 Ann Ly1,2
Ann Ly1,2 Diana S. Hansen1,2*
Diana S. Hansen1,2*- 1The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 2Department of Medical Biology, The University of Melbourne, Parkville, VIC, Australia
A single exposure to many viral and bacterial pathogens typically induces life-long immunity, however, the development of the protective immunity to Plasmodium parasites is strikingly less efficient and achieves only partial protection, with adults residing in endemic areas often experiencing asymptomatic infections. Although naturally acquired immunity to malaria requires both cell-mediated and humoral immune responses, antibodies govern the control of malarial disease caused by the blood-stage form of the parasites. A large body of epidemiological evidence described that antibodies to Plasmodium antigens are inefficiently generated and rapidly lost without continued parasite exposure, suggesting that malaria is accompanied by defects in the development of immunological B cell memory. This topic has been of focus of recent studies of malaria infection in humans and mice. This review examines the main findings to date on the processes that modulate the acquisition of memory B cell responses to malaria, and highlights the importance of closing outstanding gaps of knowledge in the field for the rational design of next generation therapeutics against malaria.
B Cell Immunological Memory
Immunological memory refers to the ability of the vertebrate immune system to remember previously encountered antigens or pathogens and evoke an enhanced immune response to control infection. The capacity of the host to generate T and B cell memory underlies the basis of protective immunity induced by vaccination or after exposure to specific pathogens. The generation of T cell-dependent humoral immune memory in secondary lymphoid organs (Figure 1) typically begins following B cell engagement with its cognate antigen, which triggers their migration to the B cell follicle border to receive T cell help (1). Activated B cells then differentiate along one of three possible routes, leading to the rapid production of short-lived plasmablasts, generating germinal center (GC)-independent memory B cells (MBCs), or formation of GCs in B cell follicles (2, 3). GCs establish within a few days of initial antigen encounter and mature into two distinct micro-anatomical compartments: the dark zone, where B cell clones undergo proliferative expansion and somatic hypermutation of their immunoglobulin (Ig) genes, and the light zone, where B cells expressing high-affinity antibodies are selected and undergo class switch recombination (4–6). The GC reaction leads to the generation of affinity-matured MBCs and long-lived plasma cells that contribute to host protection against re-infection. Plasma cells migrate to the bone marrow and provide a continuous source of circulating high-affinity antibody (7), while MBCs recirculate in the blood and secondary lymphoid tissue (8) to induce a rapid effector response upon antigen re-encounter (9, 10).
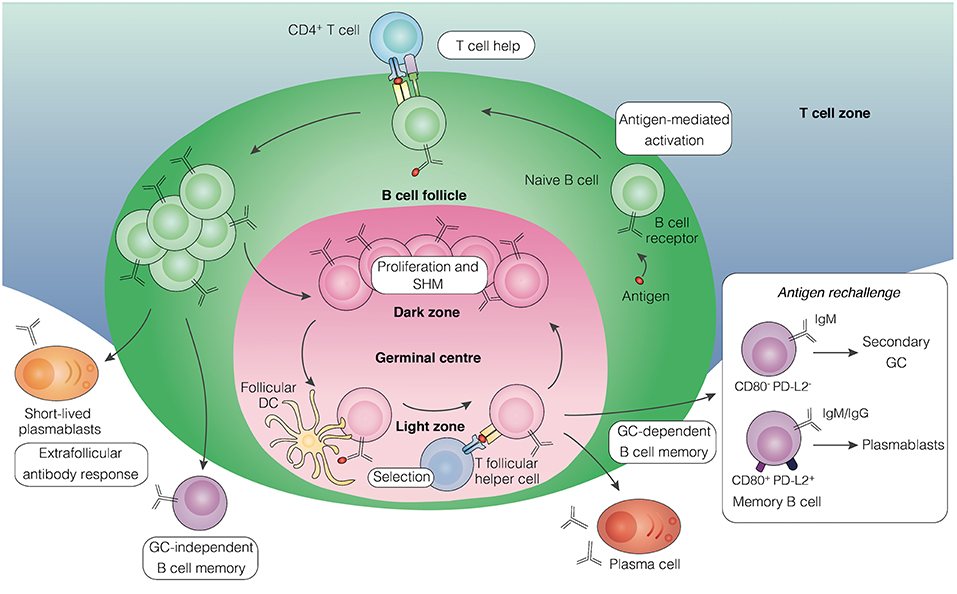
Figure 1. Pathways leading to the development of B cell memory. Upon encounter with antigen, activated B cells in secondary lymphoid tissue receive helper signals from cognate CD4+ T cells at the border of the B cell follicle and T cell zones. Some of the proliferating B cells differentiate into short-lived plasmablasts that initiate an extrafollicular antibody response, some develop into early memory B cells independently of GC formation, while others aggregate into the follicle to establish a GC. Within the GC, B cells undergo proliferation and somatic hypermutation in the dark zone, followed by affinity-based selection in the light zone with the help of T follicular helper cells and follicular dendritic cells. Long-lived plasma cells and memory B cells emerge from the GC reaction. Upon antigen rechallenge, memory B cells lacking expression of the surface molecules CD80 and PD-L2, mainly of the IgM isotype, can seed secondary GCs, whereas those expressing both molecules, comprising of IgM and IgG isotypes, predominantly generate plasmablasts. GC, germinal center; DC, dendritic cell; SHM, somatic hypermutation.
Comprehensive studies of MBC biology have led to the appreciation of substantial heterogeneity among the MBC compartment, consisting of distinct subpopulations with different effector capacity upon secondary challenge (11). In humans, the expression of unique memory-specific surface markers has been extensively used to identify and characterize MBCs. Surface expression of CD27 defines a subset of antigen-experienced MBCs in humans that are class-switched and bear Ig variable region mutations (12, 13). However, CD27 expression does not universally define all MBCs, as subsequent work identified an CD27− CD21− MBC population (14). These cells, coined as atypical MBCs in malaria, express several Fc receptor-like (FcRL) inhibitory receptors, including FcRL3 and 5 (15–17).
The development of novel labeling techniques to track antigen-specific B cells in murine models has enabled the functional characterization of MBCs expressing different antibody isotypes. Whereas, IgG MBCs have been found to preferentially differentiate into plasmablasts upon antigen rechallenge, IgM MBCs appear to have the capacity to re-enter secondary GC reactions but were not enhanced in plasmablast generation (18, 19). Recent studies have uncovered additional heterogeneity within the IgM and IgG MBC pools, proposing that MBCs lacking the memory markers CD80 and PD-L2, which primarily express IgM, are dedicated to reseeding GCs, while those expressing both markers, comprising of IgM and IgG isotypes, contribute mainly to immediate antibody-secreting function (Figure 1) (20). Consistently, IgM-expressing MBCs have recently been shown to exhibit considerable plasticity in their differentiation capacity following rechallenge (21). This division of labor in the MBC response has been proposed to support a rapid and effective response upon antigen rechallenge while concurrently permit the generation of new MBCs (11). How these findings in murine models relate to human settings is unclear, as there is still little information about the homology between mouse and humans MBC subsets with regards to their surface marker expression usage.
A Role for Memory B Cels in Naturally Acquired Immunity to Malaria
The global burden of malaria has more than halved since the turn of the century due to renewed eradication efforts, but progress has recently stalled as current intervention strategies are confronted with several major challenges, including the emergence of anti-malarial drug and insecticide resistance (22). Of the six Plasmodium species known to infect humans, P. falciparum continues to account for the majority of deaths, whereas recurrent P. vivax episodes are a significant source of morbidities. Disease syndromes of malaria range from fever to more severe complications including acute pulmonary oedema, jaundice, severe anemia, hypoglycaemia, acidosis, and cerebral malaria (23). The pathogenesis of malarial disease is thought to arise from the concerted effects of host and parasite mechanisms, including the sequestration of blood-stage parasites in microvasculature, and local and systemic inflammation induced by the parasites and their toxic products (24, 25).
