 Hao-Sen Chiang
Hao-Sen Chiang Helene Minyi Liu
Helene Minyi Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 January 2019
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.03086
This article is part of the Research TopicSTATs and IRFs in Innate Immunity: From Transcriptional Regulators to Therapeutic TargetsView all 16 articles
The antiviral innate immunity is the first line of host defense against virus infections. In mammalian cells, viral infections initiate the expression of interferons (IFNs) in the host that in turn activate an antiviral defense program to restrict viral replications by induction of IFN stimulated genes (ISGs), which are largely regulated by the IFN-regulatory factor (IRF) family and signal transducer and activator of transcription (STAT) family transcription factors. The mechanisms of action of IRFs and STATs involve several post-translational modifications, complex formation, and nuclear translocation of these transcription factors. However, many viruses, including human immunodeficiency virus (HIV), Zika virus (ZIKV), and herpes simplex virus (HSV), have evolved strategies to evade host defense, including alteration in IRF and STAT post-translational modifications, disturbing the formation and nuclear translocation of the transcription complexes as well as proteolysis/degradation of IRFs and STATs. In this review, we discuss and summarize the molecular mechanisms by which how viral components may target IRFs and STATs to antagonize the establishment of antiviral host defense. The underlying host-viral interactions determine the outcome of viral infection. Gaining mechanistic insight into these processes will be crucial in understanding how viral replication can be more effectively controlled and in developing approaches to improve virus infection outcomes.
Interferons (IFNs) were originally discovered in 1957 as proteins that interfere with virus replication (1, 2). Since then, IFNs are now divided into three sub-families: type I, II, and III with broad functions not limited to host defense against microbial infection (3–5). These secreted IFNs initiate signaling by binding distinct cell surface receptors to mount proper immune responses. Type I IFNs comprise the largest IFN family including IFN-α, IFN-β, and other subtypes. All type I IFNs bind a ubiquitously expressed heterodimeric transmembrane receptor, which is known as the IFN-α receptor (IFNAR) complex to mediate antiviral effects of type I IFNs (3). IFN-γ is the sole type II IFN largely secreted by innate lymphocytes and T cells that binds to IFN-γ receptor (IFNGR) complex and activates several immune responses to intracellular pathogens (5). Distinct from type I and type II IFNs, type III IFNs are recently discovered and consist of IFN-λ1 (interleukin-29 [IL-29]), IFN-λ2 (IL-29A), IFN-λ3 (IL-28B), and IFN-λ4 (6). They engage the mucosal surface-abundant receptor complex, IFN-λ receptor (IFNLR, also known as IL-28R) that consists of two subunits: IFNLR1 and IL10R2 in the initiation of protection against viral infection at mucosal barriers (4).
Upon virus infection, IFNs are immediately induced by the recognition of pathogen-associated molecular patterns (PAMPs) through cytoplasmic and endosomal pattern-recognition receptors (PRRs) or by cytokine-receptor binding (7). The IFN-regulatory factor (IRF) family proteins are transcription factors with critical and diverse roles that connect the sensing of microbial signatures to the expression of IFNs and pro-inflammatory cytokines as well as innate immune responses (8–10). After IFN binding and receptor dimerization, all IFNs induce IFN-stimulated gene (ISG) expression for effective antiviral responses through the activation of IFN receptor-associated Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (11). As obligate intracellular microbes, viruses must engage with the host throughout their replication; it is therefore unsurprised that pathogenic viruses often antagonize IFN responses to establish successful infections by targeting the aforementioned pathways.
In this review, we critically explore the current understanding of IRF and STAT family proteins in host antiviral immune responses activated by IFNs; we also examine how pathogenic viruses have evolved various mechanisms to suppress IRF- and STAT-mediated signaling.
The mammalian IRF family proteins are structurally related transcription factors consisting of nine members: IRF1, IRF2, IRF3, IRF4 (also called ICSAT [IFN consensus sequence-binding protein for activated T-cells], LSIRF [lymphocyte-specific IRF], PIP [PU.1-interacting protein]), IRF5, IRF6, IRF7, IRF8 (also referred to ICSBP [IFN consensus sequence-binding protein]), and IRF9 (also known as ISGF3γ [IFN-stimulated gene factor 3γ]) (9). Among nine IRFs, IRF1, IRF3, IRF5, and IRF7 play a pivotal role in the induction of IFN gene transcription during viral infection (12, 13). IRF2 and IRF4 have been implicated in the suppression of type I IFN signaling (14–16). All IRF family proteins possess two conserved functional domains: an amino (N)-terminal DNA-binding domain (DBD) and a carboxy (C)-terminal IRF-associated domain (IAD) (17). DBD is characterized by five conserved tryptophan residues that forms a helix-turn-helix structure and recognizes consensus DNA sequence known as IFN-stimulated response element (ISRE) (18). In contrast to N-terminal regions, the C-terminal regions of IRFs display a broad diversity. Two types of IAD have been identified: IAD1 and IAD2 (19). While IAD1 is conserved in all IRFs except IRF1 and IRF2, IAD2 is shared only by IRF1 and IRF2 (20). The C-terminal regions of IRFs are also involved in the interactions with other IRF family proteins or transcription factors/co-activators that are critical for the induction of IFN (21, 22). For example, IRF3 forms a complex with CREB binding protein (CBP)/p300 histone acetyltransferase (HAT) through the IAD1 domain for the induction of Ifnb1 transcription in response to virus infection (21). In the following sections, we discuss the distinct contribution of IRFs to type I IFN induction through cytoplasmic and endosomal PRR signaling cascades (Figure 1).
