 Caroline A. Jefferies
Caroline A. Jefferies
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 March 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00325
This article is part of the Research Topic STATs and IRFs in Innate Immunity: From Transcriptional Regulators to Therapeutic Targets View all 16 articles
The Interferon regulatory factors (IRFs) are a family of transcription factors that play pivotal roles in many aspects of the immune response, including immune cell development and differentiation and regulating responses to pathogens. Three family members, IRF3, IRF5, and IRF7, are critical to production of type I interferons downstream of pathogen recognition receptors that detect viral RNA and DNA. A fourth family member, IRF9, regulates interferon-driven gene expression. In addition, IRF4, IRF8, and IRF5 regulate myeloid cell development and phenotype, thus playing important roles in regulating inflammatory responses. Thus, understanding how their levels and activity is regulated is of critical importance given that perturbations in either can result in dysregulated immune responses and potential autoimmune disease. This review will focus the role of IRF family members in regulating type I IFN production and responses and myeloid cell development or differentiation, with particular emphasis on how regulation of their levels and activity by ubiquitination and microRNAs may impact autoimmune disease.
Interferon regulatory factors (IRFs) are a family of transcription factors that regulate many aspects of innate and adaptive immune responses—including driving anti-viral responses, responding to pathogens to drive pro-inflammatory responses and regulating immune cell differentiation (1). Comprised of 9 family members, the IRFs share significant homology within their N-terminal DNA-binding domain (DBD) of ~120 amino acids which forms a helix-loop-helix motif that recognizes specific DNA sequences similar to the interferon stimulated response element (ISRE). The C terminal domain is more diverse amongst family members and confers their unique function via regulating their ability to interact with each other and proteins outside of the IRF family. In general, the C terminal domain of each IRF member contains a nuclear export sequence, an autoinhibitory sequence, and an IRF-association domain which for most family members contains serine residues that are phosphorylated to regulate activity. IRF family members can both homodimerize and heterodimerize, forming both transcriptionally active or repressive complexes as discussed below [reviewed extensively elsewhere (1–3)]. Given their central role as transcriptional regulators of type I Interferon (IFN-α and -β) biology, they have been implicated in in the pathology of several autoimmune and autoinflammatory conditions, including systemic lupus erythematosus (SLE) in which overexpression of type I IFNs is thought to be a major contributor to pathology (4, 5).
This review will address the role of IRF family members in regulating type I IFN production and responses and myeloid cell development or differentiation. Specifically, it will focus on providing an update on how regulation of their levels and activity by microRNAs or ubiquitination may impact IFN-driven autoimmune disease.
The type I IFN system comprises 13 subtypes of IFN-α, in addition to IFN-β, IFN-ε, IFN-λ, and IFN-θ (6, 7). The main function of these cytokines is to direct anti-viral immunity: promoting differentiation of B cells into antibody producing plasma cells, inducing differentiation of naïve T cells to effector CD4 or CD8 T cells, reducing proliferation of Treg cells and driving the expression of MHC class I and II and costimulatory molecules on dendritic cells and monocytes (8). Under normal homeostatic conditions, IFN-α and IFN-β are produced in response to detection of viral RNA and DNA by pattern recognition receptors (PRRs). Toll like receptors 3, 7, and 9 are the canonical and best described of the PRRs that recognize viral RNA and DNA, but in more recent years cytosolic PRRs that detect intracellular RNA and DNA, such as RIG-I, c-GAS, and DDX41 have been recognized as key drivers of the antiviral response and type I IFN production [reviewed in (9)]. Both TLRs and cytosolic RNA and DNA can also recognize self RNA/DNA and drive the production of type I IFNs also. Self RNA and DNA released from dead or dying cells is detected by the endosomal TLRs, TLR3, 7, and 9, whilst damaged DNA or oxidized DNA released from damaged mitochondria is detected by cytosolic DNA sensors (10). These pathways are the primary drivers of IFN overproduction and IFN-driven pathology in SLE (11).
IRF3, IRF5, IRF7, and IRF8 have been shown to be positive regulators of type I interferon gene induction downstream of pattern recognition receptors [Figure 1, reviewed in (12)]. Whilst IRF1 was the first IRF to be identified as an inducer of type I IFNs (13, 14), subsequent analyses in Irf−/− MEFs suggested IRF1 was non-essential for induction of IFNs in response to cytosolic viruses (15). IRF3 and IRF7, the two family-members with greatest structural homology, are now known to be the principal mediators of IFN induction, acting downstream of cytosolic RNA and DNA receptors and the TLRs (TLR3, TLR4, TLR7, and TLR9) (9). IRF3 is ubiquitously expressed, whereas IRF7 is expressed only at very low levels, except in plasmacytoid DCs (pDCs) where it is relatively abundant (16). However, IRF7 expression is induced by type I IFNs, resulting in a feedforward loop that maximally drives type IFN expression (17). IRF3 is activated by phosphorylation (by kinases TBK1 and IKKε), promoting dimerization, nuclear translocation, association with the co-activator CREB-binding protein (CBP) and binding to canonical interferon response element sequence (IRES) in the promoter of IFN-β and IFN-α (18–21). Interestingly, a two-step phosphorylation of IRF3 has been proposed which involves TBK1 phosphorylation at site II (threonine 405 or serine 406) to relieve an autoinhibitory loop and promoting interaction with its co-factor Creb binding protein (CBP) and facilitating phosphorylation and full activation at site I (serine 385/386) (22–25). Activation of IRF3 occurs at intracellular vesicles via assembly of adaptor complexes, which then recruit in TBK1 and IKKε. TLR3 and TLR4 both use the adaptor protein TRIF to recruit in TBK1 to endosomes and phagosomes respectively, whereas RIG-I/MDA5 recruit the adaptor protein IPS-1 to recruit and activate TBK1 at the mitochondrial membrane. The growing number of cytosolic DNA-detecting PRRs (c-GAS, DDX41, IFI16) utilize the adaptor protein STING, found in the ER membrane, which once activated, translocates to the Golgi membrane to recruit and activate TBK1 (26). IRF3 can also directly induce the expression of cytokines in addition to type I IFNs, including CXCL10, RANTES, ISG56, IL-12p35, IL-23, and IL-15, whilst inhibiting IL-12β and TGF-β (27–33). However, it is currently unknown whether IRF3 activation can modulate the expression of these additional cytokines in all cells and downstream of all PRRs.
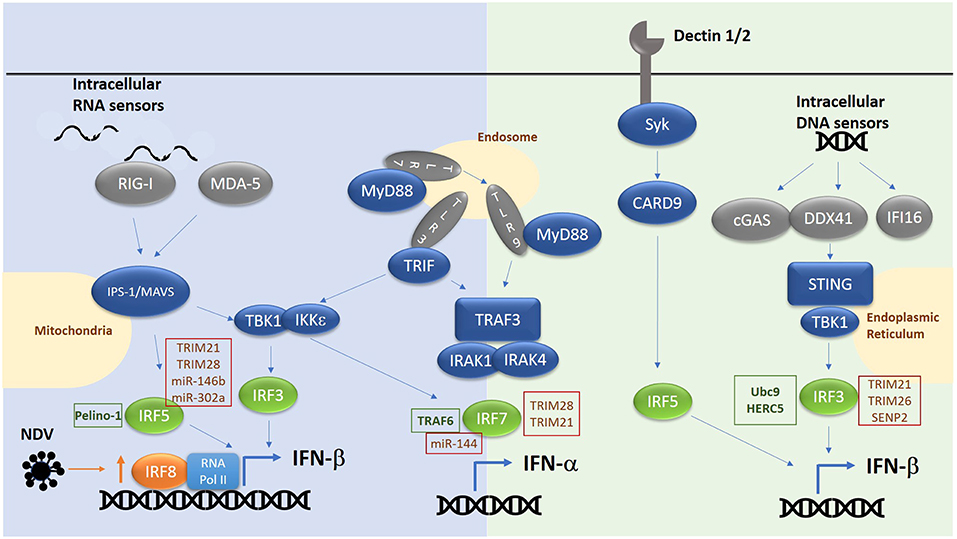
Figure 1. Overview of RNA/DNA sensing pathways. E3 ligases and microRNAs regulating IRF family members are highlighted in text boxes. Green text box for positive regulators and red for negative regulators.
In a similar manner IRF7 is activated by TBK1/IKKε downstream of cytosolic RNA/DNA sensors and TRIF dependent pathways. Here IRF7 can either homodimerize or heterodimerize with IRF3 to induce IFN-α/β expression (34). Previously it was thought that, IRF7 was not required for IFN-β expression in the early phase of a response due to its low basal level in resting cells, and that IRF3 in complex with CBP alone was required. However, consistent with a role for IRF7 as the master regulator of IFN responses (34), we now know from work in Irf7−/− MEFs that IRF7 in complex with IRF3 and CBP is essential for both the early and late phase induction of IFNs in response to single stranded RNA viruses. In pDCs in which the TLR7/TLR9 pathway is predominantly active, phosphorylation and activation of IRF7 is independent of TBK1/IKKε and instead involves recruitment of MyD88, recruitment and activation of IRAK1/2/4 signaling complex, resulting in IKKα activation and phosphorylation of IRF7, thus driving IFN-α/β expression in response to ssRNA or DNA viruses (35).
