 Ricardo Grieshaber-Bouyer
Ricardo Grieshaber-Bouyer Peter A. Nigrovic
Peter A. Nigrovic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 04 March 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00346
This article is part of the Research TopicImmunophysiology of Pediatric Rheumatic DiseasesView all 18 articles
Neutrophils are versatile innate effector cells essential for immune defense but also responsible for pathologic inflammation. This dual role complicates therapeutic targeting. However, neither neutrophils themselves nor the mechanisms they employ in different forms of immune responses are homogeneous, offering possibilities for selective intervention. Here we review heterogeneity within the neutrophil population as well as in the pathways mediating neutrophil recruitment to inflamed tissues with a view to outlining opportunities for therapeutic manipulation in inflammatory disease.
Circulating leukocytes have long been categorized by microscopic appearance as lymphocytes, monocytes, and granulocytes. In the late 1870s, Paul Ehrlich distinguished neutrophils from eosinophils and basophils using aniline stains (1). Neutrophils are diverse in phenotype, although the understanding of this heterogeneity remains relatively basic. Related challenges include the relative homogeneity of neutrophils on microscopic examination, a paucity of surface markers defining clear-cut subgroups, short in vitro lifespan, and susceptibility to activation with manipulation. Experiments using newer techniques such as mass cytometry and RNAseq often exclude neutrophils by restricting analysis to cryopreserved peripheral blood mononuclear cells (PBMC) or to cells with high mRNA content. The definition of subpopulations within neutrophils has thus lagged behind work in other lineages.
This gap does not reflect doubt about the immune importance of neutrophils. Quantitative and qualitative neutrophil defects expose patients to a high risk of infection, amply displayed in both congenital and acquired neutrophil disorders (2, 3). In mice, neutropenia resulting, for example, from congenital deficiency of the transcription factor Gfi1 translates into high mortality from bacterial pathogens (4). Safety concerns translate into an understandable reluctance to target neutrophils therapeutically.
The failure to develop such strategies passes up potential opportunities to intervene in human disease. Neutrophils feature prominently in pathogenic sterile inflammation. For example, neutrophils are ubiquitous in the inflamed joint in rheumatoid arthritis (RA), in peritonitis associated with familial Mediterranean fever, and in the neutrophilic dermatoses (5–7). Among the pediatric rheumatic diseases, neutrophils are uniformly present in inflamed juvenile idiopathic arthritis (JIA) synovial fluid and have been implicated in the pathogenesis of the childhood-restricted vasculitis Kawasaki disease (8–11) While presence alone does not establish causation, evidence for a pathogenic role is frequently compelling. For example, experimental arthritis is abrogated in mice that lack neutrophils or with impaired neutrophil migration or function (12–15). Analogous studies implicate neutrophils as key effectors in a myriad of immune mediated diseases, including neuroinflammation, colitis, and bullous pemphigoid (16, 17). Neutrophils therefore remain an interesting drug target.
The therapeutic challenge is to develop strategies that preserve the defensive contribution of neutrophils while hindering their capacity to mediate sterile inflammation. Selectivity may be achieved by leveraging differences within the neutrophil population, in the way that cancer chemotherapy for targets cells that undergo frequent mitosis or bear specific mutations. Opportunities to drive a “wedge” between protective and pathogenic functions could also arise through differences in effector pathways that neutrophils engage in responding to sterile and septic triggers. This review will explore these possibilities with a view to highlighting potential treatment targets in neutrophils.
Neutrophils arise from hematopoietic stem cells (HSCs) in bone marrow, spleen, and probably lung (Figure 1) (24, 25) HSCs give rise to multipotent progenitors (MPP), which yield common myeloid progenitors (CMP) and then granulocyte monocyte progenitors (GMP). The latter commit to a program to become monocyte/dendritic cells, mast cells, basophils, or neutrophil/monocytes (26). A proliferation-competent committed progenitor termed a preNeu develops into post-mitotic immature neutrophils (myelocytes, metamyelocytes, band cells) and finally segmented mature neutrophils (18). Immature neutrophils are also be found in peripheral blood in time of immunologic stress. Granulopoiesis is stimulated predominantly through the IL-23/IL-17/G-CSF axis and to a lesser extent by GM-CSF and M-CSF, although mice lacking all three colony stimulating factors still have ~10% of normal circulating neutrophils (19, 27). Other cytokines have also been implicated, for example IL-6, which has a special importance in emergency granulopoiesis in response to systemic infection (24, 28).
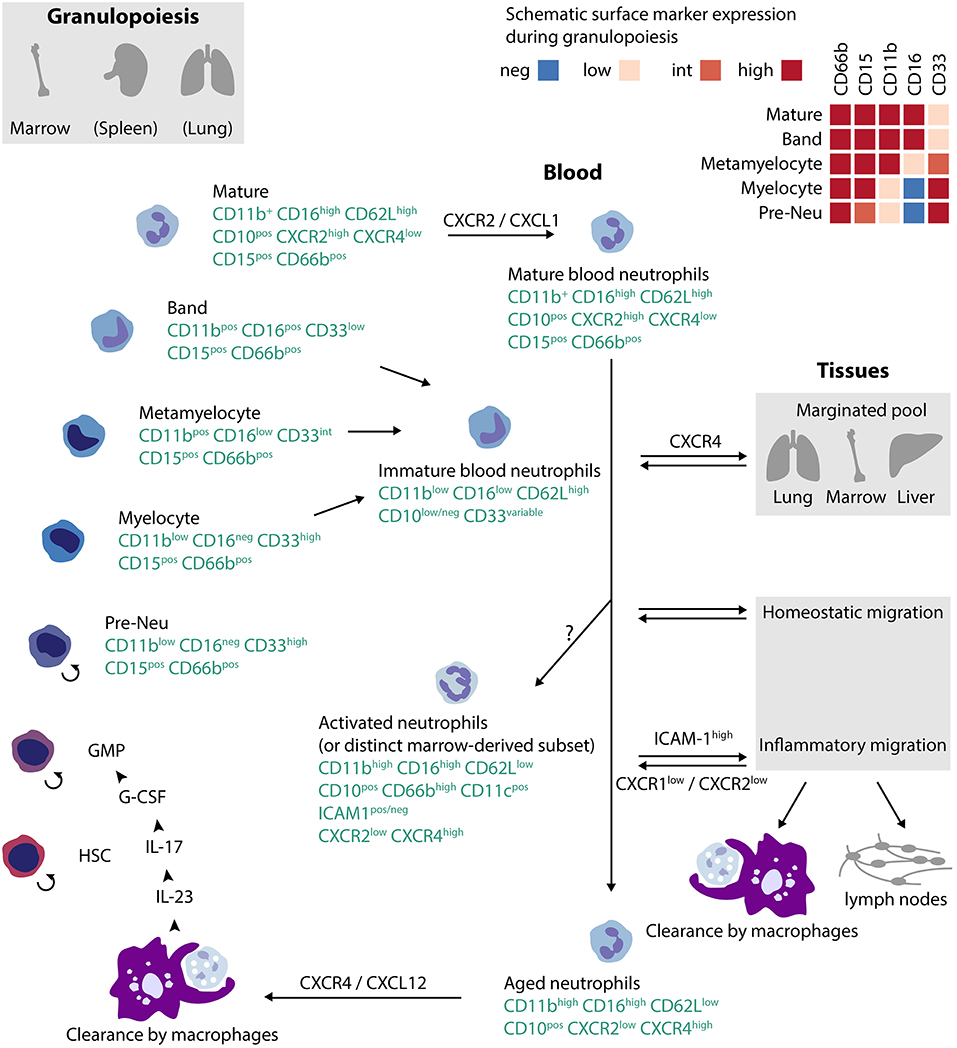
Figure 1. Lifecycle of human neutrophils. Neutrophils arise in bone marrow, spleen and (at least in mice) in lung from hematopoietic stem cells (HSC), progressing to committed granulocyte-monocyte progenitors (GMP), and then through a set of intermediate stages to mature neutrophils. Neutrophils exit to blood under the control of CXCR2, usually as mature cells but under conditions of stress also as immature cells. Over time, neutrophils age, expressing CXCR4 that mediates return to marrow. Alternate pathways for blood neutrophils include intravascular activation, intravascular margination, homeostatic migration into tissues, or migration into inflamed tissues. Clearance occurs via macrophages either in tissues or in bone marrow. The localization of the recently-defined preNeu in the previously-accepted neutrophil ontology (GMP → myeloblast → promyelocyte → myelocyte) remains uncertain; one plausible configuration is shown. The small circular arrow ↺reflects replication competence. References:(18–23).
