 Stephanie A. Legere
Stephanie A. Legere Ian D. Haidl
Ian D. Haidl Jean-François Légaré
Jean-François Légaré Jean S. Marshall
Jean S. Marshall- 1Departments of Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
- 2Department of Pathology, Dalhousie University, Halifax, NS, Canada
- 3Department of Surgery, Dalhousie Medicine New Brunswick, Saint John, NB, Canada
Mast cells (MC) are innate immune cells present in virtually all body tissues with key roles in allergic disease and host defense. MCs recognize damage-associated molecular patterns (DAMPs) through expression of multiple receptors including Toll-like receptors and the IL-33 receptor ST2. MCs can be activated to degranulate and release pre-formed mediators, to synthesize and secrete cytokines and chemokines without degranulation, and/or to produce lipid mediators. MC numbers are generally increased at sites of fibrosis. They are potent, resident, effector cells producing mediators that regulate the fibrotic process. The nature of the secretory products produced by MCs depend on micro-environmental signals and can be both pro- and anti-fibrotic. MCs have been repeatedly implicated in the pathogenesis of cardiac fibrosis and in angiogenic responses in hypoxic tissues, but these findings are controversial. Several rodent studies have indicated a protective role for MCs. MC-deficient mice have been reported to have poorer outcomes after coronary artery ligation and increased cardiac function upon MC reconstitution. In contrast, MCs have also been implicated as key drivers of fibrosis. MC stabilization during a hypertensive rat model and an atrial fibrillation mouse model rescued associated fibrosis. Discrepancies in the literature could be related to problems with mouse models of MC deficiency. To further complicate the issue, mice generally have a much lower density of MCs in their cardiac tissue than humans, and as such comparing MC deficient and MC containing mouse models is not necessarily reflective of the role of MCs in human disease. In this review, we will evaluate the literature regarding the role of MCs in cardiac fibrosis with an emphasis on what is known about MC biology, in this context. MCs have been well-studied in allergic disease and multiple pharmacological tools are available to regulate their function. We will identify potential opportunities to manipulate human MC function and the impact of their mediators with a view to preventing or reducing harmful fibrosis. Important therapeutic opportunities could arise from increased understanding of the impact of such potent, resident immune cells, with the ability to profoundly alter long term fibrotic processes.
Introduction
Mast cells (MCs) are tissue-specific innate immune cells located in sites throughout the body, including the heart (1). After differentiation from hematopoietic stem cells along the myeloid pathway, committed MC precursors which can be identified by flow cytometry transiently travel through the blood and enter into tissues to differentiate into a terminal tissue-specific MC phenotype (2). Degranulated mast cells can be identified in most species by their expression of c-Kit, FCεRI and mast cell specific proteases. MCs are known as sentinel cells, surveying the microenvironment and responding to stimuli via expression of Pattern Recognition Receptors (PRRs) that detect Pathogen and Damage-Associated Molecular Patterns (PAMPs and DAMPs) (3, 4). MCs respond in several ways: (1) they can be activated to degranulate and release stores of pre-formed mediators from their characteristic granules, (2) they can synthesize and secrete mediators de novo without degranulation, or (3) a combination of degranulation and de novo synthesis can occur.
MC degranulation occurs not only in the context of allergy (5), but also in viral infection (6, 7), skin burns (8), fractures (9), and cardiac (10) and liver ischemia reperfusion injury (11, 12). MC degranulation is associated with pro-inflammatory effects, primarily due to release of histamine, tumor necrosis factor [TNF], and proteases. MC granules contain a plethora of mediators including, but not limited to: MC-specific and non-specific proteases (tryptase, chymase, cathepsin G), lysosomal enzymes (β-hexosaminidase), biogenic amines (histamine, serotonin, dopamine), cytokines (TNF, interleukin[IL]-4, IL-5), and growth factors (stem cell factor [SCF], basic fibroblast growth factor [bFGF]) (13). Overall, MC degranulation is an important contributor to inflammatory processes in injury and infection.
MCs are multi-functional cells capable of discrete as well as overwhelming responses and have ongoing immune regulatory and sentinel roles. They can selectively secrete numerous mediators that range from pro-inflammatory (IL-1β, IL-6, interferon[IFN]-γ) to anti-inflammatory (IL-10, IL-13), as well as pro-fibrotic (transforming growth factor-β1 [TGF-β1], bFGF) and anti-fibrotic (vascular endothelial growth factor [VEGF], IL-33, prostaglandin D2 [PGD2]) (14–17). Given the potential for MCs to produce pro- and anti-fibrotic mediators, their role in tissue remodeling is controversial. Local stimuli present after tissue injury and during wound healing can result in vastly different MC responses.
After myocardial infarction (MI), wound healing restores function to damaged tissue. Fibrosis is the deposition of a collagen-based scar mediated by fibroblasts, which differentiate upon activation into myofibroblasts for collagen deposition. Normally, fibrotic deposition is essential to restore proper function, but excessive remodeling decreases contractility and cardiac function leading to chronic heart failure (18–20). Cardiac tissue resident MCs respond to DAMPs after injury to influence the progression of cardiac remodeling. Yet the exact role of MCs in cardiac fibrosis is controversial, as numerous studies have ascribed detrimental, neutral and beneficial roles (Table 1). Achieving a better understanding of how the multifaceted MC response influences post-MI healing should increase the potential to harness their activities and provide opportunities for therapy.

Table 1. The role of mast cells in animal models of cardiac fibrosis.
Mast Cells as Enhancers in Cardiac Fibrosis
MC degranulation products have important impacts on fibrosis (Figure 1A), though exact cardiac degranulation stimuli are not well-defined. MC chymase and tryptase generate the active pro-fibrotic form of TGF-β1 from latent forms released by MCs during degranulation, as well as what is present in the microenvironment (44–51). TGF-β1 is important in fibrosis through promotion of fibroblast activation, myofibroblast differentiation and collagen synthesis (18, 19). MC tryptase can directly induce these actions on fibroblasts independently of TGF-β1 (52–57). In vitro, MC chymase induces TGF-β1 production by rat cardiac fibroblasts (58). Angiotensin II (AngII) is a major mediator of fibrosis that activates fibroblasts to the myofibroblast phenotype for proliferation and collagen deposition (18, 19). MC chymase is an angiotensin converting enzyme (ACE)-independent generator of AngII in humans, dogs and mice (20, 47, 59–61). Studies employing ACE inhibition or reduction of AngII show decreased cardiac fibrosis (62–65).
