 Cuiling Zhang
Cuiling Zhang Giulietta Maruggi
Giulietta Maruggi Hu Shan
Hu Shan Junwei Li
Junwei Li- 1College of Veterinary Medicine, Qingdao Agricultural University, Qingdao, China
- 2GSK, Rockville, MD, United States
During the last two decades, there has been broad interest in RNA-based technologies for the development of prophylactic and therapeutic vaccines. Preclinical and clinical trials have shown that mRNA vaccines provide a safe and long-lasting immune response in animal models and humans. In this review, we summarize current research progress on mRNA vaccines, which have the potential to be quick-manufactured and to become powerful tools against infectious disease and we highlight the bright future of their design and applications.
Introduction
Vaccination is the most successful medical approach to disease prevention and control. The successful development and use of vaccines has saved thousands of lives and large amounts of money. In the future, vaccines have the potential to be used not only against infectious diseases but also for cancer as a prophylactic and treatment tool, and for elimination of allergens (1–3). Prior to the 1980s, vaccines were developed for protection against disease-causing microorganisms. Empirically, inactivated vaccines were produced by heat or chemical treatment, and live attenuated vaccines were generally developed in animals, cell lines or unfavorable growth conditions. During vaccine development, the mechanisms involved in conferring immunity were unknown. Nevertheless, the use of live attenuated or killed whole organism-based vaccines had enormous success in the control and eradication of a number of severe human infectious diseases, including smallpox, polio, measles, mumps, rubella, and animal infectious disease, such as classic swine fever, cattle plague, and equine infectious anemia. More recently, live attenuated (LAV), subunit and peptide based vaccines have been developed thanks to advancements in molecular biology theory and technologies. The results obtained with LAV vaccination dramatically expanded our knowledge of the mechanisms related to the immune response elicited by these vaccines. For inactivated vaccines, antigen-specific antibodies largely contribute to the prevention and control of microbe-initiated infectious disease. In addition to specific humoral immune responses. LAVs elicit strong cellular immune responses, which are critical to eradicate many intracellular pathogens. Nevertheless, the failures that are sometimes caused by inactivated vaccines are ascribed to mutation of the surface antigens of pathogens. Additional concerns about LAV applications include the potential to cause disease in immuno-compromised individuals and the possibility of reversion to a virulent form due to the back-mutation, the acquisition of compensatory mutations, or recombination with circulating transmissible wild-type strains (4, 5). Nevertheless, subunit and peptide vaccines are less effective at eliciting a robust CD8+ immune response, which is important for intracellular pathogens, including viruses and some bacteria (6, 7).
Vaccination with non-viral delivered nucleic acid-based vaccines mimics infection or immunization with live microorganisms and stimulates potent T follicular helper and germinal center B cell immune response (8, 9). Furthermore, non-viral delivered nucleic acid-based vaccine manufacturing is safe and time-saving, without the growth of highly pathogenic organisms at a large scale and less risks from contamination with live infectious reagents and the release of dangerous pathogens. Notably, for most emerging and re-emerging devastating infectious diseases, the main obstacle is obtaining a stockpile in a short timeframe (10). Non-viral delivered nucleic acid-based vaccines can fill the gap between a disease epidemic and a desperately needed vaccine (10). Non-viral delivered nucleic acids are categorized as DNA or RNA according to their type of 5-carbon sugar. From being administrated to antigen expression, DNA vaccine and RNA vaccines are processed through different pathways. In the steps between immunization with a DNA template and expression of the target antigen, the DNA has to overcome the cytoplasmic membrane and nuclear membrane, be transcribed into mRNA, and move back into the cytoplasm and initiate translation (refer to Figure 1). Although promising and with shown safety, well-tolerability and immunogenicity, DNA vaccines were characterized by suboptimal potency in early clinical trials (11). Enhanced delivery technologies, such as electroporation, have increased the efficacy of DNA vaccines in humans (12), but have not reduced the potential risk of integration of exogenous DNA into the host genome, which may cause severe mutagenesis and induced new diseases (13, 14). Since naked in vitro transcribed mRNA was found to be expressed in vivo after direct injection into mouse muscle, mRNA has been investigated extensively as a preventive and therapeutic platform (15–19). Due to the dramatic development of RNA-based vaccine studies and applications, a plethora of mRNA vaccines have entered into clinical trial (19). Comparatively, mRNA vaccines confer several advantages over viral vectored vaccines and DNA vaccines (summary in Table 1). The utilization of RNA as a therapeutic tool is not the focus of this manuscript and has been extensively reviewed elsewhere (2, 19, 20). In this review, we provide highlights on mRNA vaccines as promising tools in the prevention and control of infectious disease.
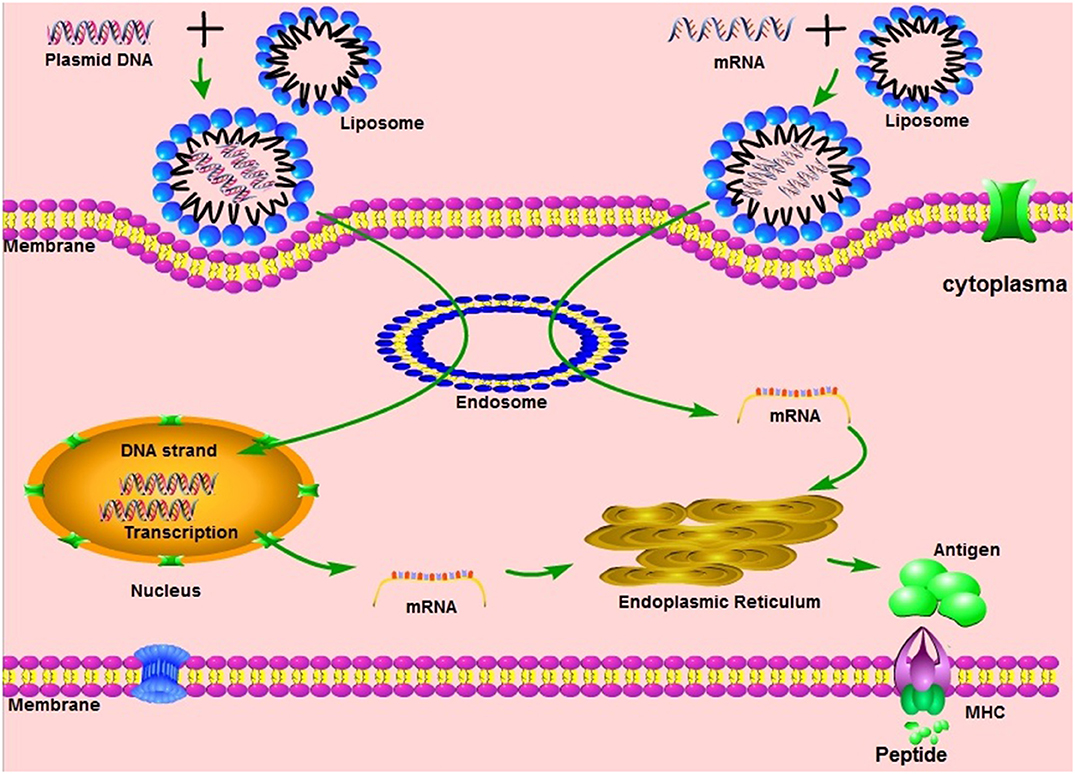
Figure 1. The mechanisms of different nucleic acid vaccines, including DNA vaccines, mRNA vaccines. MHC, Major histocompatibility complex.
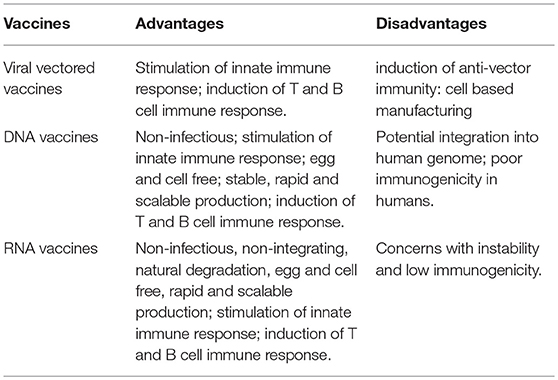
Table 1. Advantages and disadvantages of viral vectored vaccines, DNA vaccines and RNA vaccines.
Conception and Forms of mRNA Vaccines
mRNA vaccines were reported to be effective for direct gene transfer for the first time by Woff et al. (15). Currently, two forms of mRNA vaccines have been developed: conventional mRNA vaccines and self-amplifying mRNA vaccines, which are derived from positive strand RNA viruses. Although mRNA vaccines were first tested in the early 1990s, these vaccines were not initially extensively utilized due to concerns about their fragile stability caused by omnipresent ribonucleases and small-scale production. Initial demonstration that mRNA stability can be improved by optimization and formulation was published by Ross and colleagues in 1995 (21). Since that time, studies on mRNA vaccines have exploded and mRNA can now be synthetically produced, through a cell-free enzymatic transcription reaction. The in vitro transcription reaction includes a linearized plasmid DNA encoding the mRNA vaccine, as a template, a recombinant RNA polymerase, and nucleoside triphosphates as essential components. A cap structure is enzymatically added to the transcriptional product at the end of the reaction or as a synthetic cap analog in a single step procedure. Finally, a poly(A) tail will be provided to form a mature mRNA sequence.