Together with IRF3 and IRF7, IRF5 is another important member of the family involved in driving IFN production. Indeed, a risk haplotype of IRF5 is associated with SLE and results in enhanced production of type I IFN. IRF5 is expressed predominantly in B cell, monocytes, macrophages and pDCs. Activation of IRF5 involves phosphorylation by IKKβ (36, 37) at conserved residues in the IAD domain. Similar to IRF3 and IRF7, this releases an autoinhibitory loop, promoting nuclear translocation and interaction with CBP. For example, mice lacking Irf5 showed increased levels of type I IFN in their serum following infection with the RNA viruses vesicular stomatitis virus (VSV) or Newcastle disease virus (NDV) (38). This implicated the RIG-I like receptor signaling pathway in activating IRF5, which was confirmed by over-expression of MAVs inducing IRF5 activation and IFN-induction (36). In addition, bacterial sensing via nucleotide-binding oligomerization domain containing (NOD)2 has been shown to drive IRF5 phosphorylation (both via TBK1 and RIP2), leading to enhanced type I IFN expression (39, 40). In pDCs IRF5 is key to the induction of pro-inflammatory genes (IL-12, IL-6, TNF-α, and IL-23) downstream of TLR7/9-MyD88, featuring ubiquitination of IRF5 by TRAF6. Whereas, IRF7 is activated from late endosomes in response to TLR7/9 ligation to drive IFN expression, IRF5 is activated from early endosomes to drive inflammatory gene expression by binding MD88 directly, which in turn facilitates its ubiquitination and activation. Interestingly, IRF4 binds same region of MyD88 as IRF5 and negatively regulates MyD88 dependent signaling (41). IRF5 is also involved in driving IFN-β expression downstream of C type lectin receptors (CLRs) such as Dectin-1 and Dectin-2 which recognize the β-glucan cell wall of C. albicans (42). Such production requires Syk and Card9 in addition to IRF5 but is independent of other IRFs.
Thus, the co-ordinate activity of IRF3, 5, and 7 downstream of the various PRRs determines the extent of type I IFN induction and the pattern of cytokines induced. As to which IRF is activated in any given situation depends on both the initiating signal and the cell type involved. For example, in NDV-infected cells the IRF5/IRF7 heterodimer has an inhibitory effect on the IFNA1 promoter, while IRF3 and IRF5 cooperatively activate this promoter (43, 44). In addition, overexpression of IRF5 or IRF7 results in expression of a different set of IFN-α subtypes, with IRF5-overexpressing cells driving mainly IFN-α8 expression, while IRF7-overexpressing cells produce mainly IFN-α1 (45). Thus, the potential exists that different levels of expression of IRF family members in different infection and disease settings will determine the level and subtype of type I IFN being produced. Indeed, given the central role for IRF3, 5, and 7 in regulating IFN expression, it is not surprising that they have been implicated in diseases such as systemic lupus erythematosus (SLE), which are driven in part by overexpression of type I IFNs. IRF5, for example, has a strong genetic association with disease (46), and a risk haplotype which results in enhanced IRF5 expression in SLE was found to correlate with enhanced levels of proinflammatory cytokines released from monocyte-derived cells from healthy individuals stimulated with NOD2 and TLRs ligands, thus indicating the presence of a correlation between IRF5 genetic variants and IRF5-mediated transcriptional regulation of cytokine genes (47). Similarly, increased association of IRF3 with the promoter of IL-23 results in increased expression of this cytokine in SLE monocytes (33). A non-synonymous SNP in IRF7 is associated with enhanced IRF7 activity and is associated with SLE (48).
A role for IRF8 in stabilizing the basal transcription machinery at type I IFN promoters to enhance IFN expression in dendritic cells (DC) and monocytes has also been reported. Whilst principally known for its role in proinflammatory gene induction, IRF8 also reportedly takes part in a second phase of interferon induction in dendritic cells in response to Newcastle Disease virus (NDV) which triggers IFN induction via activation of RIG-I dependent pathways (49). The role of IRF8 in DC-induced IFN-β requires upregulation of IRF8 expression in a feed forward loop which then works via prolonging the recruitment of the basal transcription machinery to the promoters of IFN genes in dendritic cells. This mechanism is also at play in monocytes (50). Indeed, original investigations into a possible role for IRF8 in DC function supports a role for IRF8 in mediating the development of IFN-inducing DC subsets (51–53). However, it should be noted that the role of RIG-I in IRF8-mediated IFN induction in DCs may be indirect, driving the expression of IRF8 for example rather than directly activating this transcription factor.
Canonical type I IFN signaling occurs following binding of IFN to the ubiquitously expressed type I IFN receptor (IFNAR), comprising two transmembrane proteins, IFNAR1 and IFNAR2 (54). This results in activation of two cytoplasmic kinases JAK1 and TYK2, which subsequently phosphorylate the associated transcription factors STAT1 and STAT2 (55). Once phosphorylated STAT1 and STAT2 dimerize and interact with IRF9 to form the transcriptionally active complex, ISGF3, which binds to IFN-stimulated response elements (ISRE) in the promoter region of IFN-inducible genes (56, 57). In the ISGF3 complex, DNA binding activity is facilitated by IRF9, with STAT1 providing additional DNA contacts, thus stabilizing the complex (58). STAT2 provides a transactivation domain to enhance RNA pol II dependent gene expression but is unable to bind directly to DNA (Figure 2). In addition to ISGF3-dependent gene expression, STAT1 homodimers facilitate transcriptional responses to IFN-γ (and type I IFNs to a lesser extent) by binding to the IFN-γ activated site (GAS) DNA element. A type I IFN gene signature in the peripheral blood of SLE patients has been described which correlates with increased disease activity (59–61). This may result from enhanced levels of IFN-α or -β, or from constitutive activity of the JAK-STAT pathway, downstream of the IFNAR complex. For example, the JAK-STAT pathway has been shown to be activated in SLE patients (skin and kidney, specifically) (62–64) and in murine models (65, 66), with elevated levels of STAT1 protein detected both in monocytes and skin lesions from SLE patients. With respect to ISGF3, in a mouse model of pristane-inducible IFN-driven lupus, both IRF9 and STAT1 were shown to be required for autoantibody production and development of kidney disease (67).
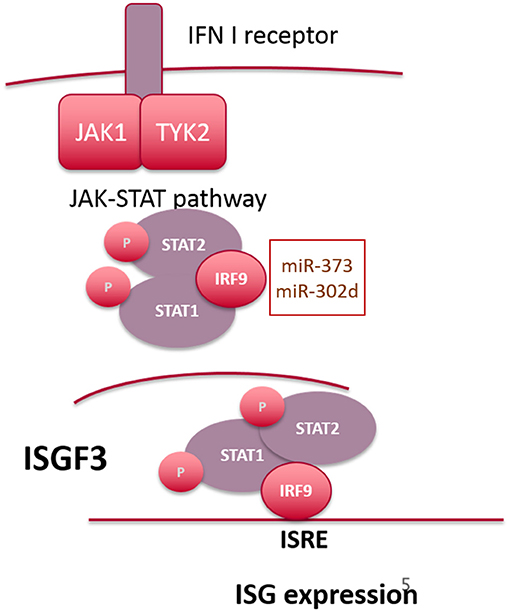
Figure 2. Overview of signaling downstream of the IFN-alpha receptor. microRNAs targeting IRF9 are highlighted in the text box.
Interestingly, the long-held paradigm that IFNα-driven tyrosine phosphorylation of both STAT1 and STAT2 is a prerequisite for interaction with IRF9 (68) has recently been challenged [reviewed in (69)]. For example, STAT2 is also capable of STAT1–independent ISRE-dependent gene expression, forming homodimers that interact with IRF9 following phosphorylation in response to IFN-α (70). However, Cheon et al. have recently demonstrated that increased expression of STAT1 and STAT2 as a result of constitutive low level IFN-β expression gives rise to a novel transcriptional complex composed of unphosphorylated STAT1 and STAT2 complexed to IRF9 (71), which drives a subset of anti-viral genes that overlap directly with the most highly expressed ISGs thus far identified in SLE patients. Although many of these studies were conducted in non-immune cells, they reveal the complexity of gene expression patterns downstream of the IFNAR receptor complex and highlight the possibility that overexpression of STAT1, STAT2, or IRF9 can have a profound effect on ISG expression and potentially allow ISG expression independent of signaling through IFNAR.
