 Vasilios A. Morikis
Vasilios A. Morikis Scott I. Simon
Scott I. Simon
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 November 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02774
This article is part of the Research Topic Leukocyte Trafficking in Homeostasis and Disease View all 12 articles
Neutrophils are the most motile of mammalian cells, a feature that enables them to protect the host against the rapid spread of pathogens from tissue into the circulatory system. A critical process is the recruitment of neutrophils to inflamed endothelium within post-capillary venules. This occurs through cooperation between at least four families of adhesion molecules and G-protein coupled signaling receptors. These adhesion molecules convert the drag force induced by blood flow acting on the cell surface into bond tension that resists detachment. A common feature of selectin-glycoprotein tethering and integrin-ICAM bond formation is the mechanics by which force acting on these specific receptor-ligand pairs influences their longevity, strength, and topographic organization on the plasma membrane. Another distinctly mechanical aspect of neutrophil guidance is the capacity of adhesive bonds to convert external mechanical force into internal biochemical signals through the transmission of force from the outside-in at focal sites of adhesive traction on inflamed endothelium. Within this region of the plasma membrane, we denote the inflammatory synapse, Ca2+ release, and intracellular signaling provide directional cues that guide actin assembly and myosin driven motive force. This review provides an overview of how bond formation and outside-in signaling controls neutrophil recruitment and migration relative to the hydrodynamic shear force of blood flow.
Leukocyte recruitment is an evolutionarily conserved process in which the target of natural selection is a fast and efficient immune system that transports neutrophils in numbers appropriate for host defense. A multi-step cascade of adhesive events, which includes ligation and signaling through selectins, integrins, and chemokine receptors, guide neutrophil recruitment to inflamed endothelium (Figure 1A). Adhesive engagement between the neutrophil and the endothelium is initiated by selectins that recognize sialylated and fucosylated carbohydrate ligands expressed on adjacent plasma membranes. E-selectin (CD62E) and P-selectin (CD62P) receptors, upregulated on inflamed endothelium, and L-selectin (CD62L), constitutively expressed on the leukocyte, are strategically positioned on the plasma membrane to form bonds that initiate cell tethering and rolling under the hydrodynamic shear exerted by flowing blood (Figure 1B). Fluid drag forces are resisted by selectin bond tension, which rapidly induces receptor redistribution and formation of focal clusters within the site of adhesive contact between adjacent plasma membranes. For instance, E-selectin and P-selectin binding to sialyl-Lewisx (sLex) on PSGL-1, or E-selectin recognition of sLex on L-selectin (only on human neutrophils), promotes neutrophil tethering and rolling that induces subsequent selectin interactions such as with glycolipids. Cell rolling allows interrogation of the vascular surface, and at optimum site density between selectins and their ligands a second event occurs that involves intracellular signaling, a process necessary to activate β2-integrins. In the absence of high affinity activation of β2-integrins, shear resistant adhesion or cell arrest is not observed; a requisite step to initiate neutrophil spreading, polarization, and transendothelial migration (1–4). The importance of β2-integrin expression and activation in innate immune function is evident in patients suffering from leukocyte adhesion deficiency 1 (LAD-I), where CD18 expression on the cell surface is lost or reduced, resulting in chronic infections, impaired wound healing, and a defect in neutrophil recruitment (5–7).
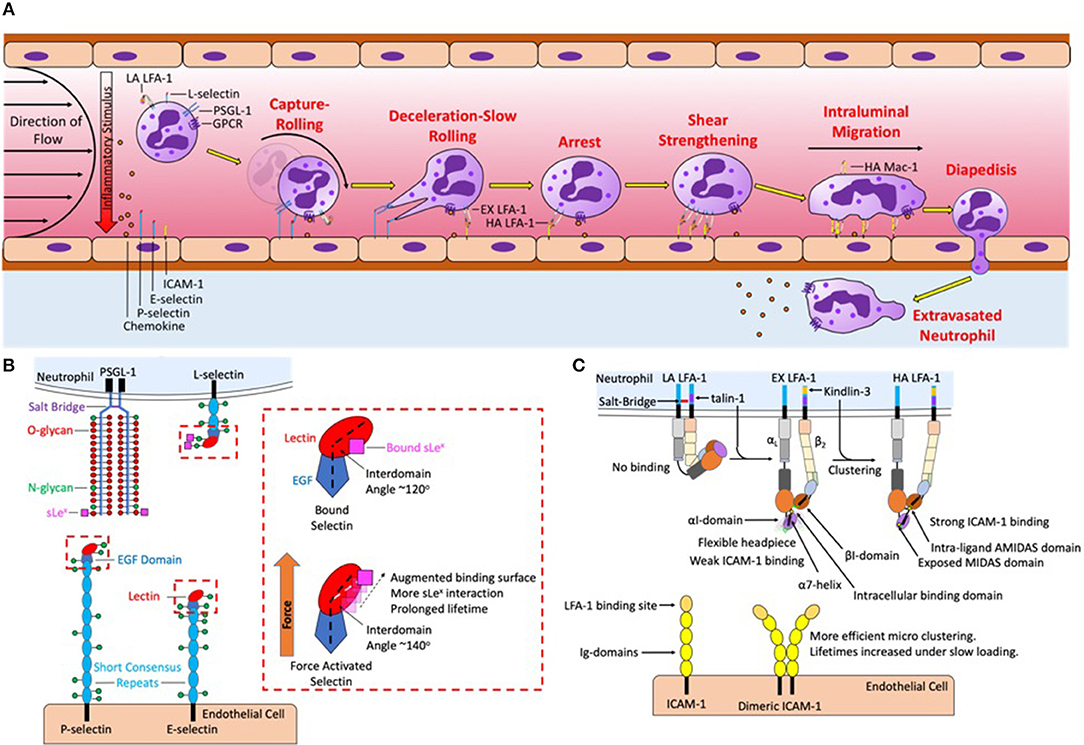
Figure 1. Neutrophil recruitment under shear flow is coordinated by selectins, chemokines, and integrin ligand binding and signaling. (A) The sequential steps of neutrophil recruitment under shear flow is initiated by hydrodynamic forces that bring L-selectin and PSGL-1 on the neutrophil surface in contact with P-selectin and E-selectin on the endothelial plasma membrane, mediating capture, and rolling. To resist shear flow selectins and PSGL-1 form transient yet strong bonds that form at the leading-edge to initiate cell rolling and break at the trailing edge under tensile force. Chemokine captured via glycosaminoglycans on the endothelial surface and force acting on L-selectin then activates LFA-1 into an extended conformation to initiate deceleration and slow rolling. A second signal is initiated by L-selectin and CXCR that upshifts integrin into a high affinity state thereby initiating arrest. Initially integrin LFA-1 is spread randomly throughout the surface of the neutrophil, force acting on high affinity LFA-1 initiates their redistribution that reinforces shear resistant cell arrest. Clusters of LFA-1 transduce calcium signaling and neutrophil shape change, leading to high affinity Mac-1 mediated intraluminal crawling along a gradient of chemokine on the endothelial surface, directing the neutrophil to the desired site of migration. Endothelial diapedesis promotes neutrophil access the site of injury or tissue infarct. (B) Mechanics of PSGL-1 and L-selectin bond formation play a key role in force transduced signaling. PSGL-1 is primarily decorated with O-glycans while L-selectin is primarily decorated with N-glycans, sLex deposited on these glycans binds to the lectin domain of P-selectin and E-selectin. Under tensile bond force catch-bonds are formed by a change in intracellular angle of selectin EGF-Lectin domain in all three selectins. Selectin catch-bonds are necessary to mechanosignal activation of LFA-1 under shear conditions in the absence of chemokine. The red box details one potential mechanism in which the selectin interdomain angle between the lectin and EGF domain shifts in response to force allowing for prolonged bond lifetimes. (C) LFA-1 is expressed predominantly on membrane microvilli in a low affinity conformation, stabilized by a salt-bridge that clasps together the intracellular αL and β2 tails. Signaling through selectin and chemokine initiates talin-1 recruitment to the proximal NPxF motif on the β2-integrin that breaks the salt-bridge and extends the integrin, revealing the ICAM-1 binding site in the headpiece. However, the headpiece remains flexible and the MIDAS binding domain remains obscured, supporting weak ICAM-1 bonds. Kindlin-3 binding to the distal NPxF motif promotes transition to a high affinity state and stabilizes microclustering of talin-1 and ICAM-1 bound LFA-1 monomers. Tensile bond force acting on high affinity LFA-1 is transduced intracellularly through a shift in the α7 helix of the αI domain. This in turn induces an allosteric shift that exposes the MIDAS domain allowing for recognition of ICAM-1 and stable bond formation. The binding between ICAM-1 and high affinity LFA-1 at low force regimes is amplified by tandem bond formation with dimeric ICAM-1.
The first β2-integrin discovered, LFA-1 (also known as CD11a/CD18 or αLβ2), is a key integrin involved in early signaling. LFA-1 converts drag forces of flowing blood into bond tension that transduces intracellular chemical signals. Once bound, high affinity LFA-1 not only acts as an adhesive anchor but also functions as a mechanosensitive receptor capable of transducing external force into internal chemical signals (8–11). The conversion of mechanical force to chemical signals at the site of contact (e.g., inflammatory synapse) can be considered a mechanism of tactile sensing through selectin and integrin bond force transduction that determines through molecular recognition where and when neutrophil emigration occurs (Figure 1C). A cooperative mechanism underlies early activation of LFA-1 that is initiated by selectin mediated capture and rolling, which facilitates G-protein coupled receptor (GPCR) binding of chemokines presented on the glycocalyx of inflamed endothelium. Rolling on E-selectin initiates the rapid extension of LFA-1 that effects deceleration of neutrophils through interaction with its endothelial ligand intracellular adhesion molecule 1 (ICAM-1) (9, 12–14). Chemokine binding of CXCR1 and CXCR2 is sufficient to initiate so called inside-out activation of β2-integrins that corresponds with a shift of LFA-1 to a high affinity conformation and promotion of tight bond formation with ICAM-1. Superposition of selectin ligand outside-in signaling during rolling via E-selectin effectively amplifies GPCR inside-out signaling, such that very low levels of chemokine engagement become stimulatory at concentrations that independently do not elicit measurable calcium flux or β2-integrin activation (7, 14–16). For example, stimulating neutrophils rolling on E-selectin under shear at a concentration of 0.05 nM IL-8, corresponding to ligation of ~10–100 CXCR receptors per cell, activates a similar level of Ca2+ release and up-shift of β2-integrin receptors to high affinity as does stimulation of cell suspensions with 5 nM IL-8 under static or very low shear conditions (9, 15). Thus, combined selectin ligand outside-in signaling via E-selectin recognition of sLex on L-selectin and LFA-1/ICAM-1 bonds effectively amplify signaling via CXCR1/2 by ~100-fold and induces inside-out activation of β2-integrin. Ligation of CXCR1 and CXCR2 also activates Mac-1 (also known as CD11b/CD18 or αMβ2) on the plasma membrane (17, 18). While LFA-1 binds the ICAM family of proteins and regulates adhesive events within seconds of cell capture and rolling, Mac-1 recognizes a wide variety of ligands including complement iC3b, fibrinogen, and fibronectin, which facilitates intravascular crawling during paracellular and transcellular migration across inflamed endothelium (19, 20). Force acting on high affinity LFA-1 induces intracellular protein assembly that provides a physical linkage between calcium release-activated channels and calcium stores associated with the endoplasmic reticulum (ER), (21–24). We propose that local regulation of intracellular calcium serves as a secondary messenger downstream of GPCR signaling to regulate neutrophil shape change during the transition from rolling to arrest. While GPCR triggers PLC-β activation of IP3 to elicit calcium release from ER stores on the order of 500 nM of Ca2+, to achieve the maximum burst in cytosolic Ca2+ flux (~1.0 μM) requires integrin activation, ligation to ICAM-1, and force transduced outside-in signaling (16, 25). In this manner, intracellular Ca2+ release functions as a gatekeeper in regulating the conversion of a passive neutrophil in circulation to one that is firmly arrested and poised to transmigrate at sites of vascular inflammation expressing appropriate levels of chemokine agonist, E-selectin, and ICAM-1.
