 Diana M. Elizondo1Nailah Z. D. Brandy1Ricardo L. L. da Silva1,2Naomi L. Haddock1,3Apollo D. Kacsinta4
Diana M. Elizondo1Nailah Z. D. Brandy1Ricardo L. L. da Silva1,2Naomi L. Haddock1,3Apollo D. Kacsinta4 Tatiana R. de Moura2
Tatiana R. de Moura2 Michael W. Lipscomb1*
Michael W. Lipscomb1*- 1Department of Biology, Howard University, Washington, DC, United States
- 2Laboratório de Imunologia e Biologia Molecular-Hospital Universitário, Universidade Federal de Sergipe, Aracaju, Brazil
- 3Immunology Program, Stanford University, Stanford, CA, United States
- 4Department of Cellular and Molecular Medicine, UCSD School of Medicine, La Jolla, CA, United States
The multistep differentiation process from hematopoietic stem cells through common myeloid progenitors into committed dendritic cell (DC) subsets remains to be fully addressed. These studies now show that Allograft Inflammatory Factor-1 (AIF1) is required for differentiation of classical DC type 1 (cDC1) subsets and monocyte-derived DC (Mo-DC). Phenotypic studies found that AIF1 expression increased in committed subsets differentiating from common myeloid progenitors (CMP). However, silencing AIF1 expression in hematopoietic stem progenitors restrained the capacity to differentiate into Mo-DC and cDC1 cell subsets under GM-CSF or Flt3-L stimuli conditions, respectively. This was further marked by restrained expression of IRF8, which is critical for development of Mo-DC and cDC1 subsets. As a result, absence of AIF1 restrained the cells at the Lin−CD117+FcγR−CD34+ CMP stage. Further biochemical studies revealed that abrogating AIF1 resulted in inhibition of the NFκB family member RelB expression and p38 MAPK phosphorylation during differentiation of Mo-DC. Lastly, protein binding studies identified that AIF1 interacts with protein kinase C (PKC) to influence downstream signaling pathways. Taken together, this is the first report showing a novel role of AIF1 as a calcium-responsive scaffold protein that supports IRF8 expression and interacts with PKC to drive NFκB-related RelB for successfully differentiating hematopoietic progenitor cells into cDC and Mo-DC subsets under Flt3-L and GM-CSF stimuli, respectively.
Introduction
Dendritic cells (DC) survey the microenvironment by capturing, processing, and presenting antigens to promote adaptive immunity (1). Hematopoietic stem cells (HSC) can be directed into either common myeloid progenitors (CMP) or common lymphoid progenitors (CLP) to give rise to diverse DC subsets (2). Furthermore, monocytes, macrophages and DC are all derived from the common monocyte-macrophage DC progenitor (MDP). Importantly, this subset can direct generation of the common DC progenitor (CDP), which can uniquely give rise to diverse DC subsets, but not to macrophages, monocytes or other granulocyte populations (3, 4). Alternatively, MDP can also direct fates toward common monocyte precursors (cMoP), which can differentially give rise to monocytes that, under diverse inflammatory stimuli, generate monocyte-derived DC (Mo-DC), or macrophages. Although DC developmental processes have been extensively studied (5), the underlying mechanisms involving other fate-determining genes remain to be delineated.
The differentiation pathway of DC subsets from hematopoietic progenitors can be driven by granulocyte macrophage-colony stimulatory factor (GM-CSF; Csf-2) (6) or Fms-Related Tyrosine Kinase 3-Ligand (Flt3-L) cytokines (7–9). In vivo and in vitro Flt3-L-derived subsets are further divided into classical (or conventional) dendritic cells (cDC), which can be distinguished by the specific expression of the Zbtb46 transcription factor (10), or plasmacytoid DC (pDC). CD8α+/CD24+/CD103+ cDC1 subsets are directed by IRF8 (11, 12), with concerted activities of the BATF3 transcription factor being required (13). Whereas, the CD11b+/SIRPα+ cDC2 are not dependent on IRF8, but still require IRF4 (14). Flt3-L stimulation in vitro can drive cDCs with profile and functions that resemble that of splenic subsets (15). GM-CSF has been shown to be crucial for promoting survival, proliferation, and homeostasis of non-lymphoid DC (6, 16). In vitro, it has been shown to expand a heterogeneous pool of Mo-DC and macrophages that expresses CD11c and MHC class II (6, 17), with Mo-DC being more proficient in cross-presenting soluble antigens than the generated macrophages (18). However, the in vivo cross-presentation role remains unclear with respect to antigen presentation and direction of cognate T cell responses.
The NFκB family signaling activities can directly regulate immune cell differentiation and responses. Activation of the NFκB pathway can be driven by the recruitment of protein kinase C (PKC) (19). Furthermore, the NFκB family member RelB directs DC development in vivo (20–22), albeit different from its observed role for in vitro generation (23). In addition to development, RelB is important in controlling MHC class II expression and maturation of DC (24). Importantly, RelB is directly suppressed by the activated states of IκB, which is an inhibitor of NF-κB proteins (24). From the MAPK pathway, the three most characterized members are ERK, JNK/SAPK, and p38 Kinase. Interestingly, p38 MAPK is important for regulating NFκB recruitment to nuclear targets (25).
Allograft Inflammatory Factor-1 (AIF1), also known as ionized-calcium binding adapter molecule 1 (Iba1), is a 17 kD interferon gamma-inducible calcium binding EF-hand protein (26). The gene has shown diverse roles in the nervous and immune systems (27, 28). In particular, AIF1 expression in activated macrophages, microglial cells and DC plays major immunomodulatory roles during inflammatory responses (26, 29, 30). Although the importance of AIF1 in antigen presentation by DC has been reported (29), no study has delineated its role in differentiation.
This report now shows that AIF1 expression in GM-CSF- or Flt3-L-stimulated hematopoietic progenitors is required for differentiation into Mo-DC and cDC1 subsets, respectively. Under Flt3-L stimuli, loss of AIF1 resulted in restrained IRF8, BATF3, RelB, and Zbtb46 expression, but not PU.1 or Id2. Interestingly, there was a greater ratio of observed cDC2 subsets. For Mo-DC, loss of AIF1 during differentiation under GM-CSF stimuli resulted in restrained CIITA, IRF8, IRF4, and RelB. Collectively, the studies revealed that absence of AIF1 alters in vitro differentiation of DC away from cDC1 and Mo-DC fates.
Materials and Methods
Animals
All animal procedures were performed in accordance and approved by the Institutional Animal Care and Use Committee. Mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in pathogen-free facilities at Howard University. C57BL/6 (wild type; WT) male and female mice 8–12 weeks of age were used as a source of bone marrow and spleen.
Generation of Monocyte-Derived Dendritic Cells (Mo-DC)
Mo-DC were generated as described by a modified protocol of Inaba et al. (17). Briefly, bone marrow cells from murine tibias and femurs were passed through a 70 μm nylon mesh to remove debris. Total isolated bone marrow cells were cultured in IMDM (Thermo Fisher; Grand Island NY) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM L-glutamine (Gibco), 100 U/mL penicillin/streptomycin (Gibco), and 20 ng/mL recombinant mouse GM-CSF (Peprotech; Rochy Hill NJ) for 7–8 days in culture.
Generation of Classical Dendritic Cells (cDC)
Briefly, bone marrow (BM) from murine tibias and femurs were passed through a 70 μm nylon mesh to remove debris. The isolated cells were treated with red blood lysis buffer. Total isolated bone marrow cells were then cultured in IMDM (Thermo Fisher; Grand Island NY) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM L-glutamine (Gibco), 100 U/mL penicillin/streptomycin (Gibco) 200 ng/mL of murine Flt3-L (Peprotech) for 8–9 days in culture.
Sorting of Lin−CD117+ Cells
Isolated bone marrow (BM) cells were incubated with a lineage (Lin) antibody cocktail conjugated to magnetic beads (Miltenyi, Auburn CA). The cocktail included: anti-mouse CD3t, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), and Ter119. After staining, cells were passed over a depletion column affixed to a magnet (Miltenyi). Flow through cells are lineage negative (Lin−). These Lin− cells were subsequently stained with CD117 antibody conjugated to magnetic microbeads (Miltenyi). After incubation, cells were passed through a collection column affixed to a magnet. The CD117 positive fraction was collected; these are the Lin−CD117+ cells.
siRNA and Electroporation
siRNA oligonucleotide (oligo) with the following sequence was used for AIF1 knockdown: 5′-CGUUCUCUAGACGGUAGAAC-3′ (siAIF1), as previously published by Elizondo et al. (29). Scrambled siRNA control oligo used was 5′-CCUAUAGAUACCGAGUGGUTT-3′. Total BM or Lin−CD117+ sorted cells were transfected with 1.5 nanomoles of siRNA on days 0, 2, and 4 of the DC differentiation process in 4 mm electroporation cuvettes using an ECM 830 square wave electroporator (Harvard Apparatus, Holliston MA) (31). The following settings were used: 310 V, 10 ms, 1 pulse.