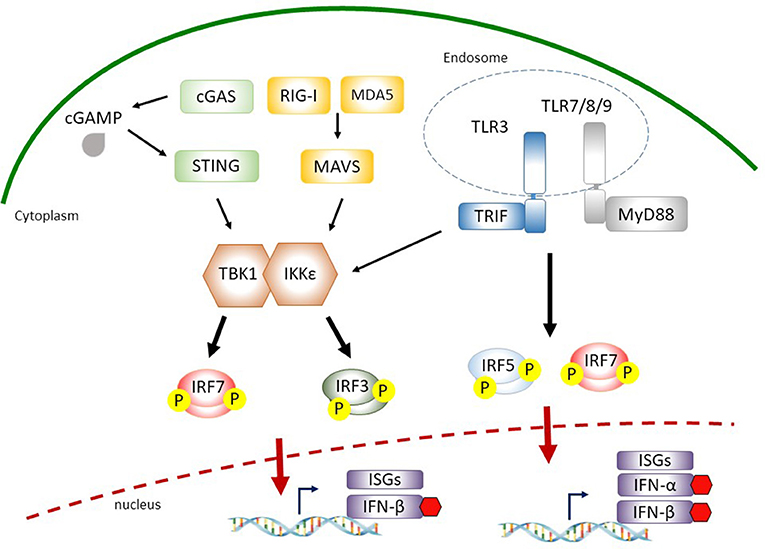
Figure 1. Interferon (IFN)-regulatory factors (IRFs) involved in cytosolic nucleic acid sensing and endosomal Toll-like receptor (TLR) signaling. During virus infection, retinoic acid-inducible gene I (RIG-I) or melanoma differentiation-associated gene 5 (MDA5) recognize cytosolic double-stranded RNA and recruit the adaptor protein mitochondria antiviral signaling protein (MAVS), which leads to the activation of TANK-binding kinase 1 (TBK1)/IκB kinase-ε (IKKε). Cytosolic double-stranded DNA is detected by cyclic-GMP-AMP (cGAMP) synthase (cGAS) or other receptors (such as DEAD-box helicase 41 (DDX41), gamma-IFN-inducible protein 16 (IFI16), not shown) to induce stimulator of IFN genes (STING)-mediated TBK1 and IKKε activation. Activated TBK1/IKKε then phosphorylate IRF3 and IRF7 that translocate into the nucleus for the induction of IFN-β. The sensing of viral pathogen-associated molecular patterns (PAMPs) by endosomal TLR3 or TLR7/8/9 leads to the phosphorylation and activation of IRF5 and IRF7 through adaptor proteins TIR-domain-containing adapter-inducing IFN (TRIF) or myeloid differentiation primary response 88 (MyD88), respectively, for the expression of type I IFNs.
During virus infection, type I IFNs are produced in infected cells via the recognition of viral PAMPs by binding to specific PRRs, such as cytosolic retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and transmembrane Toll-like receptors (TLRs) resulting in the activation of downstream IRF3 and IRF7 pathways (7, 23). Several RNA viruses directly enter the cytoplasm where they are detected by RLR family members: RIG-I and melanoma differentiation-associated gene 5 (MDA5) (24). Ligand recognition results in the recruitment of RIG-I and MDA5 to the mitochondria where they interact with mitochondria antiviral signaling protein (MAVS) through the N-terminal caspase recruitment domain (CARD) domains in RLRs and MAVS. This association relays signals to the downstream TANK-binding kinase 1 (TBK1) and IκB kinase-ε (IKKε) that phosphorylate IRF3 and IRF7 (24).
IRF3 is a constitutively expressed but tightly regulated transcription factor in the cytoplasm. It presents in an inactive form due to its auto-inhibitory mechanisms (25). Virus infections induce specific IRF3 phosphorylation that leads to its dimerization with itself or with IRF7 and forms a complex containing CBP/p300 and other coactivators followed by translocation into the nucleus for the expression of IFN-β (26). The activation process of IRF7 is similar to that of IRF3 in response to viral PAMPs. However, in contrast to constitutively expressed IRF3, the basal expression level of IRF7 is minimum but is strongly induced by type I IFN-mediated responses in an autocrine feedback loop after virus infection (discussed in detail below) (9). Moreover, a recent study from IRF3/IRF5/IRF7 triple knockout mice suggests that in addition to IRF3 and IRF7, IRF5 is also a key transcriptional factor responsible for RLR- and MAVS-mediated type I IFN expression (27).
Similar to the involvement of RLR-mediated type I IFN expression, IRF3 and IRF7 also contribute to the signaling pathways downstream of cytosolic DNA sensing and endosomal DNA/RNA recognition for the inductions of IFN-α and IFN-β during virus infection (7). Among several known cytosolic DNA sensors for the detection of viral infection, cyclic-GMP-AMP (cGAMP) synthase (cGAS) is the most recently identified (28). Upon viral DNA binding, cGAS catalyzes the production of cGAMP from ATP and GTP, a second messenger that binds and activates the endoplasmic reticulum membrane protein stimulator of IFN genes (STING) for the production of type I IFN (28, 29). STING functions as an adaptor protein that promotes TBK1-dependent IRF3/7 phosphorylation (30–33).
Transmembrane TLR3, TLR7/8, and TLR9 are the most well characterized PRRs for the recognition of viral PAMPs located in the endosomal compartments (34). TLRs initiate shared and distinct signaling pathways by recruiting different adaptor molecules for type I IFN expression. TLR3 recognizes viral dsRNA and utilizes TIR-domain-containing adapter-inducing IFN (TRIF) as an adaptor to recruit downstream TBK1, resulting in IRF3/7 phosphorylation and type I IFN production. Upon the engagements with viral ssRNA and unmethylated CpG DNA motifs by TLR7/8 and TLR9, respectively, these TLRs signal through myeloid differentiation primary response 88 (MyD88) to activate IKKα- or IKKβ-dependent phosphorylation and activation of IRF7 or IRF5, allowing the production of type I IFNs (35–37). Taken together, these studies highlight the importance of IRF3/5/7 phosphorylation and activation in the downstream of cytoplasmic/endosomal PRR signaling leading to type I IFN expression during virus infection.
Mammalian immune systems utilize IRFs, STATs and IFNs to integrate and process distinct signals to orchestrate host antiviral immunity. This has been proven in several studies utilizing genetically-modified mice that lack IFNAR, IFNGR, IFNLR, STAT1, or STAT2, respectively. These gene-knockout mice are highly susceptible to virus infections due to the impaired IFN signaling and responses (38–42). In the following sections, we examine the current understandings of how IFNs initiate antiviral immune responses via binding to their cognate heterodimeric receptors through downstream canonical JAK-STAT signaling (Figure 2).
