
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 November 2018
Sec. T Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02618
This article is part of the Research TopicTRAF Proteins in Health and DiseaseView all 12 articles
 Juan M. Zapata1,2*
Juan M. Zapata1,2* Gema Perez-Chacon1,2
Gema Perez-Chacon1,2 Pablo Carr-Baena1
Pablo Carr-Baena1 Ivan Martinez-Forero3
Ivan Martinez-Forero3 Arantza Azpilikueta3
Arantza Azpilikueta3 Itziar Otano3
Itziar Otano3 Ignacio Melero3,4,5,6,7*
Ignacio Melero3,4,5,6,7*CD137 (4-1BB, Tnsfr9) is a member of the TNF-receptor (TNFR) superfamily without known intrinsic enzymatic activity in its cytoplasmic domain. Hence, akin to other members of the TNFR family, it relies on the TNFR-Associated-Factor (TRAF) family of adaptor proteins to build the CD137 signalosome for transducing signals into the cell. Thus, upon CD137 activation by binding of CD137L trimers or by crosslinking with agonist monoclonal antibodies, TRAF1, TRAF2, and TRAF3 are readily recruited to the cytoplasmic domain of CD137, likely as homo- and/or heterotrimers with different configurations, initiating the construction of the CD137 signalosome. The formation of TRAF2-RING dimers between TRAF2 molecules from contiguous trimers would help to establish a multimeric structure of TRAF-trimers that is probably essential for CD137 signaling. In addition, available studies have identified a large number of proteins that are recruited to CD137:TRAF complexes including ubiquitin ligases and proteases, kinases, and modulatory proteins. Working in a coordinated fashion, these CD137-signalosomes will ultimately promote CD137-mediated T cell proliferation and survival and will endow T cells with stronger effector functions. Current evidence allows to envision the molecular events that might take place in the early stages of CD137-signalosome formation, underscoring the key roles of TRAFs and of K63 and K48-ubiquitination of target proteins in the signaling process. Understanding the composition and fine regulation of CD137-signalosomes assembly and disassembly will be key to improve the therapeutic activities of chimeric antigen receptors (CARs) encompassing the CD137 cytoplasmic domain and a new generation of CD137 agonists for the treatment of cancer.
TNF Receptor Associated Factors (TRAFs) are a family of 6 proteins (TRAF1 to 6) characterized for having a protein region composed by a coiled coil followed by a seven-eight anti-parallel β-sheets at the C-terminus of the protein forming what has been coined as the TRAF domain (TD) (1, 2). This domain is also known as the Meprin and TRAF-C homology domains (MATH), since meprins, a family of extracellular proteases, also have a protein domain with high homology to the TD (3). In addition, there are also 3 proteins in humans encompassing internal bona fide TRAF domains: tripartite motif (TRIM)-37, ubiquitin specific protease (USP)-7 and speckle-type POZ protein (SPOP) (4). Of note is that there is a protein known as TRAF7 that lacks a TD but has a RING and zinc finger domains similar to those of some members of the TRAF family proteins (5) and whose membership to the TRAF family is controversial.
TRAF1 to 6 were first identified as TNF-Receptor (TNFR) binding proteins, but it soon become evident that different members of the TRAF family were also involved in the regulation of pattern recognition receptors, including members of the Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and retinoic acid-inducible gene (RIG)-1-like Receptors (RLRs), thus demonstrating the key role of TRAF family proteins in the regulation of both innate and adaptive immunity [Reviewed by (6)]. Moreover, some members of the TRAF family also regulate cytokine receptors (6, 7). A role for TRAF family members in development has also been described (8–10).
TRAFs are the molecules that first engage the activated TNFR and act as scaffold proteins recruiting other proteins, including kinases, ubiquitin ligases and deubiquitinases among other regulatory proteins to conform the TNFR-signalosome. TRAF family members, with the exception of TRAF1, have a RING finger domain that endows some of them with the capacity to act as E3 ubiquitin ligases. Thus, TRAFs can ubiquitinate different components of the signalosome, including the TRAFs themselves, and modulate the activity of the complex (6).
There is a redundancy in the ability of different members of the TRAF family to interact with similar TRAF-binding peptidyl regions located in the cytosolic tails of the TNFRs [reviewed in (1, 2, 11)]. Moreover, besides this critical binding region, the surrounding amino acids to the peptide core motif in the cytosolic tail of TNFRs might also provide structural constrains that may have an effect on the binding affinity. In addition, the crystal structures of TDs bound to the cytosolic region of distinct TNFR family members have shown that particular structural features of the TD of each TRAF family member, in particular of those forming the TNFR-binding crevice, are critical in determining their specificities and binding affinities to the TNFRs [reviewed in (12)]. Altogether, these differences determine the binding specificity and affinity of the members of the TRAF family for the different TNFR family members (1, 11–14). Therefore, it is expected that a competition would be established between different TRAFs trimers to dock at the ligand-activated TNFR trimer, raising the possibility that neighboring TNFR trimers in the very same cell will hold TRAF trimers with different configurations. In addition, some TRAF family members can form heterotrimers (see below and Figure 1), adding further complexity to the system. Consequently, the composition of the signalosome mounted by each member of the TNFR family is likely to be highly influenced by the recruited TRAF family members. Besides, the signalosome composition would likely be cell type and activation state dependent, as it will be contingent on the expression levels and subcellular localization of the different proteins that could be part of this complex.
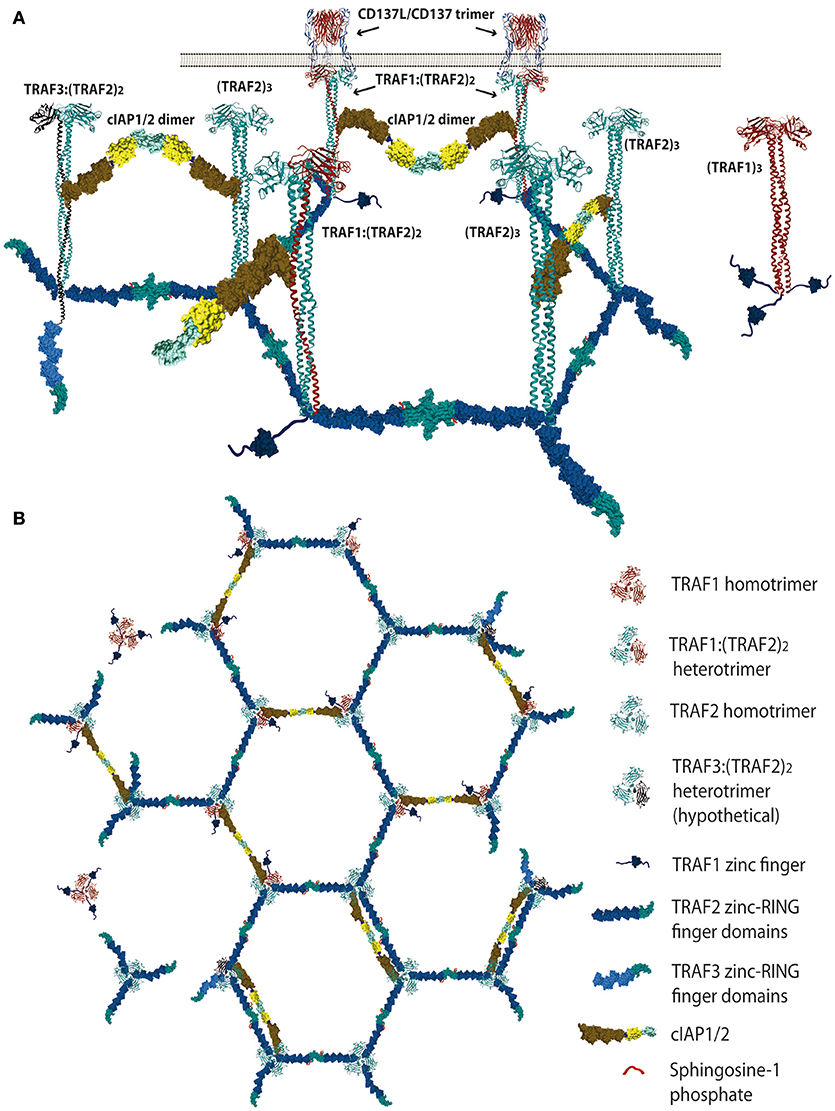
Figure 1. Schematic representation of the proposed TRAF trimer configurations and interactions in the CD137L/CD137 hexagonal lattice. (A) Lateral view representing the various TRAF-trimer configurations that could be recruited to the activated CD137 trimers. The figure also shows the TRAF2-RING finger dimers that would likely be formed between the RING finger domains of two TRAF2 molecules from adjacent trimers, which is a requirement for E3 ubiquitin ligase activity. Similar interactions between the RING domains of cIAP1/2 from contiguous trimers are also expected. (B) It is show in top view how the CD137-recruited TRAF trimers would arrange forming a large hexagonal network that would be stabilized by the establishment of RING finger domains dimers between the TRAF2 molecules from adjacent trimers or between the RING finger domains of contiguous cIAP1/2 molecules. Further explanation in the text. Protein structure coordinates were obtained from the PDB database and molecular graphics were performed with UCSF Chimera (15).