Conventional mRNA vaccines include in their simplest an ORF for the target antigen, flanked by untranslated regions (UTRs) and with a terminal poly(A) tail. After transfection, they drive transient antigen expression. In addition to conventional vaccines, there is another mRNA vaccine platform based on the genome of positive strand viruses, most commonly alphaviruses. These mRNA vaccines are based on an engineered viral genome containing the genes encoding the RNA replication machinery whereas the structural protein sequences are replaced with the gene of interest (GoI) and the resulting genomes are referred as replicons. These vaccines are named self-amplifying mRNA and are capable of directing their self-replication, through synthesis of the RNA-dependent RNA polymerase complex, generating multiple copies of the antigen-encoding mRNA, and express high levels of the heterologous gene when they are introduced into the cytoplasm of host cells, in a way that mimics production of antigens in vivo by viral pathogens, triggering both humoral and cellular immune responses (22–27). Self-amplifying mRNA can be derived from the engineered genomes of Sindbis virus, Semliki Forest virus, Kunjin virus, among others (28–30). Self-amplifying mRNAs (~9–11 kb) are generated from the DNA template with similar procedures to those previously described for conventional mRNAs and RNA molecules can be produced at a large scale in vitro. After the purified RNA replicon is delivered into host cells, either as viral particles or as synthetically formulated RNA, it is translated extensively and amplified by its encoding RNA-dependent RNA polymerase. Compared with the rapid expression of conventional mRNAs, published results have shown that vaccination with self-amplifying mRNA vaccines results in higher antigen expression levels, although delayed in time, which persist for several days in vivo. Equivalent protection is conferred but at a much lower RNA dose (31). Due to the lack of viral structural proteins, the replicon does not produce infectious viral particles. Additionally, both conventional mRNA and self-amplifying mRNAs cannot potentially integrate into the host genome and will be degraded naturally during the process of antigen expression. These characteristics indicate that mRNA vaccines have the potential to be much safer than other vaccines and are a promising vaccine platform.
Engineered mRNA With Potent Efficiency
Stability and translation of mRNA is crucial for a successful RNA vaccine (32, 33). In the process of translation, mRNA purity is critical to determine its stability and protein yield (34). Contamination with dsRNAs, derived from aberrant RNA polymerase activities, leads to the inhibition of translation and degradation of cellular mRNA and ribosomal RNA, thus decreasing protein expression by interrupting the translation machine. The removal of dsRNA can increase translation dramatically (35). Excess components and short or double strand RNAs (dsRNA) can be removed by purification. Initially, lithium chloride (LiCl) was used for this purpose, but it restricted the industrialization of mRNA vaccines and it did not remove dsRNAs. Purification via fast protein liquid chromatography (FPLC) or high-performance liquid chromatography (HPLC) could be utilized to remove any remaining product and produce mRNA at a large scale and for Good Manufacturing Practice (GMP) processes (35–37). Non-coding sequence flanking 5′ and 3′ terminal of open reading frame (ORF) is crucial for translation. The 5′ untranslated region, such as kozak sequence, or 5′ caps is required for efficient protein production (38–40). The 3′ untranslated region containing optimal poly(A) signal determined the stability of mRNA and increased protein translation (41–45). Additionally, codon optimization is a popular method to avoid rare codons with low utilization, to increase protein production, mRNA abundance and stability (46–49).
mRNA vaccines are efficient at antigen expression, but sequence and secondary structures formed by mRNAs are recognized by a number of innate immune receptors, and this recognition can inhibit protein translation. Thanks to advancement in RNA biology understanding, several methods can be employed to increase the potency of mRNA vaccines, including sequence optimization and usage of modified nucleosides. Recognition from innate immune sensors can be avoided by incorporating modified nucleosides, such as pseudouridine (Ψ), 5-methylcytidine (5 mC), cap-1 structure and optimized codons, which in turns improve translation efficiency (50–55). During the in vitro transcription of mRNA, immature mRNA would be produced as contamination which inhibited translation through stimulating innate immune activation. FPLC and HPLC purification could tackle this problem (35, 37).
Currently, most vaccines in use, with the exception of some animal vaccines, need to be transported and stored in an uninterrupted cold-chain process, which is prone to failure, especially in poor rural areas of tropical countries; these requirements are not being met by available effective vaccines to prevent and control infectious diseases. Therefore, the development of thermostable vaccines has been gaining interest. Optimization in formulation of synthetic mRNA vaccines have shown that it is possible to generate thermostable vaccines. The results described by Jones showed that freeze-dried mRNA with trehalose or naked mRNA is stable for at least 10 months at 4°C. After being transfected, these mRNAs expressed high levels of proteins and conferred highly effective and long-lasting immunity in newborn and elderly animal models (56). Another lyophilized mRNA vaccine was shown to be stable at 5–25°C for 36 months and 40°C for 6 months (57). Stitz and colleagues showed that when a protamine-encapsulated conventional mRNA-based rabies virus vaccine was subjected to oscillating temperatures between 4 and 56°C for 20 cycles and exposure 70°C, its immunogenicity and protective effects were not compromised (58). Encapsulation of mRNA with cationic liposome or cell penetrating peptide (CPP) protected mRNA from degradation by RNase. These intriguing approaches would be discussed in delivery methods.
RNA Vaccines in the Prevention of Infectious Disease
During the last two decades, mRNA vaccines have been investigated extensively for infectious disease prevention, and for cancer prophylaxis and therapy. Much progress has been made thus far (19, 20). Cancer mRNA vaccines were designed to express tumor-associated antigens that stimulate cell-mediated immune responses to clear or inhibit cancer cells (59). Most cancer vaccine are investigated more as therapeutics than prophylactics and have been reviewed elsewhere (20, 60, 61). mRNA vaccines against infectious diseases could be developed as prophylactic or therapeutic. mRNA vaccines expressing antigen of infectious pathogen induce both strong and potent T cell and humoral immune responses (8, 16, 19). As previously described the production procedure to generate mRNA vaccines is entirely cell-free, simple and rapid if compared to production of whole microbe, live attenuated and subunit vaccines. This fast and simple manufacturing process makes mRNA a promising bio-product that can potentially fill the gap between emerging infectious disease and the desperate need for effective vaccines. Producing RNA at a large scale to satisfy commercialization is the first step toward making mRNA vaccines. Currently, all components needed for mRNA production are available at the GMP grade; however, some components are supplied at a limited scale.
A great deal of research has been initially conducted on the development of cancer mRNA vaccines and has demonstrated the feasibility of producing clinical grade in vitro transcribed RNA (60). Several projects on mRNA vaccines against infectious disease have also been conducted, although clinical evaluation is still limited. For example, several RNA-based vaccine platforms have been utilized for the development of influenza vaccines. Several published results showed that RNA-based influenza vaccines induce a broadly protective immune response against not only homologous but also hetero-subtypic influenza viruses (62–66). Influenza mRNA vaccines hold great promises being an egg-free platform, and leading to production of antigen with high fidelity in mammalian cells. Recent published results demonstrated that the loss of a glycosylation site by a mutation in the hemagglutinin (HA) of the egg-adapted H3N2 vaccine strain resulted in poor neutralization of circulating H3N2 viruses in vaccinated humans and ferrets. In contrast, the process of mRNA vaccine production is egg-free, and mRNA-encoded proteins are properly folded and glycosylated in host cells after vaccine administration, thus avoiding the risk of producing incorrect antigens (67, 68).
mRNA has also been used in the veterinary field to prevent animal infectious diseases. Pulido et al. demonstrated that immunization with in vitro transcribed mRNA induced protection against foot and mouse disease virus in mice (69). Saxena and colleagues demonstrated that a self-amplifying mRNA vaccine encoding rabies virus glycoprotein induced an immune response and provided protection in mice and could potentially be used to prevent rabies in canine (70). Recently, VanBlargan et al. developed a lipid nanoparticle (LNP)-encapsulated modified mRNA vaccine encoding prM and E genes of deer powassan virus (POWV). This mRNA vaccine induced robust humoral immune response not only against POWV strains but also against the distantly related Langat virus (71). As described previously, modification of nucleosides and optimization of codons can avoid recognition by innate immune sensors to improve translation efficiency. In Table 2, studies conducted with nucleoside modified and non-modified mRNA vaccines for infectious disease are summarized (52, 58, 72–78).