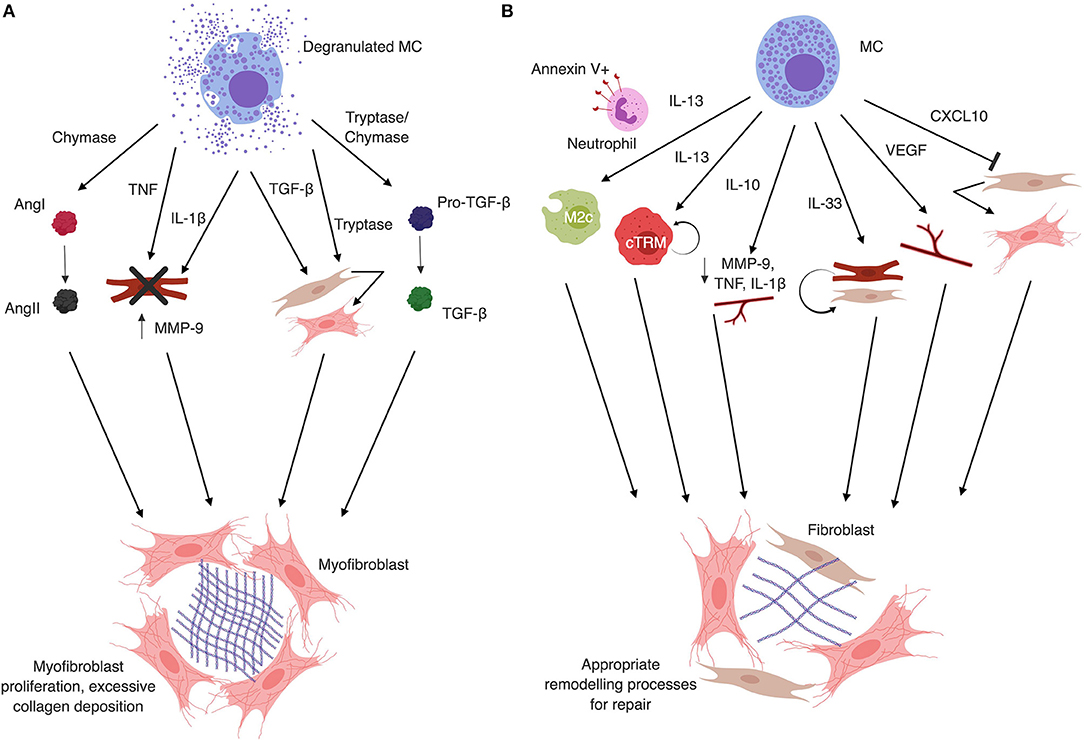
Figure 1. (A) Mast cell granule products are typically associated with fibrosis. Mast cell chymase converts Angiotensin I (AngI) to AngII independently of ACE. AngII generation directly contributes to fibrosis by inducing differentiation of fibroblasts to myofibroblasts. Mast cell degranulation-derived TNF and IL-1β induce cardiomyocyte apoptosis, MMP-9 production and inflammatory cell recruitment that enhances tissue remodeling. Mast cell tryptase can act directly on fibroblasts to induce proliferation and differentiation to the myofibroblast phenotype. Tryptase and chymase both act on latent TGF-β to convert it to the active form, which also induces fibroblast differentiation to the myofibroblast phenotype and collagen deposition. Additionally, mast cells release TGF-β upon degranulation, further contributing to the activation and differentiation of fibroblasts. (B) Mast cell secretion products can protect against fibrosis. Mast cells can produce IL-13, which in the presence of apoptotic neutrophils can induce M2c phenotype macrophages. M2c macrophages are associated with decreased fibrosis. IL-13 can also induce proliferation of local cTRM via IL-4Rα signaling, which are known to be anti-fibrotic. Mast cells can also produce IL-10, which acts in the heart to decrease IL-1β and TNF levels, reduce MMP-9 expression and activity, and increase capillary density to reduce fibrotic remodeling. IL-33, which is released by stressed cardiomyocytes and fibroblasts, but can also be produced by mast cells, has been shown to protect cardiomyocytes and fibroblasts from death under hypoxic conditions. This results in decreased inflammation and reduction in fibrosis. VEGF, which promotes angiogenesis and recapillarization of the cardiac tissue, is associated with reduced fibrosis and is another mast cell product. Finally, CXCL10 has been shown to inhibit fibroblast migration into the myocardium and delay differentiation to the pro-fibrotic myofibroblast phenotype. Figure created in BioRender.
In addition to tryptase and chymase, MCs store bFGF in their granules (3, 20, 45, 66), which, as its name suggests, is another enhancer of fibrosis. MCs also serve as sources of TNF, which is released during degranulation (13) and promotes cardiac fibrosis via induction of cardiomyocyte apoptosis, inflammation and MMP-9 production (67–70). Finally, MCs produce IL-1β during degranulation (14), which promotes fibrotic remodeling of the heart in a similar manner to TNF (70–74). Although mechanisms of action are not well-elucidated, Wang et al. found that blocking TNF and IL-1β reduced cardiac remodeling and cardiomyocyte apoptosis following AngII-induced fibrosis (70).
Numerous studies have attempted to understand MC roles in cardiac fibrosis in vivo (Table 1). Studies in rats, dogs and mice have shown that inhibition of MC degranulation or chymase activity reduces expression of fibrosis-associated genes and collagen deposition in models of dilated cardiomyopathy (DCM), ovariectomy-induced left ventricular diastolic dysfunction and MI (22–24, 26, 29). These studies are limited in their assessment of MC function exclusively through degranulation capacity, as they did not assess MC involvement in fibrosis through de novo mediator production. In a spontaneously hypertensive rat (SHR) model, degranulation inhibition increased MC number observed histologically, as well as myocardial IL-10 and IL-6 content, leading to improved outcomes and reduced fibrosis compared to untreated SHR (30). MCs are well-established sources of both IL-6 and IL-10 (14, 75). Therefore these results could suggest a potential role for MCs independent of degranulation.
Studies assessing MCs in cardiac fibrosis often analyze MC density changes that occur during remodeling, concluding a pro-fibrotic role. Studies in the mouse have demonstrated peak increases in mast cells at 7 days post-MI which result from increased infiltration of mast cell precursors identified as Lin−CD45+CD34+β7-integrin+FcγRII/III+ cells. Such mast cell increases were dependent on SCF (41), and are also associated with a degree of local mast cell precursor proliferation within the heart tissue. In canine MI and murine pulmonary artery bypass models, increases in MC density occurred alongside increases in inflammatory cell infiltration (25), fibrosis and cardiomyocyte hypertrophy (27), but no mechanistic relationships were found. Studies often only identify granulated MC populations. Common immunohistochemical (IHC) techniques for MCs identify granule-associated contents, ignoring populations of MCs that are not granulated, either due to immaturity or recent granule release. Additionally, MC degranulation releases SCF, a potent growth and chemotactic factor for MCs (13, 76), resulting in local proliferation (77) and recruitment (78). Therefore, increases in MC density may be due to activation of MCs from degranulation and not tissue damage.