The role of IRFs in infection, protective immunity and primary immunodeficiencies has been reviewed extensively elsewhere in this focused issue (72). Given the role of IRF proteins in regulating both the production and downstream signaling of type I (and type II) interferons, it is hardly surprising that they have been both genetically and biochemically shown to be important mediators of IFN driven autoimmunity (4, 73, 74). Systemic lupus erythematosus (SLE) is amongst the best characterized for the involvement of IFNs in disease pathology. For example, in SLE, elevated IFN-α is observed in over 50% of patients and correlates with disease severity, flare and tissue involvement (specifically skin, kidney, and central nervous system). In recent years a type I IFN gene signature in the peripheral blood of SLE patients has been described which correlates with increased disease activity (59–61). More recently, Rheumatoid Arthritis (RA) patients have been found to have a type I IFN signature which correlates with autoantibody production (75), indicating that type I IFNs play an important role in driving a subset of RA (75). The various effects of type I IFNs on both the innate and the adaptive immune system contribute to the breaking of immune tolerance to self, overactivation of myeloid cells, B and T lymphocytes and differentiation or polarization of myeloid cells (monocytes and neutrophils) and T cells to more pathogenic sub-types. With respect for a role for IRFs in mediating these effects, genetic association studies have identified IRF5 and IRF7 as being risk factors for developing SLE (76–80). IRF5, like IRF7, is an IFN-inducible gene and is found to be significantly upregulated in PBMCs from SLE patients compared to healthy controls. IRF5 was found to be constitutively activated in monocytes from SLE patients resulting in enhanced levels of IL-6, TNF-α, and IFN-α (81, 82). IRF5 has been shown to be critical for the development of SLE in MRL-LPR mice (80), with alteration in function or expression of IRF5 affecting both myeloid cells and B cells in SLE-like models (79, 83). The role for IRF7 has been suggested not only for is critical role in regulating IFN-a production by pDCs, but also genetic association studies showing certain SNPs in IRF7 to confer enhanced risk for developing SLE. Functionally these genetic variants were found to be associated with increased serum IFN-α in SLE patients with autoantibodies against DNA and the Smith autoantigen (84). Interestingly, IRF3 has also been shown to be associated with enhanced IFN-α levels in SLE patients, the study also identifying a novel genetic association in a Mexican cohort of SLE patients, suggesting that IRF3 may play an important but as yet underappreciated role in driving IFN expression in SLE (85). IRF3 is also strongly associated with RA—elevated levels of phosphorylated IRF3 have been identified in the synovial tissue of RA patients and IRF3 has also been strongly associated with ISG expression in RA (86, 87). Regarding a role for other IRFs in IFN-driven disease, we recently demonstrated that IRF9 expression is enhanced in SLE monocytes and positively correlates with ISG expression (88), indicating that perturbations of IRF9 levels may alter functional activity of the ISGF3 complex and potentially contribute to disease activity. The ability of IRFs to regulate IFN production and downstream signaling thus makes them important potential targets for therapeutic intervention—highlighting the importance of understanding how their activity is controlled in molecular detail. One aspect that is rarely considered in IRF biology is the effect that conventional treatments for autoimmune diseases via their ability to alter IFN expression may also affect the expression of IRFs in patients, given the fact that IRF3, 5 7, and 9 are all IFN-regulated genes. For example, glucocorticoids, the mainstay treatment for autoimmune and inflammatory disorders, inhibit the expression of IFN stimulated genes. They therefore not only alter the expression of IFN-regulated IRFs but can directly impact their activity by targeting an interaction between the glucocorticoid-sensitive coactivator GRIP1/NCOA2 and IRF family members—IRF9 and IRF3 specifically (89, 90). Thus, in IFN driven diseases glucocorticoid treatment would be expected to reduce the expression and activity of the IFN signature as has been shown for SLE (91) and RA (92). Another mainstay for treating IFN-driven diseases (particularly SLE) also has a direct effect on the expression of IFNs and can therefore affect IRF levels. These are the anti-malarial 4-aminoquinoline drugs chloroquine and hydroxychloroquine which accumulate in the endolysosomal compartment of cells and inhibit signaling of endosomal TLRs such as TLR3, 7, and 9 and hence IFN-induction. In SLE, patients on chloroquine/Plaquenil show a reduction in IFN levels and would therefore be expected to show corresponding changes in IFN-regulated IRF expression (93). Interestingly, chloroquine is implicated in directly regulating IRF3 activity via increased expression of the deubiquitinating enzyme USP25, which enhances IRF3 nuclear translocation and results in increased LPS-induced IFN-β expression (94). This raises the possibility that chloroquine can directly or indirectly affect the activity and expression of IRF proteins in SLE or other IFN-driven diseases.
Several anti-IFN therapies have been clinically evaluated in SLE in recent years with varying degrees of success. Sifalimumab improved disease in patients with moderate to severe active disease, reducing the level of IFN stimulated genes (ISGs) in patients with initially high ISG scores, whereas the effects of Rontalizumab were greatest in patients with low to moderate levels of ISGs (95, 96). Anifrolumab, a blocking antibody against the IFN receptor (as opposed to targeting IFN-α isoforms), has reportedly better efficacy, although responses are far from complete (97). Another contribution IFN-driven disease that cannot be discounted in the potential role of intracellular RNA/DNA receptors in regulating type I IFN production. The recent identification that mutations in STING or TREX1 (which both work to regulate IFN-β production) drive monogenic forms of IFN-driven disease (interferonopathies) have suggested that dysregulation of these pathways may contribute to interferon driven diseases such as SLE or Sjogren's syndrome (98). Indeed, DNA released from stressed mitochondria in SLE neutrophils has been shown to drive IFN responses via the cGAS-STING pathway (99–101). More recently the cGAS-STING pathway has been shown to contribute to ISG regulation, independent of type I signaling through the IFNAR complex, indicating that other mechanisms may be at play in driving ISG expression in cells (102). Whether cGAS-STING activation of IRF3/IRF5 drives ISG expression directly in this scenario, or whether it drives expression of type III IFNs (IL-28A, IL-28B, and IL-29) which can also drive expression of ISGs (103), remains to be fully explored. These studies highlight the need to understand these pathways in molecular detail and underscore the complexity of targeting the IFN system therapeutically.
In addition to regulating IFN production IRFs have important roles in regulating immune cell development and differentiation (Figure 3). Whilst IRFs have been shown to regulate both lymphoid and myeloid cell development and differentiation, possibly their most influential role is observed in regulating dendritic cell (DC) subset development and macrophage differentiation/polarization, with obvious consequences for inflammatory outcomes.
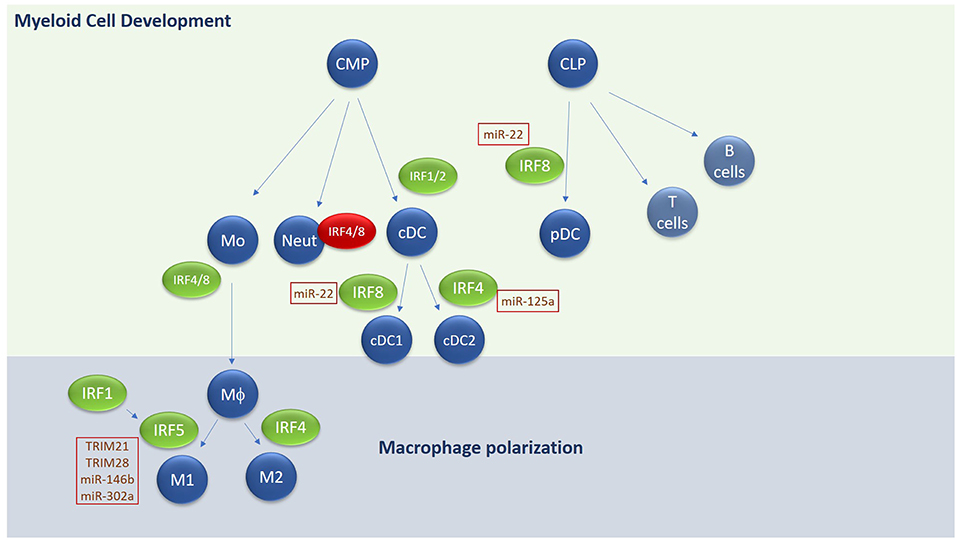
Figure 3. Overview of IRF involvement in myeloid cell development and macrophage differentiation. E3 ligases and microRNAs regulating IRF family members are highlighted in text boxes. Green text box for positive regulators and red for negative regulators. CMP, common myeloid progenitor; CLP, common lymphoid progenitor; Mo, Monocyte; Neut, Neutrophil; DC, Dendritic cell; MΦ, macrophage.
Hematopoietic stem cells give rise to both the myeloid and lymphoid arms of hematopoietic lineage. Myeloid cells derive primarily from the Common Myeloid Progenitor (CMP) whereas the lymphoid arm derive from the Common Lymphoid Progenitor (CLP). The CMP can give rise to all types of myeloid cells, including monocytes, neutrophils and most types of dendritic cells (DCs). A unique subset of DCs, termed plasmacytoid DCs derive from CLP. IRFs play an integral role in both DC and monocyte development. DCs are essential for antigen presentation and act as the bridge between innate and adaptive immune responses. They comprise four main subsets of DCs—conventional DCs (cDCs), plasmacytoid DCs (pDCs), monocyte-derived DCs, and Langerhans cells. Conventional DCs in mice are further sub-grouped into cDC1 and cDC2 subsets with different markers for human and murine counterparts (104).
Each DC subset develops under the control of differential expression of IRF4 and IRF8 in collaboration with transcription factors such as PU.1, ID2, and KLF4 (105–108). For conventional DCs, IRF8 regulates cDC1 subset development in mouse and humans, characterized by expression of CD8 or CD103 in mice or CD141 in humans and by the expression of IL-12 following TLR engagement. IRF4 on the other hand regulates cDC2 subsets, which express high levels of CD11b and CD172 in both mouse and humans and are highly efficient at inducing CD4+ T cell effector function and expansion. High expression of IRF8 in combination with E2-2 and Bcl11A are required for development of pDCs, which secrete high amounts of type I IFN in response to stimulation. IRF1 and IRF2 also appear to be important in regulating DC subset development—Irf −/− mice show a loss of splenic and epidermal DCs (due to augmented type I IFN signaling) (109, 110) whereas Irf1−/− exhibit an increase in pDCs and a decrease in CD8+ DCs in mice, along with an increase of IL-10 and TGF-β (111). In addition to regulating DC differentiation, IRF8 also promotes the commitment of myeloid progenitors to the monocyte/macrophage lineage, whilst inhibiting development of neutrophils (112). Irf8−/− mice lack bone marrow resident macrophages, in addition to CD8+ DCs and pDCs in lymphoid organs (53, 113, 114). IRF4 has also been shown to promote macrophage differentiation and impair granulocyte formation, but its role in these events is secondary to IRF8 (115). A recent role for IRF4 in negatively regulating myeloid-derived suppressor cell (MDSC) development and immunosuppressive function in tumors has recently been described (116), indicating the importance of understanding IRF-dependent regulation of myeloid cell development and function for disease.
Like DCs, macrophages play an important role in sensing pathogens, initiating innate immunity, and cross-talking with the adaptive immune system to generate an appropriate immune response. Like DCs and T cells, subsets of macrophages with differing functions have been identified [reviewed extensively in (117, 118)]. Broadly speaking they can be divided into inflammatory M1 macrophages and anti-inflammatory or resolving M2 macrophages. M2 macrophages can be further subdivided into M2a-M2d subsets. Stimuli such as GM-CSF, LPS, and IFN-γ are potent drivers of M1 polarization for example, whereas fungal products, immune complexes, M-CSF and IL-4, IL-13, IL-10, and TGF-β all promote M2 macrophages. M1 macrophages are characterized as secreting high levels of TNF-α, IFN-γ, IL-12, and IL-23, promoting strong microbiocidal functions and production of reactive nitrogen and oxygen species and promotion of Th1/Th17 responses. In contrast M2 macrophages regulate parasitic infections, promote tissue remodeling and repair and secrete immunosuppressive cytokines IL-10 and TGF-β. Regarding the different M2 subsets, M2a subtype is driven by IL-4, IL-13, and fungal and helminth infections. M2b is driven by immune complexes, IL-1/IL-18 and LPS, whilst M2c is elicited by IL-10, TGF-β and glucocorticoids. Functionally, M2a and c secrete IL-10 and TGF-β and are generally immunosuppressive, whereas M2b secrete IL-1, IL-12, and IL-10 and are implicated in inflammatory diseases such as SLE. M2d macrophages have only been identified in mice thus far and are induced from M1 macrophages following exposure to ATP (119, 120). Phenotypically they play a role in tissue remodeling and repair and have been shown to be associated with angiogenesis through secretion of VEGF (121).
