 Dirk Schlüter
Dirk Schlüter Antonio Barragan
Antonio Barragan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 February 2019
Sec. Multiple Sclerosis and Neuroimmunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00242
This article is part of the Research TopicNeurotropism of Parasites and Immune ResponsesView all 8 articles
Toxoplasma gondii is a widespread parasitic pathogen that infects over one third of the global human population. The parasite invades and chronically persists in the central nervous system (CNS) of the infected host. Parasite spread and persistence is intimately linked to an ensuing immune response, which does not only limit parasite-induced damage but also may facilitate dissemination and induce parasite-associated immunopathology. Here, we discuss various aspects of toxoplasmosis where knowledge is scarce or controversial and, the recent advances in the understanding of the delicate interplay of T. gondii with the immune system in experimental and clinical settings. This includes mechanisms for parasite passage from the circulation into the brain parenchyma across the blood-brain barrier during primary acute infection. Later, as chronic latent infection sets in with control of the parasite in the brain parenchyma, the roles of the inflammatory response and of immune cell responses in this phase of the disease are discussed. Additionally, the function of brain resident cell populations is delineated, i.e., how neurons, astrocytes and microglia serve both as target cells for the parasite but also actively contribute to the immune response. As the infection can reactivate in the CNS of immune-compromised individuals, we bring up the immunopathogenesis of reactivated toxoplasmosis, including the special case of congenital CNS manifestations. The relevance, advantages and limitations of rodent infection models for the understanding of human cerebral toxoplasmosis are discussed. Finally, this review pinpoints questions that may represent challenges to experimental and clinical science with respect to improved diagnostics, pharmacological treatments and immunotherapies.
In 1908, Nicolle and Manceaux discovered a bow (Greek: toxon) -shaped (Greek: plasma) parasite in the gundi (Ctenodactylus gundi), a North African rodent. Independently, Splendore (1908) described the parasite in a rabbit. The definitive hosts of Toxoplasma gondii are Felidae, e.g., domestic cats and other felines in which this obligate intracellular apicomplexan parasite replicates sexually in their intestine. Humans and many other warm-blooded vertebrates, e.g., rodents and birds, serve as intermediate hosts for the parasite and may become infected by oral ingestion of oocysts shed by felines or by tissue cysts that persist in the muscles and nervous system of the intermediate hosts (1).
Population structure studies of T. gondii have revealed that a few major clonal lineages predominate in different global geographical regions (2). Humans in Western Europe and North America are mainly infected by type II strains of T. gondii but type I, type III and so-called atypical strains can also infect humans and may cause disease (3). In the human host, orally ingested parasites are released from oocysts or tissue cysts (containing bradyzoite stages) and invade intestinal epithelial cells to transform into fast replicating tachyzoite stages. These tachyzoites multiply intracellularly in a parasitophorous vacuole (PV), which separates the parasite from the host cell cytoplasm. Unrestricted parasite replication results finally in the rupture of the host cell, release of tachyzoites and infection of neighboring cells. Due to the ensuing immune response and the production of anti-parasitic intracellular effector molecules by the infected cells, the tachyzoites may be either eliminated or transform into slow replicating bradyzoites, which form intracellular persisting cysts. These cysts may contain hundreds of parasites and preferentially form in neurons of the brain and retina as well as in muscle cells. It is thought that also at this chronic stage of infection, the immune system plays a critical role in parasite control but may also contribute to disease, particularly in the eye.
In healthy humans, most primary infections with T. gondii are asymptomatic or cause only mild symptoms including malaise, swelling of lymph nodes and fever (4). However, primary infection during pregnancy may be harmful or even fatal to the fetus. Bradyzoite-containing brain cysts may become reactivated, i.e., reconvert into cytotoxic tachyzoites during secondary immune deficiency and cause toxoplasmic encephalitis (TE), in particular in individuals with HIV/AIDS or organ transplants. Ocular toxoplasmosis (OT) was identified decades ago as a manifestation of congenital toxoplasmosis but in the last years it has been uncovered that OT may also develop upon primary infection of immune-competent adults and has a high risk for recrudescence even upon anti-parasitic treatment.
Here, we discuss recent advances in the understanding of the delicate interplay between T. gondii, infected cells and the immune system in experimental and clinical settings. The major focus is on parasite invasion into the brain, mechanisms of parasite control in the brain and factors leading to reactivation of chronic cerebral toxoplasmosis. In particular, we delineate similarities, differences and open questions between rodent models, and human cerebral toxoplasmosis. We briefly address some aspects on the impact of the parasite on the brain neurophysiology. However, for this specific aspect, we refer the reader primarily to recent reviews (5–8).
The mechanisms that lie behind the systemic dissemination and the parasite's penetration to the human brain are largely unknown. The concepts are thus mainly based on an extrapolation from studies in rodents, which also are natural hosts for Toxoplasma (Table 1).
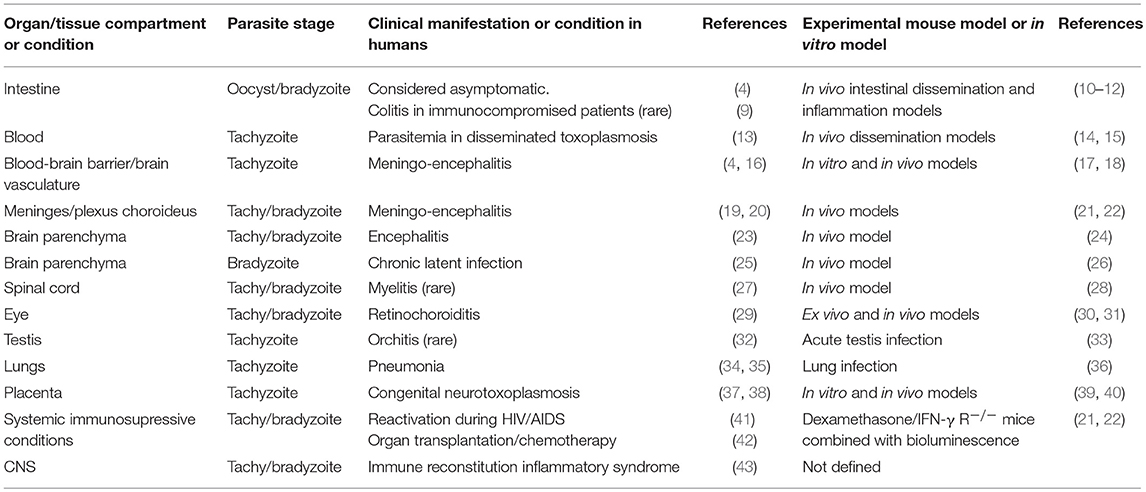
Table 1. Clinical manifestations of toxoplasmosis in humans and available mouse models, with special reference to disease in the CNS and immunoprivileged organs.
Although Toxoplasma undoubtedly causes pathology in the CNS and is often presented as a neurotropic parasite, there is some debate regarding its neurotropism, that is, whether its affinity for the CNS is superior or different to that for other organs. During acute infection, T. gondii disseminates broadly but parasite loads in mice are, in fact, substantially lower in the CNS compared with other organs. Thus, Toxoplasma can hardly be classified as a primarily neurotropic pathogen having a selective higher affinity for the CNS over other organs. However, seemingly common for human and rodent infections, the parasite overcomes the restrictiveness of the blood-brain barrier (BBB) to establish infection. In fact, T. gondii can infect virtually any nucleated cell within and outside the CNS. Yet, as chronic infection sets in, parasite cysts are found in the CNS (and musculature) and persist in the CNS while they are cleared from peripheral organs over time. In that sense, infection can be considered neurotropic because chronic reactivated infection and congenital infection manifest primarily in the CNS. Although possible associations with parasite virulence determinants such as ROP18 have recently been put forward (44), molecular evidence is missing of parasite-derived mechanisms that would define tropism for the CNS over other organs. Various aspects of the immunopathogenesis linked to the persistence of the parasite in the CNS are discussed below.
Following oral ingestion of tissue cysts (45) or oocysts (46), the parasites invade the intestinal tissue and rapidly differentiate into tachyzoites. In experimental oral or intra-peritoneal infections in mice, parasites are rapidly found in the mesenteric lymph nodes and in the blood circulation, indicating that a lymphatic-to-blood route is likely. It remains however unknown if a direct hematogenic route, i.e., from the gut tissue directly into the blood can be taken for systemic dissemination, as suggested for Salmonella (47).
How is Toxoplasma transported in the blood circulation to ultimately reach the BBB? In experimental murine models, leukocyte-associated parasites are rapidly detected in the blood while extracellular/free parasites in the blood are also present later during infection (18, 48). It remains unclear if free tachyzoites in the blood are part of the natural infection or a consequence of high parasitic loads in experimental settings. In natural infections in rodents, invasion of the CNS is accomplished with relatively low parasitemia. Regardless, given the sensitivity of free tachyzoites to neutralization by the complement system and IgM (49), leukocytes offer a safe intracellular niche for hematogenic dissemination. Also, leukocytes have been reported to deliver parasites after egress in the lung tissue (36). Different types of leukocytes can be infected and parasite transfer between leukocyte types occurs in the blood compartment and in peripheral tissues (15, 50). In this context, Toxoplasma induces a hypermigratory phenotype in human and mouse dendritic cells (DCs) (51, 52). Hypermigration of infected DCs potentiates systemic parasite dissemination in mice, including the CNS, and may cooperate with chemotactic responses of DCs (53, 54). However, hypermigration has not been linked specifically to passage across the BBB in vivo as discussed below (15).
It remains unknown if the recently discovered lymphatic system of the meningeal vessels (dura mater) in the brain (55, 56) plays a role for the systemic dissemination into the CNS or out from the CNS upon reactivated toxoplasmosis. However, the absence of dominant meningeal manifestations during cerebral toxoplasmosis advocates against a detrimental role. Similarly, no evidence indicates that the fenestrated blood vessels of the choroid plexus would be a preferred or predominant locus for CNS invasion, with penetration to the cerebrospinal fluid (CSF) and further spread to the parenchyma (22). In clinical settings, Toxoplasma is infrequently detected in the CSF by PCR and, thus, not recommended as a standard diagnostic procedure in AIDS patients with suspected reactivated TE. It is therefore likely that passage of Toxoplasma from the blood circulation is effectuated mainly across the parenchymal vasculature.
