
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 March 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00562
This article is part of the Research Topic Roles of Fc Receptors in Disease and Therapy View all 25 articles
 Tae Hyun Kang1†‡
Tae Hyun Kang1†‡ Chang-Han Lee1‡§
Chang-Han Lee1‡§ George Delidakis1
George Delidakis1 Jiwon Jung2
Jiwon Jung2 Odile Richard-Le Goff3
Odile Richard-Le Goff3 Jiwon Lee1Jin Eyun Kim2Wissam Charab1Pierre Bruhns3,4
Jiwon Lee1Jin Eyun Kim2Wissam Charab1Pierre Bruhns3,4 George Georgiou1,2,5,6*
George Georgiou1,2,5,6*IgG antibodies mediate the clearance of target cells via the engagement of Fc gamma receptors (FcγRs) on effector cells by eliciting antibody-dependent cellular cytotoxicity and phagocytosis (ADCC and ADCP, respectively). Because (i) the IgG Fc domain binds to multiple FcγRs with varying affinities; (ii) even low Fc:FcγRs affinity interactions can play a significant role when antibodies are engaged in high avidity immune complexes and (iii) most effector cells express multiple FcγRs, the clearance mechanisms that can be mediated by individual FcγR are not well-understood. Human FcγRIIIa (hFcγRIIIa; CD16a), which exists as two polymorphic variants at position 158, hFcγRIIIaV158 and hFcγRIIIaF158, is widely considered to only trigger ADCC, especially with natural killer (NK) cells as effectors. To evaluate the role of hFcγRIIIa ligation in myeloid-derived effector cells, and in particular on macrophages and monocytes which express multiple FcγRs, we engineered an aglycosylated engineered human Fc (hFc) variant, Fc3aV, which binds exclusively to hFcγRIIIaV158. Antibodies formatted with the Fc3aV variant bind to the hFcγRIIIaV158 allotype with a somewhat lower KD than their wild type IgG1 counterparts, but not to any other hFcγR. The exceptional selectivity for hFcγRIIIaV158 was demonstrated by SPR using increased avidity, dimerized GST-fused versions of the ectodomains of hFcγRs and from the absence of binding of large immune complex (IC) to CHO cells expressing each of the hFcγRs, including notably, the FcγRIIIaF158 variant or the highly homologous FcγRIIIb. We show that even though monocyte-derived GM-CSF differentiated macrophages express hFcγRIIIa at substantially lower levels than the other two major activating receptors, namely hFcγRI or hFcγRIIa, Fc3aV-formatted Rituximab and Herceptin perform ADCP toward CD20- and Her2-expressing cancer cells, respectively, at a level comparable to that of the respective wild-type antibodies. We further show that hFcγRIIIa activation plays a significant role on ADCC by human peripheral monocytes. Our data highlight the utility of Fc3aV and other similarly engineered exquisitely selective, aglycosylated Fc variants toward other hFcγRs as tools for the detailed molecular understanding of hFcγR biology.
Antibodies regulate a variety of immune responses by engaging Fcγ receptors (FcγRs) expressed on various leukocytes such as macrophages, granulocytes, dendritic cells (DCs), natural killer (NK) cells, and B cells. Immune complexes (ICs) formed by antibodies binding to multivalent antigens such as viruses and antibody-opsonized cancer or infected cells, induce activating FcγR-mediated immunoreceptor tyrosine-based activation motif (ITAM) signaling in effector cells, thereby inducing antibody-dependent cellular cytotoxicity and phagocytosis (ADCC/P) (1, 2). The activation of immune cells upon binding to ICs is regulated by the immunoreceptor tyrosine-based inhibitory motif (ITIM) signaling of the inhibitory FcγRIIb (3, 4). The ensuing signaling processes and immunological mechanisms triggered by IC binding depend on the expression level of FcγRs on various immune cell subsets and on the binding affinity of the Fc domain to the different FcγRs (2, 5). The composition of the single glycan at N297 of the human IgG1 Fc domain is known to both affect human FcγR (hFcγR) receptor affinity and, in some instances, to trigger additional signaling pathways via binding to the lectin-like type II receptors (6).
Human FcγRIIIa (hFcγRIIIa; CD16a) has been reported to be the most potent receptor for mediating ADCC (7, 8). NK cells which are considered to be the most important contributor to ADCC with peripheral blood mononuclear cells (PBMCs) as effector cells, express only hFcγRIIIa (2). hFcγRIIIa is also expressed in intermediate (CD14hi/CD16+) and non-classical (CD14dim/CD16hi) blood monocytes which are capable of inducing ADCC primarily via TNFα secretion (9, 10). Among the four human IgG subclasses, IgG1 and IgG3 have the higher affinity for hFcγRIIIa resulting in more potent ADCC in vitro (5, 11–13). A single nucleotide polymorphism of hFcγRIIIa at residue 158 which can be either Val and Phe is prevalent in humans. Earlier studies revealed that lymphoma patients expressing the higher affinity hFcγRIIIaV158 variant show improved clinical outcomes when treated with anti-CD20 (rituximab) and anti-Her2 (trastuzumab) compared to patients homozygous for the lower affinity, hFcγRIIIaF158 (14–17). The finding that higher affinity to hFcγRIIIa may lead to greater therapeutic potency stimulated extensive efforts to engineer hFc domains with improved binding hFcγRIIIa via either site-directed mutagenesis or glycoengineering, the latter accomplished primarily by completely abolishing or by reducing fucosylation (18–21). hFc defucosylated antibodies have up to 50-fold enhanced affinity to hFcγRIIIa and three defucosylated antibodies, anti-CCR4 mogamulizumab, anti-IL-5Ra benralizumab, and anti-CD20 obinutuzumab, have been evaluated for multiple therapeutic indications and approved for clinical use (21, 22).
Earlier reports had suggested that the improved therapeutic efficacy of antibodies having increased hFcγRIIIa affinity is due to more effective priming and activation of NK cells (8, 23, 24). However, in more recent studies it was observed that the depletion of macrophages, which are the predominant mediators of ADCP in tissues (25), abrogated the therapeutic efficacy of anti-CD20, anti-CD30, or anti-CD40 antibodies in mouse models whereas removal of NK cells or neutrophils did not significantly affect therapeutic efficacy (26–29). These results underline the critical role of tissue-resident macrophages and ADCP in anti-tumor antibody immunotherapy. ADCP is mainly attributed to signaling via hFcγRIIa, which is expressed at high levels on macrophages (30–32). Indeed, hFc-engineered antibodies with increased hFcγRIIa binding confer more potent ADCP (29, 31). Conversely, glycoengineered antibodies do not display higher affinity toward hFcγRIIa (33), but have been shown to increase macrophage phagocytic activity (34), potentially through hFcγRIIIa signaling.
hFcγRIIIa is expressed at much lower levels than hFcγRIIa in human in vitro monocyte-derived GM-CSF differentiated macrophages (gmMϕ) (30–32, 35). Thus, the importance of hFcγRIIIa engagement in mediating ADCP is an overlooked issue (36–38). Delineating the role of a particular hFcγR by blocking other receptors with antibodies can be problematic for two reasons: First, in vitro the cross-linking of the targeted hFcγR by blocking antibodies can alter the distribution of receptors on the membrane and also the blocking antibodies may impede the accessibility of other FcγR impacting the binding of immune complexes to other hFcγRs. Secondly, the co-administration of hFcγR blocking antibodies can complicate the design and interpretation of in vivo experiments. To circumvent these limitations our lab has been developing engineered Fc domains that have absolute selectivity for only one Fc binding protein (39, 40). The binding of IgG antibodies to hFcγRs or to the C1q component of the complement system is critically dependent on N-linked glycan at residue N297 of the Fc (41). Loss of the glycan increases the conformational flexibility of the CH2 domain resulting in very significant, albeit not complete loss of hFcγR and C1q binding and in drastically diminished effector functions (39, 42–44). Previously, our lab has developed an Fc variant, Fc5, that binds only to the high affinity hFcγRI and not to any other human FcγR. We further demonstrated that antibodies formatted with the Fc5 domain potentiate effective tumor cell death by monocyte-derived DCs via the ligation of FcγRI (39). In this work, we report on the engineering of an aglycosylated hFc domain, Fc3aV, that has essentially absolute specificity for the hFcγRIIIaV158 allotype. Using antibodies equipped with the Fc3aV variant, we showed that exclusive engagement of hFcγRIIIa results in potent ADCP with gmMϕ and also established the role of this receptor on ADCC with monocytes as effectors.
