
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 January 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00028
 Pranav Murthy1
Pranav Murthy1 Aatur D. Singhi2Mark A. Ross3
Aatur D. Singhi2Mark A. Ross3 Patricia Loughran3
Patricia Loughran3 Pedram Paragomi4Georgios I. Papachristou4
Pedram Paragomi4Georgios I. Papachristou4 David C. Whitcomb4Amer H. Zureikat1
David C. Whitcomb4Amer H. Zureikat1 Michael T. Lotze1Herbert J. Zeh III1,5
Michael T. Lotze1Herbert J. Zeh III1,5 Brian A. Boone1,6*
Brian A. Boone1,6*Background: Neutrophil extracellular traps (NETs) are generated when activated neutrophils, driven by PAD4, release their DNA, histones, HMGB1, and other intracellular granule components. NETs play a role in acute pancreatitis, worsening pancreatic inflammation, and promoting pancreatic duct obstruction. The autophagy inhibitor chloroquine (CQ) inhibits NET formation; therefore, we investigated the impact of CQ mediated NET inhibition in murine models of pancreatitis and human correlative studies.
Methods: L-arginine and choline deficient ethionine supplemented (CDE) diet models of acute pancreatitis were studied in wild type and PAD4−/− mice, incapable of forming NETs. Isolated neutrophils were stimulated to induce NET formation and visualized with fluorescence microscopy. CQ treatment (0.5 mg/ml PO) was initiated after induction of pancreatitis. Biomarkers of NET formation, including cell-free DNA, citrullinated histone H3 (CitH3), and MPO-DNA conjugates were measured in murine serum and correlative human patient serum samples.
Results: We first confirmed the role of NETs in the pathophysiology of acute pancreatitis by demonstrating that PAD4−/− mice had decreased pancreatitis severity and improved survival compared to wild-type controls. Furthermore, patients with severe acute pancreatitis had elevated levels of cell-free DNA and MPO-DNA conjugates, consistent with NET formation. Neutrophils from mice with pancreatitis were more prone to NET formation and CQ decreased this propensity to form NETs. CQ significantly reduced serum cell-free DNA and citrullinated histone H3 in murine models of pancreatitis, increasing survival in both models.
Conclusions: Inhibition of NETs with CQ decreases the severity of acute pancreatitis and improves survival. Translating these findings into clinical trials of acute pancreatitis is warranted.
Acute pancreatitis (AP) is a common, severe gastrointestinal disease, assessed as the fifth leading cause of in-hospital mortality and an annual incidence that has increased by 30% since 2000 (1, 2). AP is characterized by elevated digestive enzyme concentrations in the blood linked to altered acinar cell secretion and/or duct obstruction, which promotes autodigestive injury within the pancreas (2). These events stimulate tissue injury and activation of the innate immune system, resulting in recruitment and activation of neutrophils with subsequent release of pro-inflammatory cytokines and other substances that lead to systemic inflammation (2, 3). The pathogenesis of severe AP is still poorly understood, limiting significant treatment options outside of supportive care including bowel rest, fluid resuscitation, and pain management (1, 2).
Neutrophils play a central role in severe acute pancreatitis. In addition to release of cytokines, activated neutrophils release their DNA, histone proteins, high mobility group box 1 (HMGB1), and granule components into the extracellular space or circulation to form neutrophil extracellular traps (NETs). This process was initially considered primarily as a means for neutrophils to trap bacteria and combat infection (4–6). Although NETs are generally immuno-protective in the setting of bacterial or fungal infections (4), several studies have implicated NETs in the pathophysiology of sterile inflammatory conditions, such as lupus (7, 8), rheumatoid arthritis (9), and pancreatic cancer (10). Recent evidence demonstrates that NETs are also involved in the pathogenesis of AP by inducing trypsin activation, inflammation, and tissue damage (11) following inspissation and occlusion of pancreatic ducts (12). Moreover, NETs surround necrotic tissue in patients with severe AP and promote the subsequent systemic inflammatory response syndrome (13).
Chloroquine (CQ), is an orally available and inexpensive drug historically used to treat malaria (14) that also inhibits autophagy (15). We and others have shown that heightened autophagy promotes NET generation (16–19) and that CQ prevents NET formation (10, 20). Given the recent discovery of the role of NETs in AP, we studied whether targeted inhibition of NETs with CQ could ameliorate AP in murine models. We evaluated the role of CQ in the treatment of acute pancreatitis by observing its effect on NET formation, and impact on the severity and survival rate in murine models of pancreatitis. Furthermore, we performed correlative studies on the impact of NETs on the severity of human pancreatitis.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (Protocol # 14084123) and performed in accordance with the policies and regulations established by the University of Pittsburgh Division of Laboratory Animal Services, the Guide for the Care and Use of Laboratory Animals, and the Animal Research Reporting on in vivo Experiments (ARRIVE) guidelines. Euthanasia was performed under anesthesia using cardiac puncture resulting in exsanguination followed by cervical dislocation. Mice were housed in ventilated caging units in the Hillman Cancer Center Specific Pathogen Free (SPF) animal facility with standard housing, husbandry, and free access to food and water. C57/BL6 wild type mice (4 and 10–12 weeks) were purchased from Taconic Farms (Hudson, New York). PAD4 knockout (PAD4−/−) mice, incapable of forming NETs, were obtained as a kind gift from the late Dr. Kerri Mowen (21) and were generated on a C57/Bl6 background. Induction of AP using L-arginine (22) or choline deficient ethionine (CDE) supplemented diet (23) was performed as previously described in age and gender matched mice (24). Briefly, a sterile solution of 8% L-arginine hydrochloride (A92600, Millipore Sigma, Burlington, MA) was prepared in normal saline and adjusted to pH 7.0. Mice received 2 hourly intraperitoneal (IP) injections of L-arginine (4 g/kg), while controls were administered saline IP. Animals were treated with oral chloroquine (CQ) (0.5 mg/ml) administered in the drinking water ad libitum upon completion of second L-arginine injection. Isoflurane anesthetized mice were sacrificed via cardiac puncture at 48 or 72 h post injection. Serum was collected after blood was allowed to clot for 30 min and then spun at 10,000 g for 10 min.
