Corrigendum: Targeting Complement Pathways in Polytrauma- and Sepsis-Induced Multiple-Organ Dysfunction
 Ebru Karasu1
Ebru Karasu1 Bo Nilsson
Bo Nilsson Jörg Köhl
Jörg Köhl John D. Lambris
John D. Lambris Markus Huber-Lang
Markus Huber-Lang- 1Institute for Clinical and Experimental Trauma-Immunology, University Hospital of Ulm, Ulm, Germany
- 2Department of Immunology, Genetics and Pathology (IGP), Laboratory C5:3, Uppsala University, Uppsala, Sweden
- 3Institute for Systemic Inflammation Research (ISEF), University of Lübeck, Lübeck, Germany
- 4Division of Immunobiology, Cincinnati Children's Hospital, Cincinnati, OH, United States
- 5Department of Pathology & Laboratory Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA, United States
Exposure to traumatic or infectious insults results in a rapid activation of the complement cascade as major fluid defense system of innate immunity. The complement system acts as a master alarm system during the molecular danger response after trauma and significantly contributes to the clearance of DAMPs and PAMPs. However, depending on the origin and extent of the damaged macro- and micro -milieu, the complement system can also be either excessively activated or inhibited. In both cases, this can lead to a maladaptive immune response and subsequent multiple cellular and organ dysfunction. The arsenal of complement-specific drugs offers promising strategies for various critical conditions after trauma, hemorrhagic shock, sepsis, and multiple organ failure. The imbalanced immune response needs to be detected in a rational and real-time manner before the translational therapeutic potential of these drugs can be fully utilized. Overall, the temporal-spatial complement response after tissue trauma and during sepsis remains somewhat enigmatic and demands a clinical triad: reliable tissue damage assessment, complement activation monitoring, and potent complement targeting to highly specific rebalance the fluid phase innate immune response.
Introduction
Complement activation as a major innate defense strategy occurs early after trauma, hemorrhagic shock and during sepsis in both the experimental and clinical settings (1–6). A recent comparison of severe trauma and septic patients in an intensive care unit (ICU) showed that during sepsis, excessive activation of the complement cascade is detectable as evidenced by significantly enhanced systemic C3a concentrations, whereas during trauma, complement activation is also existent but less pronounced (7). In trauma, rapid consumption of key complement components such as C3 or C5 seems to be the primary mechanisms (7) whereas in septic conditions, consumption of complement factors may occur later (if at all), secondary to the activation. Hyper-activated and consumed defense systems including the complement and coagulation cascade can result in imbalanced immune responses, impaired clearance of tissue debris and pathogens, dysregulated coagulation, perfusion disturbances, changes in tissue and cellular microenvironment, and barrier dysfunction. All these alterations culminate in multiple signaling-, cellular-, and organ dysfunction (8, 9). Although highly specific complement inhibitors are available, translation of these observations into therapeutic strategies remains challenging and requires differential considerations (10). The immune response to damaged and infected tissue is not mono-dimensional; instead it comprises various compartmentalized responses and organ specific outcomes (11, 12). For example, in experimental sepsis there is a loss of C5aR expression on neutrophils whereas C5aR expression on various organs is significantly enhanced (13). Thus, the complement reaction to danger associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) needs to be reliably determined before any specific therapeutic intervention can be applied in the clinical setting of severe sterile or infectious insults. Furthermore, underlying triggers and mechanisms of the multiple organ dysfunction syndrome (MODS) need to be detected and further explored. Several hypotheses exist about the initiators and drivers of MODS. Main contributors seem to be barrier failure (9), electrophysiological alterations (14), microcirculatory disturbances, inflammation-induced cell dysfunction, protein alterations and microbiome shifts (15). Hibernation seems to be a contributing factor in the development of MODS, which allows the cell to shut down energy consuming functional efforts and therefore to preserve the cellular morphology (16). Although MODS can be induced by distinct injuries and infectious insults, once established, it seems to follow common pathways. For the individual and also the society, MODS remains a major burden with a high lethality rate and a high socio-economic impact and therefore requires an improved understanding, comprehensive mechanistic insights and basic as well as clinical research efforts (17).
Polytrauma-Induced MODS—Role of Complement
Polytrauma comprises life-threatening multiple injuries that activate innate and adaptive immunity with multidimensional consequences for the host (9, 18). Although some reduction in the frequency of MODS after polytrauma has been noted in the last decades, it still remains a major cause of death after severe trauma (19). Within minutes after polytrauma there is a significant increase in circulating complement activation products such as C3a, C5a, and sC5b-9 and a drop in complement hemolytic activity (2–4, 6, 20) (Figure 1). Of note, an enhanced C3a/C3 ratio in plasma early after trauma was prognostic for lethal outcome (6). Another study showed enhanced C3a levels as an indicator for MODS (20). Activation via its amplification by the alternative pathway is observed early after trauma measured by Bb plasma levels, which was additionally correlated with injury severity and the development of organ failure such as acute lung injury and acute renal failure (3). The underlying mechanisms, however, are still elusive. For example, C3a may have a direct pathophysiological impact on the lungs as an “engine” of multiple organ failure. C3a alters not only the microvascular and airway tonus (21) but induces direct pro-inflammatory effects (22) which may contribute to perfusion disturbances and cellular dysfunction. In a murine model of blunt thorax trauma, we have found enhanced C3 and heme-oxygenase-1 (HMOX1) transcriptional expression levels in the lungs early after injury (23). A recent analyses of 81 polytrauma patients also revealed an enhanced expression of HMOX1, which was associated with septic complications (24). In this context, it is noteworthy that HMOX1 is downregulated in leukemic leukocytes by C3a and C5a resulting in an enhanced cellular mobility and infectious complications (25), indicating some interaction between HMOX1 and complement activation processes. Clinical trials, evaluating the effect of targeting at the C3or C3a/C3aR are lacking. Although in a different context, a recent study showed the potential of HMOX1 to protect endothelial cells against heme-mediated complement activation. Heme activates the alternative complement pathway and also up-regulates the cyto-protective and stress-response gene HMOX1 in an organ-specific manner. While it was highly upregulated in endothelial cells of large vessels, it was poorly upregulated in the renal endothelium, which makes the renal endothelium more vulnerably for complement over-activation and showed stronger C3 deposition (26).
FIGURE 1
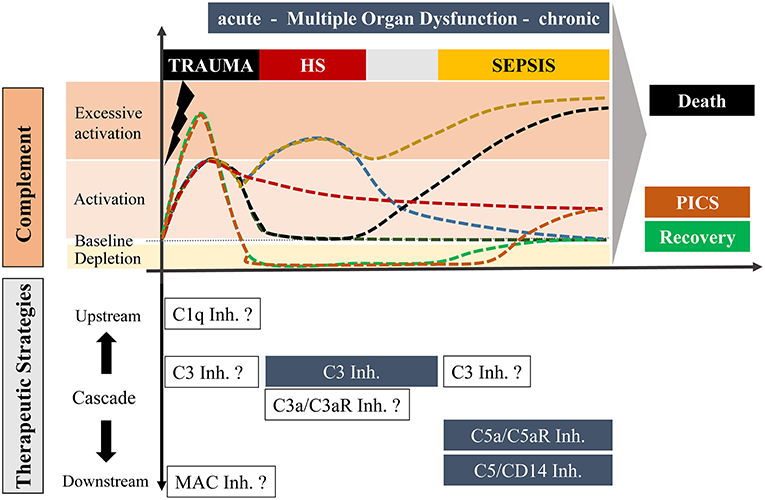
Figure 1. Overview of potential complement responses after trauma with or without hemorrhagic shock and sepsis leading to either death or recovery or “Persistent Inflammation, Immunosuppression and Catabolism Syndrome” (PICS; with a constitutively active and chronic complement state) and potential complement-targeting strategies, respectively. The upper scaling represents the temporal course of complement activation with possible scenarios. Under optimal conditions, trauma induces an acute complement activation which follows a rapid decline to a physiological state with recovery (dark green). Alternatively, complement activity can also be maintained over a long period which may be associated with PICS (red). In an unfavorable state, the acute complement activation could be followed by secondary complication/s including sepsis leading to a complement hyperactivation and death (black). Trauma with an additional hemorrhagic shock can cause excessive complement activation, which can resolve in a physiological recovery. However, an additional hemorrhagic shock with hyperactivated complement can also result in a life-threatening sepsis phase (yellow). Trauma can further cause an acute complement hyperactivation with a rapid consumption of complement which may finally be resolved in a recovery phase (green) or to complement activation at the later stage leading to chronic inflammation (orange). The downwards orientated scaling includes possible therapeutic strategies of distinct stages of complement after trauma, hemorrhagic shock and sepsis. Blue highlighted boxes include therapeutic options which already revealed beneficial effects in pre-clinical studies. Non-highlighted boxes include possible strategies which need to be investigated in future experimental approaches.
On the C5 level, the generated anaphylatoxin C5a is a potent chemoattractant that enhances surface expression of intercellular adhesion molecules on the endothelium, and thereby effectively recruits inflammatory cells to the injured site (27). The migrated cells of the first line of defense can sense, phagocyte and clear damaged tissue and induced repair processes (9). In a recent polytrauma study, leukocytes with low C5-expression on day 1 after trauma correlated with an increased risk for the development of nosocomial infections during the later course (28). The corresponding C5a receptors, C5aR1 and C5aR2, are down-regulated on leukocytes early after trauma (29), something that has been proposed as a sign of enhanced risk for infectious complications (30). Of note, no clinical trial has been proposed or designed in regard to polytrauma using downstream modulation principals of the complement cascade, e.g., modulation of the membrane attack complex (MAC). This might represent an interesting pharmaceutical target, especially since significant amounts of sC5-9 are generated early after polytrauma (2).
