 Marta Vuerich
Marta Vuerich Simon C. Robson
Simon C. Robson Maria Serena Longhi
Maria Serena Longhi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 March 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00507
This article is part of the Research Topic The Role of Ectonucleotidases in Acute and Chronic Inflammation View all 6 articles
Purinergic signaling modulates systemic and local inflammatory responses. Extracellular nucleotides, including eATP, promote inflammation, at least in part via the inflammasome upon engagement of P2 purinergic receptors. In contrast, adenosine generated during eATP phosphohydrolysis by ectonucleotidases, triggers immunosuppressive/anti-inflammatory pathways. Mounting evidence supports the role of ectonucleotidases, especially ENTPD1/CD39 and CD73, in the control of several inflammatory conditions, ranging from infectious disease, organ fibrosis to oncogenesis. Our experimental data generated over the years have indicated both CD39 and CD73 serve as pivotal regulators of intestinal and hepatic inflammation. In this context, immune cell responses are regulated by the balance between eATP and adenosine, potentially impacting disease outcomes as in gastrointestinal infection, inflammatory bowel disease, ischemia reperfusion injury of the bowel and liver, autoimmune or viral hepatitis and other inflammatory conditions, such as cancer. In this review, we report the most recent discoveries on the role of ENTPD1/CD39, CD73, and other ectonucleotidases in the regulation of intestinal and hepatic inflammation. We discuss the present knowledge, highlight the most intriguing and promising experimental data and comment on important aspects that still need to be addressed to develop purinergic-based therapies for these important illnesses.
The “purinergic signaling hypothesis” dates back to 1972 when Geoffrey Burnstock discovered that eATP and derivatives modulate gut and urinary tract neurotransmission (1). Nucleotides modulate cell responses upon binding to purinergic receptors (2–6) and also provide mediators after ectonucleotidase-mediated hydrolysis into adenosine (7, 8). Adenosine regulates cellular immune responses upon binding P1 adenosine receptors (3, 9, 10).
Ectonucleotidases include ecto-nucleoside triphosphate diphosphohydrolases (ENTPDases), ecto-5′-nucleotidase (NT5E)/CD73, ecto-nucleotide pyrophosphate phosphodiesterases (E-NPPs); CD38/NADase; NAD glycohydrolases; nucleoside diphosphate kinase; ecto-F1-F0 ATP synthases (11) and adenylate kinases. ENTPDase1, 2, 3, and 8 are surface-located enzymes that hydrolyze ATP/ADP into AMP. ENTPD2 however displays preferential ecto-ATPase activity (12). NTPDases4,5, 6, and 7 are intracellular proteins, with ENTPD5 and 6 being secreted upon heterologous expression.
NT5E/CD73, which converts AMP into adenosine, has been described both as GPI-anchored protein or soluble enzyme (13, 14). Soluble CD73 (sCD73) mainly derives from shedding of lymphocytes (13) and is present in both serum and cell-free lymph of healthy individuals (15). Increase in sCD73 levels has been reported in inflammatory conditions (16) and was negatively correlated with disease severity in patients with acute pancreatitis (17).
While being constitutively present on different immune cells, ENTPD1/CD39 and NT5E/CD73 can be further induced upon exposure to oxidative stress and hypoxia, stimulation with pro-inflammatory cytokines or following aryl hydrocarbon receptor (AhR) engagement (4, 18–21).
In this review, we focus on the role of ENTPD1/CD39 and NT5E/CD73 in gastrointestinal and hepatic inflammation.
Aberrant immune responses in gastric and intestinal disease might result in the development of chronic and progressive inflammatory statuses. In this setting, several studies emphasize the crucial role of the purinergic signaling in the modulation of GI conditions (Figure 1) (22).
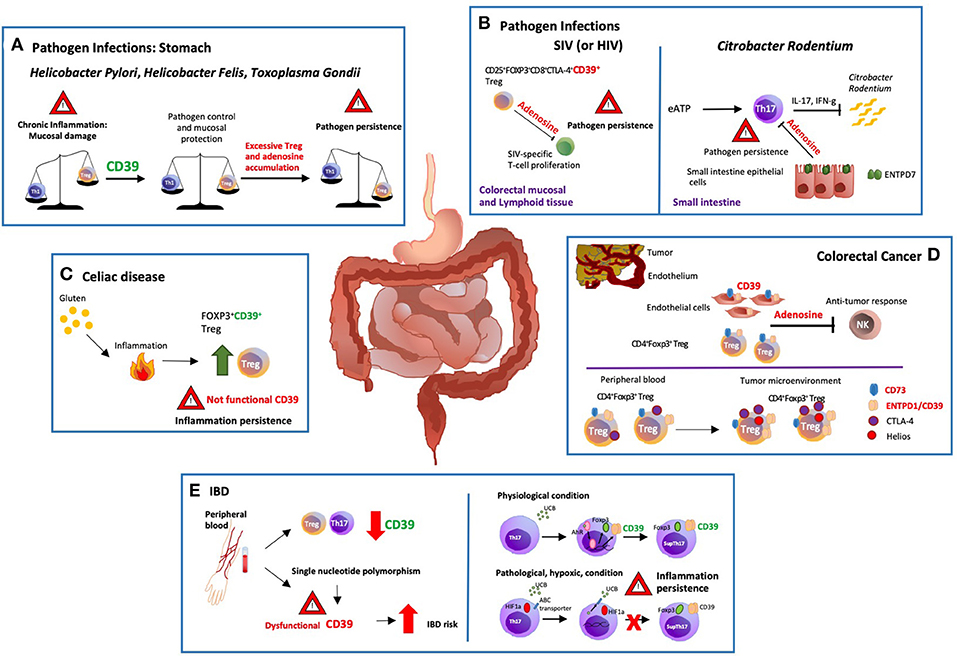
Figure 1. Modulation of gastro-intestinal disease by ectonucleotidases. (A) There are potent immune responses to gastrointestinal bacterial and parasitic infections e.g., Helicobacter pylori, Helicobacter felis and Toxoplasma gondii. The balance between pro and anti-inflammatory signals controls the development and outcome of the disease. Protracted release of Th1-related cytokines contributes to the establishment of chronic inflammation that might ultimately result in peptic ulcer disease and gastric cancer. ENTPD1/CD39 expression by regulatory T-cells (Tregs) modulates Th-cell responses; however, excessive immune regulation can also lead to pathogen persistence. (B) In a macacus rhesus model of pathogenic simian immunodeficiency virus (SIV) infection, there is rapid expansion of CD25+FOXP3+CD8+CTLA-4+CD39+ Tregs, especially in the colorectal mucosal and lymphoid tissues. This event limits anti-viral responses by suppressing the proliferation of SIV-specific T-cells. Treg accumulation is also observed in HIV patients, implicating that therapeutic strategies controlling the expansion of CD25+FOXP3+CD8+CTLA-4+ CD39+ Tregs might effectively control HIV infection restoring the anti-viral response. (C) Celiac disease is a chronic inflammatory disorder triggered by aberrant immune responses to dietary gluten. Exposure to gluten induces protective accumulation of FOXP3+CD39+ Tregs that, however, display defective suppressive function, and do not adequately control aberrant inflammatory responses. (D) ENTPD1/CD39 and CD73 are the dominant ectonucleotidases expressed by tumor endothelial cells and Tregs. Extracellular adenosine generated by CD39+ Tregs isolated from the blood of cancer patients inhibits and suppresses anti-tumor responses. Further, the tumor microenvironment impacts the phenotype and function of local cells, substantially limiting immunotherapeutic strategies. In this regard, most of the colorectal cancer-infiltrating Tregs are Helios+ and express higher levels of ENTPD1/CD39 and cytotoxic T-lymphocyte antigen 4 (CTLA-4), when compared to peripheral blood and colon-derived counterparts. (E) Low levels of ENTPD1/CD39 expression by Tregs and Th17-cells are observed in the peripheral blood of patients with inflammatory bowel disease (IBD). Moreover, single nucleotide polymorphisms associated with low ENTPD1/CD39 mRNA levels, increase susceptibility to the disease. On the other hand, in vitro exposure to unconjugated bilirubin (UCB) results in increased levels of ENTPD1/CD39 and FOXP3 expression in Th17-cells derived from healthy individuals, through a mechanism mediated by aryl hydrocarbon receptor (AhR). However, Crohn's-derived Th17-cells remain refractory to UCB immunoregulation due to altered responses to hypoxia that inhibits AhR signaling by inducing ATP-binding cassette (ABC) transporters that promote UCB efflux out of Th17-cells.