In a transverse aortic constriction model (TAC), reconstitution of irradiated WT mice with bone marrow from W/Wv MC-deficient mice led to decreased collagen content compared to WT bone marrow recipients (28). MCs are radioresistant (79), therefore efficiency of MC removal after irradiation must be assessed, and was not in this paper. In a rat cardiac allograft model, fibrosis was positively correlated with certain subsets of “mucosal” MCs (MMC) but not “connective tissue”(CTMC) as defined by expression of mouse MCP-1 and MCP-2, respectively (21). This may reflect changes in the maturity of MC populations at this site and the presence of newly recruited cells. MC activation in atherosclerosis was associated with plaque progression and destabilization (80), which implicate MCs in promoting MI but does not directly link them to later fibrotic changes. Overall, MCs have the potential to promote cardiac fibrosis and increased numbers of granulated MCs are often associated with fibrosis in animal models, but mechanistic data is lacking. Care needs to be taken in experimental design to properly assess MC contribution.
Mast Cells as Inhibitors of Cardiac Fibrosis
MCs can synthesize and secrete a wide array of proteins without degranulating, allowing them to manipulate the cardiac microenvironment after ischemic damage or reperfusion injury in the heart (Figure 1B). MCs produce a wide array of pro-inflammatory cytokines and chemokines with proven roles in the recruitment of immune cells (13, 14). Conversely, MCs produce anti-inflammatory mediators such as IL-10 (75), IL-13, and CXCL10. IL-10 is known to prevent excessive cardiac remodeling via STAT3 activation and NF-κB suppression (81–83). CXCL10 acts in the damaged myocardium independently of CXCR3 to delay fibroblast migration and differentiation (84–87). While not classically considered part of the anti-fibrotic response, MCs can produce VEGF-A (13, 14), among other important angiogenic mediators, which can increase capillary density in damaged tissues and promote proper repair in cardiac and hepatic fibrosis (88–90).
IL-13 is produced by MC in response to several stimuli (14), including IL-33 (91). MCs express the IL-33 receptor ST2 abundantly on their cell surface (92–95). IL-33 is released by cardiomyocytes and fibroblasts after damage and also produced by MCs themselves (14). IL-33 is known to promote cardiomyocyte survival and reduces fibrosis after MI (96–98). Some of these actions may be via IL-13 induction. IL-13 acts on cardiac tissue resident macrophage (cTRM) populations, which are seeded embryonically in the heart and display M2-associated and anti-fibrotic phenotypes (99–102). cTRM self-renew and expand their populations in response to sterile inflammation and IL-4Rα signaling (103). Cardiac MC IL-13 production could expand the cTRM population locally. IL-13 also reduces expression of pro-inflammatory cytokines by infiltrating cells and may impact efferocytosis, the clearance of apoptotic cells from injured or inflamed tissues (104).
Anti-fibrotic roles of MCs have also been analyzed in vivo (Table 1). MC-deficient rats and mice had reduced collagen content compared to controls in models of homocysteine induced hypertrophy and coronary artery ligation (CAL) (35), while direct MC transplantation into the murine myocardium post-CAL increased cardiac function, and capillary density and decreased scar size (36). It is important to note that traditional MC-deficient models (rat and mouse) involve mutations in the gene for c-Kit (105), which encodes the SCF receptor, a growth factor critical for MCs. This mutation also reduces hematopoietic stem cells, germ cells and melanocytes, among other effects (105). MC reconstitution experiments should be performed to confirm observations are truly MC dependent, though it is not practical in all models. Several studies have focused on MC granule (MCG) contents in fibrosis. Administration MCGS isolated from rat peritoneal MCs to the myocardium during acute MI decreased fibrosis and increased capillary density. In vitro MCG treatment of cardiomyocytes promoted survival under hypoxic conditions (37). MCG treatment of mesenchymal stem cells (MSC) in vitro prevented TGF-β1 mediated transition of MSCs to myofibroblasts in an alternative fibrotic pathway (38), even though individual MC granule products chymase and tryptase are pro-fibrotic. While there is limited evidence showing MCs are protective during cardiac fibrosis, these studies indicate that MC can have an anti-fibrotic role and could potentially be targeted therapeutically.
Mast Cells as Bystanders in Cardiac Fibrosis
Several studies suggest MCs do not influence cardiac fibrosis (Table 1). In a norepinephrine model, rats treated with degranulation inhibitor disodium cromoglycate had comparable collagen and Col1 mRNA content compared to untreated rats, therefore MCs were thought to be irrelevant (39). However, degranulation inhibition would have little impact on MC production of fibrosis regulating mediators. TAC of Wsh MC-deficient mice, another Kit-dependent deficiency model, resulted in hypertrophy and impaired cardiac function, but equivalent fibrosis compared to WT mice (40). Ngkelo et al. compared a newly developed MC-deficient mouse strain to a classical c-Kit mutation-dependent model. W/Wv mice and WT mice treated with disodium cromoglycate underwent MI, resulting in increased fibrosis and infarct size. Upon utilization of MC-deficient Cpa3Cre/+ mouse model, a more MC-specific deficiency, no difference in fibrosis was observed. Rather, MCs were important in myofilament Ca2+ sensitization and cardiac contractility (41). It remains problematic that animal models for cardiac fibrosis are limited in their ability to mimic chronic fibrotic changes seen clinically. Several potential factors in experimental design may also contribute to discrepancies in animal models that will be discussed herein.
Relevance of Research in Human Cardiac Fibrosis
Similar to animal models, data on MC involvement in human cardiac fibrosis is inconsistent (Table 1). Several human studies of cardiovascular disease have equated increases in MC density to a detrimental role in fibrotic remodeling without a clear functional relationship between the two variables (31, 32, 106). Positive correlations were observed between MC density and collagen content in human idiopathic dilated cardiomyopathy (33), end stage cardiomyopathy (31), and Chagas disease (34). It remains unclear whether this is a protective response, epiphenomenon or pathological process. Studies also indicate that MCs have no role in human cardiac fibrosis in data from patients with ischemic LV dysfunction (42) and Chagas disease (43). Overall, data varies as to the role of MCs in human cardiac fibrosis.
Human cardiac tissue is difficult to obtain and usually received as a biopsy or autopsy sample. Biopsy samples are limited in their location and tissue volume, while autopsy samples are often delayed in being treated appropriately to preserve MC. Normal control tissues are even more difficult to obtain than diseased. Future human cardiac fibrosis studies should aim to better characterize the role of MCs in disease and expand analysis beyond histological characteristics to gain mechanistic insights necessary to design new therapeutic strategies.
Confounding Factors in Mast Cell Cardiac Fibrosis Research
The role of MCs in cardiac fibrosis is contentious, although it is clear they have the potential to modify fibrotic responses and tissue repair. There are several potential reasons for observed discrepancies. First, mice are not an ideal model to study cardiac MCs. Unlike rats and dogs, mice have low heart MC content. Dogs on average have 6.8 ± 1.6 cardiac MCs/mm2, while C57BL/6 mice have 0.6 ± 0.2 cardiac MCs/mm2 (107). Data shows that MC density increases in murine hearts after damage (22, 28, 30, 33, 76, 108, 109), but it is unclear if statistically significant increases in MC content have physiological relevance, or that murine cardiac MC responses mirror those in humans. Recent evidence suggests that the distribution of mast cells in the hearts of mice also differs considerably from that in humans (110).
