 Elizabeth C. Stahl
Elizabeth C. Stahl Branimir Popovic
Branimir Popovic Bryan N. Brown
Bryan N. Brown
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 November 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02795
This article is part of the Research Topic The Role of Myeloid-Derived Cells in the Progression of Liver Disease View all 16 articles
The number of individuals aged 65 or older is projected to increase globally from 524 million in 2010 to nearly 1. 5 billion in 2050. Aged individuals are particularly at risk for developing chronic illness, while being less able to regenerate healthy tissue and tolerate whole organ transplantation procedures. In the liver, these age-related diseases include non-alcoholic fatty liver disease, alcoholic liver disease, hepatitis, fibrosis, and cirrhosis. Hepatic macrophages, a population comprised of both Kupffer cells and infiltrating monocyte derived macrophages, are implicated in several chronic liver diseases and also play important roles in the homeostatic functions of the liver. The effects of aging on hepatic macrophage population dynamics, polarization, and function are not well understood. Studies performed on macrophages derived from other aged sources, such as the bone marrow, peritoneal cavity, lungs, and brain, demonstrate general reductions in autophagy and phagocytosis, dysfunction in cytokine signaling, and altered morphology and distribution, likely mediated by epigenetic changes and mitochondrial defects, that may be applicable to hepatic macrophages. This review highlights recent findings in macrophage developmental biology and function, particularly in the liver, and discusses the role of macrophages in various age-related liver diseases. A better understanding of the biology of aging that influences hepatic macrophages and thus the progression of chronic liver disease will be crucial in order to develop new interventions and treatments for liver disease in aging populations.
The liver is an important immunological organ, serving as a surveillance system for gut-derived pathogens and producing several key immune components—complement, acute phase, and coagulation proteins (1, 2). While hepatocytes and certain non-parenchymal cells possess some inherent immunological properties [see reviews 3 and 4)], multiple populations of CD45+ immune cells are transiently or permanently located in the liver.
The healthy liver is home to several populations of lymphocytes, including natural killer (NK) cells, NK T-cells, B-cells, mucosal associated T-cells and γδ-T-cells (5–8). In humans, 40% of the resident lymphocyte population are composed of NK cells, while in mice 40% of the resident lymphocytes are NK T-cells.
Innate immune cells also have a large presence in the liver. The majority of immune cells in the liver are myeloid derived cells (9). Dendritic cell (DC) populations, including both myeloid and plasmacytoid DCs, are present in the liver and can activate T-cells under appropriate conditions (10). Myeloid-derived suppressor cells (MDSC) are also present and suppress T-cell activation (11). Macrophages derived from circulating monocytes have been identified as a motile population of myeloid cells infiltrating the liver during inflammation and at varying levels during homeostasis (12–14). The resident macrophage, known as the Kupffer cell (KC), is the most highly represented immune cell, comprising nearly one-third of non-parenchymal cells in the liver (15).
Macrophages play a key role in the homeostatic functions of the liver as well as in disease states and are among the most widely studied immune cells in the liver (15). The prevalence and severity of many chronic liver diseases increases with age, including alcoholic hepatitis, non-alcoholic steatohepatitis, cirrhosis, and hepatocellular carcinoma (16). However, mechanisms underlying changes in the liver structure and cellular function, including hepatocytes and resident immune cell populations, are not well understood. This review will cover the developmental origin and function of macrophages in the liver, as well as their implications in several age-related liver diseases, in order to better understand and expose gaps in the knowledge of the biology of liver aging and disease. As most of the experimental data has been collected from murine animal models, this review focuses largely on preclinical studies in mice and extends these findings to chronic liver disease in humans.
Macrophage development begins in the extraembryonic yolk sac from erythro-myeloid progenitors, a process known as primitive hematopoiesis, prior to the appearance of hematopoietic stem cells or monocytic precursors (17, 18). After this transient wave of primitive hematopoiesis, hematopoietic stem cells appear in the aorto-gonado-mesonephric region and migrate to the fetal liver, marking the start of definitive hematopoiesis and development of naïve macrophages through monocytic precursors (17). Definitive hematopoiesis remains in the fetal liver until approximately E18 in mice or 12 weeks post-conception in humans, after which hematopoietic stem cells migrate from the fetal liver to the bone marrow niche, where they will remain throughout adulthood (19).
Macrophages can be subdivided into tissue resident or monocyte derived populations. The number of resident macrophages varies considerably based on the tissue, and approximately 80% of total tissue resident macrophages are located in the liver (20). Resident macrophages can be colonized from a single developmental source, such as microglia and Kupffer cells, which are derived from yolk sac primitive macrophages. Alternatively, resident macrophages may be repopulated several times during development, such as Langerhans cells in the skin, which are originally derived from yolk-sac macrophages and later replaced by fetal liver monocytes (13, 21). Macrophage populations in the gut, spleen, and lungs are continuously replenished by monocyte input throughout adulthood (12, 13).
During inflammation, both resident and monocyte-derived macrophages participate in migration, expansion, and signaling depending on the tissue type and stimulus. Resident tissue macrophages, including Kupffer cells, peritoneal macrophages, and pleural macrophages, have been shown to undergo rapid proliferation in response to Th2 cytokines, such as interleukin-4 (22). Infiltrating macrophages can be derived from either classical (CD14+CD16− human, Ly6Chi mouse) or non-classical (CD14+CD16+ human, Ly6Clo mouse) monocytes, which extravasate into tissue sites under chemokine concentration gradient guidance (17, 23). Following extravasation, monocytes differentiate into macrophages whose functionality are largely based on the integration of the various signaling molecules present in the local microenvironment (24, 25).
In addition to developmental heterogeneity, macrophages are extraordinarily dynamic cells that exhibit various phenotypes ranging from a pro-inflammatory, classically activated “M1” polarization state to an anti-inflammatory, alternatively activated “M2” polarization state, as shown in Figure 1 (26, 27). M1/M2 polarization refers to extreme phenotypes that can be simulated in vitro with the addition of various stimuli (lipopolysaccharide and/or interferon-gamma for M1 activation; interleukin-4 for M2 activation), but the scenario in vivo is often more complex and thus polarization is a spectrum of phenotypes, including but not limited to M2a, M2b, and M2c sub-groups (27, 28).
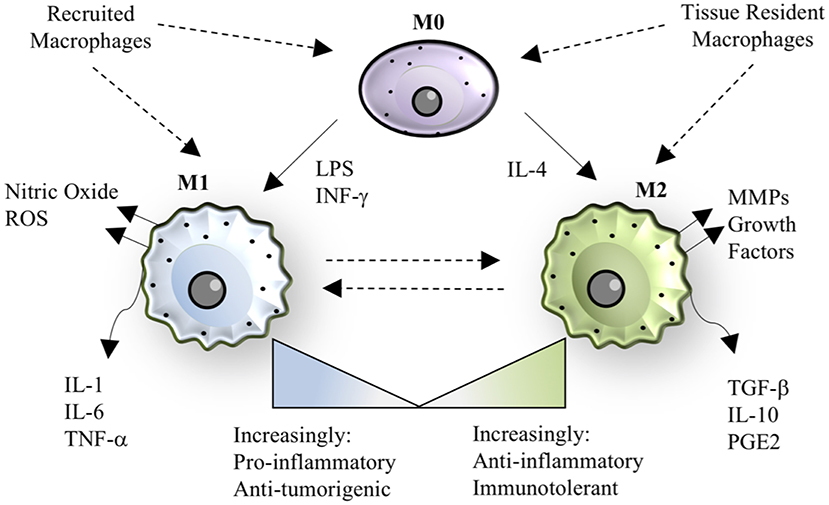
Figure 1. Macrophage polarization. The paradigm of macrophage polarization begins with a quiescent or patrolling M0 cell, that may be derived from either circulating monocytes or tissue-resident populations. The transition from M0 to M1 can be stimulated by lipopolysaccharide (LPS) or interferon-gamma, resulting in a cell that has pro-inflammatory and anti-tumorigenic properties. M1 macrophages also produce nitric oxide and other reactive oxygen species (ROS). The transition from M0 to M2 can be stimulated by interleukin-4, resulting in a cell that has anti-inflammatory or immunotolerant properties. M2 macrophages also produce matrix metalloproteinases (MMPs) and growth factors that aid in tissue regeneration. M1 and M2 phenotypes are dynamic and exist on a spectrum, where recruited macrophages tend to be more “M1-like” in the case of bacterial infection, and tissue-resident macrophages tend to be more “M2-like” during homeostasis. In specific instances, such as helminth infection, recruited macrophages take on an M2 phenotype, demonstrating the versatility and adaptability of the macrophage polarization spectrum in response to the signals in a given context.