Studies in mice suggested a circulating neutrophil half-life of 1.5 h by exogenous labeling followed by transfer and 8–10 h after in vivo labeling (29, 30). In humans, endogenous labeling raised the possibility that the neutrophil lifespan may be as long as 5.4 days (half-life 3.7 days) (20). This surprising result reflects assumptions about the relationship between marrow and circulation that have been disputed, and more recent studies suggest instead a half-life of 19 h, conforming more closely to murine data and to conventional expectations (31, 32). In vitro, human neutrophils typically undergo apoptosis within 24 h, but >90% viability even after 9 days can be achieved in the presence of GM-CSF (33). Thus, whatever the basal half-life of circulating neutrophils, it is likely that some neutrophils live for a prolonged period in vivo, especially in an inflamed context.
Neutrophils released into circulation can follow several paths (Figure 1) (34). The simplest is uneventful aging followed by return to the bone marrow, a process influenced by the microbiome and mediated through progressive expression of the SDF-1 (CXCL12) receptor CXCR4 in older neutrophils (35–38). Production of mediators by marrow macrophages phagocytizing aged neutrophils in turn regulates granulopoiesis, forming a “neutrostat” that contributes to circadian release of fresh neutrophils into blood (36). Some circulating neutrophils marginate in lung, also under the influence of CXCR4, where they can combat infection locally or be released at need into the circulation (34, 39). A marginated neutrophil population is also observed in liver and bone marrow (40).
Neutrophils recruited to inflamed tissues undergo several distinct fates. Some die locally through apoptosis or other forms of cell death, including ejection of their DNA as neutrophil extracellular traps (NETs) (41). Neutrophils can egress from tissues via lymphatics, appearing in lymph nodes loaded with antigen (42). Neutrophils from inflammatory infiltrates can return to the circulation, a possibility originally visualized in zebrafish and subsequently observed in mice (38, 43, 44). More recently, it has been recognized that neutrophils enter some tissues even without an exogenous trigger. For example, neutrophils promote angiogenesis in the regenerating uterine lining and modulate metabolism in adipose tissue, particularly in obesity (34). In healthy mice, neutrophils have also been observed in liver, skin, intestine, skeletal muscle, kidneys, and heart, although not in brain or gonads (45). While the role of these patrolling neutrophils remains to be established, one function may be to modulate the activity of local macrophages by which they are phagocytosed at the end of their lifespan (45, 46).
The neutrophil lifecycle accounts for substantial phenotypic heterogeneity (Figure 1). Mature human neutrophils exhibit a characteristic multi-lobular nucleus and high surface expression of CD16 (FcγRIII), CD62L (L-selectin), and CD10 (neutral endopeptidase), along with neutrophil lineage markers CD15 and CD66b (21, 22). By contrast, immature neutrophils released from marrow after immune stress are CD16lo and CD10lo, often but not invariably together with band nuclear morphology (22, 47, 48) Neutrophils less mature than band cells exhibit elevated CD33 (Siglec-3) and lower CXCR2, the receptor that enables mobilization out of the bone marrow niche (18, 21, 49). Immature neutrophils express more CXCR4 than mature neutrophils, likely promoting their retention in the bone marrow (18). As neutrophils age, expression of CXCR4 again increases, licensing return to the bone marrow for clearance (35). Aging is accompanied by other changes, including elevated expression of the integrins CD11b and CD11c, lower CD62L, and lower CD47, an inhibitor of phagocytosis (23). This maturation-related variation corresponds to changes in effector function. Aged neutrophils can be particularly effective in migration to sites of inflammation for immune defense, although immature neutrophils display superior bactericidal function against certain pathogens (50, 51). Murine spleen contains a mature Ly6Ghi population of motile, highly phagocytic neutrophils as well as immature Ly6Glo neutrophils with a preserved capacity for mitosis, limited mobility, and low phagocytic capacity (49). In humans receiving G-CSF, immature CD10neg neutrophils stimulate T cells while mature CD10pos neutrophils are suppressive (22).
Beyond maturational changes, neutrophils shift phenotype with activation. Mobilization of intracellular granules brings not only soluble mediators but also pre-formed membrane proteins to the neutrophil surface. These include CD66b, the α and β chains of the β2 integrin CD11b/CD18 (Mac-1), and in some individuals CD177 with its associated protease proteinase 3 (PR3) (21, 52, 53). Other surface markers are lost, including CD62L, which is rapidly shed in activated neutrophils. Activated neutrophils exhibit multiple changes in function compared with resting neutrophils. Inside-out signaling and clustering enhance integrin binding, and activated neutrophils thus exhibit enhanced mobility as well as production of lipid mediators, cytokines, chemokines, and reactive oxygen species (54).
Neutrophils acquire additional surface markers that reflect their migratory history (Figure 1). Many neutrophils that transmigrate into inflamed tissue do not die by apoptosis, as was previously assumed, but rather migrate back into the circulation (38, 43, 44). In human neutrophils, in vitro reverse transendothelial migration correlates with the appearance of surface ICAM-1 (CD54), elevation of CD18, and lower CD62L, CXCR1 and CXCR2 (55). In mice, reverse-migrated neutrophils are characterized by ICAM-1 and upregulation of CXCR4 through which they can “transplant” inflammation from the periphery to the lung before returning to the bone marrow for final clearance (38, 44, 56). ICAM-1 elevation has also been reported in human neutrophils after prolonged in vitro stimulation and in a CD16hiCD62LloCD11bhiCD11chi peripheral blood neutrophil population induced in normal donors treated with i.v. LPS, and hence is not restricted to reverse-migrated neutrophils (20). Thus healthy donors exhibit an almost uniform signature of mature neutrophils in blood, CD11b+CD16hiCD62LhiCD10hi. After administration of LPS or G-CSF, additional populations appear, including immature cells (CD11bloCD16loCD62LhiCD10lo, banded nuclear morphology) and cells with a phenotype suggestive of activated mature cells (CD11bhiCD16hiCD62LloCD10+, increased nuclear lobulation) (20, 22) Intriguingly, this latter subgroup can be found even in normal marrow; labeling studies suggest a similar age to mature neutrophils, raising the possibility that some CD62Llo cells may not be mature neutrophils activated intravascularly but rather a distinct type of neutrophil released directly from marrow under stress (57).
The broad phenotypic variability associated with maturation and activation complicates the task of discerning additional axes of heterogeneity in the form of discrete neutrophil subsets. This topic has been expertly reviewed (40, 58, 59). We will focus on targetable neutrophil heterogeneity by limiting consideration here to three areas: low-density neutrophils, immunomodulatory neutrophils, and neutrophil subgroups defined by the surface marker CD177.
The average density of neutrophils from healthy subjects is >1.080 g/ml and therefore higher than lymphocytes (1.073–1.077 g/ml) and monocytes (1.067–1.077 g/ml) (60). Density gradient centrifugation leverages these differences to separate PBMC from granulocytes. Low-density neutrophils (LDN, also termed low-density granulocytes) are neutrophils found in the PBMC layer rather than the granulocyte pellet (61, 62). On microscopic examination, many display an immature nuclear morphology, and gene expression studies suggest immaturity of the population as a whole, although expression of CD10 and other markers of maturity (e.g., CD16hi) suggest that not all LDN are neutrophils released prematurely from the marrow (22, 62, 63). Importantly, normal-density neutrophils (NDN) exposed to sera containing complement or immune complexes can also segregate with PBMC, highlighting the dynamic nature of density as a physical property of neutrophils that reflects factors such as granule content and cytoplasmic volume (22, 61). Elevated CD66b and CD11b further suggest that some LDN represent activated mature cells (62). In mice, interconversion between LDN and NDN occurs in neutrophils adoptively transferred into live animals, while ex vivo TGF-β treatment induces LDN-like features in NDN (64). Thus, LDN likely represent a diverse population of immature and activated mature neutrophils.
Elevation in peripheral blood LDN has been observed in many states of immune stress, including acute rheumatic fever, JIA, RA, systemic lupus erythematosus (SLE), autoinflammatory diseases, G-CSF administration, cancer, and sepsis (61–67). Their characteristics vary widely with context, and can for example include enhanced production of pro-inflammatory cytokines such as TNF and type I interferons, spontaneous NET generation (discussed further below), and immunosuppressive capacity (40). LDN thus reflect the phenotypic and functional plasticity of the neutrophil lineage. Whether some LDN also represent a discrete, stable neutrophil subset remains to be determined.
The view of neutrophils as simple foot-soldiers of immunity has given way to a more nuanced understanding of these cells as full participants in the immune network. Examples of the reciprocal interchange between neutrophils and adaptive immunity are abundant. Neutrophils home to lymph nodes in response to CCL19 and CCL21, carrying antigen for presentation to T cells in the context of MHC II and the canonical costimulatory molecules CD80 and CD86 (68–74). Neutrophils can differentiate into cells with surface and functional similarity to dendritic cells (75, 76). CD15int/loCD16int/lowCD11bhigh “B helper” (NBH) neutrophils have been reported in the marginal zone between lymphoid white pulp and non-lymphoid red pulp of human spleen than interact with B cells to promote IgM production and Ig class switching (77). The NBH phenotype develops under the influence of IL-10 from local cells, including splenic endothelial cells, and confers the capacity to produce mediators including APRIL, BAFF, and IL-21. Of note, not all investigators have observed these cells in human spleen, such that further exploration of NBH cells is required (78).
