 Alexia DumasDan CorralAndré ColomFlorence Levillain
Alexia DumasDan CorralAndré ColomFlorence Levillain Antonio Peixoto
Antonio Peixoto Denis HudrisierYannick Poquet†
Denis HudrisierYannick Poquet† Olivier Neyrolles*†
Olivier Neyrolles*†- Institut de Pharmacologie et de Biologie Structurale, IPBS, Université de Toulouse, CNRS, UPS, Toulouse, France
Tuberculosis (TB), caused by the airborne bacterial pathogen Mycobacterium tuberculosis, remains a major source of morbidity and mortality worldwide. So far, the study of host-pathogen interactions in TB has mostly focused on the physiology and virulence of the pathogen, as well as, on the various innate and adaptive immune compartments of the host. Microbial organisms endogenous to our body, the so-called microbiota, interact not only with invading pathogens, but also with our immune system. Yet, the impact of the microbiota on host defense against M. tuberculosis remains poorly understood. In order to address this question, we adapted a robust and reproducible mouse model of microbial dysbiosis based on a combination of wide-spectrum antibiotics. We found that microbiota dysbiosis resulted in an increased early colonization of the lungs by M. tuberculosis during the first week of infection, correlating with an altered diversity of the gut microbiota during this time period. At the cellular level, no significant difference in the recruitment of conventional myeloid cells, including macrophages, dendritic cells and neutrophils, to the lungs could be detected during the first week of infection between microbiota-competent and -deficient mice. At the molecular level, microbiota depletion did not impact the global production of pro-inflammatory cytokines, such as interferon (IFN)γ, tumor necrosis factor (TNF)α and interleukin (IL)-1β in the lungs. Strikingly, a reduced number of mucosal-associated invariant T (MAIT) cells, a population of innate-like lymphocytes whose development is known to depend on the host microbiota, was observed in the lungs of the antibiotics-treated animals after 1week of infection. These cells produced less IL-17A in antibiotics-treated mice. Notably, dysbiosis correction through the inoculation of a complex microbiota in antibiotics-treated animals reversed these phenotypes and improved the ability of MAIT cells to proliferate. Altogether, our results demonstrate that the host microbiota contributes to early protection of lung colonization by M. tuberculosis, possibly through sustaining the function(s) of MAIT cells. Our study calls for a better understanding of the impact of the microbiota on host-pathogen interactions in TB. Ultimately, this study may help to develop novel therapeutic approaches based on the use of beneficial microbes, or components thereof, to boost anti-mycobacterial immunity.
Introduction
Tuberculosis (TB) remains a major public health issue, with over 10 million cases each year and 1.7 million deaths in 2016, according to the World Health Organization (WHO Annual report 2017). Understanding the host factors that modulate susceptibility or resistance to Mycobacterium tuberculosis, the etiological agent of TB, might help develop novel intervention strategies, including a more efficacious alternative to the Bacillus Calmette-Guérin (BCG) vaccine, as well as, host-directed therapies (1).
A key parameter controlling susceptibility to TB is the balance between the virulence of the infecting M. tuberculosis strain, and the immune status of the infected host. To date, the parameters involved in this balance mostly included the host genetic background, gender, age, nutrition status, and the occurrence of human immunodeficiency virus (HIV) co-infection or diabetes co-morbidity (2). The host microbiota, which represents the entirety of the microbial communities present on the skin and the mucosal surfaces of the body, might constitute an important but yet poorly explored additional factor influencing M. tuberculosis interaction with its host, and susceptibility to TB (3, 4).
The microbiota includes mostly bacteria, and profoundly influences human health and disease (5, 6). In mice, some evidence demonstrate its beneficial role, including resistance to infection by pathogenic microorganisms (7, 8). For example, segmented filamentous bacteria (SFB) present in the gut are sufficient to induce the accumulation of T helper (Th)17 cells in the lamina propria and to protect against infection by intestinal pathogens, such as Citrobacter rodentium (9). SFB were also reported to promote host resistance to pathogens at distal tissue sites, including the lung (10), which is now commonly referred to as the “gut-lung axis” (11, 12). In addition, the host microbiota is known to promote tolerance and immune homeostasis, such as through sustaining the development of FoxP3+ regulatory T cells (Tregs) in the colon (13, 14). Although the microbiota predominates in the gut, in which it has been mostly studied, it is present in all other mucosal surfaces of the body. The lung is no exception and it has become increasingly apparent over the past years that, although considered sterile for decades, the mammalian respiratory tract harbors a genuine microbiota as well (15–17). Yet, the exact role that these microbial communities play in susceptibility or resistance to respiratory diseases still remains largely enigmatic.
In TB, variations in the composition of the host intestinal and lung microbiota have been reported in various settings, including in patients and in animal models (18–20). However, from a functional viewpoint, whether the host microbiota contributes to resistance or susceptibility to TB, and how it does so, is still poorly understood. In particular, it is not known whether the microbiota modulates lung immunity in response to M. tuberculosis infection; a question that we have addressed in the present study.
To explore the role of the microbiota in physiological processes, including immunity, two mouse models are classically used, namely germ-Free (GF) mice, which are born and raised in sterile conditions, and mice treated with various cocktails of broad-spectrum antibiotics. Because the microbiota is involved in the education of the immune system, particularly during infancy, it is well-known that GF mice display abnormal immune functions, such as an immature and underdeveloped lymphoid system (21). This is not the case in antibiotics-treated mice, which are only transiently depleted of their microbiota and possess a mature immune system (22).
Here, we adapted an antibiotic treatment-based model of microbiota dysbiosis (23) in order to study the role of the microbiota on host susceptibility to M. tuberculosis infection and anti-mycobacterial immunity. We found that dysbiosis following antibiotic treatment strongly altered the diversity of the gut microbiota, but not that of the lung community, and increased early lung colonization by the TB bacillus. These phenotypes were reversed after microbial reconstitution of antibiotics-treated mice through fecal transplantation (FT). Strikingly, impaired protection against M. tuberculosis infection in antibiotics-treated mice correlated with a decreased number of lung mucosal associated invariant T (MAIT) cells, a population of innate-like lymphocytes associated with protection against pulmonary pathogens (24, 25) and whose development is known to be microbiota-dependent (26). Moreover, MAIT cells in antibiotics-treated mice produced less interleukin (IL)-17A, and MAIT cells from FT mice proliferated more than those from antibiotics-treated mice following infection.
Altogether, our results reveal that the presence of a healthy microbiota promotes host resistance to early colonization by M. tuberculosis, possibly through sustaining the function(s) of MAIT lymphocytes.
Materials and Methods
Animal Models of Dysbiosis, Microbiota Restoration and M. tuberculosis Infection
Six-to-eight week-old female C57BL/6 mice were purchased from Charles River Laboratories. Mice were given a combination of broad-spectrum antibiotics ad libitum in drinking water. Mice were given ampicillin (1 g/L, Sigma Aldrich), vancomycin (500 mg/L, Alfa Aesar) and neomycin sulfate (1 g/L, Fisher) during 2 weeks, and this treatment was complemented with metronidazole during the third (0.5 g/L, Alfa Aesar) and fourth (1 g/L) weeks. Antibiotics-containing water was changed twice a week, and treatment was stopped 2 days before M. tuberculosis infection or fecal transplantation. Microbial depletion was confirmed by checking for the absence of live microorganisms in the feces using the colorimetric Releasat biological indicator (Mesa Labs) at 37°C, in either aerobic or anaerobic conditions. Following microbial depletion, mice were maintained in a sterile isolator during all the experiment. Recolonization with a complex microbiota was performed through a single gavage with the intestinal content from two non-treated mice. Repopulation occurred after 1 week, as assessed using the colorimetric Releasat biological indicator. Control mice received normal water during all experiment.