Hemorrhagic-Shock-Induced MODS—Role of Complement
Hemorrhagic shock is a condition of disturbed tissue perfusion, resulting in the inadequate delivery of oxygen and nutrients and inadequate clearance of waste products, all of which are vital for regular cellular function. The involvement of complement is complex.
Complement activation products have been reported to directly or indirectly alter the vascular tonus. A C3a-analog peptide was able to cause pulmonary artery constriction in a thromboxan-dependent manner (31). Similarly, C5a caused transient vasoconstriction of an isolated pulmonary artery, which was dependent on the integrity of the endothelium (32). In other reports, C5a caused a leukocyte-dependent vasospasm and triggered thromboembolic events by C5a-induced release of thromboxane-like eicosanoids (33). In line with this observation, C5a is involved in the pathogenesis of acute kidney injury in hemorrhagic shock by reducing renal blood flow by ~ 30% and the glomerular filtration rate by ~ 45%, respectively (34). Of note, when either a leukotriene antagonist was applied or neutrophils were depleted, the C5a-induced reduction in renal perfusion was not observed indicating a leukotriene-dependent (34). In contrast, systemic application of C5a in rabbits induced a reversible drop in systemic arterial pressure (MAP) and a drop in central venous pressure (CVP), decreased cardiac output (CO), and neutropenia (35). In regard to coronary artery vasotonus, conflicting data have been reported for C5a both, vasoconstrictory (36) and vasodilatory effects (37). Other studies concluded that anaphylatoxins alter the vascular tonus with a high variability depending on the localization and pre-exposure to other mediators (38). Taken together, the available data suggest that complement activation changes tissue perfusion by macro- and micro-hemodynamic effects, especially when a shock is already established.
Another central pathomechanistic driver of hemorrhagic shock is the ischemia/reperfusion injury. In this regard, several studies have shown direct activation of complement with a significant increase in MBL and C3 on the endothelium by hypoxic and reperfusion conditions (39). Of note, clinically and experimentally, hemorrhagic shock is a major driver of further organ damage and development of coagulopathy, endotheliopathy, barrier failure, immune dysfunction and MODS after polytrauma (40–43). The close relation of hemorrhagic shock and complement activation reflected by C3 consumption has been previously modeled in baboons where cobra venom factor (CVF) was injected to activate and deplete C3 before a subsequent hemorrhagic shock was induced (20). In pigs, hemorrhagic shock resulted in a drop of CH50 during the hypovolemic phase and also during early resuscitation with enhanced plasma levels of C5a and early detection of endotoxin in the blood, which contributes to MODS (44). A proteomic approach using a rodent hemorrhagic shock model revealed a few alterations of the lymphatic fluid (“toxic lymph”), which included significantly enhanced C3 precursor protein concentrations (45). Since the “toxic lymph” is considered as an important pathophysiological mechanism in the development of adult respiratory distress syndrome and MODS, these findings indicate C3 as a promising target for immune modulatory approaches (Figure 1).
Sepsis-Induced MODS—Role of Complement
Sepsis was long considered as systemic inflammatory response syndrome with evidence of pathogenic microorganisms (46). In contrast, the new definitions describe sepsis rather as organ dysfunction induced by a dysregulated host response to infection (47). This paradigm shift (48) may also change the focus of therapeutic strategies toward support of organ functions, far beyond the established eradication of the pathogens. However, it remains somehow enigmatic what “dysregulation” of the host response means or if the “inadequacy” of the host response is central for development and progression of sepsis.
Multiple experimental sepsis studies have emphasized the detrimental effects of excessive complement activation for the host (Figure 1) (49, 50). This can be considered as a “complement paradox” since complement per se is central to the innate immune defense against invading microorganisms. However, in the context of MODS development caused by sepsis, complement activation seems to enhance rather than protecting against several organ dysfunctions, especially in the heart, lungs and kidneys, representing three central organs in MODS (51–53). The multi-organ gene expression profiles in experimental sepsis seems to be either organ-specific, or common to more than one organ, or distinctly opposite in some organs (54). Furthermore, a balanced pro- and anti-inflammatory genetic response was observed and a differential gene expression for mediators responsible for preventing tissue damage, e.g., protease inhibitors, oxidant neutralizing enzymes, decoy receptors, and proteins which can protect tissue barriers (54). Concerning complement, pre-pro-complement C3 was highly expressed in all organs except in the brain during the whole course of sepsis (54). However, genetic deficiency of C3 resulted in significantly enhanced lethality in comparison to C3-sufficient mice most likely due to a loss of C3b-dependent opsonization of invaded pathogens (55). In contrast, a blockade of C5a by various strategies in sepsis models, e.g., by anti-C5a antibodies, C5aR antibodies, small peptide C5aR1 antagonists, C5a-neutralizing mirror-image (l-)aptamer C5a aptamers, was coherently protective against biochemical and histological evidence of MODS and in general improved survival of sepsis (53, 55–61). All these experimental results demonstrate that C5a-C5aR interaction is clearly involved in the pathogenesis of MODS during sepsis and represents an important therapeutic sepsis target when the novel definitions of sepsis are applied (Figure 1) (47).
In translation to the clinical setting, several non-human primate experiments and studies in humans are in line with the findings in the rodent sepsis model. Evidence of systemic complement activation with reduction of complement hemolytic activity, C3 depletion and enhanced levels of C3a and C5a and corresponding loss of C5aR on neutrophils have been described in several human studies (62–64). Of note, the reduction of C5aR1 and C5aR2 on neutrophils has been correlated with the occurrence of infectious complications in ICU patients (30) and sepsis-induced MODS (62, 65, 66). Since loss of C5aR1 and C5aR2 has been concurrently correlated to the sequential organ failure assessment (SOFA) score (65), a flow-based rapid testing, might have a bedside monitoring potential to predict infectious problems and MODS development.
MODS—Pathomechansisms Caused by Multiple Complement Dysfunctions
Several pathomechanisms contribute to the development of MODS, such as enhanced levels of DAMPs and PAMPs, reduced cytochrome P450 metabolism, macrophage activation syndrome, and cytokine-driven cellular dysfunction (67). Overall, it is clear that complement dysregulation contributes to MODS after trauma (Figure 2). Some of these aspects have been already mentioned and will be further discussed in this section.
FIGURE 2
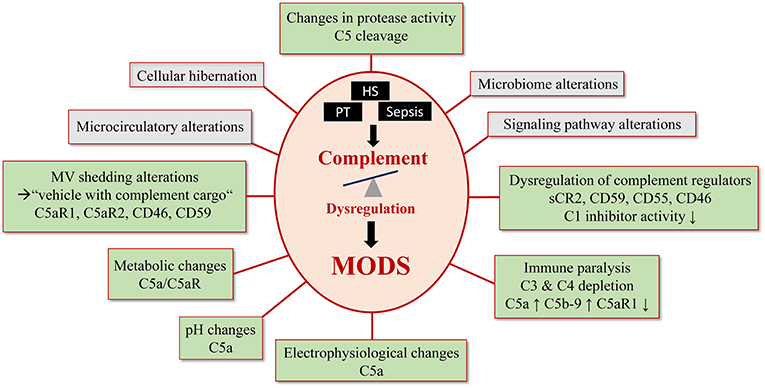
Figure 2. Complement- mediated pathomechanisms in MODS development. Polytrauma, sepsis and hemorrhagic shock cause a critical complement dysregulation, which causes dysfunction in multiple organs. This figure summarizes potential and known pathomechansims for MODS development caused by complement dysregulation. Established complement involvement is highlighted in light green and proposed complement involvement is highlighted in gray.
Immune Paralysis
Though severely injured patients receive modern ICU management, many of them show signs of immunosuppression known as persistent inflammation-immunosuppressive catabolism syndrome (PICS) (Figure 1) (68). Clinically, PICS patients suffer from persistent inflammation, immune suppression and protein catabolism, which can lead to recurrent nosocomial infections with sepsis, MODS and death (68). Severe immune suppression of the fluid-phase and cellular immune response has been proposed as “immune paralysis” of the host response to sterile and infectious insults. In a clinical case report, an inadequate response to infection with signs of systemic depletion of complement (dropping C3 and C4 levels) has been associated with the development of acute kidney injury and multiple organ failure in a 17-day old newborn (69). C5b-9 and C5a have been described as contributors to cell death, immune paralysis, cardiac dysfunction, and multiple organ failure (Figure 2) (49, 70). In support, a baboon model of Escherichia coli sepsis showed that blockade of C5 protected organs from “immune paralysis” and improved the sepsis survival rate (Table 1) (75).