Neutrophils can also suppress adaptive immunity. This capacity has gained particular attention in cancer biology, where neutrophils can promote tumor growth by inhibiting responding lymphocytes (64, 79, 80). Mechanisms include arginase-1 to deplete extracellular arginine required for T cell function, reactive oxygen and nitrogen species to impair effector T cells in favor of regulatory T cells, IL-10, and TGF-β as immunosuppressive mediators, and pathways mediated through direct cell-cell contact (20, 34, 81). Myeloid cells with the capacity to block T cell activation (and under some conditions B cells and NK cells) have been termed myeloid-derived suppressor cells (MDSC), a loosely-defined category now recognized to include both neutrophil-like and monocyte-like cells (82, 83). Neutrophilic MDSC (so-called PMN-MDSC) are typically considered relatively immature, but in G-CSF-treated donors suppressive capacity in fact resides within the mature (CD10+) fraction, both LDN and NDN (22, 83). Suppressive capacity can also be elicited ex vivo in healthy-donor neutrophils exposed to TLR ligands, consistent with in vivo LPS challenge data (20, 81). Immunosuppressive capacity is thus available to neutrophils at a range of maturational states with appropriate stimulation. It remains to be established whether this capacity represents part of a broader differentiation program in a limited group of neutrophils—i.e., whether PMN-MDSC represent one or more distinct neutrophil subsets.
Another protein expressed dichotomously in human neutrophils is CD177, originally known as NB1 (84). A glycoprotein of ~60 kD attached to the neutrophil surface via a GPI linker, CD177 is present on 40–60% of neutrophils in most donors, with a range extending from 0 to 100%; some individuals manifest a CD177int population as well (52, 85, 86). Like another dichotomously-expressed neutrophil protein olfactomedin 4 (OLFM4), CD177 is localized to the specific granules, residing in the granule membrane for rapid mobilization to the surface with cell activation; however, CD177pos and OLFM4hi subsets otherwise exhibit no interdependence (52, 87).
The function of CD177 is incompletely understood. Lacking a transmembrane domain, CD177 cannot itself transmit a signal intracellularly, but antibody ligation studies show that CD177 can signal through the β2 integrins with which it associates in cis at the neutrophil surface (88, 89). Resulting enhancement in integrin expression and affinity translate CD177 ligation into neutrophil arrest, blocking transmigration (89). CD177 thus functionally echoes murine Ly6G, a neutrophil-restricted GPI-linked protein from the same Ly6/UPAR protein family that also interacts with β2 integrins and can modulate neutrophil migration, although Ly6G ligation appears to impair rather than enhance integrin binding (15, 90, 91). The endogenous receptor for CD177 remains uncertain. In vitro data implicate the endothelial adhesion molecule PECAM-1; however CD177pos neutrophils display no particular affinity for PECAM-1-expressing platelets or in vivo migratory advantage, rendering the physiological significance of the in vitro observations uncertain (89, 92–94) Interestingly, CD177 specifically binds the neutrophil protease PR3, which is stored primarily in azurophilic and specific granules in resting neutrophils and mobilized to the membrane during activation, such that CD177pos cells are identical to PR3pos cells among activated neutrophils (95–97) Some data suggest that PR3 may promote the migration of CD177pos neutrophils, but more recent data indicate that CD177 binding impairs PR3 function, leaving the functional implications of the CD177-PR3 interaction uncertain (92, 98).
The basis for the expression of CD177 in some neutrophils but not others is partially understood. CD177 resides adjacent to a related pseudogene CD177P1 that is characterized by a stop codon in the region corresponding to CD177 exon 7. Through a process of homologous recombination (gene conversion), approximately 12% of CD177 alleles feature the CD177P1 stop codon and thus represent null variants. Accordingly, the observed allelic distribution matches that expected by Hardy-Weinberg equilibrium, with 78% WT/WT, 19% WT/null, and 3% null/null (85, 99). In subjects with 2 intact copies of CD177, the CD177pos fraction is typically 50–98%; in WT/null, 10–60%; and in null/null 0%. Why some neutrophils from WT/WT donors lack CD177 expression remains undefined, but presumably reflects epigenetic regulation (100). Interestingly, in individuals with 2 intact copies of CD177, epigenetic control enforces expression of single parental allele in all CD177-expressing neutrophils, although the purpose of such tight control is unknown (101).
Distinct immunological roles of CD177pos and CD177neg neutrophils have not yet been established. Individuals with nearly 100% CD177pos neutrophils or lacking CD177 altogether appear healthy. The proportion of neutrophils expressing CD177 in an individual typically remains stable over time but rises in pregnancy, sepsis, and pathologic conditions including polycythemia vera, vasculitis, and SLE (86, 102). Studies of circulating CD177pos and CD177neg neutrophils reveal similar expression of integrins and Fc receptors, fibronectin adhesion, in vitro migration, and reactive oxygen species production (89, 103). Gene expression profiling using microarrays identified minor differences, principally in genes encoding granule proteins, although protein levels remained similar (104). Surface PR3 has been proposed as a potential modulator of T cell proliferation (105). A recent study suggested that CD177pos cells express enhanced bactericidal capacity as well as IL-22 production, reflecting a potentially protective role in inflammatory bowel disease, including through use of a murine model (106). However, the use of an antibody clone that is known to activate neutrophils (MEM-166) to sort CD177 populations complicate the interpretation of these data, as functional measures may become confounded by the activation-mediated effect of the antibody itself (89). CD177 may be of interest in vasculitis, as it can mediate the tethering of PR3, the target of c-ANCA autoantibodies to the neutrophil surface.
Beyond population heterogeneity, opportunities for intervention in neutrophil biology could emerge through effector pathways that are employed differentially in pathogenic and defensive functions. Given the myriad effector pathways employed by neutrophils, it is likely that there are multiple such opportunities. For example, in zebrafish, H2O2 is required for initiation of neutrophil recruitment to wounding but dispensable for migration toward injected bacteria (107). The zebrafish IL-1β ortholog and its downstream signaling partner MyD88 are similarly required for neutrophil recruitment triggered by wounding but not bacteria (108). We will focus here on another intriguing discrepancy between sterile and septic neutrophil migration related to the role of neutrophil β2 integrins.
The leukocyte recruitment cascade is well-established (54, 109). Circulating neutrophils roll across the endothelial surface under the influence of adhesive interactions between endothelial P- and E-selectins and neutrophil ligands including PSGL-1, further slowed by weak, transient interactions between other receptor-ligand pairs such as endothelial ICAM-1 and low-affinity neutrophil β2 integrins. With activation, endothelial cells upregulate these adhesion molecules and neutrophils augment the quantity and affinity of surface integrins, resulting in neutrophil arrest. Further neutrophil activation via chemokines presented on the endothelial glycocalyx and/or transported by endothelial cells to the luminal surface solidifies the attachment through post-adhesion strengthening (54, 110). Adherent neutrophils crawl in an integrin-dependent manner to sites suitable for transmigration between or through endothelial cells and then along sub-endothelial pericytes to sites of eventual egress into tissue (111, 112). While much of this cascade has been defined in mice, human relevance is supported by the susceptibility to infection in patients lacking the β2 chain CD18 (113).
Yet this selectin-integrin paradigm is not the whole story. Neutrophils lacking all integrins can migrate through the 3-dimensional matrix of tissue interstitium via amoeboid motion (“flowing and squeezing”) (114) Mice lacking β2 integrins or subject to integrin blockade still mount neutrophilic infiltrates. In particular, neutrophils can enter the airway without β2 integrins, although entry into the pulmonary parenchyma exhibits partial integrin dependence, as shown in studies employing adoptive transfer of mixed wild-type and CD18–/– neutrophils (90, 115–120) Indeed, under certain circumstances integrins slow neutrophil migration into lung, such that impairing integrins actually promotes neutrophil entry (116, 120). Consistent with these murine findings, neutrophilic pneumonia is observed in humans and cows lacking CD18, although recurrent pulmonary infections remain a clinical feature of this immunodeficiency in both species (113, 121, 122). In peritoneum, migratory impairment resulting from integrin deficiency or blockade is partial, with marked variability among experimental systems (90, 115, 116, 123–125). In the liver, integrins mediate neutrophil accumulation after thermal injury but are dispensable for migration induced by live bacteria in favor of CD44-hyaluronan adhesion (126). In other sites, β2 integrins play a more clear-cut role, including skin and in joints inflamed through immune complex deposition, although at least in joints the initial dependence on integrins may become less prominent as inflammation proceeds (13, 110, 114, 116, 127–129). Thus, mechanisms employed by neutrophils to enter tissues vary with site and also with stimulus, via pathways not limited to the classic leukocyte adhesion cascade.