Selectin and integrin receptors are by nature's design mechanically tuned to function as de facto tactile sensors that convert the drag forces of flowing blood to tensile bond force that transduces biochemical signals at sites of focal adhesion. Mechanosignaling superposes with chemokine signaling to provide for precise spatiotemporal regulation of neutrophil recruitment at vascular sites proximal to tissue insult and injury. Inactive LFA-1 exists in a compact bent conformation with close association of the α and β extracellular domains that maintain a low binding affinity for ICAM-1 (Figure 1C). Rolling on E-selectin and P-selectin in the absence of chemokine induces extension of LFA-1 into an intermediate affinity conformation that supports slow neutrophil rolling at velocities of ~5 μm/s. In the extended conformation the extracellular domain of LFA-1 swings outward, but remains in a relatively closed state, such that bulkier ligands are sterically hindered from accessing the metal ion-dependent adhesion site (MIDAS) domain associated with high affinity binding (Figure 1C) (12, 26–28). This physiological mechanism for neutrophil deceleration is observed in a parallel plate flow chamber whereby the dynamics of LFA-1 extension is reported by increased binding of antibody extension reporter, KIM127 (29, 30). Inside-out stimulation elicits the release of a salt bridge between the α and β chains in the intracellular component of the integrin that triggers opening of the extracellular domain in a switchblade-like motion thereby exposing the MIDAS ligand binding domain (Figure 1C) (28). Increased binding affinity corresponds to a decrease in the koff, the rate constant for dissociation of the complex, which coincides with formation of LFA-1 bond clusters with ICAM-1 that supports shear resistant cell arrest under flow conditions (12, 31, 32). Blocking high affinity LFA-1 using a small molecule allosteric agonist that prevents the MIDAS domain from opening elicits suppression of cell arrest, while slow rolling via the extended conformation is maintained (12, 33). The increase in association rate and maintenance of slow rolling under conditions whereby the high affinity state is blocked implicates LFA-1 extension as the braking mechanism that neutrophils utilize to initiate the transition to firm arrest. Data supports the contention that intermediate-affinity of LFA-1 can participate in neutrophil capture and rolling; however, further up-regulation to high affinity by other activation mechanisms, including selectins, chemokines, and inflammatory lipids, is critical for the efficient transition to arrest and prompting of transendothelial migration. In particular, selectin ligand outside-in signaling functions to activate β2-integrins, elicit Ca2+ flux, and promote F-actin formation all of which promote cell polarization and a migratory phenotype (4, 13). In particular, L-selectin and P-selectin glycoprotein ligand 1 (PSGL1) function as mechanosensitive receptors that trigger LFA-1 extension and transition to integrin mediated activation processes.
Neutrophil rolling in humans is primarily mediated by L-selectin and PSGL-1 on the neutrophil surface and P-selectin and E-selectin on inflamed endothelium (34, 35). The minimum recognition unit of selectins is the tetrasaccharide sialyl Lewisx (sLex), a sialic acid α2-3 linked to galactose anchored by a β1-4 linked N-acetylglucosamine bearing a α1-3 linked fucose (36). sLex is expressed on glycosphingolipids (GSL), O-glycans of PSGL-1, and N-glycans of L-selectin (Figure 1B) (35, 37). In human's enzyme fucosyltransferases 4, 7, and 9, as well as sialyltransferase ST3-Gal-IV, are required to assemble sLex on N- and O-linked glycans, and GSL (38–40). Recent studies employed CRISPR-Cas9 gene editing to truncate each of the commonly expressing glycan types, reveal that O-glycans are responsible for leukocyte capture and initiation of rolling while N-glycans and GSL stabilize slow cell rolling and the transition to arrest (41, 42). Extracellular PSGL-1 is decorated in serine and threonine residues that are glycosylated to primarily bear fucosylated O-glycans capped with sLex that are capable of binding the calcium ion present within the lectin domain of all three selectins (Figure 1B) (43). Human L-selectin is decorated with N-glycans capped with sLex, which enable recognition by E-selectin (44, 45). This data implicates PSGL-1 as a ligand associated with selectin mediated capture and slow rolling, while L-selectin functions as a mechanosignaling ligand of E-selectin on inflamed endothelium. However, a number of studies using transgenic mice deficient in selectins or their ligands indicate that rolling via PSGL-1 is sufficient to mechanosignal integrin activation. One key difference is that fucosyltransferase 9 plays a key role in human, but not mouse neutrophil/E-selectin interactions (39, 40). This indicates a major difference in E-selectin ligands and their potential to signal, specifically that L-selectin in mice is not an E-selectin binding partner. Interestingly, while L-selectin is not an E-selectin binding partner in mouse, both E-selectin ligand-1 and CD44 play critical roles during murine neutrophil rolling and transition to arrest (46). Studies in mice genetic knockouts indicate that ESL-1 cooperates with PSGL-1 to maintain myeloid homeostasis and initiate neutrophil recruitment, but it is PSGL-1 that does the heavy lifting when it comes to integrin activation and slow rolling. In fact, ESL-1 is the predominant E-selectin ligand used by immature hematopoietic progenitors to home to the bone marrow. As myeloid maturation occurs a functional shift in selectin ligands from ESL-1 to PSGL-1 reduces the importance of ESL-1 in selectin signaling (47). Despite L-selectin not being a functional E-selectin ligand, inhibition of L-selectin binding in mice inhibits rolling, which was largely attributed to a loss of neutrophil-neutrophil mediated secondary capture that is L-selectin/PSGL-1 dependent (48). Further highlighting the function of L-selectin in selectin ligand outside-in signaling in mouse neutrophils, Stadtmann et al. reported that PSGL-1 ligation under shear flow precipitates membrane co-localization of PSGL-1 and L-selectin, which in turn elicits outside-in signaling of LFA-1 activation (49). One important component of L-selectin signaling is the multiple intracellular binding sites on the cytosolic domain of L-selectin for binding to tyrosine kinases and other downstream activators such as PLCγ2 (Figure 2A). The colocalization between PSGL-1 and L-selectin in mouse is induced by CD44 on the cell body engaging with E-selectin, which promotes clustering of PSGL-1 and L-selectin on the neutrophil surface in a p38 dependent manner. This was shown to promote secondary leukocyte tethering and formation of the L-selectin/PSGL-1 signaling complex (46). Murine neutrophils with down regulated L-selectin expression do not form this complex and therefore cannot signal for extension of integrin in the absence of chemokine (49). Thus, even in the absence of direct recognition of L-selectin by E-selectin on mouse neutrophils, L-selectin represents a potent selectin ligand outside-in signaling receptor.
