
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 January 2019
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.03125
This article is part of the Research Topic The Role of Complement in Health and Disease View all 50 articles
 Marcin Okrój1*
Marcin Okrój1* Jan Potempa2,3*
Jan Potempa2,3*The complement system, an evolutionarily ancient component of innate immunity, is capable of protecting hosts from invading pathogens, either directly, by lysis of target cells, or indirectly, by mobilization of host immune mechanisms. However, this potentially cytotoxic cascade must be tightly regulated, since improperly controlled complement can damage healthy cells and tissues. The practical importance of this axis is highlighted when impairment of complement regulators or bacterial mechanisms of complement evasion result in pathogenic conditions. Recognition of complement as a “double-edged sword” is widely acknowledged, but another, currently underappreciated aspect of complement function has emerged as an important player in homeostatic balance—the dual outcome of complement-mediated inflammation. In most cases, the proinflammatory properties of complement are beneficial to the host. However, certain pathogens have developed the ability to utilize local inflammation as a source of nutrients and as a way to establish a niche for further colonization. Such a strategy can be illustrated in the example of periodontitis. Interestingly, certain tumors also seem to benefit from complement activation products, which promote a proangiogenic and immunosuppressive microenvironment.
The term “inflammo-philic” (= loving or attracting inflammation) was introduced in 2014 by George Hajishengallis to describe dysbiotic microbiome on the tooth surface below the gum line, which thrive in the inflammatory environment of periodontal pockets (1). Remarkably, as described in details later in this review, bacteria responsible for initiation and progression of periodontitis (periodontopathogens) have the unique ability to manipulate the complement system to disengage bacterial clearance from inflammation.
In general, the local inflammatory response to bacterial and fungal pathogens triggered by complement activation is absolutely essential to eliminate invaders (2, 3). Therefore, all successful pathogens developed a large variety of means to interfere with complement activation and/or hinder complement-dependent bacterial clearance mechanisms (2–6) (Table 1). Unfortunately, if inflammatory reaction triggered by pathogens escape the control it becomes highly detrimental to the host as illustrated by invasive candidiasis [Candida albicans (7)], meningitis [Neisseria meningitidis (8)], and sepsis [N. meningitidis (8), Staphylococcus aureus (9), and Streptococcus pyogenes (10)]. It needs to be kept in mind that an overwhelming inflammatory response and a dysregulated immune response to these infections is by no means the manifestation of an inflammophilic character of these pathogens since the controlled, local inflammation is protective against these pathogens (9). Therefore, the pathogenic strategy to endure inflammation but in the same time to take advantage of it, seems to be limited to periodontopathogens. It is fascinating that the apparently similar strategy is employed by cancer and in both cases exploitation of the complement system underlines pathology.
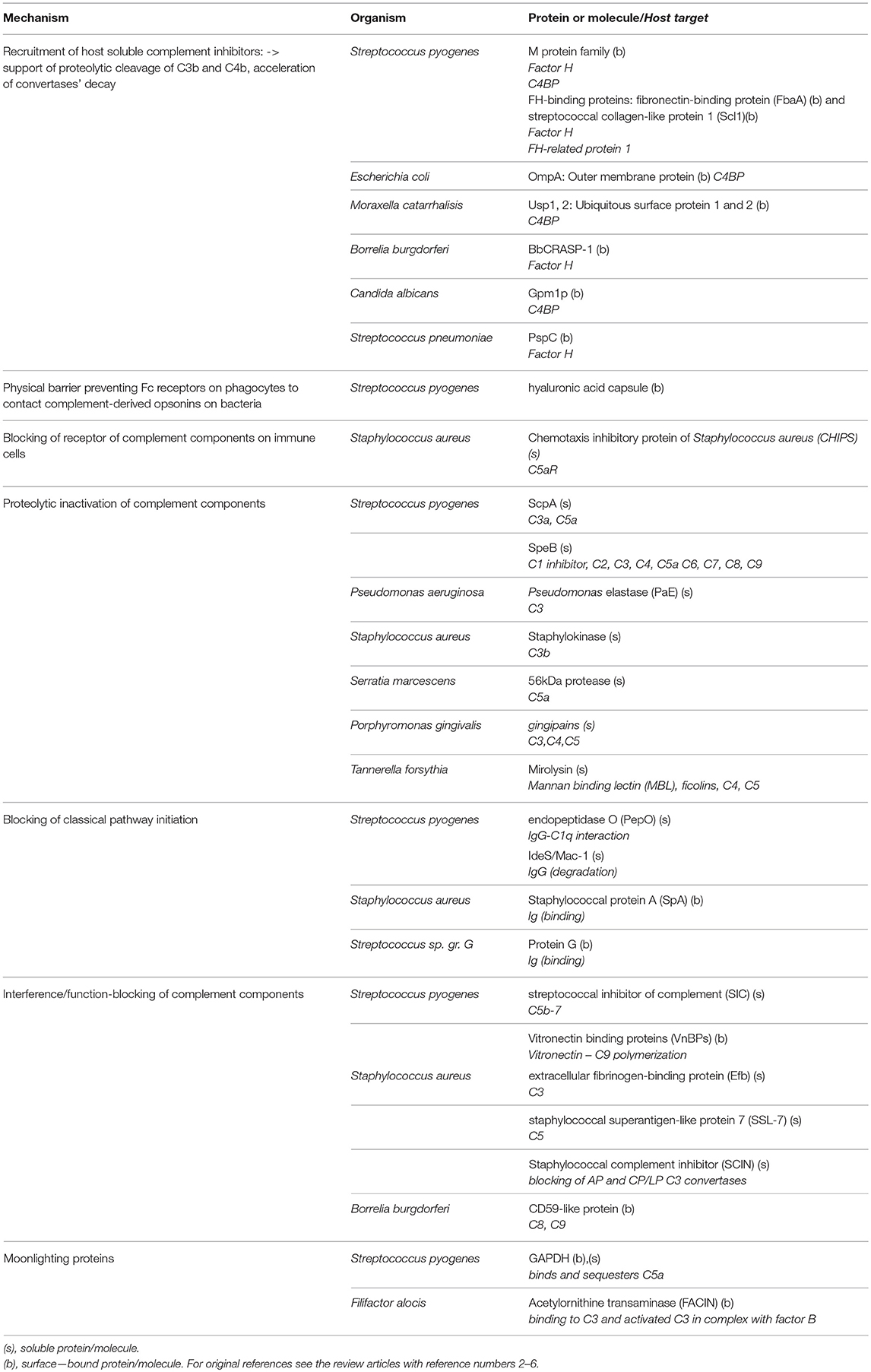
Table 1. Exemplary complement evasion strategies used by microbes.
