 Yu-Ching Su
Yu-Ching Su Farshid Jalalvand2
Farshid Jalalvand2 Kristian Riesbeck
Kristian Riesbeck
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 November 2018
Sec. Microbial Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02530
This article is part of the Research TopicUpdate on the Immune Mechanisms Against Respiratory PathogensView all 29 articles
Chronic obstructive pulmonary disease (COPD) is a debilitating respiratory disease and one of the leading causes of morbidity and mortality worldwide. It is characterized by persistent respiratory symptoms and airflow limitation due to abnormalities in the lower airway following consistent exposure to noxious particles or gases. Acute exacerbations of COPD (AECOPD) are characterized by increased cough, purulent sputum production, and dyspnea. The AECOPD is mostly associated with infection caused by common cold viruses or bacteria, or co-infections. Chronic and persistent infection by non-typeable Haemophilus influenzae (NTHi), a Gram-negative coccobacillus, contributes to almost half of the infective exacerbations caused by bacteria. This is supported by reports that NTHi is commonly isolated in the sputum from COPD patients during exacerbations. Persistent colonization of NTHi in the lower airway requires a plethora of phenotypic adaptation and virulent mechanisms that are developed over time to cope with changing environmental pressures in the airway such as host immuno-inflammatory response. Chronic inhalation of noxious irritants in COPD causes a changed balance in the lung microbiome, abnormal inflammatory response, and an impaired airway immune system. These conditions significantly provide an opportunistic platform for NTHi colonization and infection resulting in a “vicious circle.” Episodes of large inflammation as the consequences of multiple interactions between airway immune cells and NTHi, accumulatively contribute to COPD exacerbations and may result in worsening of the clinical status. In this review, we discuss in detail the interplay and crosstalk between airway immune residents and NTHi, and their effect in AECOPD for better understanding of NTHi pathogenesis in COPD patients.
The lungs are vital organs involved in gas exchange between the vascular system and the external environment, thus they are greatly exposed to the environment-derived microorganisms, including fungi, viruses, and bacteria. The bronchial tree and parenchymal tissues of the lungs, that until recently were considered as sterile, are colonized by phylogenetically-diverse microbes. The genera of Firmicutes, Bacteroidetes, and Proteobacteria are the most common phyla identified and represent 60% of the total bacterial microbiome in the healthy airway (1, 2). The majority of the lung microbiota belongs to the normal flora that play an important role in the pulmonary epithelial integrity, colonization resistance, and homeostasis of the immune system in the respiratory tract (3). A small fraction of them are, however, potentially pathogenic microorganisms that are involved in a variety of lung diseases, as exemplified by the genus Haemophilus. Non-typeable Haemophilus influenzae (NTHi) is a Gram-negative coccobacillus that are commonly residing in the human airways. Uniquely and yet unexplained, NTHi is a commensal when colonizing the nasopharynx or throat, but pathogenic in the lower airways triggering a robust inflammatory response [for reviews see (4, 5)]. NTHi is considered a potential opportunistic pathogen as it frequently infects the lower respiratory tract of lungs with structural damage as a consequence of non-infectious lung diseases or mechanical injuries. Moreover, NTHi occasionally causes bronchitis and pneumonia (6). In addition, lower airway colonization by NTHi has been associated with disease progression of several more or less non-infectious lung diseases such as bronchiectasis (7), cystic fibrosis (8), interstitial lung diseases (9, 10), but mostly in chronic obstructive pulmonary disease (COPD) (11, 12). COPD is a severe inflammatory lung disease characterized by airflow limitation with a range of pathological changes. Both genetics and environmental factors trigger the onset of COPD, however, microbes including NTHi play an important role in the acute exacerbations. This review describes the disease progression of COPD in the context of host immune-interactions linked to NTHi, and the overall impact in disease exacerbation.