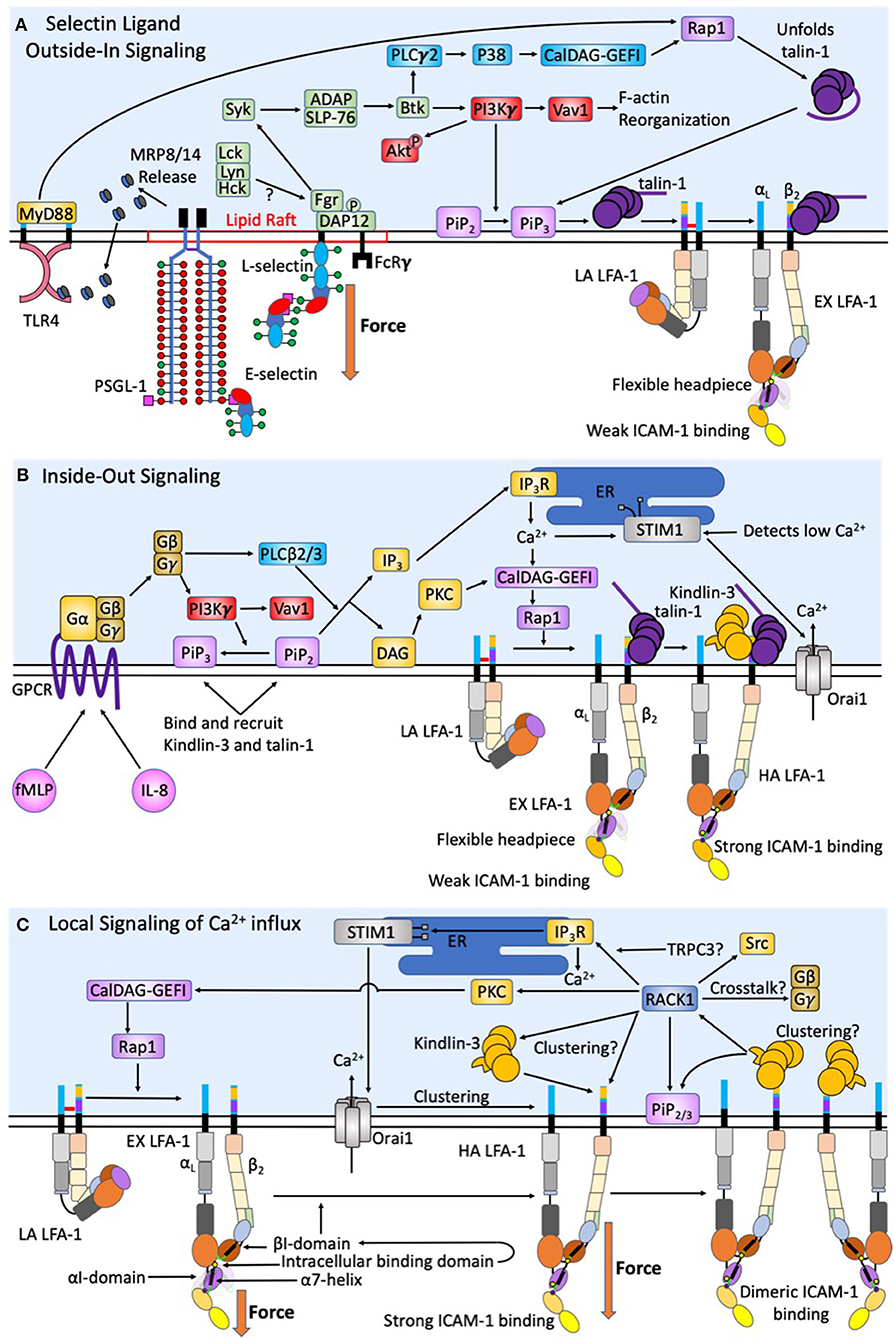
Figure 2. Intracellular signaling events act synergistically to promote human neutrophil arrest and shape change. During initial capture and rolling on inflamed endothelium a low baseline level of intracellular calcium is maintained. (A) Force acting on L-selectin and PSGL-1 induces clustering and recruitment of FcRγ and DAP-12 into lipid rafts. Phosphorylation by Fgr results in Syk activation. Other Src family kinases have also been shown to enhance selectin signaling, however only Fgr binds L-selectin cytodomain. Syk activation of SLP-76 and ADAP results in Btk activation, where signaling becomes PI3Kγ dependent. This catalyzes Vav1 activation and downstream F-actin reorganization that plays a key role in L-selectin clustering. The PLCγ2 activation of p38 and CalDAG-GEFI activate Rap1 and the unfolding of autoinhibited talin-1, which promotes recruitment to PiP3/2 and engagement and extension of LFA-1. Force acting on L-selectin catch-bonds transduces the signaling of high affinity LFA-1. Whereas, engagement of PSGL-1 and L-selectin primes MRP8/14 release. Its binding to TLR4 elicits the extension of LFA-1 and supports deceleration and cell rolling. (B) Selectin signaling is synergistic with chemokine signaling via GPCR to induce complete activation of integrin. CXCR1/2 ligation by fMLP and IL-8 elicit the dissociation of Gα from Gβγ subunits of G proteins resulting in PI3Kγ activation and PLCβ2/3, a convergence point between CXCR ligation and selectin signaling pathways. F-actin reorganization induced by PI3Kγ also results in transition of PiP2 to PiP3, which has a higher binding efficiency for Kindlin-3 and talin-1. DAG then activates PKC. Additionally, PLCβ2/3 splits PiP2 into IP3 and DAG. IP3 then binds IP3R on the ER to activate calciosome release. The gradient of intracellular calcium and activation of PKC catalyze activation of CalDAG-GEFI and Rap1 mediated integrin activation. This is a second convergence point between CXCR ligation and selectin signaling to activate LFA-1 through talin-1 recruitment. Calcium influx via Orai1 CRAC channels at focal sites of adhesion elicits the release of ER Ca2+ stores, which precipitates STIM1 association with the ER. This synergy between CRAC and the ER at the inflammatory synapse represents a positive feedback loop to enhance local calcium entry and the activation of additional LFA-1. (C) Tensile force acting on LFA-1/ICAM-1 provides for mechanotransduction of local calcium entry through Orai1 CRAC. Force acting on high affinity LFA-1 transduces from outside-in Kindlin-3 engagement. A conformational shift in the high affinity LFA-1 β2-tail exposes the Kindlin-3 binding domain. RACK-1 and Kindlin-3 both localize to the plasma membrane through its engagement with PiP2/3 to promote clustering of high affinity LFA-1. RACK1 may be a physical link between Kindlin-3, clusters of high affinity LFA-1, and TRPC3/IP3R/STIM1/Orai1; thereby completing a circuit to transduce force mediated calcium entry. RACK1 can activate PKC and CalDAG-GEFI and Rap1 providing a means of crosstalk between integrin outside-in and GPCR inside-out signaling.
Selectin engagement and mechanosignaling in neutrophils is only partially defined, and while other E-selectin ligands may play a role in capture, tethering, and signaling, we will focus on mechanosignaling through L-selectin engagement. E-selectin preferentially recognizes sLex on L-selectin and PSGL-1 in humans and signals active transport of adhesion molecules, leading to rapid cell arrest in shear flow (45). Binding of E-selectin promotes colocalization of L-selectin and PSGL-1 into membrane clusters on microvilli, an event that temporally correlates with MAPK phosphorylation and focal clustering of high-affinity CD18 (50). Genetic deletion of L-selectin in mice results in loss of phosphorylation of Akt, Syk, and phospholipase C (PLC) γ2, indicating these signaling molecules are downstream of the PSGL-1/L-selectin signaling complex (Figure 2A) (49). PSGL-1 engagement during rolling in inflamed mouse vasculature involves receptor clustering within lipid rafts that recruits membrane FcRγ and cytosolic DAP-12 (Figure 2A). Subsequent phosphorylation by Fgr then recruits Syk (51). The signaling pathway downstream of Syk includes SLP-76 and Bruton tyrosine kinase (Btk), which bifurcates the activation of PLCγ2 and phosphoinositide-3 kinase (PI3K) gamma-dependent pathways (51–53). Ligation of L-selectin and PSGL-1 enhances activation of PI3K, which acts on the effector Vav1 and results in F-actin assembly and downstream L-selectin reorganization and clustering, facilitating a feedback loop that amplifies signaling via L-selectin (54). It is known that PI3K activation results in cytoskeletal changes following leukocyte rolling, however its direct role in mechanosignaling via L-selectin outside of inducing clustering is unknown. A second pathway involving PLCγ2 activates downstream CalDAG-GEFI and p38 MAPK resulting in Rap1a activation (55). Rap1 then facilitates integrin extension and activation by recruiting Talin-1, and potentially Kindlin-3, which in turn allosterically reorients the integrin cytoplasmic domain (56). Talin-1 is an adaptor molecule that binds to the β2-integrin cytodomain to initiate extension and the high-affinity state, as well as providing a cytoskeletal anchor for vinculin (12, 56, 57). Integrin activation and clustering via talin-1 facilitates its binding to dimerized ICAM-1, this effectively prolongs the lifetime of LFA-1 bonds by ~10-fold and potentially enhances the association of adaptor molecule recruitment (27). Kindlin-3 is a similar adaptor protein to talin-1 that plays a central role in LFA-1 clustering and is critical for regulation of integrin off-rates (12, 58, 59).
It is well-established that mechanosignaling through L-selectin induces extension of β2-integrin, while the mechanism for activation of high-affinity β2-integrin independent of chemokine mediated inside-out signaling remains a point of contention. A recent discovery highlights a central role for MRP8/14 (also known as S100A8/S100A9 or calprotectin) as a signaling pathway that elicits extension of β2-integrin but not necessarily activation of the high affinity state (60). MRP8/14 is secreted by neutrophils during rolling on E-selectin where it then binds toll-like receptor 4 (TLR4) and signals in an autocrine manner activation of Rap1 and LFA-1 extension (13, 60). Ligation of PSGL-1 was sufficient to elicit release of MRP8/14 and induce extension of β2-integrin in a TLR4 dependent manner, whereas clustered L-selectin delivers a distinct signal that activates high affinity β2-integrin (13). Thus, bond tension and clustering of L-selectin cooperates with MRP8/14/TLR4 in priming LFA-1 extension, but a distinct signal is necessary for LFA-1 to achieve a high affinity state. Shear-resistant arrest is mediated in human neutrophils through clustered E-selectin/L-selectin bonds via signaling of high-affinity β2-integrin. The signaling pathway underlying MRP8/14 and TLR4 activation of Rap1 may involve PLCγ2, but this remains ill defined. Additionally, TLR4 has been shown to drive neutrophil aging which brings up a key point in neutrophil signaling, specifically dependence of senility in altering neutrophil receptor expression (61). Aged neutrophils show an enhanced expression of LFA-1, Mac-1, and CD44 and a decreased expression of L-selectin, however whether these receptor expression changes can be induced by MRP8/14 ligation of TLR4 remains unknown (62–64). Enhanced integrin expression translates to a more efficient shear-resistant recruitment of neutrophils under LPS challenge and implicates aged neutrophils as the first line of defense (65).