TABLE 1
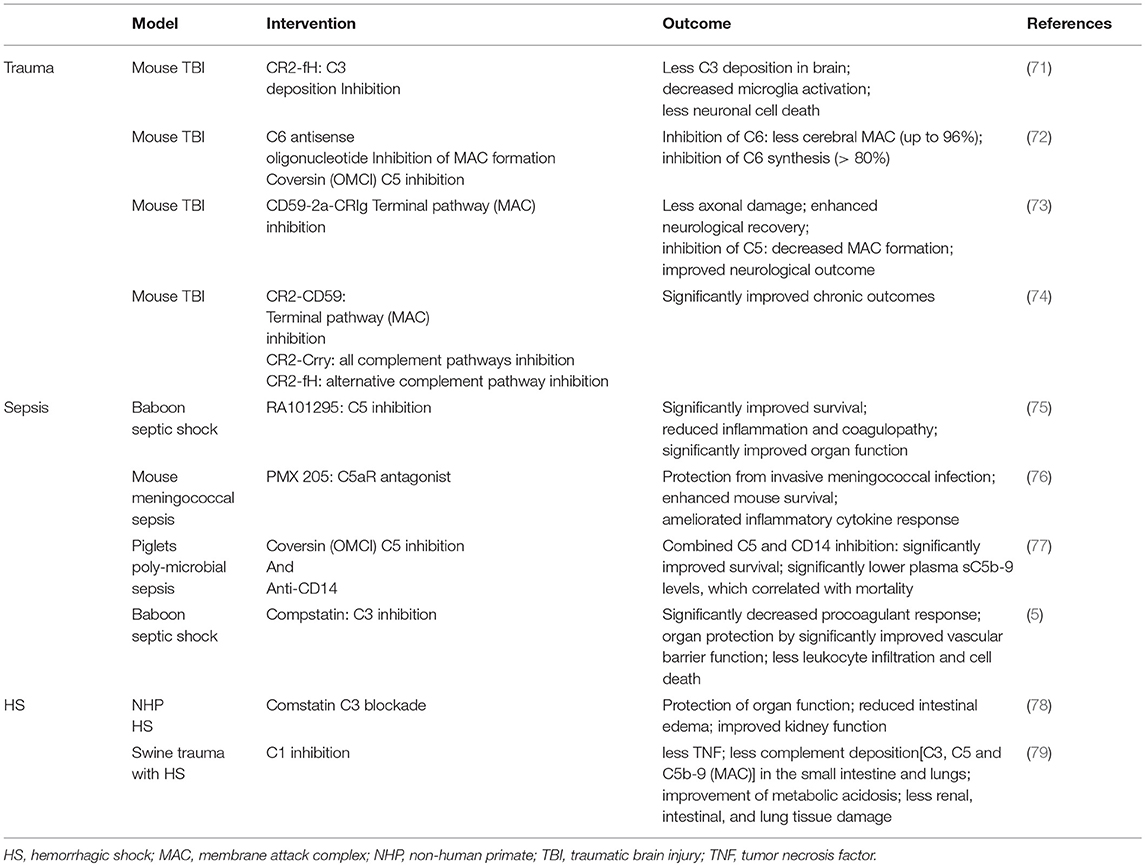
Table 1. Most representative preclinical studies addressing trauma, HS and sepsis, respectively.
Dysregulated Complement Regulators
Additional connection between MODS and signs of complementopathy are supported by the fact that soluble and membrane-bound regulators of complement activity show alterations after trauma, sepsis and hemorrhagic shock. It is well-established that severe tissue injury causes an excessive systemic intravascular activation of the complement system resulting in a loss over the control mechanisms (8). In this context, the soluble form of the complement receptor 2 (sCR2) was shown to be present after nerve injury in rodents (80). After polytrauma in humans, leukocyte expression profiles of the complement regulators (CRegs) CD55 (decay accelerating factor), CD59 (membrane attack complex inhibitor), CD46 (membrane cofactor protein), and CD35 (complement receptor 1) were cell-type- and time-dependently altered, reflecting manifestation of posttraumatic complementopathy (4, 29). Furthermore, an observational study including critically ill patients with multiple injuries and sepsis revealed that trauma patients with MODS have significantly lower C1-inhibitor activities (Figure 2) (7). A rare mutation in the complement regulatory gene factor H has been also implicated with a severe form of complement-mediated hemolytic uremic syndrome with multiple organ involvement, showing the general importance of complement regulation for organ (dys)functions (81).
Alteration of Signaling Pathway Activity
Experimental studies showed an interplay of complement with specific signaling pathways including the activation of PKC, MAPKs, and ERK (82). In a non-traumatic renal damage model, NF-κB contributed to enhanced complement activation (83), while in a similar mouse model the C5a/C5aR axis activated MAPK signaling (84). During sepsis, the activation of MAPKs and Akt signaling was complement-induced (51). These mechanistic insights require further elaboration to identify other participating mediators downstream to complement activation in trauma and hemorrhagic shock.
Changes in Protease Activity
Excessive systemic activation of proteases feed in the pathomechanisms of MODS and is known to create a vicious cycle of complement activation. Such proteases include immunoreactive trypsins and neutrophil elastase, which may directly interact with complement and have been extensively described in the case of polytrauma (9). A specific subtype of MODS has been introduced as thrombocytopenia-associated MODS, with low ADAMTS13 activity and defects in inhibitory complement regulators which may result in hyperactivation of coagulation and complement with resultant thrombosis (85). This form of MODS has been successfully treated by C5 blockade (eculizumab) and ADMTS13 reduction by plasma exchange (67). Furthermore, an unspecific and unregulated protease hyperactivity may directly cleave various complement components in a non-canonical manner, which represents a promising future research field for drug development. In this case, the intestine plays an important role in trauma and during shock, which can proceed to a proposed autodigestion phenomenon, since digestive enzymes from the pancreas are activated by enterokinases (86). Under physiological conditions, the auto-digestion is prevented by epithelial and mucosal barriers. However, during shock digestive peptides can pass the mucus and reach the epithelial cell membranes, where they disrupt junctional complexes, further activate complement and contribute barrier and organ dysfunction (87).
Microcirculatory Alteration
Patients who succumbed to traumatic-hemorrhagic shock showed an impaired microcirculation for at least 72 h, which also was a reliable predictor for a high Sequential Organ Failure Assessment (SOFA) score (88). In an ischemia rat model, application of soluble complement receptor type 1 significantly improved microvascular perfusion in the liver assessed by in vivo microscopy, suggesting an essential role of complement in microcirculatory disorders (89). Activated complement with microvascular alterations can also cause a disruption of cellular barriers leading to edema formation in lung, brain, and liver (84, 90, 91). However, hypothetical complement-caused damage of specific tight junction molecules in specific organs needs still scientific verification. Such mechanisms of MODS development may also be supported by simultaneous activation of complement and coagulation e.g., by complement-dependent generation of thrombin, which is known to efficiently break down various endothelial barriers. Furthermore, complement hyperactivation has been associated with thrombotic microangiopathies causing endothelial cell activation and thrombus formation leading to hemolytic anemia, thrombocytopenia, and organ failure (91).
Metabolic Changes
Besides microcirculatory problems, massive blood loss due to hemorrhage can cause acute circulatory failure resulting in lactic acidosis. Acute liver or renal dysfunctions are most often associated with decreased lactate clearance and a pronounced increase in blood lactate levels compared with shock patients without signs of liver or renal dysfunction (92). It is also known that shock states initiate a pronounced compensatory vasoconstriction, consequently leading to hypoxia and accumulation of metabolites associated with lactate acidosis and a low pH milieu (92, 93). In this context, complement dysfunction has a deleterious influence and correlates with a worse outcome after shock. Especially the anaphylatoxin C5a and its following signaling via the C5aR1 have been described to further enhance acidosis after septic shock. More precisely, in vitro stimulation of neutrophils from healthy donors with C5a causes C5aR1 signaling-mediated immunometabolic changes in neutrophils with an enhanced glucose uptake and enhanced glycolytic flux (Figure 2) (94). In turn, it can initiate an increased proton secretion and further lower the extracellular pH. In response to C5a exposure, intracellular pH in neutrophils significantly increases via activation of the Na+/H+ exchanger type 1 (NHE-1) leading to an impairment in neutrophil function in vitro. Supporting this evidence, neutrophils isolated from septic patients has been shown to exhibit an increased intracellular pH compared to healthy donors (94). These recent findings indicate that inflammatory processes with produced C5a is solely capable to significantly change the micro-milieu with lactate acidotic features even in the absence of an oxygen deficit. It only can be speculated on, that in the case of an additional oxygen deficit, caused by shock conditions and vascular dysfunction, the anaphylatoxins may even function as a metabolic switch toward lactate acidosis and MODS.
Microbiome Alterations
The intestinal complement is suggested to cooperate in a close relationship with the gut microbiome (95). Therefore, another contributor to MODS seems to be the alteration of the gut microbiome after trauma/hemorrhagic shock and/or sepsis. The microbiome, which is described by the phylogenetic composition and taxon relative abundance of the bacteria, is significantly altered in the first 72 h after injury. This rapid change in intestinal microbiota represents a critical phenomenon that may influence outcomes after severe trauma (15). The composition of the microbiome may influence the activation/dysregulation of complement pathways or dysregulated complement may change the microbiome composition. In the skin, complement activation modulates the inflammatory milieu by changing the cutaneous microbiota (96). Considering MODS, a mechanistic explanation for complement-microbiome interaction still remains elusive and needs further research.