M. tuberculosis (H37Rv) was grown in 7H9 liquid medium (Difco) supplemented with ADC (Middlebrook) and tyloxapol 0.05%. For infection, mice were anesthetized using isoflurane and infected intranasally with 103 colony-forming units (CFUs) of M. tuberculosis.
Preparation of Lungs Homogenates for Microbiological, Molecular, and Immunological Analyses
Mice were sacrificed, lungs were harvested aseptically, homogenized using a gentleMACS dissociator (C Tubes, Miltenyi), and incubated with DNAse I (0.1 mg/mL, Roche) and collagenase D (2 mg/mL, Roche) during 30 min at 37°C under 5% CO2. M. tuberculosis bacterial load was determined by plating serial dilutions of the lung homogenates onto 7H11 solid medium (Difco) supplemented with OADC (Middlebrook). The plates were incubated at 37°C for 3 weeks before bacterial CFUs scoring. Lungs homogenates were filtered on 40 μm cell strainers and centrifuged at 329 × g during 5 min. Supernatants were conserved for cytokine content analysis. A part of the cellular pellet was conserved in TRIzol reagent for cellular RNA analysis. In the remaining fraction, red blood cells were lysed in 150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA (pH 7.2) for immunological staining. For microbial composition analysis, lungs were harvested aseptically, homogenized using a gentleMACS dissociator (M Tubes, Miltenyi), and filtered on 40 μm cell strainers.
In vitro Stimulation, Antibody Staining, and Flow Cytometry Analysis
Single-cell suspensions from lungs homogenates were prepared as described above. In case of intracellular staining of lymphocytes, cytokine production was stimulated in RPMI (Difco) supplemented with phorbol myristate acetate (PMA) and ionomycin (50 ng/mL and 1 μg/mL, respectively, Sigma Aldrich), and blocked with brefeldin A and monensin (5 μg/mL, Becton Dickinson) for 4 h at 37°C under 5% CO2. In case of intracellular staining of myeloid cells, cytokine production was stimulated in RPMI (Difco) supplemented with lipopolysaccharide (LPS) (1 μg/mL, Sigma Aldrich), and blocked with brefeldin A (5 μg/mL, Becton Dickinson) for 4 h at 37°C under 5% CO2. Cells were stained with Zombie Aqua Viability for dead cells exclusion and non-specific binding to Fc receptors was blocked by incubating the cells with anti-CD16/32 antibodies (BioLegend). For MAIT cell staining, mouse MR1-5-OP-RU tetramer labeled with PE and MR1-Ac-6-FP tetramer labeled with APC were incubated for 45 min at room temperature in the dark. The MR1 tetramer was obtained from the NIH Tetramer Core Facility. For extracellular staining, cells were incubated with a panel of surface antibodies for 20 min at 4°C in the dark. For intracellular staining, cells were fixed and permeabilized with Fixation and Permeabilization Reagents (eBioscience) and subsequently stained with the antibodies of interest for 40 min at room temperature in the dark. All antibodies used are referenced in Supplementary Table 1. Cell staining was analyzed using LSR II or LSR Fortessa flow cytometers (BD) and FlowJo software version V10. Cells were first gated on singlets (FSC-H vs. FSC-W and SSC-H vs. SSC-W) and live cells before further analyses. An average of 10–25,000 alveolar macrophages were sorted using a FACSAria Fusion cytometer. Sorted cells were incubated in TRIzol reagent to isolate RNA.
Bacterial DNA and Cellular RNA Isolation, and Real Time-qPCR
DNA from fecal samples and lungs homogenates were extracted according to manufacturer's instructions (QIAamp Fast DNA Stool Mini Kit, Qiagen). DNA was extracted from the fecal samples, using lab tissue-specific technique. The 16S rDNA present in the samples was measured by Real Time-qPCR (RT-qPCR) in triplicate and normalized using a plasmid-based standard scale. The amount of bacterial DNA was assessed using the “Universal 16S Real Time qPCR” workflow established by Vaiomer (Vaiomer SAS, Labège, France). In the case of detection of bacterial phylums from lungs and feces DNA, RT-qPCR was performed using phylum-specific primers [Supplementary Table 2, (8, 27)] using TB green Premix Ex Taq (Takara), according to the manufacturer's protocol. All RT-qPCR reactions were carried out using a 7,500 RT-PCR System, and data were analyzed using the 7,500 Software version v2.3 (Applied Biosystems). Values were normalized using the universal 16S rRNA gene, and expressed as a fold change in experimental samples (antibiotics-treated mice or reconstituted mice) relative to control samples (non-treated mice). RNA from lungs homogenates and sorted cells were extracted using TRIzol reagent (Ambion) and RNeasy spin columns according to manufacturer's instructions (RNeasy kit, Qiagen). RNA were reverse transcribed into cDNA using M-MLV Reverse transcriptase (Invitrogen). RT-qPCR was performed using gene-targeted primers (Supplementary Table 2) as described above. Values were normalized using the Hprt housekeeping gene, and expressed as a fold change in experimental samples (antibiotics-treated mice) relative to control samples (non-treated mice).
Cytokine Quantification
Cytokines present in the supernatants from lung homogenates were quantified using a customized LEGENDplex™ Mouse Inflammation Panel and a LSRII flow cytometer, using manufacturer's instructions (BioLegend). CCL20 was detected by ELISA (Invitrogen), according to the manufacturer's instructions.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 software. An Agostino and Pearson normality test was performed to determine whether samples followed a normal distribution. Data following a normal distribution are represented as mean ± SD; data following a non-normal distribution were represented as median with interquartile range. Unpaired t-test (for normal data) or Mann-Whitney (for non-normal data) were performed when two samples were compared; ANOVA (for normal data) or Kruskal-Wallis (for non-normal data) tests were performed when more than two samples were compared. For all analyses, * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, and **** indicates P < 0.0001. Data obtained from FT mice were included in the figures only when statistical difference between antibiotics-treated and control mice were detected.
Results
Microbiota Dysbiosis Increases Early Lung Colonization by M. tuberculosis
In order to assess whether the microbiota is involved in the control of M. tuberculosis infection, we adapted a model of microbiota dysbiosis consisting in the administration of a cocktail of broad-spectrum antibiotics composed of ampicillin, neomycin sulfate, metronidazole, and vancomycin over the course of 4 weeks. This procedure has previously been shown to deplete all detectable commensals and bacterial products (23). We observed that adding metronidazole at the start of antibiotic treatment resulted in animal morbidity and mortality, presumably due to dehydration caused by a reluctance of the mice to drink metronidazole-containing water (28). Thus, the protocol was modified and metronidazole was added only during the last 2 weeks of antibiotic treatment (Figure 1A). In this way, mice did not suffer from dehydration and did not lose weight during the course of treatment (Figure 1B). Microbial depletion before M. tuberculosis infection was confirmed by quantification of total 16S rDNA in feces (Figure 1C) and by inoculation of fecal samples in reporter liquid medium incubated under aerobic (Figure 1D) or anaerobic (data not shown) conditions. Bacterial diversity was analyzed at the phylum level in control and antibiotics-treated (ABX) mice. Bacteroidetes and Firmicutes, the major phylums present in the gut microbiota (29) were strongly reduced, while Beta-proteobacteria were increased in the gut of ABX mice, compared to control animals (Figure 1E). An increase in Proteobacteria is known to be associated with gut inflammation and a bad prognostic during intestinal diseases (30, 31). We also verified if the lung community could be altered by antibiotics treatment. Colorimetric analyses did not reveal the presence of viable bacteria in the broncho-alveolar lavages (BALs) of normal, specific pathogen-free (SPF) mice (data not shown). A similar trend was observed in the lungs for the major phyla (17, 32), although statistical significance was reached only for Proteobacteria (Figure 1F).
