 Katina Schinnerling
Katina Schinnerling Carlos Rosas
Carlos Rosas Lilian Soto1,4
Lilian Soto1,4 Ranjeny Thomas
Ranjeny Thomas Juan Carlos Aguillón
Juan Carlos Aguillón
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 February 2019
Sec. Immunological Tolerance and Regulation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00203
This article is part of the Research Topic Tolerogenic Dendritic Cells and Regulatory T Cells as Therapeutics for Immune-Mediated Disorders View all 29 articles
Rodent models of rheumatoid arthritis (RA) have been used over decades to study the immunopathogenesis of the disease and to explore intervention strategies. Nevertheless, mouse models of RA reach their limit when it comes to testing of new therapeutic approaches such as cell-based therapies. Differences between the human and the murine immune system make it difficult to draw reliable conclusions about the success of immunotherapies. To overcome this issue, humanized mouse models have been established that mimic components of the human immune system in mice. Two main strategies have been pursued for humanization: the introduction of human transgenes such as human leukocyte antigen molecules or specific T cell receptors, and the generation of mouse/human chimera by transferring human cells or tissues into immunodeficient mice. Recently, both approaches have been combined to achieve more sophisticated humanized models of autoimmune diseases. This review discusses limitations of conventional mouse models of RA-like disease and provides a closer look into studies in humanized mice exploring their usefulness and necessity as preclinical models for testing of cell-based therapies in autoimmune diseases such as RA.
Rheumatoid arthritis (RA) is a chronic inflammatory disorder which affects the synovial tissue of the joints, causing articular pain and disability (1). Initially manifested locally, RA later develops into a systemic disease that involves major organ systems and reduces life expectancy (2). The disease is characterized by an infiltration of the synovium with inflammatory cells, proliferation of synovial fibroblasts, forming an invasive pannus that destroys the adjacent cartilage and bone, and progressive joint damage (3).
Rodent models have been used over decades to study the immunopathogenesis of RA and to test the efficacy of anti-rheumatic drugs (4). There are numerous rodent models of RA-like disease, each mirroring certain aspects of the disease (4–6). Important findings have emerged from studies using these models, such as the need for CD4+ T cells and B cells for the development of RA (7, 8), the importance of pro-inflammatory cytokines in RA pathogenesis (9–11), and the discovery of RA-relevant autoantigens (12, 13). Nevertheless, mouse models of RA reach their limit when it comes to testing of new therapeutic approaches such as cell-based therapies. More than 80% of potential therapeutics, which have been shown to be safe and effective in animal studies, fail when tested in humans (14, 15). One example is the lack of therapeutic efficacy of interleukin (IL-) 17 inhibitors in RA patients (16, 17), although suppression of IL-17 signaling had been shown to reduce joint inflammation as well as cartilage and bone destruction in mice (18). Similarly, inhibition of IL-1 signaling had been demonstrated to ameliorate arthritis in mice (19), while anti-IL-1 therapy displayed limited efficacy in RA patients (4, 20). Differences between the human and the murine immune system make it difficult to transfer the results from the mouse model to patients (21, 22). Consequently, translational research needs to refocus on human, and not mouse, immunology (23, 24). This is particularly important for preclinical testing of therapeutic approaches based on dendritic cells (DCs). While monocyte-derived DCs are used for clinical applications in humans (25), DCs obtained from bone marrow precursors are administered in mouse models (26, 27), which considerably affects the transferability of the results from mice to men.
Since studies of human immune responses and disease in vivo are limited by ethical and technical constraints, there is a need for animal models that on the one hand accurately mirror the pathogenesis of the autoimmune disease, and on the other allow pre-clinical testing of cell-based therapeutic approaches targeting human cells and tissues in vivo. To tackle these issues, humanized mice have been developed using two different strategies: (i) the introduction of human RA-relevant transgenes such as human leukocyte antigen (HLA) molecules, T cell receptors (TCR), or autoantigens (28), and (ii) the generation of mouse/human chimera by the engraftment of human (RA-derived) cells and/or tissues into immunodeficient mice (29) (Figure 1). Combination of both approaches will lead to more sophisticated humanized models of autoimmune diseases such as RA (32).
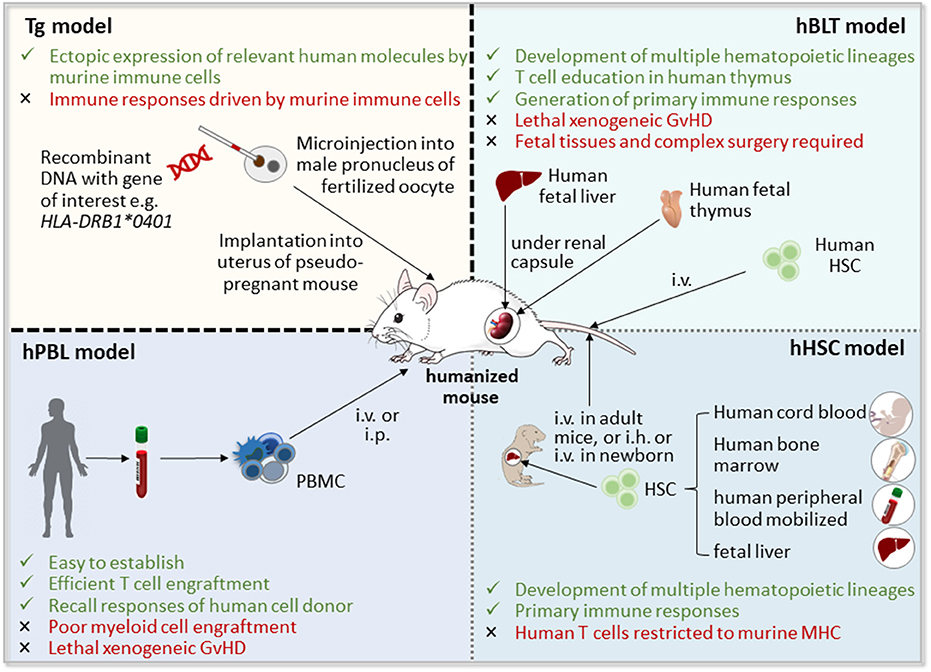
Figure 1. Strategies for the generation of humanized mice [adapted from Shultz (30) and Hahn et al. (31)]. Humanized mice can be obtained by the introduction of human transgenes, such as human leucocyte antigen (HLA) class II molecules, which will then be expressed by mouse immune cells, or by the generation of mouse/human chimera through implantation of human cells, including hematopoietic stem and progenitor cells (HSCs) and peripheral blood mononuclear cells (PBMCs), and/or human tissues, such as fetal liver and thymus, into immunodeficient mice. GvHD, graft vs. host disease; hBLT, human bone marrow-liver-thymus-engrafted; hPBL, human peripheral blood lymphocyte-engrafted; i.h., intrahepatic; i.p., intraperitoneal; i.v., intravenous; Tg, transgenic mice.
This review provides an overview of mouse models of RA-like disease and their limitations and discusses different humanization strategies for the generation of RA models, with focus on the use of humanized mice as tools for pre-clinical testing.
Although the etiology of RA is not fully known, its autoimmune nature has been widely recognized. An interplay between genetic predisposition and environmental factors, such as smoking and microbial infections, are thought to trigger the development of the disease (33). The contribution of genetic factors to RA pathogenesis is mainly attributed to certain HLA alleles of the major histocompatibility complex (MHC) class II (34). Besides presenting antigen peptides to CD4+ T cells in the periphery, HLA class II molecules are also responsible for the selection of the CD4+ TCR repertoire in the thymus and thus control the release of autoreactive cells (35, 36). A consensus amino acid motif in the P4 peptide-binding pocket of the β1 subunit of the HLA-DR molecule, denoted “shared epitope” (SE), is a major risk factor of RA (37, 38). Among HLA-DR alleles containing the SE, DRB1*0401, and *0101 have been described most extensively in the context of RA (39, 40). Various peptides derived from endogenous joint proteins, such as type II collagen (CII), cartilage proteoglycan aggrecan, and human cartilage glycoprotein (HCgp)-39, have been shown to bind to SE-containing HLA-DR molecules and to be specifically recognized by T lymphocytes from RA patients (13, 41–44). Particularly, peptides post-translationally modified by citrullination bind with high affinity to the SE, initiating citrulline-specific T and B cell responses (45, 46). Citrullination is catalyzed by peptidylarginine deiminases (PAD), which convert the amino acid arginine into citrulline, leading to a loss of positive charge that might render self-peptides immunogenic (47). Several citrullinated peptides, including fibrinogen, vimentin, α-enolase, aggrecan, and CII are present in RA joints and are targets of lymphocyte responses in RA patients carrying the SE (48, 49). Antibodies directed against citrullinated proteins/peptides (ACPAs) are specific to RA and associated with the presence of the SE and with increased disease severity (50, 51).
Antigen-presenting cells, particularly DCs, are key players in the initiation and maturation of the autoimmune response in RA (52). DCs can activate self-reactive CD4+ T cells by presenting autoantigens in the context of MHC class II molecules and providing costimulatory and pro-inflammatory signals (53–55). Activated autoreactive CD4+ T cells differentiate into inflammatory T helper (Th) cell subsets producing either interferon (IFN)-γ and tumor necrosis factor (TNF) (Th1), IL-17 and IL-21 (Th17), or a mixed cytokine profile (Th1/17), and accumulate in the inflamed joint (56–58). These autoreactive T cells drive the differentiation of B lymphocytes into plasma cells producing autoantibodies such as ACPAs (59), which in turn promote osteoclast differentiation and activation, leading to cartilage and bone erosion (33). Autoreactive CD4+ T cells also stimulate macrophages and synovial fibroblasts to secrete pro-inflammatory mediators, including TNF, IL-1, and IL-6, which contribute to synovial inflammation through recruitment of immune cells and expansion of synovial fibroblasts, forming an invasive pannus (60, 61).
Current treatment options of RA involve non-steroidal anti-inflammatory drugs, glucocorticoids and disease-modifying anti-rheumatic drugs, either conventional, such as methotrexate, targeted, like Janus kinase inhibitors, or biologics, including antagonists of proinflammatory cytokines, modifiers of T cell co-stimulation and B cell-depleting antibodies (1, 62). Besides the fact that a proportion of patients remain refractory to treatment (63), none of these drugs provides a cure of RA, requiring life-long treatment, associated with a progressive loss of efficacy, toxicity, and the appearance of serious adverse effects (64–66). A promising strategy to restore self-tolerance and thereby achieve long-term remission, is the depletion or reprogramming of autoreactive T cells by regulatory T (Treg) cells or tolerogenic DCs (tolDCs) (25, 67, 68). Naturally occurring CD4+ Treg cells are characterized by the constitutive expression of the transcription factor FOXP3 and IL-2 receptor α-chain (CD25). Treg maintain immunological tolerance and prevent autoimmune diseases by suppressing the activation and proliferation of self-reactive effector T cells (67). Although Treg cells are present in the synovial fluid of RA patients, these Treg cells fail to inhibit Th1 responses (69, 70). Strategies targeting Treg cells for the treatment of RA include (i) the expansion of autoantigen-specific naturally occurring Treg cells in vivo, (ii) their propagation in vitro and subsequent transfer back into the host, and (iii) the conversion of antigen-specific T cells into Treg cells in vitro or (iv) in vivo (67). Dendritic cells (DCs) are professional antigen-presenting cells that instruct T cells, according to the surrounding environment, to mediate immune responses or tolerance. TolDCs with immunoregulatory properties can be generated in vitro from monocytes or hematopoietic stem cells and are able to control aberrant CD4+ T cell responses through the induction of anergy, conversion of T effector into Treg cells, or deletion of autoreactive T cells (71–74). An important advantage of tolDC- or Treg-based therapy over conventional treatment of RA is its potential to modulate immune responses in an antigen-specific manner, which might permit a selective downregulation of autoreactive lymphocyte responses while avoiding a general shutdown of immunity against pathogens. Both Treg cell and tolDC-based approaches have been extensively tested in conventional mouse models of RA-like disease (75) and the safety of tolDCs has even been approved in phase I/II clinical trials (76, 77). Nevertheless, sophisticated mouse models that accurately recapitulate human RA are still missing. Humanized mouse models of RA might help to predict the efficacy and side effects of cell-based approaches in further clinical trials, as well as to adjust parameters, such as dose, injection route, and required dosing interval.
Numerous rodent models of RA are available, each of which mirrors particular aspects of the disease (4, 6). These conventional models represent classic hallmarks of RA, such as joint swelling, synovitis, pannus formation, and bone erosion, but differ in the mechanisms of induction and launched immune processes, as well as in their speed of onset, chronicity, and severity (6, 78). A distinction is made between induced and spontaneous models. In induced models, non-specific immune activation, cartilage-directed autoimmunity, or abundant exogeneous/infectious triggers cause RA-like disease, while in spontaneous models, arthritis develops without deliberate immunization and is non-limiting, providing a chronic situation like in human RA (5, 79, 80). The most frequently used models are introduced below.
Adjuvant arthritis (AA) was the first described animal model of RA and can be induced by a single intradermal injection of complete Freund's adjuvant (CFA), containing heat-inactivated mycobacteria, at the base of the tail in Lewis rats (81) or by repetitive intra-articular CFA injection in DBA/1 or C57BL/6 mice (82). The hallmark of AA is its rapid onset and progression to polyarticular inflammation, leading to a chronic erosive disease with severe joint malformation (6). The disease is driven by CD4+ T cells (83) and susceptibility to develop AA is related to MHC and non-MHC genes (84). Originally, it was assumed that mycobacterial components, such as 65k heat shock protein, cross-react with self-antigens from joint cartilage in this model (85). However, it has been shown that nonimmunogenic adjuvants such as avridine, muramyl dipeptide, pristane, and incomplete Freund's adjuvant also induce AA in many rat strains and mice, indicating that adjuvants may enhance autoreactivity to articular antigens (83, 86–88). Unlike in human RA, the AA model displays not only bone erosion, but also bone apposition at early stages of the disease with limited to no cartilage damage (79).
Collagen-induced arthritis (CIA) is the most commonly used model of RA-like disease (89). In this model, severe joint inflammation is induced through immunization with CII, a major component of hyaline cartilage, together with CFA (6, 90). Susceptibility to CIA is related to the murine MHC class II molecule H-2q whose peptide-binding pocket has a similar primary structure like the SE of RA-associated HLA-DR molecules (91, 92). Although several mouse strains are susceptible to CIA, the DBA/1 strain is the “gold standard” of this model (90). Autoreactive CD4+ T cells are required for the induction of CIA (7, 93, 94), synovial proteins are subjected to PAD-induced citrullination and an association of anti-CII antibodies and ACPA to the development of arthritis has been described (95, 96). The passive transfer of polyclonal immunoglobulin (Ig) G from sera of CIA mice or monoclonal anti-CII antibodies induces arthritis even in mouse strains that are not susceptible to CIA, indicating an important role of autoantibodies in the effector phase of the disease (97, 98). Important limitations are that CIA is an acute model, in which remission occurs at 10–14 days after disease onset (99), extra-articular manifestations are due to CFA (100) and disease severity is highly variable and dependent on environmental factors, such as grouping stress (101). Joint inflammation in CIA mice is particularly mediated by Th17 cells (102), while the pathogenesis of human RA involves Th1, Th17, and Th1/17 cells (56–58).
Proteoglycan-induced arthritis (PGIA) is induced by immunization with human cartilage proteoglycan aggrecan in susceptible BALB/c or C3H mice strains (12). The progressive chronic polyarthritis in the PGIA model shares features with human RA such as deposition of immune complexes in the joint and the presence of rheumatoid factor autoantibodies (6, 12). Susceptibility to PGIA in BALB/c mice is associated with the presentation of the dominant epitope aggrecan 89–103 by I-Ad MHC class II molecules (103). PGIA development is dependent on B cells, which present antigen and produce anti-PG antibodies, as well as on CD4+ T cells, which provide help to B cells for antibody production. Antibodies are cross-reactive between human proteoglycan, used for immunization, and self (mouse) cartilage proteoglycan (104, 105). The choice of adjuvant and route of immunization determine the prevalent Th subset in the PGIA model. Immunization with CFA and subcutaneous injection route result in predominant Th17 responses, whereas dimethyldioctadecyl-ammonium bromide (DDA) as adjuvant and intraperitoneal injection induce more Th1 cells (106, 107).
Antigen-induced arthritis (AIA) is triggered by the injection of exogeneous antigen, such as methylated bovine serum albumin (mBSA), into the joint (108). This robust model of destructive bystander arthritis develops almost independently of the genetic background of mice and leads to a moderate and limiting local disease involving both T cells and immune complexes (79, 109).
RA-like disease can also be passively transferred to naïve mice by injecting serum antibodies against endogenous CII, PG, or glucose-6-phosphate isomerase (GPI) (79, 98, 110, 111). These immune complex arthritis models are almost independent of the genetic background of mice and resemble the effector phase of the disease, in which excessive immune complex formation at joint tissues triggers complement activation and the release of inflammatory mediators through Fcγ receptor engagement on phagocytes, leading to rapid onset, but transient destructive arthritis (78). Administration of pathogenic immunoglobulins is sufficient to confer disease and does not require T or B lymphocytes, nevertheless, T cells have an enhancing effect on autoantibody-induced arthritis (112).
Spontaneous models include genetically modified mice, such as SKG, K/BxN, human TNF transgenic (Tg) and IL1ra−/− mice (5, 80). In SKG mice, arthritis development is attributed to a missense mutation in the TCR signaling adaptor molecule ZAP70, leading to a defective negative selection in the thymus and the release of autoreactive T cells (113). SKG mice establish arthritis following stimulation of the innate immune system with zymosan or other microbial triggers, while housing under pathogen-free conditions or treatment with antibiotics completely block arthritis development (114, 115).