Early epidemiological observations by Robert Koch in malaria-endemic populations described that natural immunity to malaria can be achieved, but requires years of repeated exposure to Plasmodium parasites (26). Children living in high transmission regions become immune to the most severe forms of malaria after relatively few symptomatic infections (27–29), but remain at risk of uncomplicated malaria. After years of repeated infections with age, protection from successive malaria episodes or “clinical immunity,” is acquired by the ability to substantially reduce parasite burdens (30–35). This form of protection is not paralleled by sterile immunity that prevents re-infection (36), and adults continue to be experience low-density, asymptomatic infections throughout life (37). Naturally acquired clinical immunity to malaria targets blood-stage parasites and requires antibodies, as demonstrated by studies in which the transfer of purified IgG from malaria-immune adults to children with symptomatic malaria rapidly reduced parasitemia and fever (38). Together, these observations have led to the hypothesis that the slow and imperfect acquisition of immunity to malaria reflects in the development of MBCs, and this topic has been the subject of several studies including mouse infection models as well as human settings. Here we review our current understanding on the salient features of the development of humoral immunity to malaria infection, and highlight some of the outstanding questions regarding the cellular mechanisms that underlie the slow acquisition of clinical immunity.
Antibody Responses to Blood-Stage Malaria
The paramount importance of antibodies in controlling blood-stage malaria infection was proven by seminal passive-transfer experiments, in which IgG from P. falciparum clinically immune adults protected non-immune children from high parasitemia and clinical symptoms (38, 39). Numerous immuno-epidemiological studies subsequently demonstrated that high antibody levels against specific blood-stage parasite antigens correlate with protection from disease (40–46). Antibodies may control the development of clinical symptoms by targeting the invasion and growth of the merozoite form of the blood-stage parasite and redirect their clearance by phagocytic cells via Fc and complement receptors (47). Additionally, antibodies directed against parasite antigens expressed on infected erythrocytes can promote opsonic phagocytosis, block microvasculature adherence, disrupt rosette formation with uninfected cells, and prevent erythrocyte rupture and parasite egress (47).
Antibodies may target a number of highly polymorphic and functionally redundant antigens expressed by Plasmodium parasites (48), which may represent a potential mechanism by which the parasite effectively evades the human immune system via antigenic variation (49). Asymptomatically-infected individuals who fail to mount an antibody response against P. falciparum has been shown to predict increased susceptibility to clinical disease (50, 51). In parallel, individuals detected with multi-clonal P. falciparum infections in the dry season have been associated with subsequent protection from febrile malaria (52), suggesting that the presence of persisting parasites enhance antibody recognition and enable cross-reactive responses. This supports the notion that clinical immunity may depend on the cumulative acquisition of a repertoire of antibodies to a diverse range of parasite antigens or development of cross-species antibody responses (53–55). Indeed, the breadth of parasite-specific antibody responses have been identified to increase with age in endemic populations (56–58), and the antibody repertoire diversifies rapidly during infancy but plateaus in toddlers (59). Moreover, antibodies with broad reactivity against P. falciparum that carry a gene insertion derived from the collagen-binding protein LAIR1, have been shown to undergo somatic hypermutation that increase binding to infected erythrocytes (60). LAIR1 insertions have been further revealed to represent a relatively common mechanism of antibody diversification in African individuals, and that broadly-neutralizing antibodies against Plasmodium arise from these low-affinity precursors over time (61). While antigenic variation has been proposed to explain the slow acquisition immunity to malaria, there is also an increasing body of evidence suggesting that antibody responses to malaria are poorly generated. In malaria-endemic areas, substantial declines have been reported in Plasmodium-specific antibody responses to low or undetectable levels within months, and even weeks of a clinical episode after reduced parasite exposure, despite an initial robust response (56, 57, 62–69). Studies modeling the longevity of P. falciparum merozoite-specific IgG antibodies have estimated average half-lives of <10 days in children recovering from clinical malaria (67). Similarly, short antibody half-lives ranging from only 2–7 weeks has been reported in asymptomatic children during the dry season in The Gambia (68), which contrasts dramatically with those of antibody responses to viral and bacterial antigens such as vaccinia, measles and tetanus that reportedly persist for decades following a single exposure (70–72). It is possible that antibody responses measured following a clinical malaria episode may reflect the output of short-lived antibody-secreting cells, which typically produce an immediate but transient wave of antibodies to control infection. In line with this idea, antibody-secreting cells have been detected transiently in Ugandan children immediately following acute malaria, and were found to proportionally increase again upon a second clinical episode (73). Modeling studies extended these observations, estimating the longevity of both short- and long-lived antibody-secreting cells in African children to range from 2–10 days, and 3–9 years, respectively (74). Thus, together these findings suggest that a long-lived humoral response to malaria infection can potentially be sustained after decay of transient antibody-secreting cells. More detailed mechanistic investigations are much needed to determine how parasite-specific antibody responses are sustained over time and the factors that modulate the generation and maintenance of antibody-secreting cells to infection.
The Acquisition of Memory B Cells to Natural Malaria Infection
Several studies have now shown the induction of Plasmodium-specific MBCs in response to malaria infection; although individuals exposed to high seasonal transmission have been reported to induce only low frequencies of MBCs or to lack detectable MBCs, even after exposure to parasitic loads sufficient and capable of inducing antibody responses (57, 75, 76). Consistent with the slow acquisition of antibody responses in endemic settings, the prevalence and breadth of Plasmodium-specific MBCs appear to develop incrementally with age and exposure (57, 59, 77). Longitudinal studies of children and young adults in an area of high seasonal transmission in Mali demonstrated a delayed, age-associated development of MBCs specific for P. falciparum merozoite antigens despite repeated infections annually (57). Moreover, the prevalence of MBCs acquired by children following acute malaria appeared to diminish substantially within the 6-month dry season, contrasting with the stable frequency of MBCs to tetanus toxoid vaccination in the same subjects (57). While seasonal transmission prevented the longevity of these cells to be determined beyond the dry season, this finding implies an impaired maintenance or generation of MBCs in children exposed to high transmission as they acquire clinical protection.
In contrast, individuals residing in areas of low transmission or subjected to fewer clinical episodes have been shown to generate stable Plasmodium-specific MBCs without frequent boosting (77–85). The frequencies of P. falciparum-specific MBCs detected in Thai adults that had experienced limited episodes reportedly remained stable over time, with an estimated half-life of approximately 7.5 years (81). Similarly, malaria-specific MBCs have been described to be well-maintained in individuals with a history of acute malaria but have since lived in the absence of persistent infection (83, 84). In parallel, low levels of exposure can effectively sustain parasite-specific antibody responses (79, 82, 84, 86, 87), with antibody half-lives estimated to be substantially longer than that of highly-exposed individuals, ranging from months to years (32, 81, 88), suggesting sustained antibody production from long-lived plasma cells. Collectively, these findings reveal that MBCs can be generated and sustained following a limited number of clinical episodes, while repeated infections in endemic areas could have a detrimental effect on the generation of B cell memory.
The characterization of malaria-specific MBCs to date has relied predominantly on in vitro stimulation and differentiation of circulating MBCs into antibody-secreting cells followed by detection of antigen-specific clones by ELISPOT assays. This approach precludes phenotypic analysis of the malaria-specific MBC compartment. More detailed investigations are needed to determine the ontogeny of detected MBCs and whether they contribute to effective clinical immunity in malaria-exposed individuals. Whereas, the induction of IgG-expressing MBCs has been the primary focus over the past several years, a few studies have identified IgM MBCs in malaria-exposed individuals and in malaria mouse infection models (59, 89, 90), with those induced by murine malaria found to rapidly differentiate into plasmablasts upon antigenic restimulation (89). Interestingly, MBCs isolated from malaria-exposed individuals have been described to have undergone Ig somatic hypermutations (76), and accumulate further mutations upon acute malaria, with IgM being the dominant isotype expressed prior to re-infection (59).
The use of murine infection models has also provided insight into the development of B cell memory to malaria. Although murine infection does not mirror all the features of human malaria, there are genetic and phenotypic parallels between the human parasite and rodent counterparts (91, 92). A few studies have detected the development of IgG memory B cells following non-lethal P. chabaudi infection (89, 90, 93, 94), associated with efficient generation of secondary GCs and enhanced control of re-infection (90, 95). In contrast, P. yoelii blood-stage infection has been suggested to ablate vaccination-induced MBCs (96), and further reduces the development of mature, isotype-switched MBCs against pre-erythrocytic parasite antigen, which was associated with the induction of pro-inflammatory cytokines and chemokines that may hinder effective T and B cell interactions (97).