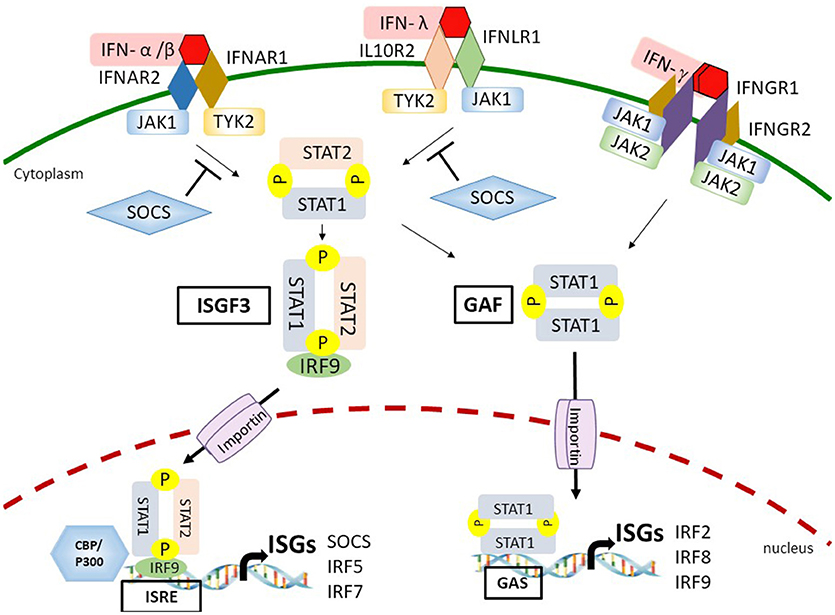
Figure 2. Interferon (IFN)-dependent IFN stimulated gene (ISG) transcription through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling. By binding to the IFN-α receptor (IFNAR) or IFN-λ receptor (IFNLR), type I/III IFNs activate the JAK-STAT pathway leading to the formation of IFN-stimulated gene factor 3 (ISGF3) and gamma-IFN activation factor (GAF) complexes. The ligation between IFN-γ and IFN-γ receptor (IFNGR) also activate the GAF complex. ISGF3 and GAF complexes then translocate into the nucleus mediated by importins and recruit additional coactivators, such as CREB binding protein (CBP)/p300 on the IFN-stimulated response element (ISRE)- or gamma IFN activated sequence (GAS)-containing promoters to stimulate expression of a distinct group of ISGs. Eventually, a set of ISGs are produced and amplify the IFN response. IFN-induced suppressor of cytokine signaling (SOCS) proteins inhibit JAK-STAT signaling by binding to phosphorylated tyrosine residues on either JAK1 or tyrosine kinase 2 (TYK2).
Almost all cell types respond to type I and type II IFNs for effective antiviral immunity (43, 44). However, the specific type III IFN receptor subunit IFNLR1 is mainly expressed on epithelial cells and immune cells, such as neutrophils that provides the first line of antiviral defense at the mucosal surfaces of gastrointestinal and respiratory tracts (42, 45). The ligation between IFN and IFNR results in receptor dimerization or oligomerization that allows the transphosphorylation of receptor-associated JAK on tyrosine residues. Subsequently, activated JAKs induce tyrosine phosphorylation of IFNR cytoplasmic tails where provides the binding sites for C-terminal Src-homology-2 (SH2) domains of STAT proteins. The STATs are then recruited to the JAKs followed by phosphorylation at a tyrosine residue (46). In the canonical pathway of type I IFN-mediated and type III IFN-mediated signaling, the phosphorylation of STAT1 on tyrosine 701 and STAT2 on tyrosine 690 leads to the formation of STAT1/STAT2 heterodimer that interacts with IRF9 to form the IFN-stimulated gene factor 3 (ISGF3) complex. ISGF3 complex then redirects and translocates into the nucleus to trigger ISG expressions by binding to ISRE and recruiting additional coactivators, such as CBP/p300 on the promoters of a distinct group of target genes (44, 47–49). In addition to forming the ISGF3 complex, all types of IFNs are able to induce a STAT1/STAT1 homodimer, known as gamma IFN–activation factor (GAF) that activates ISG transcription by direct binding to the gamma IFN activated sequence (GAS)-containing genes (4, 44). Besides the STAT1/STAT2 heterodimer and STAT1/STAT1 homodimer, type I IFNs can also activate STAT3/STAT3, STAT4/STAT4, STAT5/STAT5, and STAT6/STAT6 homodimers as well as STAT1/STAT3, STAT1/STAT4, STAT1/STAT5, STAT2/STAT3, and STAT5/STAT6 heterodimers. All of these homodimers and heterodimers can bind to GAS and drive the expression of GAS-containing ISGs (44).
ISGs are proteins present at baseline but are enhanced upon virus infection in JAK-STAT dependent pathways. A subset of ISGs are well-characterized for their direct antiviral activities. For example, IFN-induced proteins with tetratricopeptide repeat 1–3 (IFIT1–3), viperin, and myxovirus resistance 1 (Mx1) can all inhibit virus replication (50). JAK2, STAT1, STAT2, and IRF9 belong to another subset of ISGs that amplify JAK-STAT signaling to reinforce IFN responses (3). Moreover, ISGs, such as RIG-I, cGAS, IRF5, and IRF7 can further prime cells for increased detection of viral PAMPs and upregulated IFN expressions (51). Interestingly, the expression of subsets of ISGs can also be induced directly by IRFs in a pathway that is independent of JAK-STAT signaling (52). For example, IRF3 initiates the expressions of IFN-stimulated gene 15 (ISG15) and sterile alpha motif and HD domain containing protein 1 (SAMHD1) as the first responders in antiviral immunity (53, 54). Similar to IRF3, IRF1 mediates a rapid IFN-independent antiviral response downstream of RLR adaptor protein MAVS localized specifically on peroxisome (55–57). IRF7 can regulate ISG expression in the absence of IFN signaling as well (50, 58).
In addition to JAK2. STAT1, STAT2, IRF9 themselves, several other ISGs are also implicated in the regulation of JAK-STAT signaling. Suppressor of cytokine signaling (SOCS) proteins including SOCS1 and SOCS3 inhibit JAK-STAT signaling by binding to phosphorylated tyrosine residues on either JAK1 or tyrosine kinase 2 (TYK2) (59). Another ISG: the ubiquitin-specific peptidase 18 (USP18) suppresses JAK-STAT signaling induced by type I IFN at the level of IFN receptor. USP18 specifically interacts with the IFNAR2 subunit to inhibit the interaction between JAK and the IFN receptor (60). Furthermore, recent findings indicate that type I IFN-induced STAT3 cooperates with phospholipid scramblase 2 (PLSCR2) to interact with STAT2 and suppress type I IFN response (61, 62). It would be critical to further determine whether STAT family members also interact with other proteins for the regulation of JAK-STAT pathways.
As STAT family proteins are essential signaling mediators, their activation are tightly-regulated by several mechanisms in order to downregulate IFN-mediated antiviral response (63). STATs reside in an inactive form in the cytoplasm but are activated and translocate into the nucleus in response to IFN signaling. Several nucleocytoplasmic transport factors are essential for the nuclear import of phosphorylated STATs. For example, importin-α5 (also called karyopherin α1[KPNA1]) regulates nuclear import of STAT1 (64, 65). Moreover, STAT3 nuclear import is mediated by importin-α3 (66). Interestingly, the presence of phosphorylated STATs in the nucleus is transient. STAT1 can be dephosphorylated in the nucleus and actively export to the cytoplasm by the chromosome region maintenance 1 (CRM1) export receptor in a nuclear export signal (NES)-dependent manner (67). Recently, a metheyltransferase SET domain-containing protein 2 (SETD2) has been identified as a critical type I IFN signaling amplifier. Although the expression of SETD2 itself is not upregulated by type I IFN, SETD2 enhances the methylation of STAT1 on K525 as well as ISG expressions for antiviral immunity (68). However, the detail mechanism underlying the regulation and selectivity of SETD2-mediated ISG expression has not been fully explored.