CRISPR-Mediated Gene Silencing and Electroporation
The CRISPR Cas9 system was used to knockout AIF1 in Lin−CD117+ HSC or total BM. Two CRISPR DNA plasmids were created using the GeneArt CRISPR Nuclease Vector Kit (Thermo Fisher) to target AIF1. The gRNA sequences were: 5′-AGAGTAGCTGAACGTCTCCT-3′ (pAIF1-T3) and 5′-GCTGAAGAGATTAATTAGAG-3′ (pAIF1-T4). Control plasmids contained scrambled targeting sequences (pControl). Plasmids were purified using PureLink™ HiPure Plasmid Maxiprep Kit (Thermo Fisher) and cleaned with the MiraCLEAN® Endotoxin Removal Kit (Mirus, Madison WI). Twenty microgram of control or AIF1 targeting plasmids were electroporated using a square wave electroporator under the following conditions: 230 V, 4 ms, 5 pulses.
qPCR
To evaluate gene expression, cells were harvested and resuspended in Trizol (Thermo Fisher Scientific) prior to total RNA extraction. Total RNA was reverse transcribed into single-stranded cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). For quantitative PCR reactions, Gene Expression Master Mix, and the following probes purchased from Thermo Fisher Scientific were used: AIF1 (Mm00479862_g1), BATF3 (Mm01318274_m1), CD11c (ITGAX; Mm00498701_m1), CIITA (Mm00482914_m1), Id2 (Mm00711781_m1), IRF8 (Mm00492567_m1), IRF4 (Mm00516431_m1), MHC class IIα (H2-Aα; Mm00439216_m1), MHC class IIβ (H2-Aβ; Mm00439211_m1), PU.1 (SPIB1; Mm00488140_m1), RelB (Mm00485664_m1), and Zbtb46 (Mm00511327_m1). Samples were ran on the QuantStudio 5 real time-PCR system (Thermo Fisher). Expression levels of the target transcripts were calculated by the comparative Ct method (2–ΔΔCt formula) after normalization with the housekeeping genes GAPDH (Mm99999915_g1) and β-actin (Mm02619580_g1).
Antibodies and Live/Dead Staining Dye
AIF1 (EPR16588) antibody used was purchased from Abcam (Cambridge MA). CD117 (2B8), Lineage cocktail (CD3/Gr-1/CD11b/CD45R/TER-119), SCA1 (D7), CD34 (SA376A4), CD11c (N418), MHC class II (M5/114.15.2), CD8α (53–6.7), CD11b (M1/70), F4/80 (BM8), CD45R (RA3-6B2), TCRβ (H57-597), Protein Kinase C (PKC; PKC0103), CD4 (RM4-5), CD3 (17A2), IgD (11-26c.2a), CD19 (6D5), CD24 (M1/69), PDCA1 (927), CD172α (P84), IL-7Rα (A7R34), CD135 (A2F10), and CD103 (2E7) purchased from BioLegend. RelB (C1E4), NFκB p100/P52 (D7A9K), phospho-IκBα (S32), IκBα (L35A5), phosho-p38 (M139), and p38 (D13E1) purchased from Cell Signaling Technology (Danvers MA). Phospho-ERK (M206) purchased from ECM biosciences (Versailles KY) and IRF4 (343), IRF8 (V3GYWCH), NFκB p105/50 (catalog #14-6732-81), GAPDH (GA1R), and β-actin (BA3R) from Thermo Fisher. Zbtb46 (U4-1374) purchased from BD Biosciences (San Jose CA). Live/Dead fixable dead cell stain (cat# L23101) purchased from Thermo Fisher Scientific.
Western Blot Analysis
Cells were treated with NP-40 lysis buffer supplemented with a protease and phosphatase inhibitor cocktail (Amresco, Solon OH). Lysates were ran in 10–15% SDS-PAGE gels. Protein content was transferred to nitrocellulose blots by wet tank transfer. Membranes were blocked in 5% non-fat dry milk or 2% BSA prior to primary antibody staining. For phospho-specific antibodies, probing for total respective protein content was used for quantitative profiling. After primary antibody staining, secondary fluorochrome-conjugated antibodies were used for detecting protein bands on the Licor Odyssey imaging system (Licor, Lincoln NB). Datasets were analyzed using Image Studio 5.2 software (Licor).
Co-immunopreciptation
Lysates were prepared using NP-40 protein lysis buffer supplemented with a protease and phosphatase inhibitor cocktail. One hundred microgram of protein lysates were incubated with 5 μg AIF1 or IgG rabbit control antibody. Next, rec-Protein G-Sepharose® 4B conjugate beads (Thermo Fisher) were re-suspended with the protein lysates coupled with antibody. After incubation, samples were extensively washed and centrifuged at 6,000 × g for 30 s. The pelleted antibody-protein bead complex was then resuspended in 10% SDS diluted in NP-40 lysis buffer. Samples were then ran in 10% SDS-PAGE gels and transferred to a nitrocellulose membrane using wet tank transfer. Membranes were blocked with 2% BSA prior to probing with PKC antibody. Membranes were then washed prior to probing with secondary fluorochrome-conjugated antibodies and detected using the Licor Odyssey imaging system.
Cryosectioning and Fluorescence Microscopy
Mice spleens were harvested and immediately cryosectioned at 10 or 20 μm thickness prior to fixation with 3% paraformaldehyde. Samples were transferred onto glass slides prior to permeabilization using 0.3% Triton-X solution and blocking with 0.2% BSA. Sections were stained with AIF1 or IgG isotype control antibodies, followed by washing and secondary staining. Additionally, sections were co-stained with CD11c, CD4, F4/80, or CD45R/B220. DAPI (Thermo Fisher) was used as a nuclear staining dye. After incubation, slides were then washed prior to mounting with cover slips and imaging using the FSX100 fluorescence microscope (Olympus, Waltham MA). Acquired images were then analyzed using ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health).
Flow Cytometry
Cell surface staining was performed with PBS supplemented with 1 mM EDTA, 0.1% NaN3 and 2.5% bovine serum (FACS buffer). Cells were washed with FACS buffer prior to extracellular staining with fluorochrome-tagged antibodies. Dilutions were antibody specific, but roughly 10 μl of a 10 μg/mL working concentration was utilized per 2 × 105 cells. Respective isotype controls were used in all assays. Cells were then fixed with 3% PFA in PBS. For intracellular antibody labeling, fixed cells were permeabilized with 0.2% saponin in PBS with 1 mM EDTA. Next, primary antibodies or isotype controls were added at ~10 μg/mL concentrations followed by washing and subsequent staining with secondary fluorochrome-labeled antibodies. Cells were acquired on a BD FACSVerse flow cytometric analyzer (BD Biosciences). Datasets were analyzed using Flow Jo v10 (Flow Jo LLC; Ashland, OR).
Statistical Analysis
GraphPad Prism v8.0 (GraphPad Software, La Jolla CA) was used to determine statistical significance. Student unpaired two-tailed t-test was used to evaluate the significance between two groups. Error bars for all figures indicate standard errors; *< 0.05, **< 0.01 and NS = not significant.
Results
AIF1 Is Restricted to Subsets of Hematopoietic Progenitors and Myeloid-Derived Lineage Committed Cells
AIF1 expression was assessed in murine bone marrow hematopoietic progenitors. Gated populations of Lin−CD117+ and Lin+CD117− bone marrow cell populations were assessed for AIF1 in conjunction with SCA1 or CD34 co-expression. In all studies, isotype controls were used to establish gating strategies and compensate for differential levels of Fc receptors pinocytosis on the heterogenic pool of bone marrow cells. Results showed AIF1 expression at 2.96% ± 0.08 in the Lin−CD117+SCA1+ hematopoietic subsets (Figure 1A), which represents 73.4% ± 4.3 of the population positive for AIF1. Lower levels of AIF1 were found in the Lin+CD117−SCA1− bone marrow subsets at 4.64% ± 0.4. Studies found no co-expression of AIF1 with CD34 in either Lin−CD117+ or Lin+CD117− subsets (Figure 1B).