Alterations in Microvesicle (MV) Shedding
Communication is essential for cellular homeostasis and vesicle shedding has been described to play a crucial role for maintaining proper immune cell function. Extracellular vesicle shedding is altered after inflammation and is considered as a crucial contributor to MODS after multiple injury and sepsis (97). Especially shedding of microvesicles (MV) have been implicated in several inflammatory conditions including sepsis and trauma. Increased amounts of CD41+ and CD31+/CD41–/AnnexinV– MV after sepsis, released by activated platelets and leukocytes have been shown to correlate with unfavorable outcomes (98).
Furthermore, MVs from patients with multiple organ failure support the coagulation system in triggering inflammation. In respect to complement, phosphatidylserine containing MVs also serve as platform for complement activation (99). Besides activation of complement on their surfaces, MVs represent transport vehicles sending complement as cargo to neighboring as well as cells in distance (99). Hence, MV from different cellular origins may contain complement receptors including C5aR but also CRegs such as CD46 and CD59, suggesting a putative role for complement activity (99). In accordance with this, loss of C5aR1, C5aR2, and C3aR on neutrophils after multiple injury was clinically present and was correlated to infectious complications and multiple organ dysfunction (Figure 2) (18).
Electrophysiological Changes
Another concurrent theory of MODS addresses electrophysiological changes of the cellular membrane which have recently been found in neutrophils from septic pigs (14). The anaphylatoxin C5a was able to alter the membrane potential of neutrophils but not in the case of neutrophils during septic MODS where the electrophysiological response to C5a was somehow frozen (Figure 2) (14). Whether complement inhibitory strategies will stabilize the cellular membrane electrophysiology is currently under investigation.
Cellular Hibernation
Trauma causes an alarming stress situation for the whole body with an extensive inflammatory response (9). Some studies indicate that trauma especially followed by additional sepsis causes hibernation in the cellular as well as fluid phase of innate immunity including the complement system. Reflecting the evolved habit of conserving physiological resources in the event of environmental stress, with inflammation ensues a similar mechanism where energy-consuming processes are shut down in the organism. Supporting this evidence, hibernation has been observed in the septic heart with ongoing metabolic changes including the upregulation of specific glucose transporters in cardiomyocytes (100). Besides its effects on metabolism, hibernation is known to affect various immune function including leukocyte migration, as well as adaptive immune responses and interestingly complement function, by lowering complement levels and reduced expression of C3 mRNA in the liver, which depicts a suggestive link post-shock. (101). However, another study demonstrated that hibernators are protected from shock-induced injury, inflammation, and organ function (102). Strikingly, arctic ground squirrels challenged with cardiac arrest or hemorrhagic shock showed no markers of organ damage, systemic inflammation, or loss of acid/base balance as indicated by a negative base excess. Neither reduced body temperature nor hibernation season are components of this protection, indicating still unknown mechanisms involved (102).
Unfortunately, no supporting data is available indicating that future research on the complement function during hibernation, especially following trauma and shock is needed. Further, the ability to induce a fully reversible state of immune suppression in humans by artificial hibernation might aid the treatment of several inflammatory and immune-mediated diseases.
Targeting Complement Pathways in MODS
Despite improvements in trauma care, the morbidity and mortality of MODS remains very high.
Therefore, new therapeutic strategies are urgently needed. Since complement is critically involved in initiation and progression of MODS, targeting complement as well as molecules contributing to complement activation represent promising future clinical approaches. Other complement-associated inflammatory conditions already addressed such a targeting strategy. Above 20 complement-interfering drugs have been evaluated in clinical settings so far. A few of them received FDA approval for inflammatory indications including eculizumab targeting the terminal complement pathway starting from C5, which is currently used for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) (103). As existing complement therapeutics cannot target every complement-driven disease status, it is a requisite to evaluate the different complement stages, their relevance, and target them in a disease-specific manner (10).
In the context of trauma, hemorrhagic shock and sepsis a few preclinical experimental studies focus on complement-targeting studies (Table 1). Besides complement activation products, other molecules including CRP, HMGB1, or mitochondrial DNA play crucial roles in contributing to complement dysregulation after sepsis and trauma, thus profoundly influencing secondary outcomes. Therefore, a rational approach seems to address molecules, which may further boost complement activity. Likewise, HMGB1 is a relevant danger molecule and complement has been described to regulate HMGB1 release from human neutrophils (104) such that, complement inhibition has proved to be protective in blast-induced acute lung injury in rats by ameliorating HMGB1-mediated inflammation (105).
Targeting Complement After (poly)Trauma
In polytrauma patients, the only study of complement intervention early after trauma with RCT quality used C1 esterase inhibitor. However, this study has been terminated based on the heterogeneity of the patients (106). Experimentally, the majority of the complement targeting studies were performed in mouse traumatic brain injury (TBI) models. In this case, various interventions to inhibit complement activity, such as inhibitors for MAC formation and C3 deposition inhibitors have been applied, analyzed and reviewed elsewhere (9, 107, 108). To further clarify the impact of distinct complement activation pathways on neuroinflammation, a recent study compared the effect of complement inhibitors targeting upstream as well as downstream complement activity, respectively. This study brought into light that instead of downstream complement activation complexes, upstream complement activation products of the alternative pathway predominantly modulate and propagate chronic inflammation (74) (Table 1).
Targeting Complement in Hemorrhagic Shock
Based on the deduced potential of complement inhibitors to improve micro-perfusion disturbances, ischemia/reperfusion injury and the inflammatory response during hemorrhagic shock, various shock models have been tested for possible benefits of complement interventions. In a rat model of pressure controlled hemorrhagic shock, complement depletion by CVF improved the recovery of the mean arterial pressure (MAP) post shock (109). In a pig model of pressure controlled hemorrhagic shock, application of a C1-inhibitor improved the functional performance and reduced C3 deposition on a multi-organ level including liver, small intestine and lungs (Table 1) (79). Limitation of organ injury and sustained survival by C1 inhibitor therapy has also been reported for a porcine injury model mimicking battlefield injury (110). Further downstream, C3 deficient mice with bilateral femur fracture and hemorrhagic shock resulted in protective effects with reduced circulating DAMPs (e.g., dsDNA), decreased systemic inflammatory response and improved organ performance (e.g., liver enzymes) (111). In a rat model of hemorrhagic shock, C3 depletion by CVF, and the soluble form of CR1 to inhibit C3 action restored vascular reactivity to norepinephrine in the superior mesenteric artery (112). In the clinical setting, C3 inhibition has not been tested in hemorrhagic shock so far except in a recent study from our group, where non-human primates were modeled with severe hemorrhagic shock of 30 mmHg MAP for 1 h, and a delayed application of CP40 was tested (78). C3 blockade by the compstatin compound CP40 seems most promising since there was evidence of improved renal function, attenuated intestinal edema and reduced signs of systemic inflammation and coagulopathy (78). However, the transfer into clinical reality is pending although C3 inhibition seems rather safe in the case of hemorrhagic shock. Most likely, an early inhibition to avoid excessive C3 activation might be enough to improve the outcome (113).
On a C5 level, intestinal injury caused by hypovolemic shock in mice was ameliorated by application of a small peptide C5aR1 antagonist and C5 deficient littermates (114). Similarly, in a rodent model of ruptured aortic abdominal aneurysma with shock, C5 blockade by an anti-C5 antibody revealed some protective effects on remote lung injury with improvement of the bronchoalveolar permeability and myeloperoxidase (MPO) concentrations reflecting reduced neutrophil recruitment and activation (115). Combined inhibition of the complement component C5 and the Toll-like receptor co-factor CD14 in a porcine sepsis model, showed beneficial effects in regard to survival, hemodynamic parameters and systemic inflammation including complement activation (Figure 1, Table 1). First preclinical studies in non-human primates indicate appearance of sC5b-9 in the serum of traumatic-hemorrhagic shock and ex vivo effectiveness of C5 inhibitors in prevention of hemolysis, e.g., the small, inhibitory peptide RA101348, commercially-available C5 inhibitory antibodies and Quidel's A217 antibody (106). However, a RCT-clinical study with C5 inhibitory strategies in hemorrhagic shock has not been performed so far most likely based on the rareness and uncertain evidence of a great impact on MODS development in the so far performed preclinical non-human primate studies. Therefore, research providing information about the histological and biochemical data on the multi-organ level in non-human primate studies is necessary for final assessment.
Targeting Complement in Sepsis
In the case of septic shock, targeting the C5a/C5aR axis seems to be of practical importance since it correlated with the disease severity and mortality and showed promising improvements in different sepsis models (Table 1). Concerning complement-targeted therapies in sepsis, application of RA101295, a 2-kDa macrocyclic peptide inhibitor of C5 cleavage, improved organ performance and survival in an E. coli-sepsis model of baboons (75) with lessened evidence of coagulopathy and preserved endothelial and barrier functions. Furthermore, the inhibition of C5 cleavage lead to improved histomorphology of lungs, liver, kidneys, spleen, and adrenal glands suggesting improvement of sepsis-induced MODS (75).
In the same sepsis model, systemic blockade of C3 by compstatin also revealed organ protection on multiple levels. Compstatin showed evidence of sepsis-induced coagulopathy and preserved anti-coagulatory features of the endothelium. Furthermore, C3 blockade improved hemodynamics and heart function and biochemical damage markers of the kidney and liver, indicating protective effects in sepsis-induced MODS (5). For blocking the central complement component C3 during development of sepsis-caused MODS, clinical trials might be safe (113) but these trials are pending and require a special focus on the benefits or maladies for mental alterations during sepsis.