COPD is the third leading cause of morbidity and mortality worldwide expected to affect more than 210 million people by 2030 (13, 14). According to the Global Initiative for Chronic Obstructive Lung Disease (GOLD), COPD is a pulmonary disease that is manageable, but significant exacerbations and co-morbidities may, however, contribute to the overall severity in individual patients (15). COPD is characterized by chronic airflow limitation of the peripheral airways with a range of pathological changes in the lung that are not fully reversible, and usually become progressively worse over time. The progression of COPD is associated with an abnormal inflammatory response of the lung to noxious particles or gases.
From a pathological point of view, COPD comprises a group of pulmonary abnormalities related to the inflammatory reaction of the airways, alveoli, and pulmonary vessels (16–19). These include (i) pulmonary emphysema, (ii) chronic bronchitis, and (iii) disease in the small airways. The pulmonary abnormalities progressively affect all parts of the lung, resulting in increased resistance of the conducting airways and thus chronic airflow obstruction that eventually will lead to a declined lung function. Emphysema is a permanent loss of elastic lung recoil caused by elastolytic destruction and enlargement of the alveolar wall distal to the terminal bronchioles. This consequently results in the loss of alveolar attachments to the small airways and thus limitation of airflow and gaseous exchanges. Chronic bronchitis is characterized by consecutive and chronic cough with expectorations that last for more than 3 months within 2 years. It is associated with inflammation of the bronchial walls with increased inflammatory infiltrates, hyperplasia of goblet cells, hypertrophy of tracheobronchial submucosa, increased mucous secretion and, finally, dilatation of the airway ducts (airways of about 2–4 mm in internal diameter). The majority of the ciliated epithelium lining the airways are also either compromised or dysfunctionnal, and may be replaced by non-ciliated squamous epithelial cells. Small airway diseases, on the other hand, involve hyperplasia and metaplasia of mucosal glands and goblet cells, hypersecretion of intraluminal mucus, macrophage bronchiolitis, and accumulation of lymphocytes in the small bronchioles (airways of ~2 mm or less in diameter and terminal bronchioles). In addition, distortion, fibrosis, stenosis, tortuosities, hyperplasia, and hypertrophia of the small airway smooth muscles also contribute to the loss of elasticity in the lung parenchyma. Although COPD mainly affects the lungs, it also produces significant extrapulmonary consequences as a results of an escalated inflammatory response orchestrated by airway cells and immune mediators (20, 21). The co-morbidities are commonly seen in COPD patients despite the actual mechanism responsible for the systemic inflammation remains to be elucidated.
The development of COPD is multifactorial, with cigarette or tobacco smoking being the primary cause of COPD (22, 23). Other risk factors that may promote the onset and progression of COPD includes prolonged occupational exposure to particles/gases in mining and textile industries, air pollution resulting from biomass combustion, and bronchial hyperresponsiveness (16, 18, 24). The variability of COPD incidences among smokers is also explained by a genetic pre-disposition, such as α1-antitrypsin deficiency and cutis laxa [mutation of the elastin gene (ELN)] (25, 26). The α1-antitrypsin deficiency is caused by deleterious homozygous mutations in SERPINA1 which contributes to 1–2% of COPD cases. The deficiency results in increased neutrophil elastase activity that ultimately leads to the degradation and collapse of the alveoli. Importantly, meta-analyses of genome-wide association studies (GWAS) and other genotyping studies have revealed that multiple single nucleotide polymorphism (SNPs) in at least 34 genes from different pulmonary genomic loci are associated with COPD susceptibility (27–30).