In order to reach the brain parenchyma from the cerebral blood circulation, Toxoplasma has to relate to the brain endothelium, primarily to the capillary bedding. There is in vitro evidence that Toxoplasma-infected leukocytes and free tachyzoites can transmigrate across epithelial/endothelial cell monolayers (17, 51, 57, 58) and likely extravasate in vivo in mice (59). More recent studies have put forward that infection of endothelial cells occurs and is necessary for penetration to the CNS (18). One additional possibility is that leukocytes “deliver” the parasites to the endothelium (60), as recently suggested in a pulmonary infection model in mice (36). It remains unknown how Toxoplasma crosses the BBB below the endothelial cell linings, i.e., basal lamina and astrocytic end-feet. Similarly, the eye is an immune-privileged site and similar mechanisms have been put forward for passage of the blood-retina barrier, implicating both Trojan horse mechanisms and free extracellular tachyzoites (30, 61, 62).
Where and when does Toxoplasma cross the vasculature of the BBB? While Plasmodium, causing cerebral malaria, seems to have a predilection for adhesion to post-capillary venules (63) with a possible involvement of leukocytes (64), it remains unknown if Toxoplasma crosses at the level of arterioles, capillaries or post-capillary venules. There is no evidence of a hemorrhagic process upon penetration to the CNS. However, are there perturbations in the permeability of the BBB, albeit locally, upon Toxoplasma invasion to the parenchyma? Recent work suggests that vascular dysfunction persists during chronic infection (65). The inflammatory or anti-inflammatory components that may accompany or facilitate passage across the BBB during acute infection also remain enigmatic. Future research needs to address these questions in order to understand passage of Toxoplasma to the brain parenchyma.
The kinetics of passage to the parenchyma also remains poorly understood. Early histological studies detected tachyzoites in the brain parenchyma by day 5 after intravenous inoculation of mice (66). Detection of parasites in cerebral homogenate by bioassay/plaquing assays or qPCR was earliest on day 6–7 post-infection in oral experimental infections (45, 59) and by day 3–4 after intraperitoneal inoculation in blood-perfused mice (15).
Jointly, mounting evidence show that Toxoplasma utilizes combined strategies for systemic dissemination (60), by hijacking leukocytes (51, 59, 67) and, as free parasites (18, 48), with significant differences between Toxoplasma genotypes (48, 68). If these combined strategies are also utilized to enforce the BBB awaits future investigations. Also, Toxoplasma-derived factors directly implicated in cerebral toxoplasmosis remain to date unidentified.
The mechanisms leading to parasite persistence and control in the human brain are largely unknown. The concepts are therefore mainly based on extrapolations from experimental studies in rodents (Table 1).
Primary Toxoplasma infection in humans is, in general, clinically asymptomatic or accompanied by discrete symptomatology (4). Yet, based on murine models, the parasite penetrates to the brain parenchyma during this initial phase (59). At the BBB and after contact with the endothelium, the parasite must cross one additional layer of the barrier that is constituted by the basal lamina and the astrocytic end-feet. Yet, little evidence of astrocytic infection was found during this process (26), indirectly supporting the concept that carrier immune cells could mediate transportation (26, 59). One alternative possibility is that microglia that also express a hypermigratory phenotype upon Toxoplasma infection serve as “Trojan horses” for parasitic spread in the parenchyma (69).
Studies in mice have shown that Toxoplasma cysts can be retrieved from any anatomical brain area (70) and that cyst abundance can be higher in the amygdala, thalamus and striatum (71, 72). This anatomic distribution of cysts is likely an important factor contributing to the manipulation of murine behavior by the parasite (70). Murine reactivation/recrudescence models provided additional information and illustrated that reactivation occurred preferentially in the gray matter of the frontal and parietal cortex, compared to cerebellum or other cortical areas (22). This distribution pattern of cortical parasitic foci may be consistent with blood transportation by the middle cerebral artery and extravasation from its ramifications during primary infection.
Although astrocytes, microglia and neurons can readily be infected in vitro (73), a preference for parasite localization to neurons upon cyst formation has originally been identified by Ferguson and Hutchison (74) and confirmed by others (26, 72). Exactly how Toxoplasma reaches neurons is not understood and the mechanisms allowing parasite persistence in neurons, and not astrocytes remain enigmatic (75). Neurons express MHC class I under certain circumstances, including activity-dependent, long-term structural and synaptic modifications (76, 77), and in functionally inactive IFN-γ-stimulated neurons (78). However, it is at present an open question whether all neurons infected with Toxoplasma tachyzoites or cysts express MHC class I and present parasitic antigens to CD8+ T cells. Eventually, some cyst-harboring neurons remain MHC class I negative and, thus, escape elimination by CD8+ T cells, which in principle effectively remove cysts from the brain in a perforin-dependent manner (79). Additionally, genetic fate mapping studies have demonstrated that T. gondii tachyzoites infect neurons but not astrocytes in mice and, thus, the absence of infected astrocytes may not be caused by an elimination of tachyzoites by astrocytes in vivo (26).
It has been suggested that T. gondii tachyzoite to bradyzoite conversion and cyst formation primarily occurs in post-mitotic cells such as neurons and muscle cells (80), which may even induce spontaneous bradyzoite formation without stimulation by the immune system (81). However, in the retina, both neurons and glial cells, which have the capacity to proliferate, harbor Toxoplasma cysts (82). Additionally, a Toxoplasma line was reported to convert spontaneously from tachyzoites to cyst-forming bradyzoites in human cell lines including epithelial cells, fibroblasts, muscle cell, and glial cells (83). Collectively, these data indicate that depending on the parasite strain both proliferating and post-mitotic host cells allow spontaneous cyst formation in vivo and in vitro.
Without doubt, the infection has a clear predilection for the CNS, which plays an important role for the clinical manifestations of human toxoplasmosis. Even in congenital toxoplasmosis, when the parasite could disseminate to all organs of the highly immune-compromised fetus, the preferentially infected organs are the brain and the eyes. Based on data from rodent infection models, it is generally assumed that, following primary infection, T. gondii establishes a life-lasting chronic infection in the brain. But, does T. gondii always persist after primary infection in humans? This notion is based on clinical and experimental data:
(i) T. gondii seropositive AIDS patients, with low CD4+ T cell count (<200/μl) and with specific antibodies against T. gondii proving prior infection, are at risk of developing a reactivated TE due to the loss of T cell-mediated control of persisting brain cysts (23).
(ii) T. gondii seropositive patients receiving immunosuppressive therapies, e.g., upon hematopoietic stem cell transplantation (HSCT) and solid organ transplantation (SOT), are at risk of developing a reactivated toxoplasmosis (23).
(iii) In very few cases, intracerebral T. gondii cysts have been detected by immunohistochemistry in brain autopsies of immunocompetent patients, who died from unrelated diseases (25).
(iv) Experimental studies, predominantly in mice but also in other species, have shown that upon experimental infection, T. gondii cysts persist in the CNS (84).
However, it remains questionable whether T. gondii persists for a lifetime in all infected humans. In fact, only ≈30% of T. gondii seropositive AIDS patients will develop a reactivated TE, even if the CD4+ T cell count is very low (23). In addition, only a very reduced number of T. gondii seropositive patients undergoing immunosuppression due to HSCT, develop a reactivated systemic or cerebral toxoplasmosis (85). In T. gondii seropositive AIDS and HSCT patients without reactivated TE, it is unclear whether other immune mechanism(s) compensate for the T cell deficiency and control the parasite or whether the parasite failed to persist in the brain. Additionally, the number of autopsy cases identifying asymptomatically persisting T. gondii cysts in the brain is surprisingly low. It might be argued that pathologists ought to accidentally detect T. gondii cysts more frequently based on the high seropositivity rates of adults (ranging from 20 to 80% depending on the age and geographical region), though it might be difficult to detect some few cysts in the large brain volume.
Parasite virulence and persistence in mice is strongly dependent on the host genetic background (86). Certain mouse strains harbor a high intracerebral parasite load upon experimental infection and might—in contrast to humans—even succumb due to the overwhelming infection. In contrast, other mouse strains have lower cyst counts in the brain and in certain rat strains the parasite even fails to persist in the CNS. Detailed studies in mice have linked relative resistance to the infection, i.e., time of survival and intracerebral parasite numbers, to genes inside and outside the MHC locus (87). Outbred mice (e.g., NMRI) and mouse strains with the H-2d MHC haplotype (e.g., BALB/c and B6.C H-2d mice) develop a chronic latent cerebral toxoplasmosis and survive the infection with low cyst numbers and low inflammation, whereas mice with the H-2b (e.g., C57BL/6 mice) and H-2K background (e.g., CBA/Ca mice) develop a lethal chronic progressive TE with high parasite loads and extensive inflammation (88, 89). Additionally, the genotype of the parasite might also have an impact on cyst formation. Most parasite genotypes, such as type II strains that commonly infect humans, form cysts in mice and humans. In contrast, prototypic type I strains rapidly cause lethal infection in mice before cyst formation but appear to form cysts and generate chronic infection in other species, including humans (90–92). However, even among type II strains, a range of virulence is observed in mice. Finally, the number of ingested parasites impacts on the outcome of infections and severity of cerebral toxoplasmosis, adding another layer of complexity. The relative importance of host- and parasite-specific factors but also of experimental conditions on the course of murine toxoplasmosis has recently been discussed (93).
In conclusion, parasite persistence varies significantly between host species and depends on the genetic backgrounds of both host and parasite. Altogether, it is reasonable to speculate that prolonged or life-long parasite persistence in the human brain may occur only in a fraction of T. gondii seropositive individuals. Due to the difficulties in determining and quantifying parasite persistence in the human brain, the extent of cyst formation remains unresolved in humans. Here, PCR analysis of larger brain tissue samples from T. gondii positive autopsy cases might clarify the ratio of parasite persistence vs. seropositivity.