In order to establish successful infections, viruses have evolved a variety of strategies to counteract host antiviral innate immunity. The infection outcome is determined by the race between the kinetics of virus replications and the competence of antiviral gene expression levels at the early-onset of the infection. Studies of virus-host interactions have revealed infection-induced signaling pathways that result in IRF and STAT activations as the major targets of regulation and evasion of the host anti-viral responses. The molecular mechanisms by which viruses target IRFs and STATs are highly diverse, including inhibition of IRF/STAT expressions, disruption of the post-translational modifications, alterations in the localizations, prevention of transcriptional complex formation, and promoting the degradation of IRFs and STATs.
For rapid response to the viral infections, the activation of antiviral innate immunity in mammalian cells is largely controlled by the post-translational modifications of the PRR, the downstream signaling adaptor proteins, as well as the key transcription factors, IRFs and STATs. Phosphorylation of IRF3, IRF7, STAT1, and STAT2 are highly critical for their downstream transcriptional activation, and therefore these phosphorylation events are commonly targeted by viruses. By directly promoting the dephosphorylation of IRFs and STATs or indirectly inhibiting upstream kinase activities, the activation of these transcription factors are then controlled by the invading viruses.
Vaccinia virus (VACV) encodes a late gene VH1, which is packaged into the virion and is a dual-specificity phosphatase (69). VH1 was later found to have immediate effects against host antiviral activities by directly dephosphorylating STAT1 at Tyr701 and Ser727 to reduce STAT1 activation (70, 71). Besides, VACV encodes another virulence factor, C6, which binds to the IRF3/7 kinase TBK-1 and interferes the phosphorylation and activation of IRF3 and IRF7 (72). Several other DNA viruses also have similar mechanisms to target TBK1 and/or IKKε to inhibit the phosphorylation of IRF3. It has been recently reported that the VP24 of herpes simplex virus (HSV) can target TBK1/IKKε to inhibit the phosphorylation of IRF3 (73, 74). Also, several viruses have evolved viral products that may interfere the interaction between STAT1 and JAK/TyK, e.g., the NS5A protein of Hepatitis C virus can physically interact with STAT1 to interfere the phosphorylation of STAT1 at Tyr701 (75, 76). Another example is the NSP1 protein encoded by Rotavirus, of which the expression alone may block STAT1 phosphorylation and activation (77). Ebola virus VP35 prevents the TBK1-dependent phosphorylation of IRF3 (78, 79). Marburg virus, which is closely related to Ebola virus, encodes a matrix protein VP40 to antagonize the phosphorylation of both STAT1 and STAT2 (80).
In addition, viruses may regulate IRF/STAT phosphorylation indirectly by promoting the expression of the negative regulators of IRF/STAT kinases, such as suppressor of cytokine signaling (SOCS) family, to minimize the induction of ISGs. It has been reported that Hepatitis B virus (HBV) infection as well as Hepatitis C virus (HCV) core protein may induce the expression of suppressor of cytokine signaling 3 (SOCS3) (81, 82). The e antigen of HBV, HBeAg, also stimulates the expression of suppressor of cytokine signaling 2 (SOCS2) (83). The induction of SOCS family subsequently impairs IFN/JAK/STAT signaling and therefore attenuating the activation of STAT1, which may contribute to the establishment of persistent infections of HBV and HCV.
Many viruses, such as picornaviruses and flaviviruses, encode viral proteases for viral replications. In addition to their essential roles in the virus life cycles, the viral proteases often target host proteins involved in IFN induction and response pathways to escape the host antiviral innate immunity (84). The 3C proteases (3Cpro) encoded by enterovirus (EV) 71 and EV68 disturb the expression of type I IFN and ISGs by directly cleaving IRF7 (85, 86). Porcine deltacoronavirus nsp5 cleaves STAT2 to antagonize type I IFN responses (19). These proteolytic events of IRFs and STATs often lead to the degradation of these transcription factors. For example, besides cleaving IRF7, the 3Cpro of enterovirus 71 also targets IRF9 for proteolytic degradation (87).
Conversely, a variety of viral proteins may also promote the proteasome-dependent or lysosome-dependent protein degradation of IRFs and STATs. It has been shown that the Vpu accessory protein of human immunodeficiency virus (HIV) mediates the depletion of IRF3 through lysosomal degradation or caspase-dependent cleavage (88, 89). HIV YU2 mutant which lacks the expression of Vpu could not control the activation of IRF3 upon infection (89). The NSP1 of rotavirus, a putative E3 ubiquitin ligase, mediates the degradation of cellular factors, including IRF3, IRF5, IRF7, and IRF9 but not IRF1, through recognizing the common IAD1 domain of these IRFs (90, 91).
Several members of the Paramyxoviridae, such as parainfluenza viruses, have developed strategies to target either STAT1 or STAT2 for degradation. The expression of a single viral protein, human parainfluenza virus type 2 (hPIV-2) V protein, may inhibit the type I IFN response by inducing the proteolytic degradation of STAT2 (92), and Newcastle disease virus (NDV) also encodes a V protein which can target STAT1 for degradation. The V protein of canine parainfluenza virus 5 (also known as simian virus 5) degrades both STAT1 and STAT2 in a proteasome-dependent manner (93). The NS5 proteins of flaviviruses, including dengue virus (DENV) and Zika virus (ZIKV), also share the common characteristics to target STAT2 for proteasome-dependent degradation (94–97).
After phosphorylated, the nuclear translocation of the activated IRFs and STATs is another key step to induce the transcription of downstream genes, and this process is largely dependent on the cellular nuclear import and export machineries, including importin and CRM-1 proteins. Hence, viruses have developed various strategies to negatively regulate IFN induction and response pathways by altering the localizations of activated IRFs and STATs. Ebola virus VP24 binds to the α5 and α6 subunits of importin, which are the essential components of the nuclear transporter, to block the nuclear translocation of phosphorylated STAT1 (98, 99). EV71 suppresses IFN responses by blocking STAT1 signaling through inducing importin-α5 degradation in a caspase-3-dependent manner (100). As described previously, nuclear STAT1, STAT2, and IRF9 cycle back to the cytoplasm in a CRM1-dependent nuclear export manner. Certain viral proteins, such as the nsp2 of chikungunya virus, can promote the nuclear export of STATs (101, 102). During Nipah virus infection, the V protein can directly bind to STAT1 and STAT2 in the cytoplasm, and the N protein of Nipah virus restricts the complex formation of STAT1/STAT2, which along with the CRM1-dependent nuclear export of STAT1 and STAT2 additively result in the accumulation of STAT1 and STAT2 in the cytoplasm (103–105).