Airway epithelium exposed to cigarette or tobacco smoke has compromised tight junctions and delayed epithelial wound repair (31–34). Moreover, cigarette smoke alters basal cell differentiation and subepithelial extracellular matrix (ECM) composition, and thus causes airway remodeling (i.e., goblet cell hyperplasia and small airway squamous metaplasia) (35–37). This results in mucus hypersecretion, impaired mucocilliary clearance, and airway obstruction. Tobacco or cigarette smoke also enhances proliferation and ECM deposition by activating the extracellular signal related kinase (ERK) and the p38 signaling pathway (38). The alteration of major ECM components are widespread in all lung compartments in COPD patients with a total increase of type I and III collagens, fibronectin, and laminin in parallel with reduced concentrations of proteoglycans, perlecan decorin, versican, biglycan, tenascin and elastin (39, 40). Cigarette induced-overexpression of matrix metalloproteases (MMPs-1, 2, 7, 9, 12, and 28) and elastase has also been reported, and may contribute to the airway tissue destruction and fibrosis (41–43). In addition, harmful volatile chemicals derived from cigarette smoke (i.e., acetaldehyde, acrolein, and crotonaldehyde) are prone to form carcinogen adducts with DNA and various proteins (i.e., apoliprotein E and surfactant protein A). They also dysregulate airway epithelial ion transport, disrupt the phagocytic activity of airway phagocytes, and diminish the airway surface liquid volume (44–46).
Numerous proteomics and transcriptomic analyses have unveiled the crucial impact of cigarette or tobacco smoke and COPD disease progression on airway gene expression (47, 48). The differential gene expression studies were done using COPD experimental models or clinical samples [i.e., bronchial epithelial cells, sputum, plasma, blood, and bronchoalveolar lavage (BAL) fluid]. Collectively, most of the altered genes are involved in oxidative stress, xenobiotic metabolism, antioxidant responses, DNA repair, ECM remodeling, inflammatory responses, and immune defenses, which the latter two are our major interest of discussion in this review. The omics data aid in the increased knowledge of molecular mechanisms in COPD. They may reflect the dynamic response and attempts by the airway epithelial cells to repair the cytotoxic injury primarily triggered by inhaled irritants. Deleterious and irreversible alterations occurring and interfering with the airway epithelial homeostasis and immune defense may promote COPD development and