Burkitt's lymphoma Raji cells (ATCC® CCL-86™) and SK-BR-3 breast tumor cell lines (ATCC® HTB-30™) were obtained from American Type Culture Collection. Raji cells were cultured in complete RPMI with 10% fetal bovine serum (FBS) and SK-BR-3 cells were cultured in complete DMEM with 10% FBS. The collections of CHO cells expressing FLAG-tagged hFcγRs were described previously (14, 40, 45).
Human PBMCs and neutrophils were purified from anonymous healthy volunteers using Histopaque density gradient centrifugation (Sigma-Aldrich). Neutrophils were activated with 50 ng/mL hIFNγ for 24 h. NK cells were isolated by negative immunodensity isolation using the RosetteSep Human NK Cell Enrichment Cocktail (StemCell Technologies). Human GM-CSF differentiated macrophage cells were differentiated from CD14+ monocytes with 50 ng/mL GM-CSF (BioLegend®) for 7 days. Ectodomains of hFcγRI, hFcγRIIa, hFcγRIIb, and hFcγRIIIa, were produced in Expi293 cells (Invitrogen). All primers were synthesized by Integrated DNA Technologies.
As reported previously (40), E. coli cells expressing an IgG-library (E-library) were cultured overnight at 37°C with 250 rpm shaking in Terrific Broth media (TB, Difco™) with 2% (w/v) glucose, antibiotics (35 μg/ml of chloramphenicol and 50 μg/ml kanamycin). Following overnight growth, cells were diluted 1:100 in fresh TB medium and were induced with 1 mM of isopropyl-1-thio-β-D-galactopyranoside (IPTG) and 0.2% L-arabinose. Following incubation at 25°C for 20 h, the library cells were spheroplasted. as described previously (40). Spheroplasts were labeled with 10 nM dimeric hFcγRIIIaV158-GST-rPE and 100 nM of unconjugated dimeric hFcγRIIb-GST and sorted using a FACSAriaTM flow cytometer. FACS data were analyzed with FACSDiva software. For FACS screening, clones corresponding to the top 1% fluorescent events were isolated. Sorted spheroplasts were resorted immediately after initial sorting. The hFc mutant genes in the resorted spheroplasts rescued by PCR were cloned into SfiI digested pPelB-AglycoT(H)-FLAG vector, and transformed into electro-competent E. coli Jude-1 harboring pBAD-AglycoT(L)-His. The resulting transformants were subjected to a next round of sorting and resorting.
IgG1 antibodies and hFcγRs proteins were produced by transient transfection of Expi293F cells (Thermo Fisher Scientific) using the pcDNA3.4 vector (Thermo Fisher Scientific). Antibodies were purified by Protein A high capacity agarose resin (Thermo Fisher Scientific) affinity chromatography. 25 × PBS was added to filtered supernatants to a 1 × concentration, and the mixture was passed twice over the column. The column was washed with 100 ml of 1x PBS to remove nonspecifically bound proteins. Three milliliters of 100 mM glycine-HCl (pH 2.7) was added to elute the bound proteins, and the elution was immediately neutralized with 1 ml of 1 M Tris (pH 8.0). Samples were buffer-exchanged into pH7.4 PBS using Amicon Ultra-4 (Millipore) spin columns with a 10 kDa cutoff. The purity of purified samples was assessed by 4–20% gradient SDS-PAGE gel (NuSep).
Fifty microliters of 4 μg/ml of Trastuzumab IgG1 or its hFc variants were diluted in pH7.4 phosphate buffered saline (PBS) buffer and used to coat 96 well polystyrene ELISA plate (Corning) overnight at 4°C. After blocking with 1x PBS (pH 7.4) containing 0.5% BSA for 2 h at room temperature, the plate was washed 4 times with PBS containing 0.05% Tween20 (PBST), and incubated with serially diluted dimeric GST-tagged low affinity hFcγRs and monomeric his-tagged high affinity hFcγRI at room temperature for 1 h. After washing 4 times with the PBST buffer, either 1:10,000 diluted α-GST (GE Healthcare) or α-His antibody HRP conjugate were added. After washing three times with PBST, 50 μL TMB substrate was added per well (Thermo Scientific), 50 μL of 1 M H2SO4 was added to neutralize, and the absorbance at 450 nm was recorded.
For SPR analysis, Trastuzumab IgG1 or its variants were individually immobilized on CM5 sensor chips by amine coupling, as recommended by the manufacturer (GE Healthcare). Binding experiments were performed in HBS-EP buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA, and 0.005% P20 surfactant). Serially diluted dimeric GST-tagged low affinity hFcγRs or monomeric his-tagged high affinity hFcγRI were injected at a flow rate of 30 μl/min for 60 s with a dissociation time of 5 min. The chip was regenerated after each run by sequential injection of 50 mM glycine, pH 4.0, 50 mM glycine, pH 9.5, and 3 M NaCl for 1 min each. For each run, a bovine serum albumin (BSA)-coupled surface was used as reference channel. Equilibrium dissociation constants (KD) for monovalent receptor binding were determined by fitting 1:1 Langmuir model to the data using BIAevaluation 3.2 software (GE Healthcare) and the KDs were averaged from three independent experiments (n = 3 for hFcγRIIa and hFcγRIIIa, n = 2 for hFcγRI and hFcγRIIb). The resulting sensorgrams were fit with a 1:1 Langmuir isotherm model or equilibrium binding model for monomeric hFcγRs using Biaevaluation 3.0 software.
Raji cells or SK-BR-3 cells (105 cells) were incubated with various concentration of antibody variants for 30 min at 4°C and washed with 3% BSA in PBS. Raji- or SK-BR3-bound antibody levels were detected by FITC-conjugated, F(ab′)2 Fragment, goat Anti-Human IgG Fc (Jackson ImmunoResearch Laboratories).
CHO cells expressing FLAG-tagged hFcγRs were described previously (14, 40, 45). ICs were also generated by mixing 10 μg/ml of Trastuzumab or Trastuzumab-Fc3aV and 5 μg/ml of PE-conjugated F(ab′)2 goat anti-human IgG F(ab′)2 (Jackson ImmunoResearch Laboratories) for 30 min at 37°C (14, 40, 45). CHO cells expressing hFcγRs were incubated with ICs for 1 h on ice and cell-bound ICs were detected by flow cytometry on a MACSQuant (Miltenyi Biotech). Data were analyzed with Flow Cytometry Analysis Software (FlowJo). The IC-binding activities to NK cells or neutrophils were also assayed and measured using same method, but using a FACS Calibur (BD Biosciences).