Immunological Processes Modulating the Induction of Memory B Cells in Malaria
T follicular helper (Tfh) cells are a crucial subset of CD4+ T cells that orchestrate B cell memory development by providing crucial survival and differentiation signals to B cells during the GC response (98), and have been shown to be important for the control of Plasmodium infection (99–105). The development of Tfh cells has not been extensively investigated in human malaria infection, however, a recent study identified the induction of T helper 1 (Th1) cell-like CXCR3+PD-1+ Tfh cells in the circulation of Malian children following acute P. falciparum malaria (106). Circulating Tfh cells have been proposed to be a surrogate measure of GC-derived Tfh cell responses to human infection (107, 108) and thus constitutes a valuable proxy to investigate Tfh cell biology in human settings. Circulating Tfh cells in malaria-exposed children were shown to express the Th1 lineage-defining transcription factor T-bet, and had limited functional capacity to support MBC responses in vitro (106). CXCR3+PD-1+ Tfh cells have also been identified in Brazilian adults during acute P. vivax malaria (109). Interestingly, their proportions positively correlated with repeated malaria episodes (109), implying that circulating Tfh cells accumulate with sustained parasite exposure. However, whether or not the detected Tfh cells facilitate the induction of MBCs that confer protection to disease over time remains to be investigated in larger cohorts and different transmission settings. In addition, circulating follicular regulatory T (Tfr) cells have been postulated to interfere with Tfh cell responses to infection (110). Tfr cells expressing the inhibitory molecule CTLA-4, have been detected at increased frequencies after acute malaria in Malian children (110). Future studies will be important in determining if the transition between susceptibility to clinical disease and asymptomatic infection is associated with the development of functional Tfh cell responses that support the induction of B cell memory populations.
Similar to human malaria, CD4+ T cells induced by murine infection have been described to express a dual Th1- and Tfh-associated phenotype, including chemokine receptor CXCR5, and cytokines IL-21 and IFN-γ (99, 111, 112). Precursor Tfh cells induced in response to P. berghei severe malaria infection also display a Th1 cell-like phenotype, characterized by the upregulation the chemokine receptor CXCR3 and transcription factor T-bet (113). Moreover, the expression of T-bet impaired the differentiation of Tfh cell precursors into mature Tfh cells, which significantly suppressed the development of GC B cells, plasma cells and MBCs (113). Notably, genetic ablation of T-bet or neutralization of the pro-inflammatory cytokines interferon (IFN)-γ and tumor necrosis factor (TNF) promoted Tfh cell differentiation, and restored previously impaired GC responses, demonstrating that inflammatory responses associated with the induction of symptomatic malaria reduce the magnitude of MBC responses by modulating effective Tfh cell development (114). In support of this concept, exogenous IFN-γ has been shown to reduce Tfh and GC B cell responses to Plasmodium yoelii (102), and the blockade of CXCR3-mediated signals was found to enhance precursor Tfh cell accumulation in the spleen of malaria-infected mice, thereby favoring parasitic clearance (115). Other inflammatory pathways such as type I IFN signals have also been recognized to indirectly limit Tfh and GC B cell responses to infection (116, 117), although the precise mechanisms by which this occurs is not yet clear. Recent studies suggest that the damage-signal sensor P2X7 (118), IFN regulatory factor 3 (119), and changes in metabolism and proliferation, as well as altered gene expression of chemokine receptors (120) influence the bifurcation of Tfh and Th1 cell differentiation during murine infection. Whereas, inflammatory responses to malaria appear to dampen the magnitude of the MBC response to infection (114), effective control of blood-stage malaria and its associated pro-inflammatory responses appears to permits normal progression of Tfh cell development (113, 121), giving rise to T cell memory that responds to secondary infection (112).
Whereas, it is reasonable to assume that the aforementioned modulation of Tfh cell function by inflammatory pathways elicited in response to infection is responsible for the reduced B cell responses observed during acute infection, a direct effect of IFN-γ-mediated signaling or intrinsic expression of T-bet in B cells cannot be ruled out. P. yoelii infection has recently been shown to induce a subset of B cells to express the transcription factor T-bet, and deletion of B cell-specific IFN-γ receptor or T-bet deletion elevated antibody responses to infection (122, 123). Further work to establish if inflammation directly modulates the differentiation and functional capacity of B cells is needed to address this question.
Atypical Memory B Cells: Friends or Foes?
There is evidence that Plasmodium parasites can directly modulate B cell function. In vitro studies have shown that the parasite can directly activate human naive B cells via a cysteine rich inter-domain region of P. falciparum erythrocyte membrane protein 1 (124, 125) and downstream toll-like receptor signaling (126), which may lead to non-specific polyclonal activation of B cell responses. CD27-expressing B cells were observed to be the major responding population (124, 125), suggesting a potential impact on the MBC compartment. In line with this idea, acute P. falciparum infection has been reported to modulate systemic mediators of B cell activation and survival (127, 128), which has been associated with an early proliferation of MBC subsets, prior to the induction of GC responses following experimental human infection (128), supporting a potential role of bystander activation of non-specific MBC subsets that enables parasite evasion.
Insight into the notion that B cell memory induction in malaria may be dysregulated surfaced upon the characterization of an expansion of MBCs exhibiting an atypical phenotype following persistent malaria exposure (17, 84, 129, 130). Originally described as an exhausted subset of MBCs implicated in humoral deficiencies in HIV infection (131), a phenotypically similar CD27−CD21− circulating MBC subset notable for their expression of inhibitory receptors, was detected in Plasmodium-infected individuals following acute malaria in Brazil (109), Gabon (132), Ghana (85), Gambia (77), India (133), Kenya (84, 130), Mali (15, 17, 129, 134, 135), Papua New Guinea (136, 137), Thailand (78), and Uganda (16, 73, 138), and further exacerbated by HIV co-infection (139, 140). Studies to date corroborate that the accumulation of this subset is influenced by the level of parasite exposure. Higher frequencies of atypical MBCs were found in adults compared to children with shorter exposure histories (17). Similarly, Malian adults from high transmission settings were found to have higher frequencies of these cells than Peruvian individuals residing in a low transmission area (129). A decline in the atypical MBC pool was observed during 12 months without malaria transmission (84), further substantiating a role of persistent parasite exposure in the maintenance of this population.
Recent work has investigated the function of these atypical MBCs and their relationship to classical MBCs. Evaluation of the variable gene repertoires of atypical MBCs proposed that they share a common developmental precursor to classical MBCs (15, 132, 141), however, emerging work support that atypical MBCs are phenotypically and functionally different to classical MBCs. There is still conflicting evidence on the effector capacity of atypical MBCs from malaria-infected individuals. Whereas, some support the notion that atypical MBCs are capable of secreting neutralizing antibodies (132), subsequent work contend their lack of active antibody secretion (15, 16). Atypical MBCs have been reported to express lower levels of surface IgG (136), possess reduced B cell receptor signaling, as well as impaired interleukin (IL)-6, IL-8 and antibody production in vitro (15, 16). Atypical MBC frequencies in malaria-exposed individuals have been shown to be positively correlated with pro-inflammatory cytokine levels such as IL-8 (136), and their expression of the inhibitory molecule FcRL5 has been proposed to be a marker of dysfunction associated with increased malaria exposure (16). FcRL5-expressing atypical-like MBCs in healthy individuals have been distinguished by their higher expression of inhibitory receptors such as PD-1 and transcription factors Tbx21, Bcl-6, and Sox5 and blunted proliferation capacity compared to FcRL5− B cells (142). In line with these findings, recent studies revealed that atypical MBCs in Malian children express Tbx21 (134), which encodes the Th1 transcription factor T-bet (143). Emerging evidence that T-bet becomes up-regulated in various disease contexts in subsets of B cells that share many features in common with atypical MBCs found in malaria-infected individuals. T-bet-expressing B cells have been associated with both protective and pathogenic roles depending on specific settings, which include bacterial (21), parasitic (144–146) and viral infection (147–153), cancer (154–156), autoimmune conditions (157–164), and aging (165). In malaria, atypical MBCs expressing high levels of T-bet have also been found to express decreased levels of the co-stimulatory molecule CD40 and reduced phosphorylation of B cell receptor signaling molecules (134), suggesting that T-bet expression might reduce B cell effector function. In line with this concept, the frequency of malaria episodes was found to be associated with increased T-bethi MBC in a small group of children (134). On the other hand, longitudinal cohort studies have reported that low parasite density malaria is associated with persistence (78) or accumulation of atypical MBCs over time, which raises the possibility that atypical MBCs contribute to developing immunity (73, 138), although T-bet expression was not examined in these cells. Furthermore, similar atypical MBCs that express FcRL5 have been recently detected in P. chabaudi-infected mice (166, 167), but appear to display normal features of proliferation and Ig expression, representing a short-lived population of activated MBCs (167). Collectively, a causal relationship between atypical MBCs and immune protection or disease progression remains unclear, and more studies are urgently needed to determine the functional significance of atypical MBC expansion in the acquisition of humoral immunity to malaria and the precise contribution of the transcription factor T-bet in shaping atypical MBC function.