For survival experiments, age and gender matched mice underwent two intra-peritoneal L-arginine (4 g/kg) injections an hour apart once a week for a total of 3 weeks. Survival was assessed over a 6 week period. A choline deficient ethionine (CDE) supplemented diet model of AP was also utilized as previously described (23, 25). Briefly, 4 week-old female mice were fasted for 24 h and then fed a CDE diet (960214, MP Biomedicals, Solon, OH) for 6 days. For CDE experiments, animals were treated with oral CQ (0.5 mg/ml) administered in the drinking water ad libitum at the start of the CDE diet (CDE CQ).
Blood was collected from patients with acute pancreatitis as part of a protocol approved by the Institutional Review Board at the University of Pittsburgh (#PRO08010374, PRO14060166). Severity of acute pancreatitis was classified by the revised Atlanta classification (26). Blood samples were drawn within 72 h of presentation, spun at 14,000 g for 10 min and serum was collected and frozen at −80°C using strict standard operating protocols as previously described (27). Serum samples from 5 healthy volunteers were also evaluated as controls.
Trypsin and amylase activity levels, HMGB1, and interleukin-6 (IL-6) levels in murine serum diluted 1:10 were measured using ELISA and quantified using a Tecan Saphire microplate reader. The colorimetric mouse trypsin activity ELISA assay (E4362-100, BioVision, San Francisco, CA), mouse amylase assay kit (ab102523, Abcam, Cambridge, MA), human/mouse HMGB1 ELISA (ST51011, IBL International, Hamburg, Germany), and mouse IL-6 uncoated ELISA (88-7064, Invitrogen, Carlsbad, CA) were used according to manufacturer protocols.
Under sterile conditions, bone marrow neutrophils were isolated from the femur and tibia of euthanized mice by the previously described protocol (28). Neutrophils were plated in a 24-well plate at 1.5 × 104 cells per well in Hank's Balanced Salt Solution (14025076, ThermoFisher Scientific, Waltham, MA). Neutrophils were then stimulated with 40 μm, platelet activating factor (PAF) (511075, Millipore Sigma) for 120 min. Cells were fixed with 3% paraformaldehyde and stained for DNA with Hoechst 33342 (H-3570, Molecular Probes, Grand Island, NY). Representative neutrophils were stained for citrullinated histone H3 (anti-Histone H3 (citrulline 2 + 8 + 17) antibody, Abcam, Cambridge, MA) to confirm that NETs were being visualized rather than DNA released from necrosis. NETs were visualized using a Zeiss Axiovert 40 microscope under 10x−40x magnification. Supernatant was collected, spun at 14 g for 10 min and the level of DNA measured using Quant-iT Picogreen (MP07581, Invitrogen, Grand Island, NY).
After 1:10 dilution, murine serum samples were measured for cell-free DNA (cf-DNA) with Quant-iT Picogreen (MP07581, Invitrogen) and citrullinated histone H3 was measured with the citrullinated histone H3 ELISA (501620, Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocol. Cell-free DNA was measured in the patient serum with the Quant-iT Picogreen (MP07581, Invitrogen). To show that circulating nucleosomes in sera are derived from NETs, we tested myeloperoxidase, a prominent granular component of neutrophils, attached to nucleosomes as described previously (29). MPO-DNA complexes were identified using a capture ELISA (component No.1, Hycult biotech, HK210-01, Uden, Netherlands). A 1:2 dilution of sample was added to the wells and incubated for 1 h. After washing three times, 100 μl incubation buffer containing a peroxidase-labeled anti-DNA mAb (component No.2, Cell Death ELISAPLUS, Roche; Cat. No: 11774424001, Basel, Switzerland) was used according to the manufacturer's protocol.
Resected pancreatic tissue was fixed in OCT medium and stored in −80°C until further analysis. After sectioning to 6 μm, the pancreas section was permeabilized with 0.1% Triton X-100 in Phosphate Buffered Saline (PBS) for 15 min, blocked with 20% normal donkey serum (NDS) for 45 min, and washed 1x with 0.5% BSA in PBS. The sections were incubated with anti-Histone H3 (Rabbit, 1:200, citrulline R2 + R8 + R17, Abcam, ab5103) and anti-Ly6G (Rat, 1:100, RB6-8C5, Invitrogen). After three washes, donkey anti-rabbit Alexa 488 (A21206, 1:500, Jackson ImmunoResearch Laboratories, West Grove, PA) to recognize the anti-Histone H3 was combined with the donkey anti-rat Cy3 IgG-Cy3 conjugated (712-165-153, 1:1000, JacksonImmunoResearch Laboratories, West Grove, PA) that paired with the Ly6G were both incubated for 1 h. A Hoechst nuclear stain (B-2883, Millipore Sigma) was applied at room temperature for 30 s followed by a single rinse of PBS to remove excess dye. Imaging conditions were maintained at identical settings within each antibody-labeling experiment with original gating conditions compared to sample that was a “primary delete,” processed with all reagents except primary antibody. Imaging was performed using a Nikon A1 confocal microscope (purchased with 1S10OD019973-01, awarded to Dr. Simon C. Watkins) at 20x and analyzed with Nikon Elements imaging software (NIS Elements 4.4, Tokyo, Japan).
Data are expressed as mean ± standard deviation. Results are reported from at least two independent experiments performed with at least triplicate samples. Statistical analysis was performed using Student's two tailed t-test for comparisons of two groups or 1-way ANOVA with Tukey's post-hoc tests for multiple groups (GraphPad Prism, San Diego, CA, United States). Survival analyses were conducted using the Kaplan-Meier method and Grehan-Breslow-Wilcoxon test between curves. p < 0.05 were considered statistically significant.
To confirm the impact of NET formation on the pathophysiology of severe AP, we utilized mice unable to form NETs due to a genetic ablation of protein arginine deiminase, type IV (PAD4), an enzyme responsible for chromatin decondensation and subsequent expulsion during NETosis (30). PAD4−/− mice with pancreatitis had decreased trypsin (7.25 ± 1.5 vs. 15.7 ± 2.3 μU/ml, p < 0.0001) and amylase (472 ± 242 vs. 1365 ± 322 μU/ml, p < 0.0001) activity levels compared to wild-type controls (Figures 1A,B). To evaluate the impact of NETs on survival from severe acute pancreatitis, mice were injected with L-arginine weekly for 3 weeks. Survival was significantly improved in PAD4−/− mice with pancreatitis compared to wild-type controls (median survival unreached vs. 15 days, p < 0.0001, Figure 1C).