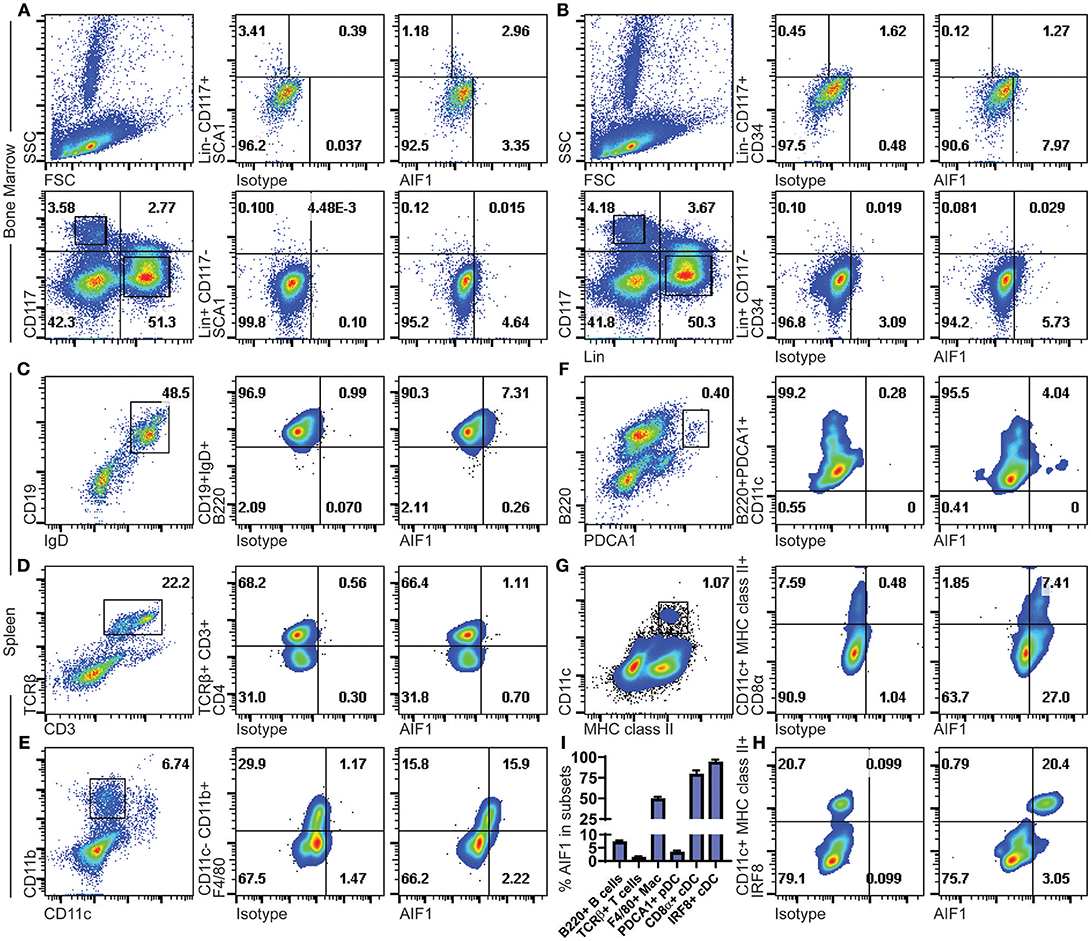
Figure 1. AIF1 is expressed in Lin−CD117+SCA1+ bone marrow hematopoietic stem cells and in spleen myeloid-derived subsets. Bone marrow cells were assessed for hematopoietic subsets. Dot plots show SSC vs. FSC and Lineage (Lin) vs. CD117 (c-Kit) populations. Expression of AIF1 was assessed in Lin−CD117+ (top row) and Lin+CD117− (bottom row) cells co-expressed with either (A) SCA1 or (B) CD34. Splenocytes were evaluated for AIF1 expression in subsets of B cells, T cells, macrophages, plasmacytoid DC, and conventional DC; cell populations were, respectively, identified by: (C) CD45R (B220)+ gated from IgD+CD19+ cells; (D) CD4+ subsets gated from CD3+TCR+ cells; (E) F4/80+ subsets gated from CD11c−CD11b+ cells; (F) CD11c+ subsets gated on B220+PDCA1+; and (G) CD8α+ gated from MHC class II+CD11c+ conventional dendritic cells. (H) IRF8+ cells gated from CD11c+MHC class II+ subsets. (I) AIF1 expression percentage in B220+ B cells gated from CD19+IgD+ subsets, TCRβ+ T cells, F4/80+ macrophages gated from CD11c−CD11b+ subsets, PDCA1+ pDC, CD8a+ cDC gated from CD11c+MHC class II+ subsets, and IRF8+ cDC gated from CD11c+MHC class II+ subsets. All gates were established using respective isotype controls. Isotype used for determining AIF1 expression is shown in all groups. Data is representative of three independent experiments.
Next, AIF1 was evaluated in murine splenocytes. For lymphocytes, B cells were assessed by gating on CD19+IgD+ subsets to evaluate CD45R(B220) co-expression with AIF1 (Figure 1C). Results revealed a small proportion of B cells that were reproducibly found to express AIF1. For T cells, splenocytes were gated on CD3+TCRβ+ to evaluate CD4+ T cells (Figure 1D). No expression was found in TCRβ+ T cells. Myeloid subsets, macrophages, plasmacytoid DC (pDC), and classical DC were assessed for AIF1 expression. Results found that the CD11b+CD11c−F4/80+ macrophage subsets at 15.9% ± 0.4, representing about 50% of the population (Figure 1E). Analysis of CD11c+B220+PDCA1+ pDC showed little-to-no expression with 4.04% ± 0.5 cells expressing AIF1 (Figure 1F), nor in the alternative gating of B220+CD11b−Gr1+CD11c+/lo pDC (Supplemental Figure 1A). However, in assessing the classical DC, results revealed high levels of AIF1 expression in the CD11c+MHC class II+ population (Figure 1G). Interestingly, the CD11c+MHC class II+CD8α+ cDC1 subsets dominantly expressed AIF1 at 7.4% ± 0.3 compared to 1.8% ± 0.2 not expressing the protein (Figure 1G). This represented 80% of the cDC1 population expressing AIF1, compared to only about 30% of AIF1 expressed in the CD8α− subsets. To further assess cDC1 profile, studies evaluated co-expression of IRF8 with AIF1 in the CD11c+MHC class II+ subsets. Results revealed that all CD11c+MHC class II+IRF8+ cDC expressed AIF1 (Figure 1H). Further analysis revealed 21.6% ± 0.5 AIF1 expression in IRF4+ cells and 21.2% ± 0.6 in Zbtb46+ cells (Supplemental Figures 1B,C). Collectively, AIF1 is predominately present in myeloid splenic subsets, with the most expression found in CD11c+MHC class II+ cDC1 population, and a lesser fraction in the CD11b+CD11c−F4/80+ macrophages (Figure 1I).
To corroborate flow cytometric analyses, spleen sections were stained to determine co-localization of AIF1 with common CD11c, F4/80, B220, and CD4 myeloid and lymphoid cell markers. Images are presented at 4X magnification, with a cropped section further magnified to 20X (Figures 2A,B). Results revealed that AIF1 was predominately co-expressed in the myeloid-associated CD11c+ and F4/80+ subsets. Little-to-no co-localization was observed in the B220+ or CD4+ cell subsets, which are markers largely associated with lymphocytes.