CD137 (4-1BB, TNFSFR9) is one of the TNFRs having a more restricted number of TRAF family members involved in its regulation, since only TRAF1, TRAF2, and TRAF3 interact with and control CD137 activity. CD137 is a member of the TNFR family whose expression is highly induced in CD8 T and NK lymphocytes upon activation, where it works as a critical costimulatory receptor (16–18). Moderate to low levels of CD137 expression could also be found in other activated immune populations, including CD4 T cells, B cells, monocytes, macrophages, granulocytes and dendritic cells and, in these cells, CD137 can also convey costimulatory signals (17, 19).
CD137 delivers potent costimulatory signals to the activated CTLs and memory T cells promoting cell proliferation and survival and also endowing CD8 T cells with CTL effector functions. As such, in the last 15 years, CD137 has become one of the most exciting targets to enhance anti-cancer immunity for its ability of boosting CTLs with anti-tumor effector functions (20–22).
CD137 binds to CD137-Ligand (CD137L, 4-1BBL, or tnfsf9), a member of the TNF superfamily (TNFSF). CD137L is mostly expressed on macrophages, activated B cells, and dendritic cells (23). In this regard, it is noteworthy that antigen presenting dendritic cells in tumors and tumor draining lymph nodes and tumor associated macrophages seem to be responsible for providing CD137L to cytotoxic T lymphocytes (CTLs) migrating to tumors (24). CD137L remains the sole intercellular ligand known for CD137, but binding of CD137 to extracellular matrix proteins, such as fibronectin, vitronectin, laminin and collagen VI (25) has been reported, albeit functional consequences of the binding to these additional putative ligands remain unknown. Interestingly, binding of CD137 to galectin-9, a member of the β-galactoside–binding family of lectins, has also been shown (26). Interestingly, galectin-9 binding to CD137 does not interfere with the binding of either CD137L or agonistic anti-CD137 mAbs to the receptor. Instead, it positively regulates CD137 function by keeping preassembled CD137 complexes together (26), which could be then further cross-linked by CD137L or by anti-CD137 mAbs.
The crystal structure of the CD137L trimer shows distinctive structural features that differ from those of other TNF family members. In this regard, CD137L trimer resembles a three-bladed propeller which is different from the cork-like shape of the trimers of other members of the family (27). This shape also confers some structural particularities to CD137/CD137L complex, which folds as a windmill-like shape structure. Despite these structural differences with other TNF and TNFR family members, these results are still fully consistent with a model for CD137/CD137L interaction similar to that of other members of the TNFR family, in which a trimeric ligand binding to three receptors conforms the basic unit of signaling (28, 29).
As for many other members of the TNFRSF, CD137 uses TRAFs as scaffold proteins to build its signalosome. CD137 has been found to bind to TRAF1, TRAF2, and TRAF3 (30–32) through two poly-acidic TRAF-binding consensus regions located in its cytosolic tail 234TTQEE238 and 246PEEEE250, which are similar to those found in other TNFR family members [reviewed in (1, 2)]. Point mutations studies showed that all three TRAFs seem to have binding preferences for the C-terminal 246PEEEE250 TRAF-binding region, suggesting that they might compete with each other for interacting with the activated receptor (31). Due to the proximity of the two TRAF binding sites, binding of one TRAF trimer to one of these regions, would render the other region unavailable by steric hindrance. However, this does not rule out the presence of different TRAFs associated to the same activated CD137 trimer, since TRAF1 and TRAF2 form heterotrimers that can associate to the activated TNFR (33).
Cross-linking of CD137 by either CD137L (30, 34) or bivalent agonistic antibodies (35) readily results in the recruitment of TRAF1 and TRAF2 to the receptor. The involvement of both TRAF family members in the regulation of CD137 signaling and function is further confirmed by several reports showing that CD137 activity is significantly affected in model systems lacking of either TRAF1 or TRAF2 (32, 36–38). However, the role of TRAF3 as a scaffold protein building the CD137 signalosome has not been confirmed and awaits further research, although the evidence indicating the induction of NF-kB2 activation by CD137 (38) implies that TRAF3 should be directly or indirectly recruited to the CD137 signalosome (see below). In addition, recent evidence shows that TRAF3, as well as TRAF1 and TRAF2, are essential for the activity of CD137-based chimeric antigen receptors (CARs) (39), further supporting TRAF3 role in CD137 function.
The absence of a RING finger domain in TRAF1 indicates that it lacks any E3 ubiquitin ligase activity and no other intrinsic enzymatic activity for TRAF1 has been identified so far (6, 40). However, TRAF1 interacts with and regulates the activity of a variety of ubiquitin ligases and proteases (33, 41, 42) and it plays critical roles in the regulation of several members of the TNFR family [Reviewed in (43)]. Initially, since TRAF1 expression is induced upon cell activation and it has similar TNFR-binding preferences than TRAF2, it was thought that TRAF1 would work toning down TNFR signaling in activated cells by outcompeting TRAF2 from binding to the TNFRs (43). Indeed, T cells from Traf1-deficient mice were hyper-responsive to TNF, supporting a role for TRAF1 as a negative TNFR2 regulator (44). However, it was soon recognized that TRAF1 was not just a TRAF2 competitor but, in some instances, rather the contrary. In this regard, TRAF1 positively modulates CD40 activity by cooperating with its activity and preventing TRAF2 degradation (45, 46). In addition, TRAF1 has been also implicated in CD137-mediated survival of activated CTL (47, 48) and of memory T cells (49).
The other TRAF family members that is critical for CD137 function is TRAF2. The RING domain that TRAF2 encompasses at its N-terminus endows it with an E3 ubiquitin ligase activity. Ubiquitin-conjugating protein (Ubc)-13 (Ube2N) is thought to be TRAF2 major E2 enzyme companion, providing TRAF2 with the capacity of mediating K63-ubiquitination and subsequent activation of itself and other target proteins (50–52). In addition, TRAF2 can also catalyze K48-ubiquitination of target proteins (53, 54). Interestingly, the crystal structure of the TRAF2 RING and the first zinc finger domains described by Wu et al. (55) revealed structural constrains that would preclude Ubc13 and other related E2 ubiquitin ligases from binding to the TRAF2 RING, raising questions on the actual ability of TRAF2 to act as an E3 ubiquitin ligase. However, these discrepancies were solved when sphingosine-1 phosphate (S1P) was identified as a cofactor required for TRAF2 E3 ubiquitin ligase activity (50). Indeed, S1P seems to act as a bridge between the RING finger domain of TRAF2 and the E2 proteins. Thus, in the presence of S1P, TRAF2 was able to ubiquitinate RIP1 and itself (and/or other TRAF2 molecules in the trimer) at K63 in the presence of Ubc13 or Ubc5 (Ube2D) (50).
While TRAF2 is expressed in resting and activated T lymphocytes, TRAF1 expression is induced upon activation (60, 61). Thus, as CD137 expression will also be induced in activated T cells (16, 17), both CD137 and TRAF1 will likely coexist in activated T cells where CD137 costimulatory activity is needed for CTL expansion and for boosting effector functions. Therefore, the composition of the CD137-TRAF signaling complexes would depends on the activation state of the cell and the relative expression levels of TRAF1 and TRAF2.
The kinetics of CD137 expression in activated CD8 T cells implies that at early activation stages low levels of CD137 will be found on the T cell surface (62, 63). However, even these low levels might be sufficient to trigger CD137 signaling upon interaction with the CD137L. In this regard, it has been proposed that the ligand-free form of TNFR family members exists on the cell surface as anti-parallel dimers arranged in a two-dimensional hexagonal lattice that brings three receptor monomers together at each lattice point [Reviewed in (28)]. This model would imply that even low level of ligand-free TNFRs might be already prearranged on the cell surface in high-density spots. In the case of CD137, galectin-9 might contribute to the maintenance of these bi-dimensional hexagonal structures (26). Assuming this model, when CD137L or other TNF family member and their corresponding TNFRs come together, the ligand trimer will shift the equilibrium from the CD137 dimeric interaction to the CD137 trimeric structure. The CD137/CD137L trimers will still occupy each lattice point preserving the hexagonal structure and maintaining neighboring activated CD137 trimers close, thus facilitating the establishment of molecular interactions between adjacent trimers. TRAF trimers will be readily recruited to the activated CD137 receptor binding to the poly-acidic TRAF-binding consensus regions located in CD137 cytosolic tail. As stated above, the composition of the TRAF trimers that would be recruited to the activated CD137 will likely depend on the expression levels of the TRAF family proteins that interact with CD137, which are TRAF1, TRAF2, and TRAF3. Since TRAF1 and TRAF2 have been shown to be critical for CD137 activity it is likely that these two TRAF family members will have a major role in building the CD137 signalosome. Wu and coworkers (33) have shown that TRAF1 and TRAF2 can associate in heterotrimers, but preferentially forming a trimer with a TRAF1:(TRAF2)2 configuration. Therefore, a mix of TRAF1 and TRAF2 homotrimers and TRAF1:(TRAF2)2 heterotrimers would be recruited to the activated CD137 in a way that would depends on their amounts and specific affinities to the TNFR.