Second, there is widespread improper use of MC stabilizing agents. Disodium cromoglycate is used to inhibit MC degranulation in mice and rats. However, while disodium cromoglycate can inhibit IgE-dependent MC degranulation in rats, it does not inhibit this response in mice at similar or higher doses (111). This calls into question the validity of studies in which disodium cromoglycate has been used to treat mice. MC stabilization drugs only prevent calcium-dependent MC degranulation, but MC secretion of mediators independently of degranulation is not impeded.
Third, mouse models of MC deficiency involving mutations in c-Kit result in a lack of hematopoietic stem cells, germ cells, and melanocytes, among others (105). The advent of several Kit-independent models of MC deficiency have allowed researchers to determine if lack of MCs impacts the pathogenesis of various diseases, or if differences are due to deficiencies in other areas. Preferable models include Cpa3Cre/+ and Cpa3-Cre; Mcl-1fl/fl mice. Discrepancies are already starting to appear (41, 112, 113), suggesting that increases or decreases in density of numerous cell types in Kit-dependent models contribute more to disease than lacking MCs. Reconstitution experiments help in this respect, but only if appropriate reconstitution can be achieved, which is not always possible.
Finally, tissue fixation for MC staining greatly impacts the ability to visualize MCs. The aldehyde tissue fixation does not allow for proper visualization of MCs, but reduces detection of MCs by 57–49% depending on the IHC method. Proper identification of MCs via IHC requires fixation with Carnoy's fixative to fully visualize MCs in tissue (114). Care needs to be taken in designing studies of MCs in cardiac fibrosis, with consideration given to the variety of actions of these cells and the difficulty of their experimental manipulation.
Therapeutic Approaches
There are discrepancies as to the exact role of MCs in cardiac fibrosis, but it is clear that these cells have the potential to promote or protect against remodeling in the myocardium. MCs have been reported in numerous studies to be increased at sites of fibrosis (21, 25, 27, 33, 41, 76) and are a rich source of selectively induced regulatory mediators, making them a powerful target for manipulation of the remodeling myocardium. Stem cell therapies are an emerging area of research to promote cardiac regeneration after damage. Adenoviral gene transfer of SCF into pig and mouse myocardium increased c-Kit+ cells following MI, and reduced fibrosis (115, 116). Direct myocardial injection of SCF following MI increased recruitment of Lin−/c-Kit+ cells to the heart and promoted wound healing (117). SCF is thought to recruit and induce proliferation of cardiac cells (CSC), c-Kit+ bone marrow cells that regenerate damaged tissue (118, 119).
However, few efforts have been made to differentiate CSCs from MCs in cardiac tissue, often only identifying Lin−/c-Kit+ cells and assessing CD45 expression (116). Recent evidence suggests that in human hearts, the vast majority of c-Kit+ cells are tryptase+ with weak/low CD45+ (120), indicating these are MCs, not CSCs. Benefits conferred by SCF expansion of c-Kit+ cells could therefore be due to expansion of MC populations instead.
MC degranulation products are pro-fibrotic through several pathways (44–49, 52–57) (Figure 1A). Inhibiting MC degranulation or actions of MC-associated proteases promotes proper wound healing after myocardial damage (23, 26, 29, 30). MC stabilizing drugs, such as ketotifen and disodium cromoglycate, have been used in human subjects (121–125). MCs express a wide array of receptors that can be targeted for activation and secretion of chemokines, growth factors, and cytokines (13, 14) without degranulation (3, 4, 126). MC activation with IL-33 via ST2 results in production of several cytokines that may protect against remodeling (91). Examples include the aforementioned beneficial roles of IL-13 and VEGF-A. Additionally, MCs could be targeted to produce IL-33, which is known to be present in the injured myocardium and is associated with improved outcomes post MI (96–98, 127). Induction of MC IL-10 production in combination with degranulation inhibition could limit excessive production of AngII and TGF-β1 while dampening excessive remodeling processes through IL-10 inhibition of NF-κB and activation of STAT3 (81, 83). Given that MCs respond to DAMPs (e.g., IL-33) by producing mediators that are beneficial in fibrosis, blocking degranulation alone could allow them to exert beneficial effects without further stimulation that has the potential to be off target. Future studies should focus on elucidating mechanisms by which cardiac MC respond to DAMPs in situ, as well as the potential of a dual function therapy that blocks MC degranulation and promotes beneficial mediator production to fully harness the power of these cells.
Conclusion
Overall, the role of MCs in cardiac fibrosis is still not well-understood. Discrepancies exist within and between animal models, and in vitro data indicates a potential for pro- and anti-fibrotic activity. Future studies into the role of MCs in cardiac fibrosis should be carefully designed to use animal models with appropriate MC content and accurate MC deficiencies with confirmation by MC reconstitution. MC stabilizing drugs should also be employed with appropriate species activity. Effort should be made wherever possible to expand on the current breadth of knowledge in human patient samples, as cardiac tissue is underused but potentially valuable. Human in vitro models could also be employed more effectively since primary human MCs can be readily generated. MCs are situated in cardiac tissue in close proximity to the remodeling myocardium and represent a valuable target for therapeutic manipulation following cardiac damage when we have the necessary information to more reliably predict the impact of such interventions in the human cardiac setting. A better understanding of their role and activities is urgently needed to move forward in this field.
Author Contributions
SL contributed to drafting and organizing the manuscript. IH provided input on the molecular and immunological aspects of the manuscript and edited the manuscript. J-FL provided input on the clinical aspects of the manuscript, assisted with writing and edited the manuscript. JM conceptualized the review, re-wrote sections of the manuscript, and edited the manuscript. All authors provided critical appraisal and approval.
Funding
This work was supported by the Canadian Institutes of Health research (CIHR) THC-135230, and also by Dalhousie Medical Research Foundation. SL—CIHR 394140, NS Heart and Stroke foundation Bright Red award, Killam Laureate.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge funding from Canadian Institutes of Health Research, Dalhousie Medical Research Foundation, and The Nova Scotia Heart and Stroke Foundation. Additionally, we would like to thank Drs. Liliana Portales-Cervantes and Dihia Meghnem for editing assistance.
References
1. Sperr WR, Bankl HC, Mundigler G, Klappacher G, Grossschmidt K, Agis H, et al. The human cardiac mast cell: localization, isolation, phenotype, and functional characterization. Blood. (1994) 84:3876–84.