Concluding Remarks and Future Perspectives
Emerging evidence both in human field studies and murine infection models are beginning to address how the induction of humoral immune responses is compromised during acute malaria, and the consequences of these processes for the establishment of long-term immunological B cell memory. There is now considerable evidence that exposure to Plasmodium parasites is associated with altered proportions of circulating memory B cells, including an expansion of atypical memory B cells. Further studies to unravel the complexity of this diverse memory B cell compartment, in terms of origin, function and protective capacity of different subpopulations are urgently needed. Information about the level of homology between human and mouse memory B cell subsets is lacking, particularly in relation to their surface marker expression patterns, which makes it difficult to infer how findings in malaria infection models translate into human settings. Gene expression analysis studies, including how transcription factors and the cytokine environment influence these processes might be required to overcome these issues and establish functional correlations between human studies and much needed mechanistic work in mice.
Given the critical importance of antibodies and long-lived humoral memory in immunity to malaria, an in-depth understanding of the factors that delay their development is undoubtedly required to inform the design of targeted therapeutic strategies to enhance immune responses to the parasite and protect against disease susceptibility. Detailed characterization of the immune processes by which B cell memory to malaria is generated and the specific effector populations required to confer protection, will undoubtedly benefit vaccine development and optimisation efforts, especially in light of the modest efficacy levels achieved to date with current vaccination regimes at a population level (168, 169).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Supported by the Australian Government National Health and Medical Research Council IRIISS, Project Grant 1058665, and the Australian Academy of Science Regional Collaboration Grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
2. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. (2016) 44:116–30. doi: 10.1016/j.immuni.2015.12.004
3. Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J Exp Med. (1992) 176:679–87.
4. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. (2007) 315:528–31. doi: 10.1126/science.1136736
5. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
6. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. (2014) 509:637–40. doi: 10.1038/nature13300
7. Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. (1998) 8:363–72.
8. Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New markers for murine memory B cells that define mutated and unmutated subsets. J Exp Med. (2007) 204:2103–14. doi: 10.1084/jem.20062571
9. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol. (2009) 182:890–901. doi: 10.4049/jimmunol.182.2.890
10. Tangye SG, Avery DT, Deenick EK, Hodgkin PD. Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J Immunol. (2003) 170:686–94. doi: 10.4049/jimmunol.170.2.686
11. Weisel F, Shlomchik M. Memory B cells of mice and humans. Ann Rev Immunol. (2017) 35:255–84. doi: 10.1146/annurev-immunol-041015-055531
12. Klein U, Kuppers R, Rajewsky K. Evidence for a large compartment of IgM-expressing memory B cells in humans. Blood. (1997) 89:1288–98.
13. Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. (1998) 188:1691–703.
14. Ehrhardt GRA, Hsu JT, Gartland L, Leu C-M, Zhang S, Davis RS, et al. Expression of the immunoregulatory molecule FcRH4 defines a distinctive tissue-based population of memory B cells. J Exp Med. (2005) 202:783–91. doi: 10.1084/jem.20050879
15. Portugal S, Tipton CM, Sohn H, Kone Y, Wang J, Li S, et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. Elife. (2015) 4:1748. doi: 10.7554/eLife.07218
16. Sullivan RT, Kim CC, Fontana MF, Feeney ME, Jagannathan P, Boyle MJ, et al. FCRL5 delineates functionally impaired memory B cells associated with Plasmodium falciparum exposure. PLoS Pathog. (2015) 11:e1004894. doi: 10.1371/journal.ppat.1004894
17. Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol. (2009) 183:2176–82. doi: 10.4049/jimmunol.0901297
18. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. (2009) 10:1292–9. doi: 10.1038/ni.1814
19. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. (2011) 331:1203–7. doi: 10.1126/science.1201730
20. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. (2014) 15:631–7. doi: 10.1038/ni.2914
21. Kenderes KJ, Levack RC, Papillion AM, Cabrera-Martinez B, Dishaw LM, Winslow GM. T-Bet+ IgM memory cells generate multi-lineage effector B cells. Cell Rep. (2018) 24:824–37.e3. doi: 10.1016/j.celrep.2018.06.074
23. White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. (2014) 383:723–35. doi: 10.1016/S0140-6736(13)60024-0
24. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. (2005) 5:722–35. doi: 10.1038/nri1686
25. Miller LH, Ackerman HC, Su X-Z, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. (2013) 19:156–67. doi: 10.1038/nm.3073
26. Koch R. Professor Koch's investigations on malaria: fourth report to the colonial department of the german colonial office. Br Med J. (1900) 1:1597–8.
27. Gupta S, Snow RW, Donnelly CA, Marsh K, Newbold C. Immunity to non-cerebral severe malaria is acquired after one or two infections. Nat Med. (1999) 5:340–3. doi: 10.1038/6560
28. Reyburn H, Mbatia R, Drakeley C, Bruce J, Carneiro I, Olomi R, et al. Association of transmission intensity and age with clinical manifestations and case fatality of severe Plasmodium falciparum malaria. JAMA. (2005) 293:1461–70. doi: 10.1001/jama.293.12.1461
29. Griffin JT, Hollingsworth TD, Reyburn H, Drakeley CJ, Riley EM, Ghani AC. Gradual acquisition of immunity to severe malaria with increasing exposure. Proc R Soc B. (2015) 282:20142657. doi: 10.1098/rspb.2014.2657
30. Bejon P, Warimwe G, Mackintosh CL, Mackinnon MJ, Kinyanjui SM, Musyoki JN, et al. Analysis of immunity to febrile malaria in children that distinguishes immunity from lack of exposure. Infect Immun. (2009) 77:1917–23. doi: 10.1128/IAI.01358-08
31. McGregor IA, Smith DA. A health, nutrition and parasitological survey in a rural village (Keneba) in west Kiang, Gambia. Trans R Soc Trop Med Hyg. (1952) 46:403–27.
32. Drakeley CJ, Carneiro I, Reyburn H, Malima R, Lusingu JPA, Cox J, et al. Altitude-dependent and -independent variations in Plasmodium falciparum prevalence in northeastern Tanzania. J Infect Dis. (2005) 191:1589–98. doi: 10.1086/429669
33. Michon P, Cole-Tobian JL, Dabod E, Schoepflin S, Igu J, Susapu M, et al. The risk of malarial infections and disease in Papua New Guinean children. Am J Trop Med Hyg. (2007) 76:997–1008. doi: 10.4269/ajtmh.2007.76.997
34. Ndungu FM, Marsh K, Fegan G, Wambua J, Nyangweso G, Ogada E, et al. Identifying children with excess malaria episodes after adjusting for variation in exposure: identification from a longitudinal study using statistical count models. BMC Med. (2015) 13:183. doi: 10.1186/s12916-015-0422-4
35. Bouchaud O, Cot M, Kony S, Durand R, Schiemann R, Ralaimazava P, et al. Do African immigrants living in France have long-term malarial immunity? Am J Trop Med Hyg. (2005) 72:21–5. doi: 10.4269/ajtmh.2005.72.21
36. Tran TM, Li S, Doumbo S, Doumtabe D, Huang C-Y, Dia S, et al. An intensive longitudinal cohort study of malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clin Infect Dis. (2013) 57:40–7. doi: 10.1093/cid/cit174
37. Okell LC, Bousema T, Griffin JT, Ouedraogo AL, Ghani AC, Drakeley CJ. Factors determining the occurrence of submicroscopic malaria infections and their relevance for control. Nat Commun. (2012) 3:1237. doi: 10.1038/ncomms2241