Cell-mediated immunity to T. gondii includes activation of innate immune cells as well as antigen-specific T and B cells. In mice, infection with T. gondii rapidly induces the MyD88-dependent production of IL-12 by DCs. In particular, T. gondii profilin activates TLR11 and TLR12 in CD8α+ DCs and also TLR12 in plasmacytoid DCs. Although humans lack TLR11 and TLR12, other TLRs may induce IL-12 production upon engagement with T. gondii, since parasite RNA and DNA is also recognized by TLR3, 7, and 9. In the early phase of toxoplasmosis, the IFN-γ production by NK cells and type I innate lymphoid cells is important for parasite control (94). Beside IL-12, presentation of parasitic antigens by DCs and macrophages is required for activation and expansion of parasite-specific CD4+ and CD8+ T cells. CD4+ and CD8+ T cells also produce IFN-γ establishing another layer of protection, which can also compensate for insufficient IFN-γ production by NK cells (95).
In parallel to the parasite spread to the brain, inflammatory leukocytes are recruited to the CNS. These inflammatory infiltrates are mainly composed of CD4+ and CD8+ T cells with the addition of F4/80+ macrophages, CD11c+ DCs, and Ly6Chigh inflammatory monocytes. CD8+ T cells recognize secreted T. gondii antigens (96) and play a key role in the control of cerebral toxoplasmosis, illustrated by the fact that resistance is linked to the Ld MHC class I gene. The parasitic GRA6 antigen is processed to a decamer epitope presented by Ld, eliciting a robust and immunodominant CD8+ T cells response (97). In contrast, an immunodominant CD8+ T cell epitope is missing in H-2b mice, which might explain why CD4+ and CD8+ T cells are both important for control of T. gondii in H-2b mice (98), whereas CD8+ T cells play a more important role in H-2d mice (99).
The major protective mechanisms of CD4+ and CD8+ T cells in cerebral toxoplasmosis are the production of protective cytokines, in particular IFN-γ and TNF. Perforin-dependent cytotoxicity seems to play some role for the control of T. gondii in the CNS but this protective pathway might be of limited importance since the majority of infected cells are MHC class I− neurons. In support, intravital microscopy studies revealed that parasite-specific CD8+ T cells transiently interact with CD11b+ cells, which might be microglia, macrophages, monocytes and DCs presenting T. gondii antigen, but not with T. gondii-infected neurons (100, 101).
In addition to T cells, several types of myeloid cells, including CD8α+CD11c+ lymphoid DCs, CD11b+CD11c+ myeloid DCs and PDCA+B220+ plasmacytoid DCs, are recruited to the T. gondii-infected brain. Also, in the CNS, DCs produce IL-12 that may contribute to the maintenance of IFN-γ production of T cells (102). In addition, inflammatory Ly6ChiCCR2+GR1+ monocytes contribute to parasite control by the production of protective cytokines (IL-1α, IL-1β, IL-6, TNF) and anti-parasitic effector molecules (reactive oxygen species (ROS) and NO) (103).
The cytokine-induced anti-parasitic effector mechanisms differ between mice and humans. Even in mice, the functional relevance of anti-parasitic effector molecules is mouse strain-dependent. Whereas, NO is essentially required for intracerebral control of T. gondii in susceptible C57BL/6 mice, neutralization of iNOS does not exacerbate chronic TE of resistant BALB/c mice (104). There is good evidence that autophagy-related molecules play a role in parasite killing (105, 106). Furthermore, small GTPases of the IRG family, in particular Irga6 and Irgb6, are crucial for intracellular control of T. gondii (107, 108). These IRGs destroy the intracellular replication niche of the parasite by disrupting the PV. Off-target damage of host cell membranes by IRGs is prevented by regulatory GMS-IRGs and mice deficient of regulatory Irgm1 and Irgm3 die from toxoplasmosis due to impaired activity of Irga6 and Irgb6 (109, 110). Parasite-derived rhoptry proteins ROP5 and ROP18 phosphorylate host IRGs such as Irga6 and can thereby inhibit membrane degradation (111). However, this virulence mechanism can be overcome by a highly polymorphic IRG protein (Irgb2-b1) depending on the genetic background of the mouse. Some of its variants can act as decoys for ROP5/18 binding, enabling other IRGs to degrade the PV (112). In addition to IRGs, guanylate-binding proteins (GBP) play an important role in the control of T. gondii in mice (113). In part in cooperation with IRGs, GBPs target the PV resulting in killing of the parasite (110). In rats, small GTPAses, NO and also reactive oxygene species contribute to the control of T. gondii (114–116).
In contrast to rodents, NO and IRGs play no role for the control of T. gondii in humans. In fact, humans are devoid of IRG genes, which are evolutionally maintained only in rodents (117). Thus, the key factors important for the control of T. gondii in mice, i.e., TLR11 and TLR12 as well as the IRGs do not exist in humans, which makes it difficult to extrapolate data from the murine model systems of toxoplasmosis to the human disease. In humans, alarmin S100A11 is released from T. gondii-infected cells and sensed by monocytes, which upregulated chemokine production to induce recruitment of additional monocytes (118). With respect to human anti-parasitic effector mechanisms, IFN-γ-induced 2,3 indoleamine dioxygenase (IDO) degrades tryptophan and plays a role for parasite control by astrocytes (119). Interestingly, it has been demonstrated that the parasite aims to restrict anti-parasitic IDO levels in the host cell: the T. gondii effector molecule TgIST inhibits IDO1 mRNA expression and T. gondii GRA15-induced NO antagonizes IFN-γ-dependent IDO levels (120, 121). Importantly, the mechanisms leading to control and elimination of T. gondii in murine and human neurons are unknown. Here, a recent study analyzing the transcriptome of T. gondii-infected murine neurons, astrocytes, fibroblasts and skeletal muscle cells might provide a source for the identification of host genes important for the control of parasite cysts in neurons (122).
Beside pro-inflammatory cytokines, immunosuppressive cytokines play an important role to prevent immunopathology in toxoplasmosis. The inflammatory monocytes produce IL-10, which prevents an overshooting and immunopathological response in the T. gondii-inflamed brain (103). In addition, microglia, macrophages, regulatory B cells, Tbet+Foxp3− Th1 cells and some CD8+ T cells produce IL-10. IL-10 protects from lethal immunopathology in acute systemic and down-regulates the immune response in chronic cerebral toxoplasmosis (123, 124). In addition, astrocytes produce IL-27, which also inhibits immunopathological Th17 responses in TE (125). In contrast to mice, the CNS immunopathology of human primary toxoplasmosis remains unexplored. In might play a role in some patient categories, e.g., those with epilepsy, since cerebral inflammation is an important trigger of epilepsy and T. gondii seropositivity is higher in patients with epilepsy (126–128). As deduced from murine cerebral toxoplasmosis, excessive inflammation might contribute to BBB dysfunction and profound changes in excitatory (in particular glutamate) and inhibitory (in particular GABA) neurotransmission paving the way to epilepsy (129, 130). Interestingly, there is also good evidence that a dysbalance of T cell subpopulations plays a major role in acute and reactivated human OT [reviewed in Maenz et al. (29)].
In murine TE, strongly activated astrocytes proliferate and form a “ring wall” around T. gondii-associated lesions. In the absence of the gp130 receptor, which is important for signaling of cytokines of the IL-6 family, astrocytes become apoptotic resulting in a failure to contain the inflammatory lesions and parasites, with widespread inflammation and finally lethal outcome of TE (131). The important bordering function of astrocytes is further exemplified in mice lacking GFAP, the major intermediary filament of astrocytes. Absence of GFAP in astrocytes results also in increased inflammation and increased parasite loads (132). Additionally, in vitro and in vivo studies have revealed that astrocytes protect from TE in a STAT1-dependent manner (133) and produce a number of cytokines (IL-1α, IL-6, GM-CSF) and chemokines (CXCL10, CCL2), all with important immunoregulatory functions in TE (134, 135). As mentioned before, astrocytic IL-27 is of key importance to limit activation of the intracerebral immune response. In parallel, suppression of astrocyte activity by TGF-β is required to limit neuroinflammation, to control parasite load and to prevent neuronal damage (125, 136). Thus, astrocytes exert important pro- and anti-inflammatory activities to balance parasite control and intracerebral immune responses. However, although astrocytes can be readily infected in vitro, in a mouse infection model neurons rather than astrocytes were targeted by Toxoplasma for infection and injection of effector molecules (26, 137).
In the CNS, neurons are the primary target cell of the parasite. However, the molecular mechanisms of neuronal parasite control remain unclear. Upon in vitro infection, neurons produce IL-6, TGF-β1, CCL3, and CCL4 (138). Interestingly, expression of the common IL6-cytokine family receptor in neurons is required to prevent hyperinflammation, neuronal loss, parasite replication and, finally, death from TE (139). Thus, the gp130 receptor mediates survival of neurons under inflammatory conditions and is further important for the production of immunosuppressive induction of TGF-β1 and IL-27 by neurons.
A third brain parenchymal cell population, strongly activated during TE is the microglia. Microglia are yolk sac-derived cells that develop independently from the bone marrow (140). In murine toxoplasmosis, microglia are strongly activated throughout the entire brain as evidenced by upregulation of MHC class I and II antigens (24, 134). In addition to de novo expression of a number of immunological important cell surface molecules, including CD200, CD11a, CD11b, microglia produce several cytokines and chemokines such as IL-6, IL-10, TNF, and GM-CSF in TE and upon infection in vitro (141). Functionally important, microglia suppress the proliferation of intracerebral T cells, most probably to prevent excessive T cell proliferation and immunopathology due to the continuous T cell stimulation with the persisting parasite antigens (138).