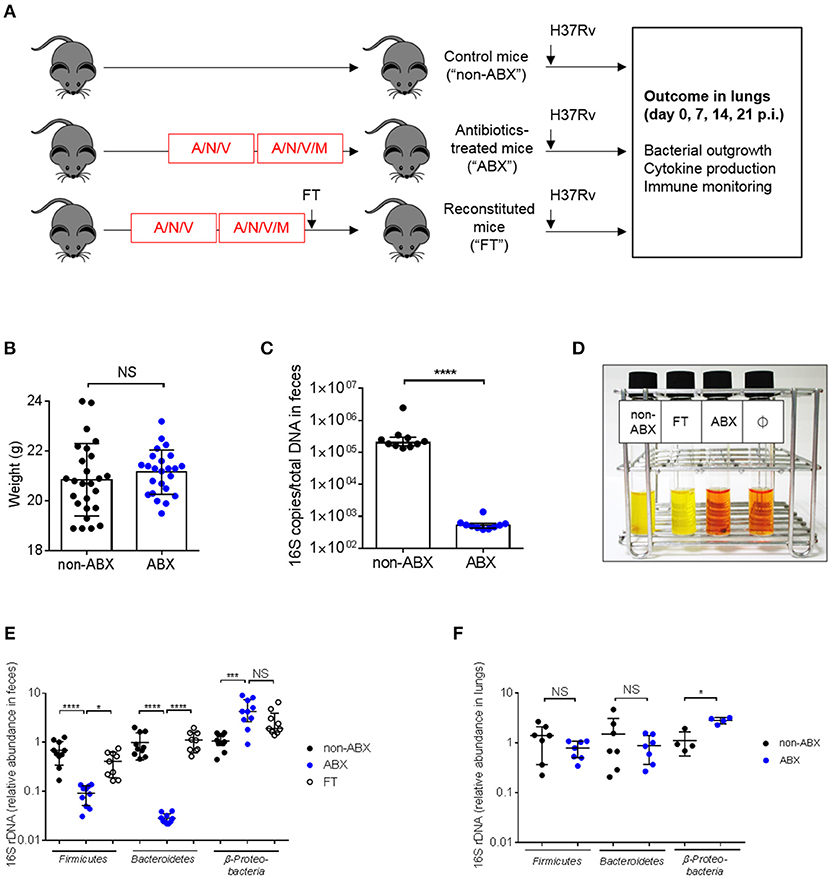
Figure 1. Mouse model to study the impact of microbiota dysbiosis during M. tuberculosis infection. (A) Experimental design. Six-to-eight weeks old C57BL/6 female mice were treated for 4 weeks with a cocktail of broad-spectrum antibiotics (ampicillin, A, neomycin sulfate, N, vancomycin, V, and metronidazole, M, which was added during the last 2 weeks) in drinking water (ABX) or were given water without antibiotics (non-ABX). Microbiota reconstitution was realized through fecal transplantation (FT) after a single oral gavage with the intestinal content from 2 control mice and occurred during 1 week. The three groups of mice received an intranasal challenge of M. tuberculosis H37Rv (103 CFUs/mouse). Bacterial outgrowth, cytokine production and immune monitoring were determined 0, 7, 14, or 21 days post-infection (p.i.) in the lungs. (B) Mouse weight in the ABX vs. non-ABX groups at the time of M. tuberculosis infection. Data from 4 independent experiments (n = 4–14 mice/group/experiment) were pooled and the graph represent mean ± SD of the pooled data. Data were analyzed using the unpaired Student's t-test. (C) The amount of bacterial rDNA was assessed using the “Universal 16S RT-qPCR” kit, calculated as 16S rDNA gene copies per total DNA extracted from feces of non-ABX and ABX groups before infection. (D) Colorimetric evaluation of the presence of microbial flora in the feces of the different groups of animals before infection; Ø indicates negative control. Yellow color indicates the presence of live microorganisms. Image depicted in (D) is representative of 3 independent experiments. Fecal (E) and lung (F) microbiota were detected by RT-qPCR using phylum-specific primers. Relative Ct value compared to universal 16S rRNA gene Ct value (ΔCt) and the mean of ΔCt values in the control group (ΔΔCt). (E, F) data from 2–3 independent experiments (n = 2–4 mice/group/experiment) were pooled. The graph (E) show mean ± SD (Firmicutes and Bacteroidetes) and median with interquartile range (Proteobacteria) of the pooled data. Data were analyzed using ANOVA (Firmicutes and Bacteroidetes) and the Kruskal-Wallis test (Proteobacteria). The graph (F) show median with interquartile range of the pooled data. Data were analyzed using the Mann-Whitney test. NS, not significant; *p < 0.05; ***p < 0.001; ****p < 0.0001.
In order to confirm the direct contribution of the host microbiota to the observed phenotypes, we generated groups of ABX mice reconstituted with the intestinal content from untreated control mice through fecal transplantation (FT). Some features of normal mice were restored in ABX mice after FT (Figures 1D,E). In particular, the diversity of the major phylums (e.g., Bacteroidetes and Firmicutes) was similar between control and FT mice (Figure 1E).
We next verified that the antibiotic treatment did not alter the level and functionality of several lung immune cell populations at the steady state (Supplementary Figure 1). The total numbers of macrophages, dendritic cells, neutrophils, natural killer (NK), CD4 T lymphocytes, and B cells were quantified by flow cytometry. We found no difference in the number of these cells (Supplementary Figure 1A), and in the ability of macrophages and CD4 T cells to produce cytokines (Supplementary Figure 1B) between control and ABX mice.
Control (non-ABX) and ABX mice were infected intranasally with M. tuberculosis H37Rv 2 days after cessation of antibiotics treatment, and the lungs of the infected animals were collected 7, 14, and 21 days after infection, homogenized and plated onto agar medium for colony-forming unit (CFUs) scoring. Compared to control mice, ABX mice contained about twice as many bacteria in their lungs at day 7 post-infection (p.i., Figure 2A). This phenotype was only transient since there was no difference in the lung bacterial load between control and ABX mice after 14 or 21 days p.i. (Figure 2B); similarly no differences in bacterial loads were observed in the spleen of control vs. ABX animals 21 days p.i. (data not shown). This phenotype correlated with a reduced amount of total gut microbial rDNA in ABX mice 7 days p.i, while this difference was abrogated 21 days p.i. (Figure 2C), indicating that ABX mice were recolonized following cessation of the antibiotic treatment. Yet, microbial recolonization over time in ABX mice was only partial, which correlated with the presence of an enlarged caecum in ABX mice, as previously observed in GF mice (21) (Figure 2D). The relative abundance of Firmicutes was no more different in infected ABX and control mice 7 days p.i., however the relative abundance of Bacteroidetes and Proteobacteria was still altered in ABX animals at this time point (Figure 2E), which likely reflected ongoing recolonization.