Hydrodynamic drag force acting on flowing neutrophils induces catch bond formation between E-selectin and L-selectin. The mechanics of selectin-sLex bonds and how they transduce outside-in signals via Src and Lck kinases are active areas of investigation (53, 66). A logical question is how catch-bond behavior of selectins dictates their capacity to engage in selectin ligand outside-in signaling. Neutrophil selectin binding kinetics have been studied using a variety of techniques including, atomic force microscopy, bioforce probes, surface plasmon resonance, and parallel plate flow channels (3, 67). Selectin bonds exhibit a triphasic adhesive response that is denoted slip-catch-slip bonds (68, 69). Slip bonds exhibit a shortened lifetime as the tensile force acting on them is increased. Catch bonds on the other hand increase lifetime as applied tensile force is increased. Neutrophils bound via PSGL-1 by P-selectin at low wall shear stress (<0.3 dynes/cm2) capture sporadically, dissociate frequently, and roll with high velocity (70). In contrast, higher shear stress (between 0.3 and 1 dynes/cm2) promotes more efficient capture and steady rolling at constant velocity, indicative of selectin catch bond behavior (69, 71–73). As wall shear stresses exceeds 1 dynes/cm2, slip bond behavior is again observed. An experiment that begins to explain the mechanism of catch-bond behavior was application of Rivipansel to antagonize L-selectin/E-selectin bonds, along with selectin ligand outside-in mechanosignaling of high affinity LFA-1 (13). Rivipansel is a rationally designed pan-selectin inhibitor that mimics the sLex tetrasacharide structure and an extended sulfate domain recognized by all three selectins lectin domain and in clinical trials for treatment of vaso-occlusive crisis in Sickle Cell disease (13, 74, 75). We recently reported that treatment with Rivipansel, blocks catch-bond formation between E-selectin and L-selectin and effectively inhibits selectin ligand outside-in signaling of integrin activation and neutrophil rolling to arrest (13). A structural explanation that begins to shed light on selectin catch-bond behavior was derived from co-crystallization between E-selectin and sLex that was imaged by small-angle X-ray scattering and modeled with molecular dynamics simulation. This analysis predicted that E-selectin bound to sLex under force caused an opening in the angle between the lectin and EGF domains from ~120 to ~141° (Figure 1B) (76). This rotation facilitates more efficient engagement of sLex expressing ligands by E-selectin resulting in an increase in bond strength and resistance to shear flow, indicative of a catch bond. Molecular dynamics simulations of the lectin-EGF domain angle between P-selectin bound with sLex is predicted to be ~114.6°, similar to the E-selectin/sLex angle. When tensile force is applied to this complex the interdomain angle opens to an extended conformation of ~140° allowing it to align in the direction of force application resulting in an enhanced off rate (43, 77). This catch-bond strengthening phenomenon has not been reported for L-selectin bound to sLex, but it is highly likely given the homology in lectin structure between the three selectins. A second mechanism that accounts for catch-bond behavior is when force induced dissociation of ligand results in rebinding by the lectin headpiece as the angle between the lectin-EGF domain increases (Figure 1B) (77). Bond lifetime is prolonged through multiple dissociations required for complete ligand detachment. Another model, known as the allosteric model, involves reorientation of the crystal structure of P-selectin when ligand binding elicits a shift in residues 83–89 in the lectin domain, thereby augmenting the binding surface and increasing the affinity between the lectin domain and sLex (78). Swapping the alanine 28 in the lectin domain with a bulky histidine to effectively open the 83–89 loop, resulted in decreased dissociation constant and slower rolling velocity on a substrate of PSGL-1 (78). It is noteworthy that the slower rolling was not observed at low shear stress regimes, corroborating the slip to catch transition induced at the higher shear stress. Taken together, modeling, and experimentation have shown that tensile force of sufficient magnitude and applied at defined rate can elicit an allosteric shift in selectins that in turn influence the strength and lifetime during bond formation with sLex presenting ligands. LFA-1 bonds also convert from a low or intermediate affinity state to a high affinity state during rolling at low shear stresses within the catch-bond regime between 0.5 and 2 dyne/cm2. This was first demonstrated by neutrophil capture on a substrate co-expressing E-selectin-IgG and high affinity β2-integrin reporter mAb24 (13). However, a different study reported that the majority of neutrophils sheared in microfluidic channels at 6 dynes/cm2, outside the catch-bond regime, did not activate and arrest on a substrate presenting mAb24, but bound efficiently on a substrate presenting the extension reporting antibody KIM127 (30). Taken together the data indicate that, while catch-bond formation via E-selectin/L-selectin engagement provides a distinct signal to activate high affinity LFA-1 and neutrophil arrest. This elucidates the importance of hydrodynamics in selectin-sLex catch-bond mechanics to provide a force sensitive mechanism for signaling optimum recruitment at appropriate shear stress.
So far, we have only focused on integrin activation via selectins, it is important to note that GPCR inside-out signaling is capable of transitioning LFA-1 from a low to high affinity state. This begs the question; how does selectin ligand outside-in signaling cooperate with inside-out signaling generated by CXCR1 and CXCR2 engagement by chemokine? While the role of GPCR signaling is well-reviewed elsewhere (79, 80), here we focus on how signaling facilitates LFA-1 mediated neutrophil arrest and a migratory phenotype (Figure 2B). Chemokine stimulation of GPCR in neutrophils results in stimulation of G proteins, separating Gα from the Gβγ subunits triggering PLC-β and PI3Kγ activation. PLC-β cleaves phosphatidylinositol 4,5 biphosphate (PIP2) into diacylglycerol (DAG) and inositol-1,4,5 triphosphate (IP3) (18, 81). IP3 then binds IP3 receptor (IP3R) localized on the membrane of the endoplasmic reticulum (ER) triggering cytosolic release of calcium (82, 83). The release of calcium within the ER is sensed by stromal interaction molecule 1 (STIM1), which functions to localize the ER to the primary calcium release activated calcium (CRAC) channel, Orai1 on the neutrophil membrane (Figure 2B) (83). Orai1 forms a hexamer that regulates influx of extracellular calcium (22, 84). PI3Kγ signals downstream of GPCR signaling by catalyzing PiP2 conversion to PiP3 and its association with the membrane near the integrin cytoplasmic domain. PiP3 functions to recruit Kindlin-3, Skap2, and other PH domain interacting molecules that are necessary for integrin transition from extension to high affinity more efficiently than PiP2 (14, 85, 86). PI3Kγ represents a convergence point between selectin ligand outside-in signaling and inside-out GPCR signaling pathways, and its activity cooperatively regulates activation of integrin. Each signaling pathway elicits a calcium influx, selectins through TRPC channels and chemokine through Orai1, and both pathways converge upon Rap1 dependent activation of integrin. It is widely known that high concentrations of chemokine can promote activation of LFA-1 and the onset of neutrophil deceleration and arrest. It appears that nature has designed a cooperative system by which very low levels of chemokine signaling can superpose with selectin catch-bond dependent signaling to amplify the response and likelihood of recruitment of surveilling neutrophils, perhaps most relevant in skin where E-selectin is expressed at low levels (14, 15). While there is lots of evidence supporting L-selectin and CXCR1/2 cooperativity in neutrophils, there has been no synergy observed between L-selectin and CCR7 signaling for enhancing LFA-1 activation in lymphocytes.
LFA-1 and Mac-1 are both involved in the transition from arrest to a migratory phenotype. However, it appears that the sequence of adhesive events is important for the precise synchronization of transendothelial migration. LFA-1 bonds function to initiate neutrophil arrest, while Mac-1 bonds provide migratory traction. Bond number and strength dictate the adhesive lifetime and translates to the amount of force that is transmitted across the membrane. Forces transmitted via LFA-1 and Mac-1 bonds in neutrophils are in part a function of their respective ligands, ICAM-1 and ICAM-2 are the main ligands on inflamed endothelium bound by activated LFA-1, while Mac-1 primarily binds to RAGE and JAM-C (87). It is noteworthy that LFA-1 can also bind to JAM-A and JAM-C, while Mac-1 recognizes the Ig-domain 3 of ICAM-1, albeit with lesser bond strength (19, 80, 88). Direct measurements of adhesion efficiency and rupture force for Mac-1 and LFA-1 bonds locked into a high or low affinity state were performed using atomic force microscopy (AFM) targeting the slip bond regime (87). An AFM tip was functionalized with LFA-1 or Mac-1 locked into specific states via allosteric antibodies or activated via manganese. Bond formation was induced by bringing this tip into contact with a surface of counter ligands ICAM-1, ICAM-2, RAGE, JAM-A, or JAM-C and then retracted at various rates. Deflection of the cantilever, as measured by a deflection of a laser beam reflected off the back of the cantilever, brought to light distinct features of bond rupture force and lifetime. The differences between mean rupture force of high affinity and low affinity LFA-1 was most pronounced when bound to ICAM-1 (56.1 ± 4.1 pN), ICAM-2 (37.7 ± 2.0 pN), JAM-A (37.4 ± 4.3 pN), and JAM-C (34.0 ± 5.9 pN) (87). The difference between high affinity and low affinity Mac-1 was most pronounced when bound to JAM-C (32.0 ± 2.8 pN) and RAGE (25.2 ± 4.2 pN) (87). When activated to high affinity, LFA-1/ICAM-1 and Mac-1/JAM-C bonds show the greatest strength. This correlates well with the observed function in the adhesion cascade of each integrin subunit, specifically LFA-1 mediates shear resistant cell arrest, while Mac-1 functions primarily in cell migration. It is important to note that the differential spatial localization of LFA-1 (on microvilli) and Mac-1 (on microvilli and cell body) on the neutrophil surface may result in each bond experiencing a distinct force regime during arrest in shear flow. This in turn can influence catch-bond behavior and result in different functional roles for each subunit during ligand binding.
Given that the magnitude of shear stress dictates the efficiency and lifetime of adhesion (89), it is critical to review how molecular mechanics are regulated and activated compared to quiescent neutrophils. Evans et al., was the first to measure off-rates and bond lifetimes between LFA-1 and ICAM-1 at the single integrin scale utilizing a bioforce probe (90). Activated LFA-1 possesses persistent mechanical strength exceeding 20 pN per bonds with lifetimes on the order of ~1 s when tensile force is applied at rates of ~10 pN/s. When force was ramped between ~10 and 1,000 pN/s it was observed that unbinding increased exponentially, indicating that LFA-1 bond lifetime is highly sensitive to force application. When locked in a high affinity state in Mn2+ enriched buffer, LFA-1 lifetime decreased to ~3 ms at a bond strength of ~64 pN (90). When these curves were extrapolated to zero force, bond lifetime increased to ~2 min corresponding to an off rate of ~0.05/s. As force ramps were increased to very high levels exceeding 7,000 pN/s, the force sensitivity of the off rates between LFA-1 and ICAM-1 disappeared, suggesting that large forces induce a change in molecular configuration of the complex (90). Locking recombinant LFA-1 at high affinity with Mn2+, or for native LFA-1 on neutrophils with allosteric activating antibody 327C or stimulation with IL-8, and testing bond formation with recombinant dimeric ICAM-1 revealed nearly identical changes in off-rates as force was ramped (91). Taken together, these studies indicate that a conformational switch elicited by either inside-out or selectin ligand mediated outside-in signaling results in LFA-1 heterodimers binding in tandem with domains 1–2 of parallel ICAM-1 molecules to establish long-lived bond formation that supports neutrophil firm arrest (Figure 2C).