2. Hallgren J, Gurish MF. Mast cell progenitor trafficking and maturation. Adv Exp Med Biol. (2011) 716:14–28. doi: 10.1007/978-1-4419-9533-9_2
3. Redegeld FA, Yu Y, Kumari S, Charles N, Blank U. Non-IgE mediated mast cell activation. Immunol Rev. (2018) 282:87–113. doi: 10.1111/imr.12629
4. Halova I, Rönnberg E, Draberova L, Vliagoftis H, Nilsson GP, Draber P. Changing the threshold—Signals and mechanisms of mast cell priming. Immunol Rev. (2018) 282:73–86. doi: 10.1111/imr.12625
5. Amin K. The role of mast cells in allergic inflammation. Respir Med. (2012) 106:9–14. doi: 10.1016/j.rmed.2011.09.007
6. Syenina A, Jagaraj CJ, Aman SA, Sridharan A, St John AL. Dengue vascular leakage is augmented by mast cell degranulation mediated by immunoglobulin Fcγ receptors. Elife. (2015) 4:1–16. doi: 10.7554/eLife.05291
7. Shirato K, Taguchi F. Mast cell degranulation is induced by A549 airway epithelial cell infected with respiratory syncytial virus. Virology. (2009) 386:88–93. doi: 10.1016/j.virol.2009.01.011
8. Bankova LG, Lezcano C, Pejler G, Stevens RL, Murphy GF, Austen KF, et al. Mouse mast cell proteases 4 and 5 mediate epidermal injury through disruption of tight junctions. J Immunol. (2014) 192:2812–20. doi: 10.4049/jimmunol.1301794
9. Li WW, Guo TZ, Liang D, Sun Y, Kingery WS, Clark JD. Substance P signaling controls mast cell activation, degranulation, and nociceptive sensitization in a rat fracture model of complex regional pain syndrome. Anesthesiology. (2012) 116:882–95. doi: 10.1097/ALN.0b013e31824bb303
10. Zheng J, Wei CC, Hase N, Shi K, Killingsworth CR, Litovsky SH, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS ONE. (2014) 9:e94732. doi: 10.1371/journal.pone.0094732
11. El-Shitany N, El-desoky K. Cromoglycate, not ketotifen, ameliorated the injured effect of warm ischemia/reperfusion in rat liver: role of mast cell degranulation, oxidative stress, proinflammatory cytokine, and inducible nitric oxide synthase. Drug Des Devel Ther. (2015) 9:5237–46. doi: 10.2147/DDDT.S88337
12. Yang M, Ma Y, Tao S, Ding J, Rao L, Jiang H, et al. Mast cell degranulation promotes ischemia–reperfusion injury in rat liver. J Surg Res. (2014) 186:170–8. doi: 10.1016/j.jss.2013.08.021
13. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. (2014) 14:478–94. doi: 10.1038/nri3690
14. Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. (2018) 282:121–50. doi: 10.1111/imr.12634
15. Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
16. Janicki JS, Brower GL, Levick SP. Chapter 8 - The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. In: Hughes MR, McNagny KM, editors. Mast Cells: Methods and Protocols. New York, NY: Springer Science+Business Media (2015). p. 121–39.
17. Overed-Sayer C, Rapley L, Mustelin T, Clarke DL. Are mast cells instrumental for fibrotic diseases? Front Pharmacol. (2014) 4:174. doi: 10.3389/fphar.2013.00174
18. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
19. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction. Circ Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
20. Bradding P, Pejler G. The controversial role of mast cells in fibrosis. Immunol Rev. (2018) 282:198–231. doi: 10.1111/imr.12626
21. Zweifel M, Matozan K, Dahinden C, Schaffner T, Mohacsi P. Eotaxin/CCL11 levels correlate with myocardial fibrosis and mast cell density in native and transplanted rat hearts. Transplant Proc. (2010) 42:2763–6. doi: 10.1016/j.transproceed.2010.05.152
22. Palaniyandi SS, Inagaki K, Mochly-Rosen D. Mast cells and εPKC: a role in cardiac remodeling in hypertension-induced heart failure. J Mol Cell Cardiol. (2008) 45:779–86. doi: 10.1016/j.yjmcc.2008.08.009
23. Kanemitsu H, Takai S, Tsuneyoshi H, Yoshikawa E, Nishina T, Miyazaki M, et al. Chronic chymase inhibition preserves cardiac function after left ventricular repair in rats. Eur J Cardio Thoracic Surg. (2008) 33:25–31. doi: 10.1016/j.ejcts.2007.09.040
24. Wang H, Da Silva J, Alencar A, Zapata-Sudo G, Lin MR, Sun X, et al. Mast cell inhibition attenuates cardiac remodeling and diastolic dysfunction in middle-aged, ovariectomized fischer 344 × brown Norway rats. J Cardiovasc Pharmacol. (2016) 68:49–57. doi: 10.1097/FJC.0000000000000385
25. Somasundaram P, Ren G, Nagar H, Kraemer D, Mendoza L, Michael LH, et al. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J Pathol. (2005) 205:102–11. doi: 10.1002/path.1690
26. Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. (2003) 107:2555–8. doi: 10.1161/01.CIR.0000074041.81728.79
27. Luitel H, Sydykov A, Schymura Y, Mamazhakypov A, Janssen W, Pradhan K, et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol Rep. (2017) 5:1–14. doi: 10.14814/phy2.13146
28. Liao C, Akazawa H, Tamagawa M, Ito K, Yasuda N, Kudo Y, et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J Clin Invest. (2010) 120:242–53. doi: 10.1172/JCI39942
29. Wei CC, Hase N, Inoue Y, Bradley EW, Yahiro E, Li M, et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest. (2010) 120:1229–39. doi: 10.1172/JCI39345
30. Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. (2009) 53:1041–7. doi: 10.1161/HYPERTENSIONAHA.108.123158
31. Akgul A, Youker KA, Noon GP, Loebe M. Quantitative changes in mast cell populations after left ventricular assist device implantation. ASAIO J. (2005) 51:275–80. doi: 10.1097/01.MAT.0000150507.61120.00
32. Dilsizian V, Eckelman WC, Loredo ML, Jagoda EM, Shirani J. Evidence for tissue angiotensin-converting enzyme in explanted hearts of ischemic cardiomyopathy using targeted radiotracer technique. J Nucl Med. (2007) 48:182–7.