Regarding IRF involvement in M1/M2 differentiation, IRF4 is strongly associated with M2 polarization, interacting with other transcription factors and chromatin remodelers to drive M2a or M2c subsets (122). The histone demethylase Jumonji domain containing-3 (Jmjd3) is involved in depressing M2-associated genes by reversing epigenetic modifications and has been shown by Satoh et al to work in concert with IRF4 to induce M2 polarization (123). Both IRF4 and Jmjd3 induce expression of M2-specific genes, arginase 1, FIZZ1, Ym1, and mannose receptor (MR) in response to IL-4 stimulation. Both Jmjd3 and IRF4 expression is driven by IL-4 in macrophages, and they in turn reciprocally regulate expression of each other (123, 124). Thus, IRF4 and Jmjd3 regulate M2a polarization downstream of IL-4 and IL-13. IRF4 also antagonizes IRF5 binding to MyD88 and in this way promotes M2 over M1 differentiation (125). Whether IRF4 is required for M2b, M2c, or M2d polarization is currently unknown.
IRF5 is the key transcription factor regulating M1 polarization (126, 127). Various inflammatory stimuli such as GM-CSF, LPS, and IFN-γ can upregulate the expression of IRF5. Enhanced expression of IRF5 in M1 macrophages is required to drive transcription of M1 markers such as IL-12, TNF-α, and IFN-γ and repress IL-10 (128). IRF5 has also been shown to regulate IL-23 secretion from macrophages, thus triggering the differentiation of Th17 cells (126). Thus, by influencing macrophage polarization toward an M1 phenotype, IRF5 plays an important role in regulating downstream adaptive immune responses and T helper cell differentiation toward a Th1 or Th17 phenotype. IRF1 seems to facilitate M1 polarization in general—priming expression of inflammatory genes associated with an M1 phenotype, such as IL-12p35 and IL-12p40 and synergizing with IRF8 to drive IL-12 production. IRF1 can also directly co-operate with IRF5 in order to drive M1 polarization in response to IFN-γ (125) and IRF1 and IFN-β work together to enhance IRF5 expression and as a consequence, M1 polarization in U937 cells. Thus, IRF1 promotes M1 polarization through its ability to enhance IRF5 levels and activity. The ability of IRF4 to compete with IRF5 for MyD88 binding and hence activation of downstream signals, suggests that relative levels of IRF4 and IRF5 in macrophages are important determinants of whether cells will polarize toward M1 or M2 phenotype.
Given the critical function of IRF family members in regulating IFN production and downstream signaling, and their role in regulating immune cell differentiation, means to regulate their activity are critical to preventing overstimulation of pathways and cells and consequent autoimmune disease. We will discuss two mechanisms to negatively regulate IRF family members—the post-translational modification of IRFs by ubiquitin and ubiquitin-like proteins and the epigenetic mechanism of microRNA (miR) targeting.
Ubiquitination, like phosphorylation, is a reversible process regulated by E3 ligases that add ubiquitin chains to targets and de-ubiquitinases that remove these chains [reviewed in (129, 130)]. Ubiquitin itself is a small, ubiquitously expressed, 76 amino acid (8.6 kDa) protein that is conjugated to an internal lysine of a target via the formation of an isopeptide bond between its C terminal glycine reside and the ε-amino residue of the lysine on the target protein. Ubiquitin chains are then formed on this initiating ubiquitin and the internal lysine targeted for polyubiquitination determines function—for example Lysine 48 (K48) linked chains target the protein for degradation, whereas K27 and K63 linked chains alter the activity of the protein target. Again, like phosphorylation, ubiquitination is a rapid method for activating or deactivating pathways. Indeed, signaling downstream of the PRRs is widely regulated by ubiquitination, both in order to activate signaling and to turn it off pathways once the response is deemed sufficient (131, 132). For example, the adaptor protein STING is regulated by multiple E3 ligases such as TRIM56, TRIM32 and AMFR, each activated by specific pathways in order to confer a specific outcome—i.e., STING activation, inactivation or relocalization. TRIM56 and TRIM32 catalyze K63-linked polyubiquitination of STING, driving dimerization and promoting its ability to interact with TBK1 and drive IFN-β expression (133, 134). K48-linked ubiquitination of STING by RNF5 and TRIM30a has also been reported, resulting in proteasomal degradation of STING and subsequent downregulation of cytosolic DNA-mediated signaling and IFN production (135, 136). AMFR on the other hand, in complex with INSIG1, catalyzes K27-linked polyubiquitination of STING, which acts as a platform to recruit in TBK1 and facilitating translocation to perinucleosomes and antiviral gene expression (137). Recently, ubiquitination of STING on K224 by the E3 ligase MUL1 has recently been shown to regulate its trafficking from the endoplasmic reticulum (ER) to the Golgi (138). In addition to ubiquitin, SUMO (small ubiquitin-like modifier) can also be covalently linked to lysine residues in target proteins, acting to regulate localization, protein-protein interactions, and activity of target proteins, a process known as SUMOylation. Indeed, TRIM38 has also been shown to regulate SUMOylation of STING during early responses to DNA virus, to promote its stability and enhance its activity (139). Thus, ubiquitination of proteins or addition of ubiquitin-like modifiers such SUMO is a highly dynamic, versatile, and effective means of regulating protein function and levels in cells.
The activity of IRF proteins is tightly controlled through both ubiquitination and SUMOylation. In general, ubiquitination and phosphorylation of IRFs are integrally linked, with one modification often being a pre-requisite for the other to take place (140). For example, ubiquitination of IRF7 by TRAF6 at lysine 444, 446, and 452 is required prior to TBK1/IKKε driven phosphorylation at serine 477 and 479 (141). The juxtaposition of both the ubiquitination site and phospho-acceptor site on IRF7 and other IRFs suggests that such these post-translational modifications work sequentially to recruit in all the players necessary for activation. And similar to STING, it appears that competing ubiquitin or ubiquitin-like modifications work to fine-tune and regulate IRF protein stability and function. For example, both IRF3 and IRF7 are negatively regulated by SUMOylation following viral infection in order to turn off and limit responses (142). TRIM28 is the E3 ligase that regulates IRF7 SUMOylation at K444 and K446 (143).
Regulation of IRF3 activity by ubiquitination or other ubiquitin like modifiers such as SUMO or ISG15, is highly complex, and most likely is highly dependent on context and cell type. IRF3 stability is regulated by K48-linked ubiquitination by TRIM21 promoting proteasomal degradation post TLR-stimulation in order to turn off and limit responses (144). Indeed, TRIM21 deficient mice develop SLE-like symptoms, accompanied by enhanced IFN levels, accompanied by sustained IRF3 levels post TLR-activation (145). TRIM21 also plays a role in autophagy and has been shown to interact with the p62 sequestersome protein, thus facilitating removal of IRF3 by targeted autophagy (146–148). In contrast, TRIM21 ubiquitination of IRF3 has also been shown to stabilize IRF3 activity via disrupting an interaction between IRF3 and Pin1, a protein that promotes IRF3 degradation (148–150). Both published and unpublished results from our group indicate that TRIM21-mediated regulation of IRF3 is complex and that it may in fact act to stabilize IRF3 in resting cells (as evidenced by decreased basal levels of IRF3 in TRIM21-deficient BMDMs) but then become activated, potentially by phosphorylation (151), to promote ubiquitination and proteolysis of IRF3 in order to limit and turn off anti-viral responses. TRIM21 also regulates IRF7 stability downstream of viral TLRs in order to limit antiviral responses (152). Like TRIM21, the E3 ligase RAUL adds K48-linked ubiquitin chains to both IRF3 and IRF7 and ultimately acts as a brake on the system in response to viral infection (153).
Similar to IRF7, IRF3 is also regulated by other ubiquitin-like modifiers: addition of SUMO and another modifier interferon stimulated gene 15 (ISG15) to on the N terminal DBD works to sustain IRF3 levels by protecting these sites from ubiquitination. Ubc9 for example SUMOylates IRF3 (142) whilst SENP2 is a deSUMOylating enzyme that removes SUMO for IRF3, presumably then allowing TRIM26 to ubiquitinate these residues with K48-linked chains, promoting IRF3 degradation (154, 155). ISGylation of IRF3 by HERC5 inhibits the interaction between IRF3 and PIN1, thus preventing Pin1-dependent IRF3 degradation (156). Thus, competing ubiquitin-like modifications on IRF3 work to either stabilize or degrade IRF3.
IRF5 stability is also regulated by ubiquitination. K63-linked ubiquitination of IRF5 by Pelino-1 for example positively regulates M1 polarization downstream of TLR4/IFN-γ. This study also linked the Pellino-1-IRF5 axis to regulation of glucose intolerance in obesity, with BMDMs from mice lacking Pellino-1 showing improved glucose intolerance when fed a high-fat diet (157). Work from our own lab has shown that TRIM21 differentially ubiquitinates different isoforms of IRF5, with IRF5-V1 and V-5 targeted or degradation by TRIM21 whereas IRF5-V2 and IRF5-V3 (IRF5-V2 linked to susceptibility to SLE) are resistant to TRIM21-mediated degradation, with obvious implications for downstream activity (158). TRIM28, a SUMO E3 ligase, is an additional negative regulator of IRF5 activity, promoting epigenetic modifications of IRF5-dependent genes (159).