Using the bioforce probe technique to ligate single LFA-1 molecules and measure bond kinetics it was subsequently reported that LFA-1/ICAM-1 bonds experience catch-slip bond behavior (92). Three states and corresponding distinct off-rates were identified at defined force profiles: At zero force, allosteric activation of LFA-1 to a high-affinity state elicited the most efficient binding. However, when force was ramped to ~10–15 pN on LFA-1/ICAM-1 bonds, lifetime increased to a maximum, revealing catch-bond behavior dependent upon the high affinity state (92). Beyond a threshold level of force, bond lifetimes monotonically decreased, indicative of slip bond behavior. Moreover, when LFA-1 was locked into a low affinity or extended conformation catch bonds were not detected, rather bond lifetime decreased monotonically. A structural model was proposed whereby pulling on extended LFA-1 anchored to the cytoskeleton elicits a shift in the α7 helix, thereby exposing the MIDAS domain and a shift to high affinity LFA-1 (Figure 1C). A structural model by which force stabilizes the high affinity conformation was proposed that involves movement of the α7 helix in the αL-I domain linking to an intracellular loop that shifts orientation of the adjacent AMIDAS domain in the β2-I domain (93). This was experimentally supported utilizing the antagonist XVA143 that binds internally to a site between the α7 helix and the AMIDAS domain on the β-I domain. This effectively blocked catch-bond behavior, reduced ligand binding affinity under force, and decreased bond lifetimes. This reveals the importance of the α7 helix and its binding to the intra-ligand AMIDAS domain in forming strong long-lasting catch-bonds (92). However, when force is applied the intra-ligand interaction is enhanced, indicating that force is necessary to precipitate the complete maturation of high affinity LFA-1. This external shift in geometry of β2-integrin suggests that uptake of tensile force can reinforce bond strength and lifetime via a shift in the angle of the transmembrane domain. As discussed below, we propose that a shift in transmembrane domain angle with force is responsible for initiating outside-in signaling by transducing a deformation in the Kindlin-3 binding domain within the β2-integrin cytoplasmic tail. This in turn may catalyze the association of PiPs to the integrin to enhance adaptor molecule recruitment to the site of high affinity LFA-1.
Kindlins are a family of proteins that are highly conserved and function as cytoplasmic adaptor proteins that bridge the cytoskeleton to integrins via their FERM domains. A rare mutation in Kindlin-3 is the culprit in leukocyte adhesion deficiency type-III, a disease characterized by defects in leukocyte and platelet β1-, β2-, and β3-integrin functions (6, 94–97). For LFA-1 to transition from low to intermediate and high affinity, engagement of the cytoplasmic domains by talin-1 and Kindlin-3 are necessary. These adaptor proteins are 4.1/ezrin/radixin/moesin (FERM) domain proteins with four subdomains (F0–3), whose F3 domains are capable of binding one of two specific NPxF/Y domains present on β-integrin cytoplasmic tails (98). Talin-1 also has an extended rod domain that binds actin, indicating a key role in F-actin association and local cytoskeletal rearrangement (56). NMR spectroscopy revealed that the talin-1 rod domain interacts with the F3 subdomain, masking its binding domain. This autoinhibition is disrupted by PiP engagement allowing the talin-1 F3 domain to bind the proximal NPxF/Y on β-integrin tails and break the salt bridge holding the α-β chains together, thereby initiating the transition to an extended and a high affinity state (99, 100). Kindlin-3 is endowed with a plexstrin homology (PH) domain embedded into its F2 subdomain. Kindlin-3 F3 domain binds to the membrane distal NPxF/Y motif on the β-integrin tail (56). Constitutive Kindlin-3 is not autoinhibited nor does it bind to low affinity LFA-1, indicating that its β-integrin binding domain is not exposed until integrin extension occurs. Transgenic mice in which talin-1 is genetically deleted lacks the capacity to both extend or activate high affinity integrin, while Kindlin-3 knockouts retain the capacity for LFA-1 extension, but not activation to high affinity (58). Remarkable was the finding that a 95% knockdown of Kindlin-3 in a mouse model, retained basal levels of integrin function in platelets (101). However, extended bleeding and impaired healing was observed when these mice were exposed to injury and infection. This indicates that a threshold level of Kindlin-3 and talin-1 are necessary to maintain normal function of LFA-1 (101). In fact, Kindlin-3 and talin-1 abundance is sufficient to occupy only ~50% of integrin cytodomain in granulocytes. By comparison, platelets contain twice as much adaptor proteins, this highlights a key difference in LFA-1 activation kinetics compared with GPIIbIIIa that also requires Kindlin3 for function (101). A stoichiometric balance exists between Kindlin-3/talin-1 and integrin in neutrophils, such that diffusion may be a limiting factor in the rate of LFA-1 activation.
Loss of Kindlin-3 function in patients suffering from LAD-III is characterized by suppression of LFA-1 functions, but not VLA-4 under shear conditions in both neutrophils and primary T cells (94). Tensile force, talin-1 and Kindlin-3 are necessary conditions to observe activation of high affinity LFA-1. One potential mode of LFA-1 activation is via talin-1 recruitment to the β-subunit tail thereby catalyzing the extended conformation and recognition of ICAM-1. As fluid drag transmits tensile force to the intermediate affinity bond, molecular deformation exposes the MIDAS and precipitates a transition to the high affinity state. Given that LFA-1 extension is observed to promote the engagement of Kindlin-3 at sites of focal adhesion, it is possible that force transmission on LFA-1 itself catalyzes increased binding of Kindlin-3 (16). However, the precise mechanism by which these adaptor proteins recruit to LFA-1 is ill defined, as is whether they simultaneously reside on a single LFA-1 cytodomain. It has been suggested that Kindlin-2 and talin-1 are capable of simultaneously binding a single β2-integrin tail, and due to the homology between Kindlin-2 and talin-1 F3 domain it is highly likely that Kindlin-3 can also bind to the integrin tail simultaneously with talin-1 (102). Adaptor protein binding occurs following phosphorylation of the tyrosine in their respective binding sites via Src family kinases. Binding is modulated by another key TTT phosphorylation site between the two binding regimes (58). Kindlins, filamin, 14-3-3 and other proteins can bind this domain and can affect the order in which binding to the other NPxF motifs occurs (103, 104). Kinases provide spatiotemporal regulation of integrin activation, but more research is required to elucidate its precise role in mechanosignaling. Given that talin-1 binding is retained in Kindlin-3 knockouts, a prevailing theory is that these adaptor proteins may serve as co-activators by removing potential competitive binding proteins such as 14-3-3 protein (103). An additional mechanism is via talin-1 induced extension to expose the binding site of Kindlin-3 on the integrin tail allowing it to then function as a mechanosensitive clutch. Experimental data indicates that talin-1 and Kindlin-3 play independent roles during signaling of neutrophil arrest and migration. Utilizing neutrophil-like HL-60 cells to knockout talin-1 or Kindlin-3, activated LFA-1 bonds under tensile force catalyzed calcium influx through the CRAC channel Orai1 only in the presence of Kindlin-3. The presence of talin-1 and absence of Kindlin-3 was insufficient to link LFA-1 to Orai1 and induce calcium influx (16). These data provide insight on mechanotransduction through LFA-1 under shear force conditions, which involves assembly of a complex via Kindlin-3-β2-integrin cytodomain and Orai1 to complete a circuit whereby force induces calcium flux.
Kindlin-3 association with LFA-1 is necessary for the rapid clustering of LFA-1, but it is unlikely to function as a scaffold protein in this process since it has only one binding site for the β2-integrin tail, unless Kindlin-3 is capable of complexing other Kindlin-3. Another adaptor protein that can enhance LFA-1 clustering is receptor of activated protein C kinase 1 (RACK1) (Figure 2C). RACK1 is a seven bladed propeller protein that can bind multiple Kindlin-3 with its domains 5 to 7. Kindlin-3 binds RACK1 through its PH domain and in cells with the PH domain deleted, LFA-1 clustering is inhibited (59). However, Kindlin-3 PH domains play a key role in binding numerous proteins such as SKAP2 or PiP2. Thus, knockout of the PH domain may suppress Kindlin-3 migration to the LFA-1 tail domains, independent of RACK1 (80, 86). Despite this, it is noteworthy that immunoprecipitation of a ternary complex between β2-integrin tail, RACK1, and Kindlin-3 is intact even when the Kindlin-3 F3 domain is genetically deleted (59). Given that RACK1 itself does not activate adaptor proteins, it may function as a chaperone for other adaptor proteins to bind the integrin cytodomain. RACK1 has also been shown to bind focal adhesion kinases (FAK) and Src via propeller domain 2 to promote IGF-1R receptor association with integrin, and in a similar way may induce LFA-1 clustering by promoting Kindlin-3 association under tensile bond force (105). RACK1 structure shares a similar homology to Gβ subunit and has been shown to form a heterodimer with it (106). While it is clear that RACK1 plays a role in membrane clustering of LFA-1, whether that is due to aggregation of Kindlin-3 bound to LFA-1 or by promoting the assembly of additional adaptors requires further study. Kindin-3 induced LFA-1 clustering correlates with enhanced calcium signaling, yet the complete signaling circuit has yet to be elucidated (16).
Hydrodynamic force acting on LFA-1 and Mac-1 regulates calcium entry, kinase activation, and cytoskeletal protein recruitment all of which are necessary to achieve a migratory state (15, 107–109). We propose that LFA-1 functions not only as a breaking mechanism to achieve neutrophil arrest, but also in the mechanotransduction signals delivered through focal sites of adhesive traction that oppose shear force gradients present on the endothelial surface (Figure 2C). Cooperativity between selectin engagement and chemokine binding of GPCRs activate the transition of LFA-1 from low to high affinity resulting in deceleration of the cell that occurs on the order of seconds (12). Neutrophil deceleration and arrest trigger a concomitant rise in intracellular calcium detected within seconds and which precipitates cell shape change and polarization within minutes (15). Coordination in signaling rolling to arrest and to a migratory state is interrupted by inhibiting CRAC channels with pharmacological inhibitors, or genetic deletions that alter calcium flux (22, 110). The precise number of LFA-1 receptors associated with signaling calcium flux is unknown, however, once sufficient numbers of LFA-1 transition to high affinity bonds (on the order of ~100 receptors) within ~2–3 submicron focal microclusters, local calcium entry via Orai1 is initiated promoting the coalescence of LFA-1 into micron sized macroclusters (16). This feedback loop between enhanced LFA-1 clustering and Orai1 mediated calcium entry results in a large local transient burst of intracellular calcium that is required to promote organization of high-affinity integrin within focal adhesions. This is in contrast to GPCR that are distributed around the neutrophil within microvilli, and upon ligand binding provide an inside-out signal that is more globally dispersed within the cell volume. This implicates integrin mediated calcium signaling as a central regulator of neutrophil migratory function beyond firm arrest (Figure 2C). Remarkably, RACK1 has been shown to regulate IP3R function in a manner dependent on TRPC3 that in turn promotes calcium release (111). Once calcium has been released through IP3R activation via RACK1, IP3R associates with activated STIM1 and subsequently binds Orai1 (111). Further, TRPC3 deletion in HELA and Hek cells, abrogates the association between Orai1 and IP3R (111). While this has yet to be shown in primary human neutrophils, these data highlight the potential for high affinity LFA-1 bonds under force to catalyze association of a complex composed of Kindlin-3/RACK1/TRPC3/IP3R/STIM1/Orai1 that effectively directs calcium influx and release of ER stores within focal adhesions in a manner that orients cytoskeletal force generation and neutrophil polarization (Figure 2C).