The K/BxN model was discovered coincidentally by crossing KRN mice, which express an transgenic TCR, specific for an epitope of bovine pancreas ribonuclease, with autoimmune-prone non-obese diabetic (NOD) mice. The offspring, referred to as K/BxN, spontaneously developed severe arthritis (116), driven by the activation of T cells expressing the KRN-derived TCR, which, due to cross-reactivity, recognize the ubiquitously expressed self-protein GPI bound to the NOD-derived MHC class II molecule I-Ag7 (117, 118). Activated autoreactive T cells promote polyclonal B cell activation and T helper cell-dependent production of GPI-specific IgG autoantibodies (110, 119). Transfer of serum IgG from arthritic K/BxN mice has been shown to induce robust and reproducible arthritis in healthy mice, indicating that autoantibodies might directly trigger joint inflammation (110, 120). However, it must be considered that GPI might not be an essential or RA-specific autoantigen.
TNF transgenic (Tg) mice, which constitutively express human TNF, spontaneously develop inflammatory, highly erosive polyarthritis, similar to human RA, which can be completely prevented by treatment with monoclonal antibodies against human TNF (121). When these TNF-overproducing mice were crossed to a severe combined immunodeficiency (SCID) background, lacking mature B and T cells, inflammatory arthritis still develops, indicating that TNF acts downstream of lymphocyte responses in this model (122). Thus, TNF Tg mice are not suitable for testing of therapeutic approaches that target lymphocyte responses in RA.
Mice overexpressing human IL-1α Tg present chronic destructive polyarthritis, characterized by hyperplasia of the synovial lining, pannus formation and cartilage destruction (123). However, pathogenesis of arthritis in hIL-1α Tg mice is mediated by monocytes/macrophages and activated neutrophils, while T and B lymphocytes are sparse (123). In an opposite approach, deletion of IL-1 receptor antagonist (IL-1ra), an endogenous inhibitor of IL-1 signaling, leads to a spontaneous arthritis model in which T cells are main actors (10, 124). Increased levels of antibodies against IgG (rheumatoid factor), CII and double-stranded DNA also point to the development of an autoimmune response in IL1ra−/− mice (124).
Cell-based therapeutic approaches have mainly been tested in induced models of RA-like disease (75). Adoptive transfer of antigen-specific Treg, either expanded from natural occurring Treg, or induced from conventional T cells, e.g., by enforcing the expression of the Treg-specific transcription factor FoxP3 or reprogramming through tolDCs, has been shown to suppress the progression of arthritis in models of CIA, PGIA and AIA (125–129). Therapeutic effects of tolDCs, differentiated in vitro from precursors and genetically modified to express IL-4, or modulated by either short stimulation with bacterial lipopolysaccharide (LPS), vasoactive intestinal peptide, a combination of dexamethasone, vitamin D3 and MPLA, or the NF-κB inhibitor Bay11-7082, have been reported in mouse models with established CIA and AIA (26, 27, 130–132). However, testing of tolDC-based therapeutic strategies in conventional mouse models is limited by the fact that, due to technical constraints, administered murine DCs are generated from bone marrow progenitors, while monocyte-derived DCs are used for clinical applications in humans (25). It is evident that bone marrow- and monocyte-derived DCs represent different DC subsets which might have an altered or even opposite effect on the disease in vivo.
A common weakness of all conventional models of RA is that inflammatory responses are completely mediated by the murine immune system, which differs in its components and organization from the human immune system. Discrepancies in innate and adaptive immune mechanisms between both species include the composition of leukocyte subsets in peripheral blood, Toll-like receptors, Fc receptors, Ig subclasses, B cell and T cell signaling pathways, γδ T cells, response to IFN-γ, cytokines and cytokine receptors, expression of costimulatory and adhesion molecules, chemokine and chemokine receptor expression, as well as expression of receptors involved in the uptake of phagocytic cargo, production of reactive oxygen species and IL-1 by monocyte subsets, among others (21, 22, 133). While Foxp3 defines the Treg population in mice, human FOXP3+ T cells are heterogeneous in their function (67). In response to stimulation, human CD4+ effector T cells express MHC class II molecules (134) and upregulate FOXP3 without acquiring immunoregulatory functions (135), while mouse T cells do not. In CIA mice, γδ T cells are the predominant source of IL-17 in affected joints, but they are nearly absent in joints of RA patients, where Th1 cells dominate (136). Another difference between conventional animal models of RA-like disease and human RA is their lack of gender bias (137).
Due to these species-specific differences, it is not surprising that several promising therapeutic principles found in mouse models do not work in humans (15, 16, 20). This hampers the translation of experimental data obtained from conventional murine models of RA-like disease to clinical applications in patients and creates a demand for humanized models which accurately mimic the human disease (Table 1).
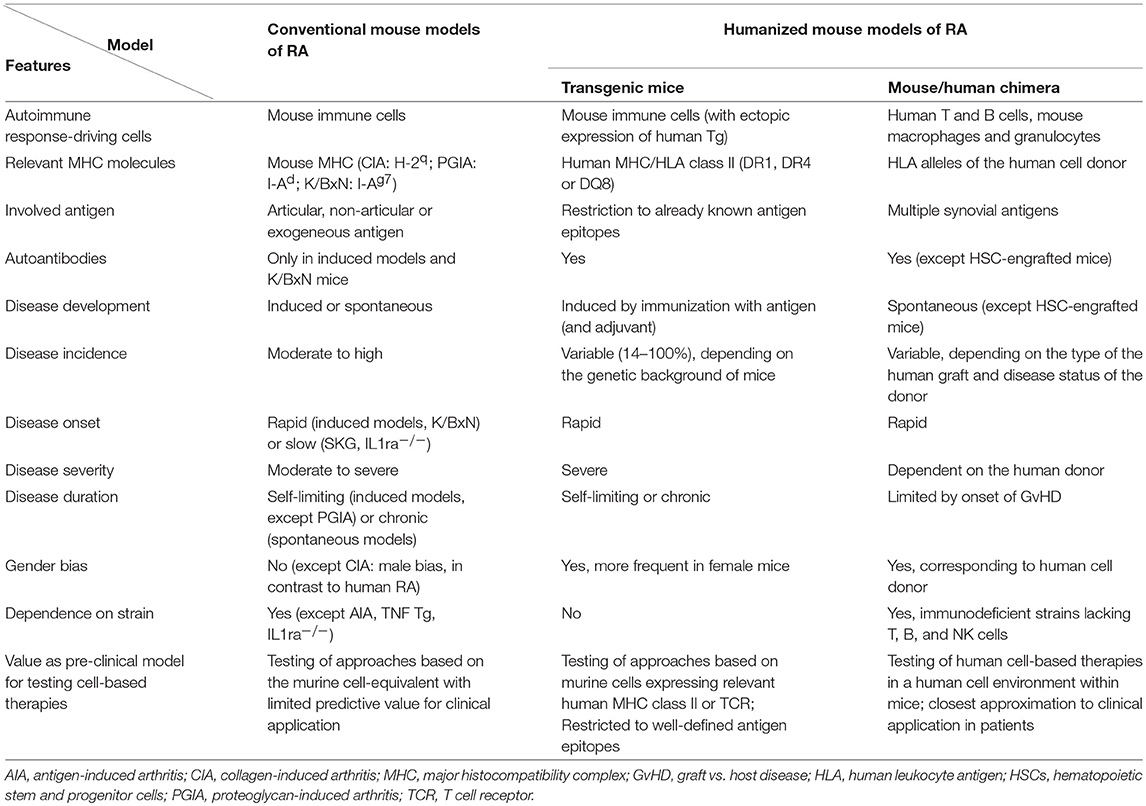
Table 1. Comparison between conventional and humanized mouse models of rheumatoid arthritis.
This humanization strategy is based on transgenic expression of human molecules, such as HLA class II, RA-associated synovial autoantigens and/or an autoantigen-specific T cell receptor, in immunocompetent mice (Figure 2).
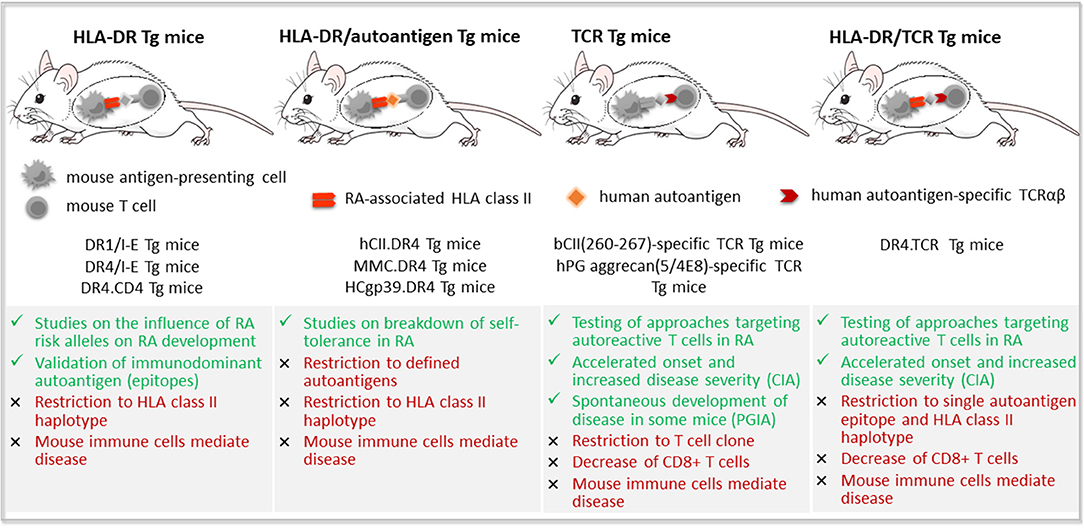
Figure 2. Transgenic mouse models of rheumatoid arthritis. Transgenic (Tg) mouse models of rheumatoid arthritis (RA) reported in the literature include mice expressing (i) RA-susceptible human leukocyte antigen (HLA) class II molecules, such as DRB1*0101 (DR1) or DRB1*0401 (DR4); (ii) human RA-associated autoantigens, such as type II collagen (CII), either complete or as mutated mouse collagen (MMC) containing the immunodominant epitope, and human cartilage glycoprotein-39 (HCgp-39); (iii) an autoantigen-specific T cell receptor (TCR) alone; or (iv) together with RA-susceptible HLA-DR molecules. Advantages are depicted in green color, limitations in red.
The advent of mice that lack endogenous MHC class II molecules and express human HLA transgenes instead has significantly advanced the understanding of the importance of certain HLA class II alleles, particular those containing the SE, in the development of RA (137). The basis for HLA class II transgenic mice was created in 1991, with the generation of a mouse with disrupted expression of the murine MHC class II genes I-A and I-E, lacking mature CD4+ T cells (138). First attempts to introduce human HLA class II transgenes in these murine MHC class II-deficient mice resulted in low CD4+ T cell selection, due to poor interaction between mouse CD4 and human HLA class II molecules (139). To circumvent this problem, two strategies have been pursued: generation of double transgenic mice expressing the human HLA gene in all antigen-presenting cells and human CD4 in all T cells (140–142), or introduction of a chimeric HLA/I-E molecule composed of the peptide-binding domain from human DR and the CD4-binding domain derived from mouse I-E (143, 144). This way, the HLA class II transgene was able to positively select CD4+ T cells expressing diverse Vβ TCRs, resulting in normal development of the CD4+ T cell compartment and maintenance of peripheral tolerance to transgenic HLA molecules (137).
The first strategy was used by Fugger and colleagues, generating HLA-DRA1*0101/-DRB1*0401 (DR4)/human CD4 Tg mice which did not develop spontaneous autoimmune diseases, but were susceptible to CIA (140, 142). Rosloniec and coworkers followed the second approach to establish Tg mice expressing chimeric HLA-DRB1*0101 (DR1)/I-E or DR4/I-E, which developed severe autoimmune arthritis following immunization with bovine or human CII, accompanied by strong DR1 and DR4-restricted T and B cell responses, respectively (13, 145). The incidence of CIA varied from 14 to 100%, according to the introduced human HLA-DR transgene, the genetic background of mice, and the applied immunization protocol (13, 142, 146). In both DR1 Tg and DR4 Tg mice, T cell response was focused to position 259–273 of human CII, suggesting that different RA-associated HLA-DR molecules containing the SE bind and present the same immunodominant epitope of CII (13, 145). Even though both HLA-DRB1*0401 (RA-susceptible) and *0402 (RA-resistant) molecules present CII and its derived peptide, only *0401 Tg mice develop a pro-inflammatory response (146). Furthermore, comparison of T cell polarization and immune responses between *0401 and *0402 Tg mice encouraged the hypothesis that the SE selects T cells with a predetermined Th17-biased cytokine profile that efficiently clear infections, but may, on the other hand, drive autoimmunity in response to pathogens or certain environmental factors (147). Interestingly, Tg mice carrying an RA-susceptible haplotype mimic the gender-bias of RA, displaying an increased susceptibility to develop the disease as well as a stronger cellular and humoral response to CII in females (146).
To date, DR4 Tg or DR1 Tg mice have been widely used to identify and validate immunodominant T cell epitopes of synovial autoantigens, such as CII, HCgp-39, proteoglycan aggrecan, fibrinogen, and vimentin, as well as to investigate the role of posttranslational modifications on epitope binding (13, 148–151). In DR4 Tg mice immunized with citrullinated (cit)-vimentin peptide aa 59-78, specific Th1 cells and strong proliferative recall responses were detected, however, mice did not show any signs of arthritis (150). In contrast, immunization of DR4 Tg mice with human cit-fibrinogen induced vigorous citrulline-specific T and B cell responses in all DR4 Tg experimental mice and triggered arthritis, characterized by synovial hyperplasia and ankylosis, in 35% of these animals (149). Transfer of splenic lymphocytes from arthritic mice induced joint swelling in the human cit-fibrinogen–injected limb of all DR4 Tg recipient mice, but not in PBS- or unmodified human fibrinogen–injected limbs (152). The fact, that administration of cit-fibrinogen to wild-type C57BL/6 mice and immunization of DR4 Tg mice with native fibrinogen failed to induce arthritis and citrulline-specific reactivity, underscores the critical role of both SE-containing HLA-DR molecules and antigen citrullination in the development of RA (149). Another study in DR4 Tg mice corroborated that immune responses to homocitrulline-containing peptides are similarly dependent on the presence of the SE and later evolve into immune responses to citrullinated antigens (153).
With the advent of tetramer technology, it has become possible to identify, track, and characterize DR-restricted autoantigen-specific T cells in DR1 Tg and DR4 Tg mice, and to corroborate their pathogenic role in the development of RA (154–156). Activated CII-specific T cells expressing high levels of Th1, Th17 and pro-inflammatory cytokines have been detected in arthritic joints of DR1 Tg and DR4 Tg mice following immunization with CII (154, 155). Although CD4+ T cells represent only a minor population of the synovial infiltrate, they were shown to display a highly restricted TCR repertoire and limited clonality and their expansion correlated with onset and severity of arthritis as well as with anti-CII antibody levels (154, 157). It is important to note, that, at the time of first clinical signs of arthritis, activated HLA-DR4:CII(158–170) tetramer-positive cells disappeared from synovial fluid and were rarely found in blood, while they persisted in lymph nodes, suggesting that autoreactive T cells play a role particularly in the early stages of arthritis, by triggering a local immune response (154, 157). This important finding, derived from studies in DR4 Tg mice also suggests a rationale for the lack of enrichment of antigen-specific T cells in the synovial fluid of patients with established RA (171).
DR1 and DR4 Tg mice have also been used to test alternative treatments for RA, such as synthetic analog peptides, which contain substitutions in critical positions of the CII immunodominant epitope and suppress inflammatory arthritis by promoting regulatory T cell responses (172, 173). Treatment of human cit-fibrinogen-immunized DR4 Tg mice with CTLA-4Ig fusion protein, a soluble form of the CTLA-4 receptor which inhibits T cell activation by competing with CD28 for binding to the costimulatory ligand CD80/CD86 (174), restrained the activation of cit-fibrinogen-specific T cell responses, and halted the progression of arthritis (152). The fact that splenocytes from cit-fibrinogen-immunized mice treated with CTLA-4Ig were unable to transfer arthritis to recipient mice supports a direct role of activated citrulline-specific T cells in arthritis development and progression (152).
The contribution of HLA-DQ polymorphism to RA susceptibility and severity has been explored by David's group, who established mice which express transgenic DQA1*0301/DQB1*0302 (DQ8) (175). In humans, HLA-DQB1*03 occurs in linkage with the RA risk locus HLA-DRB1*04 and has been suggested to affect the clinical expression of RA (176–178). Immunization with heterologous CII induced autoreactive T and B cell response and severe inflammatory arthritis in 70% of DQ8 Tg animals (179), while of mice expressing a protective DQ6 Tg, only 14% presented mild arthritis and 60% of Tg mice with mixed DQ8/DQ6 haplotype developed moderate CIA in response to CII (180). DQ8 Tg mice with deleted CD4 did not develop arthritis, while CD8 deficient DQ8 Tg mice developed severe CIA along with increased autoantibody levels, suggesting that CD4+ T cells, but not CD8+ T cells are indispensable for initiation of CIA (181). In the absence of B cells, DQ8 mice failed to develop CIA, suggesting that B cells are not only important for autoantibody production, but also for antigen presentation to autoreactive T cells (146). To simulate the situation in humans, where HLA-DR and -DQ occur in linkage, Taneja and coworkers established mice that express DR4 (either RA-susceptible *0401 or RA-resistant *0402) along with DQ8 transgene (182). Indeed, CIA incidence was increased in *0401.DQ8 double Tg mice compared to single Tg mice, while *0402.DQ8 Tg mice were resistant to CIA (182).
The link between smoking and the emergence of RA in individuals with an RA-associated HLA haplotype has been reproduced in DR4 and DQ8 Tg mice (183). Exposure to cigarette smoke augmented PAD enzyme expression and enhanced pro-inflammatory Th1 and antibody responses to native and citrullinated CII and vimentin in both DR4 and DQ8 Tg mice, while promoting Th2/Treg responses in Tg mice that expressed protective DRB1*0402 (183, 184). An association between periodontal disease and RA has been studied on DR1 Tg mice, in which oral exposure to Porphyromonas gingivalis led to increased percentage of Th17 cells in blood and lymph nodes, systemic pro-inflammatory cytokine response, loss of bone density, and generation of anti-citrullinated protein antibodies (185).