As previously described (46, 47), FCGR3A genes were specifically amplified using the following primers, 5′-TCC AAA AGC CAC ACT CAA AGA CAG CGC-3′ and 5′-GAT GGT GAT GTT CAC AGT CTC T-3′ and the sequence of amplified FCGR3A was analyzed. FCGR3A SNP was also confirmed by nested PCR using following primer sets; 5′-TCC AAA AGC CAC ACT CAA AGA CAG CGC-3′ and 5′-CTC TGA AGA CAC ATT TTT ACT CCC AAA-3′ for FCGR3A F158, and 5′-TCC AAA AGC CAC ACT CAA AGA CAG CGC-3′ and CTC TGA AGA CAC ATT TTT ACT CCC AAC for FCGR3A V158.
SK-BR-3 or Raji cells were cultured in complete DMEM or RPMI medium as above and collected by centrifugation at 300x g for 5 min. Cells were washed in PBS and labeled with 4 μM Calcein AM (Invitrogen) in PBS at 37°C with 5% CO2 for 30 min. Calcein-loaded cancer cells were washed twice, resuspended in RPMI medium, and seeded into a 96-well plate at 10,000 cells/well with various concentrations of IgG variants. As previously described (40, 48), PBMCs and PMNs were isolated from healthy anonymous donors using Histopaque (Sigma-Aldrich). CD14+ monocytes were isolated from PBMCs by magnetic bead separation (EasySep by STEMCELL Inc.) (40). Effector cells were added into a 96-well plate at 100,000 cells/well and the plates were incubated at 37°C with 5% CO2 for 4 h. Released calcein AM was detected at excitation and emission wavelengths of 485 and 535 nm, respectively. The percent of tumor cell lysis was calculated according to the following formula; 100 × (E-S)/(M-S), where E is the fluorescence of experimental well, S is that in the absence of antibody (tumor cells were incubated with medium and complement alone), and M is that of tumor cells with lysis buffer (Triton X-100 at 2% v/v, SDS 1% w/v, 100 mM NaCl, and 1 mM EDTA).
Purified monocytes were differentiated into GM-CSF differentiated macrophages (gmMϕ) by culture for 7 days in RPMI medium containing 10% FBS and 50 ng/ml GM-CSF. Harvested gmMϕ by trypsinization were seeded into 96-well plate at 105 cells/well and then cultured in RPMI medium containing 10% FBS overnight. SK-BR-3 or Raji cells, were labeled with PKH67 (Sigma-Adrich) according to the manufacturer's instructions and opsonized by serially diluted IgG variants. IgG-opsonized and PKH-67-labeled cancer cells were added into gmMϕ-attached 96-well plate at 104 cells/well. After 2 h at 37°C with 5% CO2, gmMϕ cells were detached from the plate by Trypsin-EDTA treatment for 20 min. gmMϕ were stained with anti-CD45-APC (BD bioscience) for ADCP assay with SK-BR-3 cells on ice for 15 min. For ADCP assay with Raji cells, gmMϕ were stained with anti-CD14-APC and anti-CD11b-APC (Biolegend) on ice for 15 min. Phagocytosis was evaluated by FACSAria™ (BD Bioscience), and reported as the fraction of double positive cells over the total number of tumor cells in the sample.
Blockade assays for hFcγRIIIa were performed as follows; gmMϕ were pre-incubated with 10 μg/ml of anti-CD16 mAb 3G8 F(ab′)2 (Ancell) (49–51) was pre-incubated with gmMϕ for 10 min. IgG-opsonized and PKH-67-labeled cancer cells were then incubated with anti-CD16 mAb 3G8 F(ab′)2-coated gmMϕ in RPMI1640 medium without serum. After 2 h at 37°C, the fraction of phagocytosed tumor cells was detected as described above. For all assays, an E:T ratio of 10:1 was used.
Fluorescent images of macrophages phagocytozing cancer cells were obtained by confocal microscopy using calcein AM-loaded Raji cells or SK-BR-3 cells opsonized with 20 μg/ml of antibodies and gmMϕ at 37°C for 1 h. Approximately 1 × 105 labeled cancer cells and 1 × 105 macrophages were co-incubated in 1 ml of RPMI medium. Subsequently, the co-incubated cells were labeled as described above. Phagocytosis was visualized by confocal microscopy using Zeiss LSM 710/Elyra S.1.
Aglycosylated hFc domains specific for hFcγRIIIa were engineered via the screening of combinatorial libraries expressed in E. coli using a well-established full-length IgG-display system in bacteria (36, 40). Briefly, the heavy chain and light chains of IgG (having Trastuzumab Fab arms) were separately expressed by fusing pelB and NlpA, respectively resulting in tethering of IgG on the periplasmic side of the inner membrane. In order to isolate hFcγRIIIA-selective aglycosylated hFc variants, the IgG hFc domain was mutagenized by error-prone PCR and a library of >109 E. coli transformants was generated. The bacterial cells (40) were spheroplasted and the library was screened with 10 nM of dimeric hFcγRIIIAV158-GST, conjugated with rPE in the presence of 100 nM of unconjugated hFcγRIIB-GST as a competitor (Figures 1A,B). Ninety-six individual clones from the 4th round of sorting were randomly picked and analyzed further. The 96 clones encoded 22 unique hFc variants in which S239T, V264E, V282M, T299A, L309Q, and A378V amino acid mutations were enriched (Figure S1). Interestingly, 16 out of 22 clones contained a T299A substitution in the C'E loop of CH2 domain. The T299A mutation disrupts the canonical N-X-T/S N-linked glycosylation motif on the hFc and impairs, but not completely abolishes, hFcγR binding [(24), Figure S3]. Bacteria displaying IgGs with three different mutant Fc domains, namely, TEMA (S239T, V264E, V282M, T299A), EAQ (V264E, T299A, L309Q), and Fc3aV (S239T, P248L, V264E, V282M, T299A, L309Q, A378V), were analyzed by FACS (Figure S2). Cells expressing TEMA, EAQ and Fc3aV Fc mutated IgGs showed 16-, 48.2-, and 62.4-fold increased binding activity for hFcγRIIIaV158-GST dimers, respectively, compared with wild-type (wt) aglycosylated IgG1. In addition, TEMA and Fc3aV also slightly bound to hFcγRIIa or hFcγRIIb under these experimental conditions (Figure S2).