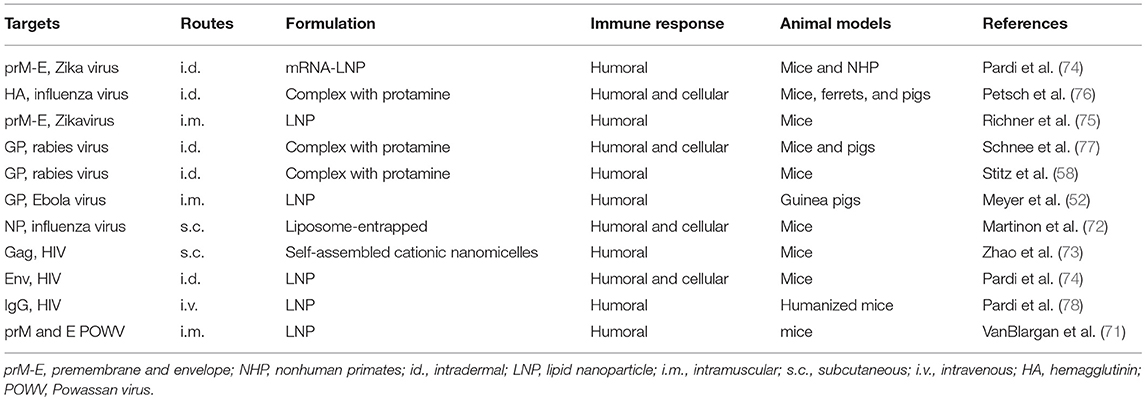
Table 2. Nucleoside modified or non-modified mRNA vaccines against infectious diseases.
Besides being used as vaccine, mRNA could also be deployed for therapeutic purposes. Interestingly, a recent publication by Pardi and colleagues showed that the adnimistration of mRNA encoding the light and heavy chains of a broadly neutralizing anti-HIV antibody encapsulated in lipid nanoparticles (LNPs) protected humanized mice from intravenous HIV challenge (79). The data suggest that the utilization of nucleoside-modified mRNA can be expanded for passive immunotherapy against HIV, cytomegalovirus (CMV), human papiloma virus, etc. Self-amplifying mRNA vaccines enable large amounts of prompt antigen expression and potent T cellular immune responses. In Table 3, we summarize publications on self-amplifying mRNA vaccines for infectious disease, delivered as viral replicon particles or synthetic formulated mRNA (80, 82–85, 87, 91, 92).
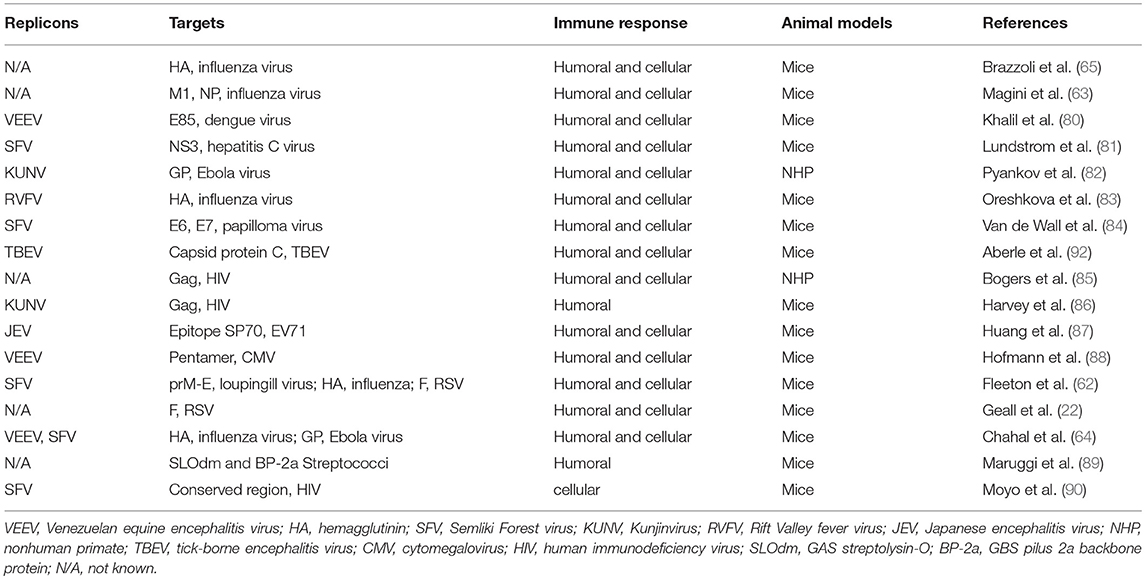
Table 3. Self-amplifying mRNA vaccines against infectious diseases.
Delivery Route and Formulation of mRNA Vaccines
The administration route and formulation of mRNA vaccines are crucial to determine the kinetics and magnitude of antigen expression as well as the potency of the immune response. For example, intravenous administration of unmodified naked mRNA resulted in rapid digestion by ribonucleases and stimulation of the innate immune response, but these limitations can be overcome by appropriate delivery systems and mRNA modifications (93). mRNA vaccines are administered via a systemic or local method based on antigen expression localization requirements. Direct intramuscular (i.m), intradermal (i.d.) or subcutaneous injection of in vitro transcribed mRNA are the main delivery routes for mRNA vaccines against infectious diseases, while intraperitoneal (i.p.) and intravenous (i.v.) administration are employed when systemic expression of antigens of interest is needed, mostly for therapeutic applications. Multiple reports have been recently published and showed that a variety of antigens can be expressed with high efficiency and induced potent humoral and cellular immune responses after mRNA vaccination. Lipid nanoparticles (LNP) loaded with nucleoside modified conventional mRNA encoding firefly luciferase have been used, for example, to examine the influence of route of administration on kinetics of antigen expression (94). i.m. and i.d. injections offered the best levels and duration of effect, with protein production peaking at 4 h and maintained locally 8–10 days post injection, depending on the dose. Both i.m. and i.d. administration in Rhesus macaques with nucleoside modified conventional mRNA encoding influenza H10 encapsulated in LNP induced protective titers, but this response occurred more rapidly by i.d. administration than by i.m. administration (95).
CV7201 is an mRNA vaccine candidate under development by CureVac AG. i.d. and i.m. injection ofCV7201 in mice and pigs induced potent humoral and T cell immune responses (77). In a phase I clinical trial, CV7201 showed long-term safety and immunogenicity against the rabies virus at alow dose. No differences were observed in terms of safety between i.d and i.m. administration or between needle-syringe or needle-free injection of CV7201. However, when neutralizing antibody titers induced by CV7201 were evaluated, needle-free administration was superior to injection with a needle (57). In an influenza vaccine test, the intranodal (i.n.) delivery of naked mRNA elicited potent CD4 and CD8 T cell immune responses in mice, and repeated i.n. injection with modified mRNA led to priming antigen-specific CD4 and CD8 T cells, whereas subcutaneous, i.d. administration did not (96). Combination with two or more delivery methods have been explored and employed in cancer mRNA vaccine development. The combination of i.v. and i.d. injection of TriMix-DC-MEL therapy showed favorable outcomes in patients with broad CD8 and CD4 T cell immune responses (97). Further studies demonstrated that i.n. and intratumor injection with TriMix mRNA into dendritic cells achieved better therapeutic outcomes than alternate injection sites (98, 99). However, i.d. administration of RNActive vaccines presented a similar immune response to the i.n. administration of conventional mRNA vaccines, which was an inconsistent result (100). Altogether, these results highlight the importance of the delivery route for effective mRNA vaccines.
Similarly, Fleeton et al. showed that i.m. injection of in vitro transcribed naked self-amplifying mRNA based on the Semliki Forest virus genome could induce a protective immune response (62). Geall and colleagues showed that i.m. administration in mice and cotton rats with very low dose of self-amplifying mRNA encoding the F protein of respiratory syncytial virus (RSV) encapsulated with a synthetic LNP induced very high titers of IgG1 and interferon (IFN)-producing CD4 and CD8 T cells (22).
Delivery tools are equally important in the effectiveness of mRNA vaccines. Ideally, the delivery vehicle should protect RNA against potential digestion by ribonuclease and confer efficient target cell uptake, easy dissociation of RNA cargo from the vehicle and escape from the endosome. Overcoming the barrier of the cytoplasmic membrane and avoiding digestion by RNases are the initial steps for efficient RNA delivery into target cells. The final important requirements for an optimal delivery vehicle are a lack of both toxicity and immune stimulation. In initial studies, mRNA synthesized in vitro was directly injected into animals. Subsequently, mRNA vaccines formulated in liposomes were confirmed to induce a virus-specific anti-influenza cytotoxic T lymphocyte (CTL) immune response in mice (72). Several methods have been explored to increase delivery efficiency and great progress has been made in the field of designing delivery vehicles form RNA vaccines (101–103). In addition to the physical methods of gene guns and electroporation, mRNA vaccines have been delivered into the cytoplasm by cationic lipids and polymers. Cationic nano-emulsion formulated mRNA was also shown to induce a potent immune response (8, 23, 85). However, several of these delivery vehicles demonstrated toxicity in vivo, which may limit their use in humans (104). New platforms were developed as transportation tools for mRNA vaccines to avoid the limitation of toxic chemical transfection reagents. Most of these platforms utilized LNPs based on modified cationic lipid or lipid polymers. LNPs facilitate the delivery of RNA and enhances antigen expression dramatically. Several groups have utilized lipids or polymers as a platform to deliver mRNA vaccines against HIV-1 by a subcutaneous route, which efficiently elicited HIV-specific CD4 and CD8 T cell responses, or by an intranasal route, which induced an antigen-specific immune response (73, 105, 106). Lipid-encapsulated mRNA of influenza HA gene segments was also tested and showed T cell activation following a single dose (107). Combining LNP technology with nucleoside modification improves the efficacy of mRNA vaccines. LNP-formulated modified mRNA of influenza virus HA from H10N8 and H7N9 induced a potent protective immune response in mice, ferrets, and cynomolgus monkeys (108).