Since the five zinc fingers and the RING finger domains of each TRAF2 molecule in the trimers will likely emanate from the intertwining coils in opposite directions (28, 64) and the active E3 ubiquitin ligase requires the formation of RING-finger dimers (65), TRAF2-RING finger dimers will likely be formed by the RING finger domains of two TRAF2 molecules from adjacent trimers, similar to what has been described for TRAF6 (66) (Figure 1). This inter-trimer bonding would help the clustering and stabilization of the two-dimensional hexagonal lattice (28, 64). In this case, since TRAF1 lacks of a RING finger domain, the presence of one TRAF1 molecule in a trimer would impede the formation of one intertrimer bonding but would not have any effect on the ability of the 2 TRAF2 molecules in the trimer to establish these TRAF2-RING dimers with neighboring TRAF2-containing trimers (Figure 1). The TRAF2-RINGs now in their active dimeric form will bind S1P and Ubc proteins (Ubc13 or UbcH5A) (Figure 2), getting ready to catalyze the K63-ubiquitination of TRAF2 itself and other target proteins (50). In addition TRAF2 trimers and TRAF1:(TRAF2)2 heterotrimers, but not TRAF1 trimers, will recruit a single cIAP1/2 (33). Indeed, cIAP1/2 will interact through its BIR1 domain (67) with the TRAF trimers by asymmetrically engaging two cIAP-interacting motifs in the coiled coil of two TRAF molecules in the trimer (33, 42). Of note is that cIAP1/2 interaction with the TRAF1:(TRAF2)2 heterotrimers is stronger than that with TRAF2 trimers and, therefore, cIAP1/2 would preferentially be bond to the TRAF1:(TRAF2)2 heterotrimers. Interestingly, TRAF1 homotrimers have a cIAP2 dissociation constant two orders of magnitude weaker than that of TRAF2 homotrimers, effectively precluding the interaction of cIAP2 with TRAF1 homotrimers (33). The interaction of cIAP1/2 BIR1 domain with TRAFs would release the cIAP-RING from its inhibitory interaction with the cIAP-CARD domain (68), allowing the formation of cIAP1/2-RING dimers and the binding of the E2 ubiquitin ligases. Since only one cIAP1/2 molecule associates to a TRAF trimer, the cIAP1/2-RING dimer would have to be formed by two cIAP1/2 molecules each one associated to adjacent TRAF trimers in the hexagonal lattice (Figures 1, 2), thus further bridging two neighbored activated TNFR complex. Altogether, these results indicate that albeit the basic signaling brick in CD137 (as well as of other TNFRs) would be a trimer, a trimer alone will not be able to signal as it seems absolutely necessary to establish inter-trimer bridging and multi-trimer clustering to build a functional signalosome (Figures 1, 2).
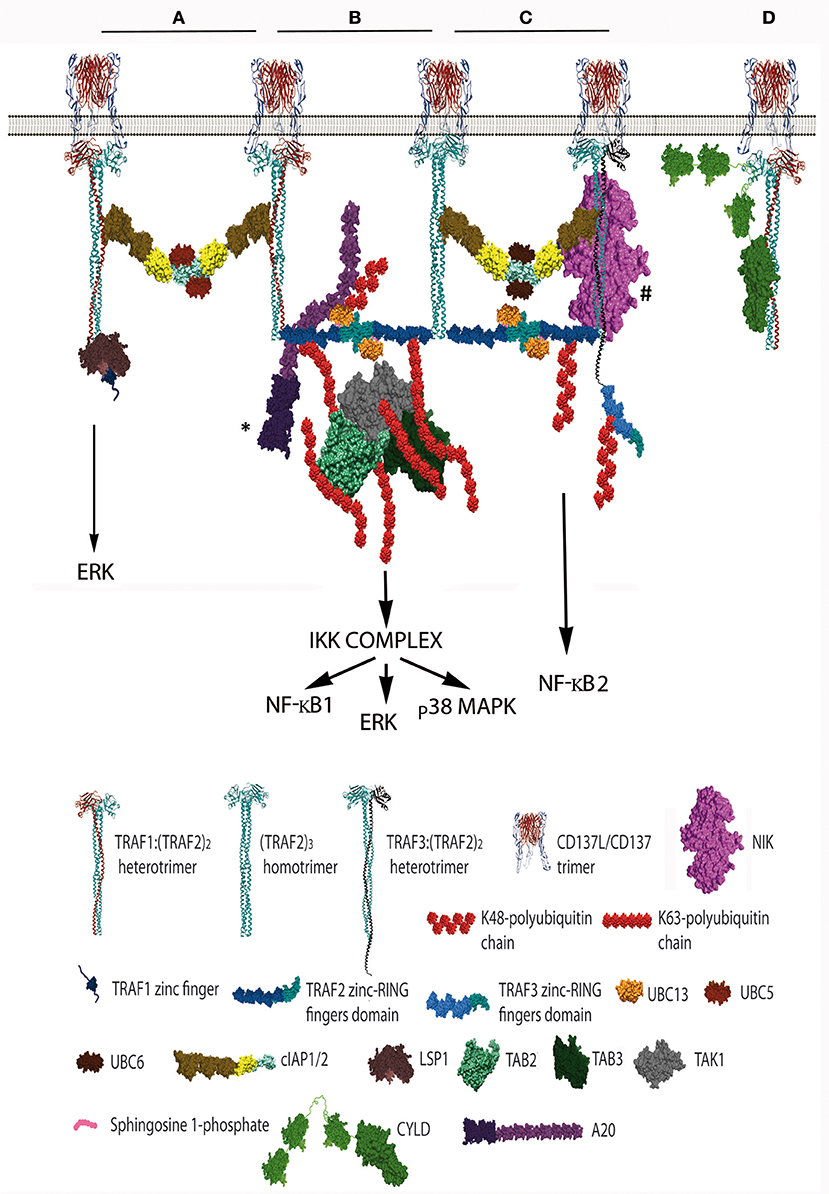
Figure 2. Schematic representation of the distinct CD137 signalosomes that would be formed upon CD137 activation. This figure illustrates the distinct signalosomes that could be formed in response to CD137 activation depending on the TRAF trimer configurations that get associated to the activated CD137. (A) cIAP1/2 bridging between 2 TRAF1(TRAF2)2 trimers. What other molecules, besides E2 proteins, would be specifically recruited to this configuration is yet unknown. The binding of Lymphocyte specific protein-1 to the N-terminal region of TRAF1 is shown. (B) The formation of a dimer between the RING finger domains of 2 TRAF2 molecules from adjacent trimers will trigger K63 ubiquitination of TRAF2 and the subsequent recruitment and activation of the TAK1/TAB1/TAB2/TAB3 complex (TAB1 is not shown). K63-TAK1-mediated IKKβ phosphorylation will activate the IKK complex activation initiating a signaling cascade that will result in NF-κB1 and ERK activation. A20 might inhibit this signaling cascade by K48-ubiquitinating Ubc13 thus inhibiting TRAF2 E3 ubiquitin ligase activity. * A20 can form dimers, but a sole A20 molecule is represented for clarity. (C) Hypothetical organization of a signalosome that includes a TRAF3:(TRAF2)2 trimer. The cIAP1/2 molecules associated either to a TRAF2 homotrimer and the hypothetical TRAF3:(TRAF2)2 trimer will form a dimer by the interaction of their RING fingers domains causing the activation of the E3 ubiquitin ligase activity. Thus, the cIAP1/2 dimer will K48-ubiquitinate TRAF3 and TRAF2 molecules targeting them for proteasome degradation and effectively releasing NIK from its interaction with TRAF3, resulting in the activation of NF-κB2 as has been observed following CD137 stimulation. # The TRAF region binding to NIK is still controversial, since reports indicating that is mediated by either the TRAF domain (56, 57) or the RING-zinc finger region (58, 59) are available. (D) CYLD interacts with the same crevice in the TRAF domain that binds to CD137 cytosolic tail. CYLD might works as a gate keeper preventing ligand-independent TRAF activation but it might also participate in the termination of CD137 signaling by outcompeting CD137 from binding to TRAF2 as shown in the figure. Further explanation in the text. Protein structure coordinates were obtained from the PDB database and molecular graphics were performed with UCSF Chimera (15). When this information was absent for a protein of interest, we modeled the proteins according to their domains using available structures of similar domains to provide an approximate representation of the protein structure and size.