The complement system is one of the oldest mechanisms of immunity. Its essential components, such as the C3 molecule, have existed through more than 500 million years of evolution (11). A primitive complement system probably appeared in the common ancestor of eumetazoa, and its original role was limited to opsonization and the induction of inflammation. Genetic events like the duplication-based appearance of pathway specific components (e.g., factor B and C2) and the gain of terminal pathway constituents (C5–C9) allowed the primodal complement system to evolve into a an advanced and complex defense system capable not only of promoting osmotic lysis of target cells, anaphylaxis, and phagocytosis, but also of crosstalk with other systems (e.g., coagulation) and signaling pathways (e.g., Toll-like receptors) involved in the maintenance of bodily homeostasis (12, 13). There are three independent complement cascades, the evolutionarily older alternative and lectin pathway (with basic elements like C3, MASPs, and factor B existing in invertebrates) and the relatively younger classical pathway developed in jawed vertebrates (11). The alternative complement pathway is constitutively active at a low level due to the spontaneous breakdown of C3 into anaphylatoxin C3a and the active C3b fragment, which activate downstream steps in the cascade. Therefore, propagation of the alternative pathway does not depend on specific activation but relies on the lack of inhibition by numerous endogenous regulators that differentiate self and non-self surfaces. This mechanism ensures constant monitoring of the body. In contrast to the alternative pathway, the classical and lectin pathways require specific stimuli, such as antibodies, C-reactive protein, phosphatydylserine, or certain sugar moieties, to be present on the surface of target cells (14–16). The upstream components of both pathways, including C1q, mannan binding protein (MBL), and the ficolins, act as sensors and thus can be considered soluble pattern recognition molecules (PRMs) (17, 18). All pathways converge at the level of the central complement molecule C3. C3 is processed by enzymatic complexes called complement convertases into C3a anaphylatoxin and the C3b fragment, which in turn forms C5 convertases. C5 convertases cleave the C5 molecule into C5a anaphylatoxin and the C5b fragment, which initiates the common terminal pathway. Binding of C6, C7, C8, and C9 leads to formation of the membrane attack complex (MAC), which targets the cell membrane and causes osmotic lysis. A schematic representation of the complement system is shown in Figure 1.
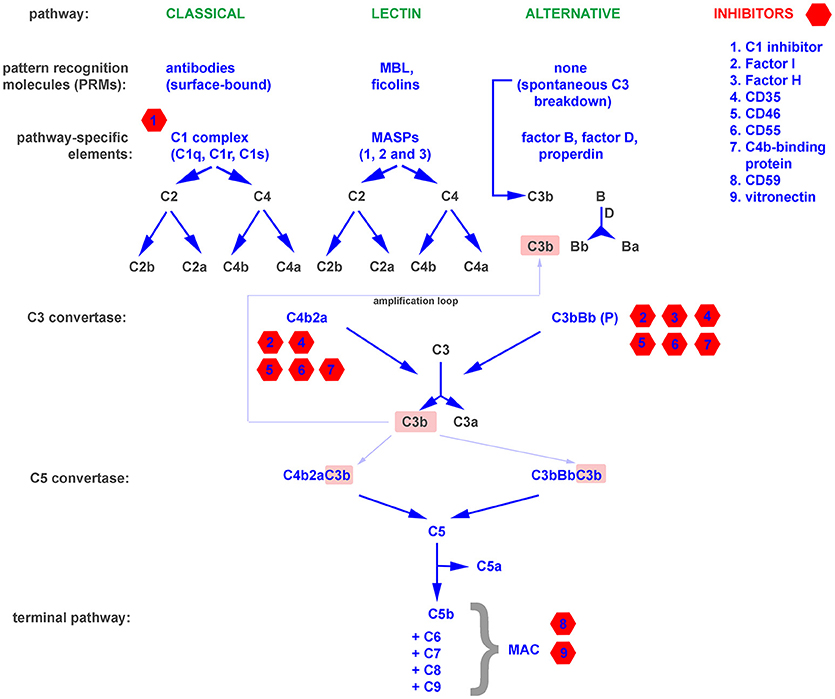
Figure 1. A simplified representation of the complement system, divided into pattern recognition molecules (PRMs), pathway specific and common components as well as inhibitors.
The binding of antibodies as a stimulus for the initiation of the complement cascade bridges the innate and adaptive immune systems. Moreover, opsonins like C3b and their degradation products act as natural adjuvants, contributing to proper presentation of antigens to lymphocytes and providing a co-stimulatory and anti-apoptotic signal for B cells (19). Deficiencies of complement, while relatively rare, emphasize the importance of this multifunctional protein cascade (20). The exact symptoms that develop depend on precisely which complement components are lacking or impaired. Deficiencies in essential components of the alternative pathway and the terminal pathway result in higher susceptibility to recurrent bacterial infections, especially these caused by Neisseriae and incidence peak up in early childhood (20). The lack of early components of the classical pathway predisposes to systemic lupus erythematosus (SLE), a disease in which the scavenging function of complement is impaired, and thus debris from dying cells persists and can act as a source of autoantigens (21). Autoimmune diseases that stem from direct damage of cells and tissues typically arise from deficiencies in complement inhibitors that normally protect the host from excessive or misguided complement attacks (22). These include C3 glomerulopathies, atypical hemolytic uremic syndrome (aHUS), age-related macular degeneration (AMD), paroxysmal nocturnal hemoglobinuria (PHN), and many more (20). Paradoxically, a deficiency of functional complement inhibitors such as factor H (the main soluble inhibitor of the alternative pathway) can also result in a deficiency of complement activation. Factor H is the main soluble inhibitor of alternative pathway, which prevents propagation of cascade beyond the spontaneous breakdown of C3 and formation of C3 convertase (Figure 1). The lack of such inhibitor fuels a positive feedback mechanism that unproductively depletes complement and leaves the host without an important line of defense (23). On the other hand, unwanted complement activation is an effector mechanism in many inflammatory diseases, including rheumatoid arthritis (24), diabetic nephropathy (25), and ischemia/reperfusion injury (26). All these examples support a perception of the complement system as a “double-edged sword,” where a proper balance is pivotal for maintaining protection while avoiding autoimmunity. Both microbial infections and tumors influence this physiological equilibrium and employ two main strategies for survival in a complement-saturated microenvironment.