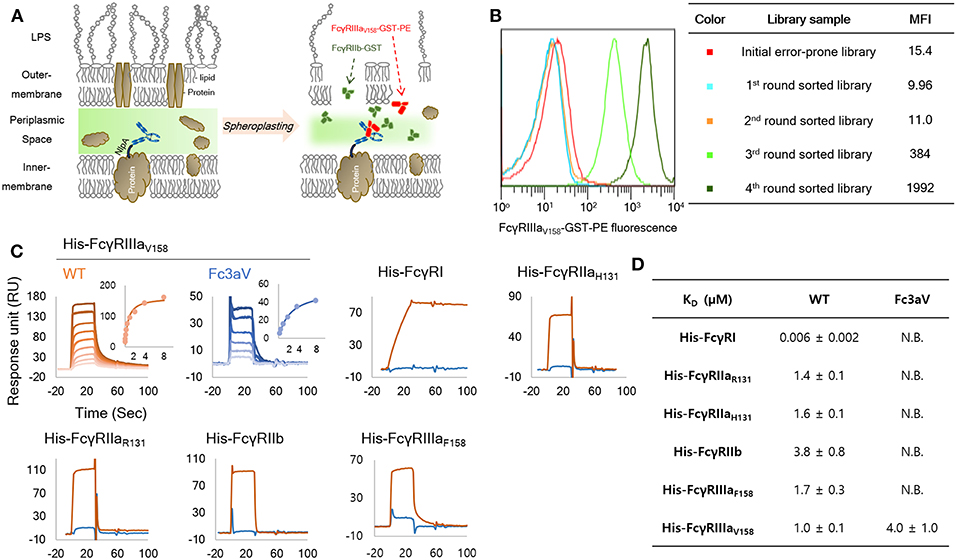
Figure 1. Engineering and biochemical characterization of a of FcγRIIIaV158-specific, Fc domain variant: (A) Schematic diagram of the high throughput screening system used for the isolation of FcγRIIIaV158−specific Fc variants from very large combinatorial libraries. Soluble fluorescent FcγRIIIaV158-GST and a large excess of non-fluorescent FcγRIIb-GST as a competitor are used for cell labeling and FACS sorting. (B) MFI (median fluorescence intensity) of initial library (red, size: 1 × 109), and 1st (light blue), 2nd (orange), 3rd (light green), and 4th (green) round sorted populations. (C,D) SPR analysis of wild-type Trastuzumab (orange) and Trastuzumab-Fc3aV (sky blue) with monomeric His-tagged FcγRs. Surface plasmon resonance (SPR) sensorgrams (C) and equilibrium dissociation constants for of Fc3aV in μM (D). N.B. means RUmax of sensorgram is lower than 5 RU with 8 μM of each His-hFcγR. All experiments were repeated three times and error corresponds to standard deviation (s.d.).
Several antibodies containing subsets of the amino acid substitutions found in TEMA, EAQ, and Fc3aV were expressed and analyzed in terms of their binding affinity and selectivity to purified monomeric, high-affinity hFcγRI-His or to GST-fused dimeric (and thus, higher avidity) versions of the low-affinity receptors, hFcγRIIa, hFcγRIIb, and hFcγRIIIa (36, 40). Three isolated Fc variants shared two mutations, V264E and T299A. First, the effect of V264E on the hFcγR-binding properties of aglycosylated Fc domain was investigated (Figure S3). Trastuzumab-V264E/T299A, showed higher hFcγRIIIa binding and decreased hFcγRIIb binding relative to the T299A variant (Figure S3). Similarly, Trastuzumab-EAQ (V264E, T299A, L309Q) showed higher affinity for hFcγRIIIa but lower hFcγRIIb binding relative to Trastuzumab-T299A/L309Q (Figure S3). Trastuzumab-Fc3aV showed selective binding to FcγRIIIaV158 by ELISA, but still displayed some binding toward FcγRIIIaF158 and FcγRIIb (Figure S3).
Due to the low sensitivity achieved by direct ELISA, we opted to measure the Fc-FcγR interactions in a highly sensitive SPR experiment. Trastuzumab-Fc3aV showed specific binding to GST-fused dimeric hFcγRIIIaV158 (KD = 0.2 ± 0.01 μM) without any significant binding to 400 nM of GST-fused dimeric hFcγRIIaH131/R131, hFcγRIIb, and hFcγRIIIaF158 by SPR (Figure S4A and Table S1). The equilibrium dissociation constants for the binding of Trastuzumab-Fc3aV to monomeric hFcγRs were also determined by SPR. Trastuzumab-Fc3aV exhibited no detectable response for any monomeric hFcγR except for hFcγRIIIaV158 to which it bound with a KD = 4 ± 1.0 μM (Figures 1C,D and Figure S4B,C).
6The binding of ICs onto cells represents an exquisitely sensitive assay for detecting physiologically relevant IgG:FcγR interactions (14, 52). Large, high avidity ICs were formed by mixing Fc3aV or other antibodies with goat F(ab′)2 anti-human F(ab′)2 and binding to CHO cells expressing the low affinity hFcγRIIaR131, hFcγRIIaH131, hFcγRIIb, hFcγRIIIaV158, and importantly hFcγRIIIaF158 which only differs from hFcγRIIIaV158 by one amino acid or the hFcγRIIIb-NA1 or hFcγRIIIb-NA2 allotype which differs from hFcγRIIIa by 6–8 amino acids at high levels was evaluated by FACS. IC formed by Trastuzumab Fc3aV bound to hFcγRIIIaV158 expressing CHO cells but did not show significant binding activities for all other hFcγRs (Figure 2A). To examine whether ICs of Trastuzumab Fc3aV bind to hFcγRI-expressing cells in a physiologically relevant context we determined binding to neutrophils from human blood, which are activated with IFN-γ. As expected, ICs formed with Trastuzumab Fc3aV did not bind to activated neutrophils which express hFcγRI, hFcγRIIa/b, and hFcγRIIIb but not hFcγRIIIa (Figure 2B) (53).
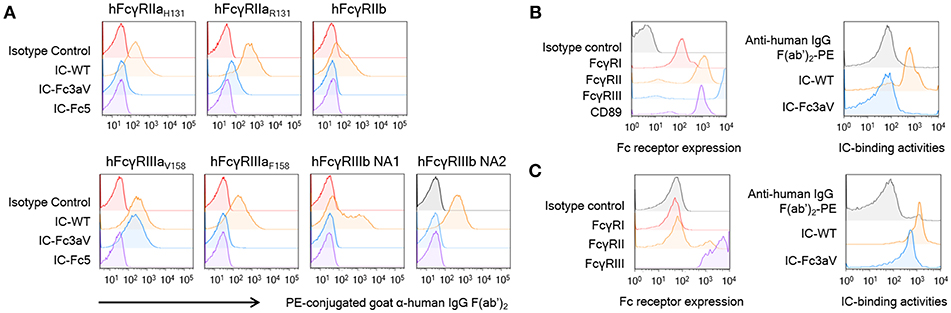
Figure 2. Fc3aV binds to FcγRIIIAV158 with exquisite selectivity (A) Binding activities of ICs (10 μg/ml), formed by mixing antibodies with F(ab′)2 anti-hIgG F(ab′)2-PE, to human FcγRs expressed by transfectant CHO cell measured by FACS, when antibodies are: isotype control (red), wt Trastuzumab (orange), Trastuzumab-Fc3aV (sky blue), and Trastuzumab-Fc5 (purple). (B,C) Binding activities of ICs on human neutrophils (B) or NK cells (C); immune complexes were formed and used at the same concentration as in (A). Left panels: Expression level of FcγRs on neutrophils activated by IFN-γ (B) or NK cells (C) detected by the respective anti-FcγR antibodies conjugated to APC; Right panels: Binding of ICs formed by wt or Fc3aV Trastuzumab, onto neutrophils or NK cells, respectively. Representative data of three independent experiments are shown.
hFcγRIIIa on the surface of NK cells has a different N-glycan composition relative to hFcγRIIIa on the surface of monocytes/macrophages or with hFcγRIIIa ectodomain produced recombinantly in Expi293F cells. Differences in receptor glycosylation can affect the affinity for IgG (54). Nonetheless, we found that, ICs formed with Trastuzumab Fc3aV show slightly lower binding to NK cells as those formed by wild-type antibodies, a finding entirely consistent with the lower affinity of Fc3aV determined by SPR (Figures 1D,2C).