Another target against which LNP delivery of formulated mRNA has shown great potential is Zika virus. No vaccine is available to prevent this mosquito-borne disease and the recent epidemic has caused worldwide concern. Richner et al. reported that two vaccinations with LNP-encapsulated modified mRNA encoding a wild-type or edited prM-E gene induced a high order neutralizing antibody titer (75, 109).
Modified mRNA-based vaccines formulated with LNPs elicited robust immune responses and protected guinea pigs from Ebola virus disease as well (52). Intravenous (i.v.) injection with modified mRNA formulated in LNPs showed maximal protein expression at 6 h post-injection (110). Both i.d. and i.m. administration with non-replicating mRNA encoding influenza H10 encapsulated in LNPs induced high protective titers, but this response occurred more rapidly by i.d. administration than by i.m. administration (95).
LNPs are a popular delivery vehicle for self-amplifying mRNA vaccines well. A plethora of studies have shown that self-amplifying mRNA encapsulated in LNP induced potent cellular and humoral immune responses by different administration routes (19, 107, 111). LNP formulated self-amplifying mRNA vaccines encoding influenza virus antigens resulted in potent T and B cell immune responses and conferred protection against homologous and heterologous influenza virus challenges as well (63, 65, 112).
Cell penetrating peptides (CPPs), a type of cationic peptide, represent promising tools for mRNA delivery into intracellular target sites. Protamine is an arginine-rich cationic peptide that can bind to mRNA and transport it into cytoplasm. Protamine was extensively used as a delivery system for cancer and viral mRNA vaccines. The self-adjuvanted RNActive vaccine platform was created with protamine and has demonstrated potency against various infectious diseases and cancers (76, 77, 100). Recently, Coolen and colleagues designed innovative delivery platform consisting of poly(lactic acid) and cationic-penetrating peptides as mRNA condensing agent. This nano-complexes were taken up by dendritic cells induced strong protein expression and innate immune response (113).
Self-amplifying mRNA coding HA [A/California/ 07/2009(H1N1)] encapsulated into oil-in-water nano-emulsion stimulated protection against homologous and heterologous influenza virus (65). Formulation in polyethylenimine (PEI) of self-amplifying mRNA encoding H1N1/PR8-HA resulted in a significantly higher antibody titer and longer durable antigen expression than using non-formulated self-amplifying mRNA (31, 114). Chitosan and PEI were also utilized to deliver self-amplifying -mRNA as nanoparticles (114, 115). Chahal et al. developed an intriguing platform consisting of a chemically modified dendrimer nanoparticle to condense self-amplifying mRNA encoding influenza HA. A single immunization in mice elicited potent CD8+ T cell and antibody responses and protected mice against a broad spectrum of lethal pathogen challenges, including H1N1 influenza, Toxoplasma gondii, and Ebola virus (64).
Additional new modified nanoparticles are currently being investigated, such as polyplexes, nanoplexes and porous polymer scaffold-mediated delivery (116–122). Although great advances have been achieved in the development of delivery tools, the ideal platform maybe a combination of different mRNA delivery tools and more efforts in understanding mechanism of action might be required.
Mechanism of Immune Response Induced by mRNA Vaccines
The immune response mechanism instigated by mRNA remains to be elucidated. The process of mRNA vaccine recognition by cellular sensors and the mechanism of sensor activation are still not clear. Intracellularly, two kinds of RNA sensors, endosomal toll-like receptors (TLRs) and the RIG-I-like receptor family, have been identified. The former set is divided into TLR-3, TLR7, TLR8, and TLR9, which are localized in the endosomal compartment of professional immune surveillance cells, such as DCs, macrophages and monocytes. TLR3 recognizes dsRNA longer than 45 base pair as well as dsRNA resulting from single strand RNA (ssRNA) forming secondary structures or derived from viral replication intermediates. TLR7 and TLR8 are activated by RNAs rich in polyuridines, guanosines and/or uridines. TLR7 can bind both dsRNA and single-stranded RNA (ssRNA), whereas TLR8 recognizes ssRNA only (123). TLR7 activation can increase antigen presentation, promote cytokine secretion and stimulate B cell responses (124). The latter family, functioning as a pattern recognition receptor (PRR), includes RIG-I, MDA5, and LGP2 (125). RIG-I preferentially recognize ssRNA and dsRNA bearing a 5′triphosphate, and stimulate IFN production (126–130). The panhandle structure in viral genome segments was directly involved IFNinduction through RIG-I activation (131). MDA5 is another cytosolic RNA sensor that detects long dsRNA generated during RNA virus replication (132) as well as RNA of synthetic origin, including poly I:C. Recognition by dsRNA induces the activation of IRF3 and NF-κB, subsequently leading to increased production of type I IFN (127, 133, 134). Sometimes, the elements of dsRNA recognized by PRR sensors can function as an adjuvant through the induction of IFN (135–137). mRNA vaccines can stimulate innate immunity through TLRs 3,7, and 8, RIG-I and MDA5 (138, 139). IFN induction by mRNA vaccines through RNA sensors is dependent on the quality of in vitro transcribed mRNA, delivery vehicle, and administration route. mRNA sensing by the innate immune system is a double-edged sword in the elimination of invading molecules. Natural exogenous mRNA stimulates strong induction of type I INFs and potent inflammatory cytokines, which instigate T and B immune responses but may negatively affect antigen expression (140–143). Interestingly, Blanchard and colleagues established a method to measure PRR activation by IVT mRNA in cells and in tissue section. In this method, proximity ligation assays (PLAs) was employed (144).
i.d. vaccination with the RNactive vaccine technology from CureVac AG, induced strong immune responses that are dependent on TLR7 signal. TLR7 activation leads to upregulation of chemokines, which in turn recruit innate immune cells such as DCs and macrophages to the site of injection (100). Activation of pro-inflammatory cytokines, such as TNF-α andIL-6 which are known to contribute to immune cells recruitment have been observed at the injection site (145).
On the other hand, an early shut-down of antigen expression after the mRNA vaccination due to PRRs activation might be detrimental. Consistently antigen expression, humoral and T cell responses to mRNA vaccination, both from conventional and amplifying mRNA, were significantly enhanced in IFNAR1/2 −/− mice (105, 146) or by co-administration of IFN antagonist (147). The negative impact from excessive IFN activation could derive not only from preventing RNA amplification, in case of self-amplifying mRNA vaccines, and expression, but also at the level of T cells. While type I IFN can determine the differentiation of antigen-primed CD8+ T cells into cytotoxic effectors, they may also promote T cell exhaustion (140). Whether type I IFN inhibits or stimulates the CD8 T cell response to mRNA vaccines might depend from the timing and intensity of type I IFN induced (140). T cell inhibition could prevail if triggering of type I IFN receptors precedes that of T cell receptors.
Modified mRNA with pseudouridines and mRNA purified with HPLC can reduce immune activation and increase antigen stability and expression (35, 148, 149). For an instance, i.p injection with mRNA containing pseudouridines induced antigen expression without the induction of cytokines in mice (150). Furthermore, some publications have shown that purity and delivery systems affect the immune response stimulated by mRNA vaccines (35). More interestingly, a recent study showed that modified mRNA encapsulated into LNPs has an adjuvant effect and induces a potent T follicular helper response and a large number of germinal center B cells with long-living, high affinity neutralizing antibodies (78).
Dentritic cell(DC) maturation is crucial to the effectiveness of mRNA-based vaccines. Generally, TLR7 was expresssed in plasmacytoid dentritic cells (pDCs) and B cells in humans, and TLR8 was expressed in conventional dentritic cells (cDCs), monocytes and macrophages. cDCs constitute the major resident DC population in normal human dermis and are characterized by CD1c expression (also known as blood dendritic cell antigen-1 (BDCA-1), whereas plasmacytoid DCs are present in the skin (129, 151, 152). The TLR7 and TLR8 locations in different DC subsets and DC locations in different organs may clarify the relationship between immune efficacy and the administration route and formulation of mRNA vaccines. Replacement of modified nucleotidesin mRNA decreased activation by binding mRNA to PRRs and reduced the innate immune response (153). mRNA vaccines not only stimulated the specific humoral immune response by the translated antigen but also the antigen-specific T cell response. Administration route and vaccine formulation determine the peak of antigen expression, which is another way to modulate the immune response (94, 154, 155). Liang et al. have shown that the kinetics of cell infiltration was largely similar between i.m. and i.d. administration in NHPs (156). The i.d. group showed stronger initial responses, probably because of rapid targeting, activation and transport to dLNs of skin DCs. Furthermore, only skin monocytes and DCs showed evidence of antigen translation at day 9, indicating prolonged antigen availability after i.d. delivery, and confirming the longer expression of mRNA-encoded antigen observed in mice (94).