In terms of applicability in the clinical setting, it has been recently shown that blocking solely C5 activation by canonical C5-convertase (e.g., by eculizumab) might not be specific enough since other serine proteases such as trypsin or thrombin can still cleave and activate C5. Therefore, a C5a blocking approach e.g., by IFX-1 anti-C5a antibody has been proposed as a targeted approach in local or systemic infection (116, 117). Overall, the specific complement targets for sepsis-induced MODS might be different than for polytrauma- or hemorrhagic shock-caused MODS especially when given early (Figure 1). The complement target might also change along the course of the disease. It is for example feasible, that early after polytrauma or during hemorrhagic shock a specific C3 inhibition is required, whereas later during development of septic complications, C5a inhibition might help against MODS development (Figure 1). And if complete depletion of a specific complement factor occurs, it might be even wise to replace it, which bolsters the importance of monitoring complement for any clinical trial.
In conclusion, MODS is a culmination of highly heterogeneous events which can involve different and complex stages of complement activation, making it nearly impossible to generate a drug addressing MODS generally. More precisely, the most prominent complement pathways and their down-stream signaling pathways involving multiple trauma with or without hemorrhagic shock and septic shock may be addressed in further preclinical studies.
Clinical Trials
Considering clinical trials of MODS caused by polytrauma, hemorrhagic shock and sepsis, three observational studies addressed complement activation after polytrauma and severe abdominal sepsis (Table 2). One study focused on the complement activation and expression on leukocytes in polytrauma patients revealing increased complement activity early after trauma and an increased shedding of complement receptors from neutrophil surfaces (NTC00710411; Table 2) (2, 62). Another observational study focuses on the molecular danger response after polytrauma. It is still recruiting patients, and the study aims to include up to 1,000 patients in a collective national data bank in collaboration with the Trauma Research Network (NTF) of the German Society for Orthopedics and Trauma (DGOU) and thus to obtain novel insight into the molecular pattern of complement activity following severe multiple injury (NTF-PT; NCT02682550; Table 2).
TABLE 2
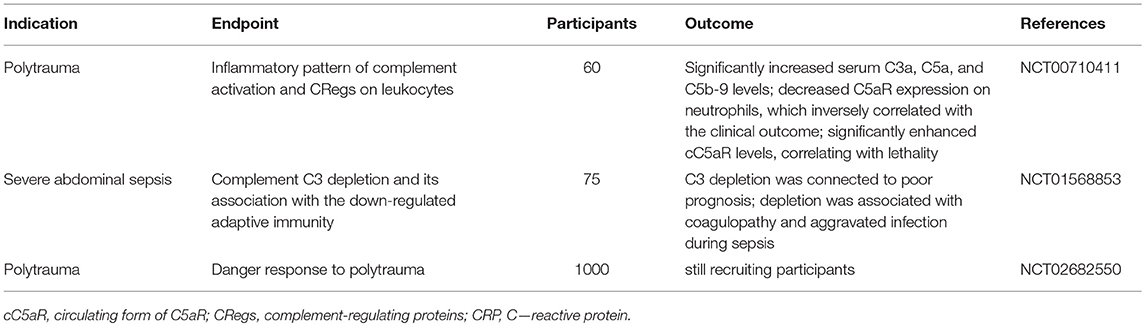
Table 2. Overview of observational clinical trials evaluating complement activation after polytrauma and sepsis.
A further observational study has set its focus on the central complement C3 by investigation of C3 alterations in patients with severe abdominal sepsis. The aim was to evaluate the impact of C3 on patients' prognosis (NCT01568853; Table 2). This study revealed that C3 depletion is associated with a poor prognosis due to dysregulated coagulation and increased susceptibility for infections (118).
In regard to human studies, three interventional trials have been initiated so far (Table 3). One study addressed the inhibition of upstream complement component (C1) by targeting C1 esterase. This mono-centered double-blind randomized placebo-controlled trial (CAESAR; NCT01275976, Table 3) has been designed using a C1-esterase inhibitor in severely injured patients with femur fracture. Although the rationale of a complement blockade on this level is very well founded because it may also reveal beneficial effects by synchronically inhibition of excessive activation of the coagulation pathway (119), the study has been terminated based on the heterogeneity of patients and challenges in recruitment. A further study has addressed the effect of the C1-esterase inhibitor on human endotoxemia by evaluating its effect on inflammation and marker of organ dysfunction (VECTOR; NCT00785018, Table 3). Detailed results of the study are still pending.
TABLE 3

Table 3. Overview of complement therapeutics for clinical trials.
Another clinical trial has been designed and performed to study complement inhibition in early, newly developing septic organ dysfunction (SCIENS; NCT02246595, Table 3) applying a monoclonal antibody against C5a, though the detailed results of this trial have not been published so far. Regarding one cardinal clinical sign of sepsis, the occult or evident alterations of the mental status, inhibitory strategies against C5a might reveal “Janus faced” effects (120). On one hand, C5a inhibition could improve sepsis-impaired blood-brain-barrier, on the other hand, neuroprotective effects by C5a might be compromised (120, 121). Therefore, alterations of the mental status need to be carefully addressed and monitored in any clinical trial using C5a inhibitory strategies.
It is important to note, that it is in general rather difficult to perform interventional studies on polytrauma patients since an informed written consent cannot be provided and the legal representatives are usually difficult to determine within the first hours after severe injury. Therefore, innovative studies early after polytrauma addressing the complement cascade are rather rare and have not been performed on the C3 or C5 level yet.
Clinical Perspectives
Although various clinically relevant models of trauma, hemorrhagic shock and sepsis have been tested already in non-human primates for the benefit of complement interventions (75, 78, 106), clinical trials in these multidimensional pathophysiologic conditions remain rare (119, 122). When complement intervention strategies are designed for the clinics, the targeted complement factor or activation product needs to be measured before the therapy can be applied. Especially when complex intensive care is necessary which can alter complement levels within a short time period, e.g., by infusion of blood products which contain highly variable concentrations of complement (activation) factors (123) or by extra-corporal circulation devices with large artificial surfaces which may deplete key complement components (124, 125), the exact status of complement activation needs to be determined. This would allow precise and timely intervention either by inhibiting or supporting the complement response after trauma or during sepsis in order to rebalance the immune response. Whereas, various highly effective and specific complement intervention strategies have been developed within the last two decades and are available now (10), in the context of the complex immune response after trauma, hemorrhagic shock and sepsis (9, 126), specific organ damage and function assessment including the immune function at bedside seems far beyond. Therefore, functional monitoring of the organ and immune systems can be considered as a prerequisite before complement interventions move into clinical routine in diseases with a complex pathophysiology. In conclusion, further scientific knowledge and translational efforts are demanded for targeting complement pathways in the setting of trauma, hemorrhagic shock and sepsis with the aim to offer causal therapy and improved outcome.
Author Contributions
EK and MH-L wrote the manuscript. All authors edited and commented on the manuscript. All authors read and approved the final manuscript.
Funding
This review was supported by grants from the German Research Foundation (DFG) (EI 866/5-1) and by grants from the DFG within the Collaborative Research Center CRC1149/2 Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma to MH-L.
Conflict of Interest Statement
JL is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs, such as AMY-101) and is the inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. JL is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals [i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives].
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CReg, complement regulator; DAMP, danger-associated molecular pattern; HMOX1, heme-oxygenase-1; HS, hemorrhagic shock; MAC, membrane attack complex; MODS, multiple organ dysfunction; MV, microvesicle; NHP, non-human primate; PAMP, pathogen-associated molecular patterns; PICS, persistent inflammation, immunosuppression and catabolism syndrome; PICS, persistent inflammation, immunosuppression and catabolism syndrome; PMN, polymorphonuclear leukocyte; sCR2, soluble complement receptor 2; TNF, tumor necrosis factor.
References
1. Fosse E, Mollnes TE, Aasen AO, Trumpy JH, Stokke T. Complement activation following multiple injuries. Acta Chir Scand. (1987) 153:325–30.
2. Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early complementopathy after multiple injuries in humans. Shock. (2012) 37:348–54. doi: 10.1097/SHK.0b013e3182471795
3. Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, et al. Role of the alternative pathway in the early complement activation following major trauma. Shock. (2007) 28:29–34. doi: 10.1097/shk.0b013e3180342439
4. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
5. Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. (2010) 116:1002–10. doi: 10.1182/blood-2010-02-269746
6. Hecke F, Schmidt U, Kola A, Bautsch W, Klos A, Kohl J. Circulating complement proteins in multiple trauma patients–correlation with injury severity, development of sepsis, and outcome. Crit Care Med. (1997) 25:2015–24. doi: 10.1097/00003246-199712000-00019
7. Helling H, Stephan B, Pindur G. Coagulation and complement system in critically ill patients. Clin Hemorheol Microcirc. (2015) 61:185–93. doi: 10.3233/CH-151993
8. Rittirsch D, Redl H, Huber-Lang M. Role of complement in multiorgan failure. Clin Dev Immunol. (2012) 2012:962927. doi: 10.1155/2012/962927
9. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. (2018) 19:327–41. doi: 10.1038/s41590-018-0064-8
10. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
11. Cavaillon JM, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. (2006) 12:151–70. doi: 10.1179/096805106X102246
12. Cavaillon JM, Giamarellos-Bourboulis EJ. << Immunosuppression >> is inappropriately qualifying the immune status of septic and sirs patients. Shock. (2018). doi: 10.1097/SHK.0000000000001266. [Epubh ahead of print].
13. Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, et al. Increased C5a receptor expression in sepsis. J Clin Invest. (2002) 110:101–8. doi: 10.1172/JCI0215409
14. Messerer DAC, Denk S, Fohr KJ, Halbgebauer R, Braun CK, Hones F, et al. Complement C5a alters the membrane potential of neutrophils during hemorrhagic shock. Mediators Inflamm. (2018) 2018:2052356. doi: 10.1155/2018/2052356
15. Howard BM, Kornblith LZ, Christie SA, Conroy AS, Nelson MF, Campion EM, et al. Characterizing the gut microbiome in trauma: significant changes in microbial diversity occur early after severe injury. Trauma Surg Acute Care Open. (2017) 2:e000108. doi: 10.1136/tsaco-2017-000108
16. Graetz TJ, Hotchkiss RS. Sepsis: preventing organ failure in sepsis - the search continues. Nat Rev Nephrol. (2017) 13:5–6. doi: 10.1038/nrneph.2016.171
17. Coopersmith CM, De BD, Deutschman CS, Ferrer R, Lat I, Machado FR, et al. Surviving sepsis campaign: research priorities for sepsis and septic shock. Crit Care Med. (2018) 46:1334–56. doi: 10.1097/CCM.0000000000003225
18. Chakraborty S, Karasu E, Huber-Lang M. Complement after trauma: suturing innate and adaptive immunity. Front Immunol. (2018) 9:2050. doi: 10.3389/fimmu.2018.02050
19. van Wessem KJP, Leenen LPH. Reduction in mortality rates of postinjury multiple organ dysfunction syndrome: a shifting paradigm? a prospective population-based cohort study. Shock. (2018) 49:33–8. doi: 10.1097/SHK.0000000000000938
20. Roumen RM, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, et al. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Crit Care Med. (1995) 23:474–80. doi: 10.1097/00003246-199503000-00010
21. Bautsch W, Hoymann HG, Zhang Q, Meier-Wiedenbach I, Raschke U, Ames RS, et al. Cutting edge: guinea pigs with a natural C3a-receptor defect exhibit decreased bronchoconstriction in allergic airway disease: evidence for an involvement of the C3a anaphylatoxin in the pathogenesis of asthma. J Immunol. (2000) 165:5401–5. doi: 10.4049/jimmunol.165.10.5401
22. Asgari E, Le FG, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
23. Ehrnthaller C, Flierl M, Perl M, Denk S, Unnewehr H, Ward PA, et al. The molecular fingerprint of lung inflammation after blunt chest trauma. Eur J Med Res. (2015) 20:70. doi: 10.1186/s40001-015-0164-y
24. Rittirsch D, Schoenborn V, Lindig S, Wanner E, Sprengel K, Gunkel S, et al. An integrated clinico-transcriptomic approach identifies a central role of the heme degradation pathway for septic complications after trauma. Ann Surg. (2016) 264:1125–34. doi: 10.1097/SLA.0000000000001553
25. Abdelbaset-Ismail A, Borkowska-Rzeszotek S, Kubis E, Bujko K, Brzezniakiewicz-Janus K, Bolkun L, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. Leukemia. (2017) 31:446–58. doi: 10.1038/leu.2016.198
26. May O, Merle NS, Grunenwald A, Gnemmi V, Leon J, Payet C, et al. Heme drives susceptibility of glomerular endothelium to complement overactivation due to inefficient upregulation of heme oxygenase-1. Front Immunol. (2018) 9:3008. doi: 10.3389/fimmu.2018.03008
27. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. (2013) 33:479–92. doi: 10.1016/j.semnephrol.2013.08.001
28. Rittirsch D, Schoenborn V, Lindig S, Wanner E, Sprengel K, Gunkel S, et al. Improvement of prognostic performance in severely injured patients by integrated clinico-transcriptomics: a translational approach. Crit Care. (2015) 19:414. doi: 10.1186/s13054-015-1127-y
29. Amara U, Kalbitz M, Perl M, Flierl MA, Rittirsch D, Weiss M, et al. Early expression changes of complement regulatory proteins and C5A receptor. (CD88) on leukocytes after multiple injury in humans. Shock. (2010) 33:568–75. doi: 10.1097/SHK.0b013e3181c799d4
30. Morris AC, Brittan M, Wilkinson TS, McAuley DF, Antonelli J, McCulloch C, et al. C5a-mediated neutrophil dysfunction is RhoA-dependent and predicts infection in critically ill patients. Blood. (2011) 117:5178–88. doi: 10.1182/blood-2010-08-304667
31. Morganroth ML, Schoeneich SO, Till GO, Ward PA, Horvath SJ, Glovsky MM. C3a57-77, a C-terminal peptide, causes thromboxane-dependent pulmonary vascular constriction in isolated perfused rat lungs. Am Rev Respir Dis. (1990) 141:296–300. doi: 10.1164/ajrccm/141.2.296
32. Crowell RE, Van Epps DE, Reed WP. Responses of isolated pulmonary arteries to the C5a anaphylatoxin. Am J Physiol. (1990) 259(5 Pt 2):H1325–9. doi: 10.1152/ajpheart.1990.259.5.H1325
33. Fortin JP, Bouthillier J, St-Pierre SA, Marceau F. Contractile effect of anaphylatoxin C5a and of a mimetic peptide on the human umbilical artery: further evidence for leukocyte-dependent vasomotion. J Cardiovasc Pharmacol. (2002) 40:815–21. doi: 10.1097/00005344-200212000-00002
34. Gulbins E, Schlottmann K, Rauterberg EW, Steinhausen M. Effects of rC5a on the circulation of normal and split hydronephrotic rat kidneys. Am J Physiol. (1993) 265(1 Pt 2):F96–103. doi: 10.1152/ajprenal.1993.265.1.F96
35. Lundberg C, Marceau F, Hugli TE. C5a-induced hemodynamic and hematologic changes in the rabbit. Role of cyclooxygenase products and polymorphonuclear leukocytes. Am J Pathol. (1987) 128:471–83.
36. Rendig SV, Gray S, Amsterdam EA. Contractile actions of C5a on isolated porcine coronary resistance and conductance arteries. Am J Physiol. (1997) 272(1 Pt 2):H12–6. doi: 10.1152/ajpheart.1997.272.1.H12
37. Schumacher WA, Fantone JC, Kunkel SE, Webb RC, Lucchesi BR. The anaphylatoxins C3a and C5a are vasodilators in the canine coronary vasculature in vitro and in vivo. Agents Actions. (1991) 34:345–9. doi: 10.1007/BF01988727
38. Marceau F, Hugli TE. effect of C3a and C5a anaphylatoxins on guinea-pig isolated blood vessels. J Pharmacol Exp Ther. (1984) 230:749–54. doi: 10.1016/0162-3109(84)90019-5
39. Collard CD, Lekowski R, Jordan JE, Agah A, Stahl GL. Complement activation following oxidative stress. Mol Immunol. (1999) 36:941–8. doi: 10.1016/S0161-5890(99)00116-9
40. Wrba L, Ohmann JJ, Eisele P, Chakraborty S, Braumuller S, Braun CK, et al. Remote intestinal injury early after experimental polytrauma and hemorrhagic shock. Shock. (2018). doi: 10.1097/SHK.0000000000001271. [Epubh ahead of print].