T-helper-cells are pivotal players in anti-bacterial responses (23). Protracted release of Th1-related cytokines, however, contributes to chronic inflammation that might ultimately result in peptic ulcer disease and gastric cancer, as in the context of Helicobacter (H.) pylori infection. However, inadequate Th1 immunity can lead to persistent infection as result of regulatory T-cell (Treg) accumulation (24–26) that supports pathogen persistence. Adenosine generation by ENTPD1/CD39 and CD73 on Treg and memory T-cells, strongly inhibits effector T-cell immunity (8, 27), as shown in vitro and in experimental models of H. felis-induced gastritis in vivo (28).
Cd73−/− mice develop a more severe gastritis, associated with heightened levels of pro-inflammatory cytokines and impaired Treg function (28). Administration of an A2A adenosine receptor (A2AR) agonist to Il-10−/− and Helicobacter-bearing mice attenuates gastritis lowering TNF-α and IFN-γ levels (29).
A comparable regulatory pathway has been observed in murine models of intestinal (and systemic) Toxoplasma gondii (T. gondii) infection. In the intestine of naive mice, conventional CD4+ T-cells and Tregs express both ENTPD1/CD39 and CD73. During acute T. gondii infection, CD73 expression is downregulated, with consequent diminished generation of immunosuppressive adenosine. As levels of the type-1 purinergic adenosine receptors are maintained, administration of receptor agonists ameliorates disease symptoms and associated dysbiosis (30).
The key role of ENTPD1/CD39 in the modulation of cellular immune response in the intestine has been suggested in a macacus rhesus model of simian immunodeficiency virus (SIV) infection. Infection with SIV results in rapid expansion of CD25+FOXP3+CD8+CTLA-4+CD39+ Tregs, especially in colorectal mucosal and lymphoid tissues, the preferential sites of virus replication. This development limits anti-viral responses by suppressing the proliferation of SIV-specific T-cells. Treg accumulation is also observed in HIV patients, implicating that therapeutic strategies aimed at containing Treg expansion might improve the control over HIV by restoring anti-viral responses (31, 32).
Purinergic signaling regulates also Th17-cell immunity (33). ENTPD7 expression in the epithelial cells of small intestine controls luminal ATP levels, therefore regulating Th17-cell development (34). In this regard, high ATP levels and Th17-cell accumulation are noted in the lamina propria of Entpd7−/− mice and homeostasis can be restored by oral administration of ATP antagonists or antibiotics (34). In the absence of ENTPD7, commensal microbiota-dependent eATP release supports Th17-cell development (34). Accordingly, Entpd7−/− mice are resistant to Citrobacter rodentium infection although suffering from severe experimental autoimmune encephalomyelitis, resulting from accumulation of IL-17 and IFN-γ (34). Control of intestinal microbiota by purinergic mediators has been also supported by recent data showing that mice deficient in the ATP-gated ionotropic P2X7 receptor display intestinal microbiotic imbalance and altered glucose metabolism (35).
Inflammatory bowel disease (IBD) is a chronic, debilitating illness characterized by excessive inflammation of the colon and small intestine that is associated with thrombophilia and heightened risk for cancer (36, 37).
Experimental and clinical evidences indicate a protective role of ENTPD1/CD39 in Crohn's disease (CD). Global Entpd1/Cd39 deletion in dextran-sulfate-sodium (DSS)-induced colitis in mice increases susceptibility to injury (38). Accordingly, high ENTPD1/CD39 expression by circulating Tregs correlates with clinical remission in IBD patients while single nucleotide polymorphisms, associated with low Entpd1/Cd39 mRNA levels, increase predisposition to Crohn's disease (39).
Crohn's patients have decreased suppressor (sup)Th17-cells, a unique effector cell subtype endowed with immunosuppressive functions. In contrast to conventional pathogenic Th17-cells, supTh17-cells express higher levels of ENTPD1/CD39 (33), more effectively generate eAMP and adenosine and hence can also potently suppress effector T-cell responses via A2A receptors.
Expression of ENTPD1/CD39 can be induced upon engagement of AhR, a mediator of toxin responses and adaptive immunity (40, 41). AhR activation induces accumulation of CD39+ and granzyme+ human Tregs in vitro and treatment with the AhR agonist 2-(1′H-indole-3′- carbonyl)-thiazole-4-carboxylic acid methyl ester has a protective effect in colitic humanized mice by increasing Foxp3+, CD39+, granzyme B+, and IL-10+ Tregs (42).
We have recently reported that in vitro exposure to unconjugated bilirubin (UCB), a product of heme oxidation that serves as AhR endogenous ligand, results in increased levels of ENTPD1/CD39 and FOXP3 in Th17-cells derived from healthy individuals but not from Crohn's disease patients (18). We have also noted that UCB treatment ameliorates DSS colitis in mice, this protective effect being dependent on ENTPD1/CD39 and AhR (18). Resistance of Crohn's-derived Th17-cells to AhR stimulation results from altered response to hypoxia that inhibits AhR signaling in IBD through induction of ABC transporters; these promoting UCB efflux out of Th17-cells (43).
Furthermore, co-expression of ENTPD1/CD39 and CD161 by T-cells supports Th17 effector phenotype through alterations in both extracellular nucleotide-mediated responses and acid sphingomyelinase catalytic bioactivity that promote IL-17 expression (44, 45). Pro-inflammatory CD4+CD39+CD161+ T-cells are increased in the blood and lamina propria of Crohn's disease patients and levels directly correlate with the disease activity (44). CD3/CD28-mediated stimulation of IFN-γ-producing CD8+ T-cells, another effector subset involved in Crohn's disease pathogenesis (46), not only increases IFN-γ production by CD8 T-cells, but also induces reactive oxygen species and ENTPD1/CD39 expression (47).