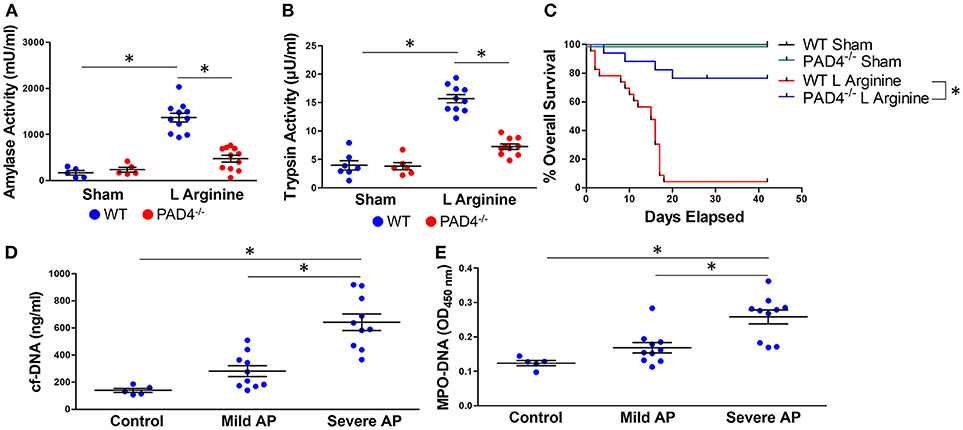
Figure 1. NETs are critical mediators of murine and human acute pancreatitis (AP). Induction of pancreatitis with L-arginine resulted in an increase in amylase (A) and trypsin (B) activity in wild-type mice that was statistically diminished in PAD4−/− mice, incapable of forming NETs. PAD4−/− mice had significantly improved survival compared with wild type mice in recurrent L-arginine induced murine acute pancreatitis (median survival unreached vs. 15 days, p < 0.0001, n = 12 per group) (C). Patients with pancreatitis had elevated levels of circulating cell-free DNA (D) and MPO-DNA conjugates (E), biomarkers of NET formation that correlated with disease severity based on the revised Atlanta classification. *p < 0.05.
We next investigated the clinical relevance of NETs in patients with AP. Serum was isolated from patients with mild or severe acute pancreatitis, as determined by the revised Atlanta classification (26). Patient demographics and clinical characteristics are reported in Supplemental Table 1. Serum cell-free DNA (642 ± 193 vs. 281 ± 127 and 140 ± 32 ng/ml, p < 0.001, Figure 1D) and MPO-DNA complexes, markers of NET formation, were significantly elevated in patients with severe AP in comparison with mild AP and healthy controls (0.258 ± 0.06 vs. 0.168 ± 0.05 and 0.123 ± 0.01 OD450, p < 0.001, Figure 1E). There was a significant correlation between levels of cf-DNA and MPO-DNA (Supplemental Figure 1, Pearson r = 0.868, 95% CI: 0.719–0.940, p < 0.0001).
Isolated bone marrow neutrophils from wild type mice were plated and stained with Hoechst to evaluate their propensity to form NETs. As previously described (10), citrullinated histone H3 was identified within NETs on this ex vivo assay (Supplemental Figure 2), confirming that DNA visualized from neutrophils was due to NET formation and not cell necrosis. At baseline, untreated neutrophils from sham control mice produced no NETs. Neutrophils from mice with L-arginine induced pancreatitis produced spontaneous NETs (Figure 2A). Isolated neutrophils were stimulated with PAF, a known inducer of NET formation. Mice with pancreatitis had a greater propensity for NET formation, demonstrated by numerous large, intricately connected NETs. Treatment with CQ markedly decreased the propensity to form NETs in response to PAF stimulation and prevented spontaneous NET formation in mice with pancreatitis.
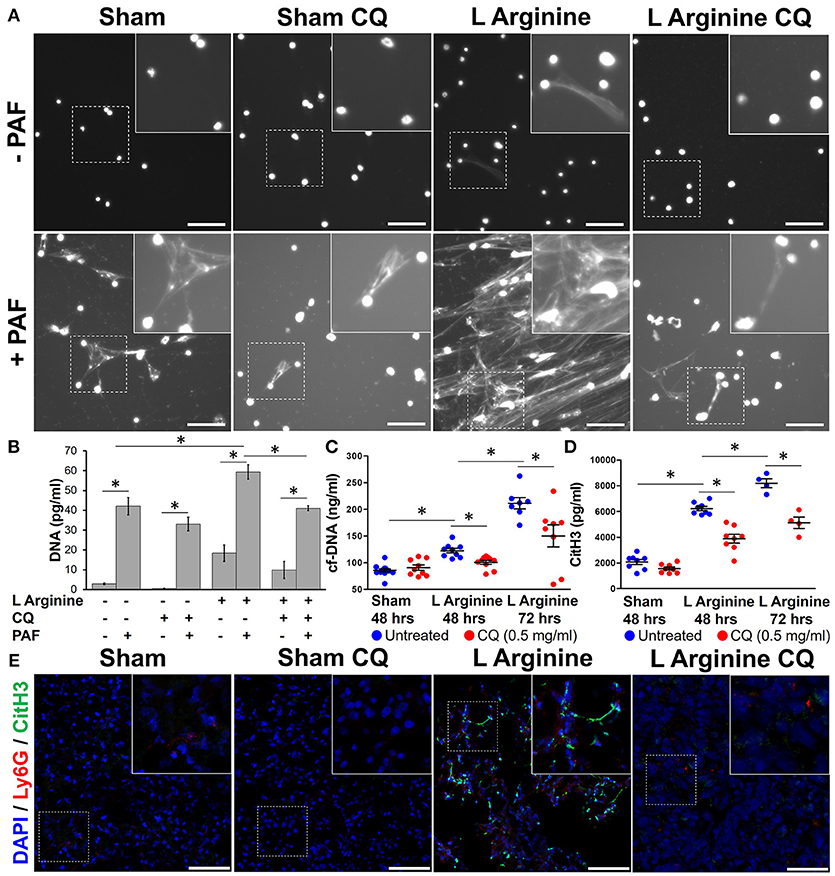
Figure 2. Chloroquine inhibits NET formation in acute pancreatitis. Neutrophils harvested from mice with pancreatitis had evidence of spontaneous NET formation and greater propensity to form NETs upon stimulation with platelet activating factor (PAF), as visualized by staining of DNA with Hoechst (A). CQ treatment dramatically reduced spontaneous and stimulated NET formation. Supernatant DNA was measured to objectively quantify NETs (B). Serum cell-free DNA (C) and citrullinated histone H3 (CitH3) (D), biomarkers of in vivo NET formation were elevated with induction of pancreatitis, but significantly reduced with chloroquine treatment. Co-localization of pancreatic CitH3 (Green), with neutrophils (Ly-6G, Red) is increased in pancreatitis mice compared to sham controls (E), but treatment with CQ reduced neutrophil CitH3 expression (20x, scale bar = 50 μm). These are representative images from at least two independent analyses. *p < 0.05.