In general, M1 polarization leads to production of canonical pro-inflammatory cytokines (IL-1, IL-6, IL-12, IL-18, TNF, IL-23, IL-27), reactive oxygen species such as nitric oxide, and chemokines (CCL2, CCL5, CXCL9, CXCL10, CXCL11, IL-8) (27). Macrophage pro-inflammatory cytokine maturation can occur through inflammasome-independent or inflammasome-dependent mechanisms (29, 30). Inflammasome formation is induced by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) binding to nod-like receptors, including NLRP3 and NLRP1, or to Pyrin receptor family members such as Pyrin and AIM2 on the surface of macrophages (31). Docking proteins, including apoptosis-associated speck-like protein, assemble and recruit pro-caspase 1, where proximity-based autoproteolytic cleavage catalyzes the activation of caspase-1 (29, 32) Caspase-1 activation subsequently catalyzes the cleavage of the pro-forms of IL-1β and IL-18 into activated forms. Caspase-1 may also induce pyroptosis, a programmed cell death pathway characterized by cellular lysis and endogenous DAMP release into the environment, to induce additional inflammatory cell recruitment (33).
M2-polarized macrophages typically appear following M1-polarized macrophages in an injury setting and serve as a counterbalance to resolve inflammation and promote tissue repair (34). The M2 polarization state leads to secretion of cytokines commonly associated with anti-inflammatory properties, such as IL-10, as well as various matrix-modulatory factors including transforming growth factor-β (TGF-β) and matrix metalloproteases (MMPs) (35). The overproduction of remodeling growth factors can lead to excessive deposition of matrix proteins (i.e., fibrosis), or excessive angiogenesis and immunosuppression in the case of tumor-associated macrophages (TAMs) (36). Therefore, the temporal regulation of M1 and M2 macrophage responses are extremely important for appropriate outcomes following injury and infection and are often dysregulated in chronic disease.
The Kupffer cell is a primitive cell, appearing early during embryogenesis (E9.5-E12.5 in mice), derived primarily from the yolk sac (14, 37, 38). Importantly, the KC population is maintained through self-proliferation, with minimal input from circulating monocytes during homeostasis (12–14, 22).
The tissue-resident Kupffer cell is the key detector of commensal or pathogenic microbial signals, danger signals, and tumor cells moving through the hepatic circulation (15, 39, 40). KCs are located along the hepatic sinusoids allowing for the low-pressure blood supply come into contact with both KCs and hepatocytes through the fenestrated endothelium (2). KCs express the complement receptor CR1g, which binds complement fragments C3b and iC3b, allowing phagocytosis of complement C3-opsonized particles even under low-pressure blood flow (41, 42). Bacterial clearance by KCs is crucial for host defense as 80% of blood-borne bacteria accumulate in the liver and are destroyed there (15, 39, 40).
PAMPs and DAMPs are present in relatively high concentrations in blood entering the liver from the gut, via the portal vein, and engage with pattern recognition receptors (PRRs), including Toll-like receptors, on the surface of macrophages and hepatocytes (43–45). With low levels of bacterial endotoxins, KCs promote immune tolerance by secreting anti-inflammatory factors including IL-10, TGF-β, and prostaglandin-E2 (PGE2), thereby inducing regulatory T-cells (46, 47). In the presence of higher concentrations of damage or pathogen-associated signaling molecules, KCs can become polarized toward an M1 phenotype and produce a variety of inflammatory cytokines including IL-1, IL-6, IL-12, TNF-α (15, 39, 40). Several liver diseases are influenced by KC activation and expansion, but their individual role has been difficult to dissect from more recently identified macrophage populations in the liver.
Within the past decade, the heterogeneity of hepatic macrophages, i.e., Kupffer cells and monocyte-derived macrophages (mMØs), has become an emerging topic in hepatology. Fate tracing experiments in the brain first determined that resident microglia are established prenatally and maintained independently from monocyte input, which was later translated to the liver (48). Holt et al. were among the first to identify two populations of macrophages in the liver (49). Through bone marrow chimera experiments, cells expressing F4/80hiCD11blo were identified as KCs, while cells expressing F4/80loCD11bhi were found to be derived from circulating monocytic progenitors (49). Shortly after, Klein et al. classified two macrophage populations in the liver: KCs as immobile, “sessile” macrophages and mMØs as motile cells (50).
In most cases, murine mMØs in the liver are derived from an influx of bone marrow derived, Ly6Chi monocytes, primarily driven by monocyte chemoattractant protein, which can be produced by KCs, stellate cells, or hepatocytes (also known as CCR2-CCL2 interactions) (51, 52). Secondary pathways of monocyte recruitment to the liver are CXCR3-CXCL10, CCR1-CCL5, and CCR8-CCL1 dependent (53–55). Murine mMØs may also be derived from Ly6Clo monocytes trafficking from the spleen, which express CD11b, but are thought to take on a more patrolling or regulatory macrophage phenotype (56). Phagocytes from the peritoneal cavity, which express F4/80, CD11b, and GATA6, have been shown to cross into the liver after subcapsular liver lesion (57), but it remains unclear if this infiltration occurs in other liver injury settings.
Kinoshita et al. determined that murine mMØs and KCs could be distinguished by CD11b and CD68 expression, respectively (40). The CD11bhi mMØs were radiosensitive and particularly efficient at producing IL-12, protecting the host against tumor xenografts, while the CD68hi KCs were radioresistant and highly efficient at phagocytosis, protecting the host against bacterial challenge (40). The use of these differentiating markers has been expanded to several disease models, identifying a damaging role of TNF/FasL production by CD11b+ mMØs in carbon-tetrachloride acute liver injury and hepatitis in hypercholesterolemic mice (58, 59). The CD11b+ mMØs were also found to accumulate in the liver following repeated lipopolysaccharide injections, but suppressed TNF efflux into the systemic circulation, thereby reducing lethal septicemia (60). In addition, the CD11b+ mMØs were recruited during diet-induced steatohepatitis in FGF5 deficient mice and following partial hepatectomy, where they were crucial for liver regeneration (61, 62). Others have observed that monocyte derived cells protect the liver from iron toxicity in hemolytic anemia by ingesting senescent and dying erythrocytes (63).
More recently, the C-type lectin, Clec4f, has been identified as a selective murine KC marker (64). Interesting, ablating KCs via Clec4f-driven diphtheria toxin causes an influx of mMØs, which can differentiate into nearly identical KCs when this “niche” is made available. Only 12 genes remained differentially expressed between monocyte derived KCs and embryonically derived KCs, including CD209f, CD163, C2, CCR3, Timd4, and Snrpn at the time points examined (64).
Taken together, KCs and mMØs play heterogenous roles in various liver disease states and cannot be considered wholly harmful or beneficial without deciphering the given context. Importantly, infiltrating monocytes may also differentiate into dendritic cells, which play distinct immunostimulatory and antigen presentation roles in the liver (65). The heterogeneity of hepatic macrophages is less well defined in humans as compared to murine animal models. The role of these diverse hepatic macrophage populations in a given disease context is just beginning to be understood and has not yet been examined in the natural aging process, a major risk factor for several chronic liver diseases.