The breakdown of immune tolerance to endogenous cartilage antigens is a central element in the development of RA. Immunization of CIA-susceptible mouse strains with non-self (e.g., human, bovine, chicken, or rat) CII induces severe arthritis whereas mice are less prone to CIA induction with self (mouse) CII (186). This is due to minor but decisive structural differences between mouse and human CII, which are located in the immunodominant T cell epitope CII(158–170, 187, 188), containing glutamic acid at position 266 in human (as well as bovine, chicken and rat) CII, whereas in mouse CII there is an aspartic acid instead (189).
In mice which express the entire human CII protein, or its immunodominant human (h)CII(158–170, 187, 188) epitope within a mutated mouse CII (MMC) protein, only autoreactive T cells should become activated upon immunization with human or rat CII. Indeed, although hCII Tg mice with cartilage-restricted expression of hCII generally mounted autoreactive T and B cell responses following immunization with hCII, only 10% of these mice developed arthritis (186). In this model, T cell tolerance was shown to depend on the expression level of hCII and its immunodominant epitope (186). In B10 mice expressing DR4 transgene together with either hCII or MMC, murine DR4-restricted T cells were tolerized against self-CII, and thus these mice were uniformly resistant to CIA (151, 190). In contrast, T cell tolerance to self-CII was incomplete in MMC.DR4 Tg mice with C3H background, rendering these mice prone to develop CIA (151). Tolerance to self-CII could be broken in B10.DR4 mice expressing MMC, but not hCII Tg, by introducing a mutation in the Ncf1 gene, which encodes a subunit of the NADPH oxidase complex, resulting in reduced ROS production (151, 191).
Mice that express a transgenic TCRαβ specific for an arthritogenic epitope of human autoantigen, such as CII or proteoglycan aggrecan, were used to gain more insight into the role of antigen-specific T cells in the development of autoimmune arthritis (192, 193). In such TCR Tg mice, more than 90% of CD4+ T cells express the antigen epitope-specific TCR transgene (192, 193).
TCR Tg mice, carrying the rearranged Vα11.1 and Vβ8.3 chain encoding genes specific to bovine CII, respond vigorously to stimulation with bovine CII or its immunodominant determinant CII(158–164, 188) in vitro, but do not develop spontaneous arthritis (192, 194). However, an accelerated onset and increased severity of arthritis was observed in CII-specific TCR Tg mice after immunization with CII in CFA, as compared to their non-Tg littermates (192, 194). Interestingly, Treg cell clones generated from splenocytes of CII-specific TCR Tg mice reduced the proliferation of CII-specific effector T cells in vivo and decreased the incidence and clinical symptoms of arthritis after adoptive transfer in CIA and collagen antibody-induced arthritis models (195).
Another TCR-Tg mouse model was generated by introducing TCR Vα1.1 and Vβ4 chains of a T cell hybridoma with MHC class II-restricted specificity for the immunodominant epitope (5/4E8) of human cartilage proteoglycan aggrecan into PGIA-susceptible BALB/c strain (193). In these TCR Tg mice, a single dose of human proteoglycan, even in the absence of adjuvant, produced disease (193), which is in contrast to the conventional PGIA model, that requires multiple immunizations with human proteoglycan in adjuvant (12).
An elegant strategy has been to combine the expression of RA-associated DR1 and a transgenic Vα2/Vβ8.1 TCR, recognizing the immunodominant determinant of bovine and human CII (196). These double Tg mice developed an accelerated and more severe form of CIA than their DR1 Tg littermates and provide a more reliable tool for testing novel therapeutic approaches (196).
TCR-Tg models support the hypothesis that antigen-specific T cells play a critical role in the initiation of arthritis. However, important limitations are the restriction to a single autoantigen epitope and the decrease of the CD8+ T cell pool in these mice (192–194).
In an ideal Tg mouse model of autoimmune arthritis, combined expression of RA-susceptible HLA-DR molecules, an RA-relevant autoantigen and a specific TCR, recognizing the immunodominant epitope of this autoantigen, would lead to spontaneous breakdown of self-tolerance and the development of arthritis. This has not been achieved so far, however, studies in Tg mouse models of RA have underscored the critical role of the SE, post-translational modification of antigen and self-reactive T cells in the development and progression of RA and contributed substantially to the identification and validation of immunodominant autoantigens and its T cell epitopes. Thus, Tg mice represent a decisive step forward in RA research and have undoubtedly enhanced the predictive value of mouse models in preclinical tests. However, it must be considered, that many of the herein presented models have been only established in one laboratory or published once. The poor reproducibility under different laboratory conditions might be at least partially explained by distinct microbial communities. Another problem is that human DR transgenes have been shown to be poorly expressed by DCs in some strains, resulting in poor antigen presentation. Finally, Tg mouse models still bear the same limitation as conventional models of RA-like disease, that is, inflammation is driven by the murine immune system that ectopically expresses SE-containing HLA class II molecules or a specific TCR but might contribute in a different way to the development of the disease.
An important advance in the generation of a preclinical mouse model of RA would be the establishment of a functional human immune system or some of its components in mice, in which they mount autoimmune responses and clinical features of RA. For this purpose, human hematopoietic stem and progenitor cells (HSC), peripheral blood mononuclear cells (PBMC), or tissue have been engrafted into immunodeficient mice, which lack the ability to reject xenografts and thus enable stable reconstitution with human cells (Figure 1).
Important achievements of the last decades have paved the way for the stable engraftment of human cells within the murine host. The Prkdcscid (protein kinase, DNA activated, catalytic polypeptide) mutation in CB17 mice, commonly denoted “SCID” (severe combined immunodeficiency) (197), results in reduced numbers of functional T and B cells and thus enables limited and transient engraftment of human PBMCs, HSCs and fetal hematopoietic tissues (198–200). However, spontaneous generation of mouse T and B cells with aging, referred to as leakiness, as well as high levels of host natural killer (NK) cells and innate immune activity impede the stable engraftment of human cells and tissues in SCID mice (29). Alternatively, targeted mutations of the recombination-activating genes 1 (Rag1) and Rag2 prevent the development of functional T and B cells in mice without causing leakiness (201, 202). Nevertheless, Rag1/2-deficient mice retain high levels of NK-cell activity, allowing only limited engraftment of human HSC (29).
An important step forward was accomplished by crossing NOD and SCID strains (203). NOD-SCID mice display additional defects in innate immunity, including the absence of complement C5, impaired macrophage cytokine production, antigen presentation and NK cell function (29, 203, 204). Although they have increased engraftment of human HSCs and PBMCs (205, 206), the limitations of NOD-SCID mice include relatively short life span due to thymic lymphomas, and residual activity of NK cells and innate immunity (29).
A decisive breakthrough was achieved by the generation of immunodeficient mice with a mutation in the Il2rg gene (207–210), encoding IL-2 receptor γ subunit (IL-2Rγ), also denominated gamma-chain, required for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 signaling (211). An absent or truncated IL-2Rγ-chain leads to defective T and B cell development, affects innate immunity and completely abolishes NK cell generation (212, 213). The main immunodeficient mouse strains bearing the IL2rg mutation are NOD-SCID Il2rgnull, including NOD-SCID Il2rgtm1Wjl (NSG) and NOD-SCID Il2rgtm1Sug (NOG), as well as NOD-Rag1null Il2rgnull (NRG) and BALB/c-Rag2null Il2rgnull (BRG) (208, 209, 214, 215). These strains support high engraftment of human tissue, HSCs and PBMCs (207, 208, 216), without the need for previous myeloablation through irradiation or drugs, thus reducing the required donor cell number and prolonging survival of humanized mice (217). Engrafted HSCs differentiate into multiple lineages of human cells, including erythrocytes, platelets, T and B lymphocytes, NK cells, DCs, monocytes/macrophages and granulocytes (207–209, 215, 218). Reconstitution of the human immune system is most efficient, when CD34+ HSCs from cord blood or fetal liver (rather than from adult peripheral blood) are injected into newborn mice, where both donor cells and recipient are set for development and expansion of the hematopoietic system (215, 219). Co-transplantation of HSCs with fetal liver and/or fetal thymus (abbreviated BLT for bone marrow, liver, thymus) further improves the systemic repopulation with multilineage human cells, by providing an autologous thymic environment for proper T cell development (220, 221). The functionality of the reestablished human immune system in immunodeficient mice has been demonstrated by the presence of lymphoid tissues and the capacity to mount adaptive immune responses, including T cell-dependent antibody responses, cell-mediated cytotoxicity and delayed type hypersensitivity reactions (207, 217–219, 222).
However, absence of the IL-2Rγ-chain results in a lack of some cytokines which are cross-reactive between human and mouse and are required for human cell differentiation and survival within the host (29, 223). Furthermore, cross-reactivity between murine and human cytokines does not necessarily imply a biological function, as demonstrated by the example of B lymphocyte survival factor/stimulator BAFF (224). To overcome this issue, human cytokines and growth factors, including IL-7, IL-6, BAFF, thrombopoietin, FLT3-ligand, IL-12, granulocyte-macrophage colony-stimulating factor and IL-3 can be either administered or provided by transgenic expression (208, 224–229).
A major obstacle to using humanized mice as a preclinical model is their susceptibility to xenogeneic graft-vs.-host disease (GvHD), which impedes the development of chronic disease (230). This applies rather to mice that have been engrafted with BLT or human PBMCs than to HSC-engrafted mice. In the first two models, mature CD4+ T cells have been educated in human thymic stroma and are therefore not tolerized to the murine antigenic environment, which leads to rapid-onset xenogeneic GvHD (231). Engraftment of human cells and GvHD onset and severity vary between donors and seem to depend on the dose of CD4+ and CD8+ T cells as well as of naïve CD4+ T cells within the transferred human cells (232, 233). In contrast, higher percentages of Treg cells delay xenogeneic GvHD (234). Importantly, human T cells in HSC-reconstituted mice are selected on murine MHC class II (H2) molecules in the mouse thymus and therefore might not be able to recognize antigens presented in the context of HLA-DR by human antigen-presenting cells in the periphery. This affects the induction of efficient immune responses, resulting in reduced Th1 activity and insufficient interactions between human T and B cells which are required for class-switch recombination (235, 236).
A significant improvement has been accomplished by the introduction of human HLA class II molecules into immunodeficient mouse strains. Transgenic expression of HLA-DR4 in NRG mice enabled proper development of CD4+ T cells and completely functional B cells from infused HSCs of HLA-DR-matched donors (237). The additional removal of murine MHC class II molecules, which are the main target of human CD4+ T cell-mediated GvHD responses further improved the generation of human antigen-specific immune responses in immunodeficient mice reconstituted with human cells (238, 239), while reducing the risk of xenogeneic GvHD (240).
While humanized mouse models based on mouse/human chimeras have been widely used for studies on cancer, human-specific infectious diseases and transplantation (216, 236, 241, 242), their great potential to recapitulate human autoimmune disorders has only recently been recognized and explored (243).
It has been shown that transfer of human HSCs with a psoriasis-prone HLA haplotype provokes a related disease in NSG mice (244). Recently it has been demonstrated that reconstitution of NSG-Ab0 DR1 mice (lacking murine MHC class II (Ab0) and expressing transgenic HLA-DR1) with HSCs from a patient with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, associated with FOXP3 dysfunction, spontaneously develop lethal autoimmune disease, involving multiple organs and the production of autoantibodies (245). In another study, autoimmune disease characterized by hepatitis, weight loss and anti-nuclear antibodies was induced in HSC-reconstituted NSG mice by inhibition of CTLA-4, a critical molecule for Treg suppressive function (246). A spontaneous humanized model of type 1 diabetes (T1D)-like disease was established by engrafting HSCs into NRG-Akita mice, which develop hyperglycemia due to a mutation in the insulin 2 gene (214). In NSG.DQ8 Tg mice reconstituted with human DQ8+ HSCs and fetal thymus, transfer of autologous human CD4+ T cells expressing an DQ8/insulin B chain peptide-specific TCR and immunization with the corresponding peptide induced hyperglycemia and diabetes (247).
Other investigators engrafted patient-derived PBMCs into immunodeficient mice to reconstitute T cell-mediated autoimmune disorders, such as Sjögren's Syndrome, Systemic Lupus Erythematosus (SLE), or T1D, involving the production of autoantibodies and tissue-specific autoreactivity (243, 248, 249). Engraftment of BRG mice with PBMC from SLE patients caused autoimmune-like disease with donor-dependent severity, characterized by nephritis, proteinuria, deposits of human IgG in kidneys and shorter life span compared to mice engrafted with PBMCs from healthy subjects (249). Similarly, engraftment of NSG.A2 mice, expressing T1D-associated HLA-A*0201 (A2) molecules, with HLA-A2 matched PBMCs from T1D patients resulted in islet infiltration by specific CD8+ T cells (243). Insulitis and pancreatic β cell death, the characteristic hallmarks of T1D, were also induced by the transfer of autoreactive CD4+ T cell lines from T1D donors into NSG.DR4 mice (250). In these models, spontaneous development and severity of autoimmune-like disease are donor-dependent and mice that have been reconstituted with PBMCs from healthy subjects fail to display an autoimmune phenotype (243, 249).
Five different approaches have been pursued to generate humanized mouse models of RA, using immunodeficient mice as host for (i) RA synovial tissue (251, 252), (ii) RA patient-derived synovial fibroblasts co-transplanted with normal human cartilage (253, 254), (iii) synovial fluid mononuclear cells (SFMCs) (255, 256), (iv) RA patient-derived PBMCs (233, 256), or (v) HSCs (257, 258) (Figure 3).
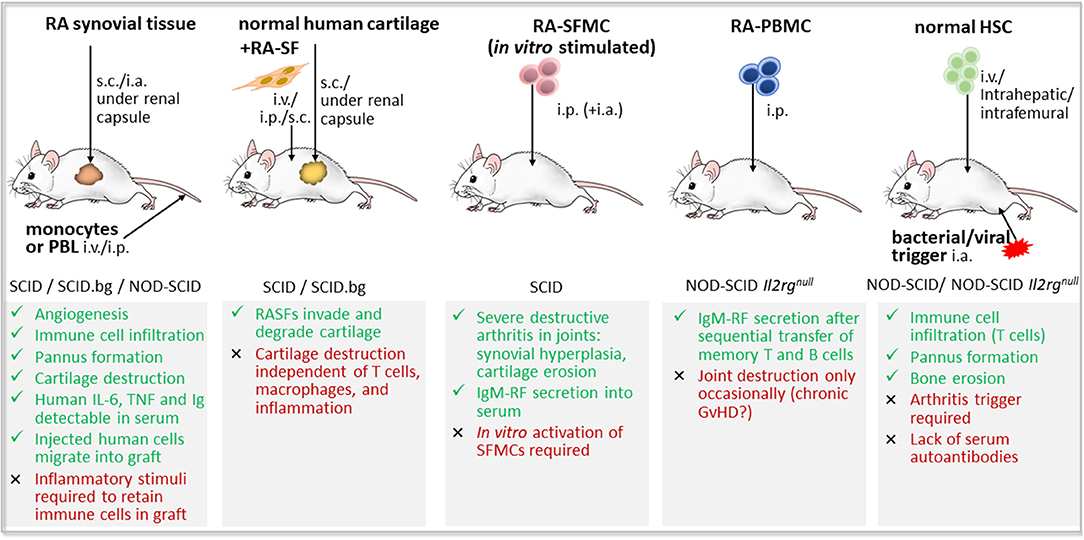
Figure 3. Humanized mouse models of rheumatoid arthritis based on the engraftment of human cells and/or tissues in immunodeficient mice. RA-like features (green color) and drawbacks (red color) of the five main approaches for the development of a humanized mouse model of rheumatoid arthritis (RA). First, transplantation of RA synovial tissue and eventual co-injection of peripheral blood lymphocytes (PBL) or monocyte cell line; Second, co-implantation of normal human cartilage and RA patient-derived synovial fibroblasts (RA-SF); Third, injection of synovial fluid mononuclear cells (SFMCs) from RA patients, either stimulated in vitro or not; Fourth, inoculation of RA patient-derived peripheral blood mononuclear cells (PBMCs); and fifth, induction of RA-like disease by viral or bacterial triggers in immunodeficient mice that have been reconstituted with human hematopoietic stem and progenitor cells (HSCs). GvHD, graft vs. host disease; i.a., intraarticular; Il2rgnull, defective interleukin 2 receptor γ chain; i.p., intraperitoneal; i.v., intravenously; NOD, non-obese diabetic mouse; s.c., subcutaneously; SCID, severe combined immunodeficiency; SCID.bg, SCID mouse with reduced NK cell activity due to beige mutation.
It has been demonstrated that implanted human rheumatoid synovium maintains its histological and phenotypic properties in SCID mice and survives for more than 6 months (251). Synovial tissue transplants become vascularized by human endothelial cells and human vessels were connected to mouse vasculature (259, 260). To generate RA synovium/SCID mouse chimeras, pieces of rheumatoid synovium were placed into SCID mice subcutaneously (260, 261), under the renal capsule (251, 262) or into joints (263, 264). As a result, RA-like disease, characterized by inflammation, angiogenesis, pannus formation and cartilage infiltration by fibroblast-like cells developed (260, 262, 263). The implanted rheumatoid synovium even invaded and destroyed co-implanted normal human cartilage (262). Importantly, only synovial tissue from RA patients, but not healthy synovium, induced pannus formation and destruction of bone and cartilage, when transplanted into joints of beige SCID mice (263). Both human and murine macrophages as well as murine granulocytes contribute to synovial inflammation in this model (263, 264). Human TNF, IL-6 and all Ig subclasses were detected in the serum of RA synovium-grafted animals (260, 265, 266). However, in the absence of inflammatory stimuli present in the RA joint, human synovium returns to a “resting state” after transplantation into immunodeficient mice, as demonstrated by the decreased expression of cell adhesion molecules ICAM-1 and VCAM-1 and emigration of mononuclear cells (259, 267). To overcome the issue of decreased lymphocyte numbers within RA synovial grafts, human allogeneic or autologous PBMCs or T cells were injected into the peritoneal cavity, tail vein, or directly into the graft of RA synovium SCID mouse chimeras (251, 268, 269). However, only 1–5% of intravenously injected peripheral blood lymphocytes (PBL) were shown to reach the graft, while most human PBL were sequestered in spleen and liver (259). Other studies demonstrated that injected T cells specifically migrate into synovial grafts, though migration was not specific to the synovial origin or inflammatory state of the human graft (251, 260). Co-administration of pro-inflammatory mediators, such as TNF, the acute-phase protein serum amyloid A and IL-15, have been shown to play a critical role in maintaining an inflammatory milieu, which retains mononuclear cells within and enhances lymphocyte and monocyte migration toward human synovial grafts (252, 267–269). Elegant studies by Weyand's group, in which either T cells or B cells were depleted from the RA synovial grafts in NOD-SCID mice, revealed that Th1 cells drive pro-inflammatory cytokine and tissue-degrading enzyme expression by synovial macrophages, and underlined a critical role of B cells in T cell activation and synovial inflammation (270, 271).