Viruses may also encode proteins which can directly bind to IRFs and STATs and maintain their cytoplasmic distribution. The C proteins of hPIV-1, which belongs to the Paramyxoviridae, blocks IFN signaling by interacting and retaining STAT1 and STAT2 in the perinuclear region in the infected cells (106). Similarly, the C protein of Sendai virus, also known as murine paramyxovirus, binds p-STAT1 to inhibit STAT1 dimerization and nuclear translocation (107, 108). Measles virus, which also belongs to the Paramyxoviridae, does not inhibit the tyrosine phosphorylation of STAT1 and STAT2 but encodes a viral protein, V protein, which directly interacts with STAT1, STAT2, and IRF9 in the cytoplasm to prevent their nuclear translocation (109). In addition to the Paramyxoviridae, the monkey rotavirus and human rotavirus Wa strain also do not restrict the activation phosphorylation of STAT1 and STAT2 but retain these transcription factors in the cytoplasm (110). HSV-1 encodes several IFN antagonists, including ICP0, which inhibits IRF3 nuclear accumulation but not IRF3 phosphorylation (111). Human papilloma virus (HPV) E7 protein interacts with IRF9 in the cytoplasm and subsequently inhibits the nuclear translocation of IRF9 as well as the formation of ISGF3 (112).
After translocation into the nucleus, the activated IRFs and STATs will then bind to the promoter region on the chromosomal DNA and recruit other transcription activators to initiate the transcription of downstream genes. A distinct group of viral proteins, viral homologs to cellular IRFs, known as vIRFs, are reported to be encoded by Kaposi's sarcoma-associated herpesvirus (KSHV) and the rhesus macaque rhadinovirus (RRV). KHSV vIRF3 interacts with host IRF5 and IRF7, and vIRF3 can suppress IRF7 DNA binding activity to inhibit IFN-α production (113, 114). Furthermore, KHSV vIRF1 binds to p300 and interferes with CBP/p300-IRF3 complex formation as well as the HAT activity of p300, and thus prevents IRF3-mediated transcriptional activation (115). HSV-1 also utilizes a similar strategy to abrogate CBP recruitment by IRF3 through the viral protein VP16 (116). RRV-encoded R6 is a virion-associated vIRF, which is capable to prevent IRF3/CBP complex docking to the IFNβ promoter region to minimize the induction of type I IFN expression upon RRV infection (117).
Besides vIRFs, other viral proteins have also been reported to regulate the DNA-binding activities and transcriptional complex formation. For example, Porcine bocavirus (PBoV) NP1 protein inhibits the DNA-binding activity of IRF3 and the DNA-binding activity ISGF3 through interactions with the DNA-binding domain of IRF9 (118, 119). The nsp1 of porcine epidemic diarrhea virus (PEDV) and the nsp1 α subunit of porcine reproductive and respiratory syndrome virus (PRRSV) both suppress the type I IFN production by promoting the proteasome dependent degradation of CBP (120, 121).
As described in ISGs and Regulation of JAK/STAT Family Proteins, the protein expression levels of various of the IRFs and STATs are upregulated in response to viral infections and IFN signaling, such as IRF7 and STAT1 (51), to form a positive feedback loop for the enhancement of antiviral activities. Therefore, disturbing the activation of basally expressed endogenous IRFs and STATs not only directly impairs the induction of the initial round of antiviral gene expressions but also prohibits the magnification of the antiviral responses against virus replication. Without sufficient and efficient antiviral gene expression in the infected cells, the viruses may competently replicate and produce viral progenies to infect neighboring cells and therefore establish a successful infection.
Some viruses directly inhibit the expression of IRFs at the transcriptional level, e.g., Epstein-Barr virus (EBV) BRLF1 inhibits the transcription of IRF3 and IRF7 (122). Another major mechanism for herpes viruses to curtail the expression of IRFs and STATs is virion host shut-off (VHS), which is mediated by the tegument protein UL41 (123). It has been shown that through its own endoribonuclease activity, HSV VHS selectively promotes the degradation of host mRNAs made before infection, including the mRNA of ISGs (124, 125). Many of the RNA viruses, including Caliciviridae, Coronaviridae, Picornaviridae, Oorthomyxoviridae, Reoviridae, and many others, have strategies to induce host translational shut-off and thus prevent the infected cells to synthesize new peptides and proteins, including those IFN-stimulated IRFs and STATs. Viruses may also upregulate certain miRNA to tune the expression of factors involved in the activation of IRFs and STATs. A recent report showed that miR-373, which reduces the expression of both JAK1 and IRF9, is upregulated by HCV infection to suppress the response to IFNs in the infected cells (126).
In the past decades, the inductions and responses of IFNs have been much revealed. The regulatory network of antiviral innate immunity including IFNs and many other cytokines is extremely complex in the mammalian cells. Since viruses carry much less genetic information than the eukaryotic cells, analyses of which cellular factors are targeted by viral products to dampen the innate immunity pathway provide us with great means to identify the crucial signaling molecules for mammalian antiviral activities. The critical role STAT1 in antiviral immunity is well-pronounced since viruses have developed numerous strategies to target STAT1 as a result of evolution. Notably, the development of STAT1 KO mice as in vivo animal models for viral infections has provided valuable tools for future virology and immunology studies (127–129).
As we reviewed in this article, all pathogenic viruses have multiple strategies to antagonize the host antiviral innate immunity. Intriguingly, several viruses selectively inhibit the type I IFN-induced but not type II IFN-induced STAT1 phosphorylation, such as ZIKV (130, 131). How this is beneficial to the virus life cycle remains to be further investigated. With better understanding the molecular mechanisms behind, future developments of antiviral agents and vaccines will be accelerated.
All authors contributed equally to this work. H-SC and HL conceptualized, wrote, and edited the manuscript.
This work was supported by grants from the Ministry of Science and Technology of Taiwan (grant # 105-2628-B-002-013-MY3 to H-SC, grant # 105-2628-B-002-014-MY3 to HL), a grant from the National Taiwan University to H-SC (grant # 107L7717), and a grant from the National Health Research Institutes of Taiwan to HL (grant # NHRI-EX107-10417S).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with the authors H-SC and HL.