Interestingly, ubiquitination of IRF1 is linked with stability and seems to be required for IL-1-induced expression of the chemokines CXCL10 and CCL5, thus promoting inflammatory cell recruitment (160). The E3 ligase responsible is the apoptosis inhibitor cIAP2, whose activity is enhanced by the sphigosphine-1-phosphate, catalyzing the addition of K63-linked chains onto IRF1. Recently Src family kinases have been shown to positively regulate K63-linked ubiquitination and accumulation of IRF1 in response to TLR7/8 signaling in monocytes and B cells (161).
As to whether other IRF proteins that are involved in regulating IFN production or downstream signaling pathways are regulated by ubiquitin-like post-translational modification remains to be determined. Given the fact that type I IFNs themselves rapidly induce expression of both E3 ligases [particularly the TRIM family (162)] that target IRFs, it is hardly surprising that many of these mechanisms are being considered as targets for therapeutic intervention in diseases driven by interferons such as SLE.
microRNAs (miRs) are important regulators of gene expression in a whole host of cellular processes and immune responses (163, 164). They are an evolutionarily conserved family of small (~22 nucleotides long) non-coding RNAs that function to bind the 3′ UTR of mRNA targets and thus regulate gene expression. Like coding RNA, non-coding RNAs such as microRNA can be either constitutively expressed or inducible—and the inducibility of these small epigenetic modifiers allows cells to exquisitely regulate and control various pathways—including those regulated by IRF proteins. Binding can trigger degradation of the target mRNA (as occurs in the majority of cases), prevent translation, or in rarer cases, stabilize the mRNA leading to positive regulation. The biogenesis and functions of microRNA have been reviewed extensively elsewhere (165–168). The focus here will be to review the role microRNAs play in regulating the levels of IRF protein members and how this contributes to both homeostasis and to disease.
There is a body of evidence to support a role for miRs in the regulation of pathways producing type I interferons and those downstream of the IFN receptor complex. For example, miRs have been implicated at all levels of TLR signaling, including manipulation of TLR levels themselves (169, 170). Downstream of the TLRs, miR-146 has been shown to target a number of signaling molecules, including IRAK1 and TRAF6 (171–173). The ability of a single miR to target multiple players on a particular pathway is a unique feature of these epigenetic regulators and suggests that they have evolved to regulate pathways and processes in the cell rather than individual players. Regarding the IRFs that regulate IFN-α and -β production, both IRF5 and IRF7 have been shown to be targeted by specific miRs. miR-302a for example is induced by influenza A and targets IRF5 directly, in order to control and limit IFN production (174). Regarding regulating IRF5 to influence M1/M2 transition, IL-10 induces miR-146b, which in turn directly targets IRF5 to promote M2 differentiation (175). microRNAs that target IRF7 on the other hand have been linked to its role in regulating oncogenesis and apoptosis rather than IFN induction per se—for example in breast cancer cells, miR-762 targets IRF7, inhibiting proliferation and invasion in a matrigel assay (176). In a separate study, miR-541 was shown to promote vascular smooth muscle cell proliferation by targeting IRF7 and thus inhibiting apoptosis (177). Regarding how microRNAs might target IRF7 in order to regulate IFN production, miR-144 was shown to target the TRAF6-IRF7 axis, targeting TRAF6 in order to attenuate attenuating the host response to influenza virus, indicating that mechanisms to regulate IRF7 activity by microRNAs exist whether direct or indirect (178).
To date however, no miR has been uncovered that specifically targets IRF3—instead many have been identified that regulate upstream adaptor proteins and hence the activity of IRF3. For example, miR-3570 targets the adaptor protein IPS-1/MAVs in order to shut-off RIG-I dependent signaling. miR-576-3p was shown to be induced in response to RNA and DNA viruses via IRF3-depependent IFN-β production, in order to shut off and limit anti-viral responses. It achieves this by targeting STING, MAVS, and TRAF3, all 3 critical players in regulating IRF3 activity or facilitating type I IFN expression. Therefore, IRF3 drives a negative regulatory loop involving miR-576-3p (179, 180).
Regarding signaling downstream of the type I IFN receptor (IFNAR), IRF9 levels and activity are critical in mediating STAT1/STAT2 driven responses. A number of microRNAs have been published that target IRF9 directly. miR-373 for example is upregulated by Hepatitis C virus (HCV) and targets IRF9 and JAK1 in order to turn off and limit anti-viral defense mechanisms (181). Our own work has shown the IRF9 is also targeted directly by miR-302d. In this study we observed that miR-302d, an estrogen regulated microRNA, is decreased in SLE monocytes, resulting in enhanced expression of IRF9 (88). The level of expression of IRF9 positively correlated with levels of interferon stimulated gene (ISG) expression and also disease activity, indicating that disruption of the microRNA balance in cells may have important consequences for immune cell function, particularly in the context of autoimmune disease.
Regarding a role for microRNAs in targeting IRFs to influence myeloid cell development or differentiation, one would expect that targeting IRF4, IRF8, or IRF5 would directly influence these events. Indeed, as mentioned above, miR-302a targets IRF5 to influence M1/M2 levels in response to viral infection (174). miR-125a has recently been shown to regulate M1/M2 differentiation and inflammation, targeting negative regulators of inflammation such as A20 and promoting an M1 or pro-inflammatory phenotype (182, 183). A recent study showed that Notch-dependent upregulation of miR-125a in tumors inhibited tumor associated macrophage function and promoted M1 macrophages via its ability to regulate HIF1-a and IRF4 (184). Regarding regulating DC development or differentiation, IRF8 is the natural target as it positively regulates pDC over cDC. In this context, miR-22 directly targets IRF8 and was shown to be highly expressed in cDCs compared with pDCs and directly influence DC differentiation (185). Thus, understanding the role of microRNAs that target IRFs involved in myeloid cell function and development may have important relevance to disease pathology.
Given the numerous roles microRNAs play in fine tuning TLR and IFN responses, it is not surprising that the dysregulation of these molecules has been implicated in SLE. To date numerous examples of dysregulated SLE associated microRNAs have been identified (186–189). Best characterized in SLE are miR-146 and miR-125, which in addition to targeting IRF5 and IRF4, also upregulate IFN-α and RANTES, respectively, thus contributing to disease activity (190, 191). miR-125a, is downregulated in SLE, has been found to negatively correlate with levels of the chemokine RANTES, a major player in organ inflammation (192) and lupus nephritis (193). Investigations into the mechanism behind this revealed a role for miR-125a in negatively regulating Kruppel-like factor 13 (KLF13) expression, a transcription factor that binds and activates the RANTES promoter, thereby inducing its expression in T cells (194). Our own work has also confirmed miR-125a expression decreased in SLE monocytes and identified a novel target, IL-16, which regulates CXCL10 expression in lung epithelial cells and helps drive lung inflammation in an autoimmune context (195). Given that monocyte and neutrophil subsets in SLE patients are key drivers of inflammation, understanding how microRNA changes in patients regulate IRF protein levels and hence contribute to myeloid cell development may be key in uncovering novel therapeutic targets.
Numerous mechanisms exist to control the innate immune response and myeloid cell differentiation in order to prevent inflammatory and autoimmune disease. As IRF family members are critical in this respect, tight regulation of their levels and activity is one mechanism of maintaining tolerance to self-antigens such as self-nucleic acids. But in different diseases it appears individual IRFs have greater or lesser involvement [reviewed in (4)]. For example—IRF3 seems to be more important in synovial inflammation in RA and responsible for ISG induction, whereas its involvement in SLE does not seem to be as important. IRF5 may perhaps be more important in SLE. So rather than targeting a single IRF for all IFN-mediated diseases, we must first understand the complex interplay between the individual IRFs in specific diseases 9 and potentially sub-types of disease in order to understand how targeting individual family members will impact the immune response as a whole.
Regarding potential targeting strategies: Ubiquitination of IRFs is a rapid and versatile way to regulate both levels and activity of IRFs, whereas epigenetic targeting of IRFs by microRNAs can fine tune IRF expression levels. Both work in concert to tailor immune responses appropriately. However, many questions remain regarding the IRFs and how they are regulated as it pertains to IFN biology: for example—what role do IRFs play in IFNAR-independent induction of ISGs? Is it possible that different combinations of STATs and IRFs can replace the canonical ISGF3 transcriptional complex? What role does regulation of availability of IRFs by microRNA targeting play in this process? And finally, can we target E3 ligases to fine tune IRF function and levels? Answering these questions will undoubtedly contribute to our understanding regarding how IRFs contribute to the pathology of autoimmune diseases such as SLE, but its biggest impact will be in explaining the following: firstly how we can improve on current IFN-targeting strategies—i.e., will JAK inhibition provide enhanced efficacy compared with IFNAR targeting strategies? And secondly, potentially uncover additional new therapeutic targets—be they modulators of E3 ligase activity or RNA-targeting strategies. As central regulators of monocytes function and IFN biology, addressing these questions promises to have a big impact in IFN-driven autoimmune disease.
The author confirms being the sole contributor of this work and has approved it for publication.