progression. Notably, gene alterations in phagosomal- and leukocyte transendothelial migration pathways (LTM) are significantly correlated with the level of T cells and airway obstruction in smokers (49). The LTM, however, were found to be further dysregulated in COPD patients. Hence, in addition to clinical/physiology variables, a number of gene products with significant differential gene expression may be targeted as specific proteomic signatures or biomarkers for early COPD detection, patient monitoring, disease subgrouping, and finally treatment selection (50, 51).
Tobacco or cigarette smoke regulates airway gene expression via two main mechanisms, by altering the status of (i) chromatin remodeling, and (ii) DNA methylation of the target genes (Figure 1) (52–54).
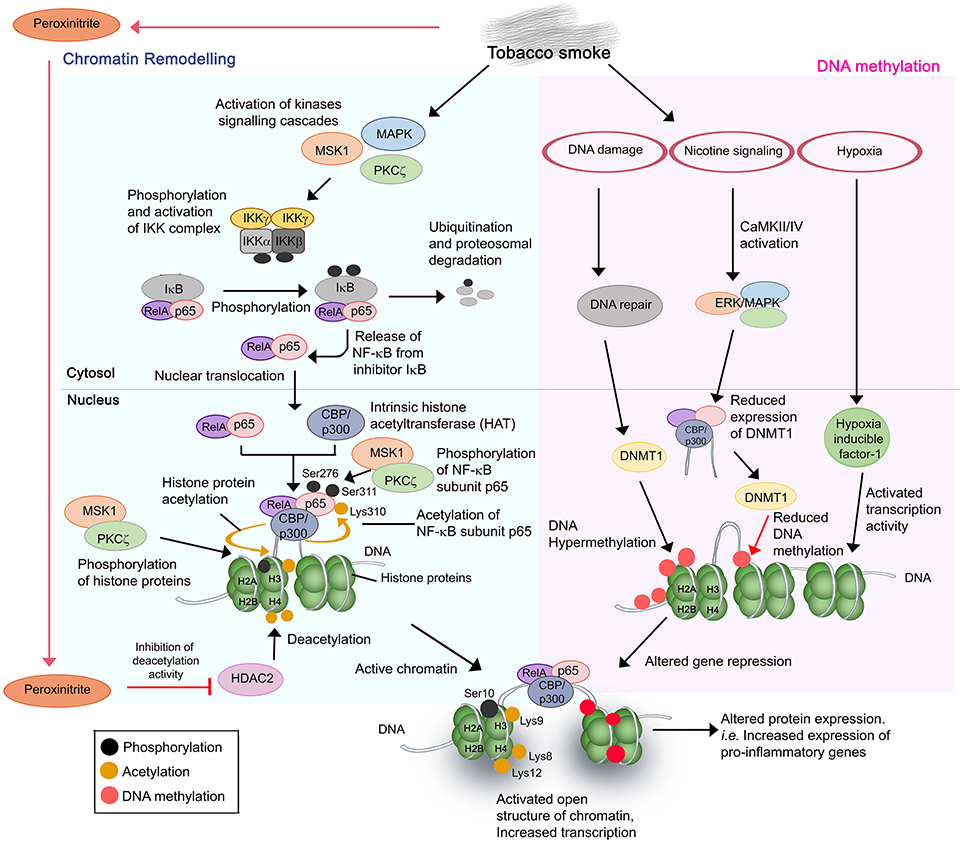
Figure 1. Cigarette and tobacco smoke has several effects on gene regulation. Nicotine and other compounds in the smoke alter gene expression by two pathways, firstly, chromatin remodeling (Left) and secondly, DNA methylation (Right). Chromatin remodeling involves activation of kinases signaling pathways, activation and nuclear translocation of transcription factor NF-κB (RelA/p65), and complex formation with CBP/p300 on specific DNA sites. CBP/p300 is intrinsically a histone acetyltransferase (HAT). Subunit p65 is further phosphorylated at Ser276 and Ser311, respectively, by MSK1 and PKCζ, whereas CBP acetylates p65 at Lys310. The phosphorylation and acetylation enhance the interaction within the NF-κB/CBP/p300 complex while stabilizing the DNA binding of NF-κB. The complex of NF-κB/CBP/p300 then modifies the histones through CBP-mediated acetylations of histone H3 (at Lys9) and H4 at Lys 8 and Lys12, and phosphorylation of H3 at Ser10 by MSK1 and PKCζ. This results in the structure change of chromatin, from a condensed structure (repressed) to an activated open conformation. The transcription of target genes is therefore increased. In the second mechanism, several side effects resulting from cigarette smoking such as DNA damage and nicotine signaling could trigger the hypermethylation or decreased methylation of target DNA. This may lead to DNA methylation anomalies and thus altered DNA expression. Resulting hypoxia due to high concentrations of carbon monoxide also contributes to altered gene expression. The aberrant gene expression by cigarette smoke mostly occurs in pro-inflammatory genes with resulting increased production of inflammatory mediators, and amplified inflammation in the COPD lung upon exposure.