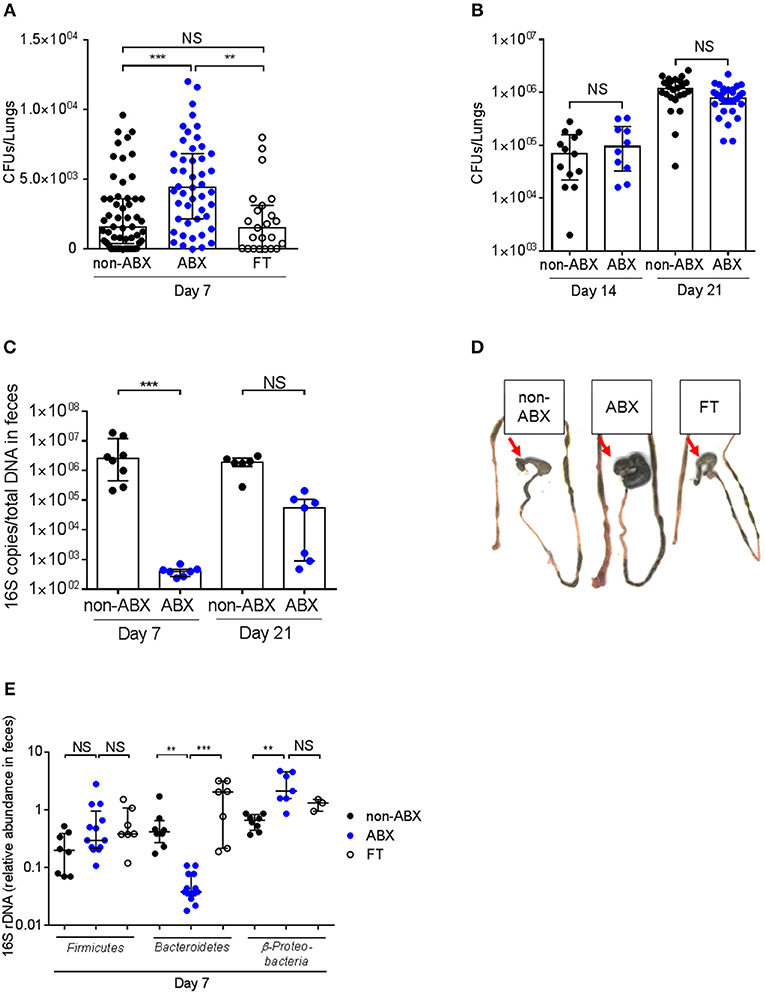
Figure 2. The host microbiota protects against early lung colonization by M. tuberculosis. (A) M. tuberculosis bacterial load (CFUs) was measured in the lungs of control (non-ABX), ABX and FT mice 7 days p.i. (B) M. tuberculosis bacterial load was measured in the lungs of control (non-ABX) and ABX mice 14 or 21 days p.i., Data from 3–6 (A) or 2 (B) independent experiments (n = 4–8 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data. Data were analyzed using the Kruskal-Wallis test. (C) The amount of bacterial rDNA was assessed using the “Universal 16S RT-qPCR” kit, calculated as 16S rDNA gene copies per total DNA extracted from feces of non-ABX, ABX, and FT groups 7 days p.i. (D) Morphology of the caecum in the non-ABX, ABX, and FT groups 7 days p.i., Red arrow indicates the caecum. Image depicted in (D) is representative of 3 independent experiments. (E) Fecal microbiota from non-ABX, ABX and FT groups was analyzed by RT-qPCR using phylum-specific primers 7 days p.i., Data were analyzed as in Figure 1. (C,E) Data from 1–4 independent experiments (n = 2–5 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data. Data were analyzed using the Kruskal-Wallis test. NS, not significant; **p < 0.01; ***p < 0.001.
Reintroduction of a complex microbiota (FT) restored all the examined phenotypes, i.e., prevented the increased lung colonization by M. tuberculosis 7 days p.i. (Figure 2A), caecum enlargement (Figure 2C), and restored, at least partially, microbial diversity (Figure 2E).
Altogether, these data demonstrate that the host microbiota contributes to early resistance against lung colonization by M. tuberculosis.
Microbiota Dysbiosis Correlates With Impaired Functions of MAIT Cells During Early Lung Colonization by M. tuberculosis
In order to understand the increased susceptibility of ABX mice to M. tuberculosis, we next quantified the early expression and production of several key inflammatory factors known to be involved in immunity to TB (33). Seven days after M. tuberculosis infection, we found no difference in the mRNA expression and/or protein production of TNFα, IL-1β, IL-6, transforming growth factor (TGF)β, interferon (IFN)γ, IL-17A and IL-12 (p70) in the lungs of ABX vs. control mice (Supplementary Figure 2). We also quantified the expression of cathelicidin-related antimicrobial peptide (CRAMP), whose production is known to be stimulated by the microbiota (34), and found no difference between ABX and control mice (Supplementary Figure 2A).
We next analyzed the early accumulation of innate myeloid and lymphoid cell populations in the lungs of infected ABX and control mice by flow cytometry. We found no difference in the number of macrophages, dendritic cells, neutrophils and natural killer (NK) cells between the two groups of animals 7 days p.i. (Supplementary Figure 3). Macrophages are known to be the main host cell target for M. tuberculosis, and are able to control mycobacterial proliferation upon activation by inflammatory cytokines, including TNFα (33). In particular, alveolar macrophages (AMs) were recently reported to sustain mycobacterial proliferation while interstitial macrophages (IMs) are more bactericidal (35). Here we found no difference in the number of AMs and IMs between ABX and control mice 7 days p.i. (Figures 3A–C); however after sorting the AM population, we found that these cells expressed less TNFα mRNA in ABX mice, as compared to in control animals (Figure 3D, left panel). Yet, the production of TNFα by AMs in ABX and control mice was similar at the protein level, as assessed by flow cytometry (Figure 3D, right panel); similar results were obtained in IMs (data not shown).
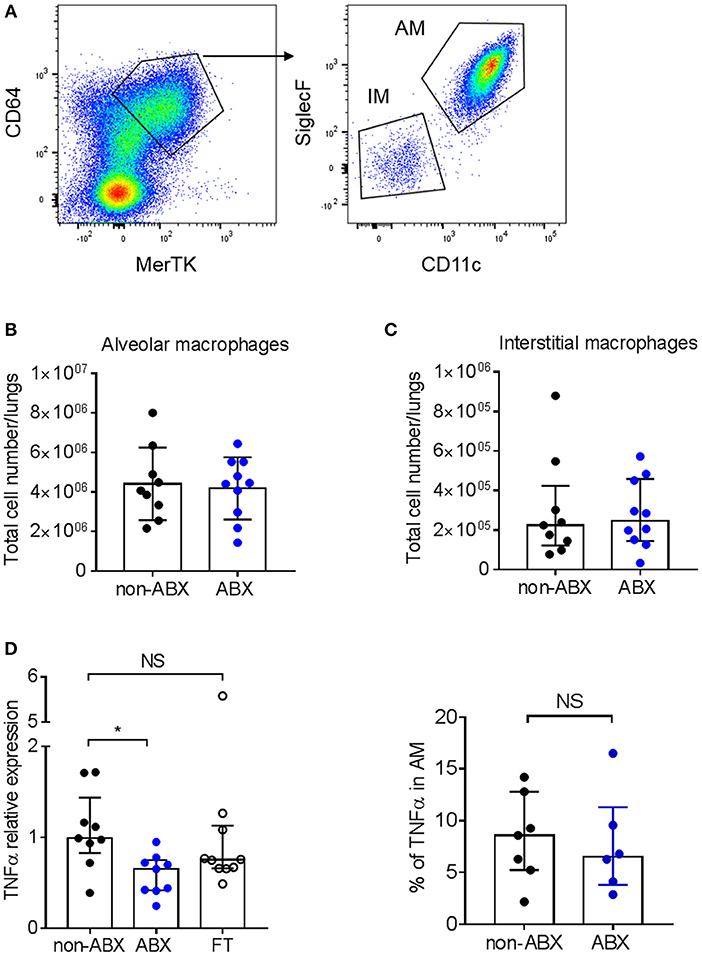
Figure 3. Microbiota dysbiosis modify TNFα mRNA expression but not protein production by alveolar macrophages. (A) Gating strategy to analyze alveolar (MerTK+CD64+SiglecF+CD11c+) and interstitial (MerTK+CD64+SiglecF−CD11c−) macrophages by flow cytometry. The total number of (B) alveolar and (C) interstitial macrophages was quantified by flow-cytometry in lung homogenates from M. tuberculosis-infected ABX vs. non-ABX mice 7 days p.i., Data from 2 independent experiments (n = 4–5 mice/group/experiment) were pooled and the graphs show mean ± SD (B) or median with interquartile range (C). Data were analyzed using the Student's t-test (B) or the Mann-Whitney test (C). (D) Left panel, MerTK+CD64+SiglecF+CD11c+ alveolar macrophages were sorted and the expression of TNFα was measured by RT-qPCR in cells from non-ABX, ABX, or FT mice 7 days p.i., Gene expression represents relative Ct value compared to Hprt Ct value (ΔCt) and the mean of ΔCt values in the control group (ΔΔCt). Right panel, cytometry analysis of the percentage of TNFα-producing MerTK+CD64+SiglecF+CD11c+ alveolar macrophages following in vitro stimulation by LPS. Data from 1–2 independent experiments (n = 4–5 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data and were analyzed using the Mann-Whitney test *p < 0.05.