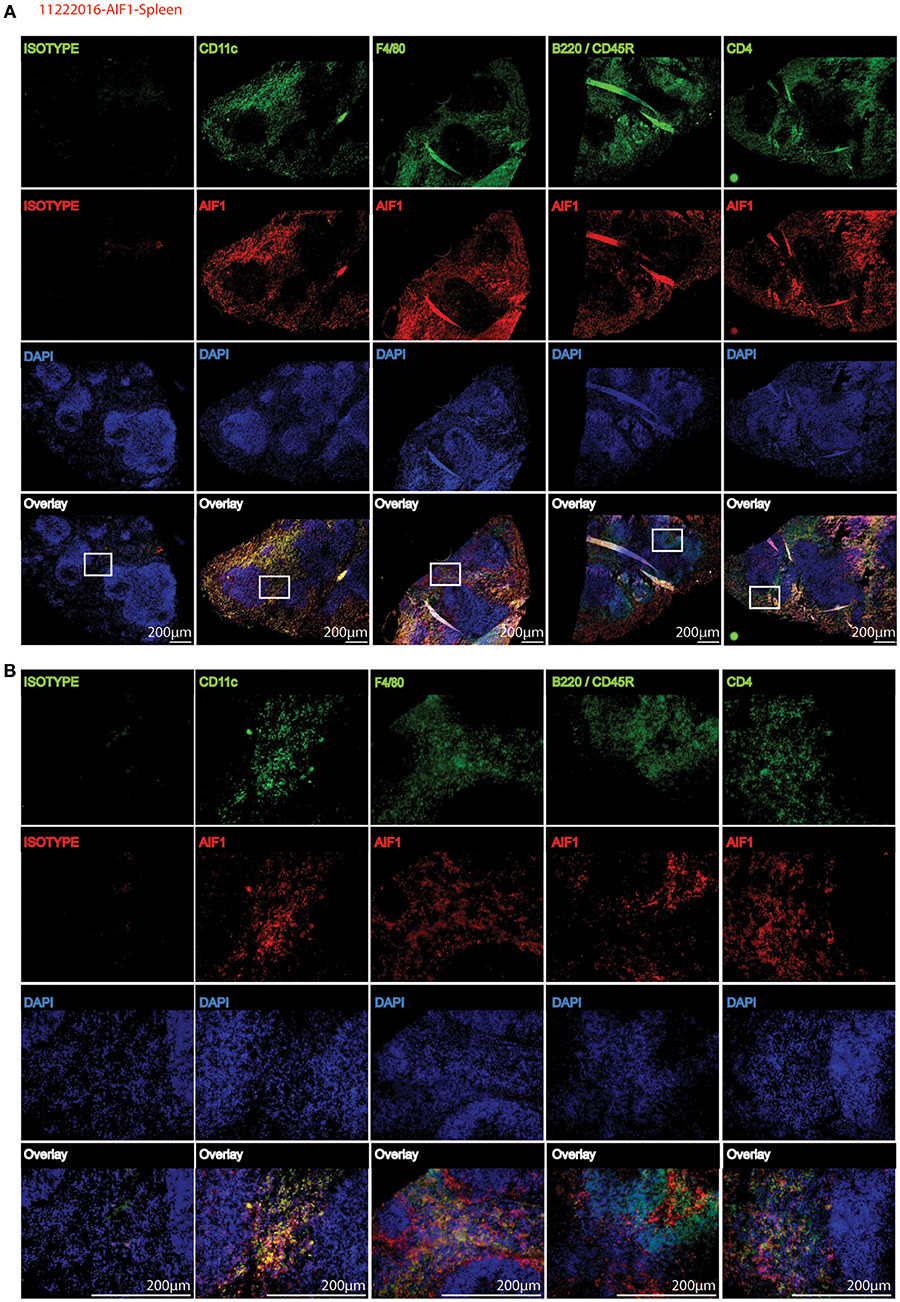
Figure 2. AIF1 is dominantly expressed in splenic myeloid subsets. Fluorescence microscopy was performed on 20 μm spleen sections evaluating AIF1 expression (red) co-stained for CD11c, F4/80, CD45R (B220), or CD4 (green) with respective isotype controls. DAPI was used to stain for nuclei (blue). Stained sections were imaged at (A) 4X and (B) 20X magnifications. Sections in the white box area of 4X images (top) represents respective magnified 20X images (below). Images were captured on an Olympus FSX100 fluorescence microscope. Data is representative of three independent experiments.
Increase in AIF1 Expression Over Time Is Directly Associated With DC Differentiation by Either GM-CSF or Flt3-L Stimulation
Having determined that AIF1 is largely present in CD11c+MHC class II+ subsets, the studies next investigated the time point(s) to which AIF1 appears during DC differentiation under GM-CSF or Flt3-L stimuli from murine bone marrow in vitro. Cells were then harvested on days 4, 6, and 8 and stained for flow cytometric analyses. Staining with live/dead exclusion dye was used to preferentially stain dead cells and identify the live population. These Live+ cells were then gated to assess AIF1 expression in CD11c+MHC class II+ subsets. Results revealed that AIF1 levels progressively increased in the DC throughout the 8-day differentiation process under GM-CSF (Figure 3A) or Flt3-L (Figure 3B). For GM-CSF stimuli, there was a day-by-day increase in AIF1 within the CD11c+MHC class II+ subsets, with day 4 at 22.8% ± 2.4, day 6 at 31.3% ± 1.7 and day 8 at 39.7% ± 2.6. Similar to GM-CSF-treated cultures, Flt3-L stimulated cultures showed that AIF1 increased throughout the differentiation, with day 4 at 4.04% ± 2.9, day 6 7.75% ± 1.6, and day 8 33.6% ± 2.4 within the CD11c+MHC class II+.
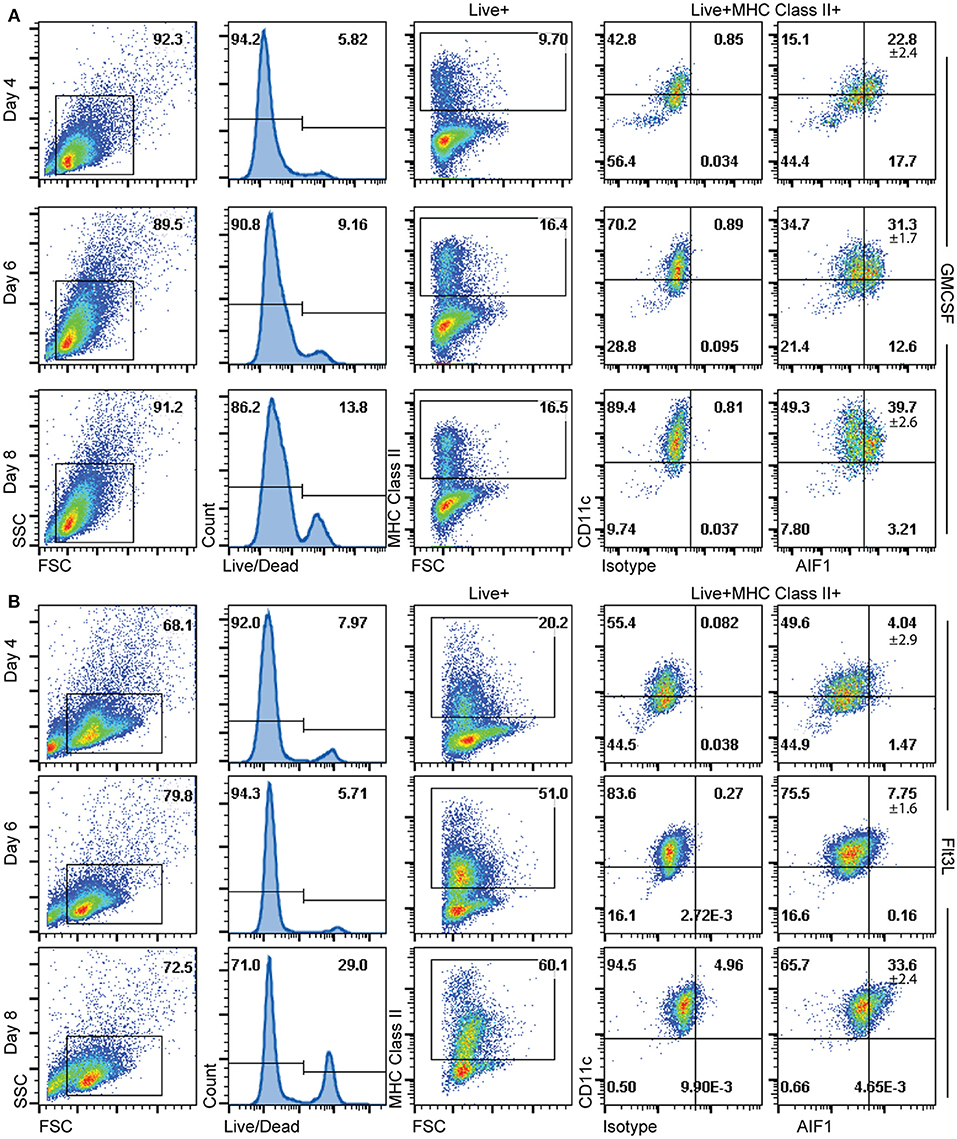
Figure 3. AIF1 expression during GM-CSF or Flt3-L DC differentiation from bone marrow-derived cells. Differentiating DC were harvested on days 4, 6, and 8 of (A) GM-CSF or (B) Flt3-L stimulated bone marrow cells. Live cells were gated out by using live/dead fixable dyes; dead cells are preferentially labeled with the staining dye to discriminate from the live population. MHC class II+ subsets were then gated from the Live+ population. AIF1 expression was then assessed in CD11c+ subsets gated from MHC class II+ groups. All gates were established using isotype controls. Data is representative of five independent experiments.
Differential Expression of AIF1 in DC Subsets Derived From Spleen or Expanded in vitro
For further analyses, myeloid subsets derived from the spleen or in vitro expanded from bone marrow precursors under either GM-CSF- or Flt3-L-stimuli were co-stained for MHC class II, CD11c, CD8α, CD11b, and CD103. Results revealed that the majority of the CD11c+MHC class II+CD8α+ cDC1 subsets express AIF1, with considerably lower levels found in CD11b+ cDC2 (Supplemental Figures 2A–C). As expected, no CD103+ myeloid DC were detected in the spleen. There was relatively low levels of AIF1 present in MHC class II+CD11c−CD11b+ subsets, whereas the highest expression was in the CD11c+CD11b− populations (Supplemental Figures 2D–F).