Chromatin remodeling is a result of a disrupted balance in histone acetylation/deacetylation (55). Excessive activation of more than 20 transcription factors including NF-κB, and lipoprotein peroxidation products (peroxinitrite, acrolein, and 4HNE from tobacco smoking) contributes to such anomaly. NF-κB is a key inflammatory and redox-sensitive transcription factor that plays a direct role in cigarette smoke-induced airway inflammation. NF-κB has been described as a “smoke-sensor” due to its sensitive activation by tobacco residues (56). Stimulation of multiple signaling cascades [p38 mitogen-activated protein (MAPK) kinases, mitogen and stress-activated kinase 1 (MSK1), protein kinase C zeta (PKCζ), and IκB kinase (IKK) complex (IKKα, IKKβ, and NEMO)] by tobacco residues promotes the activation and nuclear translocation of transcription factor NF-κB RelA/p65 (54, 57–64). This is followed by a complex formation of NF-κB/CBP-p300 [coactivator, CREB-binding protein (CBP) or CBP/p300] at target DNA sequences. It should be noted that CBP/p300 also has intrinsic histone acetyltransferase (HAT) activity. Subsequent acetylation and phosphorylation of the subunit p65 in the NF-κB/CBP-p300 complex by the activated MSK1/PKCζ-signaling pathways (and other 11 different kinases), and CBP/p300, respectively, are required for the full activation of NF-κB (57, 60, 63). This enhances the DNA binding affinity of the complex. Histones H3 and H4 in the chromatin complex of target sequences are then being acetylated (histone H3 at Lys9; H4 at Lys8 and Lys12) and phosphorylated (histone H3 at Ser10) by the subunit CBP of the NF-κB/CBP-p300 complex, and the activated MSK1 and PKCζ, respectively. The hyperacetylated core histones, however, fail to be neutralized or deacetylated by a dysfunctional histone deacetylase (HDAC2). Peroxinitrite nitrates the tyrosine residues of the HDAC2 and causes inhibition of activation and reduced expression of the protein. Of note, peroxinitrite is a by-product generated from the immune cell-derived nitrite oxide (NO) and reactive oxygen species (ROS) of cigarette smoke (65, 66).
Cigarette or tobacco smoke disturbs the DNA methylation status of target genes through several mechanisms. Firstly, DNA damage caused by cigarette smoke stimulates the DNA methyltransferase 1 (DNMT) to actively induce CpGs methylation at the damaged site (67). The hypermethylation is prone to introduce error of methylation in some target genes, resulting in reduced gene expression. Secondly, activation of nicotine signaling pathway by tobacco smoke causes CaMKII/IV and ERK/MAPK pathway activation that subsequently induces the activity of CBP to suppress the expression of DNMT1. This may result in reduced DNA methylation and thus altered level of gene repression by DNMT (68–70). Finally, enhanced activities of transcription factors such as hypoxia inducible factor 1 due to the high level of carbon monoxide and hypoxia have also been reported to influence airway gene expression (71).
Consequently, the combinatorial effect from both aberrant acetylation of histone and DNA methylation promotes the transformation of chromatin from a condensed structure to an activated open conformation. This facilitates irregular accessibility of DNA for transcription machineries, hence irregular gene expression by various cell types in the airway. The mechanisms reported are responsible for increased expression of NF-κB-dependent proinflammatory gene products [i.e., IL-1β, IL-6, IL-8, CCL-5 cyclooxygenase (COX)-2, and MIP-2/CXCL2] in both pulmonary structural cells (bronchial, small airway, and alveolar epithelial cells) and immune cells (alveolar macrophages), increased VEGF and iNOS in nasal fibroblasts and lymphocytes (Jurkat T cells), respectively, and decreased activity of antioxidant transcription factor Nrf2 and α1-antitrypsin in bronchial epithelial cells (54, 56, 57, 59, 62–64, 72–79). These may contribute to the anatomical anomalies in the airway and excessive inflammatory responses among smokers during the course of COPD.
COPD is associated with chronic inflammation in the peripheral airways orchestrated by both innate and adaptive immune responses that are interconnected via dendritic cells (80). Increasing numbers of inflammatory cells (neutrophils, macrophages, T and B lymphocytes, mast cell, eosinophils, and dendritic cells) and inflammatory mediators are accumulated in the airway lumen/wall in the lung parenchyma (19, 81). These immune cells and inflammatory mediators can hence be detected in the sputum and BAL fluid of COPD patients. The level of accumulation is positively correlated with disease severity. An increasing number of studies using animal models and clinical tissues have reported the nature of excessive airway inflammatory responses in COPD. Despite this, the heterogeneity in symptoms progression among COPD patients remain unexplained. The overall mechanism of COPD inflammatory immune response is depicted in Figure 2.