Increase in CD73+CD4+ T-cells, which are enriched in IL-17 producing lymphocytes, is detected in the lamina propria and peripheral blood of IBD patients during active inflammation. In Crohn's disease, accumulation of pathogenic Th17-cells has been also associated with heightened CD73 levels (48). Interestingly, exposure to TNF increases CD73 expression on CD4+ T-cells, while anti-TNF monoclonal antibody (infliximab) has the opposite effects, therefore implicating CD73+ Th17-cells as a surrogate marker of disease activity and response to treatment (48).
Recent data have shown a protective role for ENTPD2 and ENTPD3 in neuro-immune interactions in Crohn's disease (49). ENTPD2-3 are expressed by enteric nervous system cells in both the human and murine colon. Both Entpd2−/− and Entpd3−/− mice are more susceptible to DSS-induced colitis and Entpd2−/− colonic macrophages display a more pro-inflammatory phenotype as compared to wild type controls (49). A significant proportion of the microparticle-associated ectonucleotidase activity is sensitive to POM6, inferring the presence of NTPDases, either −2 or/and −3. Further, human plasma samples obtained from Crohn's patients, show overall decreases in ADPase activity, this alteration being directly correlated with disease activity (49).
Celiac disease is a chronic inflammatory disorder frequently associated with IBD and triggered by aberrant immune responses to dietary gluten. Recent work has shown that gluten exposure induces protective accumulation of FOXP3+CD39+ Tregs in celiac patients. These Tregs however, are dysfunctional and exhibit impaired suppression (50).
There is evidence that microparticles (MPs) released from cells in the inflammatory site can be loaded with ENTPDase mRNA (51). Such mRNA content within MPs can be taken up by incorporating cells and be subsequently translated into functional NTPDases. This phenomenon occurs between leukocytes and vascular endothelial cells (51). Our collaborators, others and we have also noted that levels of microRNAs (miRs) present in plasma MPs are modulated by CD39 expression and that one microRNA, miR-142-3p, might impacts CD39 levels per se (52, 53).
Based on this evidence, MPs could serve as biomarkers of inflammatory pathways as well as therapeutic tools to modulate the function of cells taking up these exosomes and MPs (51–54).
Colorectal cancer (CRC) is the second leading cause of tumor-related death in the United States. ENTPD1/CD39 and CD73 are the major ectonucleotidases expressed by tumor endothelial cells and Tregs. Extra-cellular adenosine generated by circulating CD39+ Tregs of cancer patients not only inhibits anti-tumor responses and stimulates vascular endothelial cell proliferation, but also reduces monocyte ability to activate the endothelium, limiting migration of effector T-cells into the tumor (55–58).
In a murine model of hepatic metastatic cancer, resulting from portal vein infusion of MCA38 colon cancer cells and melanoma B16/F10 cells, ENTPD1/CD39 expression on Tregs strongly suppresses natural killer (NK)-mediated anti-tumor immunity (59); whereas ENTPD1/CD39 inhibition restores anti-cancer responses, significantly limiting tumor growth (58, 59). The correlation between levels of ENTPD1/CD39 in the host and CRC progression has been confirmed in orthotopic transplanted murine cancer models; while in clinical samples, lower levels of ENTPD1/CD39 mRNA in malignant CRC tissues correlates with prolonged survival and less invasiveness (60).
The tumor microenvironment strongly impacts the phenotype and function of immune cells, substantially limiting immunotherapeutic strategies. In this regard, most of the CRC-infiltrating Tregs are Helios+ and express high levels of ENTPD1/CD39 and cytotoxic T-lymphocyte antigen4 (CTLA-4).
There is also evidence that CD8+ lymphocytes infiltrating human CRC recognize a wide range of epitopes unrelated to the tumor, including those recognized during previous viral infections. Such CD8+ lymphocytes display wide variability in ENTPD1/CD39 expression, which correlates with the clinical status of patients (61).
Ectonucleotidases can be also expressed in the liver in different cell populations, including resident immune cells and endothelial cells. As observed in the rat, the specific cellular localization and function are strongly affected by variations in the organ homeostasis. In healthy rat liver, CD73 expression partially overlaps with that of ENTPD1/CD39 in fibroblastic cells underneath vascular endothelial cells and smooth muscle cells, and with that of ENTPDase8 in bile canaliculi. In portal spaces, CD73 is expressed in a fibroblast subpopulation, which is adjacent to ENTPDase2+ portal fibroblasts. At variance with healthy, quiescent states, the expression, and activity of these ectonucleotidases are largely altered in fibrotic livers (62).
Below we discuss the role of ectonucleotidases, especially that of ENTPD1/CD39, in major pathological hepatic conditions (Figure 2).
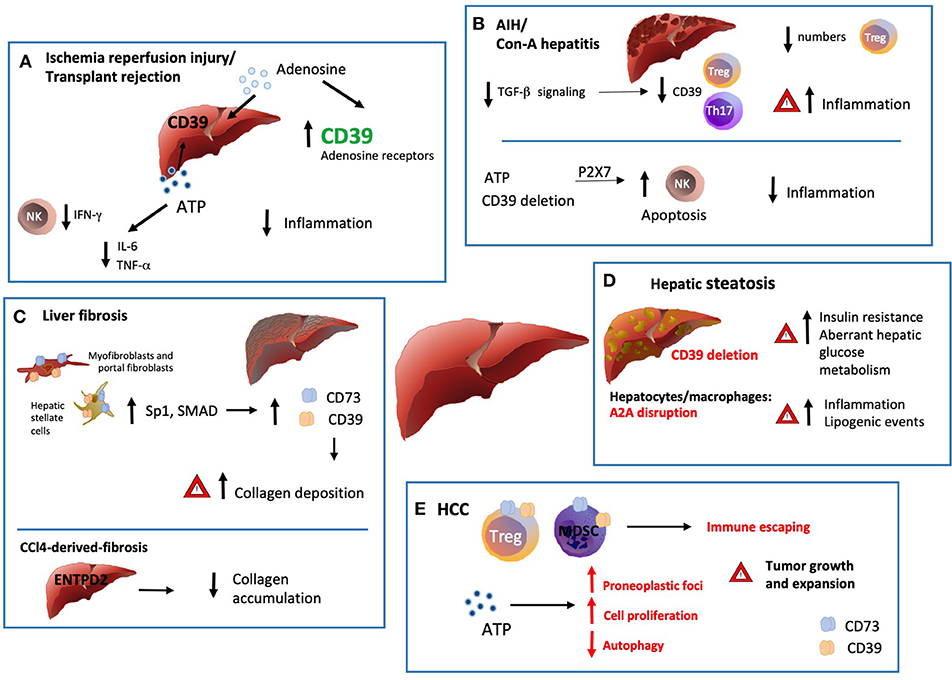
Figure 2. Ectonucleotidases in inflammatory liver conditions. (A) Ischemia/reperfusion injury (IRI) is triggered by the vascular damage consequent to blood reperfusion of oxygen deprived ischemic tissues, as with organ transplantation. ENTPD1 expression by donor livers and treatment with exogenous adenosine at high concentrations in preservation solutions protects grafts from ischemic damage with extended cold preservation times. Pharmacologic preconditioning through stimulation of adenosine receptors has been also associated with protection from ischemia by increasing ENTPD1/CD39 expression, via Sp1 transcription factor activation. This protective effect is abrogated in the absence of ENTPD1/CD39 but can be restored by adenosine administration. There is also evidence that exogenous and cautious ATP infusions can improve the hepatic function and post-ischemic clinical condition, (at least in part) by decreasing the plasma levels of IL-6 and TNF. (B) Numerical and functional impairment of Tregs contributes to immune imbalance in autoimmune hepatitis (AIH). Tregs and Th17-cells isolated from AIH patients display defective ENTPD1/CD39 expression and fail to control eATP mediated pro-inflammatory Th17 accumulation. Treg acquisition of pro-inflammatory properties together with low ENTPD1/CD39 expression might result from dysfunction in TGF-β signaling. (C) Liver fibrosis is driven by activation and accumulation of myofibroblasts and hepatic stellate cells (HSC), the predominant source of extracellular matrix and collagen in the organ. ENTPD1/CD39 