Innate immunity relies on recognition of a spectrum of pathogen-associated molecular patterns (PAMPs), invariable molecular determinants typical for the most common invaders, including lipopolysaccharide (LPS), lipoteichoic acid, flagellin, double-stranded RNA, β1-3 glucan, N-formylmethionine peptides, and many more. The constant region of the antibody heavy chain (Fc) also falls into this category of molecules. Pathogen-associated molecular patterns (PAMPs) bind to specific receptors on innate immune cells and activate effector mechanisms such as complement or antibody-dependent cell cytotoxicity (ADCC). Molecules that sense PAMPs and trigger immune system activation are called pattern recognition receptors (PRRs) and include Toll-like receptors (TLRs), C-type lectins, and NOD-like receptors. Soluble molecules, which exert an analogical role are termed as PRMs and the most upstream components of the classical and lectin pathways (C1 complex, MBL, and ficolins) belong to this group (18). Complement is therefore a multispecific and powerful defense system against pathogens that is theoretically capable of eliminating every cell unless constrained by endogenous complement inhibitors. In practice, since complement co-evolved with pathogens over millions of years, pathogens have developed various mechanisms to evade complement attack. Pathogens employ a variety of tactics for this purpose, including proteolytic cleavage of complement components, mimicking and hijacking host complement inhibitors, inactivation of the C3 molecule, preventing of complement-mediated activation of immune cells, depletion of antibodies, and unproductive exhaustion of early complement components [reviewed in (2, 4, 27–29)]. Selected examples of abovementioned strategies are given in Table 1.
Similarly to bacterial, fungal, or viral pathogens expressing PAMPs, tumor cells are visible to the immune system due to changes in their mutational or metabolic status, which is reflected by changes in the expression of cell surface molecules. The presentation of epitopes derived from mutated proteins (so-called neoantigens) within MHC I molecules (30) as well as the peroxidation of membrane lipids or changed patterns of glycosylation distinguish tumor cells from normal cells.
Spontaneous fixation of complement onto the surface of tumor cells is of low physiological relevance due to the low titer of naturally occurring antitumor antibodies and the expression of complement inhibitors by tumor cells (31). The introduction of antitumor monoclonal antibodies, which is considered a breakthrough in tumor immunology, enabled researchers to use the cytotoxic potential of the complement system to combat cancer (32). Complement-activating therapeutics like the anti-CD20 antibodies rituximab and ofatumumab are first-line therapies in the treatment of B cell malignancies. However, certain patients fail to respond or only partially respond to antitumor antibodies, and one possible explanation is the unfavorable ratio of the molecular target (e.g., CD20) to membrane-bound complement inhibitors on the surface of tumor cells (33, 34). Successful experiments in which bispecific antibodies against CD20 and CD55 were used (35) or in which complement inhibitors were silenced (36) support the theory that inhibition of complement by tumor cells is an important mechanism of cancer resistance. Expression of membrane-bound complement inhibitors like CD35 (Complement receptor 1, CR1), CD46, CD55, and CD59 is typical for nucleated cells, and the majority of cell types express at least one of these molecules. In contrast, the production of soluble complement inhibitors such as factor I, factor H, C4b-binding protein (C4BP) is usually the domain of liver hepatocytes, and there are only few extrahepatic sources of fluid-phase complement regulators (37). However, the expression of soluble complement inhibitors by tumor cells has been described, and it seems to provide an additional level of protection, as shown in an in vitro model of non-small lung cancer cell (NSCLC) lines expressing factor I, C4BP, and factor H (38). The tumor-supporting effect of endogenous factor H expressed by NSCLC cells was shown in vivo in a mouse xenograft model (39, 40). Further evidence for the pro-tumor effect of soluble complement inhibitors comes from analysis of tissue microarrays of breast cancer specimens. Expression of factor I was positively correlated with tumor size, de-differentiation score (Nottingham scale), and poor prognosis (cancer-specific survival and recurrence-free survival) (41). Other investigators reported a correlation between factor I expression and tumor aggressiveness in cutaneous squamous cell carcinoma (42). In addition to expressing soluble complement inhibitors, tumor cells can also hijack these proteins from the plasma. Horl et al. showed that blocking factor H binding to the surface of leukemia cells increased the cytotoxicity of rituximab (43) and ofatumumab (44). Although factor H is an inhibitor of the alternative complement pathway, it plays a role in enhancing the complement cascade when it is initiated via the classical pathway (such as by antitumor antibodies) at the level of C3b formation. C3b gives rise to an amplification loop (Figure 1) due to the formation of alternative convertases, which are targets for factor H. Points of action of particular complement inhibitors are indicated in Figure 1. Another possible way to increase tumor cell resistance to complement attack is removal of the MAC from the surface, a process dependent on endocytosis or active rearrangement of the cell membrane mediated by phosphorylation of essential signaling proteins (45).
The logical consequence of complement inhibition by microbes and tumor cells at various stages of the cascade is a more aggressive and more drug-resistant phenotype, as discussed above. However, the picture is not as simple as it may originally seem, and another strategy used by pathogens and tumor cells to evade complement has been described.