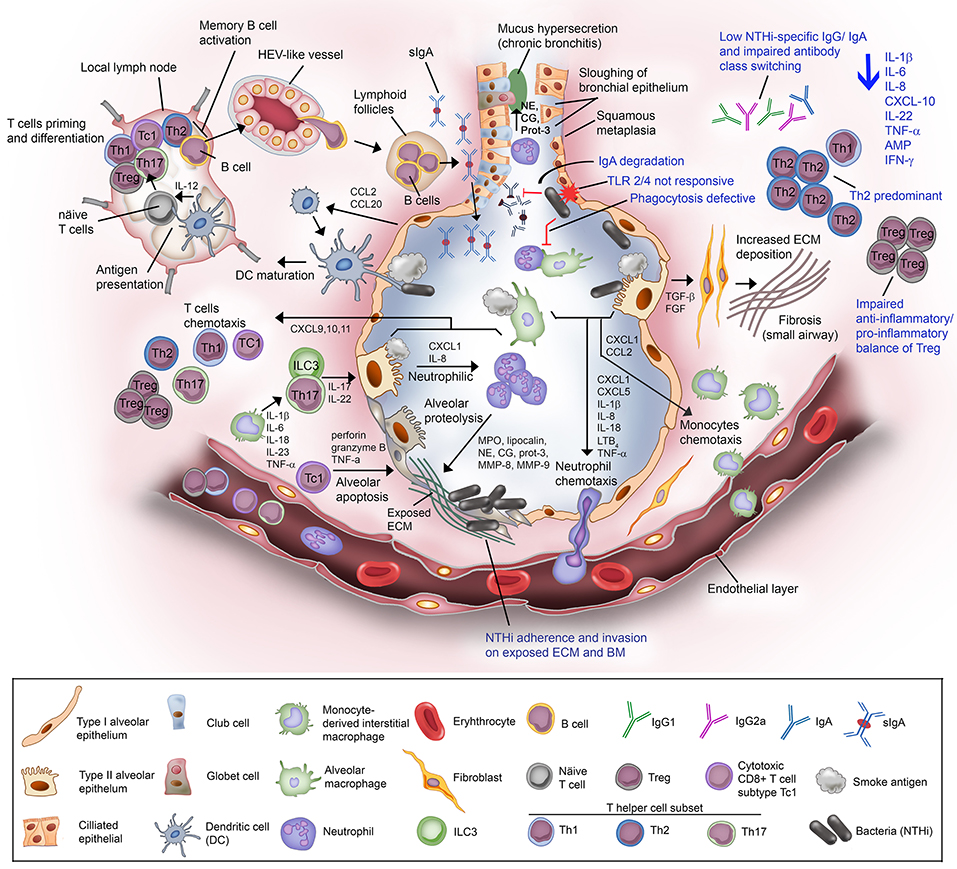
Figure 2. Non-typeable H. influenzae-dependent immune responses in the lower airway of COPD patients result in inflammation. Airway epithelium exposed to cigarette or tobacco smoke display an increased permeability with compromised tight junctions, and airway remodeling (goblet cell hyperplasia and small airway squamous metaplasia). Cigarette smoke causes the activation of airway epithelium and alveolar macrophages. The activated airway structural and resident immune cells release an array of chemotactic factors responsible for recruitment of inflammatory and immune cells to the lung. Activated epithelium produces TGF-β and FGF that triggers the production of ECM molecules by fibroblasts. Increased deposition of ECM causes progression of fibrosis and air flow limitation. The chemokines CXCL1 and IL-8, and LTB4 attract the circulating neutrophils through the receptors CXCR2 and BLT1, respectively. Meanwhile, CXCL1 and CCL2 targeting the receptors CXCR2 and CCR2 on blood monocytes are also released. Recruited blood monocytes differentiate into macrophages in the airway tissue. Activated alveolar macrophage and epithelium cell also release inflammasome (1L-1β and IL-18) for neutrophils survival and activation of helper T cells Th17. The chemokine IL-23 are released by macrophages to attract T helper cell subset Th17, and ILC3. Both Th17 and ILC3 will release IL-17 and IL-22 that will act on the alveolar epithelium to release CXCL1 and IL-8 for enhanced recruitment of neutrophils, resulting in neutrophilic inflammation. Activated neutrophils are thereafter degranulated and release myeloperoxidase (MPO), lipocalin, neutrophil elastase (NE), cathepsin-G (CG), proteinase-3 (Prot-3), and matrix metalloprotease (MMP) 8 and 9. The granulated products are proteolytic and elastilolytic to aveolar, causing alveolar destruction and emphysema. In addition, NE, CG, and Prot-3 are also targeting goblet cells and submucosal glands to induce hypersecretion of mucus. Dendritic cells carrying the receptors CCR2 and CCR6 are recruited to airway tissue via chemottractants CCL2 and CCL20. The dendritic cells uptake the antigen (smoke residues), and present the antigens to the naïve T cells