and CD73, are upregulated in HSC, portal fibroblasts and in fibrous septa. This overexpression, mediated by SP1 and SMAD promoter elements, is a direct consequence of the myofibroblastic differentiation. Interestingly, in the setting of CCl4-derived-fibrosis, ENTPD2 expression and re-distribution from the portal areas to the fibrotic septa, has a protective role against excessive collagen accumulation. (D) In hepatic steatosis and alcoholic hepatitis, Entpd1/Cd39 deletion correlates with increased insulin resistance and aberrant hepatic glucose metabolism. Accordingly, disruption of A2AR expression in hepatocytes and macrophages also directly correlates with the severity of obesity–associated non–alcoholic-fatty-liver-disease, promoting inflammation and lipogenic events. (E) ATP scavenging by ENTPD1/CD39 expressed by CD4+FOXP3+ Tregs, endothelial cells and myeloid derived suppressor cells (MDSC) promotes hepatic tumor growth in mice. Once exposed to hypoxic microenvironment, HCC upregulates ENTPD2 expression, further supporting MDSC accumulation and immunosuppressive activity. On the other hand, recent experiments have documented occurrence of liver cancer also in Entpd1/Cd39−/− mice. These latter findings would result from eATP-P2 receptor-mediated suppression of tumor cell autophagy and boosting of cell proliferation.
Ischemia/reperfusion injury (IRI) is triggered by the vascular damage consequent to blood reperfusion of oxygen deprived tissues. IRI is driven by accumulation of inflammatory mediators, including adenine nucleotides and is associated with platelet activation and, ultimately, organ rejection (63). ENTPD1 expression by donor livers and treatment with high concentration adenosine protect grafts from ischemic damage (64). Pharmacologic preconditioning through stimulation of adenosine receptors also protects from ischemia (65, 66) by increasing ENTPD1/CD39 expression via Sp1 (66). This protective effect, abrogated in Entpd1/Cd39−/− mice, can be restored in hemizygous Cd39-deficient mice following apyrase or adenosine administration.
ENTPD1/CD39 expression reduces pro-inflammatory activity and promotes protective phenotypes in conventional liver myeloid dendritic cells (mDC) in IRI and transplant models (67, 68). However, there is also evidence that ATP infusion improves hepatic function and post ischemic clinical condition by downregulating IL-6 and TNF plasma levels (69). Similar anti-inflammatory effects are achieved by specifically deleting ENTPD1/CD39 in NK cells, suggesting a regulatory role for ATP/P2 receptor axis during liver injury and subsequent regeneration (70). These results clearly show that, although adenosine and ENTPD1/CD39 are commonly known as immunosuppressive factors, the mechanisms regulating the inflammatory response are complex and markedly impacted by specific cellular conditions.
Recent studies have revealed that eATP scavenging has protective effects in sepsis-induced liver injury (71). ENTPD1/CD39 expression by macrophages strongly suppresses pro-inflammatory responses, especially those P2X7-mediated. Accordingly, in the same experimental settings, Entpd1/Cd39 genetic deletion exacerbates end-organ injury (72).
Inflammatory liver injury caused by acetaminophen (APAP) toxicity can be linked to purinergic stimulation of immune cells and vascular endothelium. Indeed, P2X7 is crucial in these responses as exposure to ATP ligands is required for manifestations of APAP-induced hepatotoxicity. APAP toxicity is very pronounced in Entpd1/Cd39−/− mice, which show hepatic hemorrhagic necrosis and high mortality. Exogenous apyrase also decreases APAP-induced mortality in wild type mice (73).
Autoimmune hepatitis (AIH) is a severe hepatopathy mediated by aberrant activation of CD8+ and CD4+ effectors, including Th17-cells. Decreased numbers and functional impairment of Tregs contribute to immune imbalance in AIH (74–76). Tregs isolated from AIH patients display decreased ENTPD1/CD39 expression and fail to control eATP-mediated Th17 accumulation (77). Low levels of and functional defects in Th17CD39+ cells have been also detected in juvenile autoimmune liver disease. Here too, the impairments in ENTPD1/CD39 and A2A expression might bolster and promote cellular effector properties. Moreover, levels of adenosine deaminase are significantly increased in AIH patients and positively correlate with inflammation and fibrosis scores (78).
Natural killer T-cells (NKT) are another cell population involved in AIH pathogenesis. In murine models of Concanavalin-A induced hepatitis, genetic deletion of Entpd1/Cd39 promotes eATP/P2X7-mediated NKT apoptosis and paradoxically provides protection from liver injury (79). Extracellular purines differentially impact different cell types (Treg vs. NKT cells) in certain pathological conditions, as previously shown in the context of hyperoxic lung injury (80). These counter intuitive findings indicate the complexity of purinergic immunomodulation in the liver, and elsewhere (80).
Hepatic fibrosis is a pathological process that develops as a response to chronic inflammation and ongoing liver injury. The pathological process is driven by activation and accumulation of myofibroblasts, a heterogeneous population of activated non-parenchymal liver cells and hepatic stellate cells (HSC). These two cell types are likely the predominant source of extracellular matrix and collagen in the liver.
Different ectonucleotidases, including CD73 and ENTPD family members, are upregulated in HSC, portal fibroblasts and in fibrous septa (62, 81, 82). Such levels of overexpression, mediated by SP1 and SMAD promoter elements, are thought to result from myofibroblastic differentiation.
In experimental models, Cd73-deficient mice are resistant to development of liver fibrosis, suggesting a pathological role for AMPase activity and adenosine generation in fibrogenesis (82).
To the contrary, ENTPD2 ATPase activity as expressed by myofibroblasts is protective in the setting of CCl4-induced fibrosis as null mice develop more liver scarring in this model. ENTPD2 expression and re-distribution from the portal areas to the fibrotic septa, has a protective role against excessive collagen accumulation in the liver. These salutary effects could be ascribed to anti-inflammatory effects of extracellular ATP scavenging by members of the ENTPD family. In contrast, after partial hepatectomy or 3,5-diethoxycarbonyl-1,4- dihydrocollidine (DDC)-induced hepatocellular injury, the Entpd2 deletion does not significantly impact the fibrotic response in mutant mice (83).