To determine how hFcγRIIIa contributes to immune effector functions, we performed ADCP and ADCC assays using anti-CD20 Rituximab-Fc3aV or anti-Her2 Trastuzumab-Fc3aV and CD20+ Raji cells or Her2high SK-BR-3 breast cancer cells as targets, respectively. As expected, Fc3aV-formatted antibodies with Trastuzumab or Rituximab Fab showed equivalent binding with their wt counterparts to CD20+ Raji cells or Her2high SK-BR-3 breast cancer cells, respectively since mutations of the Fc domain did not affect antigen binding (Figure S5). We first performed ADCP assays with gmMϕ, which had been differentiated in vitro from human peripheral blood-derived CD14+ monocytes. gmMϕ express hFcγRI at moderate, hFcγRIIa/b at high and hFcγRIIIaV158 at low levels, as determined by flow cytometry (Figure 3A) (30–32, 35). Raji cells were labeled with fluorescent PKH67 and then opsonized with Fc-engineered Rituximab variants, followed by incubation with gmMϕ, as previously reported (40, 48). The extent of phagocytosis was evaluated using flow cytometry by determining the percentage of cells staining double positive for PKH67 and anti-CD14-APC or anti-CD11b-APC over the total number of PKH67+ cancer cells. We found that Rituximab-Fc3aV, which as described above binds only to hFcγRIIIaV158, has ADCP activity that is comparable to that of wt Rituximab (Figures 3B,C).
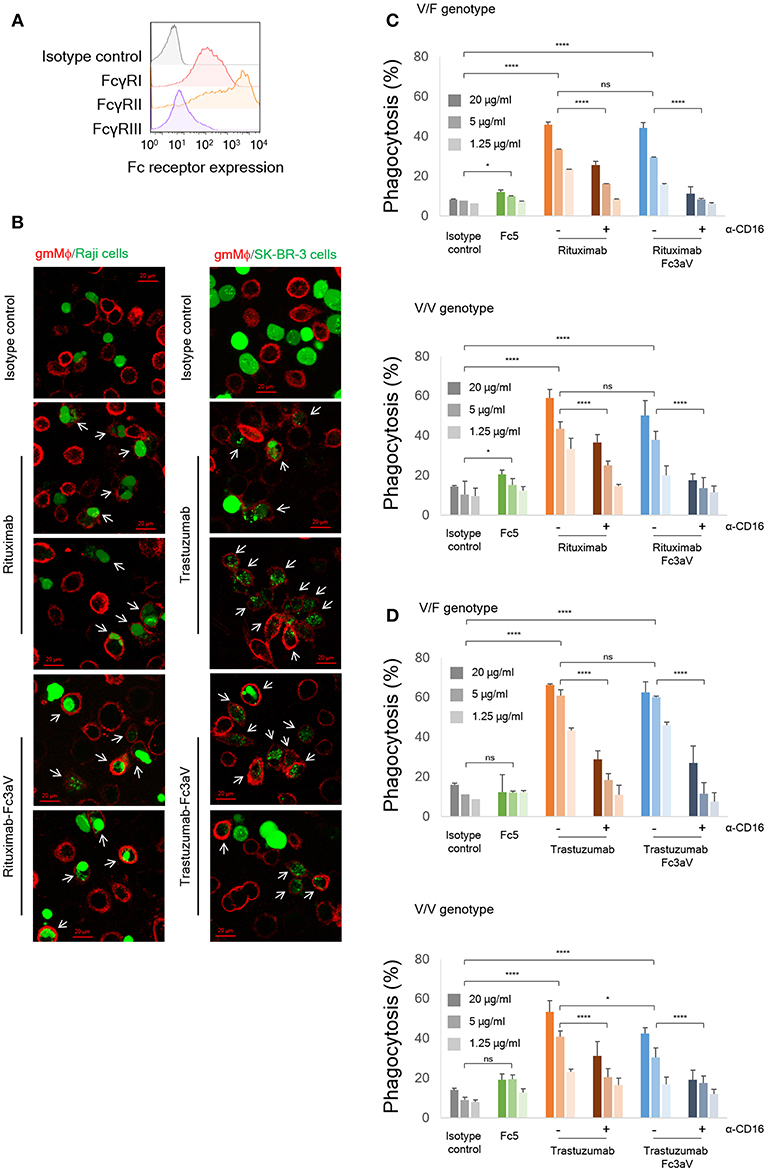
Figure 3. In vitro phagocytosis assays. (A) Expression levels of FcγRs on monocyte-derived human gmMϕ were detected by FACS using the respective anti-FcγR-APC antibodies. (B) Fluorescent images of phagocytosis of cancer cells (green, left panel: Raji and right panel: SK-BR-3) by gmMϕ (red). Macrophages were stained with anti-CD14 and anti-CD11b-APC and cancer cells were stained with Calcein-AM. White arrows indicate phagocytosed cancer cells. (C,D) ADCP assays with gmMϕ (V/F and V/V genotypes) as effectors and (C) PKH67-labeled CD20+ Raji cells or (D) PKH67-labeled Her2high SK-BR-3 cells as targets (Effector:Target ratio of 10:1). Macrophages were pre-incubated with or without 10 μg/ml of anti-CD16 mAb 3G8 F(ab′)2 for 10 min prior to the addition of wt or variants of trastuzumab. ADCP data are shown for an FcγRIIIa heterozygous donor (V/F) and one donor homozygous for the FcγRIIIaV158 allotype (V/V). Representative data from for independent experiments. Error bars are standard deviations of triplicate samples. Statistical analysis was performed by two way ANOVA with Tukey's multiple comparisons test (ns: P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
hFcγRIIIa-mediated phagocytosis by gmMϕ was also confirmed with Her2high SK-BR-3 cancer cells opsonized with Trastuzumab-Fc3aV antibodies (Figures 3B,D). ADCP by either wt IgG- or Fc3aV- were significantly inhibited by the addition of a blocking, anti-CD16 F(ab′)2 [mAb clone 3G8 (49–51)], confirming the importance of hFcγRIIIa in ADCP (Figures 3C,D). Opsonization of CD20+ Raji cells with Rituximab equipped with Fc5, an engineered hFc domain which as discussed above engages hFcγRI selectively, resulted in a low, but statistically significant level of ADCP. However, we did not detect phagocytosis of Her2+ SK-BR-3 cells using Trastuzumab-Fc5 (Figures 3C,D).
We also determined ADCC activity by detecting calcein-AM release from cancer cells (CD20+ Raji cells) with PBMC, monocytes, or NK cells, isolated from the same healthy donor (V/V or V/F genotype), as effectors (n = 3 donors). Based on the weaker binding activity of immune complexes with Fc3aV antibodies on NK cells compared to that of w.t. IgG1 antibodies (Figure 2C), the ADCC potency of PBMCs, or NK cells using Rituximab-Fc3aV was weaker than the level observed with wt glycosylated IgG1 Rituximab (Figures 4A,B). Similarly, Rituximab-Fc3aV showed significant ADCC activity with monocytes from one V/V and two V/F donors, which was weaker than w.t. Rituximab (Figure 4C). Consistent with our findings, recently Yeap et al. presented evidence that hFcγRIIIa (CD16a) ligation is important for ADCC by monocytes. We also examined whether antibodies formatted with the hFcγRI-specific Fc5 can trigger ADCC. Rituximab-Fc5 initiated a low but statistically significant activity of ADCC with PBMCs as effectors in all donors analyzed (Figure 4A). With monocytes we only observed a low level of hFcγRI mediated ADCC in two out of three donors, consistent with the high degree of hFcγRI variability from donor to donor (Figure 4C). Finally, as expected, Fc5 formatted antibodies did not trigger ADCC with NK cells which do not express any hFcγRI (Figure 4B).