Could this variability be exploited therapeutically? In liver, sterile infiltration is dependent on β2 integrins, while these integrins appear dispensable for septic infiltration (126). The generalizability of this principle was tested in murine peritoneum. Co-transfer of WT and CD18–/– neutrophils identified a markedly greater role for β2 integrins in sterile than septic peritonitis (i.p. IL-1β vs. live E. coli) (90). Correspondingly, targeting integrin-mediated neutrophil recruitment via an antibody directed against the integrin modulator Ly6G attenuated only sterile neutrophil infiltration, and then only in neutrophils expressing CD18. Consistent with the known variation in integrin dependence, ligation of Ly6G attenuated integrin-dependent arthritis and integrin-mediated post-adhesion strengthening on inflamed cremaster muscle but had no effect on integrin-independent neutrophil infiltration into lung (15, 90). These findings suggest that neutrophil migration in sterile disease could potentially be targeted without impairing antimicrobial defense. To the extent that integrin compromise can be rendered neutrophil-selective, as with Ly6G ligation, blockade is unlikely to phenocopy human CD18 deficiency, which impacts not only neutrophils but also monocytes, macrophages, and T cells. Humans do not express Ly6G, but CD177 could potentially fulfill a similar role, given its similar structure, selective expression in neutrophils, spatial association with β2 integrins, and capacity to block neutrophil migration upon ligation (88, 89, 91, 93). Anti-CD177 (clone MEM166) arrests migration by enhancing integrin-mediated adhesion via mechanisms including inside-out signaling and impaired integrin recycling (89). Since the relevant endogenous counterligands of both Ly6G and CD177 are unknown, it remains unclear if this difference in effect reflects intrinsic differences between these proteins or variability among the available targeting antibodies.
Importantly, not only integrin binding but also timely integrin release is required for successful transmigration. Interference with this step through interventions that prevent integrin affinity modulation represent a further opportunity for intervention in migration, a “leukadherin”-type mechanism (120, 130). To date, however, this approach lacks specificity for neutrophils.
Heterogeneity within the neutrophil population and in the pathways that neutrophils employ to access inflamed tissues represent opportunities for intervention in neutrophil-mediated disease. None of these have yet been explored definitively, so this discussion remains necessarily speculative. We will focus on three diseases: inflammatory arthritis, SLE, and vasculitis.
This disease family encompasses conditions including JIA, adult RA, and crystalline arthropathies such as gout (131). The presence of neutrophils in the joint fluid is the sine qua non of active inflammation across this spectrum. Experimental data across species implicate neutrophils in both initiation and perpetuation of disease (13–15, 132–134). Neutrophils stimulated via C5a arrest at the synovial endothelium in a β2 integrin-dependent manner and transmigrate under the influence of leukotriene B4 (LTB4) and other chemokines (110, 135, 136). Within the joint, activated neutrophils provide LTB4 and IL-1β that amplify the inflammatory process (13, 136, 137). Abundant in synovial fluid, neutrophils remain sparse in synovial tissues, although they can be observed in the inflamed pannus early in disease and at the cartilage-pannus junction (138, 139). Their proteases can injure cartilage, including through “frustrated phagocytosis” of embedded immune complexes (140). More recently, neutrophils have been recognized as a source of citrullinated autoantigens in seropositive RA at sites including joint, oropharynx and lung (141–147). Interestingly, not all neutrophil activity in arthritis is pathogenic. Neutrophil microvesicles can protect cartilage by promoting local production of TGF-β (148). In gout, aggregated NETs can help resolve flares through protease-mediated clearance of pro-inflammatory mediators, though the practical contribution of these mechanisms remains unclear (149, 150).
Neutrophils likely play roles in arthritis beyond their immediate impact within the joint environment. Their capacity for regulation of B cells and T cells, and for transport and presentation of antigen, has been noted above. Neutrophils provide mediators that contribute to systemic inflammation. For example, they are a major source of the pro-inflammatory calgranulins S100A8/A9 and S100A12, danger-associated molecular pattern (DAMP) proteins that can activate other cells via pathways including TLR4 ligation (151–153). In systemic JIA, a form of childhood arthritis characterized by fever and rash, concentrations of these mediators in blood are highly elevated, correlating with circulating neutrophil counts and potentially contributing to IL-1β release by monocytes and other cells (48, 154). Recent studies have identified a specific expansion of hypersegmented CD16pos CD62Ldim neutrophils in patients with systemic JIA with active, systemic symptoms compared to patients with active arthritis or inactive disease (155). These changes in phenotype and count are accompanied by a sepsis-like transcriptomic pattern in systemic JIA circulating neutrophils (67).
Points of intervention in neutrophil biology in arthritis range across a broad spectrum, including recruitment, effector pathways, and antigen generation. Mechanisms of recruitment blockade in mice include neutrophil-specific integrin blockade and chemokine antagonism (15, 90, 156). Targeting toxins and other compounds to neutrophils, e.g., via scavenger receptors, could potentially hasten resolution of inflammation (157). A similar strategy could alter the ability of neutrophils to generate citrullinated autoantigens, for example by introducing inhibitors of peptidylarginine deiminase enzymes (158). Of note, RA was one of the first diseases to be associated with LDN (61). Induction of the LDN phenotype by RA plasma, complement, or aggregated IgG suggests that LDN could represent an activated and/or degranulated cell population (61, 159). Direct ex vivo analysis of RA LDN has found markers of immaturity but failed to identify the enhanced capacity for NET formation observed in SLE LDN, such that their role in antigen generation remains uncertain (63).
The evidence for a role for neutrophils in SLE is now compelling, as has been reviewed in depth (158, 160, 161) Most of the attention has focused on LDN and NETs. The presence of neutrophils in PBMC preparations was described originally in a cohort of diseases including SLE, and it was in SLE that the term “low density granulocytes” was first applied (61, 62). LDN within SLE have several features that suggest a role in disease pathogenesis. They can elaborate type I interferons and generate NETs in vitro without exogenous stimuli, expose SLE-associated autoantigens and stimulate interferon production by plasmacytoid dendritic cells. (62, 162–164). Free DNA has been observed in lupus nephritis kidneys, potentially reflecting an impaired ability to clear NETs (165). Indeed, some SLE patients exhibit autoantibodies against DNase I, the enzyme primarily responsible for NET clearance, or against the NET themselves that block enzymatic attack (165). NETs can be elicited by anti-phospholipid antibodies, potentially contributing to elevated thrombosis risk in SLE (166). Finally, neutrophils may contribute to aberrant B cell development in SLE bone marrow through production of IFNα and B cell growth factors (167).
This substantial evidence base renders neutrophils, and particular NETosis, an intriguing therapeutic target in SLE (158). Interference with NETosis can ameliorate manifestations of experimental SLE (168, 169). Importantly, however, genetic deficiency of PAD4 and other pathways required for NETosis can have no effect or even worsen SLE-like disease in mice (170–172). Which murine studies replicate the human potential of NET blockade remains to be determined. Depleting or blocking LDN could represent an avenue forward, but will require further understanding of their origin and role in immune defense as well as in pathogenic inflammation.
Inflammatory disease of blood vessels can assume many forms, with a severity ranging from trivial to catastrophic. Neutrophilic infiltration into blood vessels is a common feature of vasculitis. For example, neutrophils are the dominant tissue leukocyte in immune complex-mediated leukocytoclastic vasculitis and in inflamed coronary arteries in Kawasaki disease (173–175). Neutrophils may mediate Henoch-Schönlein purpura (HSP), the most common childhood vasculitis, through their ability to recognize the Fc portion of IgA molecules (176). In anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis and related in vivo disease models, the neutrophil enzymes PR3, and myeloperoxidase are targeted by autoantibodies and play a potential pathogenic role, mediating neutrophil activation and β2 integrin-dependent adhesion to endothelium (177–182). For PR3, the subset of neutrophils expressing CD177 may play a particularly important role, because CD177 binds PR3 to enable surface expression at high level (92, 96). This binding enables anti-PR3 antibodies to activate neutrophils by signaling via CD177-associated integrins, potentially similar to the activation of neutrophils via anti-CD177 antibodies (88, 89, 103). Of note, PR3 may be expressed via CD177-independent pathways, and CD177 expression is not an invariable requirement for neutrophil-mediated inflammation induced via anti-PR3 antibodies (102, 183). ANCAs bound to the neutrophil surface also activate neutrophils via their surface Fc receptors and can trigger NETs that contribute to tissue injury (184–186). Detailed mechanistic understanding of the role of neutrophils in non-ANCA vasculitis remains more limited, but it is plausible to suspect that their presence in these sterile inflammatory infiltrates reflects a pathogenic role (187).