There is evidence that ENTPD1/CD39 limits hepatic accumulation of gut primed CD8 T-cells, preventing biliary injury and subsequent fibrosis (84). In this context, Entpd1/Cd39 deletion results in increased levels of hepatic CD8 T-cells following upregulation of the T-cell gut-tropism receptor, integrin α4β7. Accordingly, in Mdr2−/−Cd39−/− mice, CD8 cell depletion as well as gut decontamination and administration of stable ATP agonist or antibiotics, attenuates hepatobiliary injury and fibrosis (47, 83).
Purinergic signaling and adenosinergic effects are important modulators of metabolic disease. Entpd1/Cd39 deletion correlates with increased insulin resistance and aberrant hepatic glucose metabolism (85). Furthermore, A1 adenosine receptor expression on adipocytes impacts fatty acids metabolism, including lipolysis, diabetes, dyslipidemia, and insulin resistance (86).
Adenosine can be also generated during ethanol metabolism and the effects of ethanol-induced hepatic steatosis might be therefore mediated by adenosine receptors, especially A1 and A2B (87). Disruption of A2AR in hepatocytes and macrophages is directly linked to the severity of obesity-associated non-alcoholic-fatty-liver-disease, promoting inflammation and lipogenic events (88).
A potentially fatal consequence of liver transplantation is that of immune-mediated organ rejection. Increasing evidences reveal a protective role of ENTPD1/CD39, the upregulation and augmented activity of which, achieved also upon exogenous administration, improve liver survival in allotransplantation models (89). Further, ENTPD1/CD39 expression in liver allografts modulates the anti-donor effector T-cell responses and Treg infiltration, ameliorating organ rejection and preventing graft-vs.-host reactions (90).
Liver xenograft rejection is a consequence of vascular inflammation and thrombosis that is partially mediated by extracellular nucleotides (91, 92).
Hepatocellular carcinoma (HCC) is the most frequent type of primary liver cancer in adults and is the major cause of death in cirrhotic patients (93). HCC growth and expansion are supported by accumulation of cellular and inflammatory metabolites, including eATP that promotes the generation of preneoplastic foci via P2 receptors (94–96). Further experimentation suggests the development of autochthonous liver cancer in Entpd1/Cd39−/− mice (97), resulting from comparable eATP-P2 receptor-mediated changes: inclusive of suppression of liver cell autophagy, altered metabolism, and boosting of proliferation.
Recent work has shown that perturbations in purinergic signaling promote HCC growth, also by supporting immune escaping. ATP scavenging by ENTPD1/CD39 expressed by Tregs and endothelial cells hence facilitates metastatic and transplanted hepatic tumor growth in mice (59, 98, 99).
Generation of adenosine by ENTPD1/CD39 expressed by Tregs and myeloid derived suppressor cells (MDSC) inhibits effector cell proliferation and function (59). Interestingly, in the presence of hypoxic microenvironment, HCC cells upregulate ENTPD2 that preferentially converts extracellular ATP to ADP and little AMP, further supporting the accumulation and immune suppressive activity of MDSC (100).
ENTPD5/CD39L4, a soluble endoplasmic reticulum UDPase can also directly modulate tumor growth impacting N-glycosylation and cell metabolism and has been proposed as target for anti-cancer therapy (53). Other studies, however, have reported contrasting findings, showing increased risk of HCC in Entpd5 null mice (101).
Table 1 summarizes changes in ectonucleotidase expression and activity in GI and liver experimental models and human conditions.
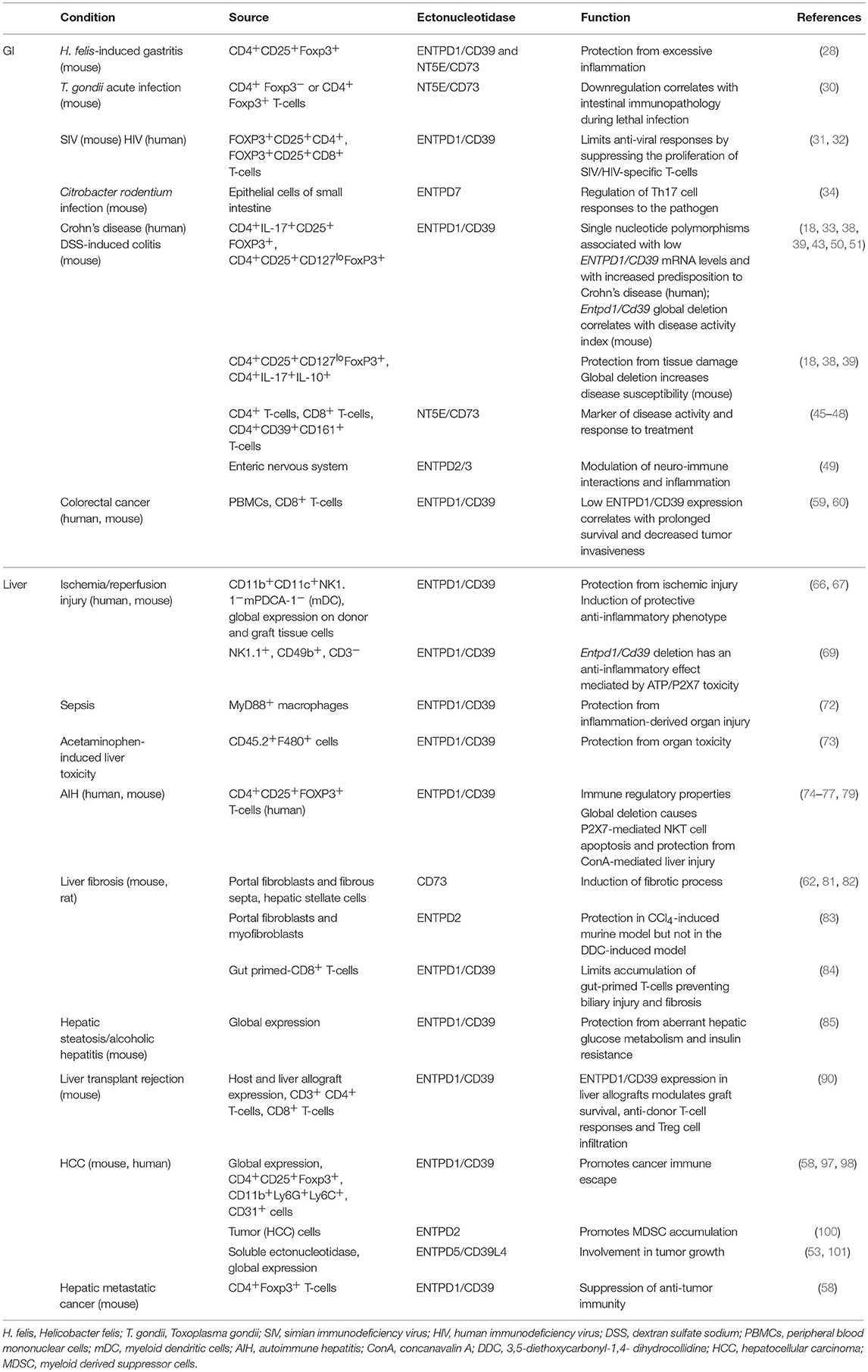
Table 1. Ectonucleotidase expression and activity in GI and liver experimental models and human diseases.