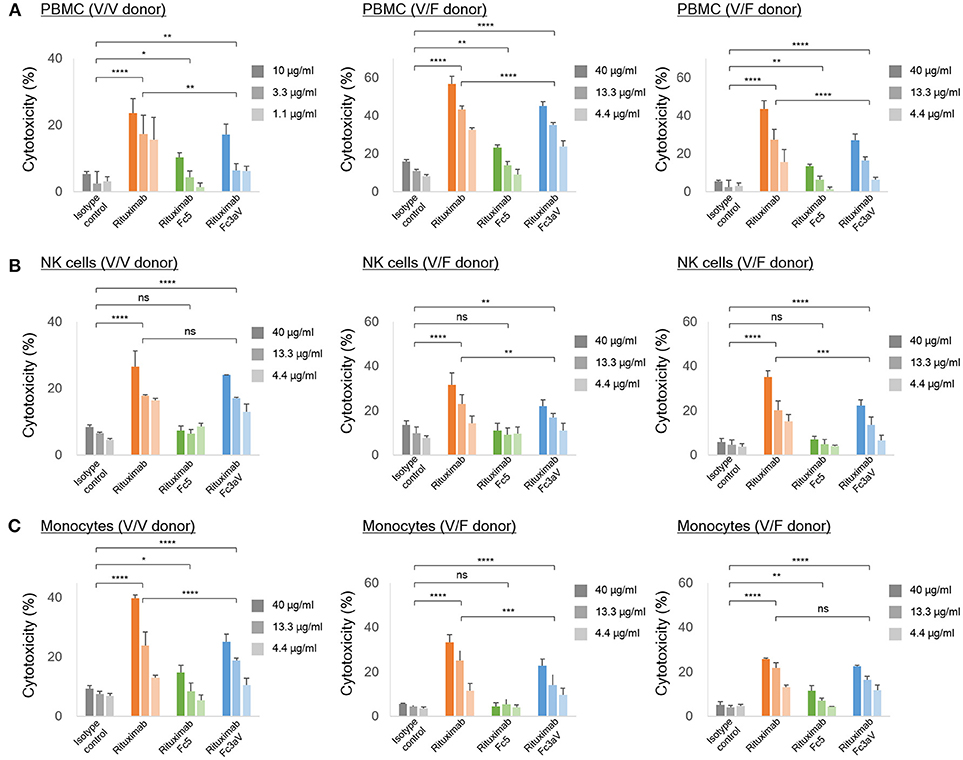
Figure 4. In vitro ADCC assays. ADCC assays with calcein-AM loaded Raji cells as targets and (A) PBMCs, (B) NK cells, and (C) freshly isolated monocytes from the same healthy donor [homozygous V/V (left) or heterozygous V/F (center and right) and E:T ratio = 10:1] as effector cells. (A–C) The level of the released calcein-AM was measured to detect target cell lysis after 4 h. Error bars correspond to standard deviations of triplicate samples. Statistical analysis was performed by two way ANOVA with Tukey's multiple comparisons test (ns: P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
A number of engineered Fc domains including the SDIE (S239D/I332E), SDALIE (S239D/A330L/I332E), S298A/E333A/K334A, GASDALIE (G236A/S239D/A330L/I332E), and GASDIE (G236A/S239D/I332E) variants showing increased hFcγRIIIa affinity had been engineered (5, 55). However, all these Fcs show detectable and, in some instances, high affinity to other hFcγR and therefore they are not suitable for mechanistic studies to selectively delineate the phenotypes induced solely via the engagement of FcγRIIIa. Removal of the single glycan in the IgG hFc domain markedly increases the flexibility of the CH2 conformation resulting in dramatically decreased binding to all hFcγRs as well as to C1q (44). We had shown earlier that mutations in aglycosylated human IgG Fc domains can stabilize a particular CH2 conformer and restore binding selectively either to a single Fc binding protein, e.g., hFcγRI or C1q or, alternatively to multiple hFcγRs (36, 39, 40). Extending these earlier studies, we have now engineered an aglycosylated hFc variant, Fc3aV, with high selectivity for hFcγRIIIaV158 and containing seven amino acids substitutions (S239T, P248L, V264E, V282M, T299A, L309Q, and A378V). Antibodies with the Fc3aV hFc domain do not show any binding to the high affinity receptor hFcγRI or to dimeric high avidity versions of the low affinity receptors hFcγRIIa/b by SPR. Consistent with this biophysical data, ICs formed by antibodies formatted with the Fc3aV domain did not bind significantly to CHO-cells expressing low-affinity hFcγRs except hFcγRIIIaV158. Since hFcγRIIIa glycoproteins expressed by HEK293 or CHO cells are exclusively modified by biantennary complex-type glycans with partial sialylation (56), and the N-glycan composition can in turn affect its affinity for IgG (54, 57) we also evaluated binding activity of ICs to primary cells. Furthermore, immune complexes of Fc3aV antibodies bind to NK cells which express hFcγRIIIa and not to activated neutrophils which express hFcγRI, hFcγRIIa, and hFcγRIIIb but not hFcγRIIIa (Figures 2B,C).
We used Fc3aV to clarify the contribution of hFcγRIIIa to ADCP. Data from clinical studies and animal models have underscored the significance of phagocytosis in the clearance of cancer cells (7, 25–29, 58). Importantly, the depletion of macrophages in mouse models was shown to abolish the therapeutic efficacy of anti-CD20, anti-CD30, or anti-CD40 antibodies in B-cell cancer models, in contrast to depletion of NK cells or neutrophils (26–29).
While earlier reports had suggested that ADCP is mediated primarily by binding to hFcγRIIa (37) here we present evidence showing that selective engagement of human hFcγRIIIa, is sufficient to trigger potent phagocytosis by gmMϕ. The ADCP activity we observed with CD20+ or Her2+ cancer cells as targets and gmMϕ as effectors is particularly noteworthy because, as had been reported earlier and confirmed here, cell surface expression of hFcγRIIIa on gmMϕ is significantly lower than that of hFcγRIIa and FcγRI [Figure 3A,(36–38)]. One reason why selective ligation of hFcγRIIIa by Fc3aV antibodies results in ADCP that is comparable to that observed with wt Rituximab which binds to all hFcγR on macrophages and notably the much more highly expressed hFcγRIIa is that, unlike wt antibodies Fc3aV does not bind to the inhibitory hFcγRIIb and therefore is unable to trigger ITIM phosphorylation and downstream signaling processes. hFcγRIIb ligation is known to inhibit the effector functions initiated by the engagement of the activating Fc receptors including ADCP (2). We note that human macrophages show a large degree of phenotypic variations, which depend on the tissue of origin and on culture method in vitro and is associated with significant differences in FcγR expression levels and especially hFcγRIIIa. For example, red pulp macrophages express significantly higher levels of FcγRIIIa relative to the other FcγRs (31). Similarly in vitro differentiated macrophages stimulated with M-CSF have generally higher levels of hFcγRIIIa compared to gmMϕ (59, 60). Moreover, donor to donor variation effects in ADCP assays are routinely observed. Along these lines, we note that the data in Figure 3D shows that with Trastuzumab-Fc3aV antibodies (but not when using Rituximab-Fc3aV and Raji cells as targets, Figure 3C), the absolute level of phagocytosis in an hFcγRIIIa V/F heterozygous donor was slightly higher than that observed in a different donor expressing the V/V allele. The fact that high ADCP, at a level comparable to that observed with w.t. antibodies, could be mediated with the Fc3aV variant in four donors, with different target cells (Raji or SK-BR-3) and furthermore, that phagocytosis by Fc3aV antibodies was inhibited by hFcγRIIIa-blocking antibodies support our conclusion that hFcγRIIIa engagement alone is sufficient to trigger significant ADCP by gmMϕ.?NK cells which only express hFcγRIIIa and can release cytotoxic granules are considered to be the most important contributor to ADCC in vitro with PBMCs as effector cells (2). As expected, opsonization of tumor cells with Fc3aV antibodies triggered potent ADCC with both PBMCs and with NK cells as effectors. Similarly we found that the selective ligation of hFcγRIIIa on monocytes strongly triggers ADCC, a finding consistent with recent reports (10). As with the ADCP results discussed above, donor to donor variability precludes a comparison of subtle differences in the absolute magnitude of ADCC among donors. For example, in Figure 4, with PBMCs but not with monocytes or NK cells as effectors, the level of ADCC in a donor that is homozygous for the expression of the high affinity 158 V allele was lower than that of the V/F heterozygous donors. This may be in part due to the fact that the level of ADCC with PBMCs as effectors depends on factors other than the hFcγRIIIa allotype including, importantly, the ratio and phenotypes of monocytes to NK cells, since cytokines or receptors expressed by monocytes can impact NK cell-mediated cytotoxicity (61).