These considerations render neutrophils an interesting target population in vasculitis. In some patients, there is an emergent need to shut down inflammation to protect affected tissues including lung, kidney, brain, nerve, and heart. In such cases, short-term infectious risk might be a tolerable exchange for rapid cessation of disease activity. In other cases, vasculitis is chronic and indolent, and a more careful balancing act is required. The neutrophil populations and pathways to be targeted vary with the disease. These include CD177pos neutrophils in anti-PR3 ANCA-associated vasculitis, which could be depleted or treated to interfere with the ability of CD177 to bind PR3, although non-CD177-mediated PR3 expression may limit the effectiveness of such a strategy (102). Blockade of neutrophil β2 integrins could attenuate vasculitis mediated through firm adhesion between neutrophil and endothelium. Intracellular activation pathways and NETs have also been proposed as targets (188, 189).
Neutrophils exhibit a broad range of phenotypes. Much of this variability reflects developmental stage and activation status, integrating both stimulatory exposures and migratory history. As a result, neutrophils diverge from one another in nuclear morphology, buoyancy, surface markers, migratory, and phagocytic capacity, NET generation, and immunomodulatory function, among other characteristics. There remains intense interest in the possibility that this diversity manifests specific developmental programs to which individual neutrophils become committed, reflecting thereby true neutrophil subsets. However, to date evidence in favor of discrete subsets is insufficient to reject the alternative hypothesis that phenotypic variation reflects the impact of diverse environments on neutrophils within a single developmental continuum. The growing capacity for single-cell analysis of immune populations will likely provide important insights into this biology in coming years.
For the purposes of therapeutic targeting, the ontogeny of neutrophils is less important than the fact of their phenotypic diversity, now well-established if still incompletely delineated. This diversity opens the possibility of targeting neutrophils engaged in disease pathogenesis without similarly perturbing neutrophils engaged in antimicrobial defense. Such “wedge opportunities” arise not only with respect to heterogeneity within neutrophil population but also, somewhat less appreciated, in the pathways employed by neutrophils to respond to different stimuli. We reviewed here the evidence in favor of a greater role for neutrophil β2 integrins in neutrophil migration toward sterile than septic triggers, at least in some sites, and the potential role for neutrophil-specific integrin modulators (Ly6G in mice, potentially CD177 in humans) to enable lineage-specific integrin targeting. Better understanding of neutrophil biology will open further possibilities for the selective manipulation of this lineage in human therapeutics.
RG-B and PAN conceptualized and wrote the review and created the figure.
RG-B was supported by an MD Fellowship from Boehringer Ingelheim Fonds, The German Academic Scholarship Foundation (Studienstiftung) and a JBC Microgrant. PAN was supported by NIH awards R01 AR065538, R01 AR073201, and P30 AR070253, the Fundación Bechara and the Arbuckle Family Fund for Arthritis Research. The authors declare no related conflicts of interest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ehrlich P. Beiträge für Theorie und Praxis der Histologischen Färbung. Leipzig (1878). Available online at: https://www.worldcat.org/title/beitrage-fur-theorie-und-praxis-der-histologischen-farbung/oclc/63372150
2. Bodey GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med. (1966) 64:328–40. doi: 10.7326/0003-4819-64-2-328
3. Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev. (2003) 16:597–621. doi: 10.1128/CMR.16.4.597-621.2003
4. Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. (2003) 18:109–20. doi: 10.1016/S1074-7613(02)00501-0
5. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
6. Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet. (1998) 351:659–64. doi: 10.1016/S0140-6736(97)09408-7
7. Callen JP. Neutrophilic dermatoses. Dermatol Clin. (2002) 20:409–19. doi: 10.1016/S0733-8635(02)00006-2
8. Biezeveld MH, van Mierlo G, Lutter R, Kuipers IM, Dekker T, Hack CE, et al. Sustained activation of neutrophils in the course of Kawasaki disease: an association with matrix metalloproteinases. Clin Exp Immunol. (2005) 141:183–8. doi: 10.1111/j.1365-2249.2005.02829.x
9. Punzi L, Ramonda R, Glorioso S, Schiavon F, Mariuz S, Gambari PF. Predictive value of synovial fluid analysis in juvenile chronic arthritis. Ann Rheum Dis. (1992) 51:522–4. doi: 10.1136/ard.51.4.522
10. Foell D, Wittkowski H, Hammerschmidt I, Wulffraat N, Schmeling H, Frosch M, et al. Monitoring neutrophil activation in juvenile rheumatoid arthritis by S100A12 serum concentrations. Arthritis Rheum. (2004) 50:1286–95. doi: 10.1002/art.20125
11. Deanehan JK, Nigrovic PA, Milewski MD, Tan Tanny SP, Kimia AA, Smith BG, et al. Synovial fluid findings in children with knee monoarthritis in lyme disease endemic areas. Pediatr Emerg Care. (2014) 30:16–9. doi: 10.1097/PEC.0000000000000028
12. Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. (2001) 167:1601–8. doi: 10.4049/jimmunol.167.3.1601
13. Monach PA, Nigrovic PA, Chen M, Hock H, Lee DM, Benoist C, et al. Neutrophils in a mouse model of autoantibody-mediated arthritis: critical producers of Fc receptor gamma, the receptor for C5a, and lymphocyte function-associated antigen 1. Arthritis Rheum. (2010) 62:753–64. doi: 10.1002/art.27238
14. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. (2011) 187:4319–30. doi: 10.4049/jimmunol.1100341
15. Wang JX, Bair AM, King SL, Shnayder R, Huang YF, Shieh CC, et al. Ly6G ligation blocks recruitment of neutrophils via a beta2-integrin-dependent mechanism. Blood. (2012) 120:1489–98. doi: 10.1182/blood-2012-01-404046
16. Németh T, Mócsai A, Lowell CA. Neutrophils in animal models of autoimmune disease. Semin Immunol. (2016) 28:174–86. doi: 10.1016/j.smim.2016.04.001
17. Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. (1997) 100:1256–63. doi: 10.1172/JCI119639
18. Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. (2018) 48:364–79. e368. doi: 10.1016/j.immuni.2018.02.002
19. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. (2005) 22:285–94. doi: 10.1016/j.immuni.2005.01.011
20. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. (2012) 122:327–36. doi: 10.1172/JCI57990
21. Elghetany MT. Surface antigen changes during normal neutrophilic development: a critical review. Blood Cells Mol Dis. (2002) 28:260–74. doi: 10.1006/bcmd.2002.0513
22. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(-) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood. (2017) 129:1343–56. doi: 10.1182/blood-2016-04-713206
23. Adrover JM, Nicolás-Ávila JA, Hidalgo A. Aging: a temporal dimension for neutrophils. Trends Immunol. (2016) 37:334–45. doi: 10.1016/j.it.2016.03.005
24. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. (2014) 14:302–14. doi: 10.1038/nri3660
25. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. (2017) 544:105–9. doi: 10.1038/nature21706
26. Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. (2007) 26:726–40. doi: 10.1016/j.immuni.2007.06.004
27. Hibbs ML, Quilici C, Kountouri N, Seymour JF, Armes JE, Burgess AW, et al. Mice lacking three myeloid colony-stimulating factors (G-CSF, GM-CSF, and M-CSF) still produce macrophages and granulocytes and mount an inflammatory response in a sterile model of peritonitis. J Immunol. (2007) 178:6435–43. doi: 10.4049/jimmunol.178.10.6435
28. King CL, Xianli J, Malhotra I, Liu S, Mahmoud AA, Oettgen HC. Mice with a targeted deletion of the IgE gene have increased worm burdens and reduced granulomatous inflammation following primary infection with Schistosoma mansoni. J Immunol. (1997) 158:294–300.
29. Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. (2002) 100:854–61. doi: 10.1182/blood.V100.3.854
30. Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L913–21. doi: 10.1152/ajplung.2001.281.4.L913
31. Tofts PS, Chevassut T, Cutajar M, Dowell NG, Peters AM. Doubts concerning the recently reported human neutrophil lifespan of 5.4 days. Blood. (2011) 117:6053–4. doi: 10.1182/blood-2010-10-310532
32. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood. (2016) 127:3431–8. doi: 10.1182/blood-2016-03-700336
33. Brach MA, deVos S, Gruss HJ, Herrmann F. Prolongation of survival of human polymorphonuclear neutrophils by granulocyte-macrophage colony-stimulating factor is caused by inhibition of programmed cell death. Blood. (1992) 80:2920–4.