38. Cohen S, Carrington S, McGregor IA. Gamma-globulin and acquired immunity to human malaria. Nature. (1961) 192:733–7.
39. McGregor IA, Carrington SP, Cohen S. Treatment of East African P. falciparum malaria with West African human γ-globulin. Trans R Soc Trop Med Hyg. (1963) 57:170–5. doi: 10.1016/0035-9203(63)90058-0
40. Chiu CYH, Hodder AN, Lin CS, Hill DL, Li Wai Suen CSN, Schofield L, et al. Antibodies to the Plasmodium falciparum proteins MSPDBL1 and MSPDBL2 opsonize merozoites, inhibit parasite growth, and predict protection from clinical malaria. J Infect Dis. (2015) 212:406–15. doi: 10.1093/infdis/jiv057
41. Chiu CYH, Healer J, Thompson JK, Chen L, Kaul A, Savergave L, et al. Association of antibodies to Plasmodium falciparum reticulocyte binding protein homolog 5 with protection from clinical malaria. Front Microbiol. (2014) 5:314. doi: 10.3389/fmicb.2014.00314
42. Richards JS, Stanisic DI, Fowkes FJI, Tavul L, Dabod E, Thompson JK, et al. Association between naturally acquired antibodies to erythrocyte-binding antigens of Plasmodium falciparum and protection from malaria and high-density parasitemia. Clin Infect Dis. (2010) 51:e50–60. doi: 10.1086/656413
43. Fowkes FJI, Richards JS, Simpson JA, Beeson JG. The relationship between anti-merozoite antibodies and incidence of Plasmodium falciparum malaria: a systematic review and meta-analysis. PLoS Med. (2010) 7:e1000218–94. doi: 10.1371/journal.pmed.1000218
44. Stanisic DI, Richards JS, McCallum FJ, Michon P, King CL, Schoepflin S, et al. Immunoglobulin G subclass-specific responses against Plasmodium falciparum merozoite antigens are associated with control of parasitemia and protection from symptomatic illness. Infect Immun. (2009) 77:1165–74. doi: 10.1128/IAI.01129-08
45. John CC, O'Donnell RA, Sumba PO, Moormann AM, de Koning-Ward TF, King CL, et al. Evidence that invasion-inhibitory antibodies specific for the 19-kDa fragment of merozoite surface protein-1 (MSP-1 19) can play a protective role against blood-stage Plasmodium falciparum infection in individuals in a malaria endemic area of Africa. J Immunol. (2004) 173:666–72. doi: 10.4049/jimmunol.173.1.666
46. Dodoo D, Staalsoe T, Giha H, Kurtzhals JA, Akanmori BD, Koram K, et al. Antibodies to variant antigens on the surfaces of infected erythrocytes are associated with protection from malaria in Ghanaian children. Infect Immun. (2001) 69:3713–8. doi: 10.1128/IAI.69.6.3713-3718.2001
47. Teo A, Feng G, Brown GV, Beeson JG, Rogerson SJ. Functional antibodies and protection against blood-stage malaria. Trends Parasitol. (2016) 32:887–98. doi: 10.1016/j.pt.2016.07.003
48. Chan J-A, Howell KB, Reiling L, Ataide R, Mackintosh CL, Fowkes FJI, et al. Targets of antibodies against Plasmodium falciparum-infected erythrocytes in malaria immunity. J Clin Invest. (2012) 122:3227–38. doi: 10.1172/JCI62182
49. Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. (2008) 62:445–70. doi: 10.1146/annurev.micro.61.080706.093134
50. Mackintosh CL, Mwangi T, Kinyanjui SM, Mosobo M, Pinches R, Williams TN, et al. Failure to respond to the surface of Plasmodium falciparum infected erythrocytes predicts susceptibility to clinical malaria amongst African children. Int J Parasitol. (2008) 38:1445–54. doi: 10.1016/j.ijpara.2008.03.009
51. Kinyanjui SM, Mwangi T, Bull PC, Newbold CI, Marsh K. Protection against clinical malaria by heterologous immunoglobulin G antibodies against malaria-infected erythrocyte variant surface antigens requires interaction with asymptomatic infections. J Infect Dis. (2004) 190:1527–33. doi: 10.1086/424675
52. Sondén K, Doumbo S, Hammar U, Homann MV, Ongoiba A, Traore B, et al. Asymptomatic multiclonal Plasmodium falciparum infections carried through the dry season predict protection against subsequent clinical malaria. J Infect Dis. (2015) 212:jiv088–616. doi: 10.1093/infdis/jiv088
53. King CL, Davies DH, Felgner P, Baum E, Jain A, Randall A, et al. Biosignatures of exposure/transmission and immunity. Am J Trop Med Hyg. (2015) 93:16–27. doi: 10.4269/ajtmh.15-0037
54. Murungi LM, Kamuyu G, Lowe B, Bejon P, Theisen M, Kinyanjui SM, et al. A threshold concentration of anti-merozoite antibodies is required for protection from clinical episodes of malaria. Vaccine. (2013) 31:3936–42. doi: 10.1016/j.vaccine.2013.06.042
55. Arama C, Skinner J, Doumtabe D, Portugal S, Tran TM, Jain A, et al. Genetic resistance to malaria is associated with greater enhancement of immunoglobulin (Ig)M than IgG responses to a broad array of Plasmodium falciparum antigens. Open Forum Infect Dis. (2015) 2:ofv118. doi: 10.1093/ofid/ofv118
56. Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci USA. (2010) 107:6958–63. doi: 10.1073/pnas.1001323107
57. Weiss GE, Traore B, Kayentao K, Ongoiba A, Doumbo S, Doumtabe D, et al. The Plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. (2010) 6:e1000912. doi: 10.1371/journal.ppat.1000912
58. Finney OC, Danziger SA, Molina DM, Vignali M, Takagi A, Ji M, et al. Predicting antidisease immunity using proteome arrays and sera from children naturally exposed to malaria. Mol Cell Proteomics. (2014) 13:2646–60. doi: 10.1074/mcp.M113.036632
59. Wendel, He C, Qu M, Di Wu, Hernandez SM, Ma K-Y, et al. Accurate immune repertoire sequencing reveals malaria infection driven antibody lineage diversification in young children. Nat Commun. (2017) 8:1–14. doi: 10.1038/s41467-017-00645-x
60. Tan J, Pieper K, Piccoli L, Abdi A, Perez MF, Geiger R, et al. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature. (2016) 529:105–9. doi: 10.1038/nature16450
61. Pieper K, Tan J, Piccoli L, Foglierini M, Barbieri S, Chen Y, et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature. (2017) 548:597–601. doi: 10.1038/nature23670
62. Ceravolo IP, Sanchez BAM, Sousa TN, Guerra BM, Soares IS, Braga EM, et al. Naturally acquired inhibitory antibodies to Plasmodium vivax Duffy binding protein are short-lived and allele-specific following a single malaria infection. Clin Exp Immunol. (2009) 156:502–10. doi: 10.1111/j.1365-2249.2009.03931.x
63. Fruh K, Doumbo O, Muller HM, Koita O, McBride J, Crisanti A, et al. Human antibody response to the major merozoite surface antigen of Plasmodium falciparum is strain specific and short-lived. Infect Immun. (1991) 59:1319–24.
64. Cavanagh DR, Dodoo D, Hviid L, Kurtzhals JA, Theander TG, Akanmori BD, et al. Antibodies to the N-terminal block 2 of Plasmodium falciparum merozoite surface protein 1 are associated with protection against clinical malaria. Infect Immun. (2004) 72:6492–502. doi: 10.1128/iai.72.11.6492-6502.2004
65. Cavanagh DR, Elhassan IM, Roper C, Robinson VJ, Giha H, Holder AA, et al. A longitudinal study of type-specific antibody responses to Plasmodium falciparum merozoite surface protein-1 in an area of unstable malaria in Sudan. J Immunol. (1998) 161:347–59.
66. Soares IS, Oliveira SG, Souza JM, Rodrigues MM. Antibody response to the N and C-terminal regions of the Plasmodium vivax Merozoite Surface Protein 1 in individuals living in an area of exclusive transmission of P-vivax malaria in the north of Brazil. Acta Trop. (1999) 72:13–24.
67. Kinyanjui SM, Conway DJ, Lanar DE, Marsh K. IgG antibody responses to Plasmodium falciparum merozoite antigens in Kenyan children have a short half-life. Malar J. (2007) 6:82. doi: 10.1186/1475-2875-6-82
68. Akpogheneta OJ, Duah NO, Tetteh KKA, Dunyo S, Lanar DE, Pinder M, et al. Duration of naturally acquired antibody responses to blood-stage Plasmodium falciparum is age dependent and antigen specific. Infect Immun. (2008) 76:1748–55. doi: 10.1128/IAI.01333-07
69. Fonjungo PN, Elhassan IM, Cavanagh DR, Theander TG, Hviid L, Roper C, et al. A longitudinal study of human antibody responses to Plasmodium falciparum rhoptry-associated protein 1 in a region of seasonal and unstable malaria transmission. Infect Immunol. (1999) 67:2975–85.