In this regard, it is worth mentioning that since TRAF1 lacks RING finger domain and TRAF1 homotrimers cannot recruit cIAP1/2 to the CD137 signalosome, CD137-associated TRAF1 homotrimers would fail on bridging adjacent trimers through the formation of RING-dimers, which might result in the disruption of the hexagonal CD137 network and the inhibition of the signaling. While this scenario might provide a rationale for the TRAF1-mediated inhibitory effects on some members of the TNFR family (43), many evidence support a positive role for TRAF1 in CD137 signaling (37, 47–49, 69). Therefore, other proteins interacting with TRAF1 might not only provide new functionality to the signalosome but also might contribute to the clustering of the activated CD137 receptors. In this regard, it has been shown that recruitment to CD137 of leukocyte-specific protein-1, a protein involved in CD137-mediated ERK activation, is mediated by its interaction with TRAF1 (70) (Figure 2A). Furthermore, Watts and coworkers (71) have shown that the TRAF-domain of TRAF1 directly interacts with three components of the linear ubiquitination (LUBAC) complex, SHARPIN, HOIP, and HOIL-1. In addition, Greenfeld and coworkers (41) have shown that TRAF1 is a key component of the Epstein-Barr virus Late Membrane Protein (LMP)-1 signaling complex, a protein that mimics TNFRs and uses TRAF proteins as scaffold (72). In this model, LMP1 promoted the association between TRAF1 and LUBAC and stimulated the linear M1-linked poly-ubiquitination of TRAF1, thus allowing TRAF1-mediated recruitment of the M1-ubiquitin binding proteins IKKγ and deubiquitinase (DUB) A20 (41) (see below). TRAF2 was essential for both LUBAC interaction and M1-polyubiquitination of TRAF1 (41), strongly suggesting the participation of TRAF1:(TRAF2)2 heterotrimers in this activity. In addition, binding of cIAP1 (73) and CYLD (74) to HOIP has also been reported. Although there is no evidence to date implicating LUBAC and M1-ubiquitination in the regulation of CD137 signalosome, research on this issue is warranted.
Soon after ligand activation, the growing CD137 signalosome gets decorated with K63-ubiquitinated proteins, mostly composed by K63-TRAF2 (36). Polyubiquitin chains linking the carboxyl terminus of ubiquitin molecules to the K63 of the next ubiquitin are well known as docking sites for downstream signaling components, and are required for building an effective signalosome (75–77). This is opposite to the polyubiquitination at K48, which in most cases targets proteins for proteosome-mediated degradation (77). TRAF2, associated to Ubc13 or UbcH5A, seems to be the main responsible of its own K63-ubiquitination (50) although cIAP1/2 associated to UbcH5A or Ubc13 also can catalyze K63-ubiquitination (78, 79). The next component of the CD137 signalosome getting recruited by K63-polyubiquitinated TRAF2 is a kinase complex composed by the transforming growth factor beta-activated kinase (TAK)-1 and TAK binding proteins (TAB)-1, 2 and 3 and (Figure 2B). Once recruited, TAK1 will get K63-ubiquitinated by TRAF2 (80). Taking lessons from the mechanism recently described for TRAF6-mediated TAK1 activation (81), efficient TRAF6-mediated TAK1 activation requires the synthesis of long K63-polyubiquitin chains by TRAF6. These long K63-polyubiquitin chains would have to be recognized by TAB2 and 3 (82) irrespective of whether they remain conjugated to TRAF6 or been unanchored (81). Interestingly, A20 which is a component of the CD137 signalosome (see below), effectively removes long K63-linked polyubiquitin chains from TRAF6 without disassembling the chains themselves (83). Once activated, TAK1 will phosphorylate the inhibitor of nuclear factor κ-B kinase (IKK)-β leading to the activation of canonical NF-κB (75) and ERK1/2 (48). TAK1 will also induce the activation of mitogen activated kinases kinases (MKK) MAP kinases, leading to p38MAPK activation (84).
While ubiquitination is a chief mechanism controlling TRAF-mediated CD137 signaling, the regulation of TRAF activity by phosphorylation has also been described. In this regard, it has been shown that TRAF2 phosphorylation at T117 by PKC promotes both K63-ubiquitination of TRAF2 and the recruitment of the IKK complex to activated TNFRs (85). Moreover, TRAF1 phosphorylation at S139 by TANK-binding kinase inhibits NF-κB activation in response to CD137 engagement (86).
Besides the induction of the canonical NF-κB pathway by CD137, the activation of the alternative NF-κB pathway by this TNFR has also been reported (38). The molecular mechanism controlling NF-κB2 is different to that controlling the canonical NF-κB1 pathway. In non-activated cells, NF-κB2 activation is prevented by continuous NF-κB-inducing kinase (NIK) degradation by a complex formed by TRAF2/cIAP and TRAF3/NIK. In non-activated cells, this complex works promoting cIAP1/2 mediated K48-ubiquitination of NIK and its subsequent proteasome-mediated degradation. However, upon TNFR activation, binding of this complex to the receptor results in cIAP-dependent degradation of TRAF3 (and often also of TRAF2), releasing NIK and allowing p100 processing to the active p52 NF-κB subunit [reviewed in (87)]. Thus, the induction of NF-κB2 by CD137 (38) implies that TRAF3 and NIK would have to be recruited to the CD137 signalosome (Figure 2C). In support of this event, it has been shown that TRAF3 is degraded upon CD137 engagement (38). However, it is still unclear whether TRAF2 and TRAF3 would be recruited as homotrimers to adjacent CD137 trimers or as TRAF2/TRAF3 heterotrimers to one ligand-activated CD137 trimer. In this regard, there is evidence suggesting the existence of TRAF2/TRAF3 heterotrimers (88). In addition, it is noteworthy that TRAF1 has been shown to directly interact with NIK, suggesting that TRAF1:(TRAF2)2-cIAP1/2 complexes can also be a component of the E3 ubiquitin ligase complex for NIK (33, 89). However, there are conflictive results on whether TRAF1 is an activator or an inhibitor of the NF-κB2 pathway. It has been proposed that the binding of TRAF1 to NIK causes the disruption of TRAF2:cIAP1/2 binding, resulting in NIK stabilization and NF-κB2 activation (89). However, studies on the role of TRAF1 in CD137-mediated NF-κB activation show that in the absence of TRAF1, NF-κB1 induction is restricted while NF-κB2 induction proceeds more efficiently (38). These results might indicate that the tighter association of cIAP1/2 to the TRAF1:(TRAF2)2 heterotrimers compared to that of the TRAF2 homotrimers might restrict the ability of cIAP1/2 to shift their targets from NIK to TRAF3. Besides, it is also conceivable that an overabundance of TRAF1 might interact with all available TRAF2 molecules, thus precluding the formation of the TRAF2:TRAF3 heterotrimers. Interestingly, and as described above, TRAF1 protects TRAF2 from degradation (45, 46). Whether this protection could be caused by the inability of cIAP1/2 in the TRAF1:(TRAF2)2 heterotrimers to K48-ubiquitinate TRAF2 while it could do it as part of the TRAF2 homotrimers deserves further investigation.
Adding just another level of complexity to an already crowded CD137 signalosome, we have recently observed the functional association of K63-DUBs A20 and CYLD to the CD137 signalosome. This interaction results in the downregulation of CD137-elicited K63-ubiquitination and signaling toward NF-κB activation in both primary T cells and transfected cell lines (90). A20 was first described as an ubiquitin-editing enzyme (91). It is composed of an N-terminal ovarian tumor (OTU) domain, which would catalyze the removal of K63-ubiquitin chains from target proteins, and a C-terminal zinc-finger domain region, which endows this protein with E3 ubiquitin ligase activity able to transfer K48-polyubiquitin chains to those target proteins, thereby promoting its proteasome-mediated degradation (92). However, recent evidence shows that A20 can efficiently remove K48-linkages but is almost inactive toward K63-linkages, raising questions on what is the actual mechanism by which A20 inhibits the NF-κB pathway [reviewed in (93)]. Interestingly, A20 is also able to inhibit K63-ubiquitination by promoting K48-ubiquitination and degradation of E2 ligases, such as Ubc13 and UbcH5C, thus effectively inhibiting the E3 ligase activity of TRAF2, cIAP1/2, and TRAF6 (94) (Figure 2B). In addition A20 can also inhibit NF-κB by interacting with and sequestering Nemo (IKKγ), thus impeding IKKβ activation without requiring the DUB and E3-ubiquitin ligase activities of A20 (95–97).