This study was supported by Lupus Research Alliance (TIL2015-332436); Arthritis Foundation Delivering on Discovery Award (AF2017-433570).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. (2008) 26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400
2. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. (2001) 19:623–55. doi: 10.1146/annurev.immunol.19.1.623
3. Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology. (2012) 1:1376–86. doi: 10.4161/onci.22475
4. Matta B, Song S, Li D, Barnes BJ. Interferon regulatory factor signaling in autoimmune disease. Cytokine. (2017) 98:15–26. doi: 10.1016/j.cyto.2017.02.006
5. Santana-de Anda K, Gomez-Martin D, Diaz-Zamudio M, Alcocer-Varela J. Interferon regulatory factors: beyond the antiviral response and their link to the development of autoimmune pathology. Autoimmun Rev. (2011) 11:98–103. doi: 10.1016/j.autrev.2011.08.006
6. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
7. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
8. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
9. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
10. Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. (2016) 44:739–54. doi: 10.1016/j.immuni.2016.04.002
11. Kanneganti TD, Kundu M, Green DR. Innate immune recognition of mtDNA–an undercover signal? Cell Metab. (2015) 21:793–4. doi: 10.1016/j.cmet.2015.05.019
12. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
13. Fujita T, Sakakibara J, Sudo Y, Miyamoto M, Kimura Y, Taniguchi T. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-beta gene regulatory elements. EMBO J. (1988) 7:3397–405. doi: 10.1002/j.1460-2075.1988.tb03213.x
14. Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. (1988) 54:903–13. doi: 10.1016/S0092-8674(88)91307-4
15. Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. (1993) 75:83–97. doi: 10.1016/S0092-8674(05)80086-8
16. Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem. (1998) 273:29210–7. doi: 10.1074/jbc.273.44.29210
17. Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. (1998) 17:6660–9. doi: 10.1093/emboj/17.22.6660
18. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. (2003) 300:1148–51. doi: 10.1126/science.1081315
19. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. (2003) 4:491–6. doi: 10.1038/ni921
20. McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA. (2004) 101:233–8. doi: 10.1073/pnas.2237236100
21. Yanai H, Chiba S, Hangai S, Kometani K, Inoue A, Kimura Y, et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc Natl Acad Sci USA. (2018) 115:5253–8. doi: 10.1073/pnas.1803936115
22. Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. (2003) 10:913–21. doi: 10.1038/nsb1002
23. Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, et al. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. (2003) 10:922–7. doi: 10.1038/nsb1001
24. Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem. (2004) 279:9698–702. doi: 10.1074/jbc.M310616200
25. Panne D, McWhirter SM, Maniatis T, Harrison SC. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J Biol Chem. (2007) 282:22816–22. doi: 10.1074/jbc.M703019200
26. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. (2016) 17:1142–9. doi: 10.1038/ni.3558
27. Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, et al. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. (2003) 306:860–6. doi: 10.1016/S0006-291X(03)01049-0
28. Goriely S, Molle C, Nguyen M, Albarani V, Haddou NO, Lin R, et al. Interferon regulatory factor 3 is involved in Toll-like receptor 4 (TLR4)- and TLR3-induced IL-12p35 gene activation. Blood. (2006) 107:1078–84. doi: 10.1182/blood-2005-06-2416
29. Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: Localization of the transactivation and autoinhibitory domains. Mol Cell Biol. (1999) 19:2465–74. doi: 10.1128/MCB.19.4.2465
30. Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M Jr., et al. Direct, interferon-independent activation of the CXCL10 promoter by NF-kappaB and interferon regulatory factor 3 during hepatitis C virus infection. J Virol. (2014) 88:1582–90. doi: 10.1128/JVI.02007-13
31. Koshiba R, Yanai H, Matsuda A, Goto A, Nakajima A, Negishi H, et al. Regulation of cooperative function of the Il12b enhancer and promoter by the interferon regulatory factors 3 and 5. Biochem Biophys Res Commun. (2013) 430:95–100. doi: 10.1016/j.bbrc.2012.11.006
32. Xu P, Bailey-Bucktrout S, Xi Y, Xu D, Du D, Zhang Q, et al. Innate antiviral host defense attenuates TGF-beta function through IRF3-mediated suppression of Smad signaling. Mol Cell. (2014) 56:723–37. doi: 10.1016/j.molcel.2014.11.027
33. Smith S, Gabhann JN, Higgs R, Stacey K, Wahren-Herlenius M, Espinosa A, et al. Enhanced interferon regulatory factor 3 binding to the interleukin-23p19 promoter correlates with enhanced interleukin-23 expression in systemic lupus erythematosus. Arthritis Rheum. (2012) 64:1601–9. doi: 10.1002/art.33494
34. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. (2005) 434:772–7. doi: 10.1038/nature03464
35. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. (2013) 78:105–16. doi: 10.1101/sqb.2013.78.020321
36. Ren J, Chen X, Chen ZJ. IKKbeta is an IRF5 kinase that instigates inflammation. Proc Natl Acad Sci USA. (2014) 111:17438–43. doi: 10.1073/pnas.1418516111
37. Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. Protein kinase IKKbeta-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc Natl Acad Sci USA. (2014) 111:17432–7. doi: 10.1073/pnas.1418399111
38. Paun A, Reinert JT, Jiang Z, Medin C, Balkhi MY, Fitzgerald KA, et al. Functional characterization of murine interferon regulatory factor 5 (IRF-5) and its role in the innate antiviral response. J Biol Chem. (2008) 283:14295–308. doi: 10.1074/jbc.M800501200
39. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. (2009) 5:e1000500. doi: 10.1371/journal.ppat.1000500
40. Chang Foreman HC, Van Scoy S, Cheng TF, Reich NC. Activation of interferon regulatory factor 5 by site specific phosphorylation. PLoS ONE. (2012) 7:e33098. doi: 10.1371/journal.pone.0033098
41. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
42. del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, et al. Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity. (2013) 38:1176–86. doi: 10.1016/j.immuni.2013.05.010
43. Cheng TF, Brzostek S, Ando O, Van Scoy S, Kumar KP, Reich NC. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J Immunol. (2006) 176:7462–70. doi: 10.4049/jimmunol.176.12.7462
44. Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem. (2003) 278:16630–41. doi: 10.1074/jbc.M212609200
45. Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. (2001) 276:23382–90. doi: 10.1074/jbc.M101216200
46. Lazzari E, Jefferies CA. IRF5-mediated signaling and implications for SLE. Clin Immunol. (2014) 153:343–52. doi: 10.1016/j.clim.2014.06.001
47. Hedl M, Abraham C. IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor-mediated cytokine secretion in human monocyte-derived cells. J Immunol. (2012) 188:5348–56. doi: 10.4049/jimmunol.1103319
48. Fu Q, Zhao J, Qian X, Wong JL, Kaufman KM, Yu CY, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum. (2011) 63:749–54. doi: 10.1002/art.30193
49. Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, et al. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity. (2007) 27:228–39. doi: 10.1016/j.immuni.2007.06.009
50. Li P, Wong JJ, Sum C, Sin WX, Ng KQ, Koh MB, et al. IRF8 and IRF3 cooperatively regulate rapid interferon-beta induction in human blood monocytes. Blood. (2011) 117:2847–54. doi: 10.1182/blood-2010-07-294272
51. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC III et al., ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. (2002) 196:1415–25. doi: 10.1084/jem.20021263
52. Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ, et al. IFN regulatory factor-4 and−8 govern dendritic cell subset development and their functional diversity. J Immunol. (2005) 174:2573–81. doi: 10.4049/jimmunol.174.5.2573
53. Tsujimura H, Nagamura-Inoue T, Tamura T, Ozato K. IFN consensus sequence binding protein/IFN regulatory factor-8 guides bone marrow progenitor cells toward the macrophage lineage. J Immunol. (2002) 169:1261–9. doi: 10.4049/jimmunol.169.3.1261
54. Ng CT, Mendoza JL, Garcia KC, Oldstone MB. Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell. (2016) 164:349–52. doi: 10.1016/j.cell.2015.12.027
55. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
56. Levy DE, Kessler DS, Pine R, Darnell JE Jr. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. (1989) 3:1362–71. doi: 10.1101/gad.3.9.1362
57. Qureshi SA, Salditt-Georgieff M, Darnell JE Jr. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci USA. (1995) 92:3829–33. doi: 10.1073/pnas.92.9.3829
58. Bluyssen HA, Levy DE. Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem. (1997) 272:4600–5. doi: 10.1074/jbc.272.7.4600
59. Kennedy WP, Maciuca R, Wolslegel K, Tew W, Abbas AR, Chaivorapol C, et al. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci Med. (2015) 2:e000080. doi: 10.1136/lupus-2014-000080
60. Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal P, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. (2009) 18:980–9. doi: 10.1177/0961203309105529
61. Bauer JW, Petri M, Batliwalla FM, Koeuth T, Wilson J, Slattery C, et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: a validation study. Arthritis Rheum. (2009) 60:3098–107. doi: 10.1002/art.24803
62. Kawasaki M, Fujishiro M, Yamaguchi A, Nozawa K, Kaneko H, Takasaki Y, et al. Possible role of the JAK/STAT pathways in the regulation of T cell-interferon related genes in systemic lupus erythematosus. Lupus. (2011) 20:1231–9. doi: 10.1177/0961203311409963
63. Li J, Zhao S, Yi M, Hu X, Li J, Xie H, et al. Activation of JAK-STAT1 signal transduction pathway in lesional skin and monocytes from patients with systemic lupus erythematosus. J Central South Univ Med Sci. (2011) 36:109–15. doi: 10.3969/j.issn.1672-7347.2011.02.003
64. Liang Y, Xu WD, Yang XK, Fang XY, Liu YY, Ni J, et al. Association of signaling transducers and activators of transcription 1 and systemic lupus erythematosus. Autoimmunity. (2014) 47:141–5. doi: 10.3109/08916934.2013.873415
65. Wang S, Yang N, Zhang L, Huang B, Tan H, Liang Y, et al. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus. (2010) 19:1171–80. doi: 10.1177/0961203310367660
66. Dong J, Wang QX, Zhou CY, Ma XF, Zhang YC. Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus. (2007) 16:101–9. doi: 10.1177/0961203306075383
67. Thibault DL, Chu AD, Graham KL, Balboni I, Lee LY, Kohlmoos C, et al. IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J Clin Invest. (2008) 118:1417–26. doi: 10.1172/JCI30065
68. Pilz A, Ramsauer K, Heidari H, Leitges M, Kovarik P, Decker T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO Rep. (2003) 4:368–73. doi: 10.1038/sj.embor.embor802
69. Au-Yeung N, Mandhana R, Horvath CM. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. Jak Stat. (2013) 2:e23931. doi: 10.4161/jkst.23931
70. Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen YL, Chmielewski S, et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem J. (2015) 466:511–24. doi: 10.1042/BJ20140644
71. Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep. (2016) 17:367–82. doi: 10.15252/embr.201540726
72. Mogensen TH. IRF and STAT Transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol. (2018) 9:3047. doi: 10.3389/fimmu.2018.03047
73. Crow MK. Type I interferon in systemic lupus erythematosus. Curr Top Microbiol Immunol. (2007) 316:359–86. doi: 10.1007/978-3-540-71329-6_17
74. Ronnblom L. The importance of the type I interferon system in autoimmunity. Clin Exp Rheumatol. (2016) 34(4 Suppl. 98):21–4.