34. Nicolás-Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. (2017) 46:15–28. doi: 10.1016/j.immuni.2016.12.012
35. Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. (2003) 19:583–93. doi: 10.1016/S1074-7613(03)00263-2
36. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
37. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
38. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–6. doi: 10.1126/science.aam9690
39. Devi S, Wang Y, Chew WK, Lima R, González AN, Mattar CN, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. (2013) 210:2321–36. doi: 10.1084/jem.20130056
40. Deniset JF, Kubes P. Neutrophil heterogeneity: Bona fide subsets or polarization states? J Leukoc Biol. (2018) 103:829–38. doi: 10.1002/JLB.3RI0917-361R
41. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
42. Hampton HR, Chtanova T. The lymph node neutrophil. Semin Immunol. (2016) 28:129–36. doi: 10.1016/j.smim.2016.03.008
43. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. (2006) 80:1281–8. doi: 10.1189/jlb.0506346
44. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity. (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
45. Casanova-Acebes M, Nicolás-Ávila JA, Li JL, García-Silva S, Balachander A, Rubio-Ponce A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. (2018) 215:2778–95. doi: 10.1084/jem.20181468
46. A-Gonzalez N, Quintana JA, García-Silva S, Mazariegos M, González de la Aleja A, Nicolás-Ávila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. (2017) 214:1281–96. doi: 10.1084/jem.20161375
47. Pillay J, Ramakers BP, Kamp VM, Loi AL, Lam SW, Hietbrink F, et al. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J Leukoc Biol. (2010) 88:211–20. doi: 10.1189/jlb.1209793
48. Ter Haar NM, Tak T, Mokry M, Scholman RC, Meerding JM, de Jager W, et al. Reversal of sepsis-like features of neutrophils by interleukin-1 blockade in patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheumatol. (2018) 70:943–56. doi: 10.1002/art.40442
49. Deniset JF, Surewaard BG, Lee WY, Kubes P. Splenic Ly6G(high) mature and Ly6G(int) immature neutrophils contribute to eradication of S. pneumoniae. J Exp Med. (2017) 214:1333–50. doi: 10.1084/jem.20161621
50. Uhl B, Vadlau Y, Zuchtriegel G, Nekolla K, Sharaf K, Gaertner F, et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood. (2016) 128:2327–37. doi: 10.1182/blood-2016-05-718999
51. Leliefeld PHC, Pillay J, Vrisekoop N, Heeres M, Tak T, Kox M, et al. Differential antibacterial control by neutrophil subsets. Blood Adv. (2018) 2:1344–55. doi: 10.1182/bloodadvances.2017015578
52. Goldschmeding R, van Dalen CM, Faber N, Calafat J, Huizinga TW, van der Schoot CE, et al. Further characterization of the NB 1 antigen as a variably expressed 56-62 kD GPI-linked glycoprotein of plasma membranes and specific granules of neutrophils. Br J Haematol. (1992) 81:336–45. doi: 10.1111/j.1365-2141.1992.tb08237.x
53. Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. (1995) 374:29–33. doi: 10.1016/0014-5793(95)01073-N
54. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
55. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
56. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. (2011) 12:761–9. doi: 10.1038/ni.2062
57. Tak T, Wijten P, Heeres M, Pickkers P, Scholten A, Heck AJR, et al. Human CD62L(dim) neutrophils identified as a separate subset by proteome profiling and in vivo pulse-chase labeling. Blood. (2017) 129:3476–85. doi: 10.1182/blood-2016-07-727669
58. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
59. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
60. Zipursky A, Bow E, Seshadri RS, Brown EJ. Leukocyte density and volume in normal subjects and in patients with acute lymphoblastic leukemia. Blood. (1976) 48:361–71.
61. Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthr Rheum. (1986) 29:1334–42. doi: 10.1002/art.1780291105
62. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
63. Wright HL, Makki FA, Moots RJ, Edwards SW. Low-density granulocytes: functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J Leukoc Biol. (2017) 101:599–611. doi: 10.1189/jlb.5A0116-022R
64. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
65. Mistry P, Carmona-Rivera C, Ombrello AK, Hoffmann P, Seto NL, Jones A, et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann Rheum Dis. (2018) 77:1825–33. doi: 10.1136/annrheumdis-2018-213746
66. Morisaki T, Goya T, Ishimitsu T, Torisu M. The increase of low density subpopulations and CD10 (CALLA) negative neutrophils in severely infected patients. Surg Today. (1992) 22:322–7. doi: 10.1007/BF00308740
67. Ramanathan K, Glaser A, Lythgoe H, Ong J, Beresford MW, Midgley A, et al. Neutrophil activation signature in juvenile idiopathic arthritis indicates the presence of low-density granulocytes. Rheumatology (Oxford, England). (2018) 57:488–98. doi: 10.1093/rheumatology/kex441
68. Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. (2011) 117:1196–204. doi: 10.1182/blood-2009-11-254490
69. Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. (2008) 29:487–96. doi: 10.1016/j.immuni.2008.07.012
70. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. (2005) 106:1843–50. doi: 10.1182/blood-2005-03-1281
71. Fanger NA, Liu C, Guyre PM, Wardwell K, O'Neil J, Guo TL, et al. Activation of human T cells by major histocompatability complex class II expressing neutrophils: proliferation in the presence of superantigen, but not tetanus toxoid. Blood. (1997) 89:4128–35.
72. Mudzinski SP, Christian TP, Guo TL, Cirenza E, Hazlett KR, Gosselin EJ. Expression of HLA-DR (major histocompatibility complex class II) on neutrophils from patients treated with granulocyte-macrophage colony-stimulating factor for mobilization of stem cells. Blood. (1995) 86:2452–3.
73. Gosselin EJ, Wardwell K, Rigby WF, Guyre PM. Induction of MHC class II on human polymorphonuclear neutrophils by granulocyte/macrophage colony-stimulating factor, IFN-gamma, and IL-3. J Immunol. (1993) 151:1482–90.
74. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood. (2017) 129:1991–2001. doi: 10.1182/blood-2016-10-744441
75. Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood. (2013) 121:1677–89. doi: 10.1182/blood-2012-07-445189
76. Fites JS, Gui M, Kernien JF, Negoro P, Dagher Z, Sykes DB, et al. An unappreciated role for neutrophil-DC hybrids in immunity to invasive fungal infections. PLoS Pathog. (2018) 14:e1007073. doi: 10.1371/journal.ppat.1007073
77. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. (2011) 13:170–80. doi: 10.1038/ni.2194
78. Nagelkerke SQ, aan de Kerk DJ, Jansen MH, van den Berg TK, Kuijpers TW. Failure to detect functional neutrophil B helper cells in the human spleen. PLoS ONE. (2014) 9:e88377. doi: 10.1371/journal.pone.0088377
79. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
80. Shaul ME, Fridlender ZG. Cancer related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J. (2018) 285:4316–42. doi: 10.1111/febs.14524
81. Aarts CEM, Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. Eur J Clin Investig. (2018) 48 (Suppl. 2):e12989. doi: 10.1111/eci.12989
82. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:426. doi: 10.1158/0008-5472.CAN-06-3037
83. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
84. Lalezari P, Murphy GB, Allen FH Jr. NB1, a new neutrophil-specific antigen involved in the pathogenesis of neonatal neutropenia. J Clin Investig. (1971) 50:1108–15. doi: 10.1172/JCI106582
85. Wu Z, Liang R, Ohnesorg T, Cho V, Lam W, Abhayaratna WP, et al. Heterogeneity of human neutrophil CD177 expression results from CD177P1 pseudogene conversion. PLoS Genet. (2016) 12:e1006067. doi: 10.1371/journal.pgen.1006067
86. Stroncek DF, Caruccio L, Bettinotti M. CD177: a member of the Ly-6 gene superfamily involved with neutrophil proliferation and polycythemia vera. J Transl Med. (2004) 2:8. doi: 10.1186/1479-5876-2-8
87. Welin A, Amirbeagi F, Christenson K, Björkman L, Björnsdottir H, Forsman H, et al. The human neutrophil subsets defined by the presence or absence of OLFM4 both transmigrate into tissue in vivo and give rise to distinct NETs in vitro. PLoS ONE. (2013) 8:e69575. doi: 10.1371/journal.pone.0069575
88. Jerke U, Rolle S, Dittmar G, Bayat B, Santoso S, Sporbert A, et al. Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3-ANCA neutrophil activation. J Biol Chem. (2011) 286:7070–81. doi: 10.1074/jbc.M110.171256
89. Bai M, Grieshaber-Bouyer R, Wang J, Schmider AB, Wilson ZS, Zeng L, et al. CD177 modulates human neutrophil migration through activation-mediated integrin and chemoreceptor regulation. Blood. (2017) 130:2092–100. doi: 10.1182/blood-2017-03-768507
90. Cunin P, Lee PY, Kim E, Schmider AB, Cloutier N, Pare A, et al. Differential attenuation of β2 integrin-dependent and -independent neutrophil migration by Ly6G ligation. Blood Adv. (2019) 3:256–67. doi: 10.1182/bloodadvances.2018026732
91. Lee PY, Wang JX, Parisini E, Dascher CC, Nigrovic PA. Ly6 family proteins in neutrophil biology. J Leukoc Biol. (2013) 94:585–94. doi: 10.1189/jlb.0113014
92. Jerke U, Marino SF, Daumke O, Kettritz R. Characterization of the CD177 interaction with the ANCA antigen proteinase 3. Sci Rep. (2017) 7:43328. doi: 10.1038/srep43328
93. Bayat B, Werth S, Sachs UJ, Newman DK, Newman PJ, Santoso S. Neutrophil transmigration mediated by the neutrophil-specific antigen CD177 is influenced by the endothelial S536N dimorphism of platelet endothelial cell adhesion molecule-1. J Immunol. (2010) 184:3889–96. doi: 10.4049/jimmunol.0903136
94. Wang L, Ge S, Agustian A, Hiss M, Haller H, von Vietinghoff S. Surface receptor CD177/NB1 does not confer a recruitment advantage to neutrophilic granulocytes during human peritonitis. Eur J Haematol. (2013) 90:436–7. doi: 10.1111/ejh.12095
95. Bauer S, Abdgawad M, Gunnarsson L, Segelmark M, Tapper H, Hellmark T. Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J Leukoc Biol. (2007) 81:458–64. doi: 10.1189/jlb.0806514
96. von Vietinghoff S, Tunnemann G, Eulenberg C, Wellner M, Cristina Cardoso M, Luft FC, et al. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. (2007) 109:4487–93. doi: 10.1182/blood-2006-10-055327
97. Witko-Sarsat V, Cramer EM, Hieblot C, Guichard J, Nusbaum P, Lopez S, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood. (1999) 94:2487–96.
98. Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 contributes to transendothelial migration of NB1-positive neutrophils. J Immunol. (2012) 188:2419–26. doi: 10.4049/jimmunol.1102540
99. Li Y, Mair DC, Schuller RM, Li L, Wu J. Genetic mechanism of human neutrophil antigen 2 deficiency and expression variations. PLoS Genet. (2015) 11:e1005255. doi: 10.1371/journal.pgen.1005255
100. Jelinek J, Li J, Mnjoyan Z, Issa JP, Prchal JT, Afshar-Kharghan V. Epigenetic control of PRV-1 expression on neutrophils. Exp Hematol. (2007) 35:1677–83. doi: 10.1016/j.exphem.2007.09.008
101. Eulenberg-Gustavus C, Bähring S, Maass PG, Luft FC, Kettritz R. Gene silencing and a novel monoallelic expression pattern in distinct CD177 neutrophil subsets. J Exp Med. (2017) 214:2089–101. doi: 10.1084/jem.20161093
102. Hu N, Westra J, Huitema MG, Bijl M, Brouwer E, Stegeman CA, et al. Coexpression of CD177 and membrane proteinase 3 on neutrophils in antineutrophil cytoplasmic autoantibody-associated systemic vasculitis: anti-proteinase 3-mediated neutrophil activation is independent of the role of CD177-expressing neutrophils. Arthr Rheum. (2009) 60:1548–57. doi: 10.1002/art.24442
103. Schreiber A, Luft FC, Kettritz R. Membrane proteinase 3 expression and ANCA-induced neutrophil activation. Kidney Int. (2004) 65:2172–83. doi: 10.1111/j.1523-1755.2004.00640.x
104. Hu N, Mora-Jensen H, Theilgaard-Mönch K, Doornbos-van der Meer B, Huitema MG, Stegeman CA, et al. Differential expression of granulopoiesis related genes in neutrophil subsets distinguished by membrane expression of CD177. PLoS ONE. (2014) 9:e99671. doi: 10.1371/journal.pone.0099671
105. Yang TH, St John LS, Garber HR, Kerros C, Ruisaard KE, Clise-Dwyer K, et al. Membrane-associated proteinase 3 on granulocytes and acute myeloid leukemia inhibits T cell proliferation. J Immunol. (2018) 201:1389–99. doi: 10.4049/jimmunol.1800324
106. Zhou G, Yu L, Fang L, Yang W, Yu T, Miao Y, et al. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. (2017) 67:1052–63. doi: 10.1136/gutjnl-2016-313535
107. Deng Q, Harvie EA, Huttenlocher A. Distinct signalling mechanisms mediate neutrophil attraction to bacterial infection and tissue injury. Cell Microbiol. (2012) 14:517–28. doi: 10.1111/j.1462-5822.2011.01738.x
108. Yan B, Han P, Pan L, Lu W, Xiong J, Zhang M, et al.IL-1beta and reactive oxygen species differentially regulate neutrophil directional migration and Basal random motility in a zebrafish injury-induced inflammation model. J Immunol. (2014) 192:5998–6008. doi: 10.4049/jimmunol.1301645
109. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
110. Miyabe Y, Miyabe C, Murooka TT, Kim EY, Newton GA, Kim ND, et al. Complement C5a receptor is the key initiator of neutrophil adhesion igniting immune complex-induced arthritis. Sci Immunol. (2017) 2:eaaj2195. doi: 10.1126/sciimmunol.aaj2195
111. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
112. Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. (2012) 209:1219–34. doi: 10.1084/jem.20111622
113. Almarza Novoa E, Kasbekar S, Thrasher AJ, Kohn DB, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. (2018) 6:1418–20 e1410. doi: 10.1016/j.jaip.2017.12.008
114. Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. (2008) 453:51–5. doi: 10.1038/nature06887
115. Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J Immunol. (1990) 144:2327–33.
116. Mizgerd JP, Kubo H, Kutkoski GJ, Bhagwan SD, Scharffetter-Kochanek K, Beaudet AL, et al. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J Exp Med. (1997) 186:1357–64. doi: 10.1084/jem.186.8.1357
117. Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM. Effects of CD18 deficiency on the emigration of murine neutrophils during pneumonia. J Immunol. (1999) 163:995–9.
118. Basit A, Reutershan J, Morris MA, Solga M, Rose CE Jr, Ley K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L200–7. doi: 10.1152/ajplung.00346.2005
119. Kadioglu A, De Filippo K, Bangert M, Fernandes VE, Richards L, Jones K, et al. The integrins Mac-1 and alpha4beta1 perform crucial roles in neutrophil and T cell recruitment to lungs during Streptococcus pneumoniae infection. J Immunol. (2011) 186:5907–15. doi: 10.4049/jimmunol.1001533
120. Wilson ZS, Ahn LB, Serratelli WS, Belley MD, Lomas-Neira J, Sen M, et al. Activated beta2 integrins restrict neutrophil recruitment during murine acute pseudomonal pneumonia. Am J Respir Cell Mol Biol. (2017) 56:620–7. doi: 10.1165/rcmb.2016-0215OC
121. Hawkins HK, Heffelfinger SC, Anderson DC. Leukocyte adhesion deficiency: clinical and postmortem observations. Pediatr Pathol. (1992) 12:119–30. doi: 10.3109/15513819209023288
122. Nagahata H, Masuyama A, Masue M, Yuki M, Higuchi H, Ohtsuka H, et al. Leukocyte emigration in normal calves and calves with leukocyte adhesion deficiency. J Vet Med Sci. (1997) 59:1143–7. doi: 10.1292/jvms.59.1143
123. Schmits R, Kündig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. (1996) 183:1415–26. doi: 10.1084/jem.183.4.1415
124. Lu H, Smith CW, Perrard J, Bullard D, Tang L, Shappell SB, et al. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Investig. (1997) 99:1340–50. doi: 10.1172/JCI119293
125. Mócsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. (2002) 16:547–58. doi: 10.1016/S1074-7613(02)00303-5
126. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. (2010) 330:362–6. doi: 10.1126/science.1195491
127. Watts GM, Beurskens FJ, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, et al. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol. (2005) 174:3668–75. doi: 10.4049/jimmunol.174.6.3668
128. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. (1999) 163:5029–38.
