonic mucosa, NKp44, myeloid dendritic cells, IL-23, IL-1β
Citation: Castleman MJ, Dillon SM, Purba CM, Cogswell AC, Kibbie JJ, McCarter MD, Santiago ML, Barker E and Wilson CC (2019) Commensal and Pathogenic Bacteria Indirectly Induce IL-22 but Not IFNγ Production From Human Colonic ILC3s via Multiple Mechanisms. Front. Immunol. 10:649. doi: 10.3389/fimmu.2019.00649
Received: 05 December 2018; Accepted: 11 March 2019;
Published: 29 March 2019.
Edited by:
Joanna Kirman, University of Otago, New ZealandReviewed by:
Carolina V. Arancibia, University of Oxford, United KingdomJorge Caamano, University of Birmingham, United Kingdom
Copyright © 2019 Castleman, Dillon, Purba, Cogswell, Kibbie, McCarter, Santiago, Barker and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cara C. Wilson, Y2FyYS53aWxzb25AdWNkZW52ZXIuZWR1