75. Castaneda-Delgado JE, Bastian-Hernandez Y, Macias-Segura N, Santiago-Algarra D, Castillo-Ortiz JD, Aleman-Navarro AL, et al. Type I interferon gene response is increased in early and established rheumatoid arthritis and correlates with autoantibody production. Front Immunol. (2017) 8:285. doi: 10.3389/fimmu.2017.00285
76. Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol. (2012) 24:530–7. doi: 10.1016/j.coi.2012.07.008
77. Byrne JC, Ni Gabhann J, Lazzari E, Mahony R, Smith S, Stacey K, et al. Genetics of SLE: functional relevance for monocytes/macrophages in disease. Clin Dev Immunol. (2012) 2012:582352. doi: 10.1155/2012/582352
78. Watkins AA, Yasuda K, Wilson GE, Aprahamian T, Xie Y, Maganto-Garcia E, et al. IRF5 deficiency ameliorates lupus but promotes atherosclerosis and metabolic dysfunction in a mouse model of lupus-associated atherosclerosis. J Immunol. (2015) 194:1467–79. doi: 10.4049/jimmunol.1402807
79. Yasuda K, Watkins AA, Kochar GS, Wilson GE, Laskow B, Richez C, et al. Interferon regulatory factor-5 deficiency ameliorates disease severity in the MRL/lpr mouse model of lupus in the absence of a mutation in DOCK2. PLoS ONE. (2014) 9:e103478. doi: 10.1371/journal.pone.0103478
80. Tada Y, Kondo S, Aoki S, Koarada S, Inoue H, Suematsu R, et al. Interferon regulatory factor 5 is critical for the development of lupus in MRL/lpr mice. Arthritis Rheum. (2011) 63:738–48. doi: 10.1002/art.30183
81. Stone RC, Feng D, Deng J, Singh S, Yang L, Fitzgerald-Bocarsly P, et al. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheum. (2012) 64:788–98. doi: 10.1002/art.33395
82. Feng D, Stone RC, Eloranta ML, Sangster-Guity N, Nordmark G, Sigurdsson S, et al. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic lupus erythematosus. Arthritis Rheum. (2010) 62:562–73. doi: 10.1002/art.27223
83. Yasuda K, Nundel K, Watkins AA, Dhawan T, Bonegio RG, Ubellacker JM, et al. Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5-deficient mice with and without a mutation in DOCK2. Int Immunol. (2013) 25:295–306. doi: 10.1093/intimm/dxs114
84. Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. (2010) 62:553–61. doi: 10.1002/art.27182
85. Santana-de Anda K, Gomez-Martin D, Monsivais-Urenda AE, Salgado-Bustamante M, Gonzalez-Amaro R, Alcocer-Varela J. Interferon regulatory factor 3 as key element of the interferon signature in plasmacytoid dendritic cells from systemic lupus erythematosus patients: novel genetic associations in the Mexican mestizo population. Clin Exp Immunol. (2014) 178:428–37. doi: 10.1111/cei.12429
86. Sweeney SE, Kimbler TB, Firestein GS. Synoviocyte innate immune responses: II. Pivotal role of IFN regulatory factor 3. J Immunol. (2010) 184:7162–8. doi: 10.4049/jimmunol.0903944
87. Sweeney SE, Mo L, Firestein GS. Antiviral gene expression in rheumatoid arthritis: role of IKKepsilon and interferon regulatory factor 3. Arthritis Rheum. (2007) 56:743–52. doi: 10.1002/art.22421
88. Smith S. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. (2017) 79:105–11. doi: 10.1016/j.jaut.2017.03.003
89. Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol. (2010) 30:4564–74. doi: 10.1128/MCB.00146-10
90. Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. (2006) 25:108–17. doi: 10.1038/sj.emboj.7600919
91. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
92. de Jong TD, Vosslamber S, Blits M, Wolbink G, Nurmohamed MT, van der Laken CJ, et al. Effect of prednisone on type I interferon signature in rheumatoid arthritis: consequences for response prediction to rituximab. Arthritis Res Ther. (2015) 17:78. doi: 10.1186/s13075-015-0564-y
93. Willis R, Seif AM, McGwin G Jr. Martinez-Martinez LA, Gonzalez EB, Dang N, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA (LXXV), a multiethnic US cohort. Lupus. (2012) 21:830–5. doi: 10.1177/0961203312437270
94. Ding C, Li F, Long Y, Zheng J. Chloroquine attenuates lipopolysaccharide-induced inflammatory responses through upregulation of USP25. Can J Physiol Pharmacol. (2017) 95:481–91. doi: 10.1139/cjpp-2016-0303
95. Kennedy JW, Khan W. Total hip arthroplasty in systemic lupus erythematosus: a systematic review. Int J Rheumatol. (2015) 2015:475489. doi: 10.1155/2015/475489
96. Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. (2013) 65:1011–21. doi: 10.1002/art.37824
97. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
98. Lee-Kirsch MA. The Type I interferonopathies. Annu Rev Med. (2017) 68:297–315. doi: 10.1146/annurev-med-050715-104506
99. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:800–7. doi: 10.1002/art.40002
100. Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. (2018) 3:e99276. doi: 10.1172/jci.insight.99276
101. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
102. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
103. Wack A, Terczynska-Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol. (2015) 16:802–9. doi: 10.1038/ni.3212
104. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
105. Chopin M, Allan RS, Belz GT. Transcriptional regulation of dendritic cell diversity. Front Immunol. (2012) 3:26. doi: 10.3389/fimmu.2012.00026
106. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol. (2012) 12:101–13. doi: 10.1038/nri3149
107. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
108. See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. (2017) 356:eaag3009. doi: 10.1126/science.aag3009
109. Honda K, Mizutani T, Taniguchi T. Negative regulation of IFN-alpha/beta signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc Natl Acad Sci USA. (2004) 101:2416–21. doi: 10.1073/pnas.0307336101
110. Ichikawa E, Hida S, Omatsu Y, Shimoyama S, Takahara K, Miyagawa S, et al. Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc Natl Acad Sci USA. (2004) 101:3909–14. doi: 10.1073/pnas.0400610101
111. Gabriele L, Fragale A, Borghi P, Sestili P, Stellacci E, Venditti M, et al. IRF-1 deficiency skews the differentiation of dendritic cells toward plasmacytoid and tolerogenic features. J Leukoc Biol. (2006) 80:1500–11. doi: 10.1189/jlb.0406246
112. Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. (2000) 13:155–65. doi: 10.1016/S1074-7613(00)00016-9
113. Scheller M, Foerster J, Heyworth CM, Waring JF, Lohler J, Gilmore GL, et al. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood. (1999) 94:3764–71.
114. Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. (2002) 22:145–52. doi: 10.1089/107999002753452755
115. Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, et al. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS ONE. (2011) 6:e25812. doi: 10.1371/journal.pone.0025812
116. Nam S, Kang K, Cha JS, Kim JW, Lee HG, Kim Y, et al. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J Leukoc Biol. (2016) 100:1273–84. doi: 10.1189/jlb.1A0215-068RR
117. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
118. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. (2015) 2015:816460. doi: 10.1155/2015/816460
119. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation. (2013) 36:921–31. doi: 10.1007/s10753-013-9621-3
120. Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am J Pathol. (2003) 163:711–21. doi: 10.1016/S0002-9440(10)63698-X
121. Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation. (2016) 23:95–121. doi: 10.1111/micc.12259
122. Gunthner R, Anders HJ. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm. (2013) 2013:731023. doi: 10.1155/2013/731023
123. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. (2010) 11:936–44. doi: 10.1038/ni.1920
124. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. (2009) 114:3244–54. doi: 10.1182/blood-2009-04-217620
125. Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. (2006) 103:15136–41. doi: 10.1073/pnas.0607181103
126. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12:231–8. doi: 10.1038/ni.1990
127. Weiss M, Blazek K, Byrne AJ, Perocheau DP, Udalova IA. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediators Inflamm. (2013) 2013:245804. doi: 10.1155/2013/245804
128. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
129. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
130. Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. (2017) 86:129–57. doi: 10.1146/annurev-biochem-060815-014922
131. Heaton SM, Borg NA, Dixit VM. Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med. (2016) 213:1–13. doi: 10.1084/jem.20151531
132. Jefferies C, Wynne C, Higgs R. Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat Rev Immunol. (2011) 11:617–25. doi: 10.1038/nri3043
133. Zhang J, Hu MM, Wang YY, Shu HB. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem. (2012) 287:28646–55. doi: 10.1074/jbc.M112.362608
134. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. (2010) 33:765–76. doi: 10.1016/j.immuni.2010.10.013
135. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
136. Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. TRIM30alpha Is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. (2015) 11:e1005012. doi: 10.1371/journal.ppat.1005012
137. Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity. (2014) 41:919–33. doi: 10.1016/j.immuni.2014.11.011
138. Ni G, Konno H, Barber GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol. (2017) 2:eaah7119. doi: 10.1126/sciimmunol.aah7119
139. Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. (2016) 45:555–69. doi: 10.1016/j.immuni.2016.08.014
140. Higgs R, Jefferies CA. Targeting IRFs by ubiquitination: regulating antiviral responses. Biochem Soc Trans. (2008) 36(Pt 3):453–8. doi: 10.1042/BST0360453
141. Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol Cell Biol. (2008) 28:6536–46. doi: 10.1128/MCB.00785-08
142. Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M, et al. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. (2008) 283:25660–70. doi: 10.1074/jbc.M804479200
143. Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, et al. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J Immunol. (2011) 187:4754–63. doi: 10.4049/jimmunol.1101704
144. Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. (2008) 181:1780–6. doi: 10.4049/jimmunol.181.3.1780
145. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med. (2009) 206:1661–71. doi: 10.1084/jem.20090585
146. Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, et al. TRIM21 Ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. (2016) 61:720–33. doi: 10.1016/j.molcel.2016.02.007
147. Kim JY, Ozato K. The sequestosome 1/p62 attenuates cytokine gene expression in activated macrophages by inhibiting IFN regulatory factor 8 and TNF receptor-associated factor 6/NF-kappaB activity. J Immunol. (2009) 182:2131–40. doi: 10.4049/jimmunol.0802755
148. Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. (2015) 210:973–89. doi: 10.1083/jcb.201503023
149. Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, et al. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. (2006) 7:598–605. doi: 10.1038/ni1347
150. Yang K, Shi HX, Liu XY, Shan YF, Wei B, Chen S, et al. TRIM21 is essential to sustain IFN regulatory factor 3 activation during antiviral response. J Immunol. (2009) 182:3782–92. doi: 10.4049/jimmunol.0803126
151. Stacey KB, Breen E, Jefferies CA. Tyrosine phosphorylation of the E3 ubiquitin ligase TRIM21 positively regulates interaction with IRF3 and hence TRIM21 activity. PLoS ONE. (2012) 7:e34041. doi: 10.1371/journal.pone.0034041
152. Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, et al. Self protection from anti-viral responses–Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS ONE. (2010) 5:e11776. doi: 10.1371/journal.pone.0011776
153. Yu Y, Hayward GS. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity. (2010) 33:863–77. doi: 10.1016/j.immuni.2010.11.027
154. Ran Y, Liu TT, Zhou Q, Li S, Mao AP, Li Y, et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J Mol Cell Biol. (2011) 3:283–92. doi: 10.1093/jmcb/mjr020
155. Wang P, Zhao W, Zhao K, Zhang L, Gao C. TRIM26 negatively regulates interferon-beta production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. (2015) 11:e1004726. doi: 10.1371/journal.ppat.1004726
156. Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, et al. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol Cell Biol. (2010) 30:2424–36. doi: 10.1128/MCB.01466-09
157. Kim D, Lee H, Koh J, Ko JS, Yoon BR, Jeon YK, et al. Cytosolic pellino-1-mediated K63-linked ubiquitination of IRF5 in M1 macrophages regulates glucose intolerance in obesity. Cell Rep. (2017) 20:832–45. doi: 10.1016/j.celrep.2017.06.088
158. Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PLoS ONE. (2014) 9:e103609. doi: 10.1371/journal.pone.0103609
159. Eames HL, Saliba DG, Krausgruber T, Lanfrancotti A, Ryzhakov G, Udalova IA. KAP1/TRIM28: an inhibitor of IRF5 function in inflammatory macrophages. Immunobiology. (2012) 217:1315–24. doi: 10.1016/j.imbio.2012.07.026
160. Harikumar KB, Yester JW, Surace MJ, Oyeniran C, Price MM, Huang WC, et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol. (2014) 15:231–8. doi: 10.1038/ni.2810
161. Tulli L, Cattaneo F, Vinot J, Baldari CT, D'Oro U. Src family kinases regulate interferon regulatory factor 1 K63 ubiquitination following activation by TLR7/8 vaccine adjuvant in human monocytes and B cells. Front Immunol. (2018) 9:330. doi: 10.3389/fimmu.2018.00330
162. Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, et al. Human TRIM gene expression in response to interferons. PLoS ONE. (2009) 4:e4894. doi: 10.1371/journal.pone.0004894
163. Mehta A, Baltimore D. microRNAs as regulatory elements in immune system logic. Nat Rev Immunol. (2016) 16:279–94. doi: 10.1038/nri.2016.40
164. Davidson-Moncada J, Papavasiliou FN, Tam W. microRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci. (2010) 1183:183–94. doi: 10.1111/j.1749-6632.2009.05121.x
165. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
166. Bartel DP. microRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
167. He L, Hannon GJ. microRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. (2004) 5:522–31. doi: 10.1038/nrg1379
168. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. (2018) 9:402. doi: 10.3389/fendo.2018.00402
169. He X, Jing Z, Cheng G. microRNAs: new regulators of Toll-like receptor signalling pathways. Biomed Res Int. (2014) 2014:945169. doi: 10.1155/2014/945169
170. Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol. (2011) 8:388–403. doi: 10.1038/cmi.2011.26
171. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. (2009) 183:2150–8. doi: 10.4049/jimmunol.0900707
172. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
173. Jurkin J, Schichl YM, Koeffel R, Bauer T, Richter S, Konradi S, et al. miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation. J Immunol. (2010) 184:4955–65. doi: 10.4049/jimmunol.0903021
174. Chen X, Zhou L, Peng N, Yu H, Li M, Cao Z, et al. MicroRNA-302a suppresses influenza A virus-stimulated interferon regulatory factor-5 expression and cytokine storm induction. J Biol Chem. (2017) 292:21291–303. doi: 10.1074/jbc.M117.805937
175. Peng L, Zhang H, Hao Y, Xu F, Yang J, Zhang R, et al. Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. EBiomedicine. (2016) 14:83–96. doi: 10.1016/j.ebiom.2016.10.041
176. Li Y, Huang R, Wang L, Hao J, Zhang Q, Ling R, et al. microRNA-762 promotes breast cancer cell proliferation and invasion by targeting IRF7 expression. Cell Prolif. (2015) 48:643–9. doi: 10.1111/cpr.12223
177. Yang F, Xu Z, Duan S, Luo M. MicroRNA-541 promotes the proliferation of vascular smooth muscle cells by targeting IRF7. Am J Transl Res. (2016) 8:506–15.
178. Rosenberger CM, Podyminogin RL, Diercks AH, Treuting PM, Peschon JJ, Rodriguez D, et al. miR-144 attenuates the host response to influenza virus by targeting the TRAF6-IRF7 signaling axis. PLoS Pathog. (2017) 13:e1006305. doi: 10.1371/journal.ppat.1006305
179. Xu T, Chu Q, Cui J, Bi D. Inducible MicroRNA-3570 feedback inhibits the RIG-I-dependent innate immune response to rhabdovirus in teleost fish by targeting MAVS/IPS-1. J Virol. (2018) 92:e01594–17. doi: 10.1128/JVI.01594-17
180. Yarbrough ML, Zhang K, Sakthivel R, Forst CV, Posner BA, Barber GN, et al. Primate-specific miR-576-3p sets host defense signalling threshold. Nat Commun. (2014) 5:4963. doi: 10.1038/ncomms5963
181. Mukherjee A, Di Bisceglie AM, Ray RB. Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway. J Virol. (2015) 89:3356–65. doi: 10.1128/JVI.03085-14
182. Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. miR-125a-5p regulates differential activation of macrophages and inflammation. J Biol Chem. (2013) 288:35428–36. doi: 10.1074/jbc.M112.426866
183. Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. microRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci USA. (2012) 109:7865–70. doi: 10.1073/pnas.1200081109
184. Zhao JL, Huang F, He F, Gao CC, Liang SQ, Ma PF, et al. Forced activation of notch in macrophages represses tumor growth by upregulating miR-125a and disabling tumor-associated macrophages. Cancer Res. (2016) 76:1403–15. doi: 10.1158/0008-5472.CAN-15-2019
185. Li HS, Greeley N, Sugimoto N, Liu YJ, Watowich SS. miR-22 controls Irf8 mRNA abundance and murine dendritic cell development. PLoS ONE. (2012) 7:e52341. doi: 10.1371/journal.pone.0052341
186. Dai Y, Sui W, Lan H, Yan Q, Huang H, Huang Y. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. (2009) 29:749–54. doi: 10.1007/s00296-008-0758-6
187. Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. (2011) 70:1496–506. doi: 10.1136/ard.2010.139857
188. Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, et al. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. (2007) 16:939–46. doi: 10.1177/0961203307084158
189. Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. (2009) 60:1065–75. doi: 10.1002/art.24436
190. Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. (2010) 184:6773–81. doi: 10.4049/jimmunol.0904060
191. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. (2011) 63:1376–86. doi: 10.1002/art.30196
192. Conti P, DiGioacchino M. MCP-1 and RANTES are mediators of acute and chronic inflammation. Allergy Asthma Proc. (2001) 22:133–7. doi: 10.2500/108854101778148737
193. Moore KJ, Wada T, Barbee SD, Kelley VR. Gene transfer of RANTES elicits autoimmune renal injury in MRL-Faslpr mice. Kidney Int. (1998) 53:1631–41. doi: 10.1046/j.1523-1755.1998.00911.x
194. Zhao X, Tang Y, Qu B, Cui H, Wang S, Wang L, et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. (2010) 62:3425–35. doi: 10.1002/art.27632
Keywords: interferon, ubiquitin, E3 ligase, microRNA, monocyte
Citation: Jefferies CA (2019) Regulating IRFs in IFN Driven Disease. Front. Immunol. 10:325. doi: 10.3389/fimmu.2019.00325
Received: 06 December 2018; Accepted: 07 February 2019;
Published: 29 March 2019.
Edited by:
Hans A. R. Bluyssen, Adam Mickiewicz University in Poznań, PolandReviewed by:
Betsy J. Barnes, Feinstein Institute for Medical Research, United StatesCopyright © 2019 Jefferies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline A. Jefferies, Y2Fyb2xpbmUuamVmZmVyaWVzQGNzaHMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.