Data on immune mechanisms leading to the control of intracerebral parasites in humans are scarce. Knowledge is mainly based on extrapolations from reactivated toxoplasmosis in AIDS patients and from severe toxoplasmosis in severely immunosuppressed patients, including HSCT, SOT, immunosuppressive therapy and primary immunodeficiency. Since some cytokines such as IFN-γ, TNF, and IL-12 are key factors for the control T. gondii in mice, a high risk for severe primary or reactivated cerebral toxoplasmosis might be expected in patients with primary deficiency for these cytokines and their receptors. However, patients with primary immunodeficiency and severe toxoplasmosis are rarely reported and found in databases. For example, only 2 cases of congenital immunodeficiency–one being primary IFN-γ receptor deficiency- were reported in a multi-center study of 180 cases of toxoplasmosis (142). Furthermore, in a series of 115 patients with IFN-γR1, IFN-γR2, and STAT1-deficiencies, respectively, only one single patient developed clinical toxoplasmosis (143) and among 141 patients with IL-12Rβ1deficiency no single patient developed toxoplasmosis (144). One might also expect that severe toxoplasmosis may manifest upon iatrogenic immunosuppression by T cell-ablating antibodies and anti-cytokine/cytokine receptor therapy. However, although anti-TNF therapy is becoming common, only very few cases of severe toxoplasmosis, reactivated TE or OT have been reported in patients undergoing anti-TNF therapy (145–147). In contrast, mice treated with anti-TNF show an impaired parasite control (148). It has been argued that an underreporting of severe toxoplasmosis in patients treated with biological therapies explains the low number of cases. However, this appears unlikely, since patients under therapy with anti-TNF and other biological therapies are continuously monitored for clinical signs and parameters of infections. In contrast to toxoplasmosis, both humans and mice have an increased risk for tuberculosis under anti-TNF treatment (149), which further highlights the discrepancies between different infections.
What causes this discrepancy between mouse and human data? As excellently delineated in Gazzinelli et al. (117), it is important to keep in mind that rodents play a crucial role in the predator-bait life cycle of the parasite, whereas humans are an accidental intermediate host of T. gondii. Therefore, the murine immune system is under continuous evolutionary pressure to develop immunity against newly emerging virulent T. gondii strains, which develop in the feline intestine and aim to produce large amounts of parasites in rodents. Consequently, the major murine molecules, i.e., TLR11, TLR12, and IRGs protecting against toxoplasmosis have developed in mice but not in humans.
Groups of patients with severe immunosuppression are at a risk of developing cerebral toxoplasmosis. These include patients with AIDS, SOT, in particular heart transplantation, and HSCT. Additionally, the developing fetus is at risk due to its immature immune system. Because animal models of immunosuppression often use dexamethasone treatment yielding a broad immunosuppression, it is difficult to draw parallels to CD4+ T cell deficiency in HIV-infected individuals (21, 22) (Table 1).
In AIDS patients, the reactivation of latent cerebral toxoplasmosis is clinically important. Approximately 30% of T. gondii seropositive AIDS patients with a CD4+ T cell count below 200/μl develop a reactivated TE, which is lethal if not treated adequately with anti-parasitic drugs (23). Why is the association between reactivated TE and AIDS so strong? The common and most accepted answer is that AIDS patients have a disturbed anti-parasitic T cell response, and, therefore, fail to control the intracellular persisting parasite. Failure to control the parasite in the CNS results in bradyzoite to tachyzoite conversion and a necrotizing TE. However, it has to be kept in mind that T. gondii persists in neurons, which can be MHC negative and, therefore, T cells cannot directly interact with neurons in this respect.
Furthermore, AIDS patients harbor a high cerebral viral load due to replication of the virus in microglia and astrocytes (150, 151). Therefore, it is tempting to speculate that (i) the virus itself and (ii) a dysregulation of microglia and astrocytes may contribute to reactivated TE. In support of this view, in T. gondii seropositive patients treated with antibodies depleting T cells or blocking the entry of T cells into the CNS, reactivation of TE does not occur. In fact, only one case of OT has been reported in a multiple sclerosis patient with anti-VLA4 (natalizumab) treatment (152).
One important but rare clinical manifestation is Toxoplasma-associated immune reconstitution inflammatory syndrome (IRIS). Clinically, IRIS can manifest in AIDS patients with reactivated TE and with recent anti-viral therapy. Upon reconstitution of the immune system, T cells may recognize the parasitic antigen and produce large amounts of cytokines, which may cause new neurological symptoms. Additionally, “unmasking” Toxoplasma-associated IRIS may develop in AIDS patients with low CD4+ T cell numbers, newly established anti-retroviral therapy and unrecognized TE at the beginning of retroviral treatment. Rarely, AIDS patients with reactivated cerebral toxoplasmosis may develop an infection of the spinal cord (153).
Whereas, reactivated TE ranks among the most common neurological opportunistic infections in AIDS patients, reactivated toxoplasmosis is relatively rare in patients undergoing HSCT and SOT (85, 142). Further, reactivation of latent toxoplasmosis in transplant patients presents preferentially as a systemic infection rather than localized CNS infection (142).
The fetus may become infected if primary infection occurs shortly before or during pregnancy. If the fetus is infected, the parasite has a high predilection for manifesting in the brain and the eyes. Infection early during pregnancy causes more severe symptoms in the fetus and eventually abortion, whereas infections at later time points of gestation cause less severe symptoms (154, 155). Inversely, the risk of infection increases with the time of gestation. The role of fetal immune responses in congenital toxoplasmosis is largely unclear. Most probably, maternal parasite-specific antibodies play a superior protective role.
T. gondii has been proposed as a causative agent manipulating the behavior of intermediate rodent hosts to facilitate its parasitic transmission to the definitive feline hosts (71, 156). Also, studies in mice have detected impacts of Toxoplasma infection on dopaminergic, GABAergic and glutamatergic neurotransmission (129, 130, 157, 158). The role of the immune response, and neuronal alterations, in the manipulation of the behavior of rodents during cerebral toxoplasmosis was recently reviewed (5, 7). It should be stressed that data on parasite-mediated modulation of the behavior of rodents cannot be automatically extrapolated to humans, because anti-T. gondii immune responses differ fundamentally between mice and men. Humans are accidental intermediate hosts of T. gondii while rodent behavior is under evolutionary pressure by the parasite in order to complete the parasitic life cycle. Nevertheless, a recent study in naturally infected chimpanzees also uncovered that T. gondii may manipulate the behavior of this primate (159).
Additionally, growing evidence indicates a correlation between T. gondii seropositivity and incidence of psychiatric diseases in humans, in particular schizophrenia and bipolar disorders (160–162). Thus, epidemiological studies indicate that carriage of T. gondii may increase the risk for some psychiatric diseases and, thus, may serve as co-risk factor. From a clinical perspective, correlative data between psychiatric diseases and toxoplasmosis (or seropositivity) are of importance but need further clinical investigation. Does the combination of T. gondii infection together with a specific host genetic profile cumulate in an increased risk of schizophrenia? Does this explain why only some T. gondii-seropositive humans suffer from schizophrenia? Are T. gondii seropositive patients with schizophrenia a distinct subgroup of patients (even requiring a different treatment regime)? In addition, some studies provide an association between T. gondii seropositivity and human behavior or neurocognitive impairment (163–165) while other studies do not find such association (166–168).
Thus, mounting evidences indicate that chronic Toxoplasma infection may not be consistently “silent” in humans, as previously thought. However, at present, compelling evidence for a direct causal link between Toxoplasma infection and mental illness is still missing in humans. It is also not clarified how or to what extent parasite persistence in the CNS may impact on neuropsychiatric disorders or modulate cognitive functions. However, the recently reported intriguing effects on primate behavior might prove useful for further investigations (159). Mechanistic studies are required to elucidate the underlying pathophysiology. Also, prospective studies may elucidate whether the parasite impacts on human behavior or whether behavioral patterns result in a higher risk for T. gondii infection.
Although treatment failures have been reported for toxoplasmosis, drug resistance does not appear to be a considerable problem in the clinical setting (169). However, a major problem is that tissue cysts are not eliminated by current drug treatments, which only effectively eliminate replicating tachyzoites. The identification of new druggable targets for the bradyzoite cysts would open new pharmacological therapeutic windows (170) and offer the chance to prevent reactivated toxoplasmosis in patients with immunodeficiencies, including HIV infection and organ transplantation, and of recurrent OT in immune-competent individuals. Additionally, T. gondii seropositivity has been epidemiologically associated with neurological and mental illnesses, and with cognitive impairment. Presumably, an anti-parasitic therapy eliminating the parasite might be beneficial for these patients and might even answer the question whether T. gondii persistence contributes to these disorders.
While certain Toxoplasma genotypes have been associated with increased disease frequency and severe manifestations, knowledge on how specific genotypes contribute to the clinical severity is missing in humans. A further understanding of the association between parasite lineages and clinical manifestations, especially severe CNS disease in humans, is therefore needed (171). Further, because the clinical spectrum is contextual, the impact of human genotype on immune-surveillance -especially in relation to mechanisms of parasite reactivation- needs to be explored. Improved diagnostics could be of benefits for risk groups, including monitoring therapy and advice in relation to congenital toxoplasmosis. In addition, the more precise identification of genetic risk factors for reactivated TE in AIDS patients and those undergoing transplantation, might allow a better risk stratification.
Abrogated cyst formation in animals used for meat production would effectively truncate foodborne transmission. Similarly, interruption of the parasitic life cycle by preventing oocyst formation in felines would significantly impact on transmission. To achieve this, a further understanding of the feline parasite stages with a perspective on developing vaccines is needed. Also, an understanding of immune responses in perspective on vaccines directed toward intermediate hosts used for meat consumption.
It is evident that major advances have been done in the understanding of cerebral toxoplasmosis over the last 3 decades. The use of experimental models has provided extensive new knowledge on mechanisms of disease at the molecular, cellular and systemic levels. Additionally, light has been shed on various aspects of the host-pathogen interplay. Yet, this knowledge needs to be further applied to the understanding of the disease in humans. This still represents a significant challenge for future research.
All authors listed have made a substantial, direct and intellectual contribution to the work and approved it for publication.