CYLD is another member of the DUB family. It contains three cytoskeletal-associated protein (CAP)-glycine conserved repeats at the N-terminus and a DUB domain at its C-terminus. CYLD has a TRAF-interacting motif and has been shown to interact with TRAF2 and to catalyze the removal of TRAF2-linked K63-ubiquitin chains, precluding IKK from being activated (98). Interestingly, phosphorylation of CYLD by IKK inhibits its DUB activity (99). This result opens the possibility that CYLD might works as a gate keeper preventing ligand-independent activation, and that once receptor signaling unlocks, CYLD would be kept inactive by the active IKK complex. Since CYLD has also been found associated to the CD137-signalosome (90), this suggests that CYLD might also participate in the termination of CD137 signaling by outcompeting CD137 from binding to TRAF2 (Figure 2D). Altogether, these results underscore the relevance of the ubiquitination and deubiquitination processes in the regulation of CD137 signaling, evidencing that the balance between K63- and K48-ubiquitination of key target proteins will determine the outcome of the response. A summary of the role of TRAF1-3 in controlling CD137 signal transduction and function is provided in Table 1.
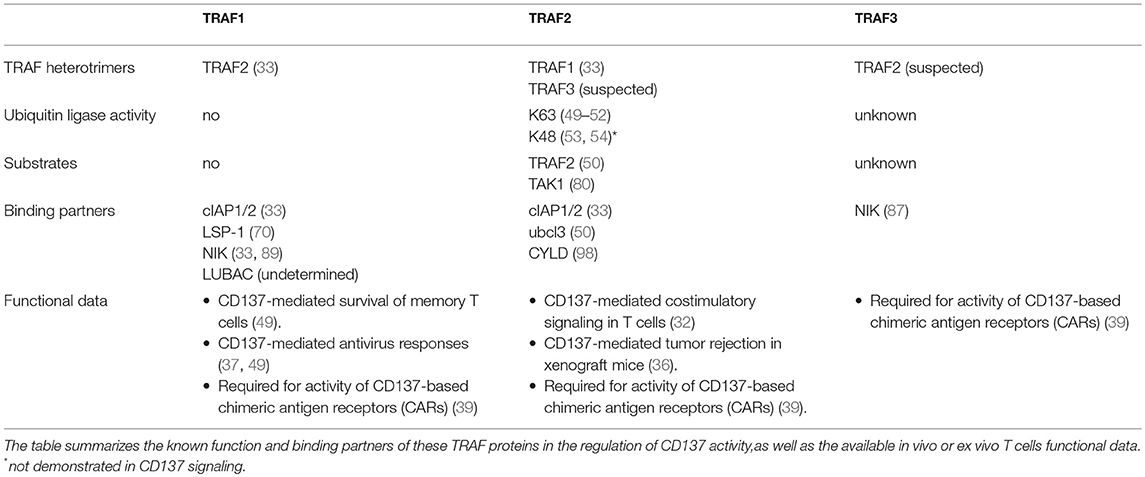
Table 1. Role of TRAF1,2 and 3 in CD137-mediated signaling.
Finally, we have observed that upon ligation with anti-CD137 antibodies, CD137 signalosome becomes internalized and is transferred to an endosomal compartment in a K63-polyubiquitin-dependent manner (36). Nam and coworkers (34) showed that CD137 engagement caused its redistribution into lipid rafts, in a process that seems to be dependent on TRAF2 binding to Caveolin (100) and Filamin A (101), which are intrinsic components of the lipid rafts. Thus, the CD137 signalosome-containing endocyted vesicles might be caveolae that later fuse with early endosomes (102), but this awaits confirmation. Interestingly, So and Croft (103) have proposed that TRAF2-dependent recruitment of activated CD137 into lipid rafts might be behind the observed activation of PI3K-AKT signaling pathway by CD137. Lipid rafts are membrane microdomains that facilitate AKT recruitment and activation upon phosphatidylinositol-3,4,5-triphosphate accumulation in the plasma membrane (104). The mechanism involved in CD137-mediated PI3K/AKT activation is still unknown, although its relevance in promoting CD137-mediated T cell proliferation and apoptosis protection seems well sustained (105–107). As there is no evidence of a direct association of PI3K and/or AKT to the CD137 signalosome, PI3K-AKT ought to be activated by other signaling complexes, such as TCR/CD28, working together with CD137 (105, 107). Since activated TCR/CD28 reside in the lipid rafts, these lipid structures might work as multi-signaling hot-spots (103). Indeed, it would be plausible that the ligand-activated CD137 hexagonal lattice keeps trapped inside (in the center of the hexagons) TCR and CD28 complexes that would move together with the activated CD137 trimers to lipid-rafts, thus facilitating the response to antigen. However, since CD137-mediated AKT activation is delayed compared to that of ERK and NF-κB, taking hours instead of minutes (107), efficient CD137-mediated triggering of PI3K/AKT activity may require additional players (whose expression might even be induced by CD137 engagement) and/or further signaling-complexes compartmentalization to proceed.
Interestingly, we have observed that endocyted CD137 signalosome-containing vesicles remain decorated with K63-polyubiquitin chains, strongly suggesting that CD137 signaling is still active during this process (36). However, it is expected that the endosomes will later fuse with lysosomes to recycle its content. Interestingly, it has been shown that A20 can target TRAF2 to the lysosome for its degradation, which is dependent on the membrane tethering activity of A20, but not of its ubiquitin-modifying function (108). These CD137-mediated endocytosis experiments (36) were performed with agonist anti-CD137 mAbs and, therefore, it is yet to be determined whether CD137 engagement with CD137L would also cause the internalization of the complex, but it is likely that this will actually occurs for various reasons. First, CD137 internalization has been already observed in dendritic cells upon binding to CD137L fusion proteins used to target antigens for vaccination (109). Second, an accumulation of CD137 on the surface of CD137L-deficient T cells has been observed, probably as a result of the impossibility of CD137 to be internalized in the absence of CD137L (110). Third, because many key molecules in CD137 signaling, such as CD137, TRAF1, TRAF2 and cIAP1/2 are readily transcriptionally activated by NF-κB and AP1 transcription factors upon CD137 activation (111–114), restocking these molecules and ensuring CTL responsiveness to new CD137 costimulatory rounds. Finally, because it has been recently described that tonic chimeric antigen receptor (CAR)-derived CD137 signaling causes T cell toxicity by the continuous TRAF2-mediated NF-κB activation and increased Fas-dependent cell death (115). This result emphasizes the deleterious effects that unrestricted CD137-signaling would have in the cells and underscores the key role of the multiple mechanisms controlling CD137 signaling described above, including CD137 internalization.
CD137 has become one of the most relevant molecular targets in cancer immunotherapy for its ability to drive CTL and NK cells anti-tumor responses. Humanized anti-CD137 mAbs have entered the clinic (21). One of those (urelumab) showed promising anti-tumor effects as a monotherapy treatment in a phase I trial. Unfortunately, a follow-up Phase II trial revealed severe liver toxicity in a significant number of patients (10%) that resulted in two fatalities (116). Consequently, trials with urelumab as a monotherapy were terminated (117). A comprehensive safety analysis of patients treated with urelumab confirmed a strong association between hepatitis and the urelumab dose and resulted in dose reductions in subsequent clinical trials (118). In this regard, ongoing clinical trials with urelumab and other anti-human CD137 mAbs used in combinatory therapies are underway. Alternative approaches are needed to circumvent the off-target toxicity associated to these treatments while preserving their efficacy, for instance, by targeting these agonist antibodies or the natural ligand to surface molecules expressed on cells present in the tumor microenvironment (21). Another important strategy to improve anti-CD137 mAb anti-tumor activity, while limiting its side effects, would be boosting CD137-mediated signal transduction. Many aspects of CD137 signaling might be of interest for drug development, including interfering with negative regulators of CD137 signaling, promoting optimal complex/scaffold formation, and keeping signaling-CD137 endosomes from lysosome degradation, among others. These approaches have been neglected so far due to our limited understanding of the different mechanisms controlling CD137 signal transduction, a limitation that could also be extended to other members of the TNFR family.