To objectively quantify the increased extracellular DNA visualized during ex vivo NET formation, supernatant cell-free DNA was measured. L-arginine pancreatitis resulted in a 6-fold increase in cell-free DNA compared to sham controls (18.4 ± 4.2 vs. 2.9 ± 0.4 ng/ml, p = 0.0004), indicative of increased NET formation during AP (Figure 2B). Treatment of CQ in pancreatitis mice decreased supernatant cell-free DNA levels in unstimulated (9.87 ± 4.3 vs. 18.37 ± 4.2 ng/ml, p = 0.03) and PAF stimulated neutrophils (40.99 ± 1.2 vs. 59.39 ± 3.6 ng/ml, p < 0.0001).
In vivo NET formation was next assessed in mice with severe acute pancreatitis. Serum levels of circulating cf-DNA were measured as a biomarker of NET formation (10). Serum cf-DNA was elevated in pancreatitis mice and was decreased with CQ treatment after 48 h (100 ± 11 vs. 122 ± 12 ng/ml, p = 0.0011, Figure 2C) and after 72 h (211 ± 28 vs. 115 ± 59 ng/mL, p < 0.001). Citrullinated histone 3 (CitH3), one of the histone proteins formed and released during NET formation, was elevated in pancreatitis at 48 h (6,228 ± 471 vs. 2,071 ± 564 pg/ml, p < 0.0001, Figure 2D) and further increased at 72 h (8,198 ± 700 vs. 6,228 ± 471 pg/ml, p = 0.0002). CitH3 levels correlated significantly with cf-DNA (Pearson r = 0.676, 95% CI 0.375–0.848, p = 0.0003). Treatment with CQ significantly reduced CitH3 levels at both time points (3,878 ± 962 vs. 6,228 ± 471 pg/ml, p < 0.0001 and 5,120 ± 876 vs. 8,198 ± 700 pg/ml, p = 0.0015). To assess for NET formation in the pancreas, CitH3 expression was evaluated in resected murine pancreatitis sections after L-arginine injection. Pancreatic neutrophil infiltration, as assessed by neutrophil Ly6g, and CitH3 was increased in pancreatitis mice compared to sham controls (Figure 2E). Unmerged images are available for review in Supplemental Figure 3. CitH3 expression co-localized with Ly6g, a neutrophil GPI-anchoring protein, suggesting that CitH3 was released from infiltrating neutrophils. Treatment of CQ markedly reduced pancreatic neutrophil infiltration and NET formation, as demonstrated by decreased CitH3/Ly-6g co-localization (Figure 2E).
Treatment with CQ in mice with pancreatitis reduced amylase (561 ± 348 vs. 2,051 ± 320 mU/ml, p < 0.0001) and trypsin activity (5.58 ± 1.5 vs. 8.44 ± 2.1 μU / ml, p = 0.006) (Figures 3A,B). As part of the systemic inflammatory response to AP, activated neutrophils and other leukocytes release pro-inflammatory compounds, further propagating local and systemic inflammation. Serum HMGB1 and IL-6 significantly increased with L-arginine induction of pancreatitis (95.9 ± 21.7 vs. 18.2 ± 9.7 ng/ml, p < 0.0001; 11.66 ± 2.82 vs. 3.85 ± 0.04 nmol) (Figures 3C,D). Administration of CQ significantly reduced serum HMGB1 (54.35 ± 14.7 vs. 95.99 ± 21.7 ng/ml, p < 0.0001, Figure 3C) and IL-6 (6.69 ± 1.07 vs. 11.66 ± 2.82 nmol, p < 0.0001, Figure 3D).
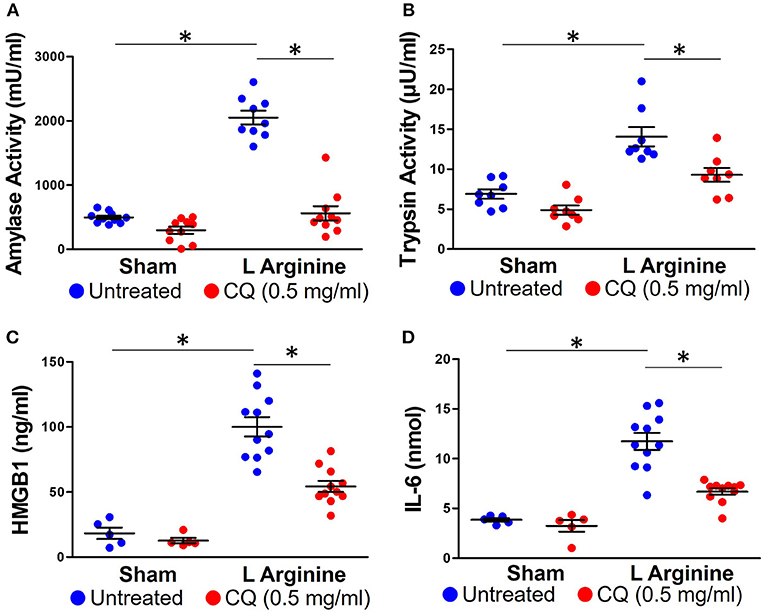
Figure 3. Chloroquine reduces local and systemic inflammation in acute pancreatitis. Chloroquine treatment led to a significant reduction in amylase (A) and trypsin (B) activity. The systemic inflammatory response to acute pancreatitis was assessed by measuring HMGB1 (C) and IL-6 (D). Chloroquine treatment led to a significant reduction in both of these circulating inflammatory markers. *p < 0.05.