41. Halbgebauer R, Braun CK, Denk S, Mayer B, Cinelli P, Radermacher P, et al. Hemorrhagic shock drives glycocalyx, barrier and organ dysfunction early after polytrauma. J Crit Care. (2018) 44:229–37. doi: 10.1016/j.jcrc.2017.11.025
42. Frith D, Goslings JC, Gaarder C, Maegele M, Cohen MJ, Allard S, et al. Definition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J Thromb Haemost. (2010) 8:1919–25. doi: 10.1111/j.1538-7836.2010.03945.x
43. Rahbar E, Cardenas JC, Baimukanova G, Usadi B, Bruhn R, Pati S, et al. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J Transl Med. (2015) 13:117. doi: 10.1186/s12967-015-0481-5
44. Szebeni J, Baranyi L, Savay S, Gotze O, Alving CR, Bunger R, et al. Complement activation during hemorrhagic shock and resuscitation in swine. Shock. (2003) 20:347–55. doi: 10.1097/01.shk.0000082444.66379.17
45. Fang JF, Shih LY, Yuan KC, Fang KY, Hwang TL, Hsieh SY. Proteomic analysis of post-hemorrhagic shock mesenteric lymph. Shock. (2010) 34:291–8. doi: 10.1097/SHK.0b013e3181ceef5e
46. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM consensus conference committee. American college of chest physicians/society of critical care medicine. Chest. (1992) 101:1644–55. doi: 10.1378/chest.101.6.1644
47. Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:775–87. doi: 10.1001/jama.2016.0289
48. Dickmann P, Scherag A, Coldewey SM, Sponholz C, Brunkhorst FM, Bauer M. [Epistemology in the intensive care unit-what is the purpose of a definition?: paradigm shift in sepsis research]. Anaesthesist. (2017) 66:622–5. doi: 10.1007/s00101-017-0315-3
50. Gerard C. Complement C5a in the sepsis syndrome–too much of a good thing? N Engl J Med. (2003) 348:167–9. doi: 10.1056/NEJMcibr022995
51. Fattahi F, Kalbitz M, Malan EA, Abe E, Jajou L, Huber-Lang MS, et al. Complement-induced activation of MAPKs and Akt during sepsis: role in cardiac dysfunction. FASEB J. (2017) 31:4129–39. doi: 10.1096/fj.201700140R
52. Kalbitz M, Fattahi F, Herron TJ, Grailer JJ, Jajou L, Lu H, et al. Complement destabilizes cardiomyocyte function in vivo after polymicrobial sepsis and in vitro. J Immunol. (2016) 197:2353–61. doi: 10.4049/jimmunol.1600091
53. Huber-Lang MS, Riedeman NC, Sarma JV, Younkin EM, McGuire SR, Laudes IJ, et al. Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. (2002) 16:1567–74. doi: 10.1096/fj.02-0209com
54. Chinnaiyan AM, Huber-Lang M, Kumar-Sinha C, Barrette TR, Shankar-Sinha S, Sarma VJ, et al. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am J Pathol. (2001) 159:1199–209. doi: 10.1016/S0002-9440(10)62505-9
55. Flierl MA, Rittirsch D, Chen AJ, Nadeau BA, Day DE, Sarma JV, et al. The complement anaphylatoxin C5a induces apoptosis in adrenomedullary cells during experimental sepsis. PLoS ONE. (2008) 3:e2560. doi: 10.1371/journal.pone.0002560
56. Flierl MA, Schreiber H, Huber-Lang MS. The role of complement, C5a and its receptors in sepsis and multiorgan dysfunction syndrome. J Invest Surg. (2006) 19:255–65. doi: 10.1080/08941930600778263
57. Flierl MA, Rittirsch D, Nadeau BA, Day DE, Zetoune FS, Sarma JV, et al. Functions of the complement components C3 and C5 during sepsis. FASEB J. (2008) 22:3483–90. doi: 10.1096/fj.08-110595
58. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. (2008) 8:776–87. doi: 10.1038/nri2402
59. Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, et al. Role of C5a in multiorgan failure during sepsis. J Immunol. (2001) 166:1193–9. doi: 10.4049/jimmunol.166.2.1193
60. Hoehlig K, Maasch C, Shushakova N, Buchner K, Huber-Lang M, Purschke WG, et al. A novel C5a-neutralizing mirror-image (l-)aptamer prevents organ failure and improves survival in experimental sepsis. Mol Ther. (2013) 21:2236–46. doi: 10.1038/mt.2013.178
61. Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, et al. Protective effects of C5a blockade in sepsis. Nat Med. (1999) 5:788–92. doi: 10.1038/10512
62. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, et al. Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol. (2013) 190:4215–25. doi: 10.4049/jimmunol.1200534
63. Solomkin JS, Jenkins MK, Nelson RD, Chenoweth D, Simmons RL. Neutrophil dysfunction in sepsis. II. Evidence for the role of complement activation products in cellular deactivation. Surgery. (1981) 90:319–27.
64. Stove S, Welte T, Wagner TO, Kola A, Klos A, Bautsch W, et al. Circulating complement proteins in patients with sepsis or systemic inflammatory response syndrome. Clin Diagn Lab Immunol. (1996) 3:175–83.
65. Xu R, Lin F, Bao C, Huang H, Ji C, Wang S, et al. Complement 5a receptor-mediated neutrophil dysfunction is associated with a poor outcome in sepsis. Cell Mol Immunol. (2016) 13:103–9. doi: 10.1038/cmi.2014.136
66. Huber-Lang M, Sarma JV, Rittirsch D, Schreiber H, Weiss M, Flierl M, et al. Changes in the novel orphan, C5a receptor (C5L2), during experimental sepsis and sepsis in humans. J Immunol. (2005) 174:1104–10. doi: 10.4049/jimmunol.174.2.1104
67. Carcillo JA, Podd B, Aneja R, Weiss SL, Hall MW, Cornell TT, et al. Pathophysiology of pediatric multiple organ dysfunction syndrome. Pediatr Crit Care Med. (2017) 18(3 Suppl. 1):S32–45. doi: 10.1097/PCC.0000000000001052
68. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin. (2017) 33:245–58. doi: 10.1016/j.ccc.2016.12.001
69. Gaudreault-Tremblay MM, Litalien C, Patey N, Merouani A. Severe acute kidney injury and multiple organ failure in a 17-day-old newborn: when pathology makes the difference. Can J Kidney Health Dis. (2018) 5:2054358118804834. doi: 10.1177/2054358118804834
70. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
71. Rich MC, Keene CN, Neher MD, Johnson K, Yu ZX, Ganivet A, et al. Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci Lett. (2016) 617:188–94. doi: 10.1016/j.neulet.2016.02.025
72. Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. (2014) 192:2339–48. doi: 10.4049/jimmunol.1302793
73. Ruseva MM, Ramaglia V, Morgan BP, Harris CL. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci USA. (2015) 112:14319–24. doi: 10.1073/pnas.1513698112
74. Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. (2018) 38:2519–2532. doi: 10.1523/JNEUROSCI.2197-17.2018
75. Keshari RS, Silasi R, Popescu NI, Patel MM, Chaaban H, Lupu C, et al. Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of Escherichia coli sepsis. Proc Natl Acad Sci USA. (2017) 114:E6390–9. doi: 10.1073/pnas.1706818114
76. Herrmann JB, Muenstermann M, Strobel L, Schubert-Unkmeir A, Woodruff TM, Gray-Owen SD, et al. Complement C5a receptor 1 exacerbates the pathophysiology of N. meningitidis sepsis and is a potential target for disease treatment. MBio. (2018) 9(1). doi: 10.1128/mBio.01755-17
77. Skjeflo EW, Sagatun C, Dybwik K, Aam S, Urving SH, Nunn MA, et al. Combined inhibition of complement and CD14 improved outcome in porcine polymicrobial sepsis. Crit Care. (2015) 19:415. doi: 10.1186/s13054-015-1129-9
78. van GM, Ricklin D, Denk S, Halbgebauer R, Braun CK, Schultze A, et al. Protective effects of the complement inhibitor compstatin CP40 in hemorrhagic shock. Shock. (2019) 51:78–87. doi: 10.1097/SHK.0000000000001127
79. Dalle Lucca JJ, Li Y, Simovic M, Pusateri AE, Falabella M, Dubick MA, et al. Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock. (2012) 38:82–91. doi: 10.1097/SHK.0b013e31825a3522
80. Lindblom RP, Aeinehband S, Strom M, Al NF, Sandholm K, Khademi M, et al. Complement receptor 2 is increased in cerebrospinal fluid of multiple sclerosis patients and regulates C3 function. Clin Immunol. (2016) 166–167:89–95. doi: 10.1016/j.clim.2016.04.003
81. Yesilbas O, Sevketoglu E, Petmezci MT, Kihtir HS, Benzer M, Berdeli A. Hemolytic uremic syndrome with multiple organ involvement secondary to complement factor H p.Arg1215X mutation. Turk J Pediatr. (2017) 59:576–80. doi: 10.24953/turkjped.2017.05.011
82. Peng H, Takano T, Papillon J, Bijian K, Khadir A, Cybulsky AV. Complement activates the c-Jun N-terminal kinase/stress-activated protein kinase in glomerular epithelial cells. J Immunol. (2002) 169:2594–601. doi: 10.4049/jimmunol.169.5.2594
83. Liu M, Wang H, Zhang J, Yang X, Li B, Wu C, et al. NF-kappaB signaling pathway-enhanced complement activation mediates renal injury in trichloroethylene-sensitized mice. J Immunotoxicol. (2018) 15:63–72. doi: 10.1080/1547691X.2017.1420712
84. Zhang C, Lee JY, Keep RF, Pandey A, Chaudhary N, Hua Y, et al. Brain edema formation and complement activation in a rat model of subarachnoid hemorrhage. Acta Neurochir Suppl. (2013) 118:157–61. doi: 10.1007/978-3-7091-1434-6_29
85. Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin. (2015) 31:661–74. doi: 10.1016/j.ccc.2015.06.004
86. DeLano FA, Hoyt DB, Schmid-Schonbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. (2013) 5:169ra11. doi: 10.1126/scitranslmed.3005046
87. Schmid-Schonbein GW. 2008 Landis award lecture. Inflammation and the autodigestion hypothesis. Microcirculation. (2009) 16:289–306. doi: 10.1080/10739680902801949
88. Tachon G, Harrois A, Tanaka S, Kato H, Huet O, Pottecher J, et al. Microcirculatory alterations in traumatic hemorrhagic shock. Crit Care Med. (2014) 42:1433–41. doi: 10.1097/CCM.0000000000000223
89. Lehmann TG, Koeppel TA, Munch S, Heger M, Kirschfink M, Klar E, et al. Impact of inhibition of complement by sCR1 on hepatic microcirculation after warm ischemia. Microvasc Res. (2001) 62:284–92. doi: 10.1006/mvre.2001.2342
90. Nuytinck JK, Goris RJ, Weerts JG, Schillings PH, Stekhoven JH. Acute generalized microvascular injury by activated complement and hypoxia: the basis of the adult respiratory distress syndrome and multiple organ failure? Br J Exp Pathol. (1986) 67:537–48.