Silencing AIF1 in Bone Marrow Cells Inhibits in vitro Differentiation of CD11c+ DC Subsets in the Presence of GM-CSF
To determine if AIF1 was responsible for controlling CD11c+ Mo-DC development under GM-CSF stimuli, expression was silenced in bone marrow cells cultured over the 8-day differentiation period. Cells were transfected with siRNA targeting AIF1 (siAIF1) or control scrambled oligos (siControl) on day 0 of the differentiation process. Western blot analysis confirmed successful knockdown of AIF1 (Figure 4A). Upon assessing generation of Mo-DC on day 4, the earliest time point to which AIF1 expression was strongly observed under GM-CSF stimulation, results revealed reduction of CD11c+AIF1+ DC from 8.55% ± 0.4 in the control group to 3.89% ± 0.3 in the AIF1 knockdown group (Figure 4B). However, less impairment in generation of CD11c+AIF1− subsets was observed, with a reduction from 9.22% ± 0.3 in the siControl compared to 7.48% ± 0.3 in the siAIF1 groups, respectively. Gene expression analyses revealed MHC class II (H2-Aα and H2-Aβ), CIITA and CD11c were all restrained upon silencing of AIF1 (Figure 4C). To corroborate transcriptional profile analyses, flow cytometry was performed. Results revealed that expression of both MHC class IIhigh and MHC class IImiddle subsets were restrained in the AIF1 silenced cohort, but less effect was seen in the MHC class IIlow population (Figure 4D). In particular, AIF1 was found to be most expressed in CD11c+MHC class IIhigh DC in comparison to the MHC class IImiddle or MHC class IIlow subsets. Taken together, these results suggest that generation of CD11c+MHC class II+ Mo-DC are dependent on AIF1 during in vitro GM-CSF differentiation from bone marrow precursors.
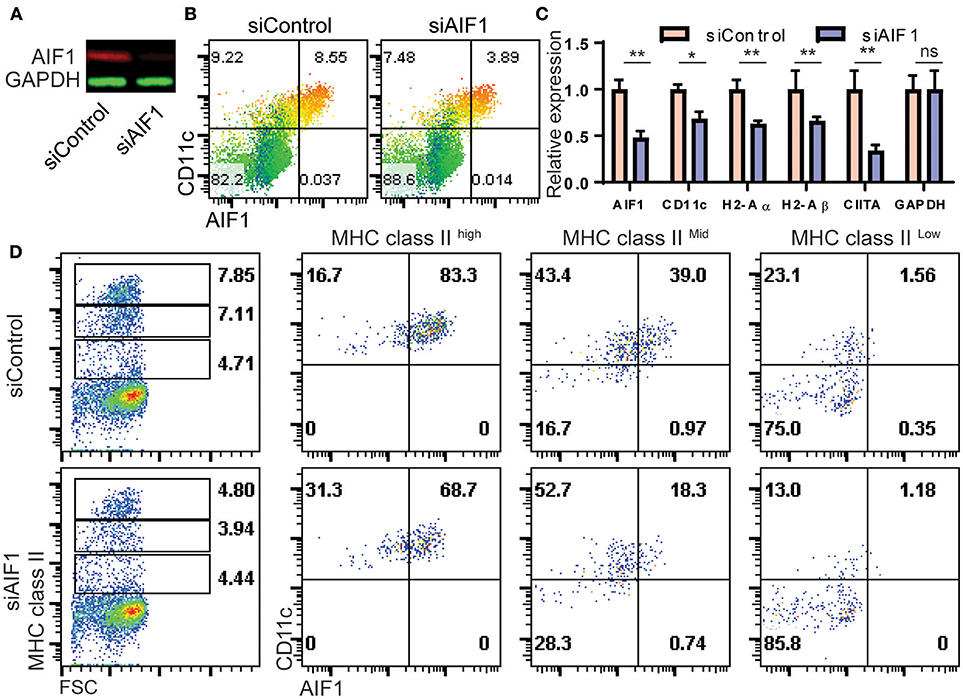
Figure 4. AIF1 silencing during GM-CSF-induced differentiation of bone marrow cells results in decreased MHC class II and CD11c expression. (A) Western blot analysis was performed on GM-CSF treated bone marrow cells silenced for AIF1 (siAIF1). Cells were collected and probed for AIF1. GAPDH served as an internal loading control. (B) Dot plots show flow cytometric analysis of AIF1 protein co-expression with CD11c using a heat map gradient relative to MHC class II. Heat-mapping is a visual technique which assigns a red-blue gradient to a range of data, varying the color based on the current cells' value relative to the distribution (with red as high and blue as low). (C) mRNA relative expression for AIF1, CD11c, H2-Aα, H2-Aβ, CIITA, and GAPDH was quantified in siControl and siAIF1 groups and plotted as a bar graph. (D) Flow cytometric analysis of AIF1 co-expression with CD11c in MHC class II high, MHC class II middle, or MHC class II low gated subsets. Data is representative of three independent experiments. Error bars for all figures indicate standard errors; *< 0.05, **< 0.01 and NS, not significant.
Loss of AIF1 Restrains Commitment of Hematopoietic Stem Cell Progenitors Into cDC1 Subsets Through Repression of IRF8 and Batf3 Under Flt3-L Stimuli
To more accurately assess cDC generation from hematopoietic precursors, expression of AIF1 was silenced in bone marrow cells by transfection with plasmids carrying the CRISPR-Cas9 with guide RNA targeting the gene at exon 7. Transfected cells were then cultured with Flt3-L for 9 days. Cells were then evaluated for expression of hematopoietic stem cell and progenitor markers by flow cytometric analyses. Results revealed a higher proportion of Lin−CD117+ in the AIF1 silenced group (pAIF1), having an increase from 14.4% ± 0.8 from the CRISPR-Cas9 control (pControl) to 29.0% ± 1.4 (Figure 5A). Next, the Lin−CD117+ cells were gated on to discretely assess levels of CD16/32 (FcγR) vs. CD34 to determine changes in proportion of CMP, megakaryocytic-erythroid progenitors, and granulocyte-monocyte progenitors (GMP). Results found no expression of FcγR associated with GMP subsets, as expected under Flt3-L treatments. However, there was a significant increase in the Lin−CD117+FcγR−CD34+ subsets from 13.9% ± 1.2 in the control group to 40.4 ± 0.8 upon AIF1 silencing (Figure 5A). Further analyses discovered increased SCA1 expression in the Lin−CD117+FcγR−CD34+ gated subset from 43.9% ± 1.4 in control to 83.1% ± 1.7 in pAIF1 transfected cohorts (Figure 5A).