Low oxygen concentration is a feature of rapidly growing solid tumors, which cannot develop the vasculature necessary for the efficient supply of nutrients to proliferating neoplastic cells. Therefore, the expanding tumor mass sooner or later develops hypoxic cores. Normal cells are equipped with a sensor of oxygen concentration that works at the transcriptional level. Hypoxia-inducible factor Iα (HIF-1α) can stabilize the p53 tumor suppressor, triggering either apoptotic signaling or metabolic reprogramming of the cell (46). Both of these processes lead to changes in the molecules expressed at the cell surface, which has the effect of making the tumor cell visible to the immune system. Previous studies with human umbilical vein endothelial cells (HUVECs) revealed that these cells activate the classical complement pathway in response to hypoxia and as well as during subsequent reoxygenation. At the same time, HUVECs increased their surface expression of two membrane-bound complement inhibitors, CD46 and CD55 (47), which induce the proteolytic cleavage of activated complement components C3b and C4b, respectively, and the dissociation of the corresponding complement convertases. Another study showed a 3.6-fold increase in HUVEC expression of complement receptor 1 (CR1 or CD35) after 48 h of hypoxia (48). These results suggest that endothelial cells actively counteract complement activation under hypoxic conditions, and therefore the expression of complement inhibitors in hypoxic NSCLC cells was studied (49). These cells not only expressed membrane-bound complement inhibitors but also produced soluble inhibitors of complement, including C4BP and factors I and H (38). In contrast to HUVECs, NSCLCs significantly downregulated the mRNA expression of all complement inhibitors tested except CD59 after 24 h of hypoxia, but a drop in the mRNA expression of soluble complement inhibitors was detected as early as 6 h after hypoxic challenge. Importantly, this rapid decrease did not correspond to the number of dying cells, which did not significantly increase in first 24 h (49). The conclusion is that unlike endothelial cells, NSCLCs do not utilize protection mechanisms that prevent the deposition of early complement components during hypoxia, but they do maintain expression of CD59, which protects from the terminal stages of complement attack (the insertion of the MAC into the membrane) (47–49).
From the research reviewed above, it has become apparent that lung cancer cells may benefit from the propagation of local inflammation mediated by C3a and C5a. Possible scenarios include the production of proangiogenic and growth factors by tumor-infiltrating lymphocytes and macrophages as well as the mobilization of immune suppressor cells that impair tumor antigen presentation (50–53). Indirect support for this hypothesis comes from studies done by Ajona et al. who reported elevated C4d deposition in lung tumors and its correlation with decreased survival (54). Moreover, high levels of soluble C4d in the plasma could discriminate between patients with benign pulmonary nodules and lung cancer (55), and were associated with reduced survival of individuals with early and advanced lung cancer. C4d levels in the plasma were also reduced after surgical removal of the tumor (54). C4d is an end degradation product of the activated C4b molecule, a hallmark of classical complement pathway activation. For that reason, one can assume that the survival and malignant potential of NSCLC cells is based on stimulation of complement. Indeed, anaphylatoxin C5a is one of the key players in complement-mediated support of lung cancer growth. Corrales et al. found that C5 deposition and subsequent C5a generation in NSCLC cells was much higher than in non-malignant bronchial epithelial cells in the presence of serum (56). Interestingly, tumor cells but not non-transformed cells produced endogenous C5, and C5a generation took place even in the absence of serum. C5a levels in the plasma of lung cancer patients were also found to be elevated, similarly to C4d levels. C5a also stimulated migration and tube formation by HUVECs in vitro. Finally, the impact of C5a was tested in a syngenic mouse model of 3LL lung cancer. Microvessel density was compared in 3LL tumors in mice treated with a C5a receptor (C5aR) antagonist. Tumors in the mice treated with the C5aR antagonist showed significantly fewer microvessels (56). Additionally, C5a signaling positively influenced the recruitment of myeloid-derived suppressor cells (MDSCs; CD11b+, Ly6c+), as blockade of C5aR reduced the number of MDSCs in tumor-bearing mice. The authors also found decreased expression of molecules associated with an immunosuppressive state and silencing of the immune response (ARG1, CTLA-4, IL-10, LAG3, and PD-L1) in C5aR antagonist-treated mice (57–59).
Importantly, the first evidence for the impact of C5a on the mobilization of MDSCs into the tumor mass was shown by Markiewski et al. in the TC-1 tumor model, a lung epithelial cell line expressing human papilloma virus (HPV) E6 and E7 antigens (60). The authors found that C5aR-deficient mice developed smaller tumors than wild-type littermates, and the same effect was observed when a C5aR antagonist was administered. However, in this model, the slower rate of tumor growth in C5aR antagonist-treated animals was not dependent on tumor cell proliferation/apoptosis or angiogenesis, as evidenced by analysis of end-point tumor specimens. Conversely, there were differences in the infiltration of tumor tissue by cytotoxic T cells, the main effectors of the antitumor immune response. Profiling of MDSCs isolated from tumors and spleens of C5aR-deficient, tumor-inoculated animals confirmed that C5a contributes to the accumulation of MDSCs in peripheral lymphoid organs and their migration into tumors. Of note, MDSCs isolated from mice with disabled C5aR signaling were less able to suppress T cell proliferation in vitro. This deficiency was linked to lower production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in mononuclear MDSC from C5aR-deficient animals.
Similarly to lung cancer cells, endogenous C5a generation by pancreatic and colon cancer cells was later reported. These cells processed C5 with a cell surface-expressed serine protease and expressed C5aR, suggesting autocrine activation of complement (61). In ovarian cancer, endogenous production of complement components and autocrine stimulation of the anaphylatoxin receptors C3aR and C5aR was suggested to be an important mechanism supporting tumor growth (62). The observed effect was independent of infiltration by cytotoxic T cells, since experiments with silenced expression of C3 yielded the same result (i.e., reduced tumor growth) in CD8 T cell-sufficient and -deficient mice. A direct effect of C3aR and C5aR agonists on proliferation, migration, and invasion of tumor cells has also been reported. Finally, quantification of C3 mRNA in tumors from patients with ovarian cancer showed that overall survival in patients with low tumor expression of C3 was more than double that of patients with high expression of C3 in the tumor (62).