Since the microbiota is known to play a part in the effector functions of innate-like lymphocytes (36), we next explored the accumulation of NKT cells, γ/δ T cells and MAIT cells in the lungs of ABX and control mice during M. tuberculosis infection. The total number and percentage of NKT (NK1.1+CD3+) and γ/δ T (TCRγ/δ+CD3+) lymphocytes were similar in the lungs of infected ABX and control mice (Supplementary Figure 4). Strikingly, the only difference we observed between the two groups of mice was related to the accumulation of mucosal-associated invariant T (MAIT) cells, characterized by their MR1-5-OP-RU tetramer+TCRβ+ phenotype (Figure 4A), with a marked decrease in the total number and percentage of these cells in ABX mice, compared to in control mice 7 days p.i. (Figures 4B,C). This phenotype was reversed in FT mice (Figures 4B,C) and was no longer observed at later time points p.i. (Figure 4B), in line with our findings for lung bacterial loads (Figures 2A,B). Altogether, these data suggest that the host microbiota promotes early resistance to M. tuberculosis infection through sustaining the accumulation of MAIT cells in the lungs.
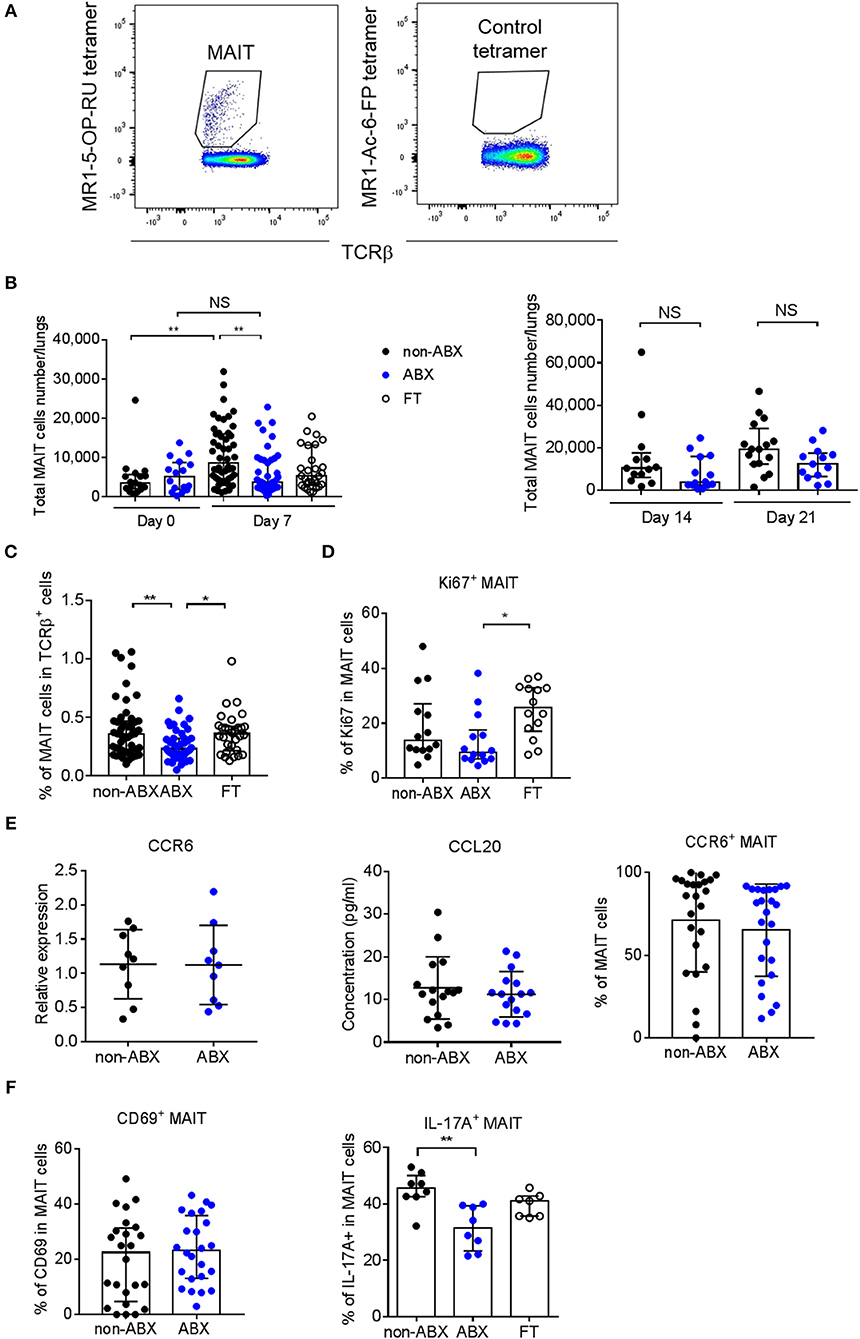
Figure 4. MAIT cell accumulation in the lungs and IL-17A-production are decreased early after M. tuberculosis infection in microbiota-altered mice. (A) Gating strategy to analyze MAIT cells (MR1-5-OP-RU tetramer+TCRβ+) by flow cytometry. A MR1–Ac-6-FP tetramer+TCRβ+ staining was used as a control. (B,C) Total number (B) and percentage (C) of MAIT cells in the lungs of control (non-ABX) ABX, and FT mice 0 and 7 (B, left panel), 14 and 21 (B, right panel), and 7 (C) days p.i., Data from 2–6 independent experiments (n = 4–8 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data. Data were analyzed using the Kruskal-Wallis test. (D) Intracellular analysis of Ki67 expression in MAIT cells from non-ABX, ABX, and FT mice 7 days p.i., by flow cytometry. Data from 2 independent experiments (n = 4–8 mice/group/experiment) were pooled and the graphs show the median with interquartile range of the pooled data. Data were analyzed using the Kruskal-Wallis test. (E) The expression of CCR6 (left) in whole lung homogenates or in MAIT cells (right), and the production of CCL20 in lung supernatant (middle), were measured by RT-qPCR (left), flow cytometry (right) and ELISA (middle) in ABX and control (non-ABX) mice 7 days p.i., RT-qPCR data were analyzed as in Supplementary Figure 2. Data from 3 (left) or 4 (middle and right) independent experiments (n = 3–7 mice/group/experiment) were pooled and the graphs show mean ± SD of the pooled data. Data were analyzed using the Student's t-test. (F) Cytometry analysis of percentage of activated (MR1-5-OP-RU tetramer+TCRβ+CD69+) MAIT cells (left), and IL-17A-producing (MR1-5-OP-RU tetramer+TCRβ+IL-17A+) MAIT cells (right) in lungs of control (non-ABX), ABX and FT mice 7 days p.i., Data from 4 (left) or 1 representative of 3 (right) independent experiments (n = 5–8 mice/group/experiment) were pooled and the graphs show mean ± SD of the pooled data. (left) or median with interquartile range (right) of the pooled data. Data were analyzed using the unpaired Student's t-test (left) or the Kruskal-Wallis test (right). NS, not significant; *p < 0.05; **p < 0.01.