129. Harding MG, Zhang K, Conly J, Kubes P. Neutrophil crawling in capillaries; a novel immune response to Staphylococcus aureus. PLoS Pathog. (2014) 10:e1004379. doi: 10.1371/journal.ppat.1004379
130. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, et al. Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal. (2011) 4:ra57. doi: 10.1126/scisignal.2001811
131. Nigrovic PA, Raychaudhuri S, Thompson SD. Genetics and the classification of arthritis in adults and children. Arthritis Rheumatol. (2018) 70:7–17. doi: 10.1002/art.40350
132. Nandakumar KS, Svensson L, Holmdahl R. Collagen type II-specific monoclonal antibody-induced arthritis in mice: description of the disease and the influence of age, sex, and genes. Am J Pathol. (2003) 163:1827–37. doi: 10.1016/S0002-9440(10)63542-0
133. Phelps P, McCarty DJ Jr. Crystal-induced inflammation in canine joints. II. Importance of polymorphonuclear leukocytes. J Exp Med. (1966) 124:115–26. doi: 10.1084/jem.124.1.115
134. Podolin PL, Bolognese BJ, Foley JJ, Schmidt DB, Buckley PT, Widdowson KL, et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J Immunol. (2002) 169:6435–44. doi: 10.4049/jimmunol.169.11.6435
135. Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med. (2006) 203:829–35. doi: 10.1084/jem.20052349
136. Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. (2010) 33:266–78. doi: 10.1016/j.immuni.2010.07.018
137. Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. (2006) 203:837–42. doi: 10.1084/jem.20052371
138. Mohr W, Westerhellweg H, Wessinghage D. Polymorphonuclear granulocytes in rheumatic tissue destruction. III. an electron microscopic study of PMNs at the pannus-cartilage junction in rheumatoid arthritis. Ann Rheum Dis. (1981) 40:396–9. doi: 10.1136/ard.40.4.396
139. Bromley M, Woolley DE. Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheum. (1984) 27:857–63. doi: 10.1002/art.1780270804
140. Henson PM. The immunologic release of constituents from neutrophil leukocytes. II. Mechanisms of release during phagocytosis, and adherence to nonphagocytosable surfaces. J Immunol. (1971) 107:1547–57.
141. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra140. doi: 10.1126/scitranslmed.3005580
142. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
143. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol. (2017) 69:1165–75. doi: 10.1002/art.40066
144. Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, et al. Antibody responses to citrullinated and noncitrullinated antigens in the sputum of subjects with rheumatoid arthritis and subjects at risk for development of rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:516–27. doi: 10.1002/art.40401
145. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
146. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med. (2013) 5:209ra150. doi: 10.1126/scitranslmed.3006869
147. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
148. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med. (2015) 7:315ra190. doi: 10.1126/scitranslmed.aac5608
149. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. (2014) 20:511–7. doi: 10.1038/nm.3547
150. Reber LL, Gaudenzio N, Starkl P, Galli SJ. Neutrophils are not required for resolution of acute gouty arthritis in mice. Nat Med. (2016) 22:1382–4. doi: 10.1038/nm.4216
151. Vogl T, Pröpper C, Hartmann M, Strey A, Strupat K, van den Bos C, et al. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J Biol Chem. (1999) 274:25291–6. doi: 10.1074/jbc.274.36.25291
152. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
153. Foell D, Wittkowski H, Kessel C, Lüken A, Weinhage T, Varga G, et al. Proinflammatory S100A12 can activate human monocytes via Toll-like receptor 4. Am J Respir Crit Care Med. (2013) 187:1324–34. doi: 10.1164/rccm.201209-1602OC
154. Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. (2009) 60:883–91. doi: 10.1002/art.24349
155. Brown RA, Henderlight M, Do T, Yasin S, Grom AA, DeLay M, et al. Neutrophils from children with systemic juvenile idiopathic arthritis exhibit persistent proinflammatory activation despite long-standing clinically inactive disease. Front Immunol. (2018) 9:2995. doi: 10.3389/fimmu.2018.02995
156. Angelini A, Miyabe Y, Newsted D, Kwan BH, Miyabe C, Kelly RL, et al. Directed evolution of broadly crossreactive chemokine-blocking antibodies efficacious in arthritis. Nat Commun. (2018) 9:1461. doi: 10.1038/s41467-018-03687-x
157. Robertson JD, Ward JR, Avila-Olias M, Battaglia G, Renshaw SA. Targeting neutrophilic inflammation using polymersome-mediated cellular delivery. J Immunol. (2017) 198:3596–604. doi: 10.4049/jimmunol.1601901
158. Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. (2016) 12:402–13. doi: 10.1038/nrneph.2016.71
159. Chang M, Nigrovic PA. Mechanisms of antibody-dependent and -independent arthritis. JCI insight. (in press).
160. Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. (2015) 27:448–53. doi: 10.1097/BOR.0000000000000197
161. Apel F, Zychlinsky A, Kenny EF.Apel F, Zychlinsky A, Kenny EF. The role of neutrophil extracellular traps in rheumatic diseases. Nat Rev Rheumatol. (2018) 14:467–75. doi: 10.1038/s41584-018-0039-z
162. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
163. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
164. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
165. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
166. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. (2015) 67:2990–3003. doi: 10.1002/art.39247
167. Palanichamy A, Bauer JW, Yalavarthi S, Meednu N, Barnard J, Owen T, et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J Immunol. (2014) 192:906–18. doi: 10.4049/jimmunol.1302112
168. Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Investig. (2013) 123:2981–93. doi: 10.1172/JCI67390
169. Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. (2015) 74:2199–206. doi: 10.1136/annrheumdis-2014-205365
170. Gordon RA, Herter JM, Rosetti F, Campbell AM, Nishi H, Kashgarian M, et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight. (2017) 2:92926. doi: 10.1172/jci.insight.92926
171. Kienhöfer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight. (2017) 2:92920. doi: 10.1172/jci.insight.92920
172. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. (2012) 4:157ra141. doi: 10.1126/scitranslmed.3004801
173. Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. (2010) 56:3–23. doi: 10.1111/j.1365-2559.2009.03443.x
174. Fujiwara H, Hamashima Y. Pathology of the heart in Kawasaki disease. Pediatrics. (1978) 61:100–7.
175. Takahashi K, Oharaseki T, Naoe S, Wakayama M, Yokouchi Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr Int. (2005) 47:305–10. doi: 10.1111/j.1442-200x.2005.02049.x
176. Heineke MH, Ballering AV, Jamin A, Ben Mkaddem S, Monteiro RC, Van Egmond M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schonlein purpura). Autoimmun Rev. (2017) 16:1246–53. doi: 10.1016/j.autrev.2017.10.009
177. Savage CO. Pathogenesis of anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis. Clin Exp Immunol. (2011) 164 (Suppl. 1):23–6. doi: 10.1111/j.1365-2249.2011.04362.x
178. Radford DJ, Savage CO, Nash GB. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum. (2000) 43:1337–45. doi: 10.1002/1529-0131(200006)43:6<1337::AID-ANR16>3.0.CO;2-M
179. Radford DJ, Luu NT, Hewins P, Nash GB, Savage CO. Antineutrophil cytoplasmic antibodies stabilize adhesion and promote migration of flowing neutrophils on endothelial cells. Arthritis Rheumat. (2001) 44:2851–61. doi: 10.1002/1529-0131(200112)44:12<2851::AID-ART473>3.0.CO;2-2
180. Dick J, Gan PY, Ford SL, Odobasic D, Alikhan MA, Loosen SH, et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. (2018) 93:615–25. doi: 10.1016/j.kint.2017.09.018
181. Kuligowski MP, Kwan RY, Lo C, Wong C, James WG, Bourges D, et al. Antimyeloperoxidase antibodies rapidly induce alpha-4-integrin-dependent glomerular neutrophil adhesion. Blood. (2009) 113:6485–94. doi: 10.1182/blood-2008-12-192617
182. Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, et al. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. (2005) 106:2050–8. doi: 10.1182/blood-2005-03-0921
183. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Investig. (2015) 125:4107–21. doi: 10.1172/JCI78182
184. Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol. (1994) 153:1271–80.
185. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine. (2009) 15:623-625. doi: 10.1038/nm.1959
186. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol. (2014) 25:990–7. doi: 10.1681/ASN.2013060606
187. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. (2016) 213:1983–98. doi: 10.1084/jem.20151853
188. Schreiber A, Rolle S, Peripelittchenko L, Rademann J, Schneider W, Luft FC, et al. Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int. (2010) 77:118–28. doi: 10.1038/ki.2009.420
Keywords: neutrophil, immune mediated disease, therapeutic opportunities, granulopoiesis, neutrophil migration, CD177, autoimmune, therapeutic targeting
Citation: Grieshaber-Bouyer R and Nigrovic PA (2019) Neutrophil Heterogeneity as Therapeutic Opportunity in Immune-Mediated Disease. Front. Immunol. 10:346. doi: 10.3389/fimmu.2019.00346
Received: 14 December 2018; Accepted: 11 February 2019;
Published: 04 March 2019.
Edited by:
Abbe N. de Vallejo, University of Pittsburgh, United StatesReviewed by:
Michael Hickey, Monash University, AustraliaCopyright © 2019 Grieshaber-Bouyer and Nigrovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter A. Nigrovic, cG5pZ3JvdmljQGJ3aC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.