The process of aging is closely associated with a number of degenerative modifications in the liver, where hepatic structure and cell function are observed to decline. Both hepatocytes and macrophages exhibit deficits in mitochondrial function, linked to a decline in autophagy and production of pro-inflammatory molecules (66, 67). While the effects of aging on hepatic macrophages have not been well characterized on a cellular and molecular level, several studies have examined macrophages from alternative tissue sources and may offer some insight about hepatic macrophages.
Several studies have shown that the volume of blood in the liver decreases in elderly individuals, leading to a total volume loss of 20–40% (68). In addition, the thickness of liver sinusoidal endothelial cells fenestrations increases, limiting the exchange of molecules to and from the liver (69). At the serum level, aging is associated with reductions in albumin and bilirubin, increases in alkaline phosphatase, and minimal changes in aminotransferase levels (70). The metabolism of cholesterol in the liver also decreases, leading to increased blood cholesterol and neutral fat levels over time (70). However, recent investigations suggest the most essential change in liver aging is loss of the functional liver cell mass (71).
Changes in the morphology of hepatocytes may be related to increased polyploidy, accumulation of lipofuscin in the cytoplasm, and declining surface area of endoplasmic reticulum and number of mitochondria, ultimately negatively affecting the function of hepatocytes (66, 72, 73). The decline in hepatocyte mitochondrial function has been suggested to enhance the vulnerability of aged livers to acute injury and to cause delays in liver regeneration (74). The oxidative capacity of the liver also declines with aging, and therefore medications that require oxidation, such as benzodiazepines, may accumulate in toxic levels and are not prescribed to elderly individuals (75). In addition, aging livers are known to accumulate a multiprotein C/EBPalpha-Brm-HDAC1 complex that silences elongation factor 2 (E2F)-dependent promoters, thereby reducing proliferation and regenerative capacity of hepatocytes (76).
Senescent cells accumulate in the liver during aging and in chronic liver disease; and may include hepatocytes, cholangiocytes, stellate cells, and immune cells [reviewed here: (77)]. Cells become senescent as a result of replicative exhaustion (telomere shortening), DNA damage, or oxidative stress, among other mechanisms (78). Senescent cells are characterized by the expression of cell cycle inhibitors p21, p16, and p53, which prevent replication and apoptosis, as well as secretion of pro-inflammatory cytokines that signal for their removal but can lead to tissue damage when chronically expressed (79, 80). Senescent hepatocytes accumulate with age and appear as an almost “universal phenomenon” in chronic liver diseases including hepatitis B and C infection, alcoholic and non-alcoholic liver disease, and genetic haemochromatosis (77). Senescent hepatocytes undergo metabolic changes, such as increased transport of conjugated bilirubin into the hepatic sinusoids and insulin resistance through dysregulated glucose transporter expression and Akt signaling among others, which dysregulates normal function (81). Interestingly, the length of telomeres was found to be conserved in aged hepatocytes and bile duct cholangiocytes but decreased in aged Kupffer cells and stellate cells, suggesting cell-specific mechanisms of senescent phenotype acquisition in the liver (82).
Overall, the effects of aging on non-parenchymal cell subsets, particularly hepatic macrophages, is an understudied area. Hilmer et al. determined that the number and basal activity of Kupffer cells was increased with old age in a rat model (83), however this work was performed before the heterogeneity of liver macrophages was fully contemplated. Similarly, Singh et al. identified a change in liver macrophage distribution with aging, where old mice had more F4/80+ macrophages located in large lymphoid structures (also containing T-cells and B-cells), and a reduction in the number of spindle shaped macrophages throughout the parenchyma (84).
Recent work by Maeso-Diaz et al. characterized molecular changes in LSECs, stellate cells, and hepatic macrophages in the livers of aged rats (85). The authors found a significant deregulation of LSEC phenotype with aging as demonstrated by downregulation of vasodilatory pathways (nitric oxide, heme oxygenase), increase in oxidative stress, and decrease in angiocrine markers (stabilin-2, CD32b, and VEGFR2), in addition to increased portal pressure and vascular resistance (85). Aged stellate cells had increased markers of activation (alpha smooth muscle actin and collagen I), increased intracellular lipid stores, and alterations in retinoid metabolism (85). Finally, the authors noted an increase in recruitment of proinflammatory cells in aged livers, including increased IL-6 expression in isolated hepatic macrophages, but no change in the traditional macrophage polarization markers (85). Human livers showed similar trends in downregulation of endothelial markers, but substantial work remains to extend and decode these findings in both aged murine and human systems.
The process of aging has been shown to affect macrophage polarization and function in multiple tissues and disease models, with compelling underlying mechanisms that may be broadly applicable to hepatic macrophages, as summarized in Figure 2.
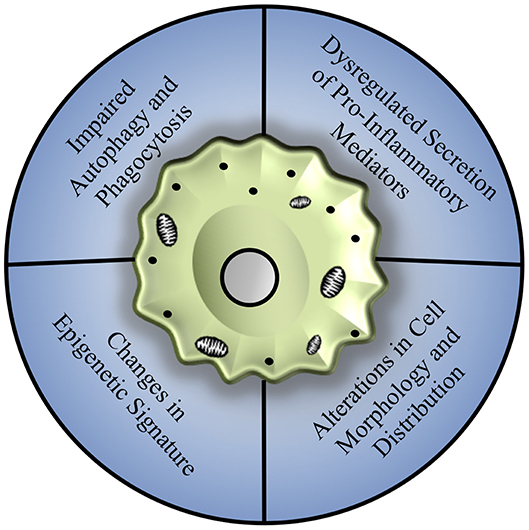
Figure 2. Mechanisms of age-associated decline in macrophage function. Macrophages from various tissue sources, including the bone marrow, peritoneal cavity, lungs, and brain, show an array of dysfunction including a general reduction in autophagy and phagocytosis, dysregulation of pro-inflammatory cytokine production associated with changes in toll-like receptor expression, and alterations in cell morphology and distribution that may be related to repopulation of tissues by infiltrating monocyte populations, largely mediated by changes in epigenetic signature and mitochondrial dysfunction.
Autophagy, or the intracellular degradation of damaged proteins and organelles in autophagosomes, is one such process which has been shown to become dysregulated with increasing age in macrophage populations (86). Deletion of autophagy genes Atg5 and Atg7 in bone marrow-derived macrophages results in decreased antigen presentation capacity, impaired maturation, altered mitochondrial metabolism, downregulation of surface receptors such as toll-like receptor 4 (TLR4), and increased secretion of pro-inflammatory cytokines (86, 87). The phenotype of autophagy deficient bone marrow macrophages closely mimics the phenotype of macrophages isolated from aged individuals (86), which may be driven by the hypermethylation of autophagy associated genes (88). It remains to be seen if restoring autophagic flux to aged macrophages can improve their function and polarization status, however caloric restriction has been shown to improve both longevity and autophagic capacity in animal models (89).
Phagocytic clearance of extracellular pathogens and antigen presentation tends to be attenuated in aged macrophage populations as well (90). While no difference in phagocytic capacity was found between young and aged bone marrow derived macrophages, peritoneal macrophages exhibit an age-associated decline in phagocytic capacity and antigen presentation (90). This reduction in phagocytic ability was driven by age-related alterations in the local microenvironment, as young peritoneal macrophages transplanted into aged peritoneal space exhibited reduced phagocytic and antigen presentation capabilities (90). Alveolar macrophages isolated from aged individuals have been shown to exhibit reduced phagocytic capacity as well as attenuated expression of genes associated with macrophage proliferation (91). This reduction both in cell number and phagocytic ability contributes to the age-related mortality risk following pathogen infection, such as in the case of an influenza lung infection model (91). In the liver, there have been mixed reports on the maintenance of Kupffer cell phagocytosis, where one study reported a deficit (92) and another reported an increase in phagocytosis with aging (83).