We have briefly discussed how the balance between ATP and adenosine dictates outcomes of inflammatory conditions of the GI tract and liver. Multiple questions, however, remain as to how this balance is regulated, and can be possibly targeted in different disease settings.
Development of purinergic-based therapies that could be used alone or in combination with already existing treatments, might be implemented to control these responses in the gut and liver. The goal of such interventions will be to re-establish and maintain immunologic tolerance and promote healing in these important acute and chronic inflammatory processes.
MV wrote the manuscript. SCR and MSL reviewed and edited the manuscript.
This work has been supported by the National Institute of Health (R01 DK108894 to MSL; P01 HL107152 and R21 CA164970 to SCR); AASLD Pilot Research Award (to MSL); Pfizer research support to SCR; the Helmsley Charitable Trust (grant 281574.5069091.0010 to SCR); and by the Department of Defense Award W81XWH-16-0464 (to SCR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol. (2014) 99:16–34. doi: 10.1113/expphysiol.2013.071951
2. Burnstock G. Introduction: P2 receptors. Curr Top Med Chem. (2004) 4:793–803. doi: 10.2174/1568026043451014
3. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
4. Bianchi G, Vuerich M, Pellegatti P, Marimpietri D, Emionite L, Marigo I, et al. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. (2014) 5:e1135. doi: 10.1038/cddis.2014.109
5. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
6. Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci. (2015) 191:117–23. doi: 10.1016/j.autneu.2015.04.011
7. Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by CD39. Trends Immunol. (2016) 37:427–39. doi: 10.1016/j.it.2016.04.009
8. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
9. Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol. (2014) 49:473–97. doi: 10.3109/10409238.2014.953627
10. Jacobson KA. Introduction to adenosine receptors as therapeutic targets. Handb Exp Pharmacol. (2009) 193:1–24. doi: 10.1007/978-3-540-89615-9_1
11. Moser TL, Kenan DJ, Ashley TA, Roy JA, Goodman MD, Misra UK, et al. Endothelial cell surface F1-F0 ATP synthase is active in ATP synthesis and is inhibited by angiostatin. Proc Natl Acad Sci USA. (2001) 98:6656–61. doi: 10.1073/pnas.131067798
12. Kukulski F, Levesque SA, Lavoie EG, Lecka J, Bigonnesse F, Knowles AF, et al. Comparative hydrolysis of P2 receptor agonists by NTPDases 1:2:3 and 8. Puriner Signal. (2005) 1:193–204. doi: 10.1007/s11302-005-6217-x
13. Airas L, Niemela J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73:a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol. (1997) 136:421–31. doi: 10.1083/jcb.136.2.421
14. Yegutkin G, Bodin P, Burnstock G. Effect of shear stress on the release of soluble ecto-enzymes ATPase and 5′-nucleotidase along with endogenous ATP from vascular endothelial cells. Br J Pharmacol. (2000) 129:921–6. doi: 10.1038/sj.bjp.0703136
15. Yegutkin GG, Auvinen K, Rantakari P, Hollmen M, Karikoski M, Grenman R, et al. Ecto-5′-nucleotidase/CD73 enhances endothelial barrier function and sprouting in blood but not lymphatic vasculature. Eur J Immunol. (2015) 45:562–73. doi: 10.1002/eji.201444856
16. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
17. Maksimow M, Kyhala L, Nieminen A, Kylanpaa L, Aalto K, Elima K, et al. Early prediction of persistent organ failure by soluble CD73 in patients with acute pancreatitis*. Crit Care Med. (2014) 42:2556–64. doi: 10.1097/CCM.0000000000000550
18. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. (2017) 2:e92791. doi: 10.1172/jci.insight.92791
19. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. (1998) 161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x
20. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
21. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. (2014) 193:3155–64. doi: 10.4049/jimmunol.1400578
22. Antonioli L, Colucci R, Pellegrini C, Giustarini G, Tuccori M, Blandizzi C, et al. The role of purinergic pathways in the pathophysiology of gut diseases: pharmacological modulation and potential therapeutic applications. Pharmacol Ther. (2013) 139:157–88. doi: 10.1016/j.pharmthera.2013.04.002
23. Bagheri N, Salimzadeh L, Shirzad H. The role of T helper 1-cell response in Helicobacter pylori-infection. Microb Pathog. (2018) 123:1–8. doi: 10.1016/j.micpath.2018.06.033
24. Enarsson K, Lundgren A, Kindlund B, Hermansson M, Roncador G, Banham AH, et al. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin Immunol. (2006) 121:358–68. doi: 10.1016/j.clim.2006.07.002
25. Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. (2005) 73:523–31. doi: 10.1128/IAI.73.1.523-531.2005
26. Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. (2006) 131:525–37. doi: 10.1053/j.gastro.2006.05.001
27. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73:which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. J Immunol. (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
28. Alam MS, Kurtz CC, Rowlett RM, Reuter BK, Wiznerowicz E, Das S, et al. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J Infect Dis. (2009) 199:494–504. doi: 10.1086/596205
29. Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol. (2009) 2:232–42. doi: 10.1038/mi.2009.4
30. Francois V, Shehade H, Acolty V, Preyat N, Delree P, Moser M, et al. Intestinal immunopathology is associated with decreased CD73-generated adenosine during lethal infection. Mucosal Immunol. (2015) 8:773–84. doi: 10.1038/mi.2014.108
31. Nigam P, Kwa S, Velu V, Amara RR. Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection. J Immunol. (2011) 186:745–53. doi: 10.4049/jimmunol.1002807
32. Nigam P, Velu V, Kannanganat S, Chennareddi L, Kwa S, Siddiqui M, et al. Expansion of FOXP3+ CD8 T cells with suppressive potential in colorectal mucosa following a pathogenic simian immunodeficiency virus infection correlates with diminished antiviral T cell response and viral control. J Immunol. (2010) 184:1690–701. doi: 10.4049/jimmunol.0902955
33. Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS ONE. (2014) 9:e87956. doi: 10.1371/journal.pone.0087956
34. Kusu T, Kayama H, Kinoshita M, Jeon SG, Ueda Y, Goto Y, et al. Ecto-nucleoside triphosphate diphosphohydrolase 7 controls Th17 cell responses through regulation of luminal ATP in the small intestine. J Immunol. (2013) 190:774–83. doi: 10.4049/jimmunol.1103067
35. Perruzza L, Gargari G, Proietti M, Fosso B, D'Erchia AM, Faliti CE, et al. T follicular helper cells promote a beneficial gut ecosystem for host metabolic homeostasis by sensing microbiota-derived extracellular ATP. Cell Rep. (2017) 18:2566–75. doi: 10.1016/j.celrep.2017.02.061
36. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. (2007) 369:1627–40. doi: 10.1016/S0140-6736(07)60750-8
37. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. (2007) 369:1641–57. doi: 10.1016/S0140-6736(07)60751-X
38. Gibson DJ, Elliott L, McDermott E, Tosetto M, Keegan D, Byrne K, et al. Heightened expression of CD39 by regulatory T lymphocytes is associated with therapeutic remission in inflammatory bowel disease. Inflamm Bowel Dis. (2015) 21:2806–14. doi: 10.1097/MIB.0000000000000566
39. Friedman DJ, Kunzli BM, A-Rahim YI, Sevigny J, Berberat PO, Enjyoji K, et al. From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. (2009) 106:16788–93. doi: 10.1073/pnas.0902869106
40. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
41. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. (2008) 453:106–9. doi: 10.1038/nature06881
42. Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. (2016) 17:1318–29. doi: 10.1016/j.celrep.2016.09.082
43. Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrugge L, Csizmadia E, et al. HIF-1alpha-induced xenobiotic transporters promote Th17 responses in Crohn's disease. J Autoimmun. (2018) 94:122–33. doi: 10.1016/j.jaut.2018.07.022
44. Bai A, Moss A, Kokkotou E, Usheva A, Sun X, Cheifetz A, et al. CD39 and CD161 modulate Th17 responses in Crohn's disease. J Immunol. (2014) 193:3366–77. doi: 10.4049/jimmunol.1400346
45. Bai A, Robson S. Beyond ecto-nucleotidase: CD39 defines human Th17 cells with CD161. Puriner Signal. (2015) 11:317–9. doi: 10.1007/s11302-015-9457-4
46. Funderburg NT, Stubblefield Park SR, Sung HC, Hardy G, Clagett B, Ignatz-Hoover J, et al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology. (2013) 140:87–97. doi: 10.1111/imm.12114
47. Bai A, Moss A, Rothweiler S, Longhi MS, Wu Y, Junger WG, et al. NADH oxidase-dependent CD39 expression by CD8(+) T cells modulates interferon gamma responses via generation of adenosine. Nat Commun. (2015) 6:8819. doi: 10.1038/ncomms9819
48. Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, et al. CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol. (2012) 42:3062–72. doi: 10.1002/eji.201242623
49. Feldbrugge L, Moss AC, Yee EU, Csizmadia E, Mitsuhashi S, Longhi MS, et al. Expression of Ecto-nucleoside triphosphate diphosphohydrolases-2 and -3 in the enteric nervous system affects inflammation in experimental colitis and Crohn's disease. J Crohns Colitis. (2017) 11:1113–23. doi: 10.1093/ecco-jcc/jjx058
50. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
51. Banz Y, Beldi G, Wu Y, Atkinson B, Usheva A, Robson SC. CD39 is incorporated into plasma microparticles where it maintains functional properties and impacts endothelial activation. Br J Haematol. (2008) 142:627–37. doi: 10.1111/j.1365-2141.2008.07230.x
52. Kuhn S, Splith K, Ballschuh C, Feldbrugge L, Krenzien F, Atanasov G, et al. Mononuclear-cell-derived microparticles attenuate endothelial inflammation by transfer of miR-142–3p in a CD39 dependent manner. Purinergic Signal. (2018) 14:423–32. doi: 10.1007/s11302-018-9624-5
53. Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, et al. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. (2010) 143:711–24. doi: 10.1016/j.cell.2010.10.010
54. Haller CA, Cui W, Wen J, Robson SC, Chaikof EL. Reconstitution of CD39 in liposomes amplifies nucleoside triphosphate diphosphohydrolase activity and restores thromboregulatory properties. J Vasc Surg. (2006) 43:816–23. doi: 10.1016/j.jvs.2005.11.057
55. Sundstrom P, Stenstad H, Langenes V, Ahlmanner F, Theander L, Ndah TG, et al. Regulatory T cells from colon cancer patients inhibit effector T-cell migration through an adenosine-dependent mechanism. Cancer Immunol Res. (2016) 4:183–93. doi: 10.1158/2326-6066.CIR-15-0050
56. Hoskin DW, Reynolds T, Blay J. Adenosine as a possible inhibitor of killer T-cell activation in the microenvironment of solid tumours. Int J Cancer. (1994) 59:854–5. doi: 10.1002/ijc.2910590625
57. Spychala J. Tumor-promoting functions of adenosine. Pharmacol Ther. (2000) 87:161–73. doi: 10.1016/S0163-7258(00)00053-X
58. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
59. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
60. Kunzli BM, Bernlochner MI, Rath S, Kaser S, Csizmadia E, Enjyoji K, et al. Impact of CD39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal. (2011) 7:231–41. doi: 10.1007/s11302-011-9228-9
61. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
62. Fausther M, Lecka J, Soliman E, Kauffenstein G, Pelletier J, Sheung N, et al. Coexpression of ecto-5'-nucleotidase/CD73 with specific NTPDases differentially regulates adenosine formation in the rat liver. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G447–59. doi: 10.1152/ajpgi.00165.2011
63. Beldi G, Enjyoji K, Wu Y, Miller L, Banz Y, Sun X, et al. The role of purinergic signaling in the liver and in transplantation: effects of extracellular nucleotides on hepatic graft vascular injury, rejection and metabolism. Front Biosci. (2008) 13:2588–603. doi: 10.2741/2868
64. Flye MW, Yu S. The synergistic effect of superoxide dismutase and adenosine triphosphate-MgCl2 on acute hepatic ischemia. Transplant Proc. (1987) 19:1324–6.