These limitations would also apply to the usage of CD137 signaling for enhancing chimeric antigen receptor (CAR) T cell effectiveness. In this regard, transducing T cells with a CAR-construct containing the CD137 cytosolic tail together with the CAR-CD3ζ proved to be effective in increasing CTL cell survival, targeting of CTLs to the tumor and boosting anti-tumor activities (119). Remarkably, it has been recently demonstrated the clinical effectiveness of this therapy in the treatment of relapsed or refractory B-cell acute lymphoblastic leukemia (120, 121).
In the case of CD137 containing CARs, recent evidence shows that the activity of CD19-targeted CAR T cells with a CD137 endodomain is dependent on TRAF1, 2 and 3 and also on NF-κB activation (39). However, little is known on whether the molecular mechanisms controlling the extent of the response are similar to those of native CD137. In this regard, and as stated above, tonic chimeric antigen receptor (CAR)-derived CD137 signaling has been shown to cause T cell toxicity by the continuous TRAF2-mediated NF-κB activation and increased Fas-dependent cell death (115), thus highlighting the need of a better understanding of the molecular mechanisms controlling CD137 signaling. Moreover, the development of CAR T cells with CD137 intracellular tail acting in tandem with the cytoplasmic domain of CD3 ζ may promote a signaling crosstalk between these 2 pathways. Interestingly, and as discussed above, this crosstalking between CD137 and the TCR might be happening at certain extent in normal CD137-signaling (103). In any event, the CD137 component in the CAR T therapy is key to ensure the functional persistence and survival of the transduced T cells (119, 120) a feature ultimately needed for clinical efficacy, but also keeping in mind that unrestrained CD137 activity might also be deleterious for the cell (115). Translational research in the signal transduction pathways controling CD137-mediated responses should focus in the identification of druggable targets that would allow toning up or toning down CD137 activity as needed. In addition, developing tools for early and reliable detection of CD137-signaling events and/or their outcome would be paramount to define pharmacodynamic biomarkers and useful parameters to optimize new generations of CD137 agonists.
JZ, GP-C, PC-B, IM-F, AA, IO, and IM critically revised the manuscript for important intellectual content. JZ, GP-C, PC-B, and IM designed the figures. JZ and IM wrote the paper.
Funding was from Instituto de Salud Carlos III (ISCIII) (PI16/00895 to JZ), Ministerio de Economía y Competitividad (MINECO) RTC-16-5118-1 to JZ and IM, SAF 2014-52361-R and SAF 2017-83267-C2-1R to IM; and Cancer Research Institute (CRI) CLIP Grant 2017, Asociación Española Contra el Cancer, Fundación BBVA and I-ON Network-BMS to IM. The cost of this publication was paid in part by funds from the European Fund for Economic and Regional Development (FEDER).
IM has served as a consultant for Bristol-Myers Squib, Roche-Genentech, Bayer, Alligator, Tusk, Bioncotech, Medimmune, Genmab, F-Star. IM receives commercial grants from Bristol Myers Squibb, Roche-Genentech and Alligator. IM-F is a full time employee for MSD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. (2002) 115:679–88.
2. Zapata JM. TNF-receptor-associated factors as targets for drug development. Expert Opin Ther Targets (2003) 7:411–25. doi: 10.1517/14728222.7.3.411
3. Zapata JM, Martinez-Garcia V, Lefebvre S. Phylogeny of the TRAF/MATH domain. Adv Exp Med Biol. (2007) 597:1–24. doi: 10.1007/978-0-387-70630-6_1
4. Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J Biol Chem. (2001) 276:24242–52. doi: 10.1074/jbc.M100354200
5. Xu LG, Li LY, Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. (2004) 279:17278–82. doi: 10.1074/jbc.C400063200
6. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. (2013) 8:7. doi: 10.1186/1750-2187-8-7
7. Wallis AM, Bishop GA. TRAF3 regulation of inhibitory signaling pathways in B and T lymphocytes by kinase and phosphatase localization. J Leukoc Biol. (2018) 103:1089–98. doi: 10.1002/JLB.2MIR0817-339RR
8. Regnier CH, Masson R, Kedinger V, Textoris J, Stoll I, Chenard MP, et al. Impaired neural tube closure, axial skeleton malformations, and tracheal ring disruption in TRAF4-deficient mice. Proc Natl Acad Sci USA. (2002) 99:5585–90. doi: 10.1073/pnas.052124799
9. Rousseau A, Rio MC, Alpy F. TRAF4, at the crossroad between morphogenesis and cancer. Cancers (2011) 3:2734–49. doi: 10.3390/cancers3022734
10. Lomaga M, Yeh W-C, Sarosi I, Duncan G, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. (1999) 13:1015–24. doi: 10.1101/gad.13.8.1015
11. Li J, Yin Q, Wu H. Structural basis of signal transduction in the TNF receptor superfamily. Adv Immunol. (2013) 119:135–53. doi: 10.1016/B978-0-12-407707-2.00005-9
12. Park HH. Structure of TRAF family: current understanding of receptor recognition. Front Immunol. (2018) 9:1999. doi: 10.3389/fimmu.2018.01999
13. Foight GW, Keating AE. Comparison of the peptide binding preferences of three closely related TRAF paralogs: TRAF2, TRAF3, and TRAF5. Protein Sci. (2016) 25:1273–89. doi: 10.1002/pro.2881
14. Kim CM, Choi JY, Bhat EA, Jeong JH, Son YJ, Kim S, et al. Crystal structure of TRAF1 TRAF domain and its implications in the TRAF1-mediated intracellular signaling pathway. Sci Rep. (2016) 6:25526. doi: 10.1038/srep25526
15. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. (2004) 25:1605–12. doi: 10.1002/jcc.20084
16. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. (1997) 186:47–55. doi: 10.1084/jem.186.1.47
17. Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol. (2001) 167:1313–24. doi: 10.4049/jimmunol.167.3.1313
18. Lin W, Voskens CJ, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood (2008) 112:699–707. doi: 10.1182/blood-2007-11-122465
19. Vinay DS, Kwon BS. 4-1BB signaling beyond T cells. Cell Mol Immunol. (2011) 8:281–4. doi: 10.1038/cmi.2010.82
20. Weigelin B, Bolanos E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci USA. (2015) 112:7551–6. doi: 10.1073/pnas.1506357112
21. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
22. Sanchez-Paulete AR, Labiano S, Rodriguez-Ruiz ME, Azpilikueta A, Etxeberria I, Bolanos E, et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur J Immunol. (2016) 46:513–22. doi: 10.1002/eji.201445388
23. Pollok KE, Kim YJ, Hurtado J, Zhou Z, Kim KK, Kwon BS. 4-1BB T-cell antigen binds to mature B cells and macrophages, and costimulates anti-mu-primed splenic B cells. Eur J Immunol. (1994) 24:367–74. doi: 10.1002/eji.1830240215
24. Kang SW, Lee SC, Park SH, Kim J, Kim HH, Lee HW, et al. Anti-CD137 Suppresses tumor growth by blocking reverse signaling by CD137 ligand. Cancer Res. (2017) 77:5989–6000. doi: 10.1158/0008-5472.CAN-17-0610
25. Chalupny NJ, Peach R, Hollenbaugh D, Ledbetter JA, Farr AG, Aruffo A. T-cell activation molecule 4-1BB binds to extracellular matrix proteins. Proc Natl Acad Sci USA. (1992) 89:10360–4 doi: 10.1073/pnas.89.21.10360
26. Madireddi S, Eun SY, Lee SW, Nemcovicova I, Mehta AK, Zajonc DM, et al. Galectin-9 controls the therapeutic activity of 4-1BB-targeting antibodies. J Exp Med. (2014) 211:1433–48. doi: 10.1084/jem.20132687
27. Won EY, Cha K, Byun JS, Kim DU, Shin S, Ahn B, et al. The structure of the trimer of human 4-1BB ligand is unique among members of the tumor necrosis factor superfamily. J Biol Chem. (2010) 285:9202–10. doi: 10.1074/jbc.M109.084442
28. Vanamee ES, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. (2018) 11:eaao4910. doi: 10.1126/scisignal.aao4910
29. Magis C, van der Sloot AM, Serrano L, Notredame C. An improved understanding of TNFL/TNFR interactions using structure-based classifications. Trends Biochem Sci. (2012) 37:353–63. doi: 10.1016/j.tibs.2012.06.002
30. Arch R, Thompson C. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclearfactor kB. Mol Cell Biol. (1998) 18:558–65. doi: 10.1128/MCB.18.1.558
31. Jang IK, Lee ZH, Kim YJ, Kim SH, Kwon BS. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem Biophys Res Commun. (1998) 242:613–20. doi: 10.1006/bbrc.1997.8016
32. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med. (1998)187:1849–62.
33. Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell (2010) 38:101–13. doi: 10.1016/j.molcel.2010.03.009
34. Nam KO, Kang H, Shin SM, Cho KH, Kwon B, Kwon BS, et al. Cross-linking of 4-1BB activates TCR-signaling pathways in CD8+ T lymphocytes. J Immunol. (2005) 174:1898–905. doi: 10.4049/jimmunol.174.4.1898
35. Cannons JL, Hoeflich KP, Woodgett JR, Watts TH. Role of the stress kinase pathway in signaling via the T cell costimulatory receptor 4-1BB. J Immunol. (1999) 163:2990–8.
36. Martinez-Forero I, Azpilikueta A, Bolanos-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol. (2013) 190:6694–706. doi: 10.4049/jimmunol.1203010
37. Wang C, McPherson AJ, Jones RB, Kawamura KS, Lin GH, Lang PA, et al. Loss of the signaling adaptor TRAF1 causes CD8+ T cell dysregulation during human and murine chronic infection. J Exp Med. (2012) 209:77–91. doi: 10.1084/jem.20110675
38. McPherson AJ, Snell LM, Mak TW, Watts TH. Opposing roles for TRAF1 in the alternative versus classical NF-kappaB pathway in T cells. J Biol Chem. (2012) 287:23010–9. doi: 10.1074/jbc.M112.350538
39. Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4-1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI Insight (2018) 3:121322. doi: 10.1172/jci.insight.121322
40. Zapata JM, Reed JC. TRAF1: lord without a RING. Science STKE (2002) 2002:pe27. doi: 10.1126/stke.2002.133.pe27
41. Greenfeld H, Takasaki K, Walsh MJ, Ersing I, Bernhardt K, Ma Y, et al. TRAF1 coordinates polyubiquitin signaling to enhance epstein-barr virus lmp1-mediated growth and survival pathway activation. PLoS Pathog. (2015) 11:e1004890. doi: 10.1371/journal.ppat.1004890
42. Mace PD, Smits C, Vaux DL, Silke J, Day CL. Asymmetric recruitment of cIAPs by TRAF2. J Mol Biol. (2010) 400:8–15. doi: 10.1016/j.jmb.2010.04.055
43. Lee SY, Choi Y. TRAF1 and its biological functions. Adv Exp Med Biol. (2007) 597:25–31. doi: 10.1007/978-0-387-70630-6_2
44. Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, et al. TRAF1 is a negative regulator of TNF signaling: enhanced TNF signaling in TRAF1-deficient mice. Immunity (2001) 15:647–57. doi: 10.1016/S1074-7613(01)00207-2
45. Arron JR, Pewzner-Jung Y, Walsh MC, Kobayashi T, Choi Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J Exp Med. (2002) 196:923–34 doi: 10.1084/jem.20020774
46. Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA. Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J Immunol. (2006) 176:5388–400 doi: 10.4049/jimmunol.176.9.5388
47. Speiser DE, Lee SY, Wong B, Arron J, Santana A, Kong YY, et al. A regulatory role for TRAF1 in antigen-induced apoptosis of T cells. J Exp Med. (1997) 185:1777–83. doi: 10.1084/jem.185.10.1777
48. Sabbagh L, Pulle G, Liu Y, Tsitsikov EN, Watts TH. ERK-dependent Bim modulation downstream of the 4-1BB-TRAF1 signaling axis is a critical mediator of CD8 T cell survival in vivo. J Immunol. (2008) 180:8093–101 doi: 10.4049/jimmunol.180.12.8093
49. Watts TH, Lin GH, Wang C, McPherson AJ, Snell LM, Sabbagh L. Role of 4-1BBL and TRAF1 in the CD8 T cell response to influenza virus and HIV. Adv Exp Med Biol. (2011) 691:177–86. doi: 10.1007/978-1-4419-6612-4_19
50. Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature (2010) 465:1084–8. doi: 10.1038/nature09128
51. Shi CS, Kehrl JH. Tumor necrosis factor (TNF)-induced germinal center kinase-related (GCKR) and stress-activated protein kinase (SAPK) activation depends upon the E2/E3 complex Ubc13-Uev1A/TNF receptor-associated factor 2 (TRAF2). J Biol Chem. (2003) 278:15429–34. doi: 10.1074/jbc.M211796200
52. Habelhah H, Takahashi S, Cho SG, Kadoya T, Watanabe T, Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. Embo J. (2004) 23:322–32. doi: 10.1038/sj.emboj.7600044
53. Gonzalvez F, Lawrence D, Yang B, Yee S, Pitti R, Marsters S, et al. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell (2012) 48:888–99. doi: 10.1016/j.molcel.2012.09.031
54. Robeson AC, Lindblom KR, Wojton J, Kornbluth S, Matsuura K. Dimer-specific immunoprecipitation of active caspase-2 identifies TRAF proteins as novel activators. EMBO J. (2018) 37:e97072. doi: 10.15252/embj.201797072
55. Yin Q, Lamothe B, Darnay BG, Wu H. Structural basis for the lack of E2 interaction in the RING domain of TRAF2. Biochemistry (2009) 48:10558–67. doi: 10.1021/bi901462e
56. Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. (2004) 279:26243–50. doi: 10.1074/jbc.M403286200
57. Sanjo H, Zajonc DM, Braden R, Norris PS, Ware CF. Allosteric regulation of the ubiquitin:NIK and ubiquitin:TRAF3 E3 ligases by the lymphotoxin-beta receptor. J Biol Chem. (2010) 285:17148–55. doi: 10.1074/jbc.M110.105874
58. He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G. Specificity of TRAF3 in its negative regulation of the noncanonical NF-kappa B pathway. J Biol Chem. (2007) 282:3688–94. doi: 10.1074/jbc.M610271200
59. Lin WW, Hildebrand JM, Bishop GA. A complex relationship between TRAF3 and non-canonical NF-kappaB2 activation in B lymphocytes. Front Immunol. (2013) 4:477. doi: 10.3389/fimmu.2013.00477
60. Zapata JM, Krajewska M, Krajewski S, Kitada S, Welsh K, Monks A, et al. TNFR-associated factor family protein expression in normal tissues and lymphoid malignancies. J Immunol. (2000) 165:5084–96. doi: 10.4049/jimmunol.165.9.5084
61. Schwenzer R, Siemienski K, Liptay S, Schubert G, Peters N, Scheurich P, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. (1999) 274:19368–74. doi: 10.1074/jbc.274.27.19368
62. Lee SW, Park Y, Song A, Cheroutre H, Kwon BS, Croft M. Functional dichotomy between OX40 and 4-1BB in modulating effector CD8 T cell responses. J Immunol. (2006) 177:4464–72. doi: 10.4049/jimmunol.177.7.4464
63. Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol. (2002) 168:4897–906. doi: 10.4049/jimmunol.168.10.4897
64. Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Ann Rev Biophy. (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
65. Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta (2014) 1843:47–60. doi: 10.1016/j.bbamcr.2013.05.026
66. Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol. (2009) 16:658–66. doi: 10.1038/nsmb.1605
67. Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. (2006) 281:1080–90. doi: 10.1074/jbc.M509381200
68. Lopez J, John SW, Tenev T, Rautureau GJ, Hinds MG, Francalanci F, et al. CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol Cell (2011) 42:569–83. doi: 10.1016/j.molcel.2011.04.008
69. Sabbagh L, Srokowski CC, Pulle G, Snell LM, Sedgmen BJ, Liu Y, et al. A critical role for TNF receptor-associated factor 1 and Bim down-regulation in CD8 memory T cell survival. Proc Natl Acad Sci USA. (2006) 103:18703–8. doi: 10.1073/pnas.0602919103
70. Sabbagh L, Andreeva D, Laramee GD, Oussa NA, Lew D, Bisson N, et al. Leukocyte-specific protein 1 links TNF receptor-associated factor 1 to survival signaling downstream of 4-1BB in T cells. J Leukoc Biol. (2013) 93:713–21. doi: 10.1189/jlb.1112579
71. Abdul-Sater AA, Edilova MI, Clouthier DL, Mbanwi A, Kremmer E, Watts TH. The signaling adaptor TRAF1 negatively regulates Toll-like receptor signaling and this underlies its role in rheumatic disease. Nat Immunol. (2017) 18:26–35. doi: 10.1038/ni.3618
72. Stunz LL, Bishop GA. Latent membrane protein 1 and the B lymphocyte-a complex relationship. Crit Rev Immunol. (2014) 34:177–98. doi: 10.1615/CritRevImmunol.2014010041
73. Borghi A, Haegman M, Fischer R, Carpentier I, Bertrand MJM, Libert C, et al. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-kappaB signaling. Biochem Pharmacol. (2018) 153:292–8. doi: 10.1016/j.bcp.2018.01.039
74. Takiuchi T, Nakagawa T, Tamiya H, Fujita H, Sasaki Y, Saeki Y, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells (2014) 19:254–72. doi: 10.1111/gtc.12128
75. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. (2012) 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x