Furthermore, the reduction in expression levels of autophagosome components prevents the endocytosis of both inflammasome components and damaged mitochondria and can thereby promote chronic activation of pro-inflammatory signals in aged cells (87). This phenomenon has been termed “inflamm-aging,” one of the hallmarks of aging, and has been strongly correlated to morbidity (93). In addition to dysregulated cytokine production, aging can promote alterations in the secretion of oxidative species (94–96). Bone marrow-derived macrophages isolated from aged murine and human donors have been shown to produce greater concentrations of nitric oxide and reactive oxidative species than macrophages isolated from young donors (96). However, this upregulation in the secretion of reactive oxidative species was not observed following pro-inflammatory stimulation of peritoneal macrophages (94, 95). Further studies will be necessary to fully understand tissue-specific changes in respiratory burst characteristics.
The morphology of macrophages can also be influenced by the aging process. Microglia have been shown to increase cell soma volume while reducing the length of cellular processes in aged brains (97). These morphological changes reduce the capability of the microglia to interact with neural cells and perform routine surveillance of the local microenvironment (97). While microglial population sizes increase to account for this reduction in cell process size (17), the population expansion tends to occur in a non-homogenous manner, disrupting the uniform microglial distributions commonly observed in young animals (97). In addition, aged microglia exhibit enhanced secretion of reactive oxidative species following central nervous system injury through upregulation of NADPH oxidase 2 (NOX2) (98), and secrete elevated concentrations of IL-1β mediated by hypomethylation of CpG sires in the IL-1β proximal promoter (99).
Infiltration of monocyte-derived macrophages to multiple tissue sites has been observed in age-related diseases, including Alzheimer's disease (100), which leads to increased inflammation and phagocytosis. In the cardiovascular system, the tissue resident macrophage population (yolk-sac derived, M2-like) is replaced by bone marrow-derived macrophages over time (101, 102). This shift in macrophage populations contributes to deleterious outcomes following cardiac injury, such as chronic inflammation, fibrotic scar deposition, and reduced re-vascularization of ischemic tissue following injury (101, 103). It remains unclear whether age-related changes in macrophage responses are a primary result of shifting cell origins and polarization states driven by the microenvironment or by cell intrinsic functions, although both are likely in play.
While cell intrinsic properties, such as mitochondrial capacity and autophagy, may drive changes in age-associated macrophage polarization, this only represents a partial view of the complex innate and adaptive immune dysfunction which occurs with age. For example, age-related alterations in the T cell compartment, which are numerous [reviewed here (104, 105)], can shift the relative Th1 and Th2 cytokine concentrations and directly impact macrophage polarization (106, 107).
Non-immune cell types can also contribute to the age-associated immune dysfunction. With age, increasingly large numbers of pre-adipocytes differentiate into mature adipocytes, with enhanced secretion of leptin, TNF-α, and IL-6, and reduced secretion of adiponectin—a factor commonly associated with M2 macrophage polarization, thereby promoting M1 phenotypes (108–110). Numerous DAMPs, which have been shown to promote inflammasome formation and activation, also have age-related increases in concentration, including cholesterol (111), amyloid-β (112, 113), hydroxyapatite crystals (114), purine catabolic end products such as uric acid (115), and ATP (116). Radical oxidative species, which tend to exhibit an increase in production and secretion with increasing age, have also been shown to play an important role in inflammasome activation through oxidation of proteins, lipids, and DNA, leading to inflammatory cytokine secretion into the aged microenvironment (29, 117, 118). More recently, the effect of the aging gut microbiome on macrophage function has been considered (119). Raising mice under germ-free conditions preserves the bactericidal capacity of alveolar macrophages and reduces secretion of IL-6 in old age, mediated by reduced dysbiosis and improved gut permeability (119).
While this is only a brief consideration of additional factors that have been observed to influence macrophage function, it serves to highlight the complexity and tissue specificity that defines age-related immunomodulation. The mechanisms governing the acquisition of age-related dysfunction in infiltrating and tissue resident macrophage populations continues to be an active area of research, and it will be essential for investigators to utilize an array of cell markers and descriptive methods to appropriately place cells along the spectrum of polarization states.
Aging is a major risk factor for the development and prognosis of several chronic liver diseases and conditions, including nonalcoholic fatty liver disease, alcoholic liver disease, hepatitis C, and an increased susceptibility to develop fibrosis and cirrhosis (16). The reduced capacity of aged livers to regenerate and to tolerate transplantation leads to increased risk of mortality from chronic liver diseases, as demonstrated in Figure 3.
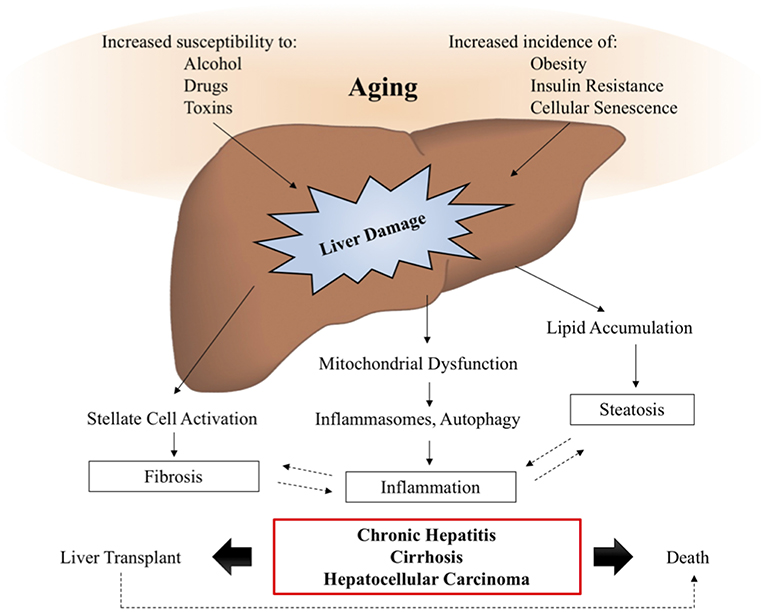
Figure 3. Aging and liver disease. With increasing age, the liver becomes more susceptible to damage from alcohol, drugs, and toxins, while the prevalence of metabolic disease, obesity, and cellular senescence are known to increase. These insults lead to the activation of signaling pathways driving liver pathology, such as inflammation, steatosis, and fibrosis, which are often involved in positive feedback loops, further exacerbating the symptoms of liver disease. In a subset of patients, liver disease will progress to chronic hepatitis, cirrhosis, and hepatocellular carcinoma, also known as end stage liver disease, for which transplant is the only viable therapy. Studies suggest that elderly patients have decreased survival post-transplantation, demonstrating the need for additional therapies to intervene and prevent the onset of age-related liver disease symptoms.
Both Kupffer cells and infiltrating monocytes are implicated to various levels in the etiology of chronic liver disease, however the distinction between these two cell populations is less clear in human clinical liver samples compared to animal studies. Importantly, most animal models of chronic liver disease are induced in young, healthy animals through genetic manipulation or by administering specific diets or chemical toxins. Findings from these animal studies are important to identify novel mechanisms regulating liver disease but may not prove relevant or all-encompassing when translating the findings to older, human populations, as evidenced by the moderate success of several anti-fibrotic therapies in clinical trials (120). Distinguishing between the biology of aging and age-associated pathologies can be a difficult task but is an important effort in the field of hepatology. Additional studies are needed to assess the effect of aging on the liver prior to the onset of age-associated pathologies in order to dissect mechanisms of aging from the manifestation of the pathology itself.
Aging is a major risk factor for the progression of liver fibrosis, particularly in hepatitis, which can advance to portal hypertension and cirrhosis (121). Fibrosis occurs when excessive connective tissue is deposited following acute or chronic liver damage, and is often the starting point of architectural distortion and dysfunction that prevents normal functioning of the liver (122). Age-related dysfunction in hepatic macrophages and stellate cells are heavily implicated in development of fibrosis and may be related to oxidative stress and macrophage polarization, but these mechanisms are not well understood (123).