The authors apologize to colleagues whose work could not be cited due to space constraints. We appreciate all the members of H-SC Laboratory and HL Laboratory for interactive discussions.
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
2. Lindenmann J, Burke DC, Isaacs A. Studies on the production, mode of action and properties of interferon. Br J Exp Pathol. (1957) 38:551–62.
3. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
4. Wack A, Terczynska-Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol. (2015) 16:802–9. doi: 10.1038/ni.3212
5. Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
6. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. (2013) 45:164–71. doi: 10.1038/ng.2521
7. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses (2011) 3:920–40. doi: 10.3390/v3060920
8. Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. (2006) 6:644–58. doi: 10.1038/nri1900
9. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. (2013) 78:105–16. doi: 10.1101/sqb.2013.78.020321
10. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. (2008) 26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400
11. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. (2007) 282:20059–63. doi: 10.1074/jbc.R700016200
12. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
13. Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol. (2015) 12:47–52. doi: 10.1016/j.coviro.2015.02.003
14. Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity (2000) 13:643–55. doi: 10.1016/S1074-7613(00)00064-9
15. Honda K, Mizutani T, Taniguchi T. Negative regulation of IFN-alpha/beta signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc Natl Acad Sci USA. (2004) 101:2416–21. doi: 10.1073/pnas.0307336101
16. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
17. Takaoka A, Tamura T, Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. (2008) 99:467–78. doi: 10.1111/j.1349-7006.2007.00720.x
18. Darnell JE Jr Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21. doi: 10.1126/science.8197455
19. Meraro D, Hashmueli S, Koren B, Azriel A, Oumard A, Kirchhoff S, et al. Protein-protein and DNA-protein interactions affect the activity of lymphoid-specific IFN regulatory factors. J Immunol. (1999) 163:6468–78.
20. Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology (2012) 1:1376–86. doi: 10.4161/onci.22475
21. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. (1998) 17:1087–95. doi: 10.1093/emboj/17.4.1087
22. Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur J Biochem. (2004) 271:3693–703. doi: 10.1111/j.1432-1033.2004.04310.x
23. Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
24. Reikine S, Nguyen JB, Modis Y. pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. (2014) 5:342. doi: 10.3389/fimmu.2014.00342
25. Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. (2003) 10:913–21. doi: 10.1038/nsb1002
26. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell (1998) 1:507–18. doi: 10.1016/S1097-2765(00)80051-9
27. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. (2013) 9:e1003118. doi: 10.1371/annotation/4de7ddfd-52df-4f87-8ca4-d48afe646ca8
28. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
29. Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe (2016) 19:150–8. doi: 10.1016/j.chom.2016.01.010
30. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
31. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
32. Zhao W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. (2013) 587:542–8. doi: 10.1016/j.febslet.2013.01.052
33. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
34. Carty M, Bowie AG. Recent insights into the role of Toll-like receptors in viral infection. Clin Exp Immunol. (2010) 161:397–406. doi: 10.1111/j.1365-2249.2010.04196.x
35. Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, et al. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature (2006) 440:949–53. doi: 10.1038/nature04641
36. Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. Protein kinase IKKbeta-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc Natl Acad Sci USA. (2014) 111:17432–7. doi: 10.1073/pnas.1418399111
37. Ren J, Chen X, Chen ZJ. IKKbeta is an IRF5 kinase that instigates inflammation. Proc Natl Acad Sci USA. (2014) 111:17438–43. doi: 10.1073/pnas.1418516111
38. Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, et al. Immune response in mice that lack the interferon-gamma receptor. Science (1993) 259:1742–5. doi: 10.1126/science.8456301
39. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell (1996) 84:431–42. doi: 10.1016/S0092-8674(00)81288-X
40. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity (2000) 13:795–804. doi: 10.1016/S1074-7613(00)00077-7
41. de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. (2007) 282:20053–7. doi: 10.1074/jbc.R700006200
42. Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. (2015) 11:e1004782. doi: 10.1371/journal.ppat.1004782
43. Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264:1918–21. doi: 10.1126/science.8009221
44. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
45. Zanoni I, Granucci F, Broggi A. Interferon (IFN)-lambda takes the Helm: immunomodulatory roles of type III IFNs. Front Immunol. (2017) 8:1661. doi: 10.3389/fimmu.2017.01661
46. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
47. Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE. Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc Natl Acad Sci USA. (1992) 89:7836–9. doi: 10.1073/pnas.89.16.7836
48. Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D'Andrea A, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature (1996) 383:344–7. doi: 10.1038/383344a0
49. Zhang JJ, Vinkemeier U, Gu W, Chakravarti D, Horvath CM, Darnell JE. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc Natl Acad Sci USA. (1996) 93:15092–6. doi: 10.1073/pnas.93.26.15092
50. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. (2011) 1:519–25. doi: 10.1016/j.coviro.2011.10.008
51. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
52. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
53. Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol. (2013) 425:4995–5008. doi: 10.1016/j.jmb.2013.09.041
54. Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel J. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep. (2016) 16:1492–1501. doi: 10.1016/j.celrep.2016.07.002
55. Schmid S, Mordstein M, Kochs G, García-Sastre A, Tenoever BR. Transcription factor redundancy ensures induction of the antiviral state. J Biol Chem. (2010) 285:42013–22. doi: 10.1074/jbc.M110.165936
56. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472:481–5. doi: 10.1038/nature09907
57. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell (2010) 141:668–81. doi: 10.1016/j.cell.2010.04.018
58. Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Diamond MS. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol. (2008) 82:8465–75. doi: 10.1128/JVI.00918-08
59. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. (2008) 19:414–22. doi: 10.1016/j.semcdb.2008.07.010
60. Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. (2006) 25:2358–67. doi: 10.1038/sj.emboj.7601149
61. Wang WB, Levy DE, Lee CK. STAT3 negatively regulates type I IFN-mediated antiviral response. J Immunol. (2011) 187:2578–85. doi: 10.4049/jimmunol.1004128
62. Tsai MH, Lee CK. STAT3 cooperates with phospholipid scramblase 2 to suppress type I interferon response. Front Immunol. (2018) 9:1886. doi: 10.3389/fimmu.2018.01886
63. Chen W, Daines MO, Khurana Hershey GK. Turning off signal transducer and activator of transcription (STAT): the negative regulation of STAT signaling. J Allergy Clin Immunol. (2004) 114:476–89; quiz 490. doi: 10.1016/j.jaci.2004.06.042
64. McBride KM, Banninger G, McDonald C, Reich NC. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. (2002) 21:1754–63. doi: 10.1093/emboj/21.7.1754
65. Fagerlund R, Mélen K, Kinnunen L, Julkunen I. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin alpha 5. J Biol Chem. (2002) 277:30072–8. doi: 10.1074/jbc.M202943200
66. Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA. (2005) 102:8150–5. doi: 10.1073/pnas.05010643102
67. McBride KM, McDonald C, Reich NC. Nuclear export signal located within theDNA-binding domain of the STAT1transcription factor. EMBO J. (2000) 19:6196–206. doi: 10.1093/emboj/19.22.6196
68. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell (2017) 170:492–506 e14. doi: 10.1016/j.cell.2017.06.042