at lymph nodes. Uncommitted T lymphocytes are thereafter primed to the presented antigen and activated by IL-12 derived from dendritic cells (professional antigen presenting cells; APC). Mature/activated T cells expressing receptor CXCR3 are chemotactic toward CXCL9, CXCL10, and CXCL11 and are recruited to the lung tissue. Cytotoxic CD8+ T cell subtype Tc1 releases perforin and granzyme B resulting in epithelial apoptosis contributing to emphysema progression. For the humoral immune response, B cells are activated by Th2, enter the circulation via high-endothelial venule (HEV)-like vessel and transported to lung tissue, and organized into lymphoid follicles at peripheral airway. B cell-derived plasma cells from lymphoid follicles release IgA, and secreted into airway lumen as secretory IgA (sIgA) via the polymeric immunoglobulin receptor. Mucosal antibodies play an important role to eradicate pathogens and noxious antigens via immune exclusion. However, the airway defense by sIgA is diminished by NTHi IgA protease that degrade the antibodies. TLR2 and TLR4 of the airway phagocytes and epithelium following exposure to cigarette smoke are not responding to P6 and LOS of NTHi. This results in defective phagocytosis and delayed bacterial clearance from the airway. The suppressed TLR4 induction in T cells has also lead to Th2 predominant immune response, with low production of IFN-γ and reduced T cell-mediated immune killing of NTHi. Moreover, NTHi downregulates Foxp3 of Tregs and thus impairs the anti-inflammatory/pro-inflammatory balance of Tregs. The extensive immunosuppressive activity by Tregs diminishes the response of effector T to NTHi stimulation. Lastly, plasma cells from COPD patients fail to produce NTHi-specific antibodies and compromised immunoglobulin class switching. The impairment of the host immune response in COPD toward NTHi infection are labeled in blue. In total, NTHi infection in COPD lung adversely reduces the production of IL-1β, IL-6, IL-8, CXCL-10, IL-22, TNF-α, antimicrobial peptide (AMP), and IFN-γ. This may explain the inefficient eradication of airway pathogens in COPD patients whereby persistent NTHi infection concomitantly escalates the inflammation and thus exacerbation in COPD.