Somewhat unexpectedly we found that antibodies with the hFcγRI-selective Fc5 domain elicited much weaker ADCP with gmMϕ opsonized with Fc3aV antibodies, even though as shown in Figure 3A FACS analysis shows a >10-fold lower signal for hFcγRIIIa on gmMϕs compared that for hFcγRI. However, it should be noted that the FACS data in Figure 3A does not reflect the absolute levels of the different receptors since the FACS data were obtained with different antibodies having different affinities for their respective hFcγR antigens. This effect is not due to poor activation of hFcγRI by Fc5 antibodies as we had established earlier that Fc5 antibodies are efficient in activating hFcγRI and initiating target cell clearance by classical dendritic cells (39). The weaker effector functions associated with ligation of hFcγRI may be due to subtle, cell-specific effects related to differences in downstream signaling following ITAM phosphorylation and the elucidation of these effects will require further investigation.
In summary, functional studies with macrophages and other myeloid-derived cells capable of clearing opsonized targets using Fc3aV formatted antibodies may prove very useful for elucidating the role of hFcγRIIIa ligation on the importance of different effector functions and leukocytes subsets for the mechanism of action of therapeutic antibodies. Myeloid cells express multiple hFcγRs and therefore when they are activated with immune complexes formed by numerous antibodies the resulting effector functions represent the integrated effect due to signaling by all the hFcγRs. The use of engineered Fc domains with absolute selectivity for only one hFcγR can be a powerful tool for elucidating the subtle signaling differences and phenotypic effects elicited by each individual Fc receptor and how these ultimately contribute to the clearance of target pathogenic cells.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
TK, C-HL, and GG conceived and designed the research. C-HL, TK, GD, JJ, OG, JL, JK, and WC performed the experiments. GG, C-HL, TK, GD, JJ, and PB analyzed the data. GG, C-HL, TK, GD, JJ, JL, and PB wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Yuri Tanno for assistance with protein expression and Anna Webb for assistance with confocal microscope. This work was supported by the Clayton Foundation (Austin, TX, USA), the Institut Pasteur and the Institut National de la Santé et de la Recherche Médicale (Paris, France).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00562/full#supplementary-material
1. Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. (2010) 236:265–75. doi: 10.1111/j.1600-065X.2010.00910.x
2. Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
3. Smith KGC, Clatworthy MR. FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. (2010) 10:328–43. doi: 10.1038/nri2762
4. Leibson PJ. The regulation of lymphocyte activation by inhibitory receptors. Curr Opin Immunol. (2004) 16:328–36. doi: 10.1016/j.coi.2004.03.006
5. Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350.
6. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. (2014) 15:707–16. doi: 10.1038/ni.2939
7. Bakema JE, van Egmond M. Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. In: Daeron M, Nimmerjahn F, editors. Fc Receptors Current Topics in Microbiology and Immunology. Springer International Publishing. (2014) 382:373–92. doi: 10.1007/978-3-319-07911-0_17
8. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. (2007) 110:2561–4. doi: 10.1182/blood-2007-01-070656
9. Ziegler-Heitbrock L. Blood monocytes and their subsets: established features and open questions. Front Immunol. (2015) 6:423. doi: 10.3389/fimmu.2015.00423
10. Yeap WH, Wong KL, Shimasaki N, Teo ECY, Quek JKS, Yong HX, et al. CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci Rep. (2016) 6:34310. doi: 10.1038/srep34310
11. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SEA, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol. (2005) 42:501–10. doi: 10.1016/j.molimm.2004.07.034
12. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. (2012) 119:5640–9. doi: 10.1182/blood-2012-01-380121
13. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. (2015) 15:361–70. doi: 10.1038/nrc3930
14. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
15. Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. (2003) 21:3940–7. doi: 10.1200/JCO.2003.05.013
16. Liu D, Tian Y, Sun D, Sun H, Jin Y, Dong M. The FCGR3A polymorphism predicts the response to rituximab-based therapy in patients with non-Hodgkin lymphoma: a meta-analysis. Ann Hematol. (2016) 95:1483–90. doi: 10.1007/s00277-016-2723-x
17. Gavin PG, Song N, Kim SR, Lipchik C, Johnson NL, Bandos H, et al. Association of polymorphisms in FCGR2A and FCGR3A with degree of trastuzumab benefit in the adjuvant treatment of ERBB2/HER2–positive breast cancer: analysis of the NSABP B-31 trial. JAMA Oncol. (2017) 3:335–41. doi: 10.1001/jamaoncol.2016.4884
18. Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol. (1999) 17:176–80.
19. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. (2006) 103:4005–10. doi: 10.1073/pnas.0508123103
20. Liu Z, Gunasekaran K, Wang W, Razinkov V, Sekirov L, Leng E, et al. Asymmetrical Fc engineering greatly enhances antibody-dependent cellular cytotoxicity (ADCC) effector function and stability of the modified antibodies. J Biol Chem. (2014) 289:3571–90. doi: 10.1074/jbc.M113.513366
21. Niwa R, Satoh M. The current status and prospects of antibody engineering for therapeutic use: focus on glycoengineering technology. J Pharm Sci. (2015) 104:930–41. doi: 10.1002/jps.24316
22. Marcus R, Davies A, Ando K, Klapper W, Opat S, Owen C, et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N Engl J Med. (2017) 377:1331–44. doi: 10.1056/NEJMoa1614598
23. Capuano C, Pighi C, Molfetta R, Paolini R, Battella S, Palmieri G, et al. Obinutuzumab-mediated high-affinity ligation of FcγRIIIA/CD16 primes NK cells for IFNγ production. Oncoimmunology. (2017) 6:e1290037. doi: 10.1080/2162402X.2017.1290037
24. Dall'Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, et al. Rituximab-dependent cytotoxicity by natural killer cells influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. (2004) 64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862
25. Gül N, Babes L, Siegmund K, Korthouwer R, Bögels M, Braster R, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. (2014) 124:812–23. doi: 10.1172/JCI66776
26. Oflazoglu E, Stone IJ, Gordon KA, Grewal IS, van Rooijen N, Law C-L, et al. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood. (2007) 110:4370–2. doi: 10.1182/blood-2007-06-097014
27. Oflazoglu E, Stone IJ, Brown L, Gordon KA, van Rooijen N, Jonas M, et al. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J Cancer. (2009) 100:113–7. doi: 10.1038/sj.bjc.6604812
28. Horikawa M, Minard-Colin V, Matsushita T, Tedder TF. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J Clin Invest. (2011) 121:4268–80. doi: 10.1172/JCI59266
29. Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, et al. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. (2008) 112:1205–13. doi: 10.1182/blood-2008-01-135160
30. Dahal LN, Dou L, Hussain K, Liu R, Earley A, Cox KL, et al. STING activation reverses lymphoma-mediated resistance to antibody immunotherapy. Cancer Res. (2017) 77:3619–31. doi: 10.1158/0008-5472.CAN-16-2784
31. Nagelkerke SQ, Bruggeman CW, den Haan JMM, Mul EPJ, van den Berg TK, van Bruggen R, et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv. (2018) 2:941–53. doi: 10.1182/bloodadvances.2017015008
32. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci J Virtual Libr. (2008) 13:453–61. doi: 10.2741/2692
33. Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. (2013) 122:3482–91. doi: 10.1182/blood-2013-05-504043
34. Herter S, Birk MC, Klein C, Gerdes C, Umana P, Bacac M. Glycoengineering of therapeutic antibodies enhances monocyte/macrophage-mediated phagocytosis and cytotoxicity. J Immunol. (2014) 192:2252–60. doi: 10.4049/jimmunol.1301249
35. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol. (2014) 14:94–108. doi: 10.1038/nri3582
36. Jung ST, Kelton W, Kang TH, Ng DTW, Andersen JT, Sandlie I, et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcγRIIa affinity and selectivity. ACS Chem Biol. (2012) 8:368–75. doi: 10.1021/cb300455f
37. Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. (2008) 7:2517–27. doi: 10.1158/1535-7163.MCT-08-0201
38. Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA. (2012) 109:2796–801. doi: 10.1073/pnas.1104303108
39. Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcγRI potentiate tumor cell killing by monocyte-dendritic cells. Proc Natl Acad Sci USA. (2010) 107:604–9. doi: 10.1073/pnas.0908590107
40. Lee C-H, Romain G, Yan W, Watanabe M, Charab W, Todorova B, et al. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat Immunol. (2017) 18:889–98. doi: 10.1038/ni.3770
41. Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. (2009) 30:356–62. doi: 10.1016/j.tips.2009.04.007
42. Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci USA. (2008) 105:20167–72. doi: 10.1073/pnas.0809257105
43. Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, et al. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J Immunol Methods. (2002) 263:133–47. doi: 10.1016/S0022-1759(02)00036-4
44. Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol. (2012) 7:1596–602. doi: 10.1021/cb300130k
45. Mancardi DA, Iannascoli B, Hoos S, England P, Daëron M, Bruhns P. FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J Clin Invest. (2008) 118:3738–50. doi: 10.1172/JCI36452
46. Erbe AK, Wang W, Gallenberger M, Hank JA, Sondel PM. Genotyping single nucleotide polymorphisms (SNPs) and copy number variability (CNV) of the FCGRs (FCGR3A and FCGR2C) expressed on NK cells. Methods Mol Biol Clifton NJ. (2016) 1441:43–56. doi: 10.1007/978-1-4939-3684-7_4
47. Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, et al. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. (2011) 11:80. doi: 10.1186/1472-6750-11-80
48. Kelton W, Mehta N, Charab W, Lee J, Lee C, Kojima T, et al. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. (2014) 21:1603–9. doi: 10.1016/j.chembiol.2014.10.017
49. Laborde EA, Vanzulli S, Beigier-Bompadre M, Isturiz MA, Ruggiero RA, Fourcade MG, et al. Immune complexes inhibit differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol. (2007) 179:673–81. doi: 10.4049/jimmunol.179.1.673
50. Choi EI, Wang R, Peterson L, Letvin NL, Reimann KA. Use of an anti-CD16 antibody for in vivo depletion of natural killer cells in rhesus macaques. Immunology. (2008) 124:215–22. doi: 10.1111/j.1365-2567.2007.02757.x
51. Tamm A, Schmidt RE. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for ligand binding. J Immunol. (1996) 157:1576–81.
52. Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcγRs. J Immunol. (2013) 190:4315–23. doi: 10.4049/jimmunol.1200501
53. Gillis C, Gouel-Chéron A, Jönsson F, Bruhns P. Contribution of human FcγRs to disease with evidence from human polymorphisms and transgenic animal studies. Front Immunol. (2014) 5:254. doi: 10.3389/fimmu.2014.00254
54. Patel KR, Roberts JT, Subedi GP, Barb AW. Restricted processing of CD16a/Fc γ receptor IIIa N-glycans from primary human NK cells impacts structure and function. J Biol Chem. (2018) 293:3477–89. doi: 10.1074/jbc.RA117.001207
55. Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. (2018) 9:63–73. doi: 10.1007/s13238-017-0473-8
56. Zeck A, Pohlentz G, Schlothauer T, Peter-Katalinić J, Regula JT. Cell type-specific and site directed N-glycosylation pattern of FcγRIIIa. J Proteome Res. (2011) 10:3031–9. doi: 10.1021/pr1012653
57. Hayes JM, Frostell A, Karlsson R, Müller S, Martín SM, Pauers M, et al. Identification of Fc gamma receptor glycoforms that produce differential binding kinetics for rituximab. Mol Cell Proteomics. (2017) 16:1770–88. doi: 10.1074/mcp.M117.066944
58. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
59. Santegoets KCM, Wenink MH, van den Berg WB, Radstake TRDJ. Fc gamma receptor IIb on GM-CSF macrophages controls immune complex mediated inhibition of inflammatory signals. PLoS ONE. (2014) 9:e110966. doi: 10.1371/journal.pone.0110966
60. Nagelkerke SQ, Dekkers G, Kustiawan I, van de Bovenkamp FS, Geissler J, Plomp R, et al. Inhibition of FcγR-mediated phagocytosis by IVIg is independent of IgG-Fc sialylation and FcγRIIb in human macrophages. Blood. (2014) 124:3709–18. doi: 10.1182/blood-2014-05-576835
Keywords: Fc engineering, ADCP, FcgR, macrophage, monocyte
Citation: Kang TH, Lee C-H, Delidakis G, Jung J, Richard-Le Goff O, Lee J, Kim JE, Charab W, Bruhns P and Georgiou G (2019) An Engineered Human Fc variant With Exquisite Selectivity for FcγRIIIaV158 Reveals That Ligation of FcγRIIIa Mediates Potent Antibody Dependent Cellular Phagocytosis With GM-CSF-Differentiated Macrophages. Front. Immunol. 10:562. doi: 10.3389/fimmu.2019.00562
Received: 03 December 2018; Accepted: 04 March 2019;
Published: 27 March 2019.
Edited by:
Mark S. Cragg, University of Southampton, United KingdomReviewed by:
Adam D. Hoppe, South Dakota State University, United StatesCopyright © 2019 Kang, Lee, Delidakis, Jung, Richard-Le Goff, Lee, Kim, Charab, Bruhns and Georgiou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Georgiou, Z2dAY2hlLnV0ZXhhcy5lZHU=
†Present Address: Tae Hyun Kang, Biopharmaceutical Research Center, CJ Healthcare R&D Center, CJ HealthCare, Icheon, South Korea
‡These authors have contributed equally to this work
§Co-first author
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.