A better elucidation of the sequence of events leading to mRNA translation and immune activation will help engineer mRNA vaccines to induce the correct balance of type I IFN induction, positively affecting vaccine outcome.
Clinical Trials
Compared with the prophylactic and therapeutic application of mRNA in cancer, clinical trials of mRNA vaccines for infectious disease are still in their early age. Pilot clinical trials with DCs transfected with mRNA encoding various HIV-1 antigens, cellular molecules, or pp65 of human cytomegalovirus showed that mRNA vaccines are safe and that they elicited antigen-specific CD4+ and CD8+ T cell immune responses; however, no reduction of viral load was observed (157–160).
In a recent clinical trial of protamine complexed mRNA vaccine against rabies virus, the results showed that RNA complexed with protamine is safe and well-tolerated in vivo, but efficacy was highly dependent on the dose and route of administration. The efficacy of administration with a needle-free device was much better than with direct needle injection (57, 161). Results of a phase I showed LNP-formulated modified H10N8 mRNA vaccine induced robust humoral immune response in volunteers with mild or moderate adverse reaction (108).
Prospective of RNA-based Vaccines
A plethora of publications have shown that mRNA-based vaccines are a promising novel platform that is high flexible, scalable, inexpensive, and cold-chain free. Most importantly, mRNA-based vaccines can fill the gap between emerging pandemic infectious disease and a bountiful supply of effective vaccines. A variety of preclinical and clinical projects have made enormous strides toward the conceivable application of mRNA vaccines and have suggested that mRNA-based prophylaxis and therapy can be translated to human applications. Although in medical application, magnitude of responses was lower than predicted from than those observed into animal models, the results of pilot clinical trials have shown good tolerability and that mRNA vaccination can induce antigen-specific T and B cell immune responses (57, 108). Therefore, mRNA holds great promises, but further insights into the mechanism of action and potency are still needed for full development of mRNA vaccines. The exploration of new strategies is needed to create applicable mRNA vaccines and to decrease the dose. As described above, the molecular impact of the innate immune response stimulated by mRNA through PAMP recognition is still not clear. Multiple efforts have been made to improve the stability and delivery efficiency of in vivo mRNA vaccine, including incorporation of 5′ and 3′ terminal untranslated regions and chemically modified nucleosides (162–164). Study demonstrated removal of dsRNA contaminants by high performance liquid chromatography purification of in vitro transcribed mRNA prolonged the translation (35). Research has demonstrated that modified nucleoside decreases the innate immune response and enhances protein expression. Optimization of the 5′-untranslated region (5′-UTR) of mRNA, whose secondary structures are recognized by cell-specific RNA binding proteins or PAMP molecules can maximize the translational yield of mRNA therapeutics and vaccines (43, 165). However, improper incorporation of modified nucleosides can have a negative impact on transcription products and increase costs.
Based on the results of the above described studies, a better understanding of the mechanism of action of mRNA vaccines, the identification and development of a new delivery system, and improvement of mRNA vaccine design will be attained (166).
mRNA vaccines have great potential and offer advantages over conventional vaccines. The growing body of preclinical and clinical results demonstrates that prophylaxis and therapy with mRNA promises to be useful for preventing infectious disease and treating tumors and that mRNA vaccines are safe and tolerated in animal models and humans. Additionally, future improvements should increase antigen-specific immune responses and the magnitude of memory immune cell responses, including memory B and T cell responses. Although mRNA vaccine technology has still not extensively tested in humans, publications of preclinical and early clinical tests have emerged in recent years, in which promising results were reported. This evoked the momentum of biocompanies to commercialize mRNA vaccines with great enthusiasm (167, 168). Some private funding resources and institutes have supported the research and development of mRNA vaccines (169, 170). Despite the need for further optimization of manufacturing processes to generate mRNA vaccines, these processes hopefully will be streamlined to be establish large-scale production. It is just a matter of time for RNA vaccines to be used in humans and animals.
Author Contributions
CZ, JL, and HS wrote this manuscript. JL and GM revised this manuscript.
Funding
Priority Academic Talent Team Cultivation Program of Shandong Colleges and Universities, Talent Program of Qingdao Agricultural University. The National Thirteen-Five Key research and development program of China (2017YFD0500805).
Conflict of Interest Statement
GM is an employee of the GSK group of companies and reports ownership of GSK shares and/or restricted GSK shares.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize for not including all of the publications by our colleagues.
References
1. Lents MP, Barbosa LP, Santana ALA, Pinheiro EEG, Mugabe LC, Biscarde CEA, et al. Immunocastration of goats using anti-gonadotrophin releasing hormone vaccine. Theriogenology. (2018) 114:7–13. doi: 10.1016/j.theriogenology.2018.03.013
2. Cuzick J. Preventive therapy for cancer. Lancet Oncol. (2017) 18:e472–e482. doi: 10.1016/S1470-2045(17)30536-3
3. Scheiblhofer S, Thalhamer J, Weiss R. DNA and mRNA vaccination against allergies. Pediatr Allergy Immunol. (2018) 29:679–88. doi: 10.1111/pai.12964
4. Li B, Fang L, Xu Z, Liu S, Gao J, Jiang Y, et al. Recombination in vaccine and circulating strains of porcine reproductive and respiratory syndrome viruses. Emerg Infect Dis. (2009) 15:2032–5. doi: 10.3201/eid1512.090390
5. Zhou B, Meliopoulos VA, Wang W, Lin X, Stucker KM, Halpin RA, et al. Wentworth. reversion of cold-adapted live attenuated influenza vaccine into a pathogenic virus. J Virol. (2016) 90:8454–63. doi: 10.1128/JVI.00163-16
6. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
7. Li J, Arevalo MT, Chen Y, Chen S, Zeng M. T-cell-mediated cross-strain protective immunity elicited by prime-boost vaccination with a live attenuated influenza vaccine. Int J Infect Dis. (2014) 27:37–43. doi: 10.1016/j.ijid.2014.05.016
8. Pardi N, Parkhouse K, Kirkpatrick E, McMahon M, Zost SJ, Mui BL, et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat Commun. (2018) 9:3361. doi: 10.1038/s41467-018-05482-0
9. Hollister K, Chen Y, Wang S, Wu H, Mondal A, Clegg N, et al. The role of follicular helper T cells and the germinal center in HIV-1 gp120 DNA prime and gp120 protein boost vaccination. Hum Vaccin Immunother. (2014) 10:1985–92. doi: 10.4161/hv.28659
10. Rauch S, Jasny E, Schmidt KE, Petsch B. New vaccine technologies to combat outbreak situations. Front Immunol. (2018) 9:1963. doi: 10.3389/fimmu.2018.01963
11. Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. (2008) 9:776–88. doi: 10.1038/nrg2432
12. Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med. (2012) 4:155ra138. doi: 10.1126/scitranslmed.3004414
13. Geall AJ, Mandl CW, Ulmer JB. RNA: the new revolution in nucleic acid vaccines. Semin Immunol. (2013) 25:152–9. doi: 10.1016/j.smim.2013.05.001
14. Faurez F, Dory D, Le Moigne V, Gravier R, Jestin A. Biosafety of DNA vaccines: New generation of DNA vectors and current knowledge on the fate of plasmids after injection. Vaccine. (2010) 28:3888–95. doi: 10.1016/j.vaccine.2010.03.040
15. Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. (1990) 247:1465–8.
16. Iavarone CT, O'Hagan D, Yu D, Delahaye NF, Ulmer JB. Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines. (2017) 16:871–81. doi: 10.1080/14760584.2017.1355245
17. Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ. Developing mRNA-vaccine technologies. RNA Biol. (2012) 9:1319–30. doi: 10.4161/rna.22269
18. Kallen KJ, Thess A. A development that may evolve into a revolution in medicine: mRNA as the basis for novel, nucleotide-based vaccines and drugs. Ther Adv Vaccines. (2014) 2:10–31. doi: 10.1177/2051013613508729
19. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. (2018) 17:261–79. doi: 10.1038/nrd.2017.243
20. Fiedler K, Lazzaro S, Lutz J, Rauch S, Heidenreich R. mRNA Cancer Vaccines. Recent Results Cancer Res. (2016) 209:61–85. doi: 10.1007/978-3-319-42934-2_5
22. Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci USA. (2012) 109:14604–9. doi: 10.1073/pnas.1209367109
23. Brito LA, Chan M, Shaw CA, Hekele A, Carsillo T, Schaefer M, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol Ther. (2014) 22:2118–29. doi: 10.1038/mt.2014.133
24. Deering RP, Kommareddy S, Ulmer JB, Brito LA, Geall AJ. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin Drug Deliv. (2014) 11:885–99. doi: 10.1517/17425247.2014.901308
25. Tews BA, Meyers G. Self-Replicating RNA. Methods Mol Biol. (2017) 1499:15–35. doi: 10.1007/978-1-4939-6481-9_2
26. Lundstrom K. Replicon RNA viral vectors as vaccines. Vaccines. (2016) 4: pii: E39. doi: 10.3390/vaccines4040039
27. Ljungberg KP. Liljestrom. Self-replicating alphavirus RNA vaccines. Expert Rev Vaccines. (2015) 14:177–94. doi: 10.1586/14760584.2015.965690
28. Anraku I, Harvey TJ, Linedale R, Gardner J, Harrich D, Suhrbier A, et al. Kunjin virus replicon vaccine vectors induce protective CD8+ T-cell immunity. J Virol. (2002) 76:3791–9. doi: 10.1128/JVI.76.8.3791-3799.2002
29. Johanning FW, Conry RM, LoBuglio AF, Wright M, Sumerel LA, Pike MJ, et al. A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res. (1995) 23:1495–501.