The RA synovium/SCID mouse chimera model has been widely used to study the properties of RA synovium in vivo, and to explore the effect of inflammatory mediators and its inhibitors on angiogenesis, cytokine secretion, and inflammatory cell infiltration (252, 266, 268, 269). The RA synovium/SCID mouse model was also used to investigate the mechanisms of action of anti-rheumatic drugs in vivo and to test novel biologic agents for the treatment of RA, such as monoclonal antibodies directed against human IL-6R, CD147 or Fas, as well as T and B cell–related therapies using CTLA-4Ig, anti-CD20, and anti–IL-17 antibodies (266, 272–275). For example, methotrexate decreased the number of inflammatory cells in RA synovial grafts through apoptosis (272), and anti-TNF antibodies, although reducing synovial inflammation, did not prevent bone and articular cartilage damage (276). This model also demonstrated inefficacy of anti-IL-1 and CTLA-4Ig therapy in the reduction of cellular infiltration and IL-6 secretion, and showed selective decreases in IL-6 secretion by anti-IL-17 only in those RA synovial tissues which contain high numbers of T cells (266).
Co-implantation of normal human cartilage with RA synovium into SCID mice resulted in pannus-like formation, cartilage invasion and perichondrocytic degradation for an extended period (261, 262). Intra-graft injection of IL-10 inhibited cartilage degradation and decreased ICAM-1 expression in and PBMC traffic toward RA synovial tissue in this model (261). In another approach, tissue derived from human RA pannus was implanted subcutaneously together with a slice of dentin into SCID mice (265). This work elucidated that only concomitant treatment with methotrexate and the TNF inhibitor infliximab suppressed pit formation in the dentin slice and thus detains bone destruction (265).
In the SCID mouse co-implantation model of RA, synovial fibroblasts from RA patients (RASF) and normal cartilage were placed together in a gel sponge either subcutaneously or under the renal capsule (253, 254). The gel sponge replaced the synovial matrix as carrier for synovial fibroblasts, providing an environment devoid of stimulatory or inhibitory effects of other cellular and matrix components (253). This allowed study of the invasion of human RASF into human cartilage in a non-inflammatory environment. Using this model, RASFs were shown to invade and destroy human articular cartilage, independent of T cells, macrophages, and inflammation (253, 277). Activated RASFs specifically migrated toward, invaded and degraded implanted human cartilage, mirroring the progression from oligo- to polyarticular disease (254). However, due to the absence of inflammation and (antigen-specific) T and B cell responses, this SCID mouse co-implantation model of RA appears to rather reflect a facet of the disease, than the complete process of RA development.
A first attempt to engraft mononuclear cells from peripheral blood, synovial fluid or synovial tissue of RA patients into immunodeficient mice was made in 1990 by Tighe and colleagues (255). Transferred PBMCs and SFMCs continued producing human IgG and IgM in SCID host mice, and IgM rheumatoid factor could be detected in mouse serum for more than 20 weeks after human cell engraftment (255). Later, Sakata and coworkers showed that previously stimulated synovial fluid-derived T cells from RA patients, simultaneously injected into knee joint and peritoneal cavity, caused severe destructive arthritis in SCID mice (256). Interestingly, arthritis occurred not only in the joint that received the cell injection but also in other joints, suggesting RA-like polyarthritis (256). In contrast, transfer of unstimulated synovial fluid-derived T cells or in vitro-activated PBMCs from RA patients failed to trigger arthritis in SCID mice (256, 263).
Almost two decades later, Ishikawa and colleagues engrafted RA patient-derived PBMCs into NOG immunodeficient mice (233). Since transfer of PBMC often causes lethal GvHD, the naïve CD4+ T cell fraction was removed from PBMCs, and RA patient-derived CD4+ memory T cells and B cells were transferred sequentially, resulting in sustained production of IgM rheumatoid factor autoantibodies in human cell-engrafted NOG mice (233). However, only some mice reconstituted with RA patient-derived PBMCs displayed histological joint destruction, which was difficult to distinguish from alterations due to chronic GvHD (233). Nonetheless, it has been proven that human T cells engrafted in NOD-scid IL2rgnull mice migrate to air pouches containing RA synovial fluid and this recruitment was abolished by CXCR3 agonist (278).
The development of RA-like disease in immunodeficient mice engrafted with human cells seems to critically depend on the origin (peripheral blood vs. synovial fluid) and previous ex vivo-activation of transferred cells through mitogen or autoantigen, as well as on cell number and route of administration (intra-articular, intraperitoneal, or intravenous). Unlike other models of autoimmune diseases (243, 248, 249), injection of PBMCs of RA patients, even when activated, was insufficient to induce arthritis (233, 256, 263), indicating that pathogenic T cells might be concentrated in synovial fluid and tissue. Previous ex vivo activation of synovial fluid-derived T cells appears to facilitate the establishment of arthritis in immunodeficient mice through expansion of antigen-specific arthritogenic T cell clones and concomitant induction of growth factors such as IL-2 (256). Sole intra-articular injection of small numbers of mitogen-stimulated SFMCs or CII-specific T cell lines was not sufficient to induce joint destruction in beige SCID mice (263), while combination of systemic and local administration of synovial fluid-derived cells appeared to concert inflammatory cell infiltration and local synovial cell proliferation and thus promote the development of RA-like lesions (256). It is of note that at 4 weeks after engraftment into NOD-scid IL2rgnull mice, almost all human cells were activated T cells and thus, it must be considered that the absence of myeloid cells may affect the development of a RA-like disease (278). The supply of antigen-presenting cells e.g., through repetitive injections of (auto-)antigen-loaded DCs or B cells, might be an option to circumvent this issue (158, 187, 188, 233).
With the advent of immunodeficient mouse models that enable the complete reconstitution of the human immune system through engraftment of HSCs, researchers have also exploited this option to establish humanized mouse models of RA.
Injection of Chlamydia trachomatis into knee joints of NOD-SCID mice, that had been previously irradiated, treated with anti-CD122 Ab to block the IL-2R and repopulated with CD34+ bone marrow-derived HSCs from osteoarthritis patients, induced synovial inflammation with a predominance of human CD68+ macrophages (159). The main limitation of this model is the absence of mature human CD4+ and CD8+ T lymphocytes (159). To overcome this issue, later models used umbilical cord blood-derived HSCs which were able to give rise to all immune cell types, instead of HSCs from adult peripheral blood with restricted hematopoietic potential (210, 219).
Kuwana and coworkers used NOG mice transplanted with cord blood-derived HSCs to investigate the contribution of Epstein-Barr virus (EBV) infection to the development of RA (257). Erosive arthritis of the major joints, histologically characterized by pannus formation, bone marrow edema, synovial membrane proliferation and infiltration of inflammatory cells, mainly T cells, occurred in 65% of low dose EBV-infected, but not in non-infected, humanized NOG mice (257). However, rheumatoid factor and ACPA autoantibodies were not detected in peripheral blood of these mice (257). In another approach, acute inflammatory arthritis was induced in HSC-reconstituted NSG mice, through intra-articular injection of CFA (258). These mice developed clinical and histological signs of arthritis, such as swelling, erythema, decrease of function, immune cell infiltration, and bone erosion (258). Elimination of murine neutrophils by pre-treatment with anti-Gr-1 antibody did not affect arthritis development and human leukocytes were detected in inflammatory infiltrates, indicating that RA-like disease was mediated by engrafted human cells infiltrating the joint (258). Treatment with the TNF inhibitor Etanercept prior to the induction of CFA-triggered arthritis was shown to decrease scores of pannus formation, inflammation and bone erosion, as well as human cell counts in joints (258).
So far only healthy donor-derived HSCs have been used to establish the human immune system in immunodeficient mice. An ideal model of RA pathogenesis would utilize HSCs derived from RA patients, which has been non-feasible. On the other hand, stem cells obtained from adult peripheral blood have proved to be unsuitable to completely reconstitute the immune system in immunodeficient mice (159, 219, 258). Induced pluripotent HSCs or cord blood-derived HSCs from relatives or offspring of RA patients, which might be equally predisposed to develop RA, or healthy individuals with RA-susceptible HLA-DR haplotype, might serve as source of HSCs for engraftment, but are difficult to obtain. Moreover, RA models based on immunodeficient mice, whose immune system has been reconstituted through engraftment of human stem cells, require a local inflammatory stimulus in the joint, such as viral or bacterial infection, to induce immigration of inflammatory cells and trigger RA-like disease.
Taken together, the ultimate humanized model of RA, mirroring all aspects of this chronic inflammatory autoimmune disease does not yet exist. Since T cells play a minor role in synovial tissue implant and RASF transfer models, these models are less suitable for testing cell-based therapeutic strategies targeting autoreactive T cells. Autoreactive lymphocytes are found in peripheral blood and, to a much higher extent, in synovial fluid and tissue of RA patients, which might therefore be the cellular source of choice to recapitulate arthritis in immunodeficient mice. However, in vitro T cell activation prior to transfer seems to be necessary, raising the question of antigen-specificity (256). Finally, development of arthritis-like symptoms in immunodeficient mice that have been engrafted with healthy donor HSCs requires pathogenic stimuli and thus does not involve RA-associated autoantigens (159, 258), unless HSC donors expressing RA risk alleles are used for the reconstitution of the human immune system.
Humanized mouse models, particularly those reconstituted with human leukocyte populations, provide a promising tool for preclinical testing of novel therapeutic approaches, such as cell-based immunotherapy (Figure 4).
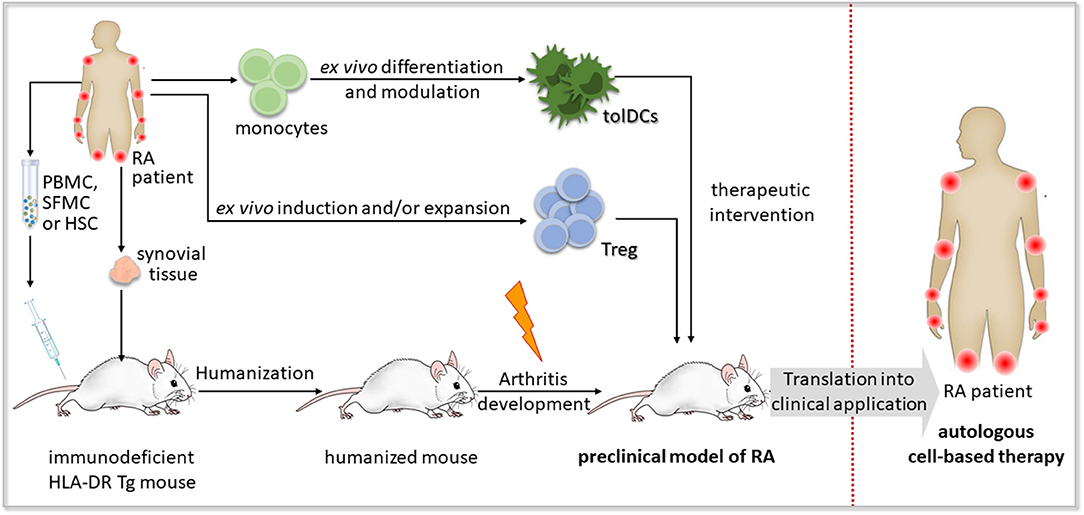
Figure 4. Concept of preclinical testing of cell-based immunotherapy for rheumatoid arthritis in humanized mice. A humanized mouse model of rheumatoid arthritis (RA) could be established by the engraftment of human synovial tissue, hematopoietic stem and progenitor cells (HSCs), peripheral blood mononuclear cells (PBMCs) or synovial fluid mononuclear cells (SFMCs) from an RA patient bearing HLA-DR risk alleles such as HLA-DR*0401 or HLA-DR*0101 into immunodeficient mice expressing the respective transgenic (Tg) HLA-DR molecules. Autoimmune-like disease, if not developed spontaneously, might be induced by an additional trigger, such as RA-associated autoantigens or autoreactive T cell clones. The obtained humanized mouse model of RA enables preclinical testing of cell-based immunotherapeutic approaches applying autologous regulatory T (Treg) cells or tolerogenic dendritic cells (tolDCs) to recover self-tolerance, before their transfer to clinical application in RA patients.
The clinical response of individual patients toward autologous cell-based immunotherapy has already been tested in humanized mouse models of diverse human cancers (160, 241). Adoptively transferred Treg cells, converted from CD4+ T cells through enforced expression of IL-10, were demonstrated to exert anti-tumor and anti-leukemic effects in NSG mice reconstituted with ALL-CM or THP-1 leukemia cell lines, through granzyme B-mediated lysis of myeloid tumor cells in a HLA class I-dependent but antigen-independent manner (161). These CD4IL10 cells were also able to contribute to graft vs. leukemia activity of injected allogeneic PBMCs, while preventing xenogeneic GvHD (161). Administration of autologous mature DCs, pulsed with tumor-associated antigen MART1 or WT1, was shown to induce antigen-specific cytotoxic T cell responses in NSG mice reconstituted with human PBL (158, 187). In a humanized mouse model of breast cancer, established by injecting breast cancer stem cells into the mammary fat pad of HSC-reconstituted NOD-SCID mice, the application of autologous DCs loaded with breast cancer stem cell antigen reduced tumor size and prolonged survival (162).
Adoptive immunotherapy with human DCs, engineered to be long-lived and to co-express high levels of human IFN-α, human GM-CSF and cytomegalovirus pp65 antigen, regenerated immunity de novo in NRG mice after human allogeneic HSC transplantation (163, 164). In vivo targeting of Epstein-Barr virus nuclear antigen 1 (EBNA1) to the endocytic receptor DEC-205 expressed by CD141+ DCs, stimulated antigen-specific CD4+ T cell responses in NSG mice reconstituted with human fetal liver-derived CD34+ HSCs (165).
Promising cell-based approaches have also been achieved in humanized mouse models of transplant rejection. Injection of freshly isolated or ex vivo-expanded human CD4+ or CD8+ Treg cells into immunodeficient mice, that had been transplanted with human skin and reconstituted with allogeneic PBMCs, supported long-term survival of skin allografts (166–168). Similarly, transfer of ex vivo expanded autologous Treg cells prevented rejection of islet xenografts in mice reconstituted with human PBMCs or CD34+ HSCs by inhibiting immune cell infiltration and T effector cell differentiation in an IL-10 dependent manner (169, 170, 279).
In a humanized model of allergic airway disease, consisting of NSG mice reconstituted with PBMC from patients with allergic asthma and sensitized to birch pollen in the presence of IL-4, administration of autologous Treg cells or in vivo Treg induction through sGARP or polyclonal activation via gp120, abrogated airway hyperresponsiveness and reduced airway inflammation in the lung in a TGF-β receptor 2-dependent manner (280, 281). In a similar model of allergen-specific gut inflammation, induced by rectal challenge with birch, grass pollen, or dust mite allergen, adoptive transfer of activated Treg cells decreased allergen-specific Th2 responses and IgE secretion (282).
Despite the great potential of humanized mice as preclinical models (241, 283, 284), there are only few studies that use mouse/human chimera to explore the efficacy of cell-based immunotherapies for autoimmune diseases in vivo, none of them in RA.
In humanized models of autoimmune T1D, autoantigen-specific vaccination strategies have been described, which aim at inducing Treg cells that prevent the destruction of insulin-producing β cells (285, 286). Systemic delivery of nanoparticles, coated with autoimmune disease-relevant peptides bound to MHC class II molecules, promoted the in vivo generation and expansion of antigen-specific FOXP3-CD49b+LAG-3+ Treg cells in different humanized mouse models, including NSG mice reconstituted with T1D patient-derived CD8+ T cell-depleted PBMCs, and ameliorated clinical and pathological signs of CIA and experimental autoimmune encephalomyelitis in HLA-DR4-IE Tg mice (285). In another study, sub-immunogenic vaccination of human HSC-engrafted NSG.DQ8 Tg mice with agonistic mimetopes of the T1D-relevant insulin B-chain epitope induced insulin-specific FOXP3+ Treg cells in vivo (286).
To explore therapeutic strategies to overcome the resistance of effector T cells toward Treg-mediated suppression in multiple sclerosis (MS) patients, a humanized model based on newborn Rag2−/−γ−/− mice engrafted with CD25-depleted PBMCs from different MS patients was applied (287). Transfer of gp120-activated Treg cells from healthy subjects into these mice prevented GvHD only when engrafted PBMCs were derived from MS patients that received disease-modifying therapy but not from those being therapy-naïve (287).
In a humanized model of myasthenia gravis (MG), generated by subcutaneous engraftment of thymic MG fragments into NSG mice, administration of in vitro-preconditioned human mesenchymal stem cells improved the disease by decreasing the serum level of acetylcholine receptor (AChR)-specific autoantibodies and restoring AChR expression at the muscle endplate (288).
Novel therapeutics for RA have been tested so far only in DR1 and DR4 Tg mice. Particularly, the capacity of CII analog peptide has been demonstrated to induce in vivo antigen-specific inhibitory T cells capable of suppressing CIA dependent on IL-10 and IL-4 secretion (173, 289). Nevertheless, it seems to be only a matter of time until humanized chimeric mouse models of RA will be available for preclinical testing of cell-based therapeutic strategies. But there are still some important obstacles to overcome (Table 2).

Table 2. Challenges of recapitulating rheumatoid arthritis in chimeric humanized mice.
Many of the RA models have been difficult to reproduce by other laboratories, requiring profound characterization of environmental und genetic susceptibility factors that interfere with, or promote, RA-like disease in humanized mice.