A lack of calcium release or entry via CRAC impairs various physiological events in immune cells, implicating calcium as a pivotal secondary messenger (23, 24, 81, 82, 107–109, 112–114). The role of calcium in T cell regulation can provide insight into calcium signaling in neutrophils. Through the use of genetically-encoded calcium indicators it has been shown that T cell interaction with antigen presenting cells in vivo results in low levels of local calcium release (115). Local calcium enhances T-cell mechanosignaling within the immune synapse by promoting T cell receptor clustering and the binding of anionic phospholipids within the plasma membrane, similar to how local calcium bursts in neutrophils regulates activation and integrin build-up within the inflammatory synapse at sites of focal adhesions. Furthermore, calcium entry via Orai1 is responsible for T cell homing to lymph nodes and is necessary for high-affinity integrin LFA-1 activation (116). The magnitude of calcium bursts builds over time and function to recruit more LFA-1, which in turn activates additional Orai1 in a feedback loop to promote adhesion and signaling. Once LFA-1 is engaged between the T cell and antigen presenting cell, external calcium concentration rises above cytosolic, lending credence to the theory that co-localization between membrane receptors and CRAC provides a spatially localized signal that is scaled by the surface area of the cluster which dictates its contribution to cell activation. Neutrophils appear to engage in a similar mechanical process in which LFA-1 bond traction provides spatiotemporal cues, but this occurs within seconds as opposed to hours for T cells and serves to synchronize the multistep process leading to transmigration.
LFA-1 bond formation provides a spatial queue, while calcium provides a temporal queue to signal cell shape change and polarization. Localized calcium flux provides a signal to initiate local cytoskeletal reorganization and subsequent cellular motility (Figure 2C). Contractile and protrusion forces created by filamentous actin (F-actin) during cytoskeletal reorganization enables the formation of pseudopods that lead migration and contractile rings that organizes formation of the uropod at the rear that generates traction force (117–119). We propose that local generation of calcium gradients generated by CRAC channels concentrated within sites of focal adhesion provides a signal to catalyze cytoskeletal actin formation and interaction with myosin to drive immune cell motility (119). In T-cells sustained calcium is necessary for continued actin polymerization and microcluster formation within the immunological synapse between the T-cell and antigen presenting cell (120). In neutrophils, deficiency of Wiskott-Aldrich syndrome protein (WASp) results in defects in β2-integrin clustering, signaling of calcium flux, and cell motility (117, 121). This implicates F-actin mediated cytoskeletal reorganization in integrin clustering and highlights the importance of calcium signaling in this process. Enhanced calcium signaling promotes additional F-actin polymerization and cell spreading through binding to gelsolin a 6-domain actin binding protein that uses calcium to regulate actin filament assembly (122, 123). Once calcium is bound, gelsolin undergoes a conformational change that exposes its actin binding site, thereby promoting cytoskeletal F-actin assembly (124–126). The asymmetry of front/back actin polymerization may be a consequence of the spatial pattern of integrin mediated calcium entry. F-actin also plays an important role in internalization of CRAC channels, providing a putative mechanism for down regulating extracellular calcium entry as neutrophils prepare to transmigrate at appropriate sites of inflammation (21). This illustrates a key feedback mechanism in which calcium entry and cytoskeletal reorganization provides feedback to organize a migratory phenotype in immune cells.
Neutrophils function as the sentinels of the innate immune system by patrolling miles of vasculature in the microcirculation. To accomplish this critical function, they have evolved adhesive mechanisms that facilitate efficient recruitment at the precise location of tissue insult through the conversion of tensile bond force into biochemical signals. This review provides a scheme by which neutrophil tethering and rolling via selectins leads to integrin activation and shear resistant arrest, a set of mechanosignaling based events necessary for subsequent generation of neutrophil protrusions and diapedesis. The latter process is thought to require a chemotactic gradient that guides neutrophils to the site of tissue insult. In a previous Frontiers of Immunity review, we detailed how cytosolic release of Ca2+ converges with influx through CRAC to dynamically modulate the number and location of β2-integrin bonds, which function to synchronize the transition from rolling to arrest and neutrophil shape polarization necessary for diapedesis (9). Recent studies have lent quantitative insight into the physical mechanisms by which L-selectin and integrin catch-bonds convert shear stress into chemical signals within distinct regions of plasma membrane enriched in kinases, phosphoinositides, and cytosolic adaptors (13, 39, 42, 49, 76). Although the specific mechanism of outside-in mechanosignaling is lacking, experimental evidence and structural models indicate that LFA-1 cytosolic domains directly complex with Kindlin-3 and Orai1 and this is regulated by the magnitude of tensile force. We propose that bond tension at durable sites of focal adhesive contact cause reorientation of the integrin headpiece with ICAM-1 and strengthening of the bond. This concomitantly elicits deformation of the LFA-1 cytodomain, thereby exposing the binding site for the PH domain of Kindlin-3 (16, 87, 96). In this review, we put forth the premise that the conversion of LFA-1 to a high affinity state capable of stable bond formation with ICAM-1 is a gatekeeper of this mechanically sensitive linkage that governs transmembrane Ca2+ influx. This in turn, facilitates recognition and binding by Kindlin-3 and talin-1 that leads to engagement with RACK1 and FAK and activation of STIM1/Orai1 channels within the focal region of contact on an arrested neutrophil. This contact-mediated circuit is triggered by tensile force conducted via LFA-1 bonds, promotes the calcium feedback loop to recruit additional high-affinity LFA-1 into macroclusters that serve as a nexus for Rho-GTPase activation and F-actin polymerization at contractile regions through which lamellipodia form (127). At sufficient levels of intracellular calcium, F-actin polymerization links to talin-1 tails that reinforce the binding of vinculin. Shape change and cell migration is then mediated by Mac-1 redistribution and bond formation at the uropod where myosins assist in contractile force generation and actin movement (128). In this manner, high affinity integrin bonds effectively function as tactile sensors of the magnitude and direction of hydrodynamic drag forces. Thus, neutrophils dynamically redistribute focal adhesions in a pattern that directs intracellular calcium flux that orients the major axis of neutrophil polarization and generation of motile force to direct innate immune cells at appropriate sites experiencing inflammation.
VM wrote the initial draft of the manuscript with the aid of SS. VM and SS both edited the manuscript to its current form. VM designed the figures and SS edited the figures.
The work was supported by grants AI047294 and AI129302 (SS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell (1991) 67:1033–6. doi: 10.1016/0092-8674(91)90279-8
2. Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev. (2002) 186:8–18. doi: 10.1034/j.1600-065X.2002.18602.x
3. Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. (2005) 7:151–85. doi: 10.1146/annurev.bioeng.7.060804.100423
4. Gong Y, Zhang Y, Feng S, Liu X, Lu S, Long M. Dynamic contributions of P- and E-selectins to beta2-integrin-induced neutrophil transmigration. Faseb J. (2017) 31:212–23. doi: 10.1096/fj.201600398RRR
5. von Andrian UH, Berger EM, Ramezani L, Chambers JD, Ochs HD, Harlan JM, et al. In vivo behavior of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J Clin Invest. (1993) 91:2893–7. doi: 10.1172/JCI116535
6. Hogg N, Stewart MP, Scarth SL, Newton R, Shaw JM, Law SKA, et al. A novel leukocyte adhesion deficiency caused by expressed but nonfunctional β2 integrins Mac-1 and LFA-1. J Clin Invest. (1999) 103:97–106. doi: 10.1172/JCI3312
7. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. (2009) 27:339–62. doi: 10.1146/annurev.immunol.021908.132554
8. Dixit N, Yamayoshi I, Nazarian A, Simon SI. Migrational guidance of neutrophils is mechanotransduced via high-affinity LFA-1 and calcium flux. J Immunol. (2011) 187:472–81. doi: 10.4049/jimmunol.1004197
9. Dixit N, Simon SI. Chemokines, selectins and intracellular calcium flux: temporal and spatial cues for leukocyte arrest. Front Immunol. (2012) 3:188. doi: 10.3389/fimmu.2012.00188
10. Moretti FA, Moser M, Lyck R, Abadier M, Ruppert R, Engelhardt B, et al. Kindlin-3 regulates integrin activation and adhesion reinforcement of effector T cells. Proc Natl Acad Sci USA. (2013) 110:17005–10. doi: 10.1073/pnas.1316032110
11. Stadtmann A, Zarbock A. The role of kindlin in neutrophil recruitment to inflammatory sites. Curr Opin Hematol. (2017) 24:38–45. doi: 10.1097/MOH.0000000000000294
12. Lefort CT, Ley K. Neutrophil arrest by LFA-1 activation. Front Immunol. (2012) 3:157. doi: 10.3389/fimmu.2012.00157
13. Morikis VA, Chase S, Wun T, Chaikof EL, Magnani JL, Simon SI. Selectin catch-bonds mechanotransduce integrin activation and neutrophil arrest on inflamed endothelium under shear flow. Blood (2017) 130:2101–10. doi: 10.1182/blood-2017-05-783027
14. Yago T, Zhang N, Zhao L, Abrams CS, McEver RP. Selectins and chemokines use shared and distinct signals to activate beta2 integrins in neutrophils. Blood Adv. (2018) 2:731–44. doi: 10.1182/bloodadvances.2017015602
15. Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, Simon SI. Calcium flux in neutrophils synchronizes beta2 integrin adhesive and signaling events that guide inflammatory recruitment. Ann Biomed Eng. (2008) 36:632–46. doi: 10.1007/s10439-008-9453-8
16. Dixit N, Kim MH, Rossaint J, Yamayoshi I, Zarbock A, Simon SI. Leukocyte function antigen-1, kindlin-3, and calcium flux orchestrate neutrophil recruitment during inflammation. J Immunol. (2012) 189:5954–64. doi: 10.4049/jimmunol.1201638
17. Kappelmayer J, Bernabei A, Gikakis N, Edmunds LH Jr, Colman RW. Upregulation of Mac-1 surface expression on neutrophils during simulated extracorporeal circulation. J Lab Clin Med. (1993) 121:118–26.