Collaborative research of DS and AB was supported by a grant from the European Community in the EU/ERA-net Neuron (VR/2014-7533 to AB and 01EW1506 to DS) network. The research was additionally supported by the Deutsche Forschungsgemeinschaft (SFB 854, TP5 to DS) and the Swedish Research Council (2018-02411 to AB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank members of the Barragan lab and Gopala Nishanth (University of Magdeburg, Germany) for careful reading of the manuscript and for helpful discussions.
1. Dubey JP. Toxoplasma Gondii. In: Baron S, editor. Medical Microbiology. 4th ed. Galveston, TX: University of Texas Medical Branch at Galveston (1996).
2. Sibley LD, Ajioka JW. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol. (2008) 62:329–51. doi: 10.1146/annurev.micro.62.081307.162925
3. Sibley LD, Khan A, Ajioka JW, Rosenthal BM. Genetic diversity of Toxoplasma gondii in animals and humans. Philos Trans R Soc Lond B Biol Sci. (2009) 364:2749–61. doi: 10.1098/rstb.2009.0087
4. Joynson DH, Wreghitt TJ. Toxoplasmosis: A Comprehensive Clinical Guide. Cambridge: Cambridge University Press (2001).
5. Parlog A, Schluter D, Dunay IR. Toxoplasma gondii -induced neuronal alterations. Parasite Immunol. (2015) 37:159–70. doi: 10.1111/pim.12157
6. Mendez OA, Koshy AA. Toxoplasma gondii: Entry, association, and physiological influence on the central nervous system. PLoS Pathog. (2017) 13:e1006351. doi: 10.1371/journal.ppat.1006351
7. Tedford E, McConkey G. Neurophysiological Changes Induced by Chronic Toxoplasma gondii Infection. Pathogens (2017) 6:2) doi: 10.20944/preprints201705.0071.v1
8. Wohlfert EA, Blader IJ, Wilson EH. Brains and brawn: toxoplasma infections of the central nervous system and skeletal muscle. Trends Parasitol. (2017) 33:519–31. doi: 10.1016/j.pt.2017.04.001
9. Pauwels A, Meyohas MC, Eliaszewicz M, Legendre C, Mougeot G, Frottier J. Toxoplasma colitis in the acquired immunodeficiency syndrome. Am J Gastroenterol. (1992) 87:518–9.
10. Gregg B, Taylor BC, John B, Tait-Wojno ED, Girgis NM, Miller N, et al. Replication and distribution of Toxoplasma gondii in the small intestine after oral infection with tissue cysts. Infect Immun. (2013) 81:1635–43. doi: 10.1128/IAI.01126-12
11. Coombes JL, Charsar BA, Han SJ, Halkias J, Chan SW, Koshy AA, et al. Motile invaded neutrophils in the small intestine of Toxoplasma gondii -infected mice reveal a potential mechanism for parasite spread. Proc Natl Acad Sci USA. (2013) 110:E1913–1922. doi: 10.1073/pnas.1220272110
12. Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. (2006) 177:8785–95. doi: 10.4049/jimmunol.177.12.8785
13. Vaughan LB, Wenzel RP. Disseminated toxoplasmosis presenting as septic shock five weeks after renal transplantation. Transpl Infect Dis. (2013) 15:E20–24. doi: 10.1111/tid.12044
14. Djurkovic-Djakovic O, Djokic V, Vujanic M, Zivkovic T, Bobic B, Nikolic A, et al. Kinetics of parasite burdens in blood and tissues during murine toxoplasmosis. Exp Parasitol. (2012) 131:372–6. doi: 10.1016/j.exppara.2012.05.006
15. Kanatani S, Fuks JM, Olafsson EB, Westermark L, Chambers B, Varas-Godoy M, et al. Voltage-dependent calcium channel signaling mediates GABAA receptor-induced migratory activation of dendritic cells infected by Toxoplasma gondii. PLoS Pathog. (2017) 13:e1006739. doi: 10.1371/journal.ppat.1006739
16. Luft BJ, Hafner R, Korzun AH, Leport C, Antoniskis D, Bosler EM, et al. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. N Engl J Med. (1993) 329:995–1000. doi: 10.1056/NEJM199309303291403
17. Lachenmaier SM, Deli MA, Meissner M, Liesenfeld O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J Neuroimmunol. (2011) 232:119–30. doi: 10.1016/j.jneuroim.2010.10.029
18. Konradt C, Ueno N, Christian DA, Delong JH, Pritchard GH, Herz J, et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat Microbiol. (2016) 1:16001. doi: 10.1038/nmicrobiol.2016.1
19. Ganiem AR, Dian S, Indriati A, Chaidir L, Wisaksana R, Sturm P, et al. Cerebral toxoplasmosis mimicking subacute meningitis in HIV-infected patients; a cohort study from Indonesia. PLoS Negl Trop Dis. (2013) 7:e1994. doi: 10.1371/journal.pntd.0001994
20. Falangola MF, Petito CK. Choroid plexus infection in cerebral toxoplasmosis in AIDS patients. Neurology (1993) 43:2035–40. doi: 10.1212/WNL.43.10.2035
21. Nicoll S, Wright S, Maley SW, Burns S, Buxton D. A mouse model of recrudescence of Toxoplasma gondii infection. J Med Microbiol. (1997) 46:263–6. doi: 10.1099/00222615-46-3-263
22. Dellacasa-Lindberg I, Hitziger N, Barragan A. Localized recrudescence of Toxoplasma infections in the central nervous system of immunocompromised mice assessed by in vivo bioluminescence imaging. Microbes Infect. (2007) 9:1291–8. doi: 10.1016/j.micinf.2007.06.003
23. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet (2004) 363:1965–76. doi: 10.1016/S0140-6736(04)16412-X
24. Schluter D, Lohler J, Deckert M, Hof H, Schwendemann G. Toxoplasma encephalitis of immunocompetent and nude mice: immunohistochemical characterisation of Toxoplasma antigen, infiltrates and major histocompatibility complex gene products. J Neuroimmunol. (1991) 31:185–98. doi: 10.1016/0165-5728(91)90040-E
25. Pusch L, Romeike B, Deckert M, Mawrin C. Persistent toxoplasma bradyzoite cysts in the brain: incidental finding in an immunocompetent patient without evidence of a toxoplasmosis. Clin Neuropathol. (2009) 28:210–2. doi: 10.5414/NPP28210
26. Cabral CM, Tuladhar S, Dietrich HK, Nguyen E, MacDonald WR, Trivedi T, et al. Neurons are the primary target cell for the brain-tropic intracellular parasite Toxoplasma gondii. PLoS Pathog. (2016) 12:e1005447. doi: 10.1371/journal.ppat.1005447
27. Garcia-Garcia C, Castillo-Alvarez F, Azcona-Gutierrez JM, Herraiz MJ, Ibarra V, Oteo JA. Spinal cord toxoplasmosis in human immunodeficiency virus infection/acquired immunodeficiency syndrome. Infect Dis. (2015) 47:277–82. doi: 10.3109/00365548.2014.993421
28. Mohle L, Parlog A, Pahnke J, Dunay IR. Spinal cord pathology in chronic experimental Toxoplasma gondii infection. Eur J Microbiol Immunol. (2014) 4:65–75. doi: 10.1556/EuJMI.4.2014.1.6
29. Maenz M, Schluter D, Liesenfeld O, Schares G, Gross U, Pleyer U. Ocular toxoplasmosis past, present and new aspects of an old disease. Prog Retin Eye Res. (2014) 39:77–106. doi: 10.1016/j.preteyeres.2013.12.005
30. Furtado JM, Ashander LM, Mohs K, Chipps TJ, Appukuttan B, Smith JR. Toxoplasma gondii migration within and infection of human retina. PLoS ONE (2013) 8:e54358. doi: 10.1371/journal.pone.0054358
31. Dukaczewska A, Tedesco R, Liesenfeld O. Experimental models of ocular infection with Toxoplasma Gondii. Eur J Microbiol Immunol. (2015) 5:293–305. doi: 10.1556/1886.2015.00045
32. Crider SR, Horstman WG, Massey GS. Toxoplasma orchitis: report of a case and a review of the literature. Am J Med. (1988) 85:421–4. doi: 10.1016/0002-9343(88)90599-2
33. Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol. (2005) 7:837–48. doi: 10.1111/j.1462-5822.2005.00517.x
34. Leal FE, Cavazzana CL, de Andrade HF Jr, Galisteo A Jr, de Mendonca JS, Kallas EG. Toxoplasma gondii pneumonia in immunocompetent subjects: case report and review. Clin Infect Dis. (2007) 44:e62–66. doi: 10.1086/511871
36. Baba M, Batanova T, Kitoh K, Takashima Y. Adhesion of Toxoplasma gondii tachyzoite-infected vehicle leukocytes to capillary endothelial cells triggers timely parasite egression. Sci Rep. (2017) 7:5675. doi: 10.1038/s41598-017-05956-z
37. Arora N, Sadovsky Y, Dermody TS, Coyne CB. Microbial vertical transmission during human pregnancy. Cell Host Microbe. (2017) 21:561–7. doi: 10.1016/j.chom.2017.04.007
38. Khan K, Khan W. Congenital toxoplasmosis: an overview of the neurological and ocular manifestations. Parasitol Int. (2018) 67:715–21. doi: 10.1016/j.parint.2018.07.004
39. Ander SE, Rudzki EN, Arora N, Sadovsky Y, Coyne CB, Boyle JP. Human placental syncytiotrophoblasts restrict Toxoplasma gondii attachment and replication and respond to infection by producing immunomodulatory chemokines. MBio (2018) 9:e01678–17. doi: 10.1128/mBio.01678-17
40. Vargas-Villavicencio JA, Cedillo-Pelaez C, Rico-Torres CP, Besne-Merida A, Garcia-Vazquez F, Saldana JI, et al. Mouse model of congenital infection with a non-virulent Toxoplasma gondii strain: vertical transmission, “sterile” fetal damage, or both? Exp Parasitol. (2016) 166:116–23. doi: 10.1016/j.exppara.2016.04.002
41. Derouin F, Pelloux H, Parasitology ESG. Prevention of toxoplasmosis in transplant patients. Clin Microbiol Infect. (2008) 14:1089–101. doi: 10.1111/j.1469-0691.2008.02091.x
42. Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. (1992) 15:211–22. doi: 10.1093/clinids/15.2.211