30. Zhou X, Berglund P, Rhodes G, Parker SE, Jondal M, Liljestrom P. Self-replicating semliki forest virus RNA as recombinant vaccine. Vaccine. (1994) 12:1510–4.
31. Vogel AB, Lambert L, Kinnear E, Busse D, Erbar S, Reuter KC, et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther. (2018) 26:446–55. doi: 10.1016/j.ymthe.2017.11.017
32. Weissman D. mRNA transcript therapy. Expert Rev Vaccines. (2015) 14:265–81. doi: 10.1586/14760584.2015.973859
33. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov. (2014) 13:759–80. doi: 10.1038/nrd4278
34. Probst J, Weide B, Scheel B, Pichler BJ, Hoerr I, Rammensee HG, et al. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. (2007) 14:1175–80. doi: 10.1038/sj.gt.3302964
35. Kariko K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. (2011) 39:e142. doi: 10.1093/nar/gkr695
37. Weissman D, Pardi N, Muramatsu H, Kariko K. HPLC purification of in vitro transcribed long RNA. Methods Mol Biol. (2013) 969:43–54. doi: 10.1007/978-1-62703-260-5_3
38. Kozak M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. (1987) 15:8125–48.
39. Fechter P, Brownlee GG. Recognition of mRNA cap structures by viral and cellular proteins. J Gen Virol. (2005) 86:1239–49. doi: 10.1099/vir.0.80755-0
40. Ramanathan, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. (2016) 44:7511–26. doi: 10.1093/nar/gkw551
41. Lee H, Bingham SE, Webber AN. Function of 3' non-coding sequences and stop codon usage in expression of the chloroplast psaB gene in Chlamydomonas reinhardtii. Plant Mol Biol. (1996) 31:337–54.
42. Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. (1991) 5:2108–16.
43. Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. (2006) 108:4009–17. doi: 10.1182/blood-2006-04-015024
44. Zohra FT, Chowdhury EH, Tada S, Hoshiba T, Akaike T. Effective delivery with enhanced translational activity synergistically accelerates mRNA-based transfection. Biochem Biophys Res Commun. (2007) 358:373–8. doi: 10.1016/j.bbrc.2007.04.059
45. Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, et al. Improving mRNA-Based therapeutic gene delivery by expression-Augmenting 3' UTRs identified by cellular library screening. Mol Ther. (2018) 18:30595–1. doi: 10.1016/j.ymthe.2018.12.011
46. Puigbo P, Guzman E, Romeu A, Garcia-Vallve S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. (2007) 35:W126–31. doi: 10.1093/nar/gkm219
47. Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. (2006) 4:e180. doi: 10.1371/journal.pbio.0040180
48. Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. (2008) 42:287–99. doi: 10.1146/annurev.genet.42.110807.091442
49. Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther. (2015) 23:1456–64. doi: 10.1038/mt.2015.103
50. Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. (2010) 38:5884–92. doi: 10.1093/nar/gkq347
51. Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release. (2015) 217:337–44. doi: 10.1016/j.jconrel.2015.08.051
52. Meyer M, Huang E, Yuzhakov O, Ramanathan P, Ciaramella G, Bukreyev A. Modified mRNA-based vaccines elicit robust immune responses and protect guinea pigs from ebola virus disease. J Infect Dis. (2018) 217:451–55. doi: 10.1093/infdis/jix592
53. Van Gulck ER, Ponsaerts P, Heyndrickx L, Vereecken K, Moerman F, De Roo A, et al. Efficient stimulation of HIV-1-specific T cells using dendritic cells electroporated with mRNA encoding autologous HIV-1 Gag and Env proteins. Blood. (2006) 107:1818–27. doi: 10.1182/blood-2005-01-0339
54. Pardi N, Weissman D. Nucleoside Modified mRNA Vaccines for Infectious Diseases. Methods Mol Biol. (2017) 1499:109–21. doi: 10.1007/978-1-4939-6481-9_6
55. Pardi N, Muramatsu H, Weissman D, Kariko K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol. (2013) 969:29–42. doi: 10.1007/978-1-62703-260-5_2
56. Jones KL, Drane D, Gowans EJ. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques. (2007) 43:675–81. doi: 10.2144/000112593
57. Alberer M, Gnad-Vogt U, Hong HS, Mehr KT, Backert L, Finak G, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. (2017) 390:1511–20. doi: 10.1016/S0140-6736(17)31665-3
58. Stitz L, Vogel A, Schnee M, Voss D, Rauch S, Mutzke T, et al. A thermostable messenger RNA based vaccine against rabies. PLoS Negl Trop Dis. (2017) 11:e0006108. doi: 10.1371/journal.pntd.0006108
59. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. (2014) 14:135–146. doi: 10.1038/nrc3670
60. Grunwitz C, Kranz LM. mRNA cancer vaccines-messages that prevail. Curr Top Microbiol Immunol. (2017) 405:145–64. doi: 10.1007/82_2017_509
61. Zhang R, Billingsley MM, Mitchell MJ. Biomaterials for vaccine-based cancer immunotherapy. J Control Release. (2018) 292:256–76. doi: 10.1016/j.jconrel.2018.10.008
62. Fleeton MN, Chen M, Berglund P, Rhodes G, Parker SE, Murphy M, et al. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J Infect Dis. (2001) 183:1395–8. doi: 10.1086/319857
63. Magini D, Giovani C, Mangiavacchi S, Maccari S, Cecchi R, Ulmer JB, et al. Self-Amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS ONE. (2016) 11:e0161193. doi: 10.1371/journal.pone.0161193
64. Chahal JS, Khan OF, Cooper CL, McPartlan JS, Tsosie JK, Tilley LD, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci USA. (2016) 113:E4133–42. doi: 10.1073/pnas.1600299113
65. Brazzoli M, Magini D, Bonci A, Buccato S, Giovani C, Kratzer R, et al. Induction of broad-based immunity and protective efficacy by self-amplifying mRNA vaccines encoding influenza virus hemagglutinin. J Virol. (2016) 90:332–44. doi: 10.1128/JVI.01786-15
66. Scorza FB, Pardi N. New kids on the block: RNA-based influenza virus vaccines. Vaccines. (2018) 6:pii: E20. doi: 10.3390/vaccines6020020
67. Zost SJ, Parkhouse K, Gumina ME, Kim K, Diaz Perez S, Wilson PC, et al. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc Natl Acad Sci USA. (2017) 114:12578–83. doi: 10.1073/pnas.1712377114
68. Wu NC, Zost SJ, Thompson AJ, Oyen D, Nycholat CM, McBride R, et al. A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine. PLoS Pathog. (2017) 13:e1006682. doi: 10.1371/journal.ppat.1006682
69. Pulido MR, Sobrino F, Borrego B, Saiz M. RNA immunization can protect mice against foot-and-mouth disease virus. Antiviral Res. (2010) 85:556–8. doi: 10.1016/j.antiviral.2009.12.005
70. Saxena S, Sonwane AA, Dahiya SS, Patel CL, Saini M, Rai A, et al. Induction of immune responses and protection in mice against rabies using a self-replicating RNA vaccine encoding rabies virus glycoprotein. Vet Microbiol. (2009) 136:36–44. doi: 10.1016/j.vetmic.2008.10.030
71. VanBlargan LA, Himansu S, Foreman BM, Ebel GD, Pierson TC, Diamond MS. An mRNA Vaccine protects mice against multiple tick-transmitted flavivirus infections. Cell Rep. (2018) 25:3382–3392 e3. doi: 10.1016/j.celrep.2018.11.082
72. Martinon F, Krishnan S, Lenzen G, Magne R, Gomard E, Guillet JG, et al., Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. (1993) 23:1719–22. doi: 10.1002/eji.1830230749
73. Zhao M, Li M, Zhang Z, Gong T, Sun X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA Drug Deliv. A. (2016) 23:2596–607. doi: 10.3109/10717544.2015.1038856
74. Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. (2017) 543:248–51. doi: 10.1038/nature21428
75. Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Diamond. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell. (2017) 168:1114–25 e10. doi: 10.1016/j.cell.2017.02.017
76. Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. (2012) 30:1210–6. doi: 10.1038/nbt.2436
77. Schnee M, Vogel AB, Voss D, Petsch B, Baumhof P, Kramps T, et al. An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLoS Negl Trop Dis. (2016) 10:e0004746. doi: 10.1371/journal.pntd.0004746
78. Pardi N, Hogan MJ, Naradikian MS, Parkhouse K, Cain DW, Jones L, et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J Exp Med. (2018). 215:1571–88. doi: 10.1084/jem.20171450
79. Pardi N, Secreto AJ, Shan X, Debonera F, Glover J, Yi Y, et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat Commun. (2017) 8:14630. doi: 10.1038/ncomms14630