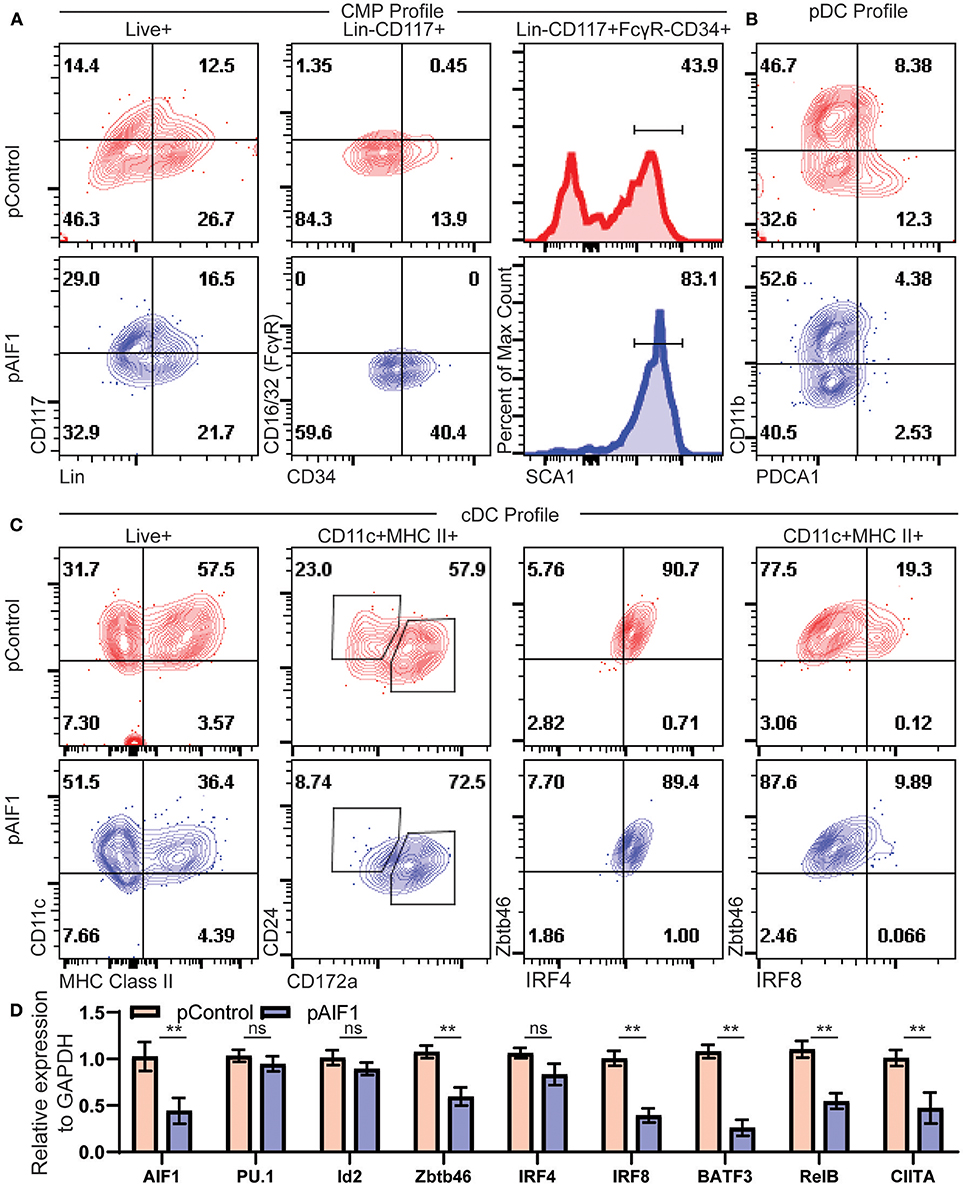
Figure 5. Absence of AIF1 restrains hematopoietic progenitor differentiation into cDC1 lineage commitment. Bone marrow cells were harvested and silenced for AIF1 by CRISPR RNAi approaches prior to differentiation into DC using Flt3-L as stimuli for 9 days. CRISPR Cas9 plasmids with either gRNA targeting AIF1 (pAIF1) or non-targeting scrambled gRNA (pControl) were used to transfect the cells. Cells were collected and stained with respective antibodies and Live/Dead viability dye to discriminate and gate on the Live+ population. (A) To assess common myeloid progenitor (CMP) profiles, dot plots show SCA1 expression in cells gated on Live+Lin−CD117+FcγR−CD34+ subset. (B) CD11b vs PDCA1 was utilized to assess pDC, where pDC are CD11b-PDCA1+. (C) cDC profile shows CD11c vs. MHC class II gated on total Live+ cells. CD24 vs CD172a were evaluated on the gated CD11c+MHC class II+ subsets. Next, cells were assessed for Zbtb46 vs. IRF4 or IRF8. (D) Real-time PCR analysis was performed on control or AIF1 silenced group to assess AIF1, PU.1, Id2, Zbtb46, IRF4, IRF8, BATF3, RelB, and CIITA. Data is representative of six independent experiments. Error bars for all figures indicate standard errors; **< 0.01 and NS, not significant.
Studies next aimed to investigate changes in classical vs. plasmacytoid DC (pDCs) generation. There was a significant decrease in CD11b−PDCA1+ subsets from 12.3% ± 2.6 to 2.53 ± 1.2, indicative of a reduction in the pDC compartment (Figure 5B). Most notably, loss of AIF1 restrained differentiation of CD11c+MHC class II+ DC from 57.5% ± 3.8 to 36.4% ± 4.1 upon AIF1 silencing (Figure 5C), recapturing observations in the Mo-DC studies. Further delineation revealed that AIF1 silencing under Flt3-L stimulation dominantly suppressed cDC1 development, but not cDC2. Evaluation of the CD11c+MHC class II+ subsets showed a decrease in the frequency of CD24+CD172α− DC (cDC1) from 23.0% ± 0.8 to 8.74% ± 1.2, with an increase of the CD24+CD172α+ population from 57.9% ± 0.8 up to 72.5% ± 2.2. Next, CD11c+MHC class II+ DC were further analyzed for Zbtb46 co-expression, which serves as a marker to clearly identify classical DC, with IRF4 or IRF8. Results revealed no changes in IRF4 expression. However, there was a significant decrease in IRF8 within the CD11c+MHC class II+Zbtb46+ subsets from 19.3% ± 0.8 to 9.89% ± 0.5. Further delineation of key transcription factors associated with cDC and pDC differentiation were assessed by quantitative PCR (qPCR). Results showed down regulation of Zbtb46, IRF8, BATF3, RelB, and CIITA, but revealed no significant changes in PU.1, IRF4, or Id2 gene expression (Figure 5D). Collectively, these results show that restrained AIF1 expression in hematopoietic progenitors suppresses differentiation into distinct cDC1 subsets, while concomitantly redirecting toward cDC2 under Flt3-L stimuli.
AIF1 Is Important for Committing Lin−CD117+ Hematopoietic Stem Cell Progenitors Into Mo-DC Subsets Under GM-CSF Stimuli
Bone marrow cells (pre-sort) were sorted for Lin−CD117+ progenitors (post-sort) to assess HSC ability to convert into Mo-DC (through the MDP-to-cMoP pathway) (Supplemental Figure 3A). Sorted Lin−CD117+ hematopoietic cells were then transfected with CRISPR-Cas9 targeting knockout of AIF1 prior to culturing with GM-CSF in vitro to generate Mo-DC. Silencing of AIF1 was successful using two different gRNAs targeting AIF1 (Supplemental Figure 3B). Results paralleled that of Flt3-L studies. The percentage of Lin−CD117+FcγR−CD34+ subsets was largely increased in absence of AIF1 during Mo-DC generation (Supplemental Figure 3C). This suggests that absence of AIF1 restrains conversion into DC subpopulations at the CMP/MDP stage during GM-CSF-stimuli conditions (Supplemental Figure 3D).
Loss of AIF1 Impairs RelB Expression and Phosphorylation of p38 MAPK
The NFκB and MAPK signaling pathways are important for DC development from hematopoietic stem cells. In particular, upregulation of the NFκB family member RelB is essential for differentiation of CD11c+ DC subsets. Loss of RelB results in deficient DC numbers and functioning in vivo (21, 22, 32). To investigate deregulated signaling events in absence of AIF1 during DC differentiation in vitro, changes in NFκB and MAPK family mRNA gene expression levels were measured. Silencing of AIF1 resulted in relative decreased expression of RelB (Figure 6A). Interestingly, there was no significant change observed in total p38 MAPK mRNA levels.
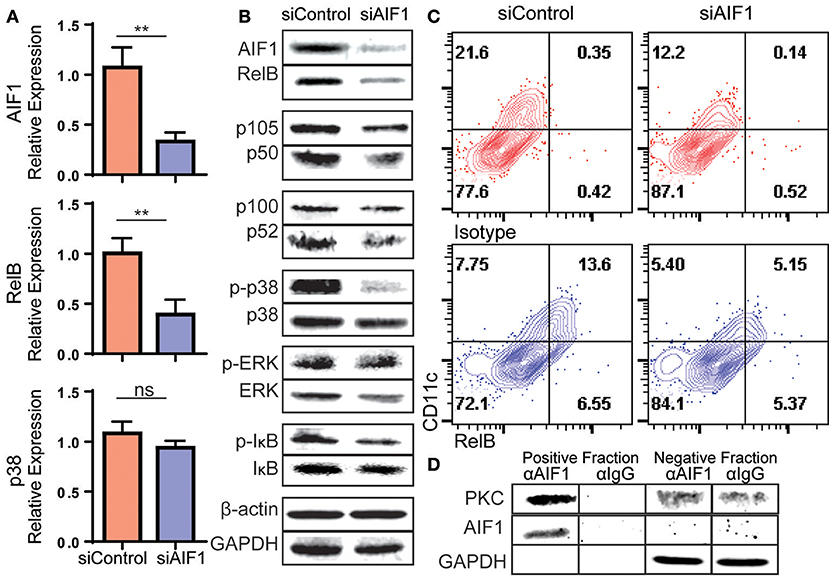
Figure 6. Loss of AIF1 inhibits RelB expression and p38 signaling during GM-CSF-induced DC differentiation. Murine bone marrow cells were sorted for Lin−CD117+ hematopoietic progenitors. These cells were silenced for AIF1 prior to stimulation with GM-CSF to generate DC. Cells harvested on day 6 were assessed by (A) qPCR for gene expression profiling of AIF1, RelB, and p38 or (B) western blot for protein expression of AIF1, RelB, p105/50, or p100/52. GAPDH and β-actin served as internal loading controls and for normalization. For evaluating phosphorylation of proteins, phosphorylated-IκB (p-IκB), -p38 (p-p38), and -ERK (p-ERK) were measured and normalized to total IκB, p38, and ERK protein, respectively. (C) Flow cytometric dot plots of control or AIF1 siRNA treated cells show RelB vs. CD11c. Gates were established using isotype controls. (D) Protein-protein interaction of AIF1 and PKC from DC were assessed. The precipitated positive fraction was pulled out with αAIF1 and further probed for PKC. IgG antibody served as isotype internal control. Negative fraction is the flow through. All data is representative of four independent experiments. Error bars for all figures indicate standard errors; **< 0.01 and NS, not significant.