In general, an increase in M1-polarized macrophages appears at the early stages of liver disease and promotes or exacerbates fibrosis, cirrhosis, and eventual liver failure. In clinical studies, the progression from hepatitis to fibrosis and eventually cirrhosis was associated with enrichment of CD14+CD16+ non-classical monocytes in the liver, which have been shown to activate stellate cells in vitro based on TGF-beta release (124). These cells may be derived from classical CD14+CD16− monocytes or infiltrate directly (125). Activation of the CCL2-CCR2 axis is associated with monocyte infiltration in both rodent models and patients with fibrosis and liver disease (126). A recent study found that the acquisition of M1 phenotypes in liver fibrosis may be regulated by interferon regulatory factor 5 (IRF5), which is significantly induced in liver macrophages in both mouse and human subjects developing fibrosis, and may represent a novel target for therapeutic intervention (127).
Elderly individuals are more likely to feel socially isolated and depressed, resulting in a rising alcohol consumption rate and prevalence of alcoholic liver disease in this population (128). Alcohol and its breakdown products are toxic to the liver, which can cause a disorder in lipid metabolism by increasing the synthesis of fatty acids and suppressing mitochondrial β-oxidation (129). The risk of alcohol toxicity is further increased in elderly populations due to alterations in metabolism or by the intake of certain medications (130).
Alcoholic liver disease can be classified into three stages: accumulation of extra fat in the liver (steatosis), alcoholic hepatitis, and cirrhosis. Macrophages were found to increase in both the early and late stages of alcoholic liver disease. Increased gut permeability and high levels of endotoxin in the portal blood results in activation of Kupffer cells (131). In addition, Ly6C+ macrophages were found to accumulate in murine models of alcoholic liver injury, regulated by CCL2 chemokine signaling (132). Clinical symptoms of alcoholic liver disease among the elderly are similar to younger counterparts, yet the prevalence of advancing to irreversible stages of the disease is greater in older individuals (133). Oxidative stress and production of TNF by hepatic macrophages, as a result of steatosis or leaky gut, causes progression to later stages (130). Furthermore, the expression of M1 pro-inflammatory genes was found to be higher in peritoneal macrophages from patients with alcohol-related cirrhosis and ascites compared to hepatitis-C related cirrhosis, suggesting systemic differences in macrophage polarization (134). Reducing leaky gut and inflammation have been identified as potential therapeutic targets for treating alcoholic liver disease (135).
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are leading causes of chronic liver disease globally and are projected to become the leading indication for liver transplantation in the United States (136–138). NAFLD often presents with metabolic syndrome, such as obesity and excessive visceral fat, hyperlipidemia, hypertension, insulin resistance and an increased secretion of pro-inflammatory cytokines (139). All of these components of metabolic syndrome are generally observed in elderly populations and many studies find increased rates of NAFLD among elderly people compared with their younger counterparts (140–142).
The mechanism for developing age-related steatosis is not fully understood but has been attributed to hepatocyte senescence driving a reduction in mitochondrial metabolism (143), decreased transport of insulin across the sinusoidal endothelium (144), reduction in autophagic flux (145), or chronic low-level inflammation (146, 147), leading to the build-up of toxic free fatty acids in the liver. Lifestyle choices, such as high fat diet (HFD), may also lead to the development of NAFLD in aged individuals. Work by Fontana et al. found that mice were equally susceptible to steatosis following HFD regardless of age. However, the older mice exhibited more severe hepatocellular injury and inflammation following administration of HFD that was attributed to increased M1 macrophage polarization in both the liver and white adipose tissues (148).
As in other chronic liver diseases, macrophages accumulate in the livers of NAFLD patients (149). Experimental mouse models have shown an accumulation of Ly6C+ monocytes is a critical step in the development of progressive fibrosis from steatohepatitis in a CCL2-dependent mechanism (131). In addition, lipids and free fatty acids contribute to DAMP-induced Kupffer cell activation (150). Patients with low-grade steatosis were found to have higher mRNA expression of M2 markers, CD206 and CD163, compared to patients with advanced steatosis. The M2 macrophages were hypothesized to promote apoptosis of M1 polarized hepatic macrophages and protect against disease progression of NAFLD or alcoholic hepatitis (149). Interestingly, soluble CD163 (sCD163), a marker of M1 macrophage polarization, increases systemically with severity of NAFLD in human patients (151), again suggesting that M1 polarization is associated with more advanced stages of the disease.
Traditional treatments for NAFLD are more challenging for elderly populations as they require lifestyle changes in exercise, diet, and medication or liver transplantation (152, 153). Findings from a recent clinical trial using a CCR2/CCR5 antagonist to block inflammation resulted in twice as many subjects achieving improvement in fibrosis and no worsening of steatohepatitis compared with placebo (154, 155). Subgroup analysis showed the therapy to be similarly effective in patients below and above 56 years of age, demonstrating promise in the context of age-associated disease.
The decline in mitochondrial capacity with aging has emerged as a key mechanism underlying several of the observed changes in the function of both hepatocytes and macrophages, likely contributing to the increased prevalence and severity of chronic liver diseases in elderly populations. In general, the accumulation of infiltrating or M1-polarized monocytes/macrophages tends to exacerbate liver disease and contribute to fibrosis, cirrhosis, and liver failure. However, very few, if any, studies have systematically identified changes in the hepatic macrophage populations of aging livers in either animal models or human patients, representing a hugely understudied area of research.
Liver transplantation is the standard treatment for patients with end stage liver disease. The number of elderly people with liver cirrhosis requiring transplantation has grown over the past 20 years and is expected to increase further (66). Overall survival rates after 1 year are approximately 90% and 10-year survival rates may be more than 70% for recipients (156). However, some studies have found a reduced survival rate in transplant recipients over 60 years of age (from 90 to 64% 1 year survival rate), primarily due to kidney dysfunction and cardiopulmonary disease complications (157). Survival rates improve for elderly patients with few comorbidities (158), thus age alone is not an exclusion criterion from liver transplantation, but all disease risk factors must be considered to devise effective treatment strategies for these age-related chronic liver diseases.
Therapies that suppress M1-polarization or infiltration of macrophages might reduce the progression of liver disease from manageable early stages to chronic end-stages requiring liver transplantation. In the context of aging specifically, promoting M2-gene expression might delay the advancement of liver disease to later stages. However, M2 macrophages have been associated with tumor progression, including clinical hepatocellular carcinoma (HCC) specimens (159). Thus, modulating macrophage polarization has been considered a double-edged sword, and the appearance of liver fibrosis or HCC would need to be closely monitored. Treating the primary drivers of age-associated liver diseases such as alcohol consumption, visceral and ectopic fat accumulation, or deficits in mitochondrial capacity and other age-related mechanisms, will be just as important as targeting the inflammatory symptoms to combat these age-related liver diseases.
Elderly populations continue to increase globally and are particularly at risk for succumbing to liver failure due to the declines in regenerative capacity, reduced survival post-liver transplantation, and tendencies to develop inflammation and fibrosis. Between 1999 and 2016, deaths in the United States from cirrhosis increased by 65% and deaths from HCC doubled (160). A better understanding of the biology of liver aging that influences the onset and progression of chronic liver disease will be crucial in order to develop new interventions and treatments for our aging populations.
ES, MH, BP, and BB contributed to the writing and editing of the manuscript and figure preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Contract grant sponsor: National Institute of Health; contract grant numbers: T32-EB001026 (ES, MH), F31 DK112633 (ES), R01 AG055564 (BB). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. Thank you also to the Pittsburgh Liver Research Center for their support.
1. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
2. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. (2016) 13:267–76. doi: 10.1038/cmi.2016.3
3. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol. (2016) 13:301–15. doi: 10.1038/cmi.2015.97
4. Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. (2016) 13:347–53. doi: 10.1038/cmi.2016.5
5. Doherty DG, Norris S, Madrigal-Estebas L, McEntee G, Traynor O, Hegarty JE, et al. The human liver contains multiple populations of NK cells, T cells, and CD3+CD56+ natural T cells with distinct cytotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J Immunol. (1999) 163:2314–21.