70. Maple PA, Jones CS, Wall EC, Vyseb A, Edmunds WJ, Andrews NJ, et al. Immunity to diphtheria and tetanus in England and Wales. Vaccine. (2000) 19:167–73. doi: 10.1016/s0264-410x(00)00184-5
71. Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med. (2003) 9:1131–7. doi: 10.1038/nm917
72. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. (2007) 357:1903–15. doi: 10.1056/NEJMoa066092
73. Sullivan RT, Ssewanyana I, Wamala S, Nankya F, Jagannathan P, Tappero JW, et al. B cell sub-types following acute malaria and associations with clinical immunity. Malar J. (2016) 15:139. doi: 10.1186/s12936-016-1190-0
74. White MT, Griffin JT, Akpogheneta O, Conway DJ, Koram KA, Riley EM, et al. Dynamics of the antibody response to Plasmodium falciparum infection in African children. J Infect Dis. (2014) 210:1115–22. doi: 10.1093/infdis/jiu219
75. Dorfman JR, Bejon P, Ndungu FM, Langhorne J, Kortok MM, Lowe BS, et al. B cell memory to 3 Plasmodium falciparum blood-stage antigens in a malaria-endemic area. J Infect Dis. (2005) 191:1623–30. doi: 10.1086/429671
76. Triller G, Scally SW, Costa G, Pissarev M, Kreschel C, Bosch A, et al. Natural parasite exposure induces protective human anti-malarial antibodies. Immunity. (2017) 47:1197–209.e10. doi: 10.1016/j.immuni.2017.11.007
77. Nogaro SI, Hafalla JC, Walther B, Remarque EJ, Tetteh KK, Conway DJ, et al. The breadth, but not the magnitude, of circulating memory B cell responses to P. falciparum increases with age/exposure in an area of low transmission. PLoS ONE. (2011) 6:e25582. doi: 10.1371/journal.pone.0025582
78. Changrob S, McHenry AM, Nyunt MH, Sattabongkot J, Han E-T, Adams JH, et al. Persistence of long-lived memory B cells specific to duffy binding protein in individuals exposed to Plasmodium vivax. Sci Rep. (2018) 8:8347. doi: 10.1038/s41598-018-26677-x
79. Min HMK, Changrob S, Soe PT, Han JH, Muh F, Lee S-K, et al. Immunogenicity of the Plasmodium vivax merozoite surface protein 1 paralog in the induction of naturally acquired antibody and memory B cell responses. Malar J. (2017) 16:354. doi: 10.1186/s12936-017-2000-z
80. Migot F, Chougnet C, Henzel D, Dubois B, Jambou R, Fievet N, et al. Anti-malaria antibody-producing B cell frequencies in adults after a Plasmodium falciparum outbreak in Madagascar. Clin Exp Immunol. (1995) 102:529–34.
81. Wipasa J, Suphavilai C, Okell LC, Cook J, Corran PH, Thaikla K, et al. Long-lived antibody and B Cell memory responses to the human malaria parasites, Plasmodium falciparum and Plasmodium vivax. PLoS Pathog. (2010) 6:e1000770. doi: 10.1371/journal.ppat.1000770
82. Clark EH, Silva CJ, Weiss GE, Li S, Padilla C, Crompton PD, et al. Plasmodium falciparum malaria in the Peruvian Amazon, a region of low transmission, is associated with immunologic memory. Infect Immun. (2012) 80:1583–92. doi: 10.1128/IAI.05961-11
83. Ndungu FM, Olotu A, Mwacharo J, Nyonda M, Apfeld J, Mramba LK, et al. Memory B cells are a more reliable archive for historical antimalarial responses than plasma antibodies in no-longer exposed children. Proc Natl Acad Sci USA. (2012) 109:8247–52. doi: 10.1073/pnas.1200472109
84. Ayieko C, Maue AC, Jura WGZO, Noland GS, Ayodo G, Rochford R, et al. Changes in B cell populations and merozoite surface protein-1-specific memory B cell responses after prolonged absence of detectable P. falciparum infection. PLoS ONE. (2013) 8:e67230. doi: 10.1371/journal.pone.0067230
85. Ampomah P, Stevenson L, Ofori MF, Barfod L, Hviid L. B-cell responses to pregnancy-restricted and -unrestricted Plasmodium falciparum erythrocyte membrane protein 1 antigens in Ghanaian women naturally exposed to malaria parasites. Infect Immunol. (2014) 82:1860–71. doi: 10.1128/IAI.01514-13
86. Moncunill G, Mayor A, Jiménez A, Nhabomba A, Casas-Vila N, Puyol L, et al. High antibody responses against Plasmodium falciparum in immigrants after extended periods of interrupted exposure to malaria. PLoS ONE. (2013) 8:e73624. doi: 10.1371/journal.pone.0073624
87. Torres KJ, Clark EH, Hernandez JN, Soto-Cornejo KE, Gamboa D, Branch OH. Antibody response dynamics to the Plasmodium falciparum conserved vaccine candidate antigen, merozoite surface protein-1 C-terminal 19kD (MSP1-19kD), in Peruvians exposed to hypoendemic malaria transmission. Malar J. (2008) 7:173. doi: 10.1186/1475-2875-7-173
88. Bousema T, Roeffen W, Meijerink H, Mwerinde H, Mwakalinga S, van Gemert G-J, et al. The dynamics of naturally acquired immune responses to Plasmodium falciparum sexual stage antigens Pfs230 & Pfs48/45 in a low endemic area in tanzania. PLoS ONE. (2010) 5:e14114. doi: 10.1371/journal.pone.0014114
89. Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, et al. Somatically hypermutated Plasmodium-specific IgM+ memory B cells are rapid, plastic, early responders upon malaria rechallenge. Immunity. (2016) 45:402–14. doi: 10.1016/j.immuni.2016.06.014
90. Stephens R, Ndungu FM, Langhorne J. Germinal centre and marginal zone B cells expand quickly in a second Plasmodium chabaudi malaria infection producing mature plasma cells. Parasite Immunol. (2009) 31:20–31. doi: 10.1111/j.1365-3024.2008.01066.x
91. Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, et al. The role of animal models for research on severe malaria. PLoS Pathog. (2012) 8:e1002401. doi: 10.1371/journal.ppat.1002401
92. Stephens R, Culleton RL, Lamb TJ. The contribution of Plasmodium chabaudi to our understanding of malaria. Trends Parasitol. (2012) 28:73–82. doi: 10.1016/j.pt.2011.10.006
93. Castillo-Méndez SI, Castillo-Méndez SI, Zago CA, Zago CA, Sardinha LR, Sardinha LR, et al. Characterization of the spleen B-cell compartment at the early and late blood-stage Plasmodium chabaudi malaria. Scand J Immunol. (2007) 66:309–19. doi: 10.1111/j.1365-3083.2007.01972.x
94. Ndungu FM, Cadman ET, Coulcher J, Nduati E, Couper E, Macdonald DW, et al. Functional memory B cells and long-lived plasma cells are generated after a single Plasmodium chabaudi infection in mice. PLoS Pathog. (2009) 5:e1000690. doi: 10.1371/journal.ppat.1000690
95. Achtman AH, Stephens R, Cadman ET, Harrison V, Langhorne J. Malaria-specific antibody responses and parasite persistence after infection of mice with Plasmodium chabaudi chabaudi. Parasite Immunol. (2007) 29:435–44. doi: 10.1111/j.1365-3024.2007.00960.x
96. Wykes MN, Zhou Y-H, Liu XQ, Good MF. Plasmodium yoelii can ablate vaccine-induced long-term protection in mice. J Immunol. (2005) 175:2510–6. doi: 10.4049/jimmunol.175.4.2510
97. Keitany GJ, Kim KS, Krishnamurty AT, Hondowicz BD, Hahn WO, Dambrauskas N, et al. Blood stage malaria disrupts humoral immunity to the pre-erythrocytic stage circumsporozoite protein. Cell Rep. (2016) 17:3193–205. doi: 10.1016/j.celrep.2016.11.060
98. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular helper T cells. Ann Rev Immun. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
99. Pérez-Mazliah D, Ng DHL, Freitas do Rosário AP, McLaughlin S, Mastelic-Gavillet B, Sodenkamp J, et al. Disruption of IL-21 signaling affects T cell-B cell interactions and abrogates protective humoral immunity to malaria. PLoS Pathog. (2015) 11:e1004715. doi: 10.1371/journal.ppat.1004715
100. Opata MM, Carpio VH, Ibitokou SA, Dillon BE, Obiero JM, Stephens R. Early Effector cells survive the contraction phase in malaria infection and generate both central and effector memory T cells. J Immunol. (2015) 194:5346–54. doi: 10.4049/jimmunol.1403216