In measuring protein levels, results corroborated successful knockdown of AIF1 (down to 47.8% ± 0.9) from controls by western blot analysis. This was concomitant with a significant decrease in RelB down to 35.9% ± 0.9 in comparison to control DC (Figure 6B). This recaptured the observed fate seen in mRNA quantitative analysis. Interestingly, there were no changes in protein levels of the NFκB family member p105/50 (NFκB1) or p100/p52 (NFκB2) upon silencing of AIF1. Next, MAPK family proteins were evaluated. In particular, prior studies have shown that AIF1 is involved in governing p38 signaling activities (33). These studies corroborated those findings, whereby phosphorylation of p38 (phos-p38) was significantly reduced down to 57.3% ± 0.8 from controls upon AIF1 silencing (Figure 6B); levels were normalized to total p38. However, studies found no significant differences in that of phosphorylated-ERK signaling. Lastly, studies found reproducibly a moderate reduction in phosphorylated-IκB (phos-IκB); levels were normalized to total IκB.
Flow cytometric analysis was utilized to corroborate the observed decrease in RelB concomitantly with CD11c in absence of AIF1. Results revealed a decrease from 13.6% ± 0.7 of CD11c+RelB+ in control DC down to 5.1% ± 0.4 upon silencing of AIF1 on day 4 (Figure 6C). Given that PKC has been shown to promote RelB expression during DC differentiation (34) and AIF1 serves as a calcium responsive scaffold protein, co-immunoprecipitation was performed to detect potential protein-protein interactions. Results revealed that PKC was found to co-precipitate with AIF1 (Figure 6D), suggesting that AIF1 may serve as a docking site for recruiting and binding the protein kinase. Taken together, this AIF1-PKC interaction may be responsible for promoting NFκB (through phosphorylation of IκB) and MAPK signaling to drive RelB expression for successful differentiation of CD11c+MHC class II+ DC from hematopoietic progenitors.
Discussion
The small proportion of AIF1 expressed in the bone marrow cells were largely restricted to the Lin−CD117+SCA1+CD34− subsets. In contrast, the spleen expressed levels of the protein in both DC and macrophage populations. AIF1 was largely found in CD11c+MHC class II+ DC, with the majority expressed in CD8α+ cDC1 subsets. Notably, a significant amount of AIF1 was also expressed in the CD8α− DC subsets. However, little-to-no expression was observed in lymphoid-derived subsets such as B and T cells or pDC, suggesting that AIF1 expression is restricted to the cDC and macrophage subsets.
Increased AIF1 levels were found in DC cultured in vitro from bone marrow cells under GM-CSF or Flt3-L stimuli conditions. Both approaches generated a population of CD11c+MHC class II+ subsets that expressed AIF1, where it was largely found in the monocyte-derived DC (expanded by GM-CSF) and cDC1 group (expanded by Flt3-L). This directly corroborates the observation of splenic cDC1 subsets dominantly expressing AIF1. Further analyses revealed that AIF1 is not present in all CD11c+MHC class II+ subsets, which supports why not all CD11c+ subsets (i.e., cDC2 expanded by the Flt3-L or the CD11b+CD11c+ monocyte-derived pool under GM-CSF stimuli) are restrained from development upon AIF1 silencing during differentiation.
Silencing AIF1 in bone marrow cells prior to in vitro differentiation by either GM-CSF or Flt3-L revealed its importance in directing DC commitment. Both siRNA and CRISPR-Cas9 approaches were used to successfully restrain AIF1 expression. To compensate for dilution of siRNA due to the accelerated proliferation rate of differentiating cells in vitro, and potentially weed out artifacts, CRISPR-Cas9 system was used to corroborate phenotypes. Both RNAi systems yielded an increase in the MDP and/or CDP in absence of AIF1 upon either GM-CSF or Flt3-L stimulation. These findings would suggest that in absence of AIF1, progenitors are partly being restrained from moving into subsets of monocyte-derived DC or cDC1 committed fates; thus, cells are being restrained at the earlier progenitor stage.
However, AIF1 seems to have a differential role in coordinating development of Mo-DC and cDC under GM-CSF- vs. Flt3-L-stimulated cultures, respectively. Under GM-CSF in vitro differentiation, absence of AIF1 was directly correlated with depressed levels of IRF4, BATF3, and CIITA. Less affect was reproducibly found on IRF8 and Zbtb46 expression. However, under Flt3-L conditions, there was largely reduced levels of Zbtb46, IRF8, BATF3, with no significant difference in IRF4 expression in cDC. This may be due to differential role and transcriptional machinery directing Mo-DC vs. cDC. For example, Mo-DC require IRF4 to induce MHC class II expression (35). Thus, AIF1 silencing could plausibly restrain IRF4 that leads to diminished MHC class II expression in the observed Mo-DC. However, the IRF8-BATF3 axis is required for coordinated cDC1, but not cDC2 fates (13, 36, 37), from Flt3-L stimulated cultures, which represent bona-fide in vivo DC counterparts. Interestingly, previous studies showed that loss of IRF8 lead to reduced expression levels of AIF1 in microglial cells (38). This further suggests bidirectional interactive roles of AIF1-IRF8 to govern myeloid differentiation and functions. However, AIF1 silencing did not perturb IRF4+ expression, which does align with no observed alteration in cDC2 fates. Therefore, AIF1 may influence/affect different transcriptional regulatory and downstream pathways that drive the respective fates of distinct DC subsets from Mo-DC vs. cDC. Coupled with literature eluding to the importance of monocytes and monocyte-derived DC as pivotal players of cross-priming events (39–41), this would suggest that AIF1 expression in myeloid cells is important for governing DC subsets involved in cross-presentation under steady and inflammatory settings.
For both cDC and Mo-DC, RelB expression was inhibited upon loss of AIF1. This would then restrict interactions with NFκB p52 complexes for downstream signaling. Given RelB is dispensable for in vitro Flt3-L cultures (23), but required for in vivo, studies focused on Mo-DC. In this respect, the proposed mechanism by which AIF1 governs DC differentiation may operate by additionally modulating RelB protein expression, as opposed to sequestering RelB activity through inhibitory protein-protein interactions with p100. Furthermore, results revealed an impaired ability to phosphorylate IκB in absence of AIF1. Reduced phosphorylation of IκB would inhibit activation and translocation of NFκB molecules for transcriptional control of RelB. Thus, it suggests that AIF1 silencing concomitantly restrains hematopoietic progenitor differentiation into Mo-DC or cDC1 subsets by disrupting NFκB transcriptional control of RelB (42).
Lastly, the studies identified that AIF1 interacts with PKC, which is an important upstream modulator of IKK phosphorylation of IκB and MAPK signaling (19). RelB activity is increased in absence of IκB-mediated activities (24). As AIF1 has been reported to function as an adaptor protein, which could explain its role in early signaling events. This correlates with prior studies that have shown that AIF1 associates with Rac2 and p38 MAPK (43), which are involved in maturation and cross-presentation (25, 44, 45). Therefore, we propose that AIF1 serves as a scaffold protein interacting with PKC to promote IRF8, BATF3, and RelB in cDC1 and RelB, CIITA, and IRF4 in Mo-DC during differentiation from HSC. In absence of AIF1, differentiation of these Mo-DC or cDC1 subsets from hematopoietic stem progenitors is abrogated.
Author Contributions
DE, NB, RdS, and ML contributed in experimental design and performing experiments. NH, TdM, and NB performed qPCR assays. AK generated approaches for siRNA and CRISPR mediated transfections. DE, NB, RdS, NH, AK, TdM, and ML were each involved in data analysis and writing of the manuscript.
Funding
Grant Support: This work was funded, in part, by the U.S. National Institutes of Health (grant# SC1GM127207), Department of Defense (grant# W911NF-14-1-0123), and National Science Foundation (grant# 1428768).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Franklin Ampy, Clarence M. Lee and Winston Anderson for assistance with statistical analyses and revision of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00173/full#supplementary-material
Abbreviations
AIF1, Allograft Inflammatory Factor-1; BM, bone marrow; cDC, classical dendritic cell; CDP, common dendritic progenitor; CMP, common myeloid progenitor; HSC, hematopoietic stem cell; LK-HSC, Lineage− CD117+ hematopoietic stem cells; MDP, macrophage, monocyte dendritic progenitors; Mo-DC, Monocyte-derived dendritic cell; PKC, protein kinase C.