We next sought to assess whether the decreased number of MAIT cells observed in the lungs of ABX mice reflected a diminished recruitment and/or proliferation of these cells following M. tuberculosis infection. We found that MAIT cells from ABX mice expressed less Ki67, an intracellular marker of cell cycling and proliferation, than their control or FT counterparts (Figure 4D). The tissue-homing chemokine receptor CCR6 and the chemokine CCL20 (referred to as the CCR6/CCL20 axis) are known to be involved in the recruitment of leukocytes, including T lymphocytes, from the intestine and other organs to the lungs (37, 38), and were found to be altered during microbiota dysbiosis in response to pulmonary infection (39). Because MAIT cells express CCR6 (40), we next determined if the CCR6/CCL20 axis was perturbed in ABX mice. We found no difference in global expression of the CCR6 transcript and production of the CCL20 protein in the lungs between M. tuberculosis-infected ABX and control mice 7 days p.i. (Figure 4E). Flow cytometry analysis revealed no difference in CCR6 expression in MAIT cells between the two groups of mice (Figure 4E). These data indicate that if microbiota dysbiosis impairs the early recruitment of MAIT cells to the lungs following M. tuberculosis infection, this does not rely on the CCR6/CCL20 axis. Moreover, they reveal that the proliferation of MAIT cells is reduced in the lungs of ABX mice.
Finally, we sought to evaluate whether the activation and effector functions of MAIT cells could be altered in the lungs of M. tuberculosis-infected ABX mice. Activated MAIT cells are known to express CD69 (25) and to produce various cytokines, including IL-17A (24, 41). In the mouse, lung MAIT cells mostly produce IL-17A (41). We found that although CD69 expression by MAIT cells was similar in ABX and control mice, MAIT cells from ABX animals produced less IL-17A, which was partially restored in FT mice 7 days p.i. (Figure 4F). The number of, and Ki67 expression by MAIT cells in the spleen did not differ between ABX and control animals. Altogether, our data reveal that a healthy microbiota is required for pulmonary MAIT cell functions, which correlates with an improved early control of M. tuberculosis growth in the lungs.
Discussion
Our results demonstrate that the microbiota contributes to early host defense against lung colonization by M. tuberculosis, possibly via effector functions MAIT lymphocytes. Several previous studies reported that M. tuberculosis infection results in a perturbation of the composition of the host microbiota in humans, as well as, in experimentally infected animals (18, 20, 42). However, the functional impact of the microbiota on host susceptibility or resistance to TB remains to be fully understood.
In a previous study using a different ABX model, Khan and colleagues reported that antibiotics-treated mice were more susceptible to M. tuberculosis, with a higher bacillary load in the lungs 21 days after infection, compared to control mice (43). This correlated with a decrease in IFNγ- and TNFα-producing CD4 T lymphocytes and an increase in FoxP3+ Tregs in the spleen of the infected ABX animals, compared to controls. At first glance, these results would appear to contradict our data since we did not observe any difference in lung bacterial load in ABX vs. control animals at 14 or 21 days p.i., However, in our study antibiotic treatment was stopped 2 days before M. tuberculosis infection, while in the study by Khan and colleagues antibiotics were maintained in the drinking water throughout the infection period. It is likely that in our experiments, animals became progressively recolonized following recovery of the endogenous microflora in an uncontrolled manner, and this may explain why we only observed an increased susceptibility of ABX mice at early time-points during infection. Thus, our study and that by Khan et al. do not appear to be contradictory but rather complementary, illustrating the overall protective function of the host microbiota against M. tuberculosis infection at early vs. late time points post-exposure. Altogether, these two studies might reveal a previously underappreciated influence of the gut microbiota on host resistance to the TB bacillus through the so-called “gut-lung axis” (44), as observed in other lung infections (11, 45). Another, yet non-mutually exclusive possibility, is that local commensals of the airways (15, 16), which are probably also affected by antibiotics treatment (46), might influence host resistance to TB, which will need further investigation (47). A limitation in our study is that the composition of the microbiota in ABX mice was not monitored over time and that animals were infected only 2 days after cessation of the antibiotics treatment. The microbial composition in ABX mice in the weeks following cessation of antibiotics treatment is unlikely to be similar to that of control animals, and monitoring the microbial composition over time following cessation of antibiotics treatment would be an important study to perform. Similarly, assessing the impact of various microbiota, in particular of a “healthy microbiota,” on susceptibility to TB is an important issue that is not addressed here and would require developing different animal models.
We found that unconventional T cells, namely MAIT cells, might be involved in microbiota-mediated early control of M. tuberculosis infection, e.g., through the production of IL-17A. MAIT cells form a population of innate-like lymphocytes expressing a semi-invariant T cell antigen receptor (TCR), activated by the MHCI–related molecule MR1 presenting microbial-derived metabolites (48, 49). MAIT cells preferentially reside in mucosal tissues, such as the lungs and the gut, and their development and maturation depend on the presence of the host microbiota (26, 50). This explains why the MAIT cell compartment is disturbed in several inflammatory disorders in which the gut microbiota is altered, such as inflammatory bowel disease and obesity (51, 52). MAIT cells were found to be involved in host protection from a variety of pathogens, including respiratory pathogens such as Klebsiella pneumoniae, and Francisella tularensis (24, 53). Reminiscent of our study, mice lacking MAIT cells also display an increased early susceptibility to mycobacterial infections (25, 26, 54). In humans, TB patients display a functionally impaired MAIT cell compartment (55, 56). In our study, we found that MAIT cells tend to proliferate less in ABX mice, which may rely on a reduced availability of MAIT ligands in this setting (49, 57, 58). Although we cannot exclude that the recruitment of these cells to the lung is also diminished in ABX mice, we provide evidence that the reduced accumulation of MAIT cells in the lungs of ABX mice is independent of the CCR6/CCL20 axis. Yet, MAIT cells express other receptors involved in cell migration, namely CCR5, CCR9, CXCR4, and CXCR6, at least in humans (59). The role played by these receptors, and possibly others, in microbiota-mediated recruitment of MAIT cells to the lungs during M. tuberculosis infection, will need to be investigated. We found that MAIT cells produce less IL-17A in ABX mice. The role of IL-17 in TB is still unclear, possibly depending on the cell type producing this cytokine and the time point at which it is produced during infection (33). Whether the host microbiota influences early control of M. tuberculosis through the modulation of the production of IL-17, and possibly other cytokines, and how it does so, will need to be addressed. In the mouse, MAIT cells mostly produce IL-17A (41) but their ability to control M. tuberculosis in infected macrophages does not seem to depend on this cytokine (54), suggesting that the possible protective effect of IL-17A we observed is not direct. The development of MAIT cells is known to depend on the microbiota, particularly on microbes synthesizing riboflavin (26, 60). Addressing the role of specific microbiota species in the MAIT-mediated early protection against M. tuberculosis infection would require recolonizing ABX mice with riboflavin-producers (e.g., Escherichia coli) and -non producers (e.g., Enterococcus faecalis), which will be of interest for future studies.
In conclusion, we show here that the host microbiota contributes to early resistance to M. tuberculosis infection in mice, possibly through stimulating the accumulation and effector functions of MAIT cells in the lungs. Whether these findings apply to humans will need further investigation. A significant fraction of those individuals in close contact to TB cases are known to remain tuberculin skin test (TST)-negative, and are thus thought to represent “innate controllers” (61). It is tempting to speculate that in addition to other factors, the host microbiota might contribute to the ability to control M. tuberculosis infection in an innate fashion, via MAIT cells and possibly other innate cells (62).
Ethics Statement
Animal care and experimentation were performed following the French regulation. All procedures were approved by the Ministry of Higher Education and Research (Agreement APAFIS 6497).
Author Contributions
AD, DC, DH, YP, and ON designed research. AD, DC, FL, AC, and AP performed research. AD, DC, FL, AP, DH, YP, and ON analyzed data. AD and ON wrote the paper with editorial help from DC, DH, and YP.