In recent years, there has also been growing evidence for the pro-tumor activity of anaphylatoxins and anaphylatoxin receptors in either tumor cells or the tumor stroma in multiple tumors types, including melanoma, breast, ovarian, cervical, colon, and intestinal cancer, as well as sarcoma [reviewed in (63)]. Interestingly, in addition to the larger body of work focusing on the role of C3a and C5a in promoting tumor growth, recent studies have described a pro-tumor effect of factor B silencing (64) as well as a complement-independent enhancement of tumor growth, adhesion, and angiogenesis by C1q produced by the tumor stroma (65). The concept of complement activation supporting tumor growth provided the rationale for combined inhibition of C5a and PD-1 (66, 67), suggesting that targeting complement may be an effective anticancer treatment. These novel discoveries may be perceived as contradictory to the acknowledged theory that complement inhibits tumor growth. For example, NSCLC cells, which have been shown to benefit from C5a generation (56), were previously shown to form smaller tumors in a mouse xenograft model when their endogenous expression of the complement inhibitor factor H was silenced (40). In addition, potent complement activators such as the anti-CD20 immunotherapeutics rituximab and ofatumumab are first-line therapies for treatment of B cell malignancies (33). Notably, solid and circulating tumors have different requirements for growth. While sold tumors are typically depend on angiogenesis, migration, degradation of extracellular matrix, liquid tumors originate in the bone marrow, peripheral blood, or lymph nodes, which are rich in both nutrients and complement. Even solid tumors of the same origin can differ one from another in their mutational status, basal expression of growth factors and metalloproteinases, and metabolic rate. All of these parameters can influence the overall effect of complement activation on tumorigenesis and/or tumor progression. Finally, tumor cells often produce both complement activators and complement inhibitors. Thus, it seems as though tumor cells actively regulate the complement system depending on microenvironmental conditions, rather than simply avoiding constitutive inhibition or activation of complement (68).
Despite the extremely long phylogenetic distance between eukaryotic cells and bacteria, some prokaryotes have acquired strategies similar to tumor cells, which utilize the host inflammatory status to create favorable survival conditions. Bacterial growth in the human body is less dependent on neovascularization than tumor growth, and in contrast to tumor cells, bacteria do not have to overcome internal mechanisms controlling proliferation. Moreover, most bacteria can stand much harsher conditions than eukaryotic cells in terms of pH, oxygen tension, temperature, concentration of metabolites, etc. Nevertheless, the common feature between bacteria and tumor cells is the demand for nutrients and certain microelements. While solid tumors induce angiogenesis to acquire a source of nutrients, bacteria can successfully utilize products from the breakdown of local tissue. Therefore, tissue-destructive processes linked to local inflammation form permissive conditions for prokaryotic pathogens, which can survive immune attack. An additional benefit of this strategy is the elimination of inflammation-sensitive bacterial species (human commensals or normal microflora) that normally compete within the same niche (69, 70).
One of the most well-documented examples of bacteria hijacking host immunity to create an environmental niche occurs in periodontitis, a chronic inflammatory disease characterized by dysbiosis that results in degradation of the gingiva and tooth-supporting bone and ultimately leads to tooth loss (1). Periodontal disease begins from dental plaque, a microbial matrix colonizing the gum line usually as a result of inefficient oral hygiene (71). The next stage, gingivitis, is characterized by local inflammatory response to microbial plaque. The switch between non-destructive gingivitis and destructive periodontitis involve dysbiosis of the normal oral microbiome. The dysbiotic process results from an imbalance of homeostasis caused by so-called keystone pathogens (72). A keystone pathogen is usually a microorganism of low abundance that induces changes in the composition of the local microflora by introducing a new selective pressure, such as inflammation. In periodontitis, the keystone pathogen is Porphyromonas gingivalis. However, as shown by studies in mice, this Gram-negative bacteria cannot establish periodontitis by itself, but requires commensal microbes. These microbes are then converted from a symbiotic into a dysbiotic community. Pivotal experiments showed that bone loss was reduced when C3aR- or C5aR-deficient mice were inoculated with P. gingivalis and that no changes in the oral microbiota were observed in these knockout mice after P. gingivalis inoculation, in contrast to wild-type mice (69). As some tumor cells generate C5a through their surface enzymes, so does P. gingivalis. It is equipped with gingipains, outer membrane-anchored bacterial surface arginine-specific proteases with C5 convertase-like activity (73, 74). Importantly, gingipains release C5a from C5, but at higher concentrations, they degrade the larger fragment (C5b), thus preventing MAC formation (75). C3 and C4 complement proteins are also degraded by high concentrations of gingipains. Thus, human serum pre-incubated with clinical strains of P. gingivalis but not mutants lacking gingipains is devoid of bactericidal activity (74, 76). Additionally, gingipains interact with the C1 complex and increase its deposition onto bacteria surfaces (74). Based on these findings, one can postulate a biphasic effect of P. gingivalis proteolytic enzymes. A low abundance of bacteria initiates the classical complement pathway, but increasing numbers of bacteria results in the degradation of crucial complement components, leading to osmotic lysis. C5 is present in gingival crevicular fluid at concentration corresponding to 70% of that in serum and the active C5a anaphylatoxin can be locally released by convertases and bacterial proteases (77). C5a is a strong inflammatory mediator that increases vascular permeability and attracts and modulates the function of neutrophils, monocytes, and mast cells. All these events are considered antimicrobial events. Paradoxically, P. gingivalis' strategy for immune subversion by proinflammatory C5a involves targeted immunosuppression of macrophages. C5a affects intracellular killing of engulfed P. gingivalis by RNS and corrupts the crosstalk between C5aR and TLR2, one of the most important PRMs in antibacterial innate immunity (73). At the same time, C5aR-TLR2 crosstalk results in release of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α, which accelerate bone resorption and thus contributes to the pathological mechanism of periodontitis. Similarly, P. gingivalis spoils intracellular killing mechanism but not proinflammatory activity of neutrophils by degradation of TLR2 adaptor molecule MyD88 provoked by concomitant activation of TLR2 and C5aR (78).
Another functional similarity between P. gingivalis and tumor cells, which produce either complement inhibitors or complement components, is the fact that P. gingivalis not only possesses the proteolytic machinery to generate C5a but also expresses a unique enzyme, peptidyl arginine deiminase (PPAD), which can citrullinate the C-terminal arginine in C5a, a modification that results in substantial loss of anaphylatoxin chemotactic activity (79). This suggests that the evolutionary goal of pathogens like P. gingivalis is not constitutive activation or inhibition of the complement system, but rather the ability to actively control complement status depending on its current needs. As a keystone pathogen, P. gingivalis is a low-abundance species that plays a major role in remodeling the local microbiota community (69). Following establishment of P. gingivalis infection, a succession of other dysbiotic species proliferates in the periodontal plaque. Some express their own complement inhibitors, such as Tannerella forsythia, which produces karilysin and mirolysin (80, 81), Filifactor alocis, which produces FACIN (82), and Prevotella intermedia, which produces interpain A (83). Of note, interpain A works in concert with gingipains in the initial stages of infection, as both proteins activate the C1 complex and increase its deposition onto the cell surface. A dysbiotic bacterial community may promote a transcriptomic response that further improves bacterial fitness by regulation of nutrient acquisition and expression of virulence factors (84). This process resembles the crosstalk between tumor cells and the stroma, at least to a certain extent. Tumor cells can drive the polarization of infiltrating immune cells (e.g., into M2 macrophages), which in turn benefit the tumor cells by, for example, expressing angiogenic cytokines (85, 86). In addition, carcinoma-associated fibroblasts (CAF), which differentiate from normal fibroblasts upon stimulation by cancer-derived cytokines such as TGF-β, have emerged as important players in cancer progression and metastasis (87, 88).