In a survival model of recurrent L-arginine induced pancreatitis, CQ treatment significantly improved survival compared to untreated mice (median survival unreached vs. 15 days, p = 0.0001, Figure 4A). To confirm these findings in another model of murine acute pancreatitis, a choline deficient, ethionine (CDE) supplemented diet was also utilized to test the efficacy of CQ treatment. Treatment with CQ also significantly improved survival in CDE diet induced pancreatitis (median survival unreached vs. 4 days, p = 0.016, Figure 4B). Importantly, CQ did not have a significant effect on survival from severe acute pancreatitis in PAD4−/− mice, suggesting that NET inhibition may be a central mechanism for CQ mediated improvement in survival (Supplemental Figure 4).
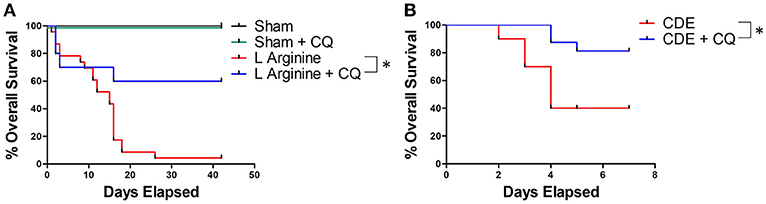
Figure 4. NET inhibition with chloroquine improves survival in murine acute pancreatitis. Chloroquine treatment led to significantly improved survival in murine severe acute pancreatitis (A, median survival unreached vs. 15 days, p = 0.0001, n = 10 per group). These profound findings were confirmed in a second model of murine pancreatitis utilizing administration of choline deficient, ethionine (CDE) supplemented diet (B, median survival unreached vs. 4 days, p = 0.016, n = 20 per group). *p < 0.05.
Acute pancreatitis (AP) is the most common disease involving the gastrointestinal tract with a mortality rate in the most severe cases, ranging from 20 to 30% (31). This mortality rate has not improved in the last three decades. No effective therapies have been developed for patients with AP outside of supportive care, highlighting the critical need for novel therapies that target the underlying pathophysiology of the disease (1). Neutrophils are critical to the development of severe AP, further promoting trypsinogen activation, recruitment of immune cells to the pancreas, and propagating tissue inflammation and damage (3). In the studies reported here, we demonstrate that NETs correlate with the severity of murine and human acute pancreatitis and that treatment with the drug chloroquine inhibits NETs and is associated with improved outcomes for severe AP.
Neutrophil extracellular traps (NETs) have emerged as a critical mediator of the innate immune response to sterile inflammatory disease (4, 7–9, 16). NETs have been identified in the pathogenesis of AP (11–13). In murine pancreatitis induced following taurocholate injection, NETs were identified within the pancreatic tissue using scanning electron microscopy to visualize co-localized DNA, histones and neutrophil elastase (11). NETs were also shown to induce trypsin activation (11). Aggregates of NET components occlude pancreatic ducts in murine IL-17 induced pancreatitis to propagate pancreatic inflammation (12). Importantly, protein arginine deiminase, type IV (PAD4), an enzyme required for NET formation that citrullinates histones to allow for decondensation, was necessary for induction of pancreatitis in this model. Herein, we expand on previous findings that NETs are a critical mediator of acute pancreatitis in two additional models of murine pancreatitis; using L-arginine (22) and a choline deficient, ethionine supplemented (CDE) diet (23) to induce pancreatitis. We found that pancreatitis primes neutrophils to increase their propensity to form NETs in an ex vivo assay. Furthermore, PAD4−/− mice, incapable of NET formation, had reduced severity and improved survival compared to wild-type controls, implicating NETs in the pathophysiology of severe pancreatitis in these models.
Because no murine model perfectly recapitulates the multiple etiologies driving human pancreatitis, establishing the relevance of NETs in human disease is critical. NET aggregates have been visualized in pancreatic ducts from human benign and malignancy associated pancreatitis (12). Patients with pancreatitis have elevated levels of NET biomarkers including cell-free DNA and DNA-histone conjugates compared with controls (11). We expanded these observations to demonstrate that NET biomarkers are not only present in human pancreatitis, but also correlate with severity of disease. While cell free DNA is non-specific for NETs and may be released from necrotic cells in necrotizing pancreatitis, MPO-DNA conjugates are a much more specific bio-marker for NET formation (32). We have also shown evidence of NETs in the pancreatic tissue during acute pancreatitis, with the immunofluorescence staining of citrullinated histone H3. While a portion of this expression does not co-localize with the neutrophil marker Ly6G, this could be due to citrullinated histone deposition from NETs in the extracellular space with clearance of Ly6G after the neutrophil dies during NETosis. Additionally, citrullination of histone H3 is a fairly specific marker of NET formation, therefore these findings do suggest NETs occur within the pancreatic tissue during acute pancreatitis. Taken together, these data suggest that NETs are a clinically relevant therapeutic target, since systemic induction of NETs are likely to contribute to the multi-system organ dysfunction that defines severe acute pancreatitis (6, 26, 33, 34).
Based on the evidence that NETs appear to be critical to the pathogenesis of murine severe acute pancreatitis and strongly associated with human severe acute pancreatitis, we evaluated NET inhibition as a novel therapeutic target to improve outcomes for patients with this disease. Strategies to deplete neutrophils or block adhesion molecules to limit neutrophil infiltration have limited tissue damage in acute pancreatitis (35–39). However, our understanding of the molecular mechanisms responsible for NET formation, allows for development of therapies to specifically target this process. Administration of DNase for NET inhibition prior to induction of acute pancreatitis resolved pancreatic tissue damage and decreased systemic inflammation in taurocholate and L-arginine induced murine acute pancreatitis (11). DNase lyses extracellular DNA released from neutrophils to prevent accumulation of NET aggregates. As a “late” inhibitor of NET formation, DNase treatment does not prevent NET formation and does little to counter-act the effects of the release of other NET components, including histones, HMGB1, elastase and other factors (40). PAD4 is an enzyme that is essential for NETosis. PAD4 inhibitors have been developed that prevent NETs and warrant further exploration in pancreatitis (41–44). Interestingly, both the receptor for advanced glycation end productions (RAGE) and HMGB1 have been studied in the development of pancreatitis (45–48) and linked to the promotion of NET formation(10, 49). This association suggests that they may exert their inflammatory effects in pancreatitis through induction of NETosis and may represent additional upstream targets for NET inhibition.