43. Martin-Blondel G, Alvarez M, Delobel P, Uro-Coste E, Cuzin L, Cuvinciuc V, et al. Toxoplasmic encephalitis IRIS in HIV-infected patients: a case series and review of the literature. J Neurol Neurosurg Psychiatry (2011) 82:691–3. doi: 10.1136/jnnp.2009.199919
44. An R, Tang Y, Chen L, Cai H, Lai DH, Liu K, et al. Encephalitis is mediated by ROP18 of Toxoplasma gondii, a severe pathogen in AIDS patients. Proc Natl Acad Sci USA. (2018) 115:E5344–52. doi: 10.1073/pnas.1801118115
45. Dubey JP. Bradyzoite-induced murine toxoplasmosis: stage conversion, pathogenesis, and tissue cyst formation in mice fed bradyzoites of different strains of Toxoplasma gondii. J Eukaryot Microbiol. (1997) 44:592–602. doi: 10.1111/j.1550-7408.1997.tb05965.x
46. Dubey JP. Distribution of tissue cysts in organs of rats fed Toxoplasma gondii oocysts. J Parasitol. (1997) 83:755–7. doi: 10.2307/3284258
47. Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature (1999) 401:804–8. doi: 10.1038/44593
48. Lambert H, Vutova PP, Adams WC, Lore K, Barragan A. The Toxoplasma gondii -shuttling function of dendritic cells is linked to the parasite genotype. Infect Immun. (2009) 77:1679–88. doi: 10.1128/IAI.01289-08
49. Couper KN, Roberts CW, Brombacher F, Alexander J, Johnson LL. Toxoplasma gondii -specific immunoglobulin M limits parasite dissemination by preventing host cell invasion. Infect Immun. (2005) 73:8060–8. doi: 10.1128/IAI.73.12.8060-8068.2005
50. Persson CM, Lambert H, Vutova PP, Dellacasa-Lindberg I, Nederby J, Yagita H, et al. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect Immun. (2009) 77:970–6. doi: 10.1128/IAI.00833-08
51. Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol. (2006) 8:1611–23. doi: 10.1111/j.1462-5822.2006.00735.x
52. Olafsson EB, Ross EC, Varas-Godoy M, Barragan A. TIMP-1 promotes hypermigration of Toxoplasma-infected primary dendritic cells via CD63/ITGB1/FAK signaling. J Cell Sci. (2019). doi: 10.1242/jcs.225193
53. Fuks JM, Arrighi RB, Weidner JM, Kumar Mendu S, Jin Z, Wallin RP, et al. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog. (2012) 8:e1003051. doi: 10.1371/journal.ppat.1003051
54. Weidner JM, Kanatani S, Hernandez-Castaneda MA, Fuks JM, Rethi B, Wallin RP, et al. Rapid cytoskeleton remodelling in dendritic cells following invasion by Toxoplasma gondii coincides with the onset of a hypermigratory phenotype. Cell Microbiol. (2013) 15:1735–52. doi: 10.1111/cmi.12145
55. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
56. Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife (2017) 6:e29738. doi: 10.7554/eLife.29738
57. Barragan A, Brossier F, Sibley LD. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell Microbiol. (2005) 7:561–8. doi: 10.1111/j.1462-5822.2005.00486.x
58. Ueno N, Harker KS, Clarke EV, McWhorter FY, Liu WF, Tenner AJ, et al. Real-time imaging of Toxoplasma-infected human monocytes under fluidic shear stress reveals rapid translocation of intracellular parasites across endothelial barriers. Cell Microbiol. (2014) 16:580–95. doi: 10.1111/cmi.12239
59. Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood (2006) 107:309–16. doi: 10.1182/blood-2005-02-0666
60. Lambert H, Barragan A. Modelling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol. (2010) 12:292–300. doi: 10.1111/j.1462-5822.2009.01417.x
61. Furtado JM, Bharadwaj AS, Ashander LM, Olivas A, Smith JR. Migration of toxoplasma gondii-infected dendritic cells across human retinal vascular endothelium. Invest Ophthalmol Vis Sci. (2012) 53:6856–62. doi: 10.1167/iovs.12-10384
62. Furtado JM, Bharadwaj AS, Chipps TJ, Pan Y, Ashander LM, Smith JR. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol Cell Biol. (2012) 90:912–5. doi: 10.1038/icb.2012.21
63. Tembo DL, Nyoni B, Murikoli RV, Mukaka M, Milner DA, Berriman M, et al. Differential PfEMP1 expression is associated with cerebral malaria pathology. PLoS Pathog. (2014) 10:e1004537. doi: 10.1371/journal.ppat.1004537
64. Nacer A, Movila A, Sohet F, Girgis NM, Gundra UM, Loke P, et al. Experimental cerebral malaria pathogenesis–hemodynamics at the blood brain barrier. PLoS Pathog. (2014) 10:e1004528. doi: 10.1371/journal.ppat.1004528
65. Estato V, Stipursky J, Gomes F, Mergener TC, Frazao-Teixeira E, Allodi S, et al. The neurotropic parasite Toxoplasma gondii induces sustained neuroinflammation with microvascular dysfunction in infected mice. Am J Pathol. (2018) 188:2674–87. doi: 10.1016/j.ajpath.2018.07.007
66. Conley FK, Jenkins KA. Immunohistological study of the anatomic relationship of toxoplasma antigens to the inflammatory response in the brains of mice chronically infected with Toxoplasma gondii. Infect Immun. (1981) 31:1184–92.
67. Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli AE, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol. (2008) 181:8485–91. doi: 10.4049/jimmunol.181.12.8485
68. Barragan A, Sibley LD. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med. (2002) 195:1625–33. doi: 10.1084/jem.20020258
69. Dellacasa-Lindberg I, Fuks JM, Arrighi RB, Lambert H, Wallin RP, Chambers BJ, et al. Migratory activation of primary cortical microglia upon infection with Toxoplasma gondii. Infect Immun. (2011) 79:3046–52. doi: 10.1128/IAI.01042-10
70. Berenreiterova M, Flegr J, Kubena AA, Nemec P. The distribution of Toxoplasma gondii cysts in the brain of a mouse with latent toxoplasmosis: implications for the behavioral manipulation hypothesis. PLoS ONE (2011) 6:e28925. doi: 10.1371/journal.pone.0028925
71. Vyas A, Kim SK, Giacomini N, Boothroyd JC, Sapolsky RM. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci USA. (2007) 104:6442–7. doi: 10.1073/pnas.0608310104
72. Haroon F, Handel U, Angenstein F, Goldschmidt J, Kreutzmann P, Lison H, et al. Toxoplasma gondii actively inhibits neuronal function in chronically infected mice. PLoS ONE (2012) 7:e35516. doi: 10.1371/journal.pone.0035516
73. Fischer HG, Nitzgen B, Reichmann G, Gross U, Hadding U. Host cells of Toxoplasma gondii encystation in infected primary culture from mouse brain. Parasitol Res. (1997) 83:637–41. doi: 10.1007/s004360050311
74. Ferguson DJP, Hutchison WM. An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice. Parasitol Res. (1987) 73:483–91. doi: 10.1007/BF00535321
75. Melzer TC, Cranston HJ, Weiss LM, Halonen SK. Host Cell Preference of Toxoplasma gondii cysts in murine brain: a confocal study. J Neuroparasitol. (2010) 1:N100505. doi: 10.4303/jnp/N100505
76. Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron (1998) 21:505–20. doi: 10.1016/S0896-6273(00)80562-0
77. Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science (2000) 290:2155–9. doi: 10.1126/science.290.5499.2155
78. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science (1995) 269:549–52. doi: 10.1126/science.7624779
79. Suzuki Y, Sa Q, Gehman M, Ochiai E. Interferon-gamma- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev Mol Med. (2011) 13:e31. doi: 10.1017/S1462399411002018
80. Luder CG, Giraldo-Velasquez M, Sendtner M, Gross U. Toxoplasma gondii in primary rat CNS cells: differential contribution of neurons, astrocytes, and microglial cells for the intracerebral development and stage differentiation. Exp Parasitol. (1999) 93:23–32. doi: 10.1006/expr.1999.4421
81. Ferreira da Silva Mda F, Barbosa HS, Gross U, Luder CG. Stress-related and spontaneous stage differentiation of Toxoplasma gondii. Mol Biosyst. (2008) 4:824–34. doi: 10.1039/b800520f
82. Song HB, Jung BK, Kim JH, Lee YH, Choi MH, Kim JH. Investigation of tissue cysts in the retina in a mouse model of ocular toxoplasmosis: distribution and interaction with glial cells. Parasitol Res. (2018) 117:2597–605. doi: 10.1007/s00436-018-5950-3
83. Paredes-Santos TC, Martins-Duarte ES, Vitor RW, de Souza W, Attias M, Vommaro RC. Spontaneous cystogenesis in vitro of a Brazilian strain of Toxoplasma gondii. Parasitol Int. (2013) 62:181–8. doi: 10.1016/j.parint.2012.12.003
84. Ferguson DJP, Hutchison WM. The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch A (1987) 411:39–43. doi: 10.1007/BF00734512
85. Robert-Gangneux F, Meroni V, Dupont D, Botterel F, Garcia JMA, Brenier-Pinchart MP, et al. Toxoplasmosis in Transplant Recipients, Europe, 2010-2014. Emerg Infect Dis. (2018) 24:1497–504. doi: 10.3201/eid2408.180045
86. Behnke MS, Dubey JP, Sibley LD. Genetic Mapping of pathogenesis determinants in Toxoplasma gondii. Annu Rev Microbiol. (2016) 70:63–81. doi: 10.1146/annurev-micro-091014-104353
87. Brown CR, Hunter CA, Estes RG, Beckmann E, Forman J, David C, et al. Definitive identification of a gene that confers resistance against Toxoplasma cyst burden and encephalitis. Immunology (1995) 85:419–28.