The role of the complement system in combating bacteria and cancer is more complicated than was initially believed. Certain pathogens have evolved the ability not only to evade complement attack but also to use it as a tool for establishing their own niche, while remaining protected from complement-mediated lysis. Such a strategy seems to be widespread in nature and has been adopted by both bacteria and tumor cells (Figure 2). It seems likely that additional pathogenic strategies remain to be discovered, and thus one must be careful when designing complement-based therapeutics. On the other hand, anti-complement approaches may be effective in the treatment of infections caused by inflammophilic microbes.
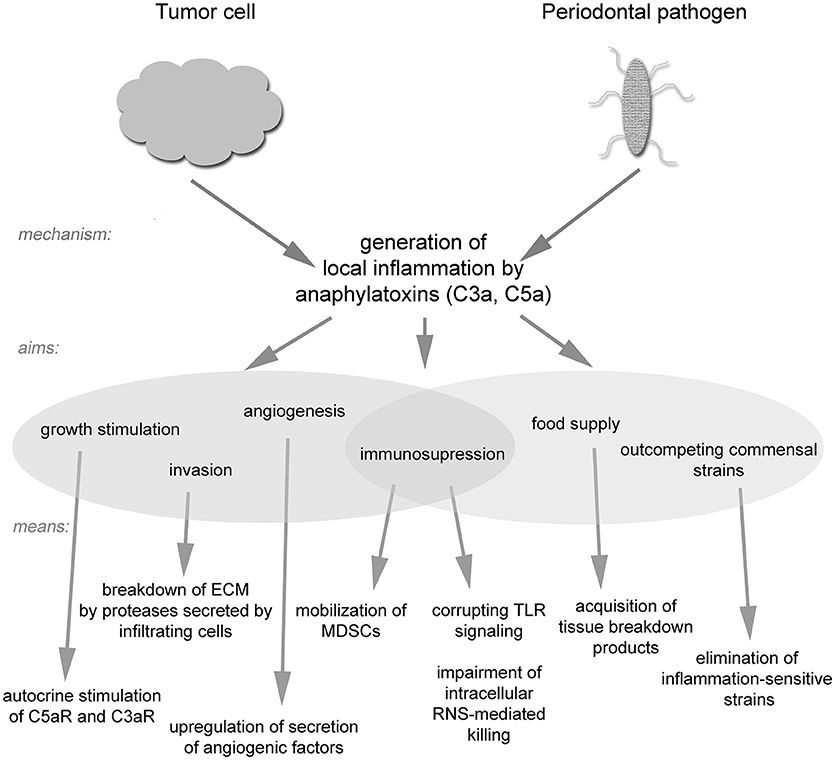
Figure 2. A schematic drawing of strategies utilized by tumor cells and inflammophilic bacteria to subvert complement activation.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
MO is supported by National Science Centre Poland, grant number 2014/14/E/NZ6/00182. JP is supported by National Science Centre Poland, grant number 2016/21/B/NZ1/00292 and NIH/NICDR grant number DE 022597.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. (2014) 29:248–57. doi: 10.1111/omi.12065
2. Ermert DML. Catch me if you can: Streptococcus pyogenes Complement Evasion Strategies. J Innate Immun. (2018) 11:3–12. doi: 10.1159/000492944
3. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. (2018) 10:455–64. doi: 10.1159/000491439
4. Blom AM, Hallstrom T, Riesbeck K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol. (2009) 46:2808–17. doi: 10.1016/j.molimm.2009.04.025
5. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. (2015) 13:529–43. doi: 10.1038/nrmicro3521
6. Pietrocola G, Nobile G, Rindi S, Speziale P. Staphylococcus aureus manipulates innate immunity through own and host-expressed proteases. Front Cell Infect Microbiol. (2017) 7:166. doi: 10.3389/fcimb.2017.00166
7. Duggan S, Leonhardt I, Hunniger K, Kurzai O. Host response to Candida albicans bloodstream infection and sepsis. Virulence (2015) 6:316–26. doi: 10.4161/21505594.2014.988096
8. Johswich K. Innate immune recognition and inflammation in Neisseria meningitidis infection. Pathog Dis. (2017) 75. doi: 10.1093/femspd/ftx022
9. Lupu F, Keshari RS, Lambris JD, Coggeshall KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res. (2014) 133 (Suppl. 1):S28–31. doi: 10.1016/j.thromres.2014.03.014
10. Reglinski M, Sriskandan S. The contribution of group A streptococcal virulence determinants to the pathogenesis of sepsis. Virulence (2014) 5:127–36. doi: 10.4161/viru.26400
11. Nonaka M. Evolution of the complement system. Subcell Biochem. (2014) 80:31–43. doi: 10.1007/978-94-017-8881-6_3
12. Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. (2010) 31:154–63. doi: 10.1016/j.it.2010.01.002
13. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
14. Biro A, Rovo Z, Papp D, Cervenak L, Varga L, Fust G, et al. Studies on the interactions between C-reactive protein and complement proteins. Immunology (2007) 121:40–50. doi: 10.1111/j.1365-2567.2007.02535.x
15. Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, et al. Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol. (2002) 32:1726–36. doi: 10.1002/1521-4141(200206)32:6<1726::AID-IMMU1726>3.0.CO;2-R
16. Kouser L, Madhukaran SP, Shastri A, Saraon A, Ferluga J, Al-Mozaini M, et al. Emerging and novel functions of complement protein C1q. Front Immunol. (2015) 6:317. doi: 10.3389/fimmu.2015.00317
17. Bohlson SS, Fraser DA, Tenner AJ. Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol Immunol. (2007) 44:33–43. doi: 10.1016/j.molimm.2006.06.021
18. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
19. Mongini PK, Jackson AE, Tolani S, Fattah RJ, Inman JK. Role of complement-binding CD21/CD19/CD81 in enhancing human B cell protection from Fas-mediated apoptosis. J Immunol. (2003) 171:5244–54. doi: 10.4049/jimmunol.171.10.5244
20. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. (2014) 61:110–7. doi: 10.1016/j.molimm.2014.06.030
21. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis. (2014) 73:1601–6. doi: 10.1136/annrheumdis-2014-205287
22. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res. (2013) 56:477–91. doi: 10.1007/s12026-013-8422-y
23. Pickering MC, Cook HT. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. (2008) 151:210–30. doi: 10.1111/j.1365-2249.2007.03574.x
24. Okroj M, Heinegard D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann Med. (2007) 39:517–30. doi: 10.1080/07853890701477546
25. Bus P, Chua JS, Klessens CQF, Zandbergen M, Wolterbeek R, van Kooten C, et al. Complement activation in patients with diabetic nephropathy. Kidney Int Rep. (2018) 3:302–13. doi: 10.1016/j.ekir.2017.10.005
26. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. (2007) 171:715–27. doi: 10.2353/ajpath.2007.070166
27. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. (2008) 6:132–42. doi: 10.1038/nrmicro1824
28. Potempa M, Potempa J. Protease-dependent mechanisms of complement evasion by bacterial pathogens. Biol Chem. (2012) 393:873–88. doi: 10.1515/hsz-2012-0174