33. Batlle M, Pérez-Villa F, Lázaro A, Garcia-Pras E, Ramirez J, Ortiz J, et al. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplant Proc. (2007) 39:2347–9. doi: 10.1016/j.transproceed.2007.06.047
34. Roldão JA, Beghini M, Ramalho LS, Porto CS, Rodrigues DBR, Teixeira VPA, et al. Comparison between the collagen intensity and mast cell density in the lingual muscles and myocardium of autopsied chronic chagasic and nonchagasic patients. Parasitol Res. (2012) 111:647–54. doi: 10.1007/s00436-012-2882-1
35. Joseph J, Kennedy RH, Devi S, Wang J, Joseph L, Hauer-jensen M, et al. Protective role of mast cells in homocysteine-induced cardiac remodeling. Am J Physiol Heart Circ Physiol. (2005) 2132:2541–5. doi: 10.1152/ajpheart.00806.2004
36. Shao Z, Nazari M, Guo L, Li SH, Sun J, Liu SM, et al. The cardiac repair benefits of inflammation do not persist: Evidence from mast cell implantation. J Cell Mol Med. (2015) 19:2751–62. doi: 10.1111/jcmm.12703
37. Kwon JS, Kim YS, Cho AS, Cho HH, Kim JS, Hong MH, et al. The novel role of mast cells in the microenvironment of acute myocardial infarction. J Mol Cell Cardiol. (2011) 50:814–25. doi: 10.1016/j.yjmcc.2011.01.019
38. Nazari M, Ni NC, Lüdke A, Li SH, Guo J, Weisel RD, et al. Mast cells promote proliferation and migration and inhibit differentiation of mesenchymal stem cells through PDGF. J Mol Cell Cardiol. (2016) 94:32–42. doi: 10.1016/j.yjmcc.2016.03.007
39. Briest W, Rabler B, Deten A, Zimmer HG. Norepinephrine-induced cardiac hypertrophy and fibrosis are not due to mast cell degranulation article. Mol Cell Biochem. (2003) 252:229–37. doi: 10.1023/A:1025596404975
40. Buckley CL, Stokes AJ. Corin-deficient W-sh mice poorly tolerate increased cardiac afterload. Regul Pept. (2011) 172:44–50. doi: 10.1016/j.regpep.2011.08.006
41. Ngkelo A, Richart A, Kirk JA, Bonnin P, Vilar J, Lemitre M, et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med. (2016) 213:1353–74. doi: 10.1084/jem.20160081
42. Frangogiannis NG, Shimoni S, Chang SM, Ren G, Shan K, Aggeli C, et al. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol. (2002) 160:1425–33. doi: 10.1016/S0002-9440(10)62568-0
43. Milei J, Fernández Alonso G, Vanzulli S, Storino R, Matturri L, Rossi L. Myocardial inflammatory infiltrate in human chronic chagasic cardiomyopathy: Immunohistochemical findings. Cardiovasc Pathol. (1996) 5:209–19.
44. Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84bond by mast cell chymase. J Biol Chem. (1994) 269:18134–40.
45. Kanbe N, Kurosawa M, Nagata H, Yamashita T, Kurimoto F, Miyachi Y. Production of fibrogenic cytokines by cord blood-derived cultured human mast cells. J Allergy Clin Immunol. (2000) 106:S85–90. doi: 10.1067/mai.2000.106777
46. Kanbe N, Kurosawa M, Nagata H, Saitoh H, Miyachi Y. Cord blood-derived human cultured mast cells produce transforming growth factor beta1. Clin Exp Allergy. (1999) 29:105–13. doi: 10.1046/j.1365-2222.1999.00459.x
47. Miyazaki M, Takai S, Jin D, Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther. (2006) 112:668–76. doi: 10.1016/j.pharmthera.2006.05.008
48. Cho SH, Lee SH, Kato A, Takabayashi T, Kulka M, Shin SC, et al. Cross-talk between human mast cells and bronchial epithelial cells in plasminogen activator inhibitor-1 production via transforming growth factor-β1. Am J Respir Cell Mol Biol. (2015) 52:88–95. doi: 10.1165/rcmb.2013-0399OC
49. Takai S, Jin D, Sakaguchi M, Katayama S, Muramatsu M, Sakaguchi M, et al. A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidine-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathic hamsters. J Pharmacol Exp Ther. (2003) 305:17–23. doi: 10.1124/jpet.102.045179
50. Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiäinen M, Kokkonen JO, et al. Activation of paracrine TGF-β1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. (2001) 15:1377–88. doi: 10.1096/fj.00-0273com
51. Tatler AL, Porte J, Knox A, Jenkins G, Pang L. Tryptase activates TGFβ in human airway smooth muscle cells via direct proteolysis. Biochem Biophys Res Commun. (2008) 370:239–42. doi: 10.1016/j.bbrc.2008.03.064
52. Akers IA, Parsons M, Hill MR, Hollenberg MD, Sanjar S, Laurent GJ, et al. Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am J Physiol Cell Mol Physiol. (2000) 278:L193–201. doi: 10.1152/ajplung.2000.278.1.L193
53. Abe M, Kurosawa M, Ishikawa O, Miyachi Y, Kido H. Mast cell tryptase stimulates both human dermal fibroblast proliferation and type I collagen production. Clin Exp. Allergy. (1998) 28:1509–17. doi: 10.1046/j.1365-2222.1998.00360.x
54. Garbuzenko E, Nagler A, Pickholtz D, Gillery P, Reich R, Maquart FX, et al. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin Exp Allergy. (2002) 32:237–46. doi: 10.1046/j.1365-2222.2002.01293.x
55. Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, et al. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. (1997) 158:2310–7.
56. Cairns JA, Walls AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest. (1997) 99:1313–21. doi: 10.1172/JCI119290
57. Gailit J, Marchese MJ, Kew RR, Gruber BL. The Differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol. (2001) 117:1113–19. doi: 10.1046/j.1523-1747.2001.15211.x
58. Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H, Shang FJ, et al. Chymase induces profibrotic response via transforming growth factor-β1/Smad activation in rat cardiac fibroblasts. Mol Cell Biochem. (2008) 310:159–66. doi: 10.1007/s11010-007-9676-2
59. Caughey GH, Raymond WW, Wolters PJ. Angiotensin II generation by mast cell alpha- and beta-chymases. Biochim Biophys Acta. (2000) 1480:245–57. doi: 10.1016/S0167-4838(00)00076-5
60. Li M, Liu K, Michalicek J, Angus JA, Hunt JE, Dell'Italia LJ, et al. Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J Clin Invest. (2004) 114:112–20. doi: 10.1172/JCI20805
61. Takai S, Jin D, Sakaguchi M, Miyazaki M. A single treatment with a specific chymase inhibitor, TY-51184, prevents vascular proliferation in canine grafted veins. J Pharmacol Sci. (2004) 94:443–8. doi: 10.1254/jphs.94.443
62. Hale TM, Robertson SJ, Burns KD, deBlois D. Short-term ACE inhibition confers long-term protection against target organ damage. Hypertens Res. (2012) 35:604–10. doi: 10.1038/hr.2012.2
63. D'Souza KM, Biwer LA, Madhavpeddi L, Ramaiah P, Shahid W, Hale TM. Persistent change in cardiac fibroblast physiology after transient ACE inhibition. Am J Physiol Circ Physiol. (2015) 309:H1346–53. doi: 10.1152/ajpheart.00615.2015
64. Liang B, Leenen FHH. Prevention of salt induced hypertension and fibrosis by angiotensin converting enzyme inhibitors in Dahl S rats. Br J Pharmacol. (2007) 152:903–14. doi: 10.1038/sj.bjp.0707472
65. Gut N, Piecha G, Aldebssi F, Schaefer S, Bekeredjian R, Schirmacher P, et al. Erythropoietin combined with ACE inhibitor prevents heart remodeling in 5/6 nephrectomized rats independently of blood pressure and kidney function. Am J Nephrol. (2013) 38:124–35. doi: 10.1159/000353106
66. Koskinen PK, Kovanen PT, Lindstedt KA, Lemström KB. Mast cells in acute and chronic rejection of rat cardiac allografts - a major source of basic fibroblast growth factor. Transplantation. (2001) 71:1741–7. doi: 10.1097/00007890-200106270-00007
67. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, et al. Tumor necrosis factor- mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. (2007) 115:1398–407. doi: 10.1161/CIRCULATIONAHA.106.643585
68. Kleinbongard P, Schulz R, Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. (2011) 16:49–69. doi: 10.1007/s10741-010-9180-8
69. Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, et al. TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol. (2013) 57:59–67. doi: 10.1016/j.yjmcc.2013.01.006
70. Wang Y, Li Y, Wu Y, Jia L, Wang J, Xie B, et al. 5TNF-α and IL-1β neutralization ameliorates angiotensin II-induced cardiac damage in male mice. Endocrinology. (2014) 155:2677–87. doi: 10.1210/en.2013-2065
71. Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. (2013) 126:1–11. doi: 10.1159/000351179
72. Fairweather DL, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Gatewood SJL, et al. Interferon-γ protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-β1, interleukin-1β, and interleukin-4 in the heart. Am J Pathol. (2004) 165:1883–94. doi: 10.1016/S0002-9440(10)63241-5
73. Coronado MJ, Brandt JE, Kim E, Bucek A, Bedja D, Abston ED, et al. Testosterone and interleukin-1β increase cardiac remodeling during coxsackievirus B3 myocarditis via serpin A 3n. Am J Physiol Circ Physiol. (2012) 302:H1726–36. doi: 10.1152/ajpheart.00783.2011
74. Frangogiannis NG. Interleukin-1 in cardiac injury, repair, and remodeling: pathophysiologic and translational concepts. Discov. (2015) 3:1–14. doi: 10.15190/d.2015.33
75. Lin TJ, Befus AD. Differential regulation of mast cell function by IL-10 and stem cell factor. J. Immunol. (1997) 159:4015–23.