88. Brown CR, McLeod R. Class I MHC genes and CD8+ T cells determine cyst number in Toxoplasma gondii infection. J Immunol. (1990) 145:3438–41.
89. Deckert-Schluter M, Schluter D, Schmidt D, Schwendemann G, Wiestler OD, Hof H. Toxoplasma encephalitis in congenic B10 and BALB mice: impact of genetic factors on the immune response. Infect Immun. (1994) 62:221–8.
90. Dubey JP. Mouse pathogenicity of Toxoplasma gondii isolated from a goat. Am J Vet Res. (1980) 41:427–9.
91. Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis. (1995) 172:1561–6. doi: 10.1093/infdis/172.6.1561
92. Grigg ME, Ganatra J, Boothroyd JC, Margolis TP. Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J Infect Dis. (2001) 184:633–9. doi: 10.1086/322800
93. Saraf P, Shwab EK, Dubey JP, Su C. On the determination of Toxoplasma gondii virulence in mice. Exp Parasitol. (2017) 174:25–30. doi: 10.1016/j.exppara.2017.01.009
94. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
95. Scharton-Kersten T, Nakajima H, Yap G, Sher A, Leonard WJ. Infection of mice lacking the common cytokine receptor gamma-chain (gamma(c)) reveals an unexpected role for CD4+ T lymphocytes in early IFN-gamma-dependent resistance to Toxoplasma gondii. J Immunol. (1998) 160:2565–9.
96. Kwok LY, Lutjen S, Soltek S, Soldati D, Busch D, Deckert M, et al. The induction and kinetics of antigen-specific CD8 T cells are defined by the stage specificity and compartmentalization of the antigen in murine toxoplasmosis. J Immunol. (2003) 170:1949–57. doi: 10.4049/jimmunol.170.4.1949
97. Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, et al. Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol. (2008) 9:937–44. doi: 10.1038/ni.1629
98. Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J Immunol. (1992) 149:175–80.
99. Lutjen S, Soltek S, Virna S, Deckert M, Schluter D. Organ- and disease-stage-specific regulation of Toxoplasma gondii -specific CD8-T-cell responses by CD4 T cells. Infect Immun. (2006) 74:5790–801. doi: 10.1128/IAI.00098-06
100. Schaeffer M, Han SJ, Chtanova T, van Dooren GG, Herzmark P, Chen Y, et al. Dynamic imaging of T cell-parasite interactions in the brains of mice chronically infected with Toxoplasma gondii. J Immunol. (2009) 182:6379–93. doi: 10.4049/jimmunol.0804307
101. Wilson EH, Harris TH, Mrass P, John B, Tait ED, Wu GF, et al. Behavior of parasite-specific effector CD8+ T cells in the brain and visualization of a kinesis-associated system of reticular fibers. Immunity (2009) 30:300–11. doi: 10.1016/j.immuni.2008.12.013
102. Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J Immunol. (2000) 164:4826–34. doi: 10.4049/jimmunol.164.9.4826
103. Biswas A, Bruder D, Wolf SA, Jeron A, Mack M, Heimesaat MM, et al. Ly6C(high) monocytes control cerebral toxoplasmosis. J Immunol. (2015) 194:3223–35. doi: 10.4049/jimmunol.1402037
104. Schluter D, Deckert-Schluter M, Lorenz E, Meyer T, Rollinghoff M, Bogdan C. Inhibition of inducible nitric oxide synthase exacerbates chronic cerebral toxoplasmosis in Toxoplasma gondii -susceptible C57BL/6 mice but does not reactivate the latent disease in T. gondii-resistant BALB/c mice. J Immunol. (1999) 162:3512–8.
105. Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity (2014) 40:924–35. doi: 10.1016/j.immuni.2014.05.006
106. Sasai M, Sakaguchi N, Ma JS, Nakamura S, Kawabata T, Bando H, et al. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat Immunol. (2017) 18:899–910. doi: 10.1038/ni.3767
107. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. (2005) 1:e24. doi: 10.1371/journal.ppat.0010024
108. Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, et al. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. (2008) 27:2495–509. doi: 10.1038/emboj.2008.176
109. Henry SC, Daniell XG, Burroughs AR, Indaram M, Howell DN, Coers J, et al. Balance of Irgm protein activities determines IFN-gamma-induced host defense. J Leukoc Biol. (2009) 85:877–85. doi: 10.1189/jlb.1008599
110. Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, et al. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog. (2013) 9:e1003414. doi: 10.1371/journal.ppat.1003414
111. Muller UB, Howard JC. The impact of Toxoplasma gondii on the mammalian genome. Curr Opin Microbiol. (2016) 32:19–25. doi: 10.1016/j.mib.2016.04.009
112. Lilue J, Muller UB, Steinfeldt T, Howard JC. Reciprocal virulence and resistance polymorphism in the relationship between Toxoplasma gondii and the house mouse. Elife (2013) 2:e01298. doi: 10.7554/eLife.01298
113. Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, et al. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity (2012) 37:302–13. doi: 10.1016/j.immuni.2012.06.009
114. Zhao ZJ, Zhang J, Wei J, Li Z, Wang T, Yi SQ, et al. Lower expression of inducible nitric oxide synthase and higher expression of arginase in rat alveolar macrophages are linked to their susceptibility to Toxoplasma gondii infection. PLoS ONE (2013) 8:e63650. doi: 10.1371/journal.pone.0063650
115. Witola WH, Kim CY, Zhang X. Inherent oxidative stress in the lewis rat is associated with resistance to toxoplasmosis. Infect Immun. (2017) 85: e00289-17. doi: 10.1128/IAI.00289-17
116. Kim CY, Zhang X, Witola WH. Small GTPase immunity-associated proteins mediate resistance to Toxoplasma gondii infection in lewis rat. Infect Immun. 86:e00582–17. doi: 10.1128/IAI.00582-17
117. Gazzinelli RT, Mendonca-Neto R, Lilue J, Howard J, Sher A. Innate resistance against Toxoplasma gondii: an evolutionary tale of mice, cats, and men. Cell Host Microbe. (2014) 15:132–8. doi: 10.1016/j.chom.2014.01.004
118. Safronova A, Araujo A, Camanzo ET, Moon TJ, Elliott MR, Beiting DP, et al. Alarmin S100A11 initiates a chemokine response to the human pathogen Toxoplasma gondii. Nat Immunol. (2019) 20:64–72. doi: 10.1038/s41590-018-0250-8
119. Daubener W, Remscheid C, Nockemann S, Pilz K, Seghrouchni S, Mackenzie C, et al. Anti-parasitic effector mechanisms in human brain tumor cells: role of interferon-gamma and tumor necrosis factor-alpha. Eur J Immunol. (1996) 26:487–92. doi: 10.1002/eji.1830260231
120. Bando H, Lee Y, Sakaguchi N, Pradipta A, Ma JS, Tanaka S, et al. Inducible nitric oxide synthase is a key host factor for toxoplasma GRA15-dependent disruption of the gamma interferon-induced antiparasitic human response. MBio (2018) 9:e01738–18. doi: 10.1128/mBio.01738-18
121. Bando H, Sakaguchi N, Lee Y, Pradipta A, Ma JS, Tanaka S, et al. Toxoplasma effector TgIST Targets Host IDO1 to antagonize the IFN-gamma-induced anti-parasitic response in human cells. Front Immunol. (2018) 9:2073. doi: 10.3389/fimmu.2018.02073
122. Swierzy IJ, Handel U, Kaever A, Jarek M, Scharfe M, Schluter D, et al. Divergent co-transcriptomes of different host cells infected with Toxoplasma gondii reveal cell type-specific host-parasite interactions. Sci Rep. (2017) 7:7229. doi: 10.1038/s41598-017-07838-w
123. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. (1996) 157:798–805.