29. Zipfel PF, Wurzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol. (2007) 44:3850–7. doi: 10.1016/j.molimm.2007.06.149
30. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature (2012) 482:405–9. doi: 10.1038/nature10803
31. Jurianz K, Ziegler S, Garcia-Schuler H, Kraus S, Bohana-Kashtan O, Fishelson Z, et al. Complement resistance of tumor cells: basal and induced mechanisms. Mol Immunol. (1999) 36:929–39. doi: 10.1016/S0161-5890(99)00115-7
32. Murawski N, Pfreundschuh M. New drugs for aggressive B-cell and T-cell lymphomas. Lancet Oncol. (2010) 11:1074–85. doi: 10.1016/S1470-2045(10)70210-2
33. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. (2013) 39:632–9. doi: 10.1016/j.ctrv.2012.10.008
34. Takei K, Yamazaki T, Sawada U, Ishizuka H, Aizawa S. Analysis of changes in CD20, CD55, and CD59 expression on established rituximab-resistant B-lymphoma cell lines. Leuk Res. (2006) 30:625–31. doi: 10.1016/j.leukres.2005.09.008
35. Macor P, Secco E, Mezzaroba N, Zorzet S, Durigutto P, Gaiotto T, et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia (2015) 29:406–14. doi: 10.1038/leu.2014.185
36. Mamidi S, Hone S, Teufel C, Sellner L, Zenz T, Kirschfink M. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology (2015) 4:e979688. doi: 10.4161/2162402X.2014.979688
37. Schlaf G, Demberg T, Beisel N, Schieferdecker HL, Gotze O. Expression and regulation of complement factors H and I in rat and human cells: some critical notes. Mol Immunol. (2001) 38:231–9. doi: 10.1016/S0161-5890(01)00045-1
38. Okroj M, Hsu YF, Ajona D, Pio R, Blom AM. Non-small cell lung cancer cells produce a functional set of complement factor I and its soluble cofactors. Mol Immunol. (2008) 45:169–79. doi: 10.1016/j.molimm.2007.04.025
39. Ajona D, Castano Z, Garayoa M, Zudaire E, Pajares MJ, Martinez A, et al. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. (2004) 64:6310–8. doi: 10.1158/0008-5472.CAN-03-2328
40. Ajona D, Hsu YF, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. (2007) 178:5991–8. doi: 10.4049/jimmunol.178.9.5991
41. Okroj M, Holmquist E, Nilsson E, Anagnostaki L, Jirstrom K, Blom AM. Local expression of complement factor I in breast cancer cells correlates with poor survival and recurrence. Cancer Immunol Immunother. (2015) 64:468–78. doi: 10.1007/s00262-015-1658-8
42. Riihila P, Nissinen L, Farshchian M, Kivisaari A, Ala-Aho R, Kallajoki M, et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J Invest Dermatol. (2014) 135:579–88. doi: 10.1038/jid.2014.376
43. Horl S, Banki Z, Huber G, Ejaz A, Windisch D, Muellauer B, et al. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia (2013) 27:2200–8. doi: 10.1038/leu.2013.169
44. Horl S, Banki Z, Huber G, Ejaz A, Mullauer B, Willenbacher E, et al. Complement factor H-derived short consensus repeat 18-20 enhanced complement-dependent cytotoxicity of ofatumumab on chronic lymphocytic leukemia cells. Haematologica (2013) 98:1939–47. doi: 10.3324/haematol.2013.089615
45. Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. (2003) 131:254–63. doi: 10.1046/j.1365-2249.2003.02066.x
46. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. (2015) 42:841–51. doi: 10.1007/s11033-015-3858-x
47. Collard CD, Vakeva A, Bukusoglu C, Zund G, Sperati CJ, Colgan SP, et al. Reoxygenation of hypoxic human umbilical vein endothelial cells activates the classic complement pathway. Circulation (1997) 96:326–33. doi: 10.1161/01.CIR.96.1.326
48. Collard CD, Bukusoglu C, Agah A, Colgan SP, Reenstra WR, Morgan BP, et al. Hypoxia-induced expression of complement receptor type 1 (CR1, CD35) in human vascular endothelial cells. Am J Physiol. (1999) 276:C450–8. doi: 10.1152/ajpcell.1999.276.2.C450
49. Okroj M, Corrales L, Stokowska A, Pio R, Blom AM. Hypoxia increases susceptibility of non-small cell lung cancer cells to complement attack. Cancer Immunol Immunother. (2009) 58:1771–880. doi: 10.1007/s00262-009-0685-8
50. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood (2004) 104:2224–34. doi: 10.1182/blood-2004-03-1109
51. Angelo LS, Kurzrock R. Vascular endothelial growth factor and its relationship to inflammatory mediators. Clin Cancer Res. (2007) 13:2825–30. doi: 10.1158/1078-0432.CCR-06-2416
52. Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. (2004) 31:2–11. doi: 10.1053/j.seminoncol.2004.03.040
53. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia (2002) 7:177–89. doi: 10.1023/A:1020304003704
54. Ajona D, Pajares MJ, Corrales L, Perez-Gracia JL, Agorreta J, Lozano MD, et al. Investigation of complement activation product c4d as a diagnostic and prognostic biomarker for lung cancer. J Natl Cancer Inst. (2013) 105:1385–93. doi: 10.1093/jnci/djt205
55. Ajona D, Okroj M, Pajares MJ, Agorreta J, Lozano MD, Zulueta JJ, et al. Complement C4d-specific antibodies for the diagnosis of lung cancer. Oncotarget (2018) 9:6346–55. doi: 10.18632/oncotarget.23690
56. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin c5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