101. Butler NS, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. (2011) 13:188–95. doi: 10.1038/ni.2180
102. Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, et al. PD-1 co-inhibitory and OX40 co-stimulatory crosstalk regulates helper T cell differentiation and anti-Plasmodium humoral immunity. Cell Host Microbe. (2015) 17:628–41. doi: 10.1016/j.chom.2015.03.007
103. Hansen DS, Obeng-Adjei N, Ly A, Ioannidis LJ, Crompton PD. Emerging concepts in T follicular helper cell responses to malaria. Int J Parasitol. (2017) 47:105–10. doi: 10.1016/j.ijpara.2016.09.004
104. Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The costimulatory molecule ICOS regulates host Th1 and follicular Th cell differentiation in response to Plasmodium chabaudi chabaudi AS infection. J Immunol. (2015) 196:778–91. doi: 10.4049/jimmunol.1403206
105. Pérez-Mazliah D, Nguyen MP, Hosking C, McLaughlin S, Lewis MD, Tumwine I, et al. Follicular helper T cells are essential for the elimination of Plasmodium infection. EBioMedicine. (2017) 24:216–30. doi: 10.1016/j.ebiom.2017.08.030
106. Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, et al. Circulating Th1-cell-type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. (2015) 13:425–39. doi: 10.1016/j.celrep.2015.09.004
107. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3−CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. (2013) 39:758–69. doi: 10.1016/j.immuni.2013.08.031
108. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7loPD-1hi CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
109. Figueiredo MM, Costa PAC, Diniz SQ, Henriques PM, Kano FS, Tada MS, et al. T follicular helper cells regulate the activation of B lymphocytes and antibody production during Plasmodium vivax infection. PLoS Pathog. (2017) 13:e1006484–23. doi: 10.1371/journal.ppat.1006484
110. Kurup SP, Obeng-Adjei N, Anthony SM, Traore B, Doumbo OK, Butler NS, et al. Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nat Med. (2017) 23:1220–5. doi: 10.1038/nm.4395
111. Carpio VH, Opata MM, Montañez ME, Banerjee PP, Dent AL, Stephens R. IFN-γ and IL-21 double producing T cells are Bcl6-independent and survive into the memory phase in Plasmodium chabaudi infection. PLoS ONE. (2015) 10:e0144654. doi: 10.1371/journal.pone.0144654
112. Zander RA, Vijay R, Pack AD, Guthmiller JJ, Graham AC, Lindner SE, et al. Th1-like Plasmodium-specific memory CD4+ T cells support humoral immunity. Cell Rep. (2017) 21:1839–52. doi: 10.1016/j.celrep.2017.10.077
113. Ryg-Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. (2016) 14:68–81. doi: 10.1016/j.celrep.2015.12.006
114. Ryg-Cornejo V, Ly A, Hansen DS. Immunological processes underlying the slow acquisition of humoral immunity to malaria. Parasitology. (2016) 143:199–207. doi: 10.1017/S0031182015001705
115. Ioannidis LJ, Nie CQ, Ly A, Ryg-Cornejo V, Chiu CY, Hansen DS. Monocyte- and neutrophil-derived CXCL10 impairs efficient control of blood-stage malaria infection and promotes severe disease. J Immunol. (2016) 196:1227–38. doi: 10.4049/jimmunol.1501562
116. Sebina I, James KR, Soon MSF, Fogg LG, Best SE, Labastida Rivera F de, et al. IFNAR1-signalling obstructs ICOS-mediated humoral immunity during non-lethal blood-stage Plasmodium infection. PLoS Pathog. (2016) 12:e1005999. doi: 10.1371/journal.ppat.1005999
117. Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJJ, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog. (2016) 12:e1005945. doi: 10.1371/journal.ppat.1005945
118. Salles ÉM de, Menezes MN de, Siqueira R, Borges da Silva H, Amaral EP, Castillo-Méndez SI, et al. P2X7 receptor drives Th1 cell differentiation and controls the follicular helper T cell population to protect against Plasmodium chabaudi malaria. PLoS Pathog. (2017) 13:e1006595. doi: 10.1371/journal.ppat.1006595
119. James KR, Soon MSF, Sebina I, Fernandez-Ruiz D, Davey G, Liligeto UN, et al. IFN regulatory factor 3 balances Th1 and T follicular helper immunity during nonlethal blood-stage Plasmodium infection. J Immunol. (2018) 200:1443–56. doi: 10.4049/jimmunol.1700782
120. Lönnberg T, Svensson V, James KR, Fernandez-Ruiz D, Sebina I, Montandon R, et al. Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol. (2017) 2:eaal2192. doi: 10.1126/sciimmunol.aal2192
121. Hahn WO, Butler NS, Lindner SE, Akilesh HM, Sather DN, Kappe SHI, et al. cGAS-mediated control of blood-stage malaria promotes Plasmodium-specific germinal center responses. JCI Insight. (2018) 3:1–18. doi: 10.1172/jci.insight.94142
122. Rivera-Correa J, Guthmiller JJ, Vijay R, Fernandez-Arias C, Pardo-Ruge MA, Gonzalez S, et al. Plasmodium DNA-mediated TLR9 activation of T-bet+ B cells contributes to autoimmune anaemia during malaria. Nat Commun. (2017) 8:584. doi: 10.1038/s41467-017-01476-6
123. Guthmiller JJ, Graham AC, Zander RA, Pope RL, Butler NS. Cutting edge: IL-10 is essential for the generation of germinal center B cell responses and anti-Plasmodium humoral immunity. J Immunol. (2017) 198:617–22. doi: 10.4049/jimmunol.1601762
124. Donati D, Zhang LP, Chêne A, Chen Q, Flick K, Nyström M, et al. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect Immun. (2004) 72:5412–8. doi: 10.1128/IAI.72.9.5412-5418.2004
125. Donati D, Mok B, Chêne A, Xu H, Thangarajh M, Glas R, et al. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J Immunol. (2006) 177:3035–44. doi: 10.4049/jimmunol.177.5.3035
126. Simone O, Bejarano MT, Pierce SK, Antonaci S, Wahlgren M, Troye-Blomberg M, et al. TLRs innate immunereceptors and Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) CIDR1 alpha-driven human polyclonal B-cell activation. Acta Trop. (2011) 119:144–50. doi: 10.1016/j.actatropica.2011.05.005
127. Nduati E, Gwela A, Karanja H, Mugyenyi C, Langhorne J, Marsh K, et al. The plasma concentration of the B cell activating factor is increased in children with acute malaria. J Infect Dis. (2011) 204:962–70. doi: 10.1093/infdis/jir438
128. Scholzen A, Teirlinck AC, Bijker EM, Roestenberg M, Hermsen CC, Hoffman SL, et al. BAFF and BAFF receptor levels correlate with B cell subset activation and redistribution in controlled human malaria infection. J Immunol. (2014) 192:3719–29. doi: 10.4049/jimmunol.1302960
129. Weiss GE, Clark EH, Li S, Traore B, Kayentao K, Ongoiba A, et al. A positive correlation between atypical memory B cells and Plasmodium falciparum transmission intensity in cross-sectional studies in Peru and Mali. PLoS ONE. (2011) 6:e15983. doi: 10.1371/journal.pone.0015983
130. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. (2013) 190:1038–47. doi: 10.4049/jimmunol.1202438
131. Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. (2008) 205:1797–805. doi: 10.1084/jem.20072683
132. Muellenbeck MF, Ueberheide B, Amulic B, Epp A, Fenyo D, Busse CE, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J Exp Med. (2013) 210:389–99. doi: 10.1084/jem.20121970
133. Patgaonkar M, Herbert F, Powale K, Gandhe P, Gogtay N, Thatte U, et al. Vivax infection alters peripheral B-cell profile and induces persistent serum IgM. Parasite Immunol. (2018) 40:e12580. doi: 10.1111/pim.12580
134. Obeng-Adjei N, Portugal S, Holla P, Li S, Sohn H, Ambegaonkar A, et al. Malaria-induced interferon-γ drives the expansion of Tbethi atypical memory B cells. PLoS Pathog. (2017) 13:e1006576. doi: 10.1371/journal.ppat.1006576