18. Lomakina EB, Waugh RE. Signaling and dynamics of activation of LFA-1 and Mac-1 by immobilized IL-8. Cell Mol Bioeng. (2010) 3:106–16. doi: 10.1007/s12195-009-0099-x
19. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. (1999) 163:5029–38.
20. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
21. Itagaki K, Kannan KB, Singh BB, Hauser CJ. Cytoskeletal reorganization internalizes multiple transient receptor potential channels and blocks calcium entry into human neutrophils. J Immunol. (2004) 172:601–7. doi: 10.4049/jimmunol.172.1.601
22. Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI. Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood (2010) 115:657–66. doi: 10.1182/blood-2009-05-224659
23. Clemens RA, Lowell CA. Store-operated calcium signaling in neutrophils. J Leukoc Biol. (2015) 98:497–502. doi: 10.1189/jlb.2MR1114-573R
24. Immler R, Simon SI, Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest. (2018) 48 (Suppl. 2):e12964. doi: 10.1111/eci.12964
25. Bei L, Hu T, Qian ZM, Shen X. Extracellular Ca2+ regulates the respiratory burst of human neutrophils. Biochim Biophys Acta (1998) 1404:475–83. doi: 10.1016/S0167-4889(98)00081-0
26. Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell (2003) 112:99–111. doi: 10.1016/S0092-8674(02)01257-6
27. Sarantos MR, Raychaudhuri S, Lum AF, Staunton DE, Simon SI. Leukocyte function-associated antigen 1-mediated adhesion stability is dynamically regulated through affinity and valency during bond formation with intercellular adhesion molecule-1. J Biol Chem. (2005) 280:28290–8. doi: 10.1074/jbc.M501662200
28. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity (2006) 25:583–94. doi: 10.1016/j.immuni.2006.07.016
29. Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood (2006) 107:2101–11. doi: 10.1182/blood-2005-06-2303
30. Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood (2010) 116:617–24. doi: 10.1182/blood-2010-01-266122
31. Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature (2004) 432:59–67. doi: 10.1038/nature02976
32. Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. (2007) 25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618
33. Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small molecule integrin antagonists that bind to the beta2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity (2003) 19:391–402. doi: 10.1016/S1074-7613(03)00238-3
34. Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell (1991) 65:859–73. doi: 10.1016/0092-8674(91)90393-D
35. Foxall C, Watson SR, Dowbenko D, Fennie C, Lasky LA, Kiso M, et al. The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J Cell Biol. (1992) 117:895–902. doi: 10.1083/jcb.117.4.895
36. Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity (1998) 9:881–90. doi: 10.1016/S1074-7613(00)80653-6
37. Nimrichter L, Burdick MM, Aoki K, Laroy W, Fierro MA, Hudson SA, et al. E-selectin receptors on human leukocytes. Blood (2008) 112:3744–52. doi: 10.1182/blood-2008-04-149641
38. Weninger W, Ulfman LH, Cheng G, Souchkova N, Quackenbush EJ, Lowe JB, et al. Specialized contributions by alpha(1,3)-fucosyltransferase-IV and FucT-VII during leukocyte rolling in dermal microvessels. Immunity (2000) 12:665–76. doi: 10.1016/S1074-7613(00)80217-4
39. Buffone A Jr, Mondal N, Gupta R, McHugh KP, Lau JT, Neelamegham S. Silencing alpha1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J Biol Chem. (2013) 288:1620–33. doi: 10.1074/jbc.M112.400929
40. Mondal N, Buffone A Jr, Neelamegham S. Distinct glycosyltransferases synthesize E-selectin ligands in human vs. mouse leukocytes Cell Adh Migr. (2013) 7:288–92. doi: 10.4161/cam.24714
41. Shao B, Yago T, Setiadi H, Wang Y, Mehta-D'souza P, Fu J, et al. O-glycans direct selectin ligands to lipid rafts on leukocytes. Proc Natl Acad Sci USA. (2015) 112:8661–6. doi: 10.1073/pnas.1507712112
42. Stolfa G, Mondal N, Zhu Y, Yu X, Buffone A Jr, Neelamegham S. Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and Glycosphingolipids to human leukocyte-endothelium adhesion. Sci Rep. (2016) 6:30392. doi: 10.1038/srep30392
43. Sundd P, Pospieszalska MK, Cheung LSL, Konstantopoulos K, Ley K. Biomechanics of leukocyte rolling. Biorheology (2011) 48:1–35. doi: 10.3233/BIR-2011-0579
44. Zollner O, Lenter MC, Blanks JE, Borges E, Steegmaier M, Zerwes HG, et al. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J Cell Biol. (1997) 136:707–16. doi: 10.1083/jcb.136.3.707
45. Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. (2000) 164:4348–58. doi: 10.4049/jimmunol.164.8.4348
46. Hidalgo A, Peired AJ, Wild M, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity (2007) 26:477–89. doi: 10.1016/j.immuni.2007.03.011
47. Sreeramkumar V, Leiva M, Stadtmann A, Pitaval C, Ortega-Rodriguez I, Wild MK, et al. Coordinated and unique functions of the E-selectin ligand ESL-1 during inflammatory and hematopoietic recruitment in mice. Blood (2013) 122:3993–4001. doi: 10.1182/blood-2013-07-514497
48. Guyer DA, Moore KL, Lynam EB, Schammel CM, Rogelj S, McEver RP, et al. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood (1996) 88:2415–21.
49. Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med. (2013) 210:2171–80. doi: 10.1084/jem.20130664
50. Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol. (2004) 172:7780–90. doi: 10.4049/jimmunol.172.12.7780
51. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med. (2008) 205:2339–47. doi: 10.1084/jem.20072660
52. Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, et al. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood (2010) 115:3118–27. doi: 10.1182/blood-2009-11-254185
53. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood (2010) 116:485–94. doi: 10.1182/blood-2009-12-259556
54. Luo J, Xu T, Wang X, Ba X, Feng X, Deepak V, et al. PI3K is involved in L-selectin- and PSGL-1-mediated neutrophil rolling on E-selectin via F-actin redistribution and assembly. J Cell Biochem. (2010) 110:910–9. doi: 10.1002/jcb.22603
55. Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur J Immunol. (2011) 41:2074–85. doi: 10.1002/eji.201041196
56. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood (2012) 119:4275–82. doi: 10.1182/blood-2011-08-373118
57. Atherton P, Stutchbury B, Wang DY, Jethwa D, Tsang R, Meiler-Rodriguez E, et al. Vinculin controls talin engagement with the actomyosin machinery. Nat Commun. (2015) 6:10038. doi: 10.1038/ncomms10038
58. Ye F, Petrich BG. Kindlin: helper, co-activator, or booster of talin in integrin activation? Curr Opin Hematol. (2011) 18:356–60. doi: 10.1097/MOH.0b013e3283497f09
59. Feng C, Li YF, Yau YH, Lee HS, Tang XY, Xue ZH, et al. Kindlin-3 mediates integrin alphaLbeta2 outside-in signaling, and it interacts with scaffold protein receptor for activated-C kinase 1 (RACK1). J Biol Chem. (2012) 287:10714–26. doi: 10.1074/jbc.M111.299594
60. Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker X, Bieber S, Nussbaum CF, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun. (2015) 6:6915. doi: 10.1038/ncomms7915
61. Adrover JM, Nicolas-Avila JA, Hidalgo A. Aging: a temporal dimension for neutrophils. Trends Immunol. (2016) 37:334–45. doi: 10.1016/j.it.2016.03.005
62. Tanji-Matsuba K, van Eeden SF, Saito Y, Okazawa M, Klut ME, Hayashi S, et al. Functional changes in aging polymorphonuclear leukocytes. Circulation (1998) 97:91–8. doi: 10.1161/01.CIR.97.1.91
63. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature (2015) 525:528–32. doi: 10.1038/nature15367
64. Kolaczkowska E. The older the faster: aged neutrophils in inflammation. Blood (2016) 128:2280–2. doi: 10.1182/blood-2016-09-739680
65. Uhl B, Vadlau Y, Zuchtriegel G, Nekolla K, Sharaf K, Gaertner F, et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood (2016) 128:2327–37. doi: 10.1182/blood-2016-05-718999
66. Xu T, Chen L, Shang X, Cui L, Luo J, Chen C, et al. Critical role of Lck in L-selectin signaling induced by sulfatides engagement. J Leukoc Biol. (2008) 84:1192–201. doi: 10.1189/jlb.0208084
67. Long M, Zhao H, Huang KS, Zhu C. Kinetic measurements of cell surface E-selectin/carbohydrate ligand interactions. Ann Biomed Eng. (2001) 29:935–46. doi: 10.1114/1.1415529
68. Beste MT, Hammer DA. Selectin catch-slip kinetics encode shear threshold adhesive behavior of rolling leukocytes. Proc Natl Acad Sci USA. (2008) 105:20716–21. doi: 10.1073/pnas.0808213105
69. Wayman AM, Chen W, McEver RP, Zhu C. Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys J. (2010) 99:1166–74. doi: 10.1016/j.bpj.2010.05.040
70. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res. (2015) 107:331–9. doi: 10.1093/cvr/cvv154
71. Taylor AD, Neelamegham S, Hellums JD, Smith CW, Simon SI. Molecular dynamics of the transition from L-selectin- to beta 2-integrin-dependent neutrophil adhesion under defined hydrodynamic shear. Biophys J. (1996) 71:3488–500. doi: 10.1016/S0006-3495(96)79544-9
72. Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature (2003) 423:190–3. doi: 10.1038/nature01605
73. Sarangapani KK, Yago T, Klopocki AG, Lawrence MB, Fieger CB, Rosen SD, et al. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J Biol Chem. (2004) 279:2291–8. doi: 10.1074/jbc.M310396200
74. Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood (2010) 116:1779–86. doi: 10.1182/blood-2009-12-260513
75. Telen MJ, Wun T, McCavit TL, De Castro LM, Krishnamurti L, Lanzkron S, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood (2015) 125:2656–64. doi: 10.1182/blood-2014-06-583351
76. Preston RC, Jakob RP, Binder FP, Sager CP, Ernst B, Maier T. E-selectin ligand complexes adopt an extended high-affinity conformation. J Mol Cell Biol. (2016) 8:62–72. doi: 10.1093/jmcb/mjv046