69. Liu K, Lemon B, Traktman P. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J Virol. (1995) 69:7823–34.
70. Koksal AC, Cingolani G. Dimerization of Vaccinia virus VH1 is essential for dephosphorylation of STAT1 at tyrosine 701. J Biol Chem. (2011) 286:14373–82. doi: 10.1074/jbc.M111.226357
71. Najarro P, Traktman P, Lewis JA. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol. (2001) 75:3185–96. doi: 10.1128/JVI.75.7.3185-3196.2001
72. Unterholzner L, Sumner RP, Baran M, Ren H, Mansur DS, Bourke NM, et al. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. (2011) 7:e1002247. doi: 10.1371/journal.ppat.1002247
73. Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. (2016) 35:1385–99. doi: 10.15252/embj.201593458
74. Zhang D, Su C, Zheng C. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol. (2016) 90:5824–5829. doi: 10.1128/JVI.00186-16
75. Cao J, Zhou Y, Gong GZ. Effect of HCV NS5A on STAT1 phosphorylation and nuclear translocation induced by IFN alpha-2b. Zhonghua Gan Zang Bing Za Zhi (2006) 14:894–7.
76. Kumthip K, Chusri P, Jilg N, Zhao L, Fusco DN, Zhao H, et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J Virol. (2012) 86:8581–91. doi: 10.1128/JVI.00533-12
77. Sen A, Rott L, Phan N, Mukherjee G, Greenberg HB. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J Virol. (2014) 88:41–53. doi: 10.1128/JVI.01501-13
78. Hartman AL, Bird BH, Towner JS, Antoniadou ZA, Zaki SR, Nichol ST. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J Virol. (2008) 82:2699–704. doi: 10.1128/JVI.02344-07
79. Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Mühlberger E, Bray M, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. (2003) 77:7945–56. doi: 10.1128/JVI.77.14.7945-7956.2003
80. Valmas C, Grosch MN, Schümann M, Olejnik J, Martinez O, Best SM, et al. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. (2010) 6:e1000721. doi: 10.1371/journal.ppat.1000721
81. Koeberlein B, zur Hausen A, Bektas N, Zentgraf H, Chin R, Nguyen LT, et al. Hepatitis B virus overexpresses suppressor of cytokine signaling-3 (SOCS3) thereby contributing to severity of inflammation in the liver. Virus Res. (2010) 148:51–9. doi: 10.1016/j.virusres.2009.12.003
82. Collins AS, Ahmed S, Napoletano S, Schroeder M, Johnston JA, Hegarty JE, et al. Hepatitis C virus (HCV)-induced suppressor of cytokine signaling (SOCS) 3 regulates proinflammatory TNF-alpha responses. J Leukoc Biol. (2014) 96:255–63. doi: 10.1189/jlb.2A1211-608RRRR
83. Kim K, Kim KH, Cheong J. Hepatitis B virus X protein impairs hepatic insulin signaling through degradation of IRS1 and induction of SOCS3. PLoS ONE (2010) 5:e8649. doi: 10.1371/journal.pone.0008649
84. Gupta SP. Viral proteases and their inhibitors. London: Elsevier; Academic Press (2017). xv, 499 pages.
85. Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol. (2013) 87:1690–8. doi: 10.1128/JVI.01855-12
86. Xiang Z, Liu L, Lei X, Zhou Z, He B, Wang J. 3C Protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J Virol. (2016) 90:1613–21. doi: 10.1128/JVI.02395-15
87. Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu JT. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis. (2011) 203:1784–90. doi: 10.1093/infdis/jir174
88. Park SY, Waheed AA, Zhang ZR, Freed EO, Bonifacino JS. HIV-1 Vpu accessory protein induces caspase-mediated cleavage of IRF3 transcription factor. J Biol Chem. (2014) 289:35102–10. doi: 10.1074/jbc.M114.5097062
89. Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M. Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J Virol. (2012) 86:8367–74. doi: 10.1128/JVI.00423-12
90. Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. (2007) 81:4473–81. doi: 10.1128/JVI.02498-06
91. Arnold MM, Barro M, Patton JT. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol. (2013) 87:9813–21. doi: 10.1128/JVI.01146-13
92. Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, et al. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology (2001) 283:230–9. doi: 10.1006/viro.2001.0856
93. Andrejeva J, Young DF, Goodbourn S, Randall RE. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J Virol. (2002) 76:2159–67. doi: 10.1128/jvi.76.5.2159-2167.2002
94. Ashour J, Laurent-Rolle M, Shi PY, García-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. (2009) 83:5408–18. doi: 10.1128/JVI.02188-08
95. Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, et al. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J Virol. (2005) 79:5414–20. doi: 10.1128/JVI.79.9.5414-5420.2005
96. Dar HA, Zaheer T, Paracha RZ, Ali A. Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infect Genet Evol. (2017) 51:143–152. doi: 10.1016/j.meegid.2017.03.027
97. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe (2016) 19:882–90. doi: 10.1016/j.chom.2016.05.009
98. Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, et al. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe (2014) 16:187–200. doi: 10.1016/j.chom.2014.07.008
99. Shabman RS, Gulcicek EE, Stone KL, Basler CF. The Ebola virus VP24 protein prevents hnRNP C1/C2 binding to karyopherin alpha1 and partially alters its nuclear import. J Infect Dis. (2011) 204 Suppl. 3:S904–10. doi: 10.1093/infdis/jir323
100. Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ, et al. Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-alpha1 degradation. J Biol Chem. (2017) 292:10262–74. doi: 10.1074/jbc.M116.745729
101. Göertz GP, McNally KL, Robertson SJ, Best SM, Pijlman GP, Fros JJ. The methyltransferase-like domain of chikungunya virus nsP2 inhibits the interferon response by promoting the nuclear export of STAT1. J Virol. (2018) 92:e01008–18. doi: 10.1128/JVI.01008-18
102. Fros JJ, Liu WJ, Prow NA, Geertsema C, Ligtenberg M, Vanlandingham DL, et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J Virol. (2010) 84:10877–87. doi: 10.1128/JVI.00949-10