6. Kenna T, Golden-Mason L, Porcelli SA, Koezuka Y, Hegarty JE, O'Farrelly C, et al. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J Immunol. (2003) 171:1775–9. doi: 10.4049/jimmunol.171.4.1775
7. Kenna T, Golden-Mason L, Norris S, Hegarty JE, O'Farrelly C, Doherty DG. Distinct subpopulations of gamma delta T cells are present in normal and tumor-bearing human liver. Clin Immunol. (2004) 113:56–63. doi: 10.1016/j.clim.2004.05.003
8. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood (2011) 117:1250–9. doi: 10.1182/blood-2010-08-303339
9. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology (2008) 47:729–36. doi: 10.1002/hep.22034
10. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. (2010) 10:753–66. doi: 10.1038/nri2858
11. Chen S, Akbar SM, Abe M, Hiasa Y, Onji M. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin Exp Immunol. (2011) 166:134–42. doi: 10.1111/j.1365-2249.2011.04445.x
12. Schulz C, Gomez Perdiguero E Chorro, L, Szabo-Rogers, H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336:86–90. doi: 10.1126/science.1219179
13. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
14. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
15. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. (2006) 26:1175–86. doi: 10.1111/j.1478-3231.2006.01342.x
16. Sheedfar F, Di Biase S, Koonen, D, Vinciguerra M. Liver diseases and aging: friends or foes? Aging Cell (2013) 12:950–4. doi: 10.1111/acel.12128
17. Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. (2014) 5:683. doi: 10.3389/fimmu.2014.00683
18. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk Sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
19. Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. (2011) 12:643–55. doi: 10.1038/nrm3184
20. Kakinuma Y, Kimura T, Watanabe Y. Possible involvement of liver resident macrophages (kupffer cells) in the pathogenesis of both intrahepatic and extrahepatic inflammation. Can J Gastroenterol Hepatol. (2017) 2017:2896809. doi: 10.1155/2017/2896809
21. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. (2012) 209:1167–81. doi: 10.1084/jem.20120340
22. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (2011) 332:1284–8. doi: 10.1126/science.1204351
23. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115:e10-9. doi: 10.1182/blood-2009-07-235028
24. Brown BN, Haschak MJ, Lopresti ST, Stahl EC. Effects of age-related shifts in cellular function and local microenvironment upon the innate immune response to implants. Semin Immunol. (2017) 29:24–32. doi: 10.1016/j.smim.2017.05.001
25. Ricard-Blum S, Vallet SD. Fragments generated upon extracellular matrix remodeling: biological regulators and potential drugs. Matrix Biol. (2017). doi: 10.1016/j.matbio.2017.11.005 [Epub ahead of print].
26. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
27. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
28. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
29. Kapetanovic R, Bokil NJ, Sweet MJ. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: a ticking time bomb in ageing. Ageing Res Rev. (2015) 24(Pt A):40–53.
30. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
31. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
32. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. (2014) 1319:82–95. doi: 10.1111/nyas.12458
33. Winkler S, Rosen-Wolff A. Caspase-1: an integral regulator of innate immunity. Semin Immunopathol. (2015) 37:419–27. doi: 10.1007/s00281-015-0494-4
34. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. (2013) 93:875–81. doi: 10.1189/jlb.1012512
35. Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci. (2015) 72:4111–26. doi: 10.1007/s00018-015-1995-y
36. Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. (2015) 12:1–4. doi: 10.1038/cmi.2014.83
37. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science (2016) 353:6304. doi: 10.1126/science.aaf4238
38. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
39. Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJ, Janssen HL, et al. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J Leukocyte Biol. (2012) 92:723–33. doi: 10.1189/jlb.1111566
40. Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. (2010) 53:903–10. doi: 10.1016/j.jhep.2010.04.037
41. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
42. Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe (2016) 20:99–106. doi: 10.1016/j.chom.2016.06.002
43. Janeway CA Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today (1992) 13:11–6. doi: 10.1016/0167-5699(92)90198-G
44. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
45. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
46. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Buschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. (1995) 22:226–9. doi: 10.1016/0168-8278(95)80433-1
47. Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. (2016) 13:316–27. doi: 10.1038/cmi.2015.104
48. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi: 10.1126/science.1194637
49. Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. (2008) 84:1410–21. doi: 10.1189/jlb.0308173
50. Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood (2007) 110:4077–85. doi: 10.1182/blood-2007-02-073841
51. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. (2015) 212:447–56. doi: 10.1084/jem.20141539
52. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology (2016) 64:1667–82. doi: 10.1002/hep.28682
53. Zhang X, Han J, Man K, Li X, Du J, Chu ES, et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol. (2016) 64:160–70. doi: 10.1016/j.jhep.2015.09.005
54. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology (2012) 55:898–909. doi: 10.1002/hep.24764
55. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. (2009) 119:1858–70. doi: 10.1172/JCI37444
56. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325:612–6. doi: 10.1126/science.1175202
57. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
58. Sato A, Nakashima H, Nakashima M, Ikarashi M, Nishiyama K, Kinoshita M, et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS ONE (2014) 9:e92515. doi: 10.1371/journal.pone.0092515
59. Nakashima H, Ogawa Y, Shono S, Kinoshita M, Nakashima M, Sato A, et al. Activation of CD11b+ Kupffer cells/macrophages as a common cause for exacerbation of TNF/Fas-ligand-dependent hepatitis in hypercholesterolemic mice. PLoS ONE (2013) 8:e49339. doi: 10.1371/journal.pone.0049339
60. Kinoshita M, Miyazaki H, Nakashima H, Nakashima M, Nishikawa M, Ishikiriyama T, et al. In vivo lipopolysaccharide tolerance recruits CD11b+ macrophages to the liver with enhanced bactericidal activity and low tumor necrosis factor-releasing capability, resulting in drastic resistance to lethal septicemia. J Innate Immun. (2017) 9:493–510. doi: 10.1159/000475931
61. Nakashima H, Nakashima M, Kinoshita M, Ikarashi M, Miyazaki H, Hanaka H, et al. Activation and increase of radio-sensitive CD11b+ recruited Kupffer cells/macrophages in diet-induced steatohepatitis in FGF5 deficient mice. Sci Rep. (2016) 6:34466. doi: 10.1038/srep34466
62. Nishiyama K, Nakashima H, Ikarashi M, Kinoshita M, Nakashima M, Aosasa S, et al. Mouse CD11b+kupffer cells recruited from bone marrow accelerate liver regeneration after partial hepatectomy. PLoS ONE (2015) 10:e0136774. doi: 10.1371/journal.pone.0136774
63. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. (2016) 22:945–51. doi: 10.1038/nm.4146
64. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
65. Kingham TP, Chaudhry UI, Plitas G, Katz SC, Raab J, DeMatteo RP. Murine liver plasmacytoid dendritic cells become potent immunostimulatory cells after Flt-3 ligand expansion. Hepatology (2007) 45:445–54. doi: 10.1002/hep.21457
66. Kim IH, Kisseleva T, Brenner DA. Aging and liver disease. Curr Opin Gastroenterol. (2015) 31:184–91. doi: 10.1097/MOG.0000000000000176
67. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) (2012) 4:166–75. doi: 10.18632/aging.100444
69. McLean AJ, Cogger VC, Chong GC, Warren A, Markus AM, Dahlstrom JE, et al. Age-related pseudocapillarization of the human liver. J Pathol. (2003) 200:112–7. doi: 10.1002/path.1328
70. Tietz NW, Shuey DF, Wekstein DR. Laboratory values in fit aging individuals–sexagenarians through centenarians. Clin Chem. (1992) 38:1167–85.