77. Lou J, Zhu C. A structure-based sliding-rebinding mechanism for catch bonds. Biophys J. (2007) 92:1471–85. doi: 10.1529/biophysj.106.097048
78. Waldron TT, Springer TA. Transmission of allostery through the lectin domain in selectin-mediated cell adhesion. Proc Natl Acad Sci USA. (2009) 106:85–90. doi: 10.1073/pnas.0810620105
79. Kehrl JH. Heterotrimeric G protein signaling: roles in immune function and fine-tuning by RGS proteins. Immunity (1998) 8:1–10. doi: 10.1016/S1074-7613(00)80453-7
80. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
81. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. (2015) 33:291–353. doi: 10.1146/annurev-immunol-032414-112212
82. Demaurex N, Nunes P. The role of STIM and ORAI proteins in phagocytic immune cells. Am J Physiol Cell Physiol. (2016) 310:C496–508. doi: 10.1152/ajpcell.00360.2015
83. Demaurex N, Saul S. The role of STIM proteins in neutrophil functions. J Physiol. (2018) 596:2699–708. doi: 10.1113/JP275639
84. Cai X, Zhou Y, Nwokonko RM, Loktionova NA, Wang X, Xin P, et al. The orai1 store-operated calcium channel functions as a hexamer. J Biol Chem. (2016) 291:25764–75. doi: 10.1074/jbc.M116.758813
85. Boettner B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol. (2009) 21:684–93. doi: 10.1016/j.ceb.2009.06.004
86. Boras M, Volmering S, Bokemeyer A, Rossaint J, Block H, Bardel B, et al. Skap2 is required for beta2 integrin-mediated neutrophil recruitment and functions. J Exp Med. (2017) 214:851–74. doi: 10.1084/jem.20160647
87. Li N, Yang H, Wang M, Lu S, Zhang Y, Long M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol Biol Cell (2018) 29:408–18. doi: 10.1091/mbc.E16-12-0827
88. Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. (2002) 3:151–8. doi: 10.1038/ni755
89. Neelamegham S, Taylor AD, Hellums JD, Dembo M, Smith CW, Simon SI. Modeling the reversible kinetics of neutrophil aggregation under hydrodynamic shear. Biophys J. (1997) 72:1527–40. doi: 10.1016/S0006-3495(97)78801-5
90. Evans E, Kinoshita K, Simon S, Leung A. Long-lived, high-strength states of ICAM-1 bonds to beta2 integrin, I: lifetimes of bonds to recombinant alphaLbeta2 under force. Biophys J. (2010) 98:1458–66. doi: 10.1016/j.bpj.2009.09.067
91. Kinoshita K, Leung A, Simon S, Evans E. Long-lived, high-strength states of ICAM-1 bonds to beta2 integrin, II: lifetimes of LFA-1 bonds under force in leukocyte signaling. Biophys J. (2010) 98:1467–75. doi: 10.1016/j.bpj.2009.12.4316
92. Chen W, Lou J, Zhu C. Forcing switch from short- to intermediate- and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem. (2010) 285:35967–78. doi: 10.1074/jbc.M110.155770
93. Chen J, Yang W, Kim M, Carman CV, Springer TA. Regulation of outside-in signaling and affinity by the beta2 I domain of integrin alphaLbeta2. Proc Natl Acad Sci USA. (2006) 103:13062–7. doi: 10.1073/pnas.0605666103
94. Manevich-Mendelson E, Feigelson SW, Pasvolsky R, Aker M, Grabovsky V, Shulman Z, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood (2009) 114:2344–53. doi: 10.1182/blood-2009-04-218636
95. Feigelson SW, Grabovsky V, Manevich-Mendelson E, Pasvolsky R, Shulman Z, Shinder V, et al. Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood (2011) 117:7042–52. doi: 10.1182/blood-2010-12-322859
96. Hart R, Stanley P, Chakravarty P, Hogg N. The kindlin 3 pleckstrin homology domain has an essential role in lymphocyte function-associated antigen 1 (LFA-1) integrin-mediated B cell adhesion and migration. J Biol Chem. (2013) 288:14852–62. doi: 10.1074/jbc.M112.434621
97. Rognoni E, Ruppert R, Fassler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. (2016) 129:17–27. doi: 10.1242/jcs.161190
98. Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science (2009) 324:895–9. doi: 10.1126/science.1163865
99. Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. (1999) 274:28071–4. doi: 10.1074/jbc.274.40.28071
100. Goksoy E, Ma YQ, Wang X, Kong X, Perera D, Plow EF, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell (2008) 31:124–33. doi: 10.1016/j.molcel.2008.06.011
101. Klapproth S, Moretti FA, Zeiler M, Ruppert R, Breithaupt U, Mueller S, et al. Minimal amounts of kindlin-3 suffice for basal platelet and leukocyte functions in mice. Blood (2015) 126:2592–600. doi: 10.1182/blood-2015-04-639310
102. Bledzka K, Bialkowska K, Nie H, Qin J, Byzova T, Wu C, et al. Tyrosine phosphorylation of integrin beta3 regulates kindlin-2 binding and integrin activation. J Biol Chem. (2010) 285:30370–4. doi: 10.1074/jbc.C110.134247
103. Takala H, Nurminen E, Nurmi SM, Aatonen M, Strandin T, Takatalo M, et al. Beta2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood (2008) 112:1853–62. doi: 10.1182/blood-2007-12-127795
104. Lim J, Hotchin NA. Signalling mechanisms of the leukocyte integrin alphaMbeta2: current and future perspectives. Biol Cell (2012) 104:631–40. doi: 10.1111/boc.201200013
105. Zhang W, Zong CS, Hermanto U, Lopez-Bergami P, Ronai Z, Wang LH. RACK1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol Cell Biol. (2006) 26:413–24. doi: 10.1128/MCB.26.2.413-424.2006
106. Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun Signal. (2011) 9:22. doi: 10.1186/1478-811X-9-22
107. Marks PW, Maxfield FR. Local and global changes in cytosolic free calcium in neutrophils during chemotaxis and phagocytosis. Cell Calcium (1990) 11:181–90. doi: 10.1016/0143-4160(90)90069-7
108. Jaconi ME, Theler JM, Schlegel W, Appel RD, Wright SD, Lew PD. Multiple elevations of cytosolic-free Ca2+ in human neutrophils: initiation by adherence receptors of the integrin family. J Cell Biol. (1991) 112:1249–57. doi: 10.1083/jcb.112.6.1249
109. Hellberg C, Eierman D, Sjolander A, Andersson T. The Ca2+ signaling capacity of the beta 2-integrin on HL60-granulocytic cells is abrogated following phosphorylation of its CD18-chain: relation to impaired protein tyrosine phosphorylation. Exp Cell Res. (1995) 217:140–8. doi: 10.1006/excr.1995.1073
110. Dewitt S, Francis RJ, Hallett MB. Ca2? and calpain control membrane expansion during the rapid cell spreading of neutrophils. J Cell Sci. (2013) 126(Pt 20):4627–35. doi: 10.1242/jcs.124917
111. Woodard GE, Lopez JJ, Jardin I, Salido GM, Rosado JA. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J Biol Chem. (2010) 285:8045–53. doi: 10.1074/jbc.M109.033605
112. Hellberg C, Molony L, Zheng L, Andersson T. Ca2+ signalling mechanisms of the beta 2 integrin on neutrophils: involvement of phospholipase C gamma 2 and Ins(1,4,5)P3. Biochem J. (1996) 317:403–9.
113. Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. (2009) 10:21–7. doi: 10.1038/ni.f.220
114. Beliveau E, Lessard V, Guillemette G. STIM1 positively regulates the Ca2+ release activity of the inositol 1,4,5-trisphosphate receptor in bovine aortic endothelial cells. PLoS ONE (2014) 9:e114718. doi: 10.1371/journal.pone.0114718
115. Le Borgne M, Raju S, Zinselmeyer BH, Le VT, Li J, Wang Y, et al. Real-time analysis of calcium signals during the early phase of T cell activation using a genetically encoded calcium biosensor. J Immunol. (2016) 196:1471–9. doi: 10.4049/jimmunol.1502414
116. Greenberg ML, Yu Y, Leverrier S, Zhang SL, Parker I, Cahalan MD. Orai1 function is essential for T cell homing to lymph nodes. J Immunol. (2013) 190:3197–206. doi: 10.4049/jimmunol.1202212
117. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell (2003) 112:453–65. doi: 10.1016/S0092-8674(03)00120-X
118. Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. (2007) 9:1110–21. doi: 10.1038/ncb1007-1110
119. Joseph N, Reicher B, Barda-Saad M. The calcium feedback loop and T cell activation: how cytoskeleton networks control intracellular calcium flux. Biochim Biophys Acta (2014) 1838:557–68. doi: 10.1016/j.bbamem.2013.07.009
120. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi: 10.1016/j.immuni.2006.04.010
121. Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, Hu Y, et al. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity (2006a) 25:285–95. doi: 10.1016/j.immuni.2006.06.014
122. Howard T, Chaponnier C, Yin H, Stossel T. Gelsolin-actin interaction and actin polymerization in human neutrophils. J Cell Biol. (1990) 110:1983–91. doi: 10.1083/jcb.110.6.1983
123. Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein. J Biol Chem. (1999) 274:33179–82. doi: 10.1074/jbc.274.47.33179
124. Kiselar JG, Janmey PA, Almo SC, Chance MR. Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci USA. (2003) 100:3942–7. doi: 10.1073/pnas.0736004100
125. Nag S, Ma Q, Wang H, Chumnarnsilpa S, Lee WL, Larsson M, et al. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc Natl Acad Sci USA. (2009) 106:13713–8. doi: 10.1073/pnas.0812374106
126. Li GH, Arora PD, Chen Y, McCulloch CA, Liu P. Multifunctional roles of gelsolin in health and diseases. Med Res Rev. (2012) 32:999–1025. doi: 10.1002/med.20231
127. Simon SI, Schmid-Schonbein GW. Kinematics of cytoplasmic deformation in neutrophils during active motion. J Biomech Eng. (1990) 112:303–10. doi: 10.1115/1.2891188
Keywords: neutrophil recruitment, mechanosignaling, selectin, integrin, outside-in signaling
Citation: Morikis VA and Simon SI (2018) Neutrophil Mechanosignaling Promotes Integrin Engagement With Endothelial Cells and Motility Within Inflamed Vessels. Front. Immunol. 9:2774. doi: 10.3389/fimmu.2018.02774
Received: 03 October 2018; Accepted: 12 November 2018;
Published: 28 November 2018.
Edited by:
Andres Hidalgo, Centro Nacional de Investigaciones Cardiovasculares (CNIC), SpainReviewed by:
Ronen Alon, Weizmann Institute of Science, IsraelCopyright © 2018 Morikis and Simon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott I. Simon, c2lzaW1vbkB1Y2RhdmlzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.