One major problem in humanized mice is that long-term evaluation of transferred human cells and the establishment of chronic disease is hampered by the development of xenogeneic GvHD, induced by cross-species interactions between mouse macrophages and human CD4+ T cells in skin and lymphatic tissues (290). HSC-reconstituted mice are less susceptible to GvHD than PBMC-engrafted mice, since T cell selection in this model occurs in the murine thymus. However, PBMCs might be the better choice for the establishment of an RA model, as autoimmunity develops due to the loss of peripheral but not central tolerance. Since it has been postulated that GvHD in human PBMC-engrafted SCID mice results from anti-mouse MHC class II reactivity of human CD4+ T cells (291), a possible solution would be to use immunodeficient mice that lack murine MHC class II molecules as platform for the engraftment of human cells (238). Additional expression of transgenic human RA-associated DR4 or DR1 molecules might further improve long-term engraftment of cells and tissues from HLA-DR-matched human donors (237, 239, 245). Alternatively, removal of the naïve CD4+ T cell fraction from transferred PBMC might reduce xenogeneic GvHD (233). Additional administration of purified autologous B cells might ensure efficient autoantibody production (233).
Another important issue is the rapid disappearance of myeloid antigen presenting cells after engraftment of PBMC into immunodeficient mice (29). Since almost all transferred T lymphocytes express HLA-DR after activation, it is conceivable that activated T cells could assume the role of APCs in humanized models (134). However, additional supply of survival factors or repeated administration of myeloid antigen presenting cells such as DCs pulsed with autoantigen might improve the propagation of autoantigen-specific T cell responses. Removal of Treg cells from the human cell graft might further facilitate the establishment of autoimmune responses in humanized mice (292).
A variety of potential RA-associated autoantigens have been described and T cell responses directed against them are extremely heterogeneous among different RA patients, requiring personalized models of RA. These can be easily established in immunodeficient mice by engrafting RA patient-derived PBMCs or SFMCs, which contain autoreactive T cells and are easier to obtain than patient-derived HSCs. Pathogenic T cell clones may be isolated from peripheral blood or synovial fluid of RA patients and expanded ex vivo before transferring them into mice. Eventually, it might be necessary to trigger arthritis by activation of human lymphocytes before transfer into mice or by co-administration of arthritogenic antigen.
Since conventional mouse models of RA are only partially suited to preclinical testing of cell-based therapies, translational research requires a humanized mouse model that accurately mirrors autoimmune processes of human RA and permits its modulation by the transfer of human immunoregulatory cells. Various approaches have been followed during the past decades to establish a humanized model of RA, with only partial success and poor reproducibility. Valuable lessons learnt from translational research models of other autoimmune diseases might help to improve current attempts. A combination of transgenic expression of RA risk alleles and the engraftment of RA patient-derived immune cells and/or RA synovial tissue seems a promising strategy to avoid GvHD and establish chronic autoimmune responses. Improved humanized mouse models of RA would provide a powerful tool for preclinical evaluation of cell-based immunotherapies.
The manuscript has been jointly written by KS and JA, and was critically revised by RT, CR, and LS.
This work was funded by CONICYT-Chile FONDECYT-Postdoctorado N°3150453 (KS), FONDECYT-Chile N°1181853 (JA), Fondef IDeA-Chile I+D ID18I10243 (JA+LS), and PCI-CONICYT REDES180028 from Chile (JA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank CONICYT-Chile for financial support.
AA, adjuvant-induced arthritis; ACPAs, anti-citrullinated protein/peptide antibodies; AIA, antigen-induced arthritis; BLT, bone marrow-liver-thymus transplant; BRG, BALB/c-Rag2null IL2rgnull; CII, type II collagen; CFA, complete Freund's adjuvant; CIA, collagen-induced arthritis; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; FOXP3, forkhead-box-protein P3; gp, glycoprotein; GvHD, graft vs. host disease; HLA, human leukocyte antigen; HSCs, hematopoietic stem and progenitor cells; Ig, immunoglobulin; IL, interleukin; IFN, interferon; MHC, major histocompatibility complex; MMC, mutated mouse collagen; NK, natural killer; NOD, non-obese diabetic; NOG, NOD-SCID Il2rgtm1Sug; NRG, NOD-Rag1null Il2rgnull; NSG, NOD-SCID Il2rgtm1Wjl; PAD, peptidylarginine deiminase; PBMCs, peripheral blood mononuclear cells; PG, proteoglycan; PGIA, Proteoglycan-induced arthritis; RA, rheumatoid arthritis; Rag1/2, recombination-activating genes 1/2; SCID, severe combined immunodeficiency; SE, shared epitope; SFMCs, synovial fluid mononuclear cells; T1D, type 1 diabetes; TCR, T cell receptor; Tg, transgenic; Th, T helper; TNF, tumor necrosis factor; tolDC, tolerogenic dendritic cell; Treg, regulatory T cell.
1. Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet (2017) 389:2338–48. doi: 10.1016/S0140-6736(17)31491-5
2. Turesson C, O'Fallon WM, Crowson CS, Gabriel SE, Matteson EL. Occurrence of extraarticular disease manifestations is associated with excess mortality in a community based cohort of patients with rheumatoid arthritis. J Rheumatol. (2002) 29:62–7. Available online at: http://www.jrheum.org/content/29/1/62.long
3. Karouzakis E, Neidhart M, Gay RE, Gay S. Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett. (2006) 106:8–13. doi: 10.1016/j.imlet.2006.04.011
4. Hegen M, Keith JC, Collins M, Nickerson-Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis. (2008) 67:1505–15. doi: 10.1136/ard.2007.076430
5. Vincent TL, Williams RO, Maciewicz R, Silman A, Garside P, Bevan S, et al. Mapping pathogenesis of arthritis through small animal models. Rheumatology (2012) 51:1931–41. doi: 10.1093/rheumatology/kes035
6. Bevaart L, Vervoordeldonk MJ, Tak PP. Evaluation of therapeutic targets in animal models of arthritis: how does it relate to rheumatoid arthritis? Arthritis Rheum. (2010) 62:2192–205. doi: 10.1002/art.27503
7. Kadowaki KM, Matsuno H, Tsuji H, Tunru I. CD4+ T cells from collagen-induced arthritic mice are essential to transfer arthritis into severe combined immunodeficient mice. Clin Exp Immunol. (1994) 97:212–8. doi: 10.1111/j.1365-2249.1994.tb06070.x
8. Taylor PC, Plater-Zyberk C, Maini RN. The role of the B cells in the adoptive transfer of collagen-induced arthritis from DBA/1 (H-2q) to SCID (H-2d) mice. Eur J Immunol. (1995) 25:763–9. doi: 10.1002/eji.1830250321
9. Saijo S, Asano M, Horai R, Yamamoto H, Iwakura Y. Suppression of autoimmune arthritis in interleukin-1-deficient mice in which T cell activation is impaired due to low levels of CD40 ligand and OX40 expression on T cells. Arthritis Rheum. (2002) 46:533–44. doi: 10.1002/art.10172
10. Horai R, Nakajima A, Habiro K, Kotani M, Nakae S, Matsuki T, et al. TNF-α is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist – deficient mice. J Clin Invest. (2004) 114:1603–11. doi: 10.1172/JCI20742
11. Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acade Sci USA. (2003) 100:5986–90. doi: 10.1073/pnas.1035999100
12. Glant TT, Mikecz K, Arzoumanian A, Poole AR. Proteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathology. Arthritis Rheum. (1987) 30:201–12. doi: 10.1002/art.1780300211
13. Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, et al. An HLA-DR1 Transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J Exp Med. (1997) 185:1113–22. doi: 10.1084/jem.185.6.1113
15. Perel P, Roberts I, Sena E, Wheble P, Briscoe C, Sandercock P, et al. Comparison of treatment effects between animal experiments and clinical trials: systematic review. Br Med J. (2007) 334:197–200. doi: 10.1136/bmj.39048.407928.BE
16. Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, Erondu N. A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J Rheumatol. (2015) 42:912–9. doi: 10.3899/jrheum.141271
17. Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis. (2013) 72:863–9. doi: 10.1136/annrheumdis-2012-201601
18. Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. (2004) 50:650–9. doi: 10.1002/art.20001
19. Joosten LA, Helsen MM, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: a comparative study using anti-TNFα, anti-IL-1α/β, and IL-1Ra. Arthritis Rheum. (1996) 39:797–809. doi: 10.1002/art.1780390513
20. Gabay C, Lamacchia C, Palmer G. IL-1 Pathways in inflammation and human diseases. Nat Rev Rheumatol. (2010) 6:232–41. doi: 10.1038/nrrheum.2010.4
21. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
22. Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Critic Rev TM Immunol. (2014) 34:433–54. doi: 10.1615/CritRevImmunol.2014011600
23. Sabroe I, Dockrell DH, Vogel SN, Renshaw SA, Whyte MK, Dower SK. Identifying and hurdling obstacles to translational research. Nat Rev Immunol. (2007) 7:77–82. doi: 10.1038/nri1999
24. Akhtar A. The flaws and human harms of animal experimentation. Cambridge Q Healthcare Ethics (2015) 24:407–19. doi: 10.1017/S0963180115000079
25. Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N. Clinical tolerogenic dendritic cells: exploring therapeutic impact on human autoimmune disease. Front Immunol. (2017) 8:1279. doi: 10.3389/fimmu.2017.01279
26. Salazar L, Aravena O, Abello P, Escobar A, Contreras-Levicoy J, Rojas-Colonelli N, et al. Modulation of established murine collagen-induced arthritis by a single inoculation of short-term lipopolysaccharide-stimulated dendritic cells. Ann Rheum Dis. (2008) 67:1235–41. doi: 10.1136/ard.2007.072199
27. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Care Res. (2010) 62:3656–65. doi: 10.1002/art.27756
28. Eming R, Visconti K, Hall F, Sekine C, Kobayashi K, Chen Q, et al. Humanized mice as a model for rheumatoid arthritis. Arthritis Res. (2002) 4:133–40. doi: 10.1186/ar580
29. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. (2007) 7:118–30. doi: 10.1038/nri2017
30. Shultz L. Next Generation Humanized Mouse Models for Human Infectious Diseases. Fondation Merieux (2017). Available online at: https://www.fondation-merieux.org/wp-content/uploads/2017/04/humanized-models-to-study-immunity-and-to-accelerate-the-development-of-new-solutions-for-human-health-2017-lenny-shultz.pdf
31. Hahn SA, Bellinghausen I, Trinschek B, Becker C. Translating treg therapy in humanized mice. Front Immunol. (2015) 6:623. doi: 10.3389/fimmu.2015.00623
32. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL, Shultz LD. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
33. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. (2017) 17:60–75. doi: 10.1038/nri.2016.124
34. de Vries RR, Huizinga TW, Toes RE. Redefining the HLA and RA association: to be or not to be anti-CCP positive. J Autoimmun (2005) 25(Suppl.):21–5. doi: 10.1016/j.jaut.2005.09.005
35. Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, et al. A molecular basis for MHC class II–associated autoimmunity. Science (1988) 240:1003–9. doi: 10.1126/science.3368786
36. Penzotti JE, Doherty D, Lybrand TP, Nepom GT. A structural model for TCR recognition of the HLA class II shared epitope sequence implicated in susceptibility to rheumatoid arthritis. J Autoimmun. (1996) 9:287–93. doi: 10.1006/jaut.1996.0037
37. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. Arthritis Rheum. (1987) 30:1205–12. doi: 10.1002/art.1780301102
38. Meyer JM, Evans TI, Small RE, Redford TW, Han J, Singh R, et al. HLA-DRB1 genotype influences risk for and severity of rheumatoid arthritis. J Rheumatol. (1999) 26:1024–34.
39. Weyand CM, McCarthy TG, Goronzy JJ. Correlation between disease phenotype and genetic heterogeneity in rheumatoid arthritis. J Clin Invest. (1995) 95:2120–6. doi: 10.1172/JCI117900
41. Kim HY, Kim WU, Cho ML, Lee SK, Youn J, Kim SI, et al. Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255-274) in patients with rheumatoid arthritis. Arthritis Rheum. (1999) 42:2085–93.
42. Boots AM, Verheijden GF, Schöningh R, van Staveren CJ, Bos E, Elewaut D, et al. Selection of self-reactive peptides within human aggrecan by use of a HLA-DRB1*0401 peptide binding motif. J Autoimmun. (1997) 10:569–78. doi: 10.1006/jaut.1997.0164
43. Szántó S, Bárdos T, Szab ó Z, David CS, Buzás EI, Mikecz K, et al. Induction of arthritis in HLA-DR4-humanized and HLA-DQ8-humanized mice by human cartilage proteoglycan aggrecan but only in the presence of an appropriate (Non-MHC) genetic background. Arthritis Rheum. (2004) 50:1984–95. doi: 10.1002/art.20285
44. Verheijden GF, Rijnders AW, Bos E, Coenen-de Roo CJ, van Staveren CJ, Miltenburg AM, et al. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis Rheum. (1997) 40:1115–25. doi: 10.1002/art.1780400616
45. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. (2003) 171:538–41. doi: 10.4049/jimmunol.171.2.538
46. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
47. Suzuki A, Yamada R, Yamamoto K. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann N Y Acad Sci. (2007) 1108:323–39. doi: 10.1196/annals.1422.034
48. Snir O, Widhe M, von Spee C, Lindberg J, Padyukov L, Lundberg K, et al. Multiple antibody reactivities to citrullinated antigens in sera from patients with rheumatoid arthritis: association with HLA-DRB1 alleles. Ann Rheum Dis. (2009) 68:736–43. doi: 10.1136/ard.2008.091355
49. Law SC, Street S, Yu CH, Capini C, Ramnoruth S, Nel HJ, et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Res Ther 14:R118. doi: 10.1186/ar3848
50. van der Helm-van Mil AH, van Gaalen FA, Kloppenburg M, de Vries RR, le Cessie S, et al. The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum. (2006). 54:1117–21. doi: 10.1002/art.21739
51. Avouac J, Gossec L, and Dougados M, Avouac J, Gossec L, Dougados M. Diagnostic and predictive value of anti-cyclic citrullinated protein antibodies in rheumatoid arthritis: a systematic literature review. Ann Rheum Dis. (2006) 65:845–51. doi: 10.1136/ard.2006.051391
52. García-González P, Ubilla-Olguín G, Catalán D, Schinnerling K, Aguillón JC. Tolerogenic dendritic cells for reprogramming of lymphocyte responses in autoimmune diseases. Autoimmun Rev. (2016) 15:1071–80. doi: 10.1016/j.autrev.2016.07.032
53. Tsark EC, Wang W, Teng YC, Arkfeld D, Dodge GR, Kovats S. Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J Immunol. (2002) 169:6625–33. doi: 10.4049/jimmunol.169.11.6625
54. Thomas R, Quinn C. Functional differentiation of dendritic cells in rheumatoid arthritis: role of CD86 in the synovium. J Immunol. (1996) 156:3074–86.
55. Schinnerling K, Soto L, García-González P, Catalán D, Aguillón JC. Skewing dendritic cell differentiation towards a tolerogenic state for recovery of tolerance in rheumatoid arthritis. Autoimmun Rev. (2015) 14:517–27. doi: 10.1016/j.autrev.2015.01.014
56. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, et al. Th1 but Not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. (2008). 67:1299–304. doi: 10.1136/ard.2007.080341
57. Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. (2009) 60:1647–56. doi: 10.1002/art.24568
58. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA. (2010) 107:14751–6. doi: 10.1073/pnas.1003852107
59. Scherer HU, Huizinga TWJ, Krönke G, Schett G, Toes REM. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol. (2018) 14:157–69. doi: 10.1038/nrrheum.2018.10
60. Schett G, Elewaut D, Mcinnes IB, Dayer J-M, Neurath MF. How cytokine networks fuel inflammation. Nat Med. (2013) 19:822–6. doi: 10.1038/nm.3260
61. Schinnerling K, Aguillón JC, Catalán D, Soto L. The role of interleukin-6 signalling and its therapeutic blockage in skewing the T cell balance in rheumatoid arthritis. Clin Exp Immunol. (2017) 189:12–20. doi: 10.1111/cei.12966
62. Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity (2017) 46:183–96. doi: 10.1016/j.immuni.2017.02.006
63. Vander Cruyssen B, Van Looy S, Wyns B, Westhovens R, Durez P, Van den Bosch F, et al. Four-year follow-up of infliximab therapy in rheumatoid arthritis patients with long-standing refractory disease: attrition and long-term evolution of disease activity. Arthritis Res Ther. (2006) 8:R112. doi: 10.1186/ar2001
64. Aletaha D, Smolen JS. The rheumatoid arthritis patient in the clinic: comparing more than 1300 consecutive DMARD courses. Rheumatology (2002) 41:1367–74. doi: 10.1093/rheumatology/41.12.1367
65. Winthrop KL, Iseman M. Bedfellows: mycobacteria and rheumatoid arthritis in the era of biologic therapy. Nat Rev Rheumatol. (2013) 9:524–31. doi: 10.1038/nrrheum.2013.82
66. Curtis JR, Lee EB, Kaplan IV, Kwok K, Geier J, Benda B, et al. Tofacitinib, an oral janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. (2016) 75:831–41. doi: 10.1136/annrheumdis-2014-205847
67. Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol. (2014) 10:543–51. doi: 10.1038/nrrheum.2014.105
68. Gregori S, Roncarolo MG. Engineered T regulatory type 1 cells for clinical application. Front Immunol. (2018) 9:233. doi: 10.3389/fimmu.2018.00233
69. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J Exp Med. (2004) 200:277–85. doi: 10.1084/jem.20040165
70. van Amelsfort JM, van Roon JA, Noordegraaf M, Jacobs KM, Bijlsma JW, Lafeber FP, et al. Proinflammatory mediator-induced reversal of CD4+,CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum. (2007) 56:732–42. doi: 10.1002/art.22414
71. Torres-Aguilar H, Aguilar-Ruiz SR, González-Pérez G, Munguía R, Bajaña S, Meraz-Ríos MA, et al. Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4 + T cells. J Immunology. (2010) 184:1765–75. doi: 10.4049/jimmunol.0902133
72. Maggi J, Schinnerling K, Pesce B, Hilkens CM, Catalán D, Aguillón JC. Dexamethasone and monophosphoryl lipid a-modulated dendritic cells promote antigen-specific tolerogenic properties on naive and memory CD4+T cells. Front Immunol. (2016) 7:359. doi: 10.3389/fimmu.2016.00359
73. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. (2009) 39:3147–59. doi: 10.1002/eji.200839103
74. Orange DE, Blachere NE, Fak J, Parveen S, Frank MO, Herre M, et al. Dendritic cells loaded with FK506 kill T cells in an antigen-specific manner and prevent autoimmunity in vivo. ELife (2013) 2013:1–13. doi: 10.7554/eLife.00105
75. Stojanovic I, Dimitrijevic M, Vives-Pi M, Mansilla MJ, Pujol-Autonell I, Rodríguez-Fernandez S, et al. Cell-based tolerogenic therapy, experience from animal models of multiple sclerosis, type 1 diabetes and rheumatoid arthritis. Curr Pharmaceut Design (2017) 23:2623–43. doi: 10.2174/1381612823666170214120708
76. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype – positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:1–12. doi: 10.1126/scitranslmed.aaa9301
77. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
78. McNamee K, Williams R, Seed M. Animal models of rheumatoid arthritis: how informative are they? Eur J Pharmacol. (2015) 759:278–86. doi: 10.1016/j.ejphar.2015.03.047
79. van den Berg WB. Lessons from animal models of arthritis over the past decade. Arthritis Res Ther. (2009) 11:250. doi: 10.1186/ar2803
80. Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. (2009) 39:2040–4. doi: 10.1002/eji.200939578
81. Pearson CM. Development of arthritis, periarthritis and periostitis in rats given adjuvants. Exp Biol Med. (1956) 91:95–101. doi: 10.3181/00379727-91-22179
82. Gauldie SD, McQueen DS, Clarke CJ, Chessell IP. A robust model of adjuvant-induced chronic unilateral arthritis in two mouse strains. J Neurosci Methods (2004) 139:281–91. doi: 10.1016/j.jneumeth.2004.05.003
83. Holmdahl R, Goldschmidt TJ, Kleinau S, Kvick C, Jonsson R. Arthritis induced in rats with adjuvant oil is a genetically restricted, dependent autoimmune disease. Immunology (1992) 76:197–202.