124. Deckert-Schluter M, Buck C, Weiner D, Kaefer N, Rang A, Hof H, et al. Interleukin-10 downregulates the intracerebral immune response in chronic Toxoplasma encephalitis. J Neuroimmunol. (1997) 76:167–76. doi: 10.1016/S0165-5728(97)00047-7
125. Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. (2006) 7:937–45. doi: 10.1038/ni1376
126. Marchi N, Granata T, Janigro D. Inflammatory pathways of seizure disorders. Trends Neurosci. (2014) 37:55–65. doi: 10.1016/j.tins.2013.11.002
127. Ngoungou EB, Bhalla D, Nzoghe A, Darde ML, Preux PM. Toxoplasmosis and epilepsy–systematic review and meta analysis. PLoS Negl Trop Dis. (2015) 9:e0003525. doi: 10.1371/journal.pntd.0003525
128. Uzorka JW, Arend SM. A critical assessment of the association between postnatal toxoplasmosis and epilepsy in immune-competent patients. Eur J Clin Microbiol Infect Dis. (2017) 36:1111–7. doi: 10.1007/s10096-016-2897-0
129. Brooks JM, Carrillo GL, Su J, Lindsay DS, Fox MA, Blader IJ. Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous system. MBio (2015) 6:e01428–e01415. doi: 10.1128/mBio.01428-15
130. David CN, Frias ES, Szu JI, Vieira PA, Hubbard JA, Lovelace J, et al. GLT-1-Dependent disruption of CNS glutamate homeostasis and neuronal function by the protozoan parasite Toxoplasma gondii. PLoS Pathog. (2016) 12:e1005643. doi: 10.1371/journal.ppat.1005643
131. Drogemuller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, et al. Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol. (2008) 181:2683–93. doi: 10.4049/jimmunol.181.4.2683
132. Stenzel W, Soltek S, Schluter D, Deckert M. The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol. (2004) 63:631–40. doi: 10.1093/jnen/63.6.631
133. Hidano S, Randall LM, Dawson L, Dietrich HK, Konradt C, Klover PJ, et al. STAT1 signaling in astrocytes is essential for control of infection in the central nervous system. MBio (2016) 7:e01881–16. doi: 10.1128/mBio.01881-16
134. Fischer HG, Nitzgen B, Reichmann G, Hadding U. Cytokine responses induced by Toxoplasma gondii in astrocytes and microglial cells. Eur J Immunol. (1997) 27:1539–48. doi: 10.1002/eji.1830270633
135. Strack A, Asensio VC, Campbell IL, Schluter D, Deckert M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. (2002) 103:458–68. doi: 10.1007/s00401-001-0491-7
136. Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, et al. Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol. (2014) 193:139–49. doi: 10.4049/jimmunol.1303284
137. Halonen SK, Weiss LM, Chiu FC. Association of host cell intermediate filaments with Toxoplasma gondii cysts in murine astrocytes in vitro. Int J Parasitol. (1998) 28:815–23. doi: 10.1016/S0020-7519(98)00035-6
138. Schluter D, Deckert M, Hof H, Frei K. Toxoplasma gondii infection of neurons induces neuronal cytokine and chemokine production, but gamma interferon- and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and growth of T. gondii Infect Immun. (2001) 69:7889–93. doi: 10.1128/IAI.69.12.7889-7893.2001
139. Handel U, Brunn A, Drogemuller K, Muller W, Deckert M, Schluter D. Neuronal gp130 expression is crucial to prevent neuronal loss, hyperinflammation, and lethal course of murine Toxoplasma encephalitis. Am J Pathol. (2012) 181:163–73. doi: 10.1016/j.ajpath.2012.03.029
140. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. (2017) 18:385–92. doi: 10.1038/ni.3703
141. Deckert M, Sedgwick JD, Fischer E, Schluter D. Regulation of microglial cell responses in murine Toxoplasma encephalitis by CD200/CD200 receptor interaction. Acta Neuropathol. (2006) 111:548–58. doi: 10.1007/s00401-006-0062-z
142. Robert-Gangneux F, Sterkers Y, Yera H, Accoceberry I, Menotti J, Cassaing S, et al. Molecular diagnosis of toxoplasmosis in immunocompromised patients: a 3-year multicenter retrospective study. J Clin Microbiol. (2015) 53:1677–84. doi: 10.1128/JCM.03282-14
143. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. (2014) 26:454–70. doi: 10.1016/j.smim.2014.09.008
144. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (2010) 89:381–402. doi: 10.1097/MD.0b013e3181fdd832
145. Gonzalez-Vicent M, Diaz MA, Sevilla J, Madero L. Cerebral toxoplasmosis following etanercept treatment for idiophatic pneumonia syndrome after autologous peripheral blood progenitor cell transplantation (PBPCT). Ann Hematol. (2003) 82:649–53. doi: 10.1007/s00277-003-0705-2
146. Lassoued S, Zabraniecki L, Marin F, Billey T. Toxoplasmic chorioretinitis and antitumor necrosis factor treatment in rheumatoid arthritis. Semin Arthritis Rheum. (2007) 36:262–3. doi: 10.1016/j.semarthrit.2006.08.004
147. Rodrigues KF, Faria e Arantes TE, Muccioli C, Neto JL, Pinheiro MM. Incidence of Toxoplasma retinochoroiditis in patients with ankylosing spondylitis after using TNF-alpha blockers. Parasitol Int. (2013) 62:272–5. doi: 10.1016/j.parint.2013.02.003
148. Gazzinelli RT, Eltoum I, Wynn TA, Sher A. Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF-a and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J Immunol. (1993) 151:3672–81.
149. Murdaca G, Spano F, Contatore M, Guastalla A, Penza E, Magnani O, et al. Infection risk associated with anti-TNF-alpha agents: a review. Expert Opin Drug Saf. (2015) 14:571–82. doi: 10.1517/14740338.2015.1009036
150. Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system Curr Opin HIV AIDS (2013) 8:165–9. doi: 10.1097/COH.0b013e32835fc601
151. Bednar MM, Sturdevant CB, Tompkins LA, Arrildt KT, Dukhovlinova E, Kincer LP, et al. Compartmentalization, viral evolution, and viral latency of HIV in the CNS. Curr HIV AIDS Rep. (2015) 12:262–71. doi: 10.1007/s11904-015-0265-9
152. Zecca C, Nessi F, Bernasconi E, Gobbi C. Ocular toxoplasmosis during natalizumab treatment. Neurology (2009) 73:1418–9. doi: 10.1212/WNL.0b013e3181bd114f
153. Vyas R, Ebright JR. Toxoplasmosis of the spinal cord in a patient with AIDS: case report and review. Clin Infect Dis. (1996) 23:1061–5. doi: 10.1093/clinids/23.5.1061
154. group SS, Thiebaut R, Leproust S, Chene G, Gilbert R. Effectiveness of prenatal treatment for congenital toxoplasmosis: a meta-analysis of individual patients' data. Lancet (2007) 369:115–22. doi: 10.1016/S0140-6736(07)60072-5
155. Cortina-Borja M, Tan HK, Wallon M, Paul M, Prusa A, Buffolano W, et al. Prenatal treatment for serious neurological sequelae of congenital toxoplasmosis: an observational prospective cohort study. PLoS Med. (2010) 7:e1000351. doi: 10.1371/journal.pmed.1000351
156. Berdoy M, Webster JP, Macdonald DW. Fatal attraction in rats infected with Toxoplasma gondii. Proc R Soc Lond B Biol Sci. (2000) 267:1591–4. doi: 10.1098/rspb.2000.1182
157. Babaie J, Sayyah M, Fard-Esfahani P, Golkar M, Gharagozli K. Contribution of dopamine neurotransmission in proconvulsant effect of Toxoplasma gondii infection in male mice. J Neurosci Res. (2017) 95:1894–905. doi: 10.1002/jnr.24036
158. Stibbs HH. Changes in brain concentrations of catecholamines and indoleamines in Toxoplasma gondii infected mice. Ann Trop Med Parasitol. (1985) 79:153–7. doi: 10.1080/00034983.1985.11811902
159. Poirotte C, Kappeler PM, Ngoubangoye B, Bourgeois S, Moussodji M, Charpentier MJ. Morbid attraction to leopard urine in Toxoplasma-infected chimpazees. Curr Biol. (2016) 26:R98–9. doi: 10.1016/j.cub.2015.12.020
160. Sutterland AL, Fond G, Kuin A, Koeter MW, Lutter R, van Gool T, et al. Beyond the association. Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: systematic review and meta-analysis. Acta Psychiatr Scand. (2015) 132:161–79. doi: 10.1111/acps.12423
161. de Barros J, Barbosa IG, Salem H, Rocha NP, Kummer A, Okusaga OO, et al. Is there any association between Toxoplasma gondii infection and bipolar disorder? A systematic review and meta-analysis. J Affect Disord. (2017) 209:59–65. doi: 10.1016/j.jad.2016.11.016
162. Xiao J, Prandovszky E, Kannan G, Pletnikov MV, Dickerson F, Severance EG, et al. Toxoplasma gondii: biological parameters of the connection to schizophrenia. Schizophr Bull. (2018) 44:983–92. doi: 10.1093/schbul/sby082
163. Flegr J. Effects of toxoplasma on human behavior. Schizophr Bull. (2007) 33:757–60. doi: 10.1093/schbul/sbl074
164. Ene L, Marcotte TD, Umlauf A, Grancea C, Temereanca A, Bharti A, et al. Latent toxoplasmosis is associated with neurocognitive impairment in young adults with and without chronic HIV infection. J Neuroimmunol. (2016) 299:1–7. doi: 10.1016/j.jneuroim.2016.08.003
165. Stock AK, Dajkic D, Kohling HL, von Heinegg EH, Fiedler M, Beste C. Humans with latent toxoplasmosis display altered reward modulation of cognitive control. Sci Rep. (2017) 7:10170. doi: 10.1038/s41598-017-10926-6
166. Sugden K, Moffitt TE, Pinto L, Poulton R, Williams BS, Caspi A. Is Toxoplasma gondii infection related to brain and behavior impairments in humans? Evidence from a Population-Representative Birth Cohort. PLoS ONE (2016) 11:e0148435. doi: 10.1371/journal.pone.0148435
167. Wyman CP, Gale SD, Hedges-Muncy A, Erickson LD, Wilson E, Hedges DW. Association between Toxoplasma gondii seropositivity and memory function in nondemented older adults. Neurobiol Aging (2017) 53:76–82. doi: 10.1016/j.neurobiolaging.2017.01.018
168. Torniainen-Holm M, Suvisaari J, Lindgren M, Harkanen T, Dickerson F, Yolken RH. The lack of association between herpes simplex virus 1 or Toxoplasma gondii infection and cognitive decline in the general population: an 11-year follow-up study. Brain Behav Immun. (2018) 76:159–64. doi: 10.1016/j.bbi.2018.01.006
169. Montazeri M, Mehrzadi S, Sharif M, Sarvi S, Tanzifi A, Aghayan SA, et al. Drug Resistance in Toxoplasma gondii. Front Microbiol. (2018) 9:2587. doi: 10.3389/fmicb.2018.02587
170. Di Cristina M, Dou Z, Lunghi M, Kannan G, Huynh MH, McGovern OL, et al. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat Microbiol. (2017) 2:17096. doi: 10.1038/nmicrobiol.2017.96
Keywords: MeSH, neurotropic agents, neuroparasite, neuroinflammation, CNS infection, congenital infection
Citation: Schlüter D and Barragan A (2019) Advances and Challenges in Understanding Cerebral Toxoplasmosis. Front. Immunol. 10:242. doi: 10.3389/fimmu.2019.00242
Received: 11 September 2018; Accepted: 28 January 2019;
Published: 14 February 2019.
Edited by:
Martin Rottenberg, Karolinska Institute (KI), SwedenReviewed by:
Michael Fox, Virginia Tech Carilion Research Institute, United StatesCopyright © 2019 Schlüter and Barragan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Schlüter, c2NobHVldGVyLmRpcmtAbWgtaGFubm92ZXIuZGU=
Antonio Barragan, YW50b25pby5iYXJyYWdhbkBzdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.