Chloroquine (CQ) is an inexpensive drug with an established safety and toxicity profile that has been discovered to inhibit NETs in pancreatic cancer, most likely through the inhibition of neutrophil autophagy, which is required for NET formation (10, 50). Chloroquine has been utilized for many years to treat patients with malaria, systemic lupus erythematosus, and rheumatoid arthritis, but has also been evaluated previously as a treatment for acute pancreatitis (51–55). Leach et al. investigated the treatment of CQ in a choline-deficient ethionine-supplemented (CDE) diet model of severe acute murine pancreatitis. CQ pre-treatment neutralized the subcellular pH of lysosomes and decreased 72 h serum amylase and trypsin activity associated with the diet-induced pancreatitis (54). These findings were supported by the work of Guillaumes et al. who showed that high-dose CQ administration improved 1-week survival in murine CDE diet induced pancreatitis from 40 to 70% (53). Despite these studies supporting the benefit of CQ in acute pancreatitis, there have also been contradictory findings (55). Our findings validate the efficacy of CQ as a treatment for AP in CDE diet induced pancreatitis and another murine model, L-arginine induced pancreatitis. CQ reduced the serum biochemical activity and levels of amylase and trypsin and also reduced systemic levels of inflammatory cytokines HMGB1 and IL-6. Importantly, CQ improved survival in two different murine models of severe acute pancreatitis.
Previously, CQ efficacy in AP was thought to be mediated by stabilization of lysosome function or by inhibiting acidification of the lysosomes to prevent activation of trypsinogen and digestive zymogens (52–54). However, controversy regarding the precise mechanism of CQ in pancreatitis remains (56). We identified inhibition of NET formation as another potential mechanism for the beneficial effects of CQ in pancreatitis. In the ex vivo NET assay, CQ treatment notably prevented spontaneous NET formation, and reduced the propensity to form NETs upon neutrophil stimulation. CQ treatment also lowered in vivo NET formation, reducing serum cell-free DNA and CitH3, a NET biomarker that allows for unwinding and expulsion of neutrophil DNA during NET formation. Immunolabeling of murine pancreatitis specimens showed that CQ treatment also decreased infiltration of neutrophils in the pancreas and pancreatic neutrophil-CitH3 co-localization. Importantly, CQ inhibits autophagy, a cell survival mechanism that utilizes degradation and recycling of damaged intracellular materials and is required for NET formation (17, 57–60), possibly by alkalinization of lysosomes (54). More recently, autophagy itself has been implicated in the pathogenesis of AP (61, 62). Other studies suggest that it is impaired, blocked, or disordered autophagy within acinar cells that initiates pancreatitis in multiple models of AP, and that restoration of autophagic flux and clearance of protein aggregates abrogates pancreatitis in these models (63). CQ also inhibits the function of activated platelets (64, 65), which are a critical mediator of the inflammatory response during acute pancreatitis (66–69). Taken together, these finding suggests that CQ, in adequate doses, minimizes the activation of trypsinogen to trypsin in the pancreatic acinar cell during cell stress (initiation of AP), and also limits progression to severe acute pancreatitis through effects on neutrophils and/or other inflammatory cells. While we demonstrate that CQ reduces NETs and that CQ treatment is associated with improved inflammatory outcomes and survival in murine acute pancreatitis, there are multiple other potential mechanisms through which CQ may be functioning, including those outlined above. In an attempt to demonstrate NET inhibition as the primary mechanism for CQ in pancreatitis, we treated PAD4−/− mice with CQ and did not observe any difference in survival as compared to wild type mice treated with CQ. This strengthens the association between CQ induced improvement in survival and NET inhibition as there is less impact for CQ when NETs are genetically diminished, however a direct causal mechanism is difficult to demonstrate. Therefore, we must interpret our results on the mechanism of CQ in acute pancreatitis and association with NET inhibition with caution.
This work is limited by the fact that no murine model of experimental pancreatitis perfectly recapitulates human disease. In consideration of this, we utilized two different murine models to demonstrate the survival benefit associated with CQ treatment. Additionally, we evaluated patient correlates to demonstrate that NETs are associated with human severe acute pancreatitis. In the current study, we initiated CQ shortly after initiation of pancreatitis to explore it as a rescue strategy for ill patients, however given the evidence that NETs may contribute to the early development of acute pancreatitis, NET inhibition should also be explored as a possible prophylactic therapeutic treatment strategy for patients at high risk for AP.
In conclusion, NETs have been identified as a key component in the pathophysiology of murine and human acute pancreatitis, driving severity and mortality, and serve as a therapeutic target to improve outcomes in AP. Chloroquine inhibits NET formation and ameliorates murine acute pancreatitis. Further study into the targeted inhibition of NETs with CQ in patients with severe acute pancreatitis or in patients at risk for recurrent pancreatitis is warranted, as well as studies designed to develop parenterally administered congeners.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
PM, ML, HZ, BB, GP, DW, and PP: study concept and design. PM, AS, MR, PL, PP, DW, GP, and BB: acquisition, analysis, and interpretation of data. PM, BB, PL, and MR: drafting of the manuscript. PM, AS, MR, PL, PP, GP, DW, AZ, ML, HZ, and BB: critical revision of the manuscript. AS, GP, DW, AZ, ML, HZ, and BB: funding, administrative, or material support.
This work was supported in part by grants from the National Institutes of Health and the Department of Defense. The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health or Department of Defense. Support for this research was provided by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 5U54GM104942-03 through the West Virginia Clinical and Translational Science Institute (BB). R01CA181450 (HZ and ML), R01CA206012-02 (HZ and ML) and the Emma Clyde Hodge Memorial Foundation. DW was supported by the U.S. Department of Defense (Award No. W81XWH-14-1-0376 and W81XWH-17-1-0502). The Nikon A1 confocal microscope used for these studies was purchased with 1S10OD019973-01, awarded to Dr. Simon C. Watkins.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00028/full#supplementary-material
AP, acute pancreatitis; CQ, chloroquine; NETs, neutrophil extracellular traps; CitH3, citrullinated histone H3; cf-DNA, cell free-DNA; IL-6, interleukin-6; PAF, platelet activating factor.