84. Kim EY, Moudgil KD. The determinants of susceptibility/resistance to adjuvant arthritis in rats. Arthritis Res Ther. (2009) 11:239. doi: 10.1186/ar2755
85. van Eden W, Thole JE, van der Zee R, Noordzij A, van Embden JD, Hensen EJ, et al. Cloning of the mycobacterial epitope recognized Ba T lymphocytes in adjuvant arthritis. Nature (1988) 331:171–3. doi: 10.1038/331171a0
86. Chang YH, Pearson CM, Abe C. Adjuvant polyarthritis. IV Induction by a synthetic adjuvant: immunologic, histopathologic, and other studies. Arthritis Rheum. (1980) 23:62–71. doi: 10.1002/art.1780230111
87. Kohashi O, Aihara K, Ozawa A, Kotani S, Azuma I. New model of a synthetic adjuvant, N-acetylmuramyl-L-alanyl-D-isoglutamine- induced arthritis: clinical and histologic studies in athymic nude and euthymic rats. Lab Invest. (1982) 47:27–36.
88. Bedwell AE, Elson CJ, Hinton CE. Immunological involvement in the pathogenesis of pristane-induced arthritis. Scand J Immunol. (1987) 25:393–8. doi: 10.1111/j.1365-3083.1987.tb02205.x
89. Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med. (1977) 146:857–68. doi: 10.1084/jem.146.3.857
90. Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. (2007) 2:1269–75. doi: 10.1038/nprot.2007.173
91. Brunsberg U, Gustafsson K, Jansson L, Michaëlsson E, Ahrlund-Richter L, Pettersson S, et al. Expression of a transgenic class II Ab gene confers susceptibility to collagen-induced arthritis. Eur J Immunol. (1994) 24:1698–702. doi: 10.1002/eji.1830240736
92. Holmdahl R. Dissection of the genetic complexity of arthritis using animal models. Immunol Lett. (2006) 103:86–91. doi: 10.1016/j.imlet.2005.10.025
93. Ranges GE, Sriram S, Cooper SM. Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J Exp Med (1985) 162:1105–10.
94. Holmdahl R, Klareskog L, Rubin K, Larsson E, Wigzell H. T lymphocytes in collagen II-induced arthritis in mice: characterization of arthritogenic collagen II-specific T-cell lines and clones. Scand J Immunol. (1985) 22:295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x
95. Vossenaar ER, Nijenhuis S, Helsen MM, van der Heijden A, Senshu T, van den Berg WB, et al. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. (2003) 48:2489–500. doi: 10.1002/art.11229
96. Förster M, Raposo B, Ekman D, Klaczkowska D, Popovic M, Nandakumar KS, et al. Genetic control of antibody production during collagen-induced arthritis development in heterogeneous stock mice. Arthritis Rheum. (2012) 64:3594–603. doi: 10.1002/art.34658
97. Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J Exp Med. (1983) 158:378–92. doi: 10.1084/jem.158.2.378
98. Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM. Induction of arthritis with monoclonal antibodies to collagen. J Immunol. (1992) 148:2103–8.
99. Mauri C, Williams RO, Walmsley M, Feldmann M. Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur J Immunol. (1996) 26:1511–8. doi: 10.1002/eji.1830260716
100. Schurgers E, Mertens F, Vanoirbeek JA, Put S, Mitera T, De Langhe E, et al. Pulmonary inflammation in mice with collagen-induced arthritis is conditioned by complete Freund's adjuvant and regulated by endogenous IFN-γ. Eur J Immunol. (2012) 42:3223–34. doi: 10.1002/eji.201242573
101. Holmdahl R, Jansson L, Andersson M, Jonsson R. Genetic, hormonal and behavioural influence on spontaneously developing arthritis in normal mice. Clin Exp Immunol. (1992) 88:467–72. doi: 10.1111/j.1365-2249.1992.tb06473.x
102. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
103. Hanyecz A, Berlo SE, Szánt ó S, Broeren CP, Mikecz K, Glant TT. Achievement of a synergistic adjuvant effect on arthritis induction by activation of innate immunity and forcing the immune response toward the Th1 phenotype. Arthritis Rheum. (2004) 50:1665–76. doi: 10.1002/art.20180
104. Mikecz K, Glant TT. Migration and homing of lymphocytes to lymphoid and synovial tissues in proteoglycan–induced murine arthritis. Arthritis Rheum. (1994) 37:1395–403. doi: 10.1002/art.1780370919
105. O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. (2005) 174:3781–8. doi: 10.4049/jimmunol.174.6.3781
106. Stoop JN, Tibbitt CA, van Eden W, Robinson JH, Hilkens CM. The choice of adjuvant determines the cytokine profile of T cells in proteoglycan-induced arthritis but does not influence disease severity. Immunology (2013) 138:68–75. doi: 10.1111/imm.12019
107. Rodeghero R, Cao Y, Olalekan SA, Iwakua Y, Glant TT, Finnegan A. Location of CD4+ T cell priming regulates the differentiation of Th1 and Th17 cells and their contribution to arthritis. J Immunol. (2013) 190:5423–35. doi: 10.4049/jimmunol.1203045
108. van den Berg WB, van Beusekom HJ, van de Putte LB, Zwarts WA, van der Sluis M. Antigen handling in antigen-induced arthritis in mice. Am J Pathol. (1982) 108:9–16.
109. Brackertz D, Mitchell GF, Vadas MA, Mackay IR. Studies on antigen-induced arthritis in mice : III. Cell and serum transfer experiments. J Immunol. (1977) 118:1645–8.
110. Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity (1999) 10:451–46. doi: 10.1016/S1074-7613(00)80045-X
111. Wipke BT, Wang Z, Nagengast W, Reichert DE, Allen PM. Staging the initiation of autoantibody-induced arthritis: a critical role for immune complexes. J Immunol. (2004) 172:7694–702. doi: 10.4049/jimmunol.172.12.7694
112. Jacobs JP, Wu HJ, Benoist C, Mathis D. (2009). IL-17-producing T cells can augment autoantibody-induced arthritis. Proc Natl Acad Sci USA. 106:21789–94. doi: 10.1073/pnas.0912152106
113. Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature (2003) 426:454–60. doi: 10.1038/nature02119
114. Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, et al. A role for fungal β-glucans and their receptor dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med. (2005) 201:949–60. doi: 10.1084/jem.20041758
115. Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, et al. Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol. (2016) 68:2646–61. doi: 10.1002/art.39783
116. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific autoimmune disease provoked by systemic autoimmunity. Cell (1996) 87:811–22. doi: 10.1016/S0092-8674(00)81989-3
117. Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science (1999) 286:1732–5. doi: 10.1126/science.286.5445.1732
118. Mangialaio S, Ji H, Korganow AS, Kouskoff V, Benoist C, Mathis D. The arthritogenic T cell receptor and its ligand in a model of spontaneous arthritis. Arthritis Rheum. (1999) 42:2517–23.
119. Christensen AD, Haase C, Cook AD, Hamilton JA. K/BxN serum-transfer arthritis as a model for human inflammatory arthritis. Front Immunol. (2016) 7:213. doi: 10.3389/fimmu.2016.00213
120. Ji H, Gauguier D, Ohmura K, Gonzalez A, Duchatelle V, Danoy P, et al. Genetic influences on the end-stage effector phase of arthritis. J Exp Med. (2001) 194:321–30. doi: 10.1084/jem.194.3.321
121. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. (1991) 10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x
122. Butler DM, Malfait AM, Mason LJ, Warden PJ, Kollias G, Maini RN, et al. DBA/1 mice expressing the human TNF-alpha transgene develop a severe, erosive arthritis: characterization of the cytokine cascade and cellular composition. J Immunol. (1997) 159:2867–76.
123. Niki Y, Yamada H, Seki S, Kikuchi T, Takaishi H, Toyama Y, et al. Macrophage- and neutrophil-dominant arthritis in human IL-1 alpha transgenic mice. J Clin Invest. (2001) 107:1127–35. doi: 10.1172/JCI11530
124. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. (2000) 191:313–20. doi: 10.1084/jem.191.2.313
125. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. (2005) 52:2212–21. doi: 10.1002/art.21195
126. Ohata J, Miura T, Johnson TA, Hori S, Ziegler SF, Kohsaka H. Enhanced efficacy of regulatory T cell transfer against increasing resistance, by elevated Foxp3 expression induced in arthritic murine hosts. Arthritis Rheum. (2007) 56:2947–56. doi: 10.1002/art.22846
127. Charbonnier LM, Han WG, Quentin J, Huizinga TW, Zwerina J, Toes RE, et al. Adoptive transfer of IL-10-secreting CD4+CD49b+regulatory T cells suppresses ongoing arthritis. J Autoimmun. (2010) 34:390–9. doi: 10.1016/j.jaut.2009.10.003
128. van Herwijnen MJ, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, et al. Regulatory T cells that recognize a ubiquitous suppressors of autoimmune arthritis. Proc Natl Acad Sci USA. (2012) 109:14134–9. doi: 10.1073/pnas.1206803109
129. Wright GP, Notley CA, Xue SA, Bendle GM, Holler A, Schumacher TN, et al. Adoptive therapy with redirected primary regulatory t cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci USA. (2009) 106:19078–83. doi: 10.1073/pnas.0907396106
130. Kim SH, Kim S, Evans CH, Ghivizzani SC, Oligino T, Robbins PD. Effective treatment of established murine collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express IL-4. J Immunol. (2001) 166:3499–505. doi: 10.4049/jimmunol.166.5.3499
131. Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci USA. (2005) 102:13562–7. doi: 10.1073/pnas.0504484102
132. Martin E, Capini C, Duggan E, Lutzky VP, Stumbles P, Pettit AR, et al. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-kappaB. Arthritis Rheum. (2007) 56:2255–66. doi: 10.1002/art.22655
133. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115:10–20. doi: 10.1182/blood-2009-07-235028
134. Holling TM, Schooten E, van Den Elsen PJ. Function and regulation of MHC class II molecules in T-lymphocytes: of mice and men. Hum Immunol. (2004) 65:282–90. doi: 10.1016/j.humimm.2004.01.005
135. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. (2007) 19:345–54. doi: 10.1093/intimm/dxm014
136. Ito Y, Usui T, Kobayashi S, Iguchi-Hashimoto M, Ito H, Yoshitomi H, et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis and Rheumatism (2009) 60:2294–303. doi: 10.1002/art.24687
137. Taneja V, David CS. Role of HLA class II gene in susceptibility/ resistance to inflammatory arthritis: studies with humanized mice. Immunol Rev. (2010) 233:62–78. doi: 10.1111/j.0105-2896.2009.00858.x
138. Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, Benoist C, et al. Mice lacking MHC class II molecules. Cell (1991) 66:1051–66. doi: 10.1016/0092-8674(91)90448-8
139. Vignali DAA, Moreno J, Schiller D, Hammerlin GJ. Species-specific binding of CD4 to the beta 2 domain of major histocompatibility complex class II molecules. J Exp Med. (1992) 175:925–32.
140. Fugger L, Michie SA, Rulifson I, Lock CB, McDevitt GS. Expression of HLA-DR4 and human CD4 transgenes in mice determines the variable region beta-chain T-cell repertoire and mediates an HLA-DR-restricted immune response. Proc Natl Acade Sci USA. (1994) 91:6151–5. doi: 10.1073/pnas.91.13.6151
141. Altmann DM, Douek DC, Frater AJ, Hetherington CM, Inoko H, Elliott JI. The T cell response of HLA-DR transgenic mice to human myelin basic protein and other antigens in the presence and absence of human CD4. J Exp Med. (1995) 181:867–75. doi: 10.1084/jem.181.3.867
142. Andersson EC, Hansen BE, Jacobsen H, Madsen LS, Andersen CB, Engberg J, et al. Definition of MHC and T cell receptor contacts in the HLA-DR4restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. Proc Natl Acade Sci USA. (1998) 95:7574–9. doi: 10.1073/pnas.95.13.7574
143. Woods A, Chen HY, Trumbauer ME, Sirotina A, Cummings R, Zaller DM. Human major histocompatibility complex class II-restricted T cell responses in transgenic mice. J Exp Med. (1994) 180:173–81.
144. Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E, et al. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J Exp Med. (1996) 183:2635–44. doi: 10.1084/jem.183.6.2635
145. Rosloniec EF, Brand DD, Myers LK, Esaki Y, Whittington KB, Zaller DM, et al. Induction of autoimmune arthritis in HLA-DR4 (DRB1*0401) transgenic mice by immunization with human and bovine type II collagen. J Immunol. (1998) 160:2573–8.
146. Taneja V, Behrens M, Mangalam A, Griffiths MM, Luthra HS, David CS. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis Rheum. (2007) 56:69–78. doi: 10.1002/art.22213
147. Luckey D, Behrens M, Smart M, Luthra H, David CS, Taneja V. DRB1*0402 may influence arthritis by promoting naive CD4 T cell differentiation in to T regulatory cells. Eur J Immunol. (2015) 44:3429–38. doi: 10.1002/eji.201344424
148. Cope AP, Patel SD, Hall F, Congia M, Hubers HAJM, Verheijden GF, et al. T cell responses to a human cartilage autoantigen in the context of rheumatoid arthritis – associated and nonassociated Hla – Dr4 alleles. Arthritis Rheum. (1999) 42:1497–507.