76. Li J, Lu H, Plante E, Meléndez GC, Levick SP, Janicki JS. Stem cell factor is responsible for the rapid response in mature mast cell density in the acutely stressed heart. J Mol Cell Cardiol. (2012) 53:469–74. doi: 10.1016/j.yjmcc.2012.07.011
77. Ito T, Smrz D, Jung MY, Bandara G, Desai A, Smrzova S, et al. Stem cell factor programs the mast cell activation phenotype. J Immunol. (2012) 188:5428–37. doi: 10.4049/jimmunol.1103366
78. Faber TW, Pullen NA, Fernando JFA, Kolawole EM, McLeod JJA, Taruselli M, et al. ADAM10 is required for SCF-induced mast cell migration. Cell Immunol. (2014) 290:80–8. doi: 10.1016/j.cellimm.2014.05.005
79. Oldford SA, Haidl ID, Howatt MA, Leiva CA, Johnston B, Marshall JS. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J Immunol. (2010) 185:7067–76. doi: 10.4049/jimmunol.1001137
80. Bot I, Shi GP, Kovanen PT. Mast cells as effectors in atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:265–71. doi: 10.1161/ATVBAHA.114.303570
81. Verma SK, Garikipati VNS, Krishnamurthy P, Schumacher SM, Grisanti LA, Cimini M, et al. Interleukin-10 inhibits bone marrow fibroblast progenitor cell-mediated cardiac fibrosis in pressure-overloaded myocardium. Circulation. (2017) 136:940–53. doi: 10.1161/CIRCULATIONAHA.117.027889
82. Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor- B. Circulation. (2012) 126:418–29. doi: 10.1161/CIRCULATIONAHA.112.112185
83. Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. (2009) 104:9–19. doi: 10.1161/CIRCRESAHA.108.188243
84. Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, et al. Induction of the CXC chemokine interferon-γ-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. (2009) 105:973–83. doi: 10.1161/CIRCRESAHA.109.199471
85. Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez-Quesada C, Lu B, et al. CXCR3-independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans. Cardiovasc Res. (2014) 103:217–27. doi: 10.1093/cvr/cvu138
86. Shiraha H, Glading A, Gupta K, Wells A. IP-10 inhibits epidermal growth factor-induced motility by decreasing epidermal growth factor receptor-mediated calpain activity. J Cell Biol. (1999) 146:243–54.
87. Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GSV, Leary CP, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. (2004) 31:395–404. doi: 10.1165/rcmb.2004-0175OC
88. Nako H, Kataoka K, Koibuchi N, Dong YF, Toyama K, Yamamoto E, et al. Novel mechanism of angiotensin II-induced cardiac injury in hypertensive rats: the critical role of ASK1 and VEGF. Hypertens Res. (2012) 35:194–200. doi: 10.1038/hr.2011.175
89. Tang JM, Luo B, Xiao JH, Lv YX, Li XL, Zhao JH, et al. VEGF-A promotes cardiac stem cell engraftment and myocardial repair in the infarcted heart. Int J Cardiol. (2015) 183:221–31. doi: 10.1016/j.ijcard.2015.01.050
90. Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. (2014) 146:1339–50.e1. doi: 10.1053/j.gastro.2014.01.061
91. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. (2007) 179:2051–4. doi: 10.4049/jimmunol.179.4.2051
92. Saluja R, Khan M, Church MK, Maurer M. The role of IL-33 and mast cells in allergy and inflammation. Clin Transl Allergy. (2015) 5:1–8. doi: 10.1186/s13601-015-0076-5
93. Saluja R, Zoltowska A, Ketelaar ME, Nilsson G. IL-33 and thymic stromal lymphopoietin in mast cell functions. Eur J Pharmacol. (2016) 778:68–76. doi: 10.1016/j.ejphar.2015.04.047
94. Motakis E, Guhl S, Ishizu Y, Itoh M, Kawaji H, Hoon MD, et al. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood. (2014) 123:e58–67. doi: 10.1182/blood-2013-02-483792
95. Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol. (2015) 45:3034–44. doi: 10.1002/eji.201545501
96. Yin H, Li P, Hu F, Wang Y, Chai X, Zhang Y. IL-33 attenuates cardiac remodeling following myocardial infarction via inhibition of the p38 MAPK and NF-κB pathways. Mol Med Rep. (2014) 9:1834–8. doi: 10.3892/mmr.2014.2051
97. Ruisong M, Xiaorong H, Gangying H, Chunfeng Y, Changjiang Z, Xuefei L, et al. The protective role of interleukin-33 in myocardial ischemia and reperfusion is associated with decreased HMGB1 expression and up-regulation of the P38 MAPK signaling pathway. PLoS ONE. (2015) 10:e0143064. doi: 10.1371/journal.pone.0143064
98. Rui T, Zhang J, Xu X, Yao Y, Kao R, Martin CM. Reduction in IL-33 expression exaggerates ischaemia/reperfusion-induced myocardial injury in mice with diabetes mellitus. Cardiovasc Res. (2012) 94:370–8. doi: 10.1093/cvr/cvs015
99. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA. (2016) 113:E1414. doi: 10.1073/pnas.1602039113
100. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
101. Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. (2014) 211:2151–8. doi: 10.1084/jem.20140639
102. Falkenham A, De Antueno R, Rosin N, Betsch D, Lee TDG, Duncan R, et al. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol. (2015) 185:927–42. doi: 10.1016/j.ajpath.2014.11.027
103. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. (2013) 210:2477–91. doi: 10.1084/jem.20121999
104. Bosurgi L, Cao YGG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. (2017) 356:1072–6. doi: 10.1126/science.aai8132
105. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol. (2012) 33:613–25. doi: 10.1016/j.it.2012.09.008
106. Petrovic D, Zorc M, Zorc-Pleskovic R, Vraspir-Porenta O. Morphometrical and stereological analysis of myocardial mast cells in myocarditis and dilated cardiomyopathy. Folia Biol. (1999) 45:63–6.
107. Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG. Mast cells and macrophages in normal C57/BL/6 mice. Histochem. Cell Biol. (2002) 118:41–9. doi: 10.1007/s00418-002-0425-z
108. Huang ZG, Jin Q, Fan M, Cong XL, Han SF, Gao H, et al. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS ONE. (2013) 8:22–4. doi: 10.1371/journal.pone.0060827
109. Zhang W, Chancey AL, Tzeng HP, Zhou Z, Lavine KJ, Gao F, et al. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation. (2011) 124:2106–16. doi: 10.1161/CIRCULATIONAHA.111.052399
110. Ingason AB, Mechmet F, Atacho DAM, Steingrímsson E, Petersen PH. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol Immunol. (2019) 105:9–15. doi: 10.1016/j.molimm.2018.11.009
111. Oka T, Kalesnikoff J, Starkl P, Tsai M, Galli SJ. Evidence questioning cromolyn's effectiveness and selectivity as a mast cell stabilizer in mice. Lab Invest. (2012) 92:1472–82. doi: 10.1038/labinvest.2012.116
112. Reber LL, Daubeuf F, Pejler G, Abrink M, Frossard N. Mast cells contribute to bleomycin-induced lung inflammation and injury in mice through a chymase/mast cell protease 4-dependent mechanism. J Immunol. (2014) 192:1847–54. doi: 10.4049/jimmunol.1300875
113. Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald HR. Hematopoietic kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab. (2015) 21:678–91. doi: 10.1016/j.cmet.2015.04.013
114. Marshall JS, Ford GP, Bell EB. Formalin sensitivity and differential staining of mast cells in human dermis. Br J Dermatol. (1987) 117:29–36. doi: 10.1111/j.1365-2133.1987.tb04087.x
115. Ishikawa K, Fish K, Aguero J, Yaniz-Galende E, Jeong D, Kho C, et al. Stem cell factor gene transfer improves cardiac function after myocardial infarction in swine. Circ Hear Fail. (2015) 8:167–74. doi: 10.1161/CIRCHEARTFAILURE.114.001711
116. Yaniz-Galende E, Chen J, Chemaly E, Liang L, Hulot JS, McCollum L, et al. Stem cell factor gene transfer promotes cardiac repair after myocardial infarction via in situ recruitment and expansion of c-kit+ cells. Circ Res. (2012) 111:1434–45. doi: 10.1161/CIRCRESAHA.111.263830
117. Lutz M, Rosenberg M, Kiessling F, Eckstein V, Heger T, Krebs J, et al. Local injection of stem cell factor (SCF) improves myocardial homing of systemically delivered c-kit + bone marrow-derived stem cells. Cardiovasc Res. (2008) 77:143–50. doi: 10.1093/cvr/cvm027
118. Reinecke H, Minami E, Zhu WZ, Laflamme MA. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ Res. (2008) 103:1058–71. doi: 10.1161/CIRCRESAHA.108.180588
119. Martin-Puig S, Wang Z, Chien KR. Lives of a heart cell: tracing the origins of cardiac progenitors. Cell Stem Cell. (2008) 2:320–31. doi: 10.1016/j.stem.2008.03.010
120. Zhou Y, Pan P, Yao L, Su M, He P, Niu N, et al. CD117-positive cells of the heart: progenitor cells or mast cells? J. Histochem. Cytochem. (2010) 58:309–16. doi: 10.1369/jhc.2009.955146
121. Klooker TK, Braak B, Koopman KE, Welting O, Wouters MM, van der Heide S, et al. The mast cell stabiliser ketotifen decreases visceral hypersensitivity and improves intestinal symptoms in patients with irritable bowel syndrome. Gut. (2010) 59:1213–21. doi: 10.1136/gut.2010.213108
122. Okayama Y, Benyon RC, Rees PH, Lowman MA, Hillier K, Church MK. Inhibition profiles of sodium cromoglycate and nedocromil sodium on mediator release from mast cells of human skin, lung, tonsil, adenoid and intestine. Clin. Exp. Allergy. (1992) 22:401–9. doi: 10.1111/j.1365-2222.1992.tb03102.x
123. Church MK, Hiroi J. Inhibition of IgE-dependent histamine release from human dispersed lung mast cells by anti-allergic drugs and salbutamol. Br J Pharmacol. (1987) 90:421–9. doi: 10.1111/j.1476-5381.1987.tb08972.x
124. Okayama Y, Church MK. Comparison of the modulatory effect of ketotifen, sodium cromoglycate, procaterol and salbutamol in human skin, lung and tonsil mast cells. Int Arch Allergy Immunol. (1992) 97:216–25. doi: 10.1159/000236122
125. Brannan JD, Gulliksson M, Anderson SD, Chew N, Seale JP, Kumlin M. Inhibition of mast cell PGD2 release protects against mannitol-induced airway narrowing. Eur Respir J. (2006) 27:944–50. doi: 10.1183/09031936.06.00078205
126. Draber P, Halova I, Polakovicova I, Kawakami T. Signal transduction and chemotaxis in mast cells. Eur J Pharmacol. (2016) 778:11–23. doi: 10.1016/j.ejphar.2015.02.057
Keywords: mast cell, cardiac fibrosis, inflammation, tissue remodeling, immunology
Citation: Legere SA, Haidl ID, Légaré J-F and Marshall JS (2019) Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front. Immunol. 10:580. doi: 10.3389/fimmu.2019.00580
Received: 01 October 2018; Accepted: 04 March 2019;
Published: 28 March 2019.
Edited by:
Robert Murray Hamilton, Hospital for Sick Children, CanadaReviewed by:
Silvia Brunelli, University of Milano-Bicocca, ItalyAntonio Riva, Foundation for Liver Research, United Kingdom
Copyright © 2019 Legere, Haidl, Légaré and Marshall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean S. Marshall, amVhbi5tYXJzaGFsbEBkYWwuY2E=