57. Pakkala S, Owonikoko TK. Immune checkpoint inhibitors in small cell lung cancer. J Thorac Dis. (2018) 10:S460–7. doi: 10.21037/jtd.2017.12.51
58. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4:e954829. doi: 10.4161/21624011.2014.954829
59. Okamura T, Fujio K, Sumitomo S, Yamamoto K. Roles of LAG3 and EGR2 in regulatory T cells. Ann Rheum Dis. (2012) 71 (Suppl. 2):i96–100. doi: 10.1136/annrheumdis-2011-200588
60. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
61. Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. (2014) 32:1715–9. doi: 10.3892/or.2014.3341
62. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. (2014) 6:1085–95. doi: 10.1016/j.celrep.2014.02.014
63. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. (2018) 18:5–18. doi: 10.1038/nri.2017.97
64. Riihila P, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, Meri S, et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am J Pathol. (2017) 187:1186–97. doi: 10.1016/j.ajpath.2017.01.006
65. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. (2016) 7:10346. doi: 10.1038/ncomms10346
66. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. (2017) 7:694–703. doi: 10.1158/2159-8290.CD-16-1184
67. Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. (2016) 6:1022–35. doi: 10.1158/2159-8290.CD-15-1412
68. Stasilojc G, Osterborg A, Blom AM, Okroj M. New perspectives on complement mediated immunotherapy. Cancer Treat Rev. (2016) 45:68–75. doi: 10.1016/j.ctrv.2016.02.009
69. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe (2011) 10:497–506. doi: 10.1016/j.chom.2011.10.006
70. Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. (2011) 11:7. doi: 10.1186/1471-2180-11-7
71. Kinane DF, Peterson M, Stathopoulou PG. Environmental and other modifying factors of the periodontal diseases. Periodontol (2006) 40:107–19. doi: 10.1111/j.1600-0757.2005.00136.x
72. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. (2012) 10:717–25. doi: 10.1038/nrmicro2873
73. Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. (2010) 3:ra11. doi: 10.1126/scisignal.2000697
74. Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. (2007) 178:7242–50. doi: 10.4049/jimmunol.178.11.7242
75. Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem. (1992) 267:18902–7.
76. Okuda K, Kato T, Naito Y, Ono M, Kikuchi Y, Takazoe I. Susceptibility of Bacteroides gingivalis to bactericidal activity of human serum. J Dent Res. (1986) 65:1024–7. doi: 10.1177/00220345860650070601
77. Schenkein HA, Genco RJ. Gingival fluid and serum in periodontal diseases. I. Quantitative study of immunoglobulins, complement components, and other plasma proteins. J Periodontol. (1977) 48:772–7. doi: 10.1902/jop.1977.48.12.772
78. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe (2014) 15:768–78. doi: 10.1016/j.chom.2014.05.012
79. Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, et al. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem. (2014) 289:32481–7. doi: 10.1074/jbc.C114.617142
80. Jusko M, Potempa J, Mizgalska D, Bielecka E, Ksiazek M, Riesbeck K, et al. A metalloproteinase mirolysin of Tannerella forsythia inhibits all pathways of the complement system. J Immunol. (2015) 195:2231–40. doi: 10.4049/jimmunol.1402892
81. Jusko M, Potempa J, Karim AY, Ksiazek M, Riesbeck K, Garred P, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol. (2012) 188:2338–49. doi: 10.4049/jimmunol.1101240
82. Jusko M, Miedziak B, Ermert D, Magda M, King BC, Bielecka E, et al. FACIN, a double-edged sword of the emerging periodontal pathogen filifactor alocis: a metabolic enzyme moonlighting as a complement inhibitor. J Immunol. (2016) 197:3245–59. doi: 10.4049/jimmunol.1600739
83. Potempa M, Potempa J, Kantyka T, Nguyen KA, Wawrzonek K, Manandhar SP, et al. Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog. (2009) 5:e1000316. doi: 10.1371/journal.ppat.1000316
84. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15:30–44. doi: 10.1038/nri3785
85. Santoni M, Massari F, Amantini C, Nabissi M, Maines F, Burattini L, et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. (2013) 62:1757–68. doi: 10.1007/s00262-013-1487-6
86. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. (2018) 9:793. doi: 10.1038/s41419-018-0818-0
87. Bremnes RM, Donnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. (2011) 6:209–17. doi: 10.1097/JTO.0b013e3181f8a1bd
Keywords: inflamation, periodontits, cancer, Porphyromonas gingivalis, complement activation
Citation: Okrój M and Potempa J (2019) Complement Activation as a Helping Hand for Inflammophilic Pathogens and Cancer. Front. Immunol. 9:3125. doi: 10.3389/fimmu.2018.03125
Received: 21 August 2018; Accepted: 18 December 2018;
Published: 10 January 2019.
Edited by:
Maciej Cedzynski, Institute for Medical Biology (PAN), PolandReviewed by:
George Hajishengallis, University of Pennsylvania, United StatesCopyright © 2019 Okrój and Potempa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcin Okrój, bWFyY2luLm9rcm9qQGd1bWVkLmVkdS5wbA==
Jan Potempa, anNwb3RlMDFAbG91aXN2aWxsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.