References
1. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. Morphology I, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
2. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol. (2012) 12:101–13. doi: 10.1038/nri3149
3. Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. (2007) 8:1217–26. doi: 10.1038/ni1522
4. Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. (2007) 8:1207–16. doi: 10.1038/ni1518
5. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327:656–61. doi: 10.1126/science.1178331
6. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity (2012) 36:1031–46. doi: 10.1016/j.immuni.2012.03.027
7. Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. (1996) 184:1953–62. doi: 10.1084/jem.184.5.1953
8. Shurin MR, Pandharipande PP, Zorina TD, Haluszczak C, Subbotin VM, Hunter O, et al. FLT3 ligand induces the generation of functionally active dendritic cells in mice. Cell Immunol. (1997) 179:174–84. doi: 10.1006/cimm.1997.1152
9. McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood (2000) 95:3489–97.
10. Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. (2012) 209:1135–52. doi: 10.1084/jem.20120030
11. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC III, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. (2002) 196:1415–25. doi: 10.1084/jem.20021263
12. Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa C, Ozato K, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood (2003) 101:305–10. doi: 10.1182/blood-2002-04-1088
13. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol. (2015) 16:708–17. doi: 10.1038/ni.3197
14. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci USA. (2004) 101:8981–6. doi: 10.1073/pnas.0402139101
15. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. (2005) 174:6592–7. doi: 10.4049/jimmunol.174.11.6592
16. Metcalf D, Johnson GR, Burgess AW. Direct stimulation by purified GM-CSF of the proliferation of multipotential and erythroid precursor cells. Blood (1980) 55:138–47.
17. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
18. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
19. Steffan NM, Bren GD, Frantz B, Tocci MJ, O'Neill EA, Paya CV. Regulation of IkB alpha phosphorylation by PKC- and Ca(2+)-dependent signal transduction pathways. J Immunol. (1995) 155:4685–91.
20. Neumann M, Fries H, Scheicher C, Keikavoussi P, Kolb-Maurer A, Brocker E, et al. Differential expression of Rel/NF-kappaB and octamer factors is a hallmark of the generation and maturation of dendritic cells. Blood (2000) 95:277–85.
21. Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature (1995) 373:531–6. doi: 10.1038/373531a0
22. Platzer B, Jorgl A, Taschner S, Hocher B, Strobl H. RelB regulates human dendritic cell subset development by promoting monocyte intermediates. Blood (2004) 104:3655–63. doi: 10.1182/blood-2004-02-0412
23. Briseno CG, Gargaro M, Durai V, Davidson JT, Theisen DJ, Anderson DA III, et al. Deficiency of transcription factor RelB perturbs myeloid and DC development by hematopoietic-extrinsic mechanisms. Proc Natl Acad Sci USA. (2017) 114:3957–62. doi: 10.1073/pnas.1619863114
24. Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nat Immunol. (2012) 13:1162–70. doi: 10.1038/ni.2446
25. Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. (2002) 3:69–75. doi: 10.1038/ni748
26. Utans U, Quist WC, McManus BM, Wilson JE, Arceci RJ, Wallace AF, et al. Allograft inflammatory factory-1. A cytokine-responsive macrophage molecule expressed in transplanted human hearts. Transplantation (1996) 61:1387–92. doi: 10.1097/00007890-199605150-00018
27. Schluesener HJ, Seid K, Kretzschmar J, Meyermann R. Allograft-inflammatory factor-1 in rat experimental autoimmune encephalomyelitis, neuritis, and uveitis: expression by activated macrophages and microglial cells. Glia (1998) 24:244–51.
28. Chen ZW, Ahren B, Ostenson CG, Cintra A, Bergman T, Moller C, et al. Identification, isolation, and characterization of daintain (allograft inflammatory factor 1), a macrophage polypeptide with effects on insulin secretion and abundantly present in the pancreas of prediabetic BB rats. Proc Natl Acad Sci USA. (1997) 94:13879–84. doi: 10.1073/pnas.94.25.13879
29. Elizondo DM, Andargie TE, Yang D, Kacsinta AD, Lipscomb MW. Inhibition of allograft inflammatory factor-1 in dendritic cells restrains CD4+ T cell effector responses and induces CD25+Foxp3+ T regulatory subsets. Front Immunol. (2017) 8:1502. doi: 10.3389/fimmu.2017.01502
30. Elizondo DM, Andargie TE, Haddock NL, da Silva RLL, de Moura TR, Lipscomb MW. IL-10 producing CD8+ CD122+ PD-1+ regulatory T cells are expanded by dendritic cells silenced for Allograft Inflammatory Factor-1. J Leukoc Biol. (2018) 105:123–30. doi: 10.1002/JLB.1A0118-010RR
31. Elizondo DM, Andargie TE, Marshall KM, Zariwala AM, Lipscomb MW. Dendritic cell expression of ADAM23 governs T cell proliferation and cytokine production through the alpha(v)beta(3) integrin receptor. J Leukocyte Biol. (2016) 100:855–64. doi: 10.1189/jlb.2HI1115-525R
32. Pettit AR, Quinn C, MacDonald KP, Cavanagh LL, Thomas G, Townsend W, et al. Nuclear localization of RelB is associated with effective antigen-presenting cell function. J Immunol. (1997) 159:3681–91.
33. Sommerville LJ, Xing C, Kelemen SE, Eguchi S, Autieri MV. Inhibition of allograft inflammatory factor-1 expression reduces development of neointimal hyperplasia and p38 kinase activity. Cardiovasc Res. (2009) 81:206–15. doi: 10.1093/cvr/cvn242
34. Cejas PJ, Carlson LM, Kolonias D, Zhang J, Lindner I, Billadeau DD, et al. Regulation of RelB expression during the initiation of dendritic cell differentiation. Mol Cell Biol. (2005) 25:7900–16. doi: 10.1128/MCB.25.17.7900-7916.2005
35. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep. (2016) 15:2462–74. doi: 10.1016/j.celrep.2016.05.025
36. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi: 10.1126/science.1164206
37. Jaiswal H, Kaushik M, Sougrat R, Gupta M, Dey A, Verma R, et al. Batf3 and Id2 have a synergistic effect on Irf8-directed classical CD8alpha+ dendritic cell development. J Immunol. (2013) 191:5993–6001. doi: 10.4049/jimmunol.1203541
38. Horiuchi M, Wakayama K, Itoh A, Kawai K, Pleasure D, Ozato K, et al. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J Neuroinflamm. (2012) 9:227. doi: 10.1186/1742-2094-9-227
39. Le Borgne M, Etchart N, Goubier A, Lira SA, Sirard JC, van Rooijen N, et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity (2006) 24:191–201. doi: 10.1016/j.immuni.2006.01.005
40. Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science (2008) 319:198–202. doi: 10.1126/science.1151869
41. Segura E, Albiston AL, Wicks IP, Chai SY, Villadangos JA. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc Natl. Acad Sci USA. (2009) 106:20377–81. doi: 10.1073/pnas.0910295106
42. Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV. RelB cellular regulation and transcriptional activity are regulated by p100. J Biol Chem. (2002) 277:1405–18. doi: 10.1074/jbc.M109619200
43. Tian Y, Autieri MV. Cytokine expression and AIF-1-mediated activation of Rac2 in vascular smooth muscle cells: a role for Rac2 in VSMC activation. Am J Physiol Cell Physiol. (2007) 292:C841–9. doi: 10.1152/ajpcell.00334.2006
44. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity (2009) 30:544–55. doi: 10.1016/j.immuni.2009.01.013
Keywords: dendritic cells, allograft inflammatory factor 1, hematopoietic stem cells, cDC1 dendritic cells, monocyte-derived DC, RelB/NFκB, IRF8 transcriptional coactivator, protein kinase C (PKC)
Citation: Elizondo DM, Brandy NZD, da Silva RLL, Haddock NL, Kacsinta AD, de Moura TR and Lipscomb MW (2019) Allograft Inflammatory Factor-1 Governs Hematopoietic Stem Cell Differentiation Into cDC1 and Monocyte-Derived Dendritic Cells Through IRF8 and RelB in vitro. Front. Immunol. 10:173. doi: 10.3389/fimmu.2019.00173
Received: 26 April 2018; Accepted: 21 January 2019;
Published: 08 February 2019.
Edited by:
Pierre Guermonprez, King's College London, United KingdomReviewed by:
Alexander Mildner, Helmholtz Association of German Research Centers (HZ), GermanyVeronika Lukacs-Kornek, Saarland University, Germany
Copyright © 2019 Elizondo, Brandy, da Silva, Haddock, Kacsinta, de Moura and Lipscomb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael W. Lipscomb, bWljaGFlbC5saXBzY29tYkBob3dhcmQuZWR1