135. Portugal S, Doumtabe D, Traore B, Miller LH, Troye-Blomberg M, Doumbo OK, et al. B cell analysis of ethnic groups in Mali with differential susceptibility to malaria. Malar J. (2012) 11:162. doi: 10.1186/1475-2875-11-162
136. Requena P, Campo JJ, Umbers AJ, Ome M, Wangnapi R, Barrios D, et al. Pregnancy and malaria exposure are associated with changes in the B cell pool and in plasma eotaxin levels. J Immunol. (2014) 193:2971–83. doi: 10.4049/jimmunol.1401037
137. Ubillos I, Campo JJ, Requena P, Ome-Kaius M, Hanieh S, Rose H, et al. Chronic exposure to malaria is associated with inhibitory and activation markers on atypical memory B cells and marginal zone-like B cells. Front Immunol. (2017) 8:966. doi: 10.3389/fimmu.2017.00966
138. Lugaajju A, Reddy SB, Wahlgren M, Kironde F, Persson KEM. Development of Plasmodium falciparum specific naïve, atypical, memory and plasma B cells during infancy and in adults in an endemic area. Malar J. (2017) 16:37. doi: 10.1186/s12936-017-1697-z
139. Subramaniam KS, Skinner J, Ivan E, Mutimura E, Kim RS, Feintuch CM, et al. HIV malaria co-infection is associated with atypical memory B cell expansion and a reduced antibody response to a broad array of Plasmodium falciparum antigens in rwandan adults. PLoS ONE. (2015) 10:e0124412. doi: 10.1371/journal.pone.0124412
140. Frosch AE, Odumade OA, Taylor JJ, Ireland K, Ayodo G, Ondigo B, et al. Decrease in numbers of naive and resting B cells in HIV-infected kenyan adults leads to a proportional increase in total and Plasmodium falciparum-specific atypical memory B cells. J Immunol. (2017) 198:4629–38. doi: 10.4049/jimmunol.1600773
141. Zinöcker S, Schindler CE, Skinner J, Rogosch T, Waisberg M, Schickel J-N, et al. The V gene repertoires of classical and atypical memory B cells in malaria-susceptible West African children. J Immunol. (2015) 194:929–39. doi: 10.4049/jimmunol.1402168
142. Li H, Borrego F, Nagata S, Tolnay M. Fc receptor-like 5 expression distinguishes two distinct subsets of human circulating tissue-like memory B cells. J Immunol. (2016) 196:4064–74. doi: 10.4049/jimmunol.1501027
143. Szabo SJ, Kim ST, Costa GL, Zhang XK, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
144. Jöhrens K, Shimizu Y, Anagnostopoulos I, Schiffmann S, Tiacci E, Falini B, et al. T-bet-positive and IRTA1-positive monocytoid B cells differ from marginal zone B cells and epithelial-associated B cells in their antigen profile and topographical distribution. Haematologica. (2005) 90:1070–7.
145. Johrens K, Anagnostopoulos I, Durkop H, Stein H. Different T-bet expression patterns characterize particular reactive lymphoid tissue lesions. Histopathology. (2006) 48:343–52. doi: 10.1111/j.1365-2559.2005.02305.x
146. Jöhrens K, Moos V, Schneider T, Stein H, Anagnostopoulos I. Interferon-gamma and T-bet expression in a patient with toxoplasmic lymphadenopathy. Hum Immunol. (2010) 71:366–71. doi: 10.1016/j.humimm.2010.01.005
147. Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, et al. T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight. (2017) 2:92943. doi: 10.1172/jci.insight.92943
148. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci USA. (2013) 110:E3216–24. doi: 10.1073/pnas.1312348110
149. Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, et al. Cutting edge: B cell-intrinsic T-bet expression is required to control chronic viral infection. J Immunol. (2016) 197:1017–22. doi: 10.4049/jimmunol.1500368
150. Rubtsova K, Rubtsov AV, Halemano K, Li SX, Kappler JW, Santiago ML, et al. T cell production of IFNγ in response to TLR7/IL-12 stimulates optimal B cell responses to viruses. PLoS ONE. (2016) 11:e0166322. doi: 10.1371/journal.pone.0166322
151. Chang L-Y, Li Y, Kaplan DE. Hepatitis C viraemia reversibly maintains subset of antigen-specific T-bet+ tissue-like memory B cells. J Viral Hepat. (2017) 24:389–96. doi: 10.1111/jvh.12659
152. Papillion AM, Kenderes KJ, Yates JL, Winslow GM. Early derivation of IgM memory cells and bone marrow plasmablasts. PLoS ONE. (2017) 12:e0178853. doi: 10.1371/journal.pone.0178853
153. Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, et al. Cutting edge: IL-4, IL-21, and IFN-γ interact to govern T-bet and CD11c expression in TLR-activated B cells. J Immunol. (2016) 197:1023–8. doi: 10.4049/jimmunol.1600522
154. Bürgler S, Gimeno A, Parente-Ribes A, Wang D, Os A, Devereux S, et al. Chronic lymphocytic leukemia cells express CD38 in response to Th1 cell-derived IFN-γ by a T-bet-dependent mechanism. J Immunol. (2015) 194:827–35. doi: 10.4049/jimmunol.1401350
155. Jöhrens K, Dietel M, Anagnostopoulos I. Three different expression patterns of T-bet in angioimmunoblastic T-cell lymphoma. Leuk Lymphoma. (2012) 53:152–5. doi: 10.3109/10428194.2011.606941
156. Jöhrens K, Moos V, Schneider T, Anagnostopoulos I. T-box-expressed-in-T-cells (T-bet) expression by the tumor cells of hairy-cell leukemia correlates with interferon-gamma production. Leuk Lymphoma. (2009) 50:1687–92. doi: 10.1080/10428190903161042
157. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest. (2017) 127:1392–404. doi: 10.1172/JCI91250
158. Wang Z, Wang Z, Wang J, Diao Y, Qian X, Zhu N. T-bet-expressing B cells are positively associated with Crohn's disease activity and support Th1 inflammation. DNA Cell Biol. (2016) 35:628–35. doi: 10.1089/dna.2016.3304
159. Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS ONE. (2013) 8:e67003. doi: 10.1371/journal.pone.0067003
160. Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
161. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. (2016) 213:715–32. doi: 10.1084/jem.20151722
162. Frisullo G, Nociti V, Iorio R, Patanella AK, Marti A, Cammarota G, et al. Increased expression of T-bet in circulating B cells from a patient with multiple sclerosis and celiac disease. Hum Immunol. (2008) 69:837–9. doi: 10.1016/j.humimm.2008.09.008
163. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med. (2016) 213:733–50. doi: 10.1084/jem.20151724
164. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
165. Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol Res. (2013) 55:210–6. doi: 10.1007/s12026-012-8365-8
166. Fontana MF, Baccarella A, Craft JF, Boyle MJ, McIntyre TI, Wood MD, et al. A novel model of asymptomatic Plasmodium parasitemia that recapitulates elements of the human immune response to chronic infection. PLoS ONE. (2016) 11:e0162132. doi: 10.1371/journal.pone.0162132
167. Pérez-Mazliah D, Gardner PJ, Schweighoffer E, McLaughlin S, Hosking C, Tumwine I, et al. Plasmodium-specific atypical memory B cells are short-lived activated B cells. Elife. (2018) 7:e39800. doi: 10.7554/eLife.39800
168. RTS S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. (2015) 386:31–45. doi: 10.1016/S0140-6736(15)60721-8
Keywords: malaria, immunity, antibodies, memory B cells, inflammation
Citation: Ly A and Hansen DS (2019) Development of B Cell Memory in Malaria. Front. Immunol. 10:559. doi: 10.3389/fimmu.2019.00559
Received: 06 November 2018; Accepted: 04 March 2019;
Published: 02 April 2019.
Edited by:
Ian Marriott, University of North Carolina at Charlotte, United StatesReviewed by:
Julius Clemence Hafalla, London School of Hygiene and Tropical Medicine (LSHTM), United KingdomRobin Stephens, The University of Texas Medical Branch at Galveston, United States
Copyright © 2019 Ly and Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana S. Hansen, aGFuc2VuQHdlaGkuZWR1LmF1