1. Tenner S, Baillie J, DeWitt J, Vege SS, American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. (2013) 108:1400–15. doi: 10.1038/ajg.2013.218
2. Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet (2015) 386:85–96. doi: 10.1016/S0140-6736(14)60649-8
3. Yang ZW, Meng XX, Xu P. Central role of neutrophil in the pathogenesis of severe acute pancreatitis. J Cell Mol Med. (2015) 19:2513–20. doi: 10.1111/jcmm.12639
4. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2017) 18:134–47. doi: 10.1038/nri.2017.105
5. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
6. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PloS ONE (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
7. Yu Y, Su K. Neutrophil extracellular traps and systemic lupus erythematosus. J Clin Cell Immunol. (2013) 4:139. doi: 10.4172/2155-9899.1000139
8. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
9. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. (2014) 16:R122. doi: 10.1186/ar4579
10. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. (2015) 22:326–34. doi: 10.1038/cgt.2015.21
11. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renstrom E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology (2015) 149:1920–31 e8. doi: 10.1053/j.gastro.2015.08.026
12. Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. (2016) 7:10973. doi: 10.1038/ncomms10973
13. Bilyy R, Fedorov V, Vovk V, Leppkes M, Dumych T, Chopyak V, et al. Neutrophil extracellular traps form a barrier between necrotic and viable areas in acute abdominal inflammation. Front Immunol. (2016) 7:424. doi: 10.3389/fimmu.2016.00424
14. Espina V, Liotta LA. Chloroquine enjoys a renaissance as an antineoplastic therapy. Clin Investig. (2013) 3:743–61. doi: 10.4155/cli.13.58
15. Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig. Drugs (2009) 10:1269–79.
16. Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, et al. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy (2017) 47:57–70. doi: 10.1111/cea.12859
17. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
18. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PloS ONE (2012) 7:e45427. doi: 10.1371/journal.pone.0045427
19. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
20. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer (2018) 18:678. doi: 10.1186/s12885-018-4584-2
21. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PloS ONE (2011) 6:e22043. doi: 10.1371/journal.pone.0022043
22. Dawra R, Sharif R, Phillips P, Dudeja V, Dhaulakhandi D, Saluja AK. Development of a new mouse model of acute pancreatitis induced by administration of L-arginine. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G1009–18. doi: 10.1152/ajpgi.00167.2006
23. Niederau C, Luthen R, Niederau MC, Grendell JH, Ferrell LD. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res. (1992) 24 (Suppl. 1):40–54. doi: 10.1159/000129238
24. Hyun JJ, Lee HS. Experimental models of pancreatitis. Clin Endosc. (2014) 47:212–6. doi: 10.5946/ce.2014.47.3.212
25. Ida S, Ohmuraya M, Hirota M, Ozaki N, Hiramatsu S, Uehara H, et al. Chronic pancreatitis in mice by treatment with choline-deficient ethionine-supplemented diet. Exp Anim. (2010) 59:421–9. doi: 10.1538/expanim.59.421
26. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, et al. Classification of acute pancreatitis−2012: revision of the Atlanta classification and definitions by international consensus. Gut (2013) 62:102–11. doi: 10.1136/gutjnl-2012-302779
27. Whitcomb DC, Muddana V, Langmead CJ, Houghton FD Jr, Guenther A, Eagon PK, et al. Angiopoietin-2, a regulator of vascular permeability in inflammation, is associated with persistent organ failure in patients with acute pancreatitis from the United States and Germany. Am J Gastroenterol. (2010) 105:2287–92. doi: 10.1038/ajg.2010.183
28. Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. (2013) e50586. doi: 10.3791/50586
29. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
30. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. (2010) 207:1853–62. doi: 10.1084/jem.20100239
31. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology (2013) 144:1252–61. doi: 10.1053/j.gastro.2013.01.068
32. Maruchi Y, Tsuda M, Mori H, Takenaka N, Gocho T, Huq MA, et al. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit Care (2018) 22:176. doi: 10.1186/s13054-018-2109-7
33. Biron BM, Chung CS, Chen Y, Wilson Z, Fallon EA, Reichner JS, et al. PAD4 deficiency leads to decreased organ dysfunction and improved survival in a dual insult model of hemorrhagic shock and sepsis. J Immunol. (2018) 200:1817–28. doi: 10.4049/jimmunol.1700639
34. Gao X, Hao S, Yan H, Ding W, Li K, Li J. Neutrophil extracellular traps contribute to the intestine damage in endotoxemic rats. J Surg Res. (2015) 195:211–8. doi: 10.1016/j.jss.2014.12.019
35. Frossard JL, Saluja A, Bhagat L, Lee HS, Bhatia M, Hofbauer B, et al. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology (1999) 116:694–701. doi: 10.1016/S0016-5085(99)70192-7
36. Hartman H, Abdulla A, Awla D, Lindkvist B, Jeppsson B, Thorlacius H, et al. P-selectin mediates neutrophil rolling and recruitment in acute pancreatitis. Br J Surg. (2012) 99:246–55. doi: 10.1002/bjs.7775
37. Awla D, Abdulla A, Syk I, Jeppsson B, Regner S, Thorlacius H. Neutrophil-derived matrix metalloproteinase-9 is a potent activator of trypsinogen in acinar cells in acute pancreatitis. J Leukoc Biol. (2012) 91:711–9. doi: 10.1189/jlb.0811443
38. Abdulla A, Awla D, Thorlacius H, Regner S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol. (2011) 90:975–82. doi: 10.1189/jlb.0411195
39. Sandoval D, Gukovskaya A, Reavey P, Gukovsky S, Sisk A, Braquet P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology (1996) 111:1081–91. doi: 10.1016/S0016-5085(96)70077-X
40. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. (2015) 6:6673. doi: 10.1038/ncomms7673
41. Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. (2015) 74:2199–206. doi: 10.1136/annrheumdis-2014-205365
42. Slack JL, Causey CP, Thompson PR. Protein arginine deiminase 4: a target for an epigenetic cancer therapy. Cell Mol Life Sci. (2011) 68:709–20. doi: 10.1007/s00018-010-0480-x
43. Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, et al. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry (2006) 45:11727–36. doi: 10.1021/bi061180d
44. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. (2015) 11:189–91. doi: 10.1038/nchembio.1735
45. Kang R, Chen R, Xie M, Cao L, Lotze MT, Tang D, et al. The receptor for advanced glycation end products activates the AIM2 inflammasome in acute pancreatitis. J Immunol. (2016) 196:4331–7. doi: 10.4049/jimmunol.1502340
46. Kang R, Lotze MT, Zeh HJ, Billiar TR, Tang D. Cell death and DAMPs in acute pancreatitis. Mol Med. (2014) 20:466–77. doi: 10.2119/molmed.2014.00117
47. Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology (2014) 146:1097–107. doi: 10.1053/j.gastro.2013.12.015
48. Yu C, Yu X, Zhu HW, Li X, Huang LH, Li ZQ, et al. Expression pattern of HMGB1 and its association with autophagy in acute necrotizing pancreatitis. Mol Med Rep. (2016) 14:5507–13. doi: 10.3892/mmr.2016.5945
49. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep. (2018) 8:2068. doi: 10.1038/s41598-018-20479-x
50. Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:2532–44. doi: 10.1002/art.38703
51. Zhang L, Chen Y, Wang L, Chen XP, Zhang WG, Wang CY, et al. Chloroquine relieves acute lung injury in rats with acute hemorrhagic necrotizing pancreatitis. J Huazhong Univ Sci Technol Med Sci. (2013) 33:357–60. doi: 10.1007/s11596-013-1124-9
52. Seyama Y, Otani T, Matsukura A, Makuuchi M. The pH modulator chloroquine blocks trypsinogen activation peptide generation in cerulein-induced pancreatitis. Pancreas (2003) 26:15–7. doi: 10.1097/00006676-200301000-00003
53. Guillaumes S, Blanco I, Villanueva A, Sans MD, Clave P, Chabas A, et al. Chloroquine stabilizes pancreatic lysosomes and improves survival of mice with diet-induced acute pancreatitis. Pancreas (1997) 14:262–6. doi: 10.1097/00006676-199704000-00007
54. Leach SD, Bilchik AJ, Karapetian O, Gorelick FS, Modlin IM. Influence of chloroquine on diet-induced pancreatitis. Pancreas (1993) 8:64–9. doi: 10.1097/00006676-199301000-00013
55. Lerch MM, Saluja AK, Dawra R, Saluja M, Steer ML. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology (1993) 104:1768–79. doi: 10.1016/0016-5085(93)90658-Y
56. Otani T. Chloroquine improves survival of mice with diet-induced acute pancreatitis. Pancreas (1998) 17:318–20. doi: 10.1097/00006676-199810000-00017
57. Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. (2015) 22:4402–10. doi: 10.1245/s10434-015-4566-4
58. Kang R, Tang D. Autophagy in pancreatic cancer pathogenesis and treatment. Am J Cancer Res. (2012) 2:383–96.
59. Sha LL, Wang H, Wang C, Peng HY, Chen M, Zhao MH. Autophagy is induced by anti-neutrophil cytoplasmic Abs and promotes neutrophil extracellular traps formation. Innate Immun. (2016) 22:658–65. doi: 10.1177/1753425916668981
60. Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol. (2016) 7:879. doi: 10.3389/fmicb.2016.00879
61. Liu X, Guo X, Li J, Wu M, Zhan X. Interferon-gamma aggravated L-arginine-induced acute pancreatitis in sprague-dawley rats and its possible mechanism: trypsinogen activation and autophagy up-regulation. Pancreas (2017) 46:619–25. doi: 10.1097/MPA.0000000000000826
62. Ji L, Li L, Qu F, Zhang G, Wang Y, Bai X, et al. Hydrogen sulphide exacerbates acute pancreatitis by over-activating autophagy via AMPK/mTOR pathway. J Cell Mol Med. (2016) 20:2349–61. doi: 10.1111/jcmm.12928
63. Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E, et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology (2018) 154:689–703. doi: 10.1053/j.gastro.2017.10.012
64. Espinola RG, Pierangeli SS, Gharavi AE, Harris EN. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost. (2002) 87:518–22. doi: 10.1055/s-0037-1613033
65. Nosal R, Jancinova V, Danihelova E. Chloroquine: a multipotent inhibitor of human platelets in vitro. Thromb Res. (2000) 98:411–21. doi: 10.1016/S0049-3848(00)00200-0
66. Mimidis K, Papadopoulos V, Kartasis Z, Baka M, Tsatlidis V, Bourikas G, et al. Assessment of platelet adhesiveness and aggregation in mild acute pancreatitis using the PFA-100 system. JOP (2004) 5:132–7.
67. Mimidis K, Papadopoulos V, Kotsianidis J, Filippou D, Spanoudakis E, Bourikas G, et al. Alterations of platelet function, number and indexes during acute pancreatitis. Pancreatology (2004) 4:22–7. doi: 10.1159/000077024
68. Abdulla A, Awla D, Hartman H, Rahman M, Jeppsson B, Regner S, et al. Role of platelets in experimental acute pancreatitis. Br J Surg. (2011) 98:93–103. doi: 10.1002/bjs.7271
Keywords: neutrophil extracellular traps, pancreatitis, chloroquine, autophagy, systemic inflammatory response, citrullinated histone
Citation: Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI, Whitcomb DC, Zureikat AH, Lotze MT, Zeh HJ III and Boone BA (2019) Enhanced Neutrophil Extracellular Trap Formation in Acute Pancreatitis Contributes to Disease Severity and Is Reduced by Chloroquine. Front. Immunol. 10:28. doi: 10.3389/fimmu.2019.00028
Received: 02 October 2018; Accepted: 08 January 2019;
Published: 24 January 2019.
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Elmar Pieterse, Radboud University Nijmegen Medical Centre, NetherlandsCopyright © 2019 Murthy, Singhi, Ross, Loughran, Paragomi, Papachristou, Whitcomb, Zureikat, Lotze, Zeh and Boone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian A. Boone, YnJpYW4uYm9vbmVAaHNjLnd2dS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.