Funding
This work was supported by CNRS, University of Toulouse, Agence Nationale de la Recherche (www.anr.fr)/Programme d'Investissements d′Avenir (ANR-11-EQUIPEX-0003 to ON), The Ministry of Higher Education, Research, and Innovation (PhD fellowship to AD), Fondation pour la Recherche Médicale (www.frm.org, grant DEQ20160334902 to ON, and fellowship to AD), the Bettencourt-Schueller Foundation (https://www.fondationbs.org, Prix Coup d′Elan to ON).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The MR1 tetramer technology was developed jointly by Dr. James McCluskey, Dr. Jamie Rossjohn, and Dr. David Fairlie, and the material was produced by the NIH Tetramer Core Facility as permitted to be distributed by the University of Melbourne. We thank the members of the Genotoul core facilities ANEXPLO (IPBS, Toulouse), in particular F. Moreau for animal experiments, and TRI, in particular Emmanuelle Naser for cytometry analysis. We thank Benjamin Raymond for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02656/full#supplementary-material
Supplementary Figure 1. Microbiota dysbiosis does not alter immune cell populations in the lungs. (A) The total numbers of F4/80+CD11c+ macrophages, CD11c+MHCII+ dendritic cells, CD11b+GR1hi neutrophils, CD3−NK1.1+ NK cells, CD3+CD4+ T lymphocytes and CD19+ B cells were quantified by flow cytometry in lung homogenates from uninfected ABX vs. non-ABX mice. (B) Cytometry analysis of percentage of TNFα-producing F4/80+CD11c+ macrophages, TNFα-producing CD3+CD4+ T lymphocytes and IL-17A-producing CD3+CD4+ T lymphocytes in lungs from uninfected ABX vs. non-ABX mice. Data from 1–3 independent experiments (n = 4–5 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data (B cells, TNFα+ macrophages, TNFα+ CD4 T cells, IL-17A+ CD4 T cells) or mean ± SD (macrophages, dendritic cells, neutrophils, NK cells, CD4 T cells). Data were analyzed using the Mann-Whitney test (B cells, TNFα+ macrophages, TNFα+ CD4 T cells, IL-17A+ CD4 T cells) or using the unpaired Student's t-test (macrophages, dendritic cells, neutrophils, NK cells, CD4 T cells).
Supplementary Figure 2. Microbiota dysbiosis does not impact early inflammatory response to M. tuberculosis infection in lungs. (A) The expression of inflammatory cytokines and the antimicrobial peptide CRAMP was measured by RT-qPCR in the lungs of non-ABX vs. ABX mice 7 days p.i., Gene expression represents relative Ct value compared to Hprt Ct value (ΔCt) and the mean of ΔCt values in the control group (ΔΔCt). (B) The production of TNFα, IFNγ, and IL-12(p70) was measured in lung homogenates from control (non-ABX) and ABX mice 7 days p.i., Data from 2–3 independent experiments (n = 2–3 mice/group/experiment) were pooled and the graphs show mean ± SD (TNFα, TGFβ, CRAMP in A) or median with interquartile range (IL-1β, IL-6, IFNγ, IL-17A in A; TNFα, IFNγ, IL-12 in B) of the pooled data. Data were analyzed using the Student's t-test (TNFα, TGFβ, CRAMP in A) or the Mann-Whitney test (IL-1β, IL-6, IFNγ, IL-17A in A; TNFα, IFNγ, IL-12 in B).
Supplementary Figure 3. Microbiota dysbiosis does not alter innate myeloid and lymphoid cell populations during early lung colonization by M. tuberculosis. The total number of F4/80+CD11c+ macrophages, CD11c+MHCII+ dendritic cells, CD11b+GR1hi neutrophils, and CD3−NK1.1+ NK cells were quantified by flow-cytometry in lung homogenates from M. tuberculosis-infected ABX vs. non-ABX mice 7 days p.i., Data from 4 independent experiments (n = 6–8 mice/group/experiment) were pooled and the graphs show median with interquartile range of the pooled data (macrophages, neutrophils) or mean ± SD (dendritic cells, NK cells). Data were analyzed using the Mann-Whitney test (macrophages, neutrophils) or using the unpaired Student's t-test (dendritic cells, NK cells).
Supplementary Figure 4. NKT and γ/δ T cells are not modified in infected microbiota-altered mice. (A) Gating strategy to analyze unconventional innate-like lymphocytes, namely NKT cells (CD3+NK1.1+) and γ/δ T cells (CD3+TCRγ/δ+) by flow cytometry. (B,C) (B) Total NKT cells (left) and γ/δ T cells (right) and (C) percentages in the lungs of control (non-ABX) and ABX mice 7 days p.i., Data from 2 independent experiments (n = 8 mice/group/experiment) were pooled and the graphs show the mean ± SD of the pooled data. Data were analyzed using the unpaired Student's t-test.
Supplementary Table 1. Primers used in the study for RT-qPCR.
Supplementary Table 2. Antibodies used in the study for flow cytometry.
References
1. Tiberi S, Du Plessis N, Walzl G, Vjecha MJ, Rao M, Ntoumi F, et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect Dis. (2018) 18:e183–98. doi: 10.1016/S1473-3099(18)30110-5
2. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers (2016) 2:16076. doi: 10.1038/nrdp.2016.76
3. Adami AJ, Cervantes JL. The microbiome at the pulmonary alveolar niche and its role in Mycobacterium tuberculosis infection. tuberculosis (2015) 95:651–8. doi: 10.1016/j.tube.2015.07.004
4. Namasivayam S, Sher A, Glickman MS, Wipperman MF. The microbiome and tuberculosis: early evidence for cross talk. mBio (2018) 9:e01420–18. doi: 10.1128/mBio.01420-18
5. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature (2012) 489:220–30. doi: 10.1038/nature11550
6. Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. (2013) 14:676–84. doi: 10.1038/ni.2640
7. Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe (2014) 15:374–81. doi: 10.1016/j.chom.2014.02.006
8. Brown RL, Sequeira RP, Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. (2017) 8:1512. doi: 10.1038/s41467-017-01803-x
9. Ivanov Ii, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
10. Gauguet S, D'ortona S, Ahnger-Pier K, Duan B, Surana NK, Lu R, et al. Intestinal microbiota of mice influences resistance to Staphylococcus aureus pneumonia. Infect Immun. (2015) 83:4003–14. doi: 10.1128/IAI.00037-15
11. Schuijt TJ, Lankelma JM, Scicluna BP, De Sousa E, Melo F, Roelofs JJ, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut (2016) 65:575–83. doi: 10.1136/gutjnl-2015-309728
12. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev. Microbiol. (2017) 15:55–63. doi: 10.1038/nrmicro.2016.142
13. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
14. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
15. Dickson RP, Erb-Downward JR, Freeman CM, Mccloskey L, Falkowski NR, Huffnagle GB, et al. Bacterial topography of the healthy human lower respiratory tract. mBio (2017) 8:e02287-16. doi: 10.1128/mBio.02287-16
16. Man WH, De Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. (2017) 15:259–70. doi: 10.1038/nrmicro.2017.14
17. Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Resp Crit Care Med. (2018) 198:497–508. doi: 10.1164/rccm.201711-2180OC
18. Winglee K, Eloe-Fadrosh E, Gupta S, Guo H, Fraser C, Bishai W. Aerosol Mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PloS ONE (2014) 9:e97048. doi: 10.1371/journal.pone.0097048
19. Luo M, Liu Y, Wu P, Luo DX, Sun Q, Zheng H, et al. Alternation of gut microbiota in patients with pulmonary tuberculosis. Front Physiol. (2017) 8:822. doi: 10.3389/fphys.2017.00822
20. Hong BY, Paulson JN, Stine OC, Weinstock GM, Cervantes JL. Meta-analysis of the lung microbiota in pulmonary tuberculosis. tuberculosis (2018) 109:102–8. doi: 10.1016/j.tube.2018.02.006
21. Al-Asmakh M, Zadjali F. Use of germ-free animal models in microbiota-related research. J Microbiol Biotechnol. (2015) 25:1583–8. doi: 10.4014/jmb.1501.01039