80. Khalil SM, Tonkin DR, Mattocks MD, Snead AT, Johnston RE, White LJ. A tetravalent alphavirus-vector based dengue vaccine provides effective immunity in an early life mouse model. Vaccine. (2014) 32:4068–74. doi: 10.1016/j.vaccine.2014.05.053
81. Lundstrom K. In vivo administration of recombinant alphavirus into rodents. Cold Spring Harb Protoc. (2012) 2012:pdb.prot070581. doi: 10.1101/pdb.prot070581
82. Pyankov OV, Bodnev SA, Pyankova OG, Solodkyi VV, Pyankov SA, Setoh YX, et al. A kunjin replicon virus-like particle vaccine provides protection against ebola virus infection in nonhuman primates. J Infect Dis. (2015) 212(Suppl 2):S368–71. doi: 10.1093/infdis/jiv019
83. Oreshkova N, Cornelissen LA, de Haan CA, Moormann RJ, Kortekaas J. Evaluation of nonspreading Rift Valley fever virus as a vaccine vector using influenza virus hemagglutinin as a model antigen. Vaccine. (2014) 32:5323–9. doi: 10.1016/j.vaccine.2014.07.051
84. Van de Wall S, Walczak M, van Rooij N, Hoogeboom BN, Meijerhof T, Nijman HW, et al. Tattoo delivery of a semliki forest virus-based vaccine encoding human papillomavirus E6 and E7. Vaccines. (2015) 3:221–38. doi: 10.3390/vaccines3020221
85. Bogers WM, Oostermeijer H, Mooij P, Koopman G, Verschoor EJ, Davis D, et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J Infect Dis. (2015) 211:947–55. doi: 10.1093/infdis/jiu522
86. Harvey TJ, Anraku I, Linedale R, Harrich D, Mackenzie J, Suhrbier A, et al. Kunjin virus replicon vectors for human immunodeficiency virus vaccine development. J Virol. (2003) 77:7796–803. doi: 10.1128/JVI.77.14.7796-7803.2003
87. Huang YT, Liao JT, Yen LC, Chang YK, Lin YL, Liao CL. Japanese encephalitis virus replicon-based vaccine expressing enterovirus-71 epitope confers dual protection from lethal challenges. J Biomed Sci. (2015) 22:74. doi: 10.1186/s12929-015-0181-8
88. Hofmann I, Wen Y, Ciferri C, Schulze A, Fuhner V, Leong M, et al. Expression of the human cytomegalovirus pentamer complex for vaccine use in a CHO system. Biotechnol Bioeng. (2015) 112:2505–15. doi: 10.1002/bit.25670
89. Maruggi G, Chiarot E, Giovani C, Buccato S, Bonacci S, Frigimelica E, et al. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccin. (2017) 35:361–8. doi: 10.1016/j.vaccine.2016.11.040
90. Moyo N, Vogel AB, Buus S, Erbar S, Wee EG, Sahin U, et al. Efficient induction of T cells against conserved HIV-1 regions by mosaic vaccines delivered as self-amplifying mRNA. Mol Ther Methods Clin Dev. (2019) 12:32–46. doi: 10.1016/j.omtm.2018.10.010
91. Ip PP, Boerma A, Regts J, Meijerhof T, Wilschut J, Nijman HW, et al. Alphavirus-based vaccines encoding nonstructural proteins of hepatitis C virus induce robust and protective T-cell responses. Mol Ther. (2014) 22:881–90. doi: 10.1038/mt.2013.287
92. Aberle JH, Aberle SW, Kofler RM, Mandl CW. Humoral and cellular immune response to RNA immunization with flavivirus replicons derived from tick-borne encephalitis virus. J Virol. (2005) 79:15107–13. doi: 10.1128/JVI.79.24.15107-15113.2005
93. Whitehead KA, Dahlman JE, Langer RS, Anderson DG. Silencing or stimulation? siRNA delivery and the immune system. Annu Rev Chem Biomol Eng. (2011) 2:77–96. doi: 10.1146/annurev-chembioeng-061010-114133
94. Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. (2015) 217:345–51. doi: 10.1016/j.jconrel.2015.08.007
95. Lindgren G, Ols S, Liang F, Thompson EA, Lin A, Hellgren F, et al. Induction of robust b cell responses after influenza mrna vaccination is accompanied by circulating hemagglutinin-specific ICOS+ PD-1+ CXCR3+ T follicular helper cells. Front Immunol. (2017) 8:1539. doi: 10.3389/fimmu.2017.01539
96. Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. (2010) 70:9031–40. doi: 10.1158/0008-5472.CAN-10-0699
97. Van Nuffel AM, Benteyn D, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol Immunother. (2012) 61:1033–43. doi: 10.1007/s00262-011-1176-2
98. Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, et al. Thielemans. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. (2012) 72:1661–71. doi: 10.1158/0008-5472.CAN-11-2957
99. Van Lint S, Renmans D, Broos K, Goethals L, Maenhout S, Benteyn D, et al. Intratumoral delivery of TriMix mRNA results in T-cell activation by cross-presenting dendritic cells. Cancer Immunol Res. (2016) 4:146–56. doi: 10.1158/2326-6066.CIR-15-0163
100. Kallen KJ, Heidenreich R, Schnee M, Petsch B, Schlake T, Thess A, et al. A novel, disruptive vaccination technology: self-adjuvanted RNActive((R)) vaccines. Hum Vaccin Immunother. (2013) 9:2263–76. doi: 10.4161/hv.25181
101. Kauffman KJ, Webber MJ, Anderson DG. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J Control Release. (2016) 240:227–34. doi: 10.1016/j.jconrel.2015.12.032
102. Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. (2017) 24:133–43. doi: 10.1038/gt.2017.5
103. Reichmuth AM, Oberli MA, Jeklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. (2016) 7:319–34. doi: 10.4155/tde-2016-0006
104. Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. (2006) 114:100–9. doi: 10.1016/j.jconrel.2006.04.014
105. Pollard C, Rejman J, De Haes W, Verrier B, Van Gulck E, Naessens T, et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther. (2013) 21:251–9. doi: 10.1038/mt.2012.202
106. Li M, Zhao M, Fu Y, Li Y, Gong T, Zhang Z, et al. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J Control Release. (2016) 228:9–19. doi: 10.1016/j.jconrel.2016.02.043
107. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. (2016) 534:396–401. doi: 10.1038/nature18300
108. Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther. (2017) 25:1316–27. doi: 10.1016/j.ymthe.2017.03.035
109. Scott JM, Lebratti TJ, Richner JM, Jiang X, Fernandez E, Zhao H, et al. Cellular and humoral immunity protect against vaginal zika virus infection in mice. J Virol. (2018) 92:JVI.00038-18. doi: 10.1128/JVI.00038-18
110. Sedic M, Senn JJ, Lynn A, Laska M, Smith M, Platz SJ, et al. Safety evaluation of lipid nanoparticle-formulated modified mRNA in the sprague-dawley rat and cynomolgus monkey. Vet Pathol. (2018) 55:341–354. doi: 10.1177/0300985817738095
111. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. (2017) 17:1326–35. doi: 10.1021/acs.nanolett.6b03329
112. Hekele A, Bertholet S, Archer J, Gibson DG, Palladino G, Brito LA, et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg Microbes Infect. (2013) 2:e52. doi: 10.1038/emi.2013.54
113. Coolen AL, Lacroix C, Mercier-Gouy P, Delaune E, Monge C, Exposito JY, et al. Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials. (2019) 195:23–37. doi: 10.1016/j.biomaterials.2018.12.019
114. Demoulins T, Milona P, Englezou PC, Ebensen T, Schulze K, Suter R, et al. Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines. Nanomedicine. (2016) 12:711–22. doi: 10.1016/j.nano.2015.11.001
115. McCullough KC, Bassi I, Milona P, Suter R, Thomann-Harwood L, Englezou P, et al. Self-replicating replicon-RNA delivery to dendritic cells by chitosan-nanoparticles for translation In Vitro and In Vivo. Mol Ther Nucleic Acids. (2014) 3:e173. doi: 10.1038/mtna.2014.24
116. Lai WF, Wong WT. Design of polymeric gene carriers for effective intracellular delivery. Trends Biotechnol. (2018) 36:713–28. doi: 10.1016/j.tibtech.2018.02.006
117. Mandal H, Katiyar SS, Swami R, Kushwah V, Katare PB, Kumar Meka A, et al. epsilon-Poly-l-Lysine/plasmid DNA nanoplexes for efficient gene delivery in vivo. Int J Pharm. (2018) 542:142–52. doi: 10.1016/j.ijpharm.2018.03.021
118. Chen R, Zhang H, Yan J, Bryers JD. Scaffold-mediated delivery for non-viral mRNA vaccines. Gene Ther. (2018) 25:556–67. doi: 10.1038/s41434-018-0040-9
119. Fornaguera C, Guerra-Rebollo M, Angel Lazaro M, Castells-Sala C, Meca-Cortes O, Ramos-Perez V, et al. mRNA delivery system for targeting antigen-presenting cells in vivo. Adv Healthc Mater. (2018) 7:e1800335. doi: 10.1002/adhm.201800335
120. Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P, et al. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther. (2013) 21:358–67. doi: 10.1038/mt.2012.250