76. Chen J, Chen ZJ. Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol. (2013) 25:4–12. doi: 10.1016/j.coi.2012.12.005
77. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
78. Mace PD, Linke K, Feltham R, Schumacher FR, Smith CA, Vaux DL, et al. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J Biol Chem. (2008) 283:31633–40. doi: 10.1074/jbc.M804753200
79. Vandenabeele P, Bertrand MJ. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat Rev Immunol. (2012) 12:833–44. doi: 10.1038/nri3325
80. Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem. (2010) 285:5347–60. doi: 10.1074/jbc.M109.076976
81. Hu L, Xu J, Xie X, Zhou Y, Tao P, Li H, et al. Oligomerization-primed coiled-coil domain interaction with Ubc13 confers processivity to TRAF6 ubiquitin ligase activity. Nat Commun. (2017) 8:814. doi: 10.1038/s41467-017-01290-0
82. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
83. Lin SC, Chung JY, Lamothe B, Rajashankar K, Lu M, Lo YC, et al. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol. (2008) 376:526–40. doi: 10.1016/j.jmb.2007.11.092
84. Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol. (2000) 165:6193–204. doi: 10.4049/jimmunol.165.11.6193
85. Li S, Wang L, Dorf ME. PKC phosphorylation of TRAF2 mediates IKKalpha/beta recruitment and K63-linked polyubiquitination. Mol Cell (2009) 33:30–42. doi: 10.1016/j.molcel.2008.11.023
86. Oussa NA, Soumounou Y, Sabbagh L. TRAF1 phosphorylation on Serine 139 modulates NF-kappaB activity downstream of 4-1BB in T cells. Biochem Biophys Res Commun. (2013) 432:129–34. doi: 10.1016/j.bbrc.2013.01.073
87. Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. (2011) 21:71–85. doi: 10.1038/cr.2010.177
88. He L, Grammer AC, Wu X, Lipsky PE. TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-kB activation. J Biol Chem. (2004) 279:55855–65. doi: 10.1074/jbc.M407284200
89. Choudhary S, Kalita M, Fang L, Patel KV, Tian B, Zhao Y, et al. Inducible tumor necrosis factor (TNF) receptor-associated factor-1 expression couples the canonical to the non-canonical NF-kappaB pathway in TNF stimulation. J Biol Chem. (2013) 288:14612–23. doi: 10.1074/jbc.M113.464081
90. Azpilikueta A, Bolanos E, Lang V, Labiano S, Aznar MA, Etxeberria I, et al. Deubiquitinases A20 and CYLD modulate costimulatory signaling via CD137 (4-1BB). Oncoimmunology (2017) 7:e1368605. doi: 10.1080/2162402X.2017.1368605
91. Evans PC, Smith TS, Lai MJ, Williams MG, Burke DF, Heyninck K, et al. A novel type of deubiquitinating enzyme. J Biol Chem. (2003) 278:23180–6. doi: 10.1074/jbc.M301863200
92. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430:694–9. doi: 10.1038/nature02794
93. Kupka S, Reichert M, Draber P, Walczak H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS J. (2016) 283:2626–39. doi: 10.1111/febs.13644
94. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science (2010) 327:1135–9. doi: 10.1126/science.1182364
95. Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell (2011) 44:559–71. doi: 10.1016/j.molcel.2011.09.015
96. Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-kappaB regulation. EMBO J. (2012) 31:3856–70. doi: 10.1038/emboj.2012.241
97. Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, et al. A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J. (2012) 31:3845–55. doi: 10.1038/emboj.2012.240
98. Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature (2003) 424:801–5. doi: 10.1038/nature01802
99. Reiley W, Zhang M, Wu X, Granger E, Sun SC. Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol Cell Biol. (2005) 25:3886–95. doi: 10.1128/MCB.25.10.3886-3895.2005
100. Feng X, Gaeta ML, Madge LA, Yang JH, Bradley JR, Pober JS. Caveolin-1 associates with TRAF2 to form a complex that is recruited to tumor necrosis factor receptors. J Biol Chem. (2001) 276:8341–9. doi: 10.1074/jbc.M007116200
101. Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J Biol Chem. (2000) 275:271–8. doi: 10.1074/jbc.275.1.271
102. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. (2013) 14:98–112. doi: 10.1038/nrm3512
103. So T, Croft M. Regulation of PI-3-kinase and Akt signaling in T lymphocytes and other cells by TNFR family molecules. Front Immunol. (2013) 4:139. doi: 10.3389/fimmu.2013.00139
104. Lasserre R, Guo XJ, Conchonaud F, Hamon Y, Hawchar O, Bernard AM, et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat Chem Biol. (2008) 4:538–47. doi: 10.1038/nchembio.103
105. Starck L, Scholz C, Dorken B, Daniel PT. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIP(short) via phosphatidylinositol 3-kinase and AKT/protein kinase B. Eur J Immunol. (2005) 35:1257–66. doi: 10.1002/eji.200425686
106. Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol. (2003) 33:2133–41. doi: 10.1002/eji.200323996
107. Lee DY, Choi BK, Lee DG, Kim YH, Kim CH, Lee SJ, et al. 4-1BB signaling activates the t cell factor 1 effector/beta-catenin pathway with delayed kinetics via ERK signaling and delayed PI3K/AKT activation to promote the proliferation of CD8+ T Cells. PLoS ONE (2013) 8:e69677. doi: 10.1371/journal.pone.0069677
108. Li L, Soetandyo N, Wang Q, Ye Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim Biophys Acta (2009) 1793:346–53. doi: 10.1016/j.bbamcr.2008.09.013
109. Sharma RK, Schabowsky RH, Srivastava AK, Elpek KG, Madireddi S, Zhao H, et al. 4-1BB ligand as an effective multifunctional immunomodulator and antigen delivery vehicle for the development of therapeutic cancer vaccines. Cancer Res. (2010) 70:3945–54. doi: 10.1158/0008-5472.CAN-09-4480
110. Eun SY, Lee SW, Xu Y, Croft M. 4-1BB ligand signaling to T cells limits T cell activation. J Immunol. (2015) 194:134–41. doi: 10.4049/jimmunol.1401383
111. Kim JO, Kim HW, Baek KM, Kang CY. NF-kappaB and AP-1 regulate activation-dependent CD137 (4-1BB) expression in T cells. FEBS Lett. (2003) 541:163–70. doi: 10.1016/S0014-5793(03)00326-0
112. Dunn IF, Sannikova TY, Geha RS, Tsitsikov EN. Identification and characterization of two CD40-inducible enhancers in the mouse TRAF1 gene locus. Mol Immunol. (2000) 37:961–73. doi: 10.1016/S0161-5890(01)00015-3
113. Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF. NF-kappaB2 mutation targets TRAF1 to induce lymphomagenesis. Blood (2007) 110:743–51. doi: 10.1182/blood-2006-11-058446
114. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science (1998) 281:1680–3.
115. Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. (2017) 21:17–26. doi: 10.1016/j.celrep.2017.09.015
116. Melero I, Hirschhorn-Cymerman D, Morales-Kastresana A, Sanmamed MF, Wolchok JD. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin Cancer Res. (2013) 19:1044–53. doi: 10.1158/1078-0432.CCR-12-2065
117. Bartkowiak T, Curran MA. 4-1BB Agonists: multi-potent potentiators of tumor immunity. Front Oncol. (2015) 5:117. doi: 10.3389/fonc.2015.00117
118. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist Anti-CD137 monoclonal antibody. Clin Cancer Res. (2017) 23:1929–36. doi: 10.1158/1078-0432.CCR-16-1272
119. Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. (2011) 71:4617–27. doi: 10.1158/0008-5472.CAN-11-0422
120. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
Keywords: CD137, 4-1BB, TNFR, TRAF1, TRAF2, TRAF3, Immunotherapy, cytotoxic T lymphocytes (CTL)
Citation: Zapata JM, Perez-Chacon G, Carr-Baena P, Martinez-Forero I, Azpilikueta A, Otano I and Melero I (2018) CD137 (4-1BB) Signalosome: Complexity Is a Matter of TRAFs. Front. Immunol. 9:2618. doi: 10.3389/fimmu.2018.02618
Received: 03 September 2018; Accepted: 24 October 2018;
Published: 15 November 2018.
Edited by:
Tania H. Watts, University of Toronto, CanadaReviewed by:
Shao-Cong Sun, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2018 Zapata, Perez-Chacon, Carr-Baena, Martinez-Forero, Azpilikueta, Otano and Melero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan M. Zapata, am16YXBhdGFAaWliLnVhbS5lcw==
Ignacio Melero, aW1lbGVyb0B1bmF2LmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.