149. Hill JA, Bell DA, Brintnell W, Yue D, Wehrli B, Jevnikar AM, et al. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J Exp Med. (2008) 205:967–79. doi: 10.1084/jem.20072051
150. Snir O, Rieck M, Gebe JA, Yue BB, Rawlings CA, Nepom G, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. (2011) 63:2873–83. doi: 10.1002/art.30445
151. Batsalova T, Dzhambazov B, Merky P, Bäcklund A, Bäcklund J. Breaking T cell tolerance against self type II collagen in HLA-DR4-transgenic mice and development of autoimmune arthritis. Arthritis Rheum. (2010) 62:1911–20. doi: 10.1002/art.27460
152. Yue D, Brintnell W, Mannik LA, Christie DA, Haeryfar SM, Madrenas J, et al. CTLA-4Ig blocks the development and progression of citrullinated fibrinogen-induced arthritis in DR4-transgenic mice. Arthritis Rheum. (2010) 62:2941–52. doi: 10.1002/art.27597
153. Lac P, Saunders S, Tutunea-Fatan E, Barra L, Bell DA, Cairns E. Immune responses to peptides containing homocitrulline or citrulline in the DR4-transgenic mouse model of rheumatoid arthritis. J Autoimmun. (2018) 89:75–81. doi: 10.1016/j.jaut.2017.12.002
154. Svendsen P, Andersen CB, Willcox N, Coyle AJ, Holmdahl R, Kamradt T, et al. Tracking of proinflammatory collagen-specific T cells in early and late collagen-induced arthritis in humanized mice. J Immunol. (2004)173:7037–45. doi: 10.4049/jimmunol.173.11.7037
155. Latham KA, Whittington KB, Zhou R, Qian Z, Rosloniec EF. Ex vivo Characterization of the autoimmune t cell response in the hla-dr1 mouse model of collagen-induced arthritis reveals long-term activation of type ii collagen-specific cells and their presence in arthritic joints. J Immunol. (2005) 174:3978–85. doi: 10.4049/jimmunol.174.7.3978
156. Miller DC, Whittington KB, Brand DD, Hasty KA, Rosloniec EF. The CII-specific autoimmune T-cell response develops in the presence of FTY720 but is regulated by enhanced treg cells that inhibit the development of autoimmune arthritis. Arthritis Res Ther. (2016) 18:1–16. doi: 10.1186/s13075-015-0909-6
157. Qian Z, Latham KA, Whittington KB, Miller DC, Brand DD, Rosloniec EF. An autoantigen-specific, highly restricted T cell repertoire infiltrates the arthritic joints of mice in an HLA-DR1 humanized mouse model of autoimmune arthritis. J Immunol. (2010) 185:110–8. doi: 10.4049/jimmunol.1000416
158. Spranger S, Frankenberger B, Schendel DJ. NOD/scid IL-2Rgnull mice: a preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J Transl Med. (2012) 10:30. doi: 10.1186/1479-5876-10-30
159. Chang NH, Inman RD, Dick JE, Wither JE. Bone marrow-derived human hematopoietic stem cells engraft NOD/SCID mice and traffic appropriately to an inflammatory stimulus in the joint. J Rheumatol. (2010) 37:496–502. doi: 10.3899/jrheum.090317
160. Jespersen H, Lindberg MF, Donia M, Söderberg EMV, Andersen R, Keller U, et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun. (2017) 8:707. doi: 10.1038/s41467-017-00786-z
161. Locafaro G, Andolfi G, Russo F, Cesana L, Spinelli A, Camisa B, et al. IL-10-engineered human CD4+Tr1 cells eliminate myeloid leukemia in an HLA class I-dependent mechanism. Mol Ther. (2017) 25:2254–69. doi: 10.1016/j.ymthe.2017.06.029
162. Pham PV, Le HT, Vu BT, Pham VQ, Le PM, Phan NL, et al. Targeting breast cancer stem cells by dendritic cell vaccination in humanized mice with breast tumor: preliminary results. OncoTargets Ther. (2016) 9:4441–51. doi: 10.2147/OTT.S105239
163. Salguero G, Daenthanasanmak A, Münz C, Raykova A, Guzmán CA, Riese P, et al. Dendritic cell-mediated immune humanization of mice: implications for allogeneic and xenogeneic stem cell transplantation. J Immunol. (2014) 192:4636–47. doi: 10.4049/jimmunol.1302887
164. Volk V, Reppas AI, Robert PA, Spineli LM, Sundarasetty BS, Theobald SJ, et al. Multidimensional analysis integrating human T-cell signatures in lymphatic tissues with sex of humanized mice for prediction of responses after dendritic cell immunization. Front Immunol. (2017) 8:1709. doi: 10.3389/fimmu.2017.01709
165. Meixlsperger S, Leung CS, Rämer PC, Pack M, Vanoaica LD, Breton G, et al. CD141+dendritic cells produce prominent amounts of IFN-α after DsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood (2013) 121:5034–44. doi: 10.1182/blood-2012-12-473413
166. Issa F, Hester J, Goto R, Nadig S, Goodacre T. Ex vivo -expanded human regulatory T cells prevent the rejection of skin allografts in a humanised mouse model 1. Transplantation (2013) 90:1321–27. doi: 10.1097/TP.0b013e3181ff8772
167. Bézie S, Meistermann D, Boucault L, Kilens S, Zoppi J, Autrusseau E, et al. Ex vivo expanded human non-cytotoxic CD8+ CD45rclow/– tregs efficiently delay skin graft rejection and GVhD in humanized mice. Front Immunol. (2018) 8:2014. doi: 10.3389/fimmu.2017.02014
168. Landman S, de Oliveira VL, van Erp PEJ, Fasse E, Bauland SCG, Joosten I, et al. Intradermal injection of low dose human regulatory T cells inhibits skin inflammation in a humanized mouse model. Sci Rep. 8:10044. doi: 10.1038/s41598-018-28346-5
169. Yi S, Ji M, Wu J, Ma X, Phillips P, Hawthorne W, et al. Adoptive transfer with in vitro expanded human regulatory T cells protects against porcine islet xenograft rejection via interleukin-10 in humanized mice. Diabetes (2012) 61:1180–91. doi: 10.2337/db11-1306
170. Wu DC, Hester J, Nadig SN, Zhang W, Trzonkowski P, Gray D, et al. Ex vivo expanded human regulatory T cells can prolong survival of a human islet allograft in a humanized mouse model. Transplantation (2013) 96:707–16. doi: 10.1097/TP.0b013e31829fa271
171. Kotzin BL, Falta MT, Crawford F, Rosloniec EF, Bill J, Marrack P, et al. Use of soluble peptide-DR4 tetramers to detect synovial t cells specific for cartilage antigens in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. (2000) 97:291–6. doi: 10.1073/pnas.97.1.291
172. Myers LK, Sakurai Y, Tang B, He X, Rosloniec EF, Stuart JM, et al. Peptide-induced suppression of collagen-induced arthritis in HLA-DR1 transgenic mice. Arthritis Rheum. (2002) 46:3369–77. doi: 10.1002/art.10687
173. Sakurai Y, Brand DD, Tang B, Rosloniec EF, Stuart JM, Kang AH, et al. Analog peptides of type II collagen can suppress arthritis in HLA-DR4 (DRB1*0401) transgenic mice. Arthritis Res Ther. (2006) 8:1–7. doi: 10.1186/ar2043
174. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. (1991) 174:561–9. doi: 10.1084/jem.174.3.561
175. Cheng S, Baisch J, Krco C, Savarirayan S, Hanson J, Hodgson K, et al. Expression and function of HLA-DQ8 (DQA1*0301/DQB1*0302) genes in transgenic mice. Eur J Immunogenet. (1996). 23:15–20.
176. Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol. (2013) 190:513–9. doi: 10.4049/jimmunol.1201891
177. van der Horst-Bruinsma IE, Visser H, Hazes JM, Breedveld FC, Verduyn W, Schreuder GM, et al. HLA-DQ-associated predisposition to and dominant HLA-DR-associated protection against rheumatoid arthritis. Hum Immunol. (1999) 60:152–8. doi: 10.1016/S0198-8859(98)00101-3
178. Milicic A, Lee D, Brown MA, Darke C, Wordsworth BP. HLA-DR/DQ haplotype in rheumatoid arthritis: novel allelic associations in UK Caucasians. J Rheumatol. (2002) 29:1821–6. Available online at: http://www.jrheum.org/content/29/9/1821.long
179. Nabozny GH, Baisch JM, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, et al. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. (1996) 183:27–37. doi: 10.1084/jem.183.1.27
180. Bradley DS, Nabozny GH, Cheng S, Zhou P, Griffiths MM, Luthra HS, et al. HLA-DQB1 polymorphism determines incidence, onset, and severity of collagen-induced arthritis in transgenic mice: implications in human rheumatoid arthritis. J Clin Investig. (1997) 100:2227–34. doi: 10.1172/JCI119760
181. Taneja V, Taneja N, Paisansinsup T, Behrens M, Griffiths M, Luthra H, et al. CD4 and CD8 T cells in susceptibility/protection to collagen-induced arthritis in HLA-DQ8-transgenic mice: implications for rheumatoid arthritis. J Immunol. (2002) 168:5867–75. doi: 10.4049/jimmunol.168.11.5867
182. Taneja V, Behrens M, Basal E, Sparks J, Griffiths MM, Luthra H, et al. Delineating the role of the HLA-DR4 ‘shared epitope' in susceptibility versus resistance to develop arthritis. J Immunol. (2008) 181:2869–77. doi: 10.4049/jimmunol.181.4.2869
183. Vassallo R, Luckey D, Behrens M, Madden B, Luthra H, David C, et al. Cellular and humoral immunity in arthritis are profoundly influenced by the interaction between cigarette smoke effects and host HLA-DR and DQ genes. Clin Immunol. (2014) 152:25–35. doi: 10.1016/j.clim.2014.02.002
184. Bidkar M, Vassallo R, Luckey D, Smart M, Mouapi K, Taneja V. Cigarette smoke induces immune responses to vimentin in both, arthritis-susceptible and -resistant humanized mice. PLoS ONE (2016) 11:1–15. doi: 10.1371/journal.pone.0162341
185. Sandal I, Karydis A, Luo J, Prislovsky A, Whittington KB, Rosloniec EF, et al. Bone loss and aggravated autoimmune arthritis in HLA-DRβ1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res Ther. (2016) 18:1–13. doi: 10.1186/s13075-016-1143-6
186. Malmström V, Ho KK, Lun J, Tam PP, Cheah KS, Holmdahl R. Arthritis susceptibility in mice expressing human type II collagen in cartilage. Scand J Immunol. (1997) 45:670–7.
187. Inoue M, Senju S, Hirata S, Irie A, Baba H, Nishimura Y. An in vivo model of priming of antigen-specific human CTL by Mo-DC in NOD/Shi-Scid IL2rγnull (NOG) mice. Immunol Lett. (2009) 126:67–72. doi: 10.1016/j.imlet.2009.08.001
188. Harui A, Kiertscher SM, Roth MD. Reconstitution of HuPBL-NSG mice with donor-matched dendritic cells enables antigen-specific T-cell activation. J Neuroimmune Pharmacol. (2011) 6:148–57. doi: 10.1007/s11481-010-9223-x
189. Michaëlsson E, Andersson M, Engström A, Holmdahl R. Identification of an immunodominant type-II collagen peptide recognized by T cells in H-2qmice: self tolerance at the level of determinant selection. Eur J Immunol. (1992) 22:1819–25. doi: 10.1002/eji.1830220722
190. Bäcklund J, Carlsen S, Höger T, Holm B, Fugger L, Kihlberg J, et al. Predominant selection of T cells specific for the glycosylated collagen type II epitope (263–270) in humanized transgenic mice and in rheumatoid arthritis. Proc Natl Acad Sci USA. (2002) 99:9960–5. doi: 10.1073/pnas.132254199
191. Hultqvist M, Bäcklund J, Bauer K, Gelderman KA, Holmdahl R. Lack of reactive oxygen species breaks T cell tolerance to collagen type, I. I, and allows development of arthritis in mice. J Immunol. (2007) 179:1431–7. doi: 10.4049/jimmunol.179.3.1431
192. Brand DD, Myers LK, Whittington KB, Latham KA, Stuart JM, Kang AH, et al. Detection of early changes in autoimmune T cell phenotype and function following intravenous administration of type II collagen in a TCR-transgenic model. J Immunol. (2002) 168:490–8. doi: 10.4049/jimmunol.168.1.490
193. Berlo SE, Guichelaar T, Ten Brink CB, van Kooten PJ, Hauet-Broeren F, Ludanyi K, et al. Increased arthritis susceptibility in cartilage proteoglycan-specific T cell receptor-transgenic mice. Arthritis Rheum. (2006) 54:2423–33. doi: 10.1002/art.22013
194. Osman GE, Cheunsuk S, Allen SE, Chi E, Liggitt HD, Hood LE, et al. Expression of a type II collagen-specific TCR transgene accelerates the onset of arthritis in mice. Int Immunol. (1998). 10:1613–22. doi: 10.1093/intimm/10.11.1613
195. Asnagli H, Martire D, Belmonte N, Quentin J, Bastian H, Boucard-Jourdin M, et al. Type 1 regulatory T cells specific for collagen type II as an efficient cell-based therapy in arthritis. Arthritis Res Ther. (2014) 16:R115. doi: 10.1186/ar4567
196. Tang B, Kim S, Hammond S, Cullins DL, Brand DD, Rosloniec EF, et al. Characterization of T cell phenotype and function in a double transgenic (collagen-specific TCR/HLA-DR1) humanized model of arthritis. Arthritis Res Ther. (2014) 16:1–13. doi: 10.1186/ar4433
197. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983). 301:527–30. doi: 10.1038/301527a0
198. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature (1988) 335:256–9. doi: 10.1038/335256a0
199. Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science (1992) 255:1137–41.
200. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-Hu mouse: murine model for the analysis of human hematolymphoid differentiation and function, (severe combined immunodeficiency). Science (1988) 143:1632–9. Available online at https://www.highbeam.com/doc/1G1-6698154.html
201. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell (1992) 68:869–77. doi: 10.1016/0092-8674(92)90030-G
202. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell (1992) 68:855–67. doi: 10.1016/0092-8674(92)90029-C
203. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Tennent B, Mckenna S, et al. Multiple defects in innate and adaptive immunologic function in NOD / LtSz-scid mice. J Immunol. (1995) 154:180–91.
204. Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J. Immunol. (1993) 150:2534–43. http://www.jimmunol.org/content/150/6/2534.abstract
205. Lowry PA, Shultz LD, Greiner DL, Hesselton RM, Kittler EL, Tiarks CY, et al. Improved engraftment of human cord blood stem cells in NOD/LtSz-Scid/Scid mice after irradiation or multiple-day injections into unirradiated recipients (1996) Biol Blood Marrow Transplant. 2:15–23.
206. Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-Scid/Scid Mice. J Infect Dis. (1995) 172:974–82. doi: 10.1093/infdis/172.4.974
207. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD / SCID / IL2 receptor γ chain null mice. Blood (2005) 106:1565–73. doi: 10.1182/blood-2005-02-0516
208. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz- Scid IL2R γ null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. (2005) 174:6477–89. doi: 10.4049/jimmunol.174.10.6477
209. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/γ formula mouse: an excellent recipient mouse model for engraftment of human cells. Blood (2002) 100:3175–82. doi: 10.1182/blood-2001-12-0207
210. Yahata T, Ando K, Nakamura Y, Ueyama Y, Shimamura K, Tamaoki N, et al. Functional human T lymphocyte development from cord blood CD34 + cells in nonobese diabetic/Shi- Scid , IL-2 receptor γ null mice. J Immunol. (2002) 169:204–9. doi: 10.4049/jimmunol.169.1.204
211. Liao W, Lin JX, Leonard WJ. IL-2 Family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. (2011) 23:598–604. doi: 10.1016/j.coi.2011.08.003
212. Cao X, Shores EW, Hu-Li J, Anver MR, Kelsail BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity (1995) 2:223–38. doi: 10.1016/1074-7613(95)90047-0
213. DiSanto JP, Müller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci USA. (1995) 92:377–81. doi: 10.1073/pnas.92.2.377
214. Brehm MA, Cuthbert A, Yang C, Miller D, Diiorio P, Laning J, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the Il2r γ null mutation. Clin Immunol. (2010) 135:84–98. doi: 10.1016/j.clim.2009.12.008
215. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG. Development of a human adaptive immune system in cord blood cell–transplanted mice. Science (2004) 304:104–7. doi: 10.1126/science.1093933
216. King M, Pearson T, Shultz LD, Leif J, Bottino R, Trucco M, et al. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin Immunol. (2008) 126:303–14. doi: 10.1016/j.clim.2007.11.001
217. Watanabe S, Ohta S, Yajima M, Terashima K, Ito M, Mugishima H, et al. Humanized NOD/SCID/IL2R γ null mice transplanted with hematopoietic stem cells under nonmyeloablative conditions show prolonged life spans and allow detailed analysis of human immunodeficiency virus type 1 pathogenesis. J Virol. (2007) 81:13259–64. doi: 10.1128/JVI.01353-07
218. Hiramatsu H, Nishikomori R, Heike T, Ito M, Kobayashi K, Katamura K, et al. Complete reconstitution of human lymphocytes from cord blood CD34+ cells using the NOD/SCID/gammacnull mice model. Blood (2003) 102:873–80. doi: 10.1182/blood-2002-09-2755
219. Lepus CM, Gibson TF, Gerber SA, Kawikova I, Szczepanik M, Hossain J, et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-Scid/Gc-/-, Balb/c-Rag1-/- Gc-/-, and C.B-17-Scid/Bg immunodeficient mice. Hum Immunol. (2009) 70:790–802. doi: 10.1016/j.humimm.2009.06.005
220. Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus / liver and CD34+ cell transplantation. Blood (2006) 108:487–92. doi: 10.1182/blood-2005-11-4388
221. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. (2006) 12:1316–22. doi: 10.1038/nm1431
222. Tonomura N, Habiro K, Shimizu A, Sykes M, Yong-guang Y. Brief report antigen-specific human T-cell responses and T cell–dependent production of human antibodies in a humanized mouse model. Blood (2008) 111:4293–97. doi: 10.1182/blood-2007-11-121319
223. Manz MG. Commentary human-hemato-lymphoid-system mice : opportunities and challenges. Immunity (2007) 26:537–41. doi: 10.1016/j.immuni.2007.05.001
224. Schmidt MR, Appel MC, Giassi LJ, Greiner DL, Shultz LD, Woodland RT. Human BLyS facilitates engraftment of human PBL derived B cells in immunodeficient mice. PLoS ONE (2008) 3:e3192. doi: 10.1371/journal.pone.0003192
225. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood (2017) 129:959–69. doi: 10.1182/blood-2016-04-709584
226. Rongvaux A, Willinger T, Takizawa H, Rathinam C, Auerbach W. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc Natl Acad Sci USA. (2011) 108:2378–83. doi: 10.1073/pnas.1019524108.