22. Lundberg R, Toft MF, August B, Hansen AK, Hansen CH. Antibiotic-treated versus germ-free rodents for microbiota transplantation studies. Gut Microbes (2016) 7:68–74. doi: 10.1080/19490976.2015.1127463
23. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41. doi: 10.1016/j.cell.2004.07.002
24. Meierovics A, Yankelevich WJ, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci USA. (2013) 110:E3119–28. doi: 10.1073/pnas.1302799110
25. Sakala IG, Kjer-Nielsen L, Eickhoff CS, Wang X, Blazevic A, Liu L, et al. Functional heterogeneity and antimycobacterial effects of mouse mucosal-associated invariant T cells specific for riboflavin metabolites. J Immunol. (2015) 195:587–601. doi: 10.4049/jimmunol.1402545
26. Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. (2010) 11:701–8. doi: 10.1038/ni.1890
27. Yang YW, Chen MK, Yang BY, Huang XJ, Zhang XR, He LQ, et al. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in mouse feces. Appl Environ. Microbiol. (2015) 81:6749–56. doi: 10.1128/AEM.01906-15
28. Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PloS ONE (2011) 6:e17996. doi: 10.1371/journal.pone.0017996
29. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
30. Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe (2012) 12:139–52. doi: 10.1016/j.chom.2012.07.004
31. Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. (2015) 33:496–503. doi: 10.1016/j.tibtech.2015.06.011
32. Kostric M, Milger K, Krauss-Etschmann S, Engel M, Vestergaard G, Schloter M, et al. Development of a stable lung microbiome in healthy neonatal mice. Microbiol Ecol. (2018) 75:529–42. doi: 10.1007/s00248-017-1068-x
33. O'garra A, Redford PS, Mcnab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Ann Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
34. Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med. (2015) 21:808–14. doi: 10.1038/nm.3871
35. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
36. Constantinides MG. Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol. (2018) 103:409–19. doi: 10.1002/JLB.3RI0917-378R
37. Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE, et al. Segmented filamentous bacteria provoke lung autoimmunity by inducing gut-lung axis Th17 cells expressing dual TCRs. Cell Host Microbe (2017) 22:697–704 e694. doi: 10.1016/j.chom.2017.10.007
38. Hernandez-Santos N, Wiesner DL, Fites JS, Mcdermott AJ, Warner T, Wuthrich M, et al. Lung epithelial cells coordinate innate lymphocytes and immunity against pulmonary fungal infection. Cell Host Microbe (2018) 23:511–22 e515. doi: 10.1016/j.chom.2018.02.011
39. Samuelson DR, Shellito JE, Maffei VJ, Tague ED, Campagna SR, Blanchard EE, et al. Alcohol-associated intestinal dysbiosis impairs pulmonary host defense against Klebsiella pneumoniae. PLoS Pathog. (2017) 13:e1006426. doi: 10.1371/journal.ppat.1006426
40. Cui Y, Franciszkiewicz K, Mburu YK, Mondot S, Le Bourhis L, Premel V, et al. Mucosal-associated invariant T cell-rich congenic mouse strain allows functional evaluation. J Clin Investig. (2015) 125:4171–85. doi: 10.1172/JCI82424
41. Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp. Med. (2015) 212:1095–108. doi: 10.1084/jem.20142110
42. Hong BY, Maulen NP, Adami AJ, Granados H, Balcells ME, Cervantes J. Microbiome changes during tuberculosis and antituberculous therapy. Clin Microbiol Rev. (2016) 29:915–26. doi: 10.1128/CMR.00096-15
43. Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, Agrewala JN. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol. (2016) 7:529. doi: 10.3389/fimmu.2016.00529
44. Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc. (2015) 12 (Suppl 2):S150–6. doi: 10.1513/AnnalsATS.201503-133AW
45. Gray J, Oehrle K, Worthen G, Alenghat T, Whitsett J, Deshmukh H. Intestinal commensal bacteria mediate lung mucosal immunity and promote resistance of newborn mice to infection. Sci Transl Med. (2017) 9:eaaf9412. doi: 10.1126/scitranslmed.aaf9412
46. Cheng M, Chen Y, Wang L, Chen W, Yang L, Shen G, et al. Commensal microbiota maintains alveolar macrophages with a low level of CCL24 production to generate anti-metastatic tumor activity. Sci Rep. (2017) 7:7471. doi: 10.1038/s41598-017-08264-8
47. Dickson RP, Cox MJ. Gut microbiota and protection from pneumococcal pneumonia. Gut (2017) 66:384. doi: 10.1136/gutjnl-2016-311823
48. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422:164–9. doi: 10.1038/nature01433
49. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature (2012) 491:717–23. doi: 10.1038/nature11605
50. Ghazarian L, Caillat-Zucman S, Houdouin V. Mucosal-associated invariant T cell interactions with commensal and pathogenic bacteria: potential role in antimicrobial immunity in the child. Front Immunol. (2017) 8:1837. doi: 10.3389/fimmu.2017.01837
51. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol. (2015) 194:5775–80. doi: 10.4049/jimmunol.1402945
52. Treiner E. Mucosal-associated invariant T cells in inflammatory bowel diseases: bystanders, defenders, or offenders? Front Immunol. (2015) 6:27. doi: 10.3389/fimmu.2015.00027
53. Georgel P, Radosavljevic M, Macquin C, Bahram S. The non-conventional MHC class I MR1 molecule controls infection by Klebsiella pneumoniae in mice. Mol Immunol. (2011) 48:769–75. doi: 10.1016/j.molimm.2010.12.002
54. Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun. (2012) 80:3256–67. doi: 10.1128/IAI.00279-12
55. Jiang J, Wang X, An H, Yang B, Cao Z, Liu Y, et al. Mucosal-associated invariant T-cell function is modulated by programmed death-1 signaling in patients with active tuberculosis. Am J Resp Crit Care Med. (2014) 190:329–39. doi: 10.1164/rccm.201401-0106OC
56. Kwon YS, Cho YN, Kim MJ, Jin HM, Jung HJ, Kang JH, et al. Mucosal-associated invariant T cells are numerically and functionally deficient in patients with mycobacterial infection and reflect disease activity. tuberculosis (2015) 95:267–74. doi: 10.1016/j.tube.2015.03.004
57. Patel O, Kjer-Nielsen L, Le Nours J, Eckle SB, Birkinshaw R, Beddoe T, et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun. (2013) 4:2142. doi: 10.1038/ncomms3142
58. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509:361–5. doi: 10.1038/nature13160
59. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood (2011) 117:1250–9. doi: 10.1182/blood-2010-08-303339
60. Tastan C, Karhan E, Zhou W, Fleming E, Voigt AY, Yao X, et al. Tuning of human MAIT cell activation by commensal bacteria species and MR1-dependent T-cell presentation. Mucosal Immunol. (2018) doi: 10.1038/s41385-018-0072-x. [Epub ahead of print].
61. Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nature Rev Immunol. (2018) 8:575–89. doi: 10.1038/s41577-018-0025-3
Keywords: microbiota, MAIT cells, macrophage, tuberculosis, IL-17
Citation: Dumas A, Corral D, Colom A, Levillain F, Peixoto A, Hudrisier D, Poquet Y and Neyrolles O (2018) The Host Microbiota Contributes to Early Protection Against Lung Colonization by Mycobacterium tuberculosis. Front. Immunol. 9:2656. doi: 10.3389/fimmu.2018.02656
Received: 30 June 2018; Accepted: 29 October 2018;
Published: 14 November 2018.
Edited by:
Robert Wilkinson, Francis Crick Institute, United KingdomReviewed by:
Rachel Lai, Imperial College London, United KingdomSivaranjani Namasivayam, National Institutes of Health (NIH), United States
Copyright © 2018 Dumas, Corral, Colom, Levillain, Peixoto, Hudrisier, Poquet and Neyrolles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Neyrolles, b2xpdmllci5uZXlyb2xsZXNAaXBicy5mcg==
†These authors have contributed equally to this work