71. Wakabayashi H, Nishiyama Y, Ushiyama T, Maeba T, Maeta H. Evaluation of the effect of age on functioning hepatocyte mass and liver blood flow using liver scintigraphy in preoperative estimations for surgical patients: comparison with CT volumetry. J Surg Res. (2002) 106:246–53. doi: 10.1006/jsre.2002.6462
72. Sastre J, Pallardo FV, Pla R, Pellin A, Juan G, O'Connor JE, et al. Aging of the liver: age-associated mitochondrial damage in intact hepatocytes. Hepatology (1996) 24:1199–205. doi: 10.1002/hep.510240536
73. Hohn A, Grune T. Lipofuscin: formation, effects and role of macroautophagy. Redox Biol. (2013) 1:140–4. doi: 10.1016/j.redox.2013.01.006
74. Poulose N, Raju R. Aging and injury: alterations in cellular energetics and organ function. Aging Dis. (2014) 5:101–8. doi: 10.14336/AD.2014.0500101
75. American Geriatrics Society Beers Criteria Update Expert P. American geriatrics society updated beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. (2012) 60:616–31. doi: 10.1111/j.1532-5415.2012.03923.x
76. Wang GL, Salisbury E, Shi X, Timchenko L, Medrano EE, Timchenko NA. HDAC1 cooperates with C/EBPalpha in the inhibition of liver proliferation in old mice. J Biol Chem. (2008) 283:26169–78. doi: 10.1074/jbc.M803544200
77. Aravinthan AD, Alexander GJM. Senescence in chronic liver disease: Is the future in aging? J Hepatol. (2016) 65:825–34. doi: 10.1016/j.jhep.2016.05.030
78. Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. (2005) 37:961-76. doi: 10.1016/j.biocel.2004.10.013
79. Campisi J. Replicative senescence: an old lives' tale? Cell (1996) 84:497–500. doi: 10.1016/S0092-8674(00)81023-5
80. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
81. Aravinthan A, Challis B, Shannon N, Hoare M, Heaney J, Alexander GJ. Selective insulin resistance in hepatocyte senescence. Exp Cell Res. (2015) 331:38–45. doi: 10.1016/j.yexcr.2014.09.025
82. Verma S, Tachtatzis P, Penrhyn-Lowe S, Scarpini C, Jurk D, Von Zglinicki T, et al. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology (2012) 56:1510–20. doi: 10.1002/hep.25787
83. Hilmer SN, Cogger VC, Le Couteur DG. Basal activity of Kupffer cells increases with old age. J Gerontol A Biol Sci Med Sci. (2007) 62:973–8. doi: 10.1093/gerona/62.9.973
84. Singh P, Coskun ZZ, Goode C, Dean A, Thompson-Snipes L, Darlington G. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology (2008) 47:1680–90. doi: 10.1002/hep.22224
85. Maeso-Diaz R, Ortega-Ribera M, Fernandez-Iglesias A, Hide D, Munoz L, Hessheimer AJ, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell (2018) 8:e12829. doi: 10.1111/acel.12829
86. Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJ, Puleston DJ, et al. Autophagy Controls acquisition of aging features in macrophages. J Innate Immun. (2015) 7:375–91. doi: 10.1159/000370112
87. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell (2011) 146:682–95. doi: 10.1016/j.cell.2011.07.030
88. Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, et al. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics (2016) 11:381–8. doi: 10.1080/15592294.2016.1144007
89. Trepanowski JF, Canale RE, Marshall KE, Kabir MM, Bloomer RJ. Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: a summary of available findings. Nutr J. (2011) 10:107. doi: 10.1186/1475-2891-10-107
90. Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell (2014) 13:699–708. doi: 10.1111/acel.12223
91. Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol. (2017) 199:1060–8. doi: 10.4049/jimmunol.1700397
92. Brouwer A, Knook DL. The reticuloendothelial system and aging: a review. Mech Ageing Dev. (1983) 21:205–28. doi: 10.1016/0047-6374(83)90042-8
93. Puzianowska-Kuznicka M, Owczarz M, Wieczorowska-Tobis K, Nadrowski P, Chudek J, Slusarczyk P, et al. Interleukin-6 and C-reactive protein, successful aging, and mortality: the PolSenior study. Immun Ageing. (2016) 13:21. doi: 10.1186/s12979-016-0076-x
94. Dimitrijevic M, Stanojevic S, Kustrimovic N, Mitic K, Vujic V, Aleksic I, et al. The influence of aging and estradiol to progesterone ratio on rat macrophage phenotypic profile and NO and TNF-alpha production. Exp Gerontol. (2013) 48:1243–54. doi: 10.1016/j.exger.2013.07.001
95. Cecilio CA, Costa EH, Simioni PU, Gabriel DL, Tamashiro WM. Aging alters the production of iNOS, arginase and cytokines in murine macrophages. Braz J Med Biol Res. (2011) 44:671–81. doi: 10.1590/S0100-879X2011007500067
96. Suchy D, Labuzek K, Buldak L, Szkudlapski D, Okopien B. Comparison of chosen activation markers of human monocytes/macrophages isolated from the peripheral blood of young and elderly volunteers. Pharmacol Rep. (2014) 66:759–65. doi: 10.1016/j.pharep.2014.04.008
97. Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell (2014) 13:60–9. doi: 10.1111/acel.12149
98. von Leden RE Khayrullina, G Moritz, KE, Byrnes KR. Age exacerbates microglial activation, oxidative stress, inflammatory and NOX2 gene expression, and delays functional recovery in a middle-aged rodent model of spinal cord injury. J Neuroinflammation (2017) 14:161. doi: 10.1186/s12974-017-0933-3
99. Cho SH, Chen JA, Sayed F, Ward ME, Gao F, Nguyen TA, et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1beta. J Neurosci. (2015) 35:807–18. doi: 10.1523/JNEUROSCI.2939-14.2015
100. Malm T, Koistinaho M, Muona A, Magga J, Koistinaho J. The role and therapeutic potential of monocytic cells in Alzheimer's disease. Glia (2010) 58:889–900. doi: 10.1002/glia.20973
101. Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, et al. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (2014) 6:399–413. doi: 10.18632/aging.100669
102. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
103. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl. Acad. Sci USA (2014) 111:16029–34. doi: 10.1073/pnas.1406508111
104. Stahl EC, Brown BN. Cell therapy strategies to combat immunosenescence. Organogenesis (2015) 11:159–72. doi: 10.1080/15476278.2015.1120046
105. Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol. (2015) 194:4073–80. doi: 10.4049/jimmunol.1500046
106. Onyema OO, Njemini R, Forti LN, Bautmans I, Aerts JL, De Waele M, et al. Aging-associated subpopulations of human CD8+T-lymphocytes identified by their CD28 and CD57 phenotypes. Arch Gerontol Geriat. (2015) 61:494–502. doi: 10.1016/j.archger.2015.08.007
107. van der Geest KSM, Abdulahad WH, Tete SM, Lorencetti PG, Horst G, Bos NA, et al. Aging disturbs the balance between effector and regulatory CD4+T cells. Exp Gerontol. (2014) 60:190–6. doi: 10.1016/j.exger.2014.11.005
108. Garg SK, Delaney C, Shi H, Yung R. Changes in adipose tissue macrophages and T cells during aging. Crit Rev Immunol. (2014) 34:1–14. doi: 10.1615/CritRevImmunol.2013006833
109. Urs S, Smith C, Campbell B, Saxton AM, Taylor J, Zhang B, et al. Gene expression profiling in human preadipocytes and adipocytes by microarray analysis. J Nutr. (2004) 134:762–70. doi: 10.1093/jn/134.4.762
110. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat tissue, aging, and cellular senescence. Aging Cell (2010) 9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x
111. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
112. Wang K, Yao Y, Zhu X, Zhang K, Zhou F, Zhu L. Amyloid beta induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. J Biochem Mol Toxicol. (2017) 31. [Epub ahead of print].
113. Murphy N, Grehan B, Lynch MA. Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin-dependent degradation of NLRP10. Neuromolecular Med. (2014) 16:205–15. doi: 10.1007/s12017-013-8274-6
114. Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci USA (2011) 108:14867–72. doi: 10.1073/pnas.1111101108
115. Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Investigat. (2006) 116:2262–71. doi: 10.1172/JCI28075
116. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi: 10.1038/nature04515
117. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. (2014) 20:8082–91. doi: 10.3748/wjg.v20.i25.8082
118. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. (2015) 22:1111–29. doi: 10.1089/ars.2014.5994
119. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-associated microbial dysbiosis promotes intestinal permeability systemic inflammation, and macrophage dysfunction. Cell Host Microbe (2017) 21:455-66e4.
120. Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. (2011) 4:391–417. doi: 10.1177/1756283X11413002
121. Iwaisako K, Brenner DA, Kisseleva T. What's new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J Gastroenterol Hepatol. (2012) 27(Suppl. 2):65–8. doi: 10.1111/j.1440-1746.2011.07002.x
122. Poynard T, Ratziu V, Charlotte F, Goodman Z, McHutchison J, Albrecht J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis c. J Hepatol. (2001) 34:730–9. doi: 10.1016/S0168-8278(00)00097-0
123. Mahrouf-Yorgov M, Collin de l'Hortet A, Cosson C, Slama A, Abdoun E, Guidotti JE, et al. Increased susceptibility to liver fibrosis with age is correlated with an altered inflammatory response. Rejuvenation Res. (2011) 14:353–63. doi: 10.1089/rej.2010.1146
124. Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE (2010) 5:e11049. doi: 10.1371/journal.pone.0011049
125. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology (2013) 57:385–98. doi: 10.1002/hep.26016
126. Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, et al. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. (1998) 152:423–30.
127. Alzaid F, Lagadec F, Albuquerque M, Ballaire R, Orliaguet L, Hainault I, et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight. (2016) 1:e88689. doi: 10.1172/jci.insight.88689
128. Blazer DG, Wu LT. The epidemiology of at-risk and binge drinking among middle-aged and elderly community adults: National Survey on Drug Use and Health. Am J Psychiatry (2009) 166:1162–9. doi: 10.1176/appi.ajp.2009.09010016
129. Kim IH, Xu J, Liu X, Koyama Y, Ma HY, Diggle K, et al. Aging increases the susceptibility of hepatic inflammation, liver fibrosis and aging in response to high-fat diet in mice. Age (Dordr). (2016) 38:291–302. doi: 10.1007/s11357-016-9938-6
130. Meier P, Seitz HK. Age, alcohol metabolism and liver disease. Current Opinion in Clinical Nutr Metab Care (2008) 11:21-6. doi: 10.1097/MCO.0b013e3282f30564
131. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
132. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. (2014) 96:657–65. doi: 10.1189/jlb.6A0114-004RR
133. Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem. (2006) 387:349–60. doi: 10.1515/BC.2006.047
134. Tapia-Abellan A, Martinez-Esparza M, Ruiz-Alcaraz AJ, Hernandez-Caselles T, Martinez-Pascual C, Miras-Lopez M, et al. The peritoneal macrophage inflammatory profile in cirrhosis depends on the alcoholic or hepatitis C viral etiology and is related to ERK phosphorylation. BMC Immunol. (2012) 13:42. doi: 10.1186/1471-2172-13-42
135. Hartmann P, Chen WC, Schnabl B. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front Physiol. (2012) 3:402. doi: 10.3389/fphys.2012.00402
136. Mikolasevic I, Filipec-Kanizaj T, Mijic M, Jakopcic I, Milic S, Hrstic I, et al. Nonalcoholic fatty liver disease and liver transplantation - Where do we stand? World J Gastroenterol. (2018) 24:1491–506. doi: 10.3748/wjg.v24.i14.1491
137. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology (2015) 148:547–55. doi: 10.1053/j.gastro.2014.11.039
138. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi: 10.1002/hep.28431
139. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes (2012) 61:1315–22. doi: 10.2337/db11-1300
140. Koehler EM, Schouten JN, Hansen BE, van Rooij FJ, Hofman A, Stricker BH, et al. Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: results from the Rotterdam study. J Hepatol. (2012) 57:1305–11. doi: 10.1016/j.jhep.2012.07.028
141. Frith J, Day CP, Henderson E, Burt AD, Newton JL. Non-alcoholic fatty liver disease in older people. Gerontology (2009) 55:607–13. doi: 10.1159/000235677
142. Noureddin M, Yates KP, Vaughn IA, Neuschwander-Tetri BA, Sanyal AJ, McCullough A, et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology (2013) 58:1644–54. doi: 10.1002/hep.26465
143. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. (2017) 8:15691. doi: 10.1038/ncomms15691
144. Mohamad M, Mitchell SJ, Wu LE, White MY, Cordwell SJ, Mach J, et al. Ultrastructure of the liver microcirculation influences hepatic and systemic insulin activity and provides a mechanism for age-related insulin resistance. Aging Cell (2016) 15:706–15. doi: 10.1111/acel.12481
145. Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. (2014) 5:e1179. doi: 10.1038/cddis.2014.162
146. Duval C, Thissen U, Keshtkar S, Accart B, Stienstra R, Boekschoten MV, et al. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57BL/6 mice. Diabetes (2010) 59:3181–91. doi: 10.2337/db10-0224
147. Kumar A, Sharma A, Duseja A, Das A, Dhiman RK, Chawla YK, et al. Patients with nonalcoholic fatty liver disease (NAFLD) have higher oxidative stress in comparison to chronic viral hepatitis. J Clin Exp Hepatol. (2013) 3:12–8. doi: 10.1016/j.jceh.2012.10.009
148. Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology (2013) 57:995–1004. doi: 10.1002/hep.26099
149. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology (2014) 59:130–42. doi: 10.1002/hep.26607
150. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer cells undergo fundamental changes during the development of experimental nash and are critical in initiating liver damage and inflammation. PLoS ONE (2016) 11:e0159524. doi: 10.1371/journal.pone.0159524
151. Kazankov K, Moller HJ, Lange A, Birkebaek NH, Holland-Fischer P, Solvig J, et al. The macrophage activation marker sCD163 is associated with changes in NAFLD and metabolic profile during lifestyle intervention in obese children. Pediatr Obes. (2015) 10:226–33. doi: 10.1111/ijpo.252
152. Shah K, Stufflebam A, Hilton TN, Sinacore DR, Klein S, Villareal DT. Diet and exercise interventions reduce intrahepatic fat content and improve insulin sensitivity in obese older adults. Obesity (Silver Spring) (2009) 17:2162–8. doi: 10.1038/oby.2009.126
153. Villareal DT, Chode S, Parimi N, Sinacore DR, Hilton T, Armamento-Villareal R, et al. Weight loss exercise, or both and physical function in obese older adults. N Engl J Med. (2011) 364:1218–29. doi: 10.1056/NEJMoa1008234
154. Friedman S, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials (2016) 47:356–65. doi: 10.1016/j.cct.2016.02.012
155. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67:1754–67. doi: 10.1002/hep.29477
156. Åberg F, Isoniemi H, and Höckerstedt K. Long-term results of liver transplantation. Scand J Surg. (2011) 100:14–21. doi: 10.1177/145749691110000104
157. Randall HB, Cao S, deVera ME. Transplantation in elderly patients. Arch Surg. (2003) 138:1089–92. doi: 10.1001/archsurg.138.10.1089
158. Adani GL, Baccarani U, Lorenzin D, Rossetto A, Nicolini D, Vecchi A, et al. Elderly versus young liver transplant recipients: patient and graft survival. Transplant Proc. (2009) 41:1293–4. doi: 10.1016/j.transproceed.2009.03.080
159. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. (2015) 62:607–16. doi: 10.1016/j.jhep.2014.10.029
Keywords: Kupffer cell, monocyte-derived macrophage, chronic liver disease, inflammation, fibrosis, senescence, mitochondria
Citation: Stahl EC, Haschak MJ, Popovic B and Brown BN (2018) Macrophages in the Aging Liver and Age-Related Liver Disease. Front. Immunol. 9:2795. doi: 10.3389/fimmu.2018.02795
Received: 31 August 2018; Accepted: 13 November 2018;
Published: 30 November 2018.
Edited by:
Hannelie Korf, KU Leuven, BelgiumReviewed by:
Joan Clària, Hospital Clínic de Barcelona, SpainCopyright © 2018 Stahl, Haschak, Popovic and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan N. Brown, YnJvd25iQHVwbWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.