91. Hofer J, Rosales A, Fischer C, Giner T. Extra-renal manifestations of complement-mediated thrombotic microangiopathies. Front Pediatr. (2014) 2:97. doi: 10.3389/fped.2014.00097
92. Kimmoun A, Novy E, Auchet T, Ducrocq N, Levy B. Hemodynamic consequences of severe lactic acidosis in shock states: from bench to bedside. Crit Care. (2015) 19:175. doi: 10.1186/s13054-015-0896-7
93. Bonanno FG. Physiopathology of shock. J Emerg Trauma Shock. (2011) 4:222–32. doi: 10.4103/0974-2700.82210
94. Denk S, Neher MD, Messerer DAC, Wiegner R, Nilsson B, Rittirsch D, et al. Complement C5a functions as a master switch for the pH balance in neutrophils exerting fundamental immunometabolic effects. J Immunol. (2017) 198:4846–54. doi: 10.4049/jimmunol.1700393
95. Sina C, Kemper C, Derer S. The intestinal complement system in inflammatory bowel disease: shaping intestinal barrier function. Semin Immunol. (2018) 37:66–73. doi: 10.1016/j.smim.2018.02.008
96. Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci USA. (2013) 110:15061–6. doi: 10.1073/pnas.1307855110
97. Kuravi SJ, Yates CM, Foster M, Harrison P, Hazeldine J, Hampson P, et al. Changes in the pattern of plasma extracellular vesicles after severe trauma. PLoS ONE. (2017) 12:e0183640. doi: 10.1371/journal.pone.0183640
98. Lehner GF, Harler U, Haller VM, Feistritzer C, Hasslacher J, Dunzendorfer S, et al. Characterization of microvesicles in septic shock using high-sensitivity flow cytometry. Shock. (2016) 46:373–81. doi: 10.1097/SHK.0000000000000657
99. Karasu E, Eisenhardt SU, Harant J, Huber-Lang M. Extracellular vesicles: packages sent with complement. Front Immunol. (2018) 9:721. doi: 10.3389/fimmu.2018.00721
100. Levy RJ, Piel DA, Acton PD, Zhou R, Ferrari VA, Karp JS, et al. Evidence of myocardial hibernation in the septic heart. Crit Care Med. (2005) 33:2752–6. doi: 10.1097/01.CCM.0000189943.60945.77
101. Maniero GD. Classical pathway serum complement activity throughout various stages of the annual cycle of a mammalian hibernator, the golden-mantled ground squirrel, Spermophilus lateralis. Dev Comp Immunol. (2002) 26:563–74. doi: 10.1016/S0145-305X(02)00006-X
102. Bogren LK, Drew KL. Ischemia/reperfusion injury resistance in hibernators is more than an effect of reduced body temperature or winter season. Temperature. (2014) 1:87–8. doi: 10.4161/temp.29761
103. Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist. (2008) 13:993–1000. doi: 10.1634/theoncologist.2008-0086
104. Wang C, Wang H, Chang DY, Hao J, Zhao MH, Chen M. High mobility group box 1 contributes to anti-neutrophil cytoplasmic antibody-induced neutrophils activation through receptor for advanced glycation end products (RAGE) and Toll-like receptor 4. Arthritis Res Ther. (2015) 17:64. doi: 10.1186/s13075-015-0587-4
105. Li Y, Yang Z, Chavko M, Liu B, Aderemi OA, Simovic MO, et al. Complement inhibition ameliorates blast-induced acute lung injury in rats: potential role of complement in intracellular HMGB1-mediated inflammation. PLoS ONE. (2018) 13:e0202594. doi: 10.1371/journal.pone.0202594
106. Paredes RM, Reyna S, Vernon P, Tadaki DK, Dallelucca JJ, Sheppard F. Generation of complement molecular complex C5b-9 (C5b-9) in response to poly-traumatic hemorrhagic shock and evaluation of C5 cleavage inhibitors in non-human primates. Int Immunopharmacol. (2018) 54:221–5. doi: 10.1016/j.intimp.2017.10.033
107. Roselli F, Karasu E, Volpe C, Huber-Lang M. Medusa's head: the complement system in traumatic brain and spinal cord injury. J Neurotrauma. (2018) 35:226–40. doi: 10.1089/neu.2017.5168
108. Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: a review. J Neuroinflammation. (2018) 15:24. doi: 10.1186/s12974-018-1066-z
109. Younger JG, Sasaki N, Waite MD, Murray HN, Saleh EF, Ravage ZB, et al. Detrimental effects of complement activation in hemorrhagic shock. J Appl Physiol (1985). (2001) 90:441–6. doi: 10.1152/jappl.2001.90.2.441
110. Campbell JC, Li Y, van AE, Relan A, Dubick M, Sheppard F, et al. C1 Inhibitor limits organ injury and prolongs survival in swine subjected to battlefield simulated injury. Shock. (2016) 46(3 Suppl. 1): 177–88. doi: 10.1097/SHK.0000000000000677
111. Cai C, Gill R, Eum HA, Cao Z, Loughran PA, Darwiche S, et al. Complement factor 3 deficiency attenuates hemorrhagic shock-related hepatic injury and systemic inflammatory response syndrome. Am J Physiol Regul Integr Comp Physiol. (2010) 299:R1175–82. doi: 10.1152/ajpregu.00282.2010
112. Chen D, Song MQ, Liu YJ, Xue YK, Cheng P, Zheng H, et al. Inhibition of complement C3 might rescue vascular hyporeactivity in a conscious hemorrhagic shock rat model. Microvasc Res. (2016) 105:23–9. doi: 10.1016/j.mvr.2015.12.006
113. Reis ES, Berger N, Wang X, Koutsogiannaki S, Doot RK, Gumas JT, et al. Safety profile after prolonged C3 inhibition. Clin Immunol. (2018) 197:96–106. doi: 10.1016/j.clim.2018.09.004
114. Fleming SD, Phillips LM, Lambris JD, Tsokos GC. Complement component C5a mediates hemorrhage-induced intestinal damage. J Surg Res. (2008) 150:196–203. doi: 10.1016/j.jss.2008.02.010
115. Harkin DW, Marron CD, Rother RP, Romaschin A, Rubin BB, Lindsay TF. C5 complement inhibition attenuates shock and acute lung injury in an experimental model of ruptured abdominal aortic aneurysm. Br J Surg. (2005) 92:1227–34. doi: 10.1002/bjs.4938
116. Riedemann NC, Habel M, Ziereisen J, Hermann M, Schneider C, Wehling C, et al. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin Immunol. (2017) 180:25–32. doi: 10.1016/j.clim.2017.03.012
117. Kanni T, Zenker O, Habel M, Riedemann N, Giamarellos-Bourboulis EJ. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol. (2018) 179:413–9. doi: 10.1111/bjd.16428
118. Ren J, Zhao Y, Yuan Y, Han G, Li W, Huang Q, et al. Complement depletion deteriorates clinical outcomes of severe abdominal sepsis: a conspirator of infection and coagulopathy in crime? PLoS ONE. (2012) 7:e47095. doi: 10.1371/journal.pone.0047095
119. Heeres M, Visser T, van Wessem KJ, Koenderman AH, Strengers PF, Koenderman L, et al. The effect of C1-esterase inhibitor on systemic inflammation in trauma patients with a femur fracture - the CAESAR study: study protocol for a randomized controlled trial. Trials. (2011) 12:223. doi: 10.1186/1745-6215-12-223
120. Annane D. Sepsis-associated delirium: the pro and con of C5a blockade. Crit Care. (2009) 13:135. doi: 10.1186/cc7754
121. Mukherjee P, Thomas S, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J Neuroinflammation. (2008) 5:5. doi: 10.1186/1742-2094-5-5
122. Zimmerman JL, Dellinger RP, Straube RC, Levin JL. Phase I trial of the recombinant soluble complement receptor 1 in acute lung injury and acute respiratory distress syndrome. Crit Care Med. (2000) 28:3149–54. doi: 10.1097/00003246-200009000-00004
123. Norda R, Schott U, Berseus O, Akerblom O, Nilsson B, Ekdahl KN, et al. Complement activation products in liquid stored plasma and C3a kinetics after transfusion of autologous plasma. Vox Sang. (2012) 102:125–33. doi: 10.1111/j.1423-0410.2011.01522.x
124. Ekdahl KN, Soveri I, Hilborn J, Fellstrom B, Nilsson B. Cardiovascular disease in haemodialysis: role of the intravascular innate immune system. Nat Rev Nephrol. (2017) 13:285–96. doi: 10.1038/nrneph.2017.17
125. Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. (2007) 44:82–94. doi: 10.1016/j.molimm.2006.06.020
Keywords: trauma, sepsis, hemorrhagic shock, MODS, complement activation, complement dysregulation, complement therapeutics, clinical trial
Citation: Karasu E, Nilsson B, Köhl J, Lambris JD and Huber-Lang M (2019) Targeting Complement Pathways in Polytrauma- and Sepsis-Induced Multiple-Organ Dysfunction. Front. Immunol. 10:543. doi: 10.3389/fimmu.2019.00543
Received: 11 January 2019; Accepted: 28 February 2019;
Published: 21 March 2019.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Lubka T. Roumenina, INSERM U1138 Centre de Recherche des Cordeliers, FranceMichael Kirschfink, Universität Heidelberg, Germany
Copyright © 2019 Karasu, Nilsson, Köhl, Lambris and Huber-Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Huber-Lang, markus.huber-lang@uniklinik-ulm.de