121. O.Haabeth AW, Blake TR, McKinlay CJ, Waymouth RM, Wender PA, Levy R. mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc Natl Acad Sci USA. (2018) 115:E9153–61. doi: 10.1073/pnas.1810002115
122. McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci USA. (2018) 115:E5859–66. doi: 10.1073/pnas.1805358115
123. Ablasser, Poeck H, Anz D, Berger M, Schlee M, Kim S, et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J Immunol. (2009) 182:6824–33. doi: 10.4049/jimmunol.0803001
124. Hua Z, Hou B. TLR signaling in B-cell development and activation. Cell Mol Immunol. (2013) 10:103–6. doi: 10.1038/cmi.2012.61
125. Kato H, Oh SW, Fujita T. RIG-I-like receptors and Type I interferonopathies. J Interferon Cytokine Res. (2017) 37:207–13. doi: 10.1089/jir.2016.0095
126. Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. (2010) 140:397–408. doi: 10.1016/j.cell.2010.01.020
127. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. (2014) 514:372–375. doi: 10.1038/nature13590
128. Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology. (2013) 218:1322–35. doi: 10.1016/j.imbio.2013.06.007
129. Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. (2010) 207:2921–30. doi: 10.1084/jem.20101102
130. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. (2013) 38:855–69. doi: 10.1016/j.immuni.2013.05.007
131. Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. Influenza A virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol. (2015) 89:6067–79. doi: 10.1128/JVI.00232-15
132. Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. (2012) 2:1187–96. doi: 10.1016/j.celrep.2012.10.005
133. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. (2001) 413:732–8. doi: 10.1038/35099560
134. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. (2007) 13:460–9. doi: 10.1016/j.molmed.2007.09.002
135. Martinez-Gil L, Goff PH, Hai R, Garcia-Sastre A, Shaw ML, Palese P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J Virol. (2013) 87:1290–300. doi: 10.1128/JVI.02338-12
136. Xu J, Mercado-Lopez X, Grier JT, Kim WK, Chun LF, Irvine EB, et al. Identification of a Natural Viral RNA Motif That Optimizes Sensing of Viral RNA by RIG-I. MBio. (2015) 6:e01265–15. doi: 10.1128/mBio.01265-15
137. Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. (2011) 208:2357–66. doi: 10.1084/jem.20111171
138. Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. (2004) 279:12542–50. doi: 10.1074/jbc.M310175200
139. Tatematsu M, Funami K, Seya T, Matsumoto M. Extracellular RNA Sensing by Pattern Recognition Receptors. J Innate Immun. (2018) 10:1–9. doi: 10.1159/000494034
140. De Beuckelaer, Grooten J, De Koker S. Type I interferons modulate CD8(+) T cell immunity to mRNA Vaccines. Trends Mol Med. (2017) 23:216–26. doi: 10.1016/j.molmed.2017.01.006
141. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. (2015) 15:231–42. doi: 10.1038/nri3806
142. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
143. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. (2005) 5:675–87. doi: 10.1038/nri1684
144. Blanchard EL, Loomis KH, Bhosle SM, Vanover D, Baumhof P, Pitard B, et al. Proximity ligation assays for in situ detection of innate immune activation: focus on in vitro-transcribed mRNA. Mol Ther Nucleic Acids. (2018) 14:52–66. doi: 10.1016/j.omtn.2018.11.002
145. Lutz J, Lazzaro S, Habbeddine M, Schmidt KE, Baumhof P, Mui BL, et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines. (2017) 2:29. doi: 10.1038/s41541-017-0032-6
146. Pepini T, Pulichino AM, Carsillo T, Carlson AL, Sari-Sarraf F, Ramsauer K, et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J Immunol. (2017) 198:4012–24. doi: 10.4049/jimmunol.1601877
147. Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. (2006) 26:804–19. doi: 10.1089/jir.2006.26.804
148. Avci-Adali M, Behring A, Steinle H, Keller T, Krajeweski S, Schlensak C, Wendel HP. In vitro synthesis of modified mRNA for induction of protein expression in human cells. J Vis Exp. (2014):e51943. doi: 10.3791/51943
149. Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. (2008) 16:1833–40. doi: 10.1038/mt.2008.200
150. Kariko K, Muramatsu H, Keller JM, Weissman D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther. (2012) 20:948–53. doi: 10.1038/mt.2012.7
151. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52.
152. Zaba LC, Krueger JG, Lowes MA, Resident and “inflammatory” dendritic cells in human skin. J Invest Dermatol. (2009) 129:302–8. doi: 10.1038/jid.2008.225
153. Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. (2011) 29:154–7. doi: 10.1038/nbt.1733
154. Broos K, Van der Jeught K, Puttemans J, Goyvaerts C, Heirman C, Dewitte H, et al. Particle-mediated intravenous delivery of antigen mRNA results in strong antigen-specific t-cell responses despite the induction of type I interferon. Mol Ther Nucleic Acids. (2016) 5:e326. doi: 10.1038/mtna.2016.38
155. Kowalczyk A, Doener F, Zanzinger K, Noth J, Baumhof P, Fotin-Mleczek M, et al. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine. (2016) 34:3882–93. doi: 10.1016/j.vaccine.2016.05.046
156. Liang F, Ploquin A, Hernandez JD, Fausther-Bovendo H, Lindgren G, Stanley D, et al. Dissociation of skeletal muscle for flow cytometric characterization of immune cells in macaques. J Immunol Methods. (2015) 425:69–78. doi: 10.1016/j.jim.2015.06.011
157. Routy JP, Boulassel MR, Yassine-Diab B, Nicolette C, Healey D, Jain R, et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin Immunol. (2010) 134:140–7. doi: 10.1016/j.clim.2009.09.009
158. Allard SD, De Keersmaecker B, de Goede AL, Verschuren EJ, Koetsveld J, Reedijk ML, et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin Immunol. (2012) 142:252–68. doi: 10.1016/j.clim.2011.10.010
159. Gandhi RT, Kwon DS, Macklin EA, Shopis JR, McLean AP, McBrine N, et al. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mrna encoding HIV-1 Gag and Nef: results of a randomized, placebo-controlled clinical trial. J Acquir Immune Defic Syndr. (2016) 71:246–53. doi: 10.1097/QAI.0000000000000852
160. Van Craenenbroeck AH, Smits EL, Anguille S, Van de Velde A, Stein B, Braeckman T, et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation. (2015) 99:120–7. doi: 10.1097/TP.0000000000000272
161. Kubler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, Vom Dorp F, et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J Immunother Cancer. (2015) 3:26. doi: 10.1186/s40425-015-0068-y
162. Burgess DJ. RNA stability: remember your driver. Nat Rev Genet. (2012) 13:72. doi: 10.1038/nrg3159
163. Lundstrom K. Latest development on RNA-based drugs and vaccines. Future Sci OA. (2018) 4:FSO300. doi: 10.4155/fsoa-2017-0151
164. Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. (2008) 180:309–18.
165. Trepotec Z, Aneja M, Geiger J, Hasenpusch G, Plank C, Rudolph C. Maximizing the translational yield of mRNA therapeutics by minimizing 5'-UTRs. Tissue Eng Part A. (2018) 25:69–79. doi: 10.1089/ten.TEA.2017.0485
166. Li J, Zeng M, Shan H, Tong C. Microneedle patches as drug and vaccine delivery platform. Curr Med Chem. (2017) 24:2413–22. doi: 10.2174/0929867324666170526124053
167. DeFrancesco L. The ‘anti-hype’ vaccine. Nat Biotechnol. (2017) 35:193–97. doi: 10.1038/nbt.3812
169. Hoerr A. successful founder off the beaten path. Nat Biotechnol. (2017) 35:900–3. doi: 10.1038/nbt.3975
Keywords: mRNA vaccine, infectious disease, delivery, mechanism, application
Citation: Zhang C, Maruggi G, Shan H and Li J (2019) Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 10:594. doi: 10.3389/fimmu.2019.00594
Received: 04 October 2018; Accepted: 05 March 2019;
Published: 27 March 2019.
Edited by:
Karl Ljungberg, Karolinska Institutet (KI), SwedenReviewed by:
Oystein Evensen, Norwegian University of Life Sciences, NorwayJacek Jemielity, University of Warsaw, Poland
Copyright © 2019 Zhang, Maruggi, Shan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junwei Li, andsaUBxYXUuZWR1LmNu