103. Sugai A, Sato H, Takayama I, Yoneda M, Kai C. Nipah and hendra virus nucleoproteins inhibit nuclear accumulation of signal transducer and activator of transcription 1 (STAT1) and STAT2 by interfering with their complex formation. J Virol. (2017) 91:e01136–17. doi: 10.1128/JVI.001136-17
104. Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. (2004) 78:5358–67. doi: 10.1128/JVI.78.10.5358-5367.2004
105. Rodriguez JJ, Parisien JP, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol. (2002) 76:11476–83. doi: 10.1128/JVI.76.22.11476-11483.2002
106. Schomacker H, Hebner RM, Boonyaratanakornkit J, Surman S, Amaro-Carambot E, Collins PL, et al. The C proteins of human parainfluenza virus type 1 block IFN signaling by binding and retaining Stat1 in perinuclear aggregates at the late endosome. PLoS ONE (2012) 7:e28382. doi: 10.1371/journal.pone.0028382
107. Oda K, Oda T, Matoba Y, Sato M, Irie T, Sakaguchi T. Structural analysis of the STAT1:STAT2 heterodimer revealed the mechanism of Sendai virus C protein-mediated blockade of type 1 interferon signaling. J Biol Chem. (2017) 292:19752–66. doi: 10.1074/jbc.M117.786285
108. Oda K, Matoba Y, Irie T, Kawabata R, Fukushi M, Sugiyama M, et al. Structural basis of the inhibition of STAT1 activity by sendai virus C protein. J Virol. (2015) 89:11487–99. doi: 10.1128/JVI.01887-15
109. Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. (2003) 77:7635–44. doi: 10.1128/JVI.77.13.7635-7644.2003
110. Holloway G, Truong TT, Coulson BS. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-kappaB. J Virol. (2009) 83:4942–51. doi: 10.1128/JVI.01450-08
111. Paladino P, Collins SE, Mossman KL. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE (2010) 5:e10428. doi: 10.1371/journal.pone.0010428
112. Melroe GT, DeLuca NA, Knipe DM. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol. (2004) 78:8411–20. doi: 10.1128/JVI.78.16.8411-8420.2004
113. Bi X, Yang L, Mancl ME, Barnes BJ. Modulation of interferon regulatory factor 5 activities by the Kaposi sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3 contributes to immune evasion and lytic induction. J Interferon Cytokine Res. (2011) 31:373–82. doi: 10.1089/jir.2010.0084
114. Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. (2007) 81:8282–92. doi: 10.1128/JVI.00235-07
115. Seo T, Lee D, Lee B, Chung JH, Choe J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) binds to, and inhibits transactivation of, CREB-binding protein. Biochem Biophys Res Commun. (2000) 270:23–7. doi: 10.1006/bbrc.2000.2393
116. Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol. (2013) 87:9788–801. doi: 10.1128/JVI.01440-13
117. Morin G, Robinson BA, Rogers KS, Wong SW. A Rhesus Rhadinovirus Viral Interferon (IFN) Regulatory factor is virion associated and inhibits the early IFN antiviral response. J Virol. (2015) 89:7707–21. doi: 10.1128/JVI.01175-15
118. Zhang R, Fang L, Wang D, Cai K, Zhang H, Xie L, et al. Porcine bocavirus NP1 negatively regulates interferon signaling pathway by targeting the DNA-binding domain of IRF9. Virology (2015) 485:414–21. doi: 10.1016/j.virol.2015.08.005
119. Zhang R, Fang L, Wu W, Zhao F, Song T, Xie L, et al. Porcine bocavirus NP1 protein suppresses type I IFN production by interfering with IRF3 DNA-binding activity. Virus Genes (2016) 52:797–805. doi: 10.1007/s11262-016-1377-z
120. Zhang Q, Shi K, Yoo D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology (2016) 489:252–68. doi: 10.1016/j.virol.2015.12.010
121. Han M, Du Y, Song C, Yoo D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1alpha subunit. Virus Res. (2013) 172:54–65. doi: 10.1016/j.virusres.2012.12.012
122. Bentz GL, Liu R, Hahn AM, Shackelford J, Pagano JS. Epstein-Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-beta. Virology (2010) 402:121–8. doi: 10.1016/j.virol.2010.003.014
123. Read GS, Karr BM, Knight K. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J Virol. (1993) 67:7149–60.
124. Chee AV, Roizman B. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J Virol. (2004) 78:4185–96. doi: 10.1128/JVI.78.8.4185-4196.2004
125. Taddeo B, Zhang W, Roizman B. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J Virol. (2013) 87:4516–22. doi: 10.1128/JVI.00005-13
126. Mukherjee A, Di Bisceglie AM, Ray RB. Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway. J Virol. (2015) 89:3356–65. doi: 10.1128/JVI.03085-14
127. Kuo YP, Tsai KN, Luo YC, Chung PJ, Su YW, Teng Y, et al. Establishment of a mouse model for the complete mosquito-mediated transmission cycle of Zika virus. PLoS Negl Trop Dis. (2018) 12:e0006417. doi: 10.1371/journal.pntd.0006417
128. Bradfute SB, Stuthman KS, Shurtleff AC, Bavari S. A STAT-1 knockout mouse model for Machupo virus pathogenesis. Virol J. (2011) 8:300. doi: 10.1186/1743-422X-8-300
129. Wan SW, Chen PW, Chen CY, Lai YC, Chu YT, Hung CY, et al. Therapeutic effects of monoclonal antibody against dengue virus NS1 in a STAT1 knockout mouse model of dengue infection. J Immunol. (2017) 199:2834–44. doi: 10.4049/jimmunol.1601523
130. Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, et al. Selective activation of type II interferon signaling by zika virus NS5 protein. J Virol. (2017) 91:e00163–17. doi: 10.1128/JVI.00163-17
Keywords: interferon, interferon-regulatory factor, signal transducer and activator of transcription signaling, interferon-stimulated gene, antiviral response, viral attenuation, viral antagonism
Citation: Chiang H-S and Liu HM (2019) The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Front. Immunol. 9:3086. doi: 10.3389/fimmu.2018.03086
Received: 03 September 2018; Accepted: 13 December 2018;
Published: 08 January 2019.
Edited by:
Chien-Kuo Lee, National Taiwan University, TaiwanReviewed by:
Junji Xing, Houston Methodist Research Institute, United StatesCopyright © 2019 Chiang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao-Sen Chiang, aHNjaGlhbmdAbnR1LmVkdS50dw==
Helene Minyi Liu, bWxpdUBudHUuZWR1LnR3
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.