227. Ding Y, Wilkinson A, Idris A, Fancke B, O'Keeffe M, Khalil D, et al. FLT3-ligand treatment of humanized mice results in the generation of large numbers of CD141 + and CD1c + dendritic cells in vivo. J Immunol. (2014) 192:1982–9. doi: 10.4049/jimmunol.1302391
228. Billerbeck E, Labitt RN, Vega K, Frias-Staheli N, Dorner M, Xiao JW, et al. Insufficient interleukin-12 signalling favours differentiation of human CD4 + and CD8 + T cells into GATA-3 + and GATA-3 + T-Bet + subsets in humanized mice. Immunology (2014) 143:202–18. doi: 10.1111/imm.12304
229. Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc Natl Acad Sci USA. (2011) 108:2390–5. doi: 10.1073/pnas.1019682108
230. Greenblatt MB, Vrbanac V, Tivey T, Tsang K, Tager AM, Aliprantis AO. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE (2012) 7:e44664. doi: 10.1371/journal.pone.0044664
231. Lockridge JL, Zhou Y, Becker YA, Ma S, Kenney SC, Hematti P, et al. Hematopoietic stem cells develop pathology resembling chronic graft-versus-host disease. Biol Blood Marrow Transplant. (2013) 19:1310–22. doi: 10.1016/j.bbmt.2013.06.007
232. van Rijn RS, Simonetti ER, Hagenbeek A, Hogenes MCH, de Weger R, Canninga-van Dijk MR, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2-/- gammac-/- double-mutant mice. Blood (2003) 102:2522–31. doi: 10.1182/blood-2002-10-3241
233. Ishikawa Y, Usui T, Shiomi A, Shimizu M, Murakami K, Mimori T. Functional engraftment of human peripheral T and B cells and sustained production of autoantibodies in NOD/LtSzscid/IL-2Rγ-/-Mice. Eur J Immunol. (2014) 44:3453–63. doi: 10.1002/eji.201444729
234. Mutis T, Van Rijn RS, Simonetti ER, Aarts-Riemens T, Emmelot ME, Van Bloois L, et al. Cancer therapy : preclinical human regulatory t cells control xenogeneic graft-versus-host disease induced by autologousT cells in RAG2-/-gammac-/- immunodeficient mice. Clin Cancer Res. (2006) 12:5520–6. doi: 10.1158/1078-0432.CCR-06-0035
235. Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, et al. The analysis of the functions of human B and T cells in humanized NOD/Shi-Scid/g c null (NOG) Mice (Hu-HSC NOG mice). Int Immunol. (2009) 21:843–58. doi: 10.1093/intimm/dxp050
236. Lee J, Brehm MA, Greiner D, Shultz LD, Kornfeld H. Engrafted human cells generate adaptive immune responses to Mycobacterium Bovis BCG infection in humanized mice. BMC Immunol. (2013) 14:53. doi: 10.1186/1471-2172-14-53
237. Danner R, Chaudhari SN, Rosenberger J, Surls J, Richie TL, Brumeanu TD, et al. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B Cells. PLoS ONE (2011) 6:e19826. doi: 10.1371/journal.pone.0019826
238. Pino S, Brehm MA, Covassin-Barberis L, King M, Gott B, Chase TH, et al. Development of novel major histocompatibility complex class I and class II-deficient NOD-SCID IL2R gamma chain knockout mice for modeling human xenogeneic graft-versus-host disease steve. Methods Mol Biol. (2010) 602:105–17. doi: 10.1007/978-1-60761-058-8_7
239. Covassin L, Laning J, Abdi R, Langevin DL, Phillips NE, Shultz LD, et al. Human peripheral blood CD4 T cell-engrafted non-obese diabetic-Scid IL2rγnullH2-Ab1tm1GruTg (human leucocyte antigen d-related 4) mice: a mouse model of human allogeneic graft-versus-host disease. Clin. Exp. Immunol. (2011) 166:269–80. doi: 10.1111/j.1365-2249.2011.04462.x
240. Ali R, Babad J, Follenzi A, Gebe JA, Brehm MA, Nepom GT, et al. Genetically modified human CD4 + T cells can be evaluated in vivo without lethal graft-versus-host disease. Immunology (2016) 148:339–51. doi: 10.1111/imm.12613
241. Wege AK. Humanized mouse models for the preclinical assessment of cancer immunotherapy. BioDrugs (2018) 32:245–66. doi: 10.1007/s40259-018-0275-4
242. Van Duyne R, Pedati C, Guendel I, Carpio L, Kehn-Hall K, Saifuddin M, et al. The Utilization of humanized mouse models for the study of human retroviral infections. Retrovirology (2009) 6:76. doi: 10.1186/1742-4690-6-76
243. Whitfield-Larry F, Young EF, Talmage G, Fudge E, Azam A, Patel S, et al. HLA-A2 – Matched peripheral blood mononuclear cells from type 1 diabetic patients, but not nondiabetic donors, transfer insulitis to NOD- Scid/Gcnull/HLA-A2 transgenic mice concurrent with the expansion of islet-specific CD8+ T cells. Diabetes (2011) 60:1726–33. doi: 10.2337/db10-1287
244. Sonntag K, Eckert F, Welker C, Müller H, Müller F, Zips D, et al. Chronic graft-versus-host-disease in CD34+-humanized NSG mice is associated with human susceptibility HLA haplotypes for autoimmune disease. J Autoimmun. (2015) 62:55–66. doi: 10.1016/j.jaut.2015.06.006
245. Goettel JA, Biswas S, Lexmond WS, Yeste A, Passerini L, Patel B, et al. Fatal autoimmunity in mice reconstituted with human hematopoietic stem cells encoding defective FOXP3. Blood (2015) 125:3886–95. doi: 10.1182/blood-2014-12-618363
246. Vudattu NK, Waldron-Lynch F, Truman LA, Deng S, Preston-Hurlburt P, Torres R, et al. Humanized mice as a model for aberrant responses in human t cell immunotherapy. J Immunol. (2014) 193:587–96. doi: 10.4049/jimmunol.1302455
247. Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci USA. (2017) 114:10954–9. doi: 10.1073/pnas.1710415114
248. Young N, Wu LC, Bruss M, Kaffenberger BH, Hampton J, Bolon B, et al. A chimeric human – mouse model of sjögren ' s syndrome. Clin Immunol. (2015) 156:1–8. doi: 10.1016/j.clim.2014.10.004
249. Andrade D, Redecha PB, Vukelic M, Qing X, Perino G, Salmon JE, et al. Engraftment of peripheral blood mononuclear cells from systemic lupus erythematosus and antiphospholipid syndrome patient donors into BALB-RAG-2−/− IL-2Rγ−/− mice a promising model for studying human disease. Arthritis Rheum. (2011) 63:2764–73. doi: 10.1002/art.30424
250. Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, et al. A humanized mouse model of autoimmune insulitis. Diabetes (2014) 63:1712–24. doi: 10.2337/db13-1141
251. Rendt KE, Barry TS, Jones DM, Richter CB, McCachren SS, Haynes BF. Engraftment of human synovium into severe combined immune deficient mice. J Immunol. (1993) 151:7324–36.
252. Connolly M, Marrelli A, Blades M, McCormick J, Maderna P, Godson C, et al. Acute serum amyloid a induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/scid mouse chimera model. J Immunol. (2010) 184:6427–37. doi: 10.4049/jimmunol.0902941
253. Müller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. (1996) 149:1607–15.
254. Lefèvre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. (2009) 15:1414–20. doi: 10.1038/nm.2050
255. Tighe H, Silverman GJ, Kozin F, Tucker R, Gulizia R, Peebles C, et al. Autoantibody production by severe combined immunodeficient (SCID) mice reconstituted with synovial cells from rheumatoid arthritis patients. Eur J Immunol. (1990) 20:1843–8. doi: 10.1002/eji.1830200832
256. Sakata A, Sakata K, Ping H, Ohmura T, Tsukano M, Kakimoto K. Successful induction of severe destructive arthritis by the transfer of in vitro-activated synovial fluid T cells from patients with Rheumatoid Arthritis (RA) in Severe Combined Immunodeficient (SCID) mice. Clin Exp Immunol. (1996) 104:247–54. doi: 10.1046/j.1365-2249.1996.979670.x
257. Kuwana Y, Takei M, Yajima M, Imadome K, Inomata H, Shiozaki M, et al. Epstein-barr virus induces erosive arthritis in humanized mice. PLoS ONE (2011) 6:e26630. doi: 10.1371/journal.pone.0026630
258. Misharin AV, Haines GK, Rose S, Gierut AK, Hotchkiss RS, Perlman H. Development of a new humanized mouse model to study acute inflammatory arthritis. J Transl Med. 10:190. doi: 10.1186/1479-5876-10-190
259. Wahid S, Blades MC, De Lord D, Brown I, Blake G, Yanni G, et al. Tumour necrosis factor-alpha (TNF-Alpha) enhances lymphocyte migration into rheumatoid synovial tissue transplanted into severe combined immunodeficient (SCID) mice. Clin Exp Immunol. (2000) 122:133–42. doi: 10.1046/j.1365-2249.2000.01342.x
260. Davis LS, Sackler M, Brezinschek RI, Lightfoot E, Bailey JL, Oppenheimer-Marks N, et al. Inflammation, immune reactivity, and angiogenesis in a severe combined immunodeficiency model of rheumatoid arthritis. Am J Pathol. (2002) 160:357–67. doi: 10.1016/S0002-9440(10)64379-9
261. Jorgensen C, Apparailly F, Couret I, Canovas F, Jacquet C, Sany J. Interleukin-4 and interleukin-10 are chondroprotective and decrease mononuclear cell recruitment in human rheumatoid synovium in vivo. Immunology (1998) 93:518–23. doi: 10.1046/j.1365-2567.1998.00457.x
262. Geiler T, Kriegsmann J, Keyszer GM, Gay RE, Gay S. A new model for rheumatoid arthritis generated by engraftment of rheumatoid synovial tissue and normal human cartilage into scid mice. Arthritis Rheumat. (1994) 37:1664–71. doi: 10.1002/art.1780371116
263. Sack U, Kuhn H, Ermann J, Kinne RW, Vogt S, Jungmichel D, et al. Synovial tissue implants from patients with rheumatoid arthritis cause cartilage destruction in knee joints of SCID.bg mice. J Rheumatol. (1994) 21:10–6.
264. Schädlich H, Ermann J, Biskop M, Falk W, Sperling F, Jüngel A, et al. Anti-inflammatory effects of systemic anti-tumour necrosis factor alpha treatment in human/murine SCID arthritis. Ann. Rheum. Dis. (1999) 58:428–34. doi: 10.1136/ard.58.7.428
265. Matsuno H, Yoshida K, Ochiai A, Okamoto M. Requirement of methotrexate in combination with anti-tumor necrosis factor-α therapy for adequate suppression of osteoclastogenesis in rheumatoid arthritis. J. Rheumatol. (2007) 34:2326–33. Available online at: http://www.jrheum.org/content/34/12/2326.long
266. Koenders MI, Marijnissen RJ, Joosten LA, Abdollahi-Roodsaz S, Di Padova FE, van de Loo FA, et al. T cell lessons from the rheumatoid arthritis synovium SCID mouse model: CD3-rich synovium lacks response to CTLA-4ig but is successfully treated by interleukin-17 neutralization. Arthritis Rheum. (2012) 64:1762–70. doi: 10.1002/art.34352
267. Jorgensen C, Couret I, Canovas F, Bologna C, Brochier J, Reme T, et al. Mononuclear cell retention in rheumatoid synovial tissue engrafted in Severe Combined Immunodeficient (SCID) mice is up-regulated by tumour necrosis factor-alpha (TNF-Alpha) and mediated through Intercellular Adhesion Molecule-1 (ICAM-1). Clin Exp Immunol. (1996) 106:20–5.
268. Oppenheimer-Marks N, Brezinschek RI, Mohamadzadeh M, Vita R, Lipsky PE. Interleukin 15 is produced by endothelial cells and increases the transendothelial migration of T cells in vitro and in the SCID mouse- human rheumatoid arthritis model in vivo. J Clin Invest. (1998) 101:1261–72. doi: 10.1172/JCI1986
269. Proudman SM, Cleland LG, Mayrhofer G. Effects of tumor necrosis factor-alpha, interleukin 1beta, and activated peripheral blood mononuclear cells on the expression of adhesion molecules and recruitment of leukocytes in rheumatoid synovial xenografts in SCID mice. J Rheumatol. (1999) 26:1877–89.
270. Klimiuk PA, Yang H, Goronzy JJ, Weyand CM. Production of cytokines and metalloproteinases in rheumatoid synovitis is T cell dependent. Clin Immunol. (1999) 90:65–78. doi: 10.1006/clim.1998.4618
271. Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. (2001) 167:4710–8. doi: 10.4049/jimmunol.167.8.4710
272. Nakazawa F, Matsuno H, Yudoh K, Katayama R, Sawai T, Uzuki M, et al. Methotrexate inhibits rheumatoid synovitis by inducing apoptosis. J Rheumatol. (2001) 28:1800–8. Available online at: http://www.jrheum.org/content/28/8/1800.long
273. Matsuno H, Sawai T, Nezuka T, Uzuki M, Tsuji H, Nishimoto N, et al. Treatment of rheumatoid synovitis with anti-reshaping human interleukin-6 receptor monoclonal antibody: use of rheumatoid arthritis tissue implants in the SCID mouse model. Arthritis Rheum. (1998) 41:2014–21.
274. Jia J, Wang C, Shi Z, Zhao J, Jia Y, Zhao-Hui Z, et al. Inhibitory effect of CD147/HAb18 monoclonal antibody on cartilage erosion and synovitis in the SCID mouse model for rheumatoid arthritis. Rheumatology (2009) 48:721–6. doi: 10.1093/rheumatology/kep099
275. Matsuno H, Yudoh K, Nakazawa F, Sawai T, Uzuki M, Nishioka K, et al. Antirheumatic effects of humanized anti-fas monoclonal antibody in human rheumatoid arthritis/SCID mouse chimera. J Rheumatol. (2002) 29:1609–14. doi: 10.1186/ar930
276. Matsuno H, Yudoh K, Katayama R, Nakazawa F, Uzuki M, Sawai T, et al. The role of TNF-α in the pathogenesis of inflammation and joint destruction in Rheumatoid Arthritis (RA): a study using a human RA/SCID mouse chimera. Rheumatology (2002) 41:329–37. doi: 10.1093/rheumatology/41.3.329
277. Seemayer CA, Kuchen S, Kuenzler P, Rihošková V, and Rethage J, Aicher WK, et al. Cartilage destruction mediated by synovial fibroblasts does not depend on proliferation in rheumatoid arthritis. Am J Pathol. (2003) 162:1549–57. doi: 10.1016/S0002-9440(10)64289-7
278. O'Boyle G, Fox CR, Walden HR, Willet JD, Mavin ER, Hine DW, et al. Chemokine receptor CXCR3 agonist prevents human T-cell migration in a humanized model of arthritic inflammation. Proc Natl Acad Sci USA. (2012) 109:4598–603. doi: 10.1073/pnas.1118104109
279. Xiao F, Ma L, Zhao M, Huang G, Mirenda V, Dorling A, et al. Ex vivo expanded human regulatory T cells delay islet allograft rejection via inhibiting islet-derived monocyte chemoattractant protein-1 production in CD34+ stem cells-reconstituted NOD-Scid IL2rγnull mice. PLoS ONE (2014) 9:e90387. doi: 10.1371/journal.pone.0090387
280. Martin H, Reuter S, Dehzad N, Heinz A, Bellinghausen I, Saloga J, et al. CD4-Mediated regulatory T-cell activation inhibits the development of disease in a humanized mouse model of allergic airway disease. J Allergy Clin Immunol. (2012) 129 521–8, 528.e1–7. doi: 10.1016/j.jaci.2011.09.038
281. Meyer-Martin H, Hahn SA, Beckert H, Belz C, Heinz A, Jonuleit H, et al. GARP inhibits allergic airway inflammation in a humanized mouse model. Allergy (2016) 71:1274–83. doi: 10.1111/all.12883
282. Eschborn M, Weigmann B, Reissig S, Waisman A, Saloga J, Bellinghausen I. Activated Glycoprotein A Repetitions Predominant (GARP)-Expressing regulatory T cells inhibit allergen-induced intestinal inflammation in humanized mice. J Allergy Clin Immunol. (2015) 136:159–68. doi: 10.1016/j.jaci.2015.04.020
283. Koboziev I, Jones-Hall Y, Valentine JF, Webb CR, Furr KL, Grisham MB. Use of humanized mice to study the pathogenesis of autoimmune and inflammatory diseases. Inflamm Bowel Dis. (2015) 21:1652–73. doi: 10.1097/MIB.0000000000000446
284. Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Ann Rev. Pathol. (2017) 24:187–215. doi: 10.1146/annurev-pathol-052016-100332
285. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature (2016) 530:434–40. doi: 10.1038/nature16962
286. Serr I, Fürst RW, Achenbach P, Scherm MG, Gökmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3+Treg induction in humanized mice. Nat Commun. (2016) 7:10991. doi: 10.1038/ncomms10991
287. Schlöder J, Berges C, Luessi F, Jonuleit H. Dimethyl fumarate therapy significantly improves the responsiveness of T cells in multiple sclerosis patients for immunoregulation by regulatory T cells. Int J Mol Sci. (2017) 18:e271. doi: 10.3390/ijms18020271
288. Sudres M, Maurer M, Robinet M, Bismuth J, Truffault F, Girard D, et al. Preconditioned mesenchymal stem cells treat myasthenia gravis in a humanized preclinical model. JCI Insight (2017) 2:e89665. doi: 10.1172/jci.insight.89665
289. Kimata M, Cullins DL, Brown ML, Brand DD, Rosloniec EF, Myers LK, et al. Characterization of inhibitory T cells induced by an analog of type II collagen in an HLA-DR1 humanized mouse model of autoimmune arthritis. Arthritis Res Ther. (2012) 14:R107. doi: 10.1186/ar3832
290. Sundarasetty B, Volk V, Theobald SJ, Rittinghausen S, Schaudien D, Neuhaus V, et al. Human effector memory T helper cells engage with mouse macrophages and cause graft-versus-host–like pathology in skin of humanized mice used in a nonclinical immunization study. Am J Pathol. (2017) 187:1380–98. doi: 10.1016/j.ajpath.2017.02.015
291. Tary-Lehmann M, Lehmann PV, Schols D, Roncarolo MG, Saxon A. Anti-SCID mouse reactivity shapes the human CD4+ T cell repertoire in Hu-PBL-SCID chimeras. J Exp Med. (1994) 180:1817–27. doi: 10.1084/jem.180.5.1817
292. Presa M, Chen YG, Grier AE, Leiter EH, Brehm MA, Greiner DL, et al. The presence and preferential activation of regulatory t cells diminish adoptive transfer of autoimmune diabetes by polyclonal Nonobese Diabetic (NOD) T cell effectors into NSG versus NOD-scid mice. J Immunol. (2015) 195:3011–9. doi: 10.4049/jimmunol.1402446
Keywords: rheumatoid arthritis, humanized mice, transgenic mice, mouse/human chimera, preclinical model, cell-based immunotherapy
Citation: Schinnerling K, Rosas C, Soto L, Thomas R and Aguillón JC (2019) Humanized Mouse Models of Rheumatoid Arthritis for Studies on Immunopathogenesis and Preclinical Testing of Cell-Based Therapies. Front. Immunol. 10:203. doi: 10.3389/fimmu.2019.00203
Received: 30 October 2018; Accepted: 23 January 2019;
Published: 19 February 2019.
Edited by:
Djordje Miljkovic, University of Belgrade, SerbiaReviewed by:
Femke Broere, Utrecht University, NetherlandsCopyright © 2019 Schinnerling, Rosas, Soto, Thomas and Aguillón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katina Schinnerling, a2F0aW5hLnNjaGlubmVybGluZ0B1bmFiLmNs
Juan Carlos Aguillón, amFndWlsbG9AbWVkLnVjaGlsZS5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.