65. Ohana G, Cohen S, Rath-Wolfson L, Fishman P. A3 adenosine receptor agonist, CF102:protects against hepatic ischemia/reperfusion injury following partial hepatectomy. Mol Med Rep. (2016) 14:4335–41. doi: 10.3892/mmr.2016.5746
66. Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol. (2010) 184:4017–24. doi: 10.4049/jimmunol.0901851
67. Yoshida O, Kimura S, Jackson EK, Robson SC, Geller DA, Murase N, et al. CD39 expression by hepatic myeloid dendritic cells attenuates inflammation in liver transplant ischemia-reperfusion injury in mice. Hepatology. (2013) 58:2163–75. doi: 10.1002/hep.26593
68. Sun X, Imai M, Nowak-Machen M, Guckelberger O, Enjyoji K, Wu Y, et al. Liver damage and systemic inflammatory responses are exacerbated by the genetic deletion of CD39 in total hepatic ischemia. Purinergic Signal. (2011) 7:427–34. doi: 10.1007/s11302-011-9239-6
69. Wang P, Ba ZF, Morrison MH, Ayala A, Dean RE, Chaudry IH. Mechanism of the beneficial effects of ATP-MgCl2 following trauma-hemorrhage and resuscitation: downregulation of inflammatory cytokine (TNF, IL-6) release. J Surg Res. (1992) 52:364–71. doi: 10.1016/0022-4804(92)90117-I
70. Beldi G, Banz Y, Kroemer A, Sun X, Wu Y, Graubardt N, et al. Deletion of CD39 on natural killer cells attenuates hepatic ischemia/reperfusion injury in mice. Hepatology. (2010) 51:1702–11. doi: 10.1002/hep.23510
71. Li X, Kondo Y, Bao Y, Staudenmaier L, Lee A, Zhang J, et al. Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med. (2017) 45:e97–104. doi: 10.1097/CCM.0000000000002052
72. Savio LEB, de Andrade Mello P, Figliuolo VR, de Avelar Almeida TF, Santana PT, Oliveira SS, et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol. (2017) 67:716–26. doi: 10.1016/j.jhep.2017.05.021
73. Hoque R, Sohail MA, Salhanick S, Malik AF, Ghani A, Robson SC. Mehal WZ. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1171–9. doi: 10.1152/ajpgi.00352.2011
74. Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. (2004) 41:31–7. doi: 10.1016/j.jhep.2004.03.008
75. Longhi MS, Ma Y, Mitry RR, Bogdanos DP, Heneghan M, Cheeseman P, et al. Effect of CD4+ CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun. (2005) 25:63–71. doi: 10.1016/j.jaut.2005.05.001
76. Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, et al. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology. (2006) 130:868–82. doi: 10.1053/j.gastro.2005.12.020
77. Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC, et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology. (2014) 59:1007–15. doi: 10.1002/hep.26583
78. Torgutalp M, Efe C, Babaoglu H, Kav T. Relationship between serum adenosine deaminase levels and liver histology in autoimmune hepatitis. World J Gastroenterol. (2017) 23:3876–82. doi: 10.3748/wjg.v23.i21.3876
79. Beldi G, Wu Y, Banz Y, Nowak M, Miller L, Enjyoji K, et al. Natural killer T cell dysfunction in CD39-null mice protects against concanavalin A-induced hepatitis. Hepatology. (2008) 48:841–52. doi: 10.1002/hep.22401
80. Nowak-Machen M, Schmelzle M, Hanidziar D, Junger W, Exley M, Otterbein L, et al. Pulmonary natural killer T cells play an essential role in mediating hyperoxic acute lung injury. Am J Respir Cell Mol Biol. (2013) 48:601–9. doi: 10.1165/rcmb.2012-0180OC
81. Fausther M, Gonzales E, Dranoff JA. Role of purinergic P2X receptors in the control of liver homeostasis. Wiley Interdiscip Rev Membr Transp Signal. (2012) 1:341–8. doi: 10.1002/wmts.3210.1002/wmts.32
82. Fausther M, Sheung N, Saiman Y, Bansal MB, Dranoff JA. Activated hepatic stellate cells upregulate transcription of ecto-5′-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G904–14. doi: 10.1152/ajpgi.00015.2012
83. Feldbrugge L, Jiang ZG, Csizmadia E, Mitsuhashi S, Tran S, Yee EU, et al. Distinct roles of ecto-nucleoside triphosphate diphosphohydrolase-2 (NTPDase2) in liver regeneration and fibrosis. Purinergic Signal. (2018) 14:37–46. doi: 10.1007/s11302-017-9590-3
84. Peng ZW, Rothweiler S, Wei G, Ikenaga N, Liu SB, Sverdlov DY, et al. The ectonucleotidase ENTPD1/CD39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatol Commun. (2017) 1:957–72. doi: 10.1002/hep4.1084
85. Enjyoji K, Kotani K, Thukral C, Blumel B, Sun X, Wu Y, et al. Deletion of cd39/entpd1 results in hepatic insulin resistance. Diabetes. (2008) 57:2311–20. doi: 10.2337/db07-1265
86. Leiva A, Guzman-Gutierrez E, Contreras-Duarte S, Fuenzalida B, Cantin C, Carvajal L, et al. Adenosine receptors: modulators of lipid availability that are controlled by lipid levels. Mol Aspects Med. (2017) 55:26–44. doi: 10.1016/j.mam.2017.01.007
87. Robson SC, Schuppan D. Adenosine: tipping the balance towards hepatic steatosis and fibrosis. J Hepatol. (2010) 52:941–3. doi: 10.1016/j.jhep.2010.02.009
88. Cai Y, Li H, Liu M, Pei Y, Zheng J, Zhou J, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology. (2018) 68:48–61. doi: 10.1002/hep.29777
89. Imai M, Takigami K, Guckelberger O, Lin Y, Sevigny J, Kaczmarek E, et al. CD39/vascular ATP diphosphohydrolase modulates xenograft survival. Transplant Proc. (2000) 32:969. doi: 10.1016/S0041-1345(00)01065-4
90. Yoshida O, Dou L, Kimura S, Yokota S, Isse K, Robson SC, et al. CD39 deficiency in murine liver allografts promotes inflammatory injury and immune-mediated rejection. Transpl Immunol. (2015) 32:76–83. doi: 10.1016/j.trim.2015.01.003
91. Bach FH, Robson SC, Winkler H, Ferran C, Stuhlmeier KM, Wrighton CJ, et al. Barriers to xenotransplantation. Nat Med. (1995) 1:869–73. doi: 10.1038/nm0995-869
92. Cowan PJ, Robson SC. Progress towards overcoming coagulopathy and hemostatic dysfunction associated with xenotransplantation. Int J Surg. (2015) 23:296–300. doi: 10.1016/j.ijsu.2015.07.682
93. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. (2012) 379:1245–55. doi: 10.1016/S0140-6736(11)61347-0
94. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, et al. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. (2012) 72:2957–69. doi: 10.1158/0008-5472.CAN-11-1947
95. Khalid M, Brisson L, Tariq M, Hao Y, Guibon R, Fromont G, et al. Carcinoma-specific expression of P2Y11 receptor and its contribution in ATP-induced purinergic signalling and cell migration in human hepatocellular carcinoma cells. Oncotarget. (2017) 8:37278–90. doi: 10.18632/oncotarget.16191
96. Maynard JP, Lee JS, Sohn BH, Yu X, Lopez-Terrada D, Finegold MJ, et al. P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients. Oncotarget. (2015) 6:41162–79. doi: 10.18632/oncotarget.6240
97. Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, et al. Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/ENTPD1 null mice. Hepatology. (2013) 57:205–16. doi: 10.1002/hep.25989
98. Cai XY, Ni XC, Yi Y, He HW, Wang JX, Fu YP, et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine. (2016) 95:e4989. doi: 10.1097/MD.0000000000004989
99. Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, et al. Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia. (2011) 13:206–16. doi: 10.1593/neo.101332
100. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
Keywords: ectonucleotidase, ATP, adenosine, T-cell, intestine, liver
Citation: Vuerich M, Robson SC and Longhi MS (2019) Ectonucleotidases in Intestinal and Hepatic Inflammation. Front. Immunol. 10:507. doi: 10.3389/fimmu.2019.00507
Received: 17 November 2018; Accepted: 25 February 2019;
Published: 19 March 2019.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur and Ingénierie Cellulaire et Génique, FranceCopyright © 2019 Vuerich, Robson and Longhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon C. Robson, c3JvYnNvbkBiaWRtYy5oYXJ2YXJkLmVkdQ==
Maria Serena Longhi, bWxvbmdoaUBiaWRtYy5oYXJ2YXJkLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.