Lung structural cells (epithelial and endothelial cells, fibroblasts, and airway smooth muscle cells) are activated by inhaled irritants through the stimulation of several pattern recognitions receptors (PRRs), with Toll-like receptor (TLR)-4 being reported as the key player in most of the inflammatory responses (82–85). This causes an increased expression and release of an array of pro-inflammatory mediators and chemokines through the oxidative pathway by the activated bronchial epithelial cells and immune cells (alveolar macrophages). The inflammatory mediators [(interleukin (IL)-1β, IL-6, IL-8, IL-33, C-X-C motif chemokine ligand (CXCL) 10, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte-colony stimulating factor (G-CSF), tumor necrosis factor (TNF)-α, fibroblast growth factor 1 and 2 (FGF1/2), transforming growth factor (TGF)-β1, C-C motif chemokine ligand (CCL) 2, CCL20, and thymic stromal lymphopoietin (TSLP)] act on recruited immune cells and resident cells to initiate a series of innate immune responses (23, 86–90). Meanwhile, activated alveolar macrophages, which are usually patrolling the lung parenchyma, further release more pro-inflammatory mediators and chemokines [IL-1β, IL-6, IL-8, IL-23, TNF-α, CCL1, CXCL1, CXCL5 (ENA-78), CXCL9, CXCL10, CXCL11, CCL2, leukotriene B4 (LTB4)], ROS, elastolytic enzyme [matrix metalloprotease protein (MMP)-2,−9, and−12; and cathepsin-K,- L, and -S], GM-CSF, and G-CSF (23, 91, 92). The enhanced levels of CCL2 and CXCL1 result in recruitement of blood monocytes expressing CCR2 and CXCR2 (receptors for CCL2 and CXCL1, respectively), to the lung and differentiate locally into macrophages. Interestingly, there are higher expression levels of the CCR2 and CXCR2 found on blood monocytes in COPD subjects (93). This may explain the rapid recruitment and excessive accumulation of monocyte-derived interstitial macrophages in the lung tissue of COPD patients (94, 95).
Upregulation of neutrophil chemoattractors (LTB4, CXCL1, CXCL5, IL-8, and TNF-α) induces a massive migration of circulating neutrophils into the lung parenchyma (96). The transmigration of blood neutrophils occurs through adherence of the granulocytes to E-selectin of endothelial cells that is found to be upregulated in COPD (97). This results in airway neutrophilia in several COPD patients (96, 98, 99). The recruited neutrophils (to the lung) are then activated to secrete granule proteins [myeloperoxidase (MPO) and neutrophil lipocalin] while releasing its own IL-8 for further neutrophilic recruitment and amplification of the inflammation (100). In addition to the macrophage-derived proteases, neutrophils also secrete serine proteases [neutrophil elastase (NE), cathepsin G, proteinase-3, MMP-8, and MMP-9] that are associated with serious alveolar destruction in emphysema (101). The protease activity may be further enhanced in conditions with genetic deficiencies or suppressed expression of α1-antitrypsin by tobacco smoke. In addition, NE, cathepsin G, and proteinase-3 are involved in the stimulation of mucus secretion from submucosal glands and goblet cells, resulting in airway mucus hypersecretion and airway obstruction in COPD (101).
The NLRP3 (NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 OR Nod-like receptor protein 3) inflammasome is a cytosolic multi-protein complex (consisting of the inflammation sensor protein NLRP3, adapter protein ASC, and the effector protein caspase-1) (102). The NLRP3 inflammasomes are involved in the COPD airway inflammation by regulating the production of pro-inflammatory cytokines IL-1α, IL-1β, and IL-18. These cytokines are important for neutrophil survival and activation of T helper (Th) 17 cells (103). Interestingly, local airway NLRP3 inflammasome activation is positively correlated with acute exacerbations and lower airway microbial colonization in COPD patients (103, 104). Moreover, in an elastase-induced emphysema model, the NLRP3 inflammasome is activated in addition to hyperproduction of mucin MUC5AC by diesel extract particles, extracellular ATP, and inflammatory protein S100 (105, 106).
The adaptive immunity is initiated at a later stage, and is recognized by the increased number of T and B lymphocytes and pulmonary dendritic cells. Dendritic cells are the major antigen-presenting cells (APC) in the airways, and link the innate and adaptive immunity. Circulating dendritic cells (expressing receptors CCR2 and CCR6) are recruited to the airway via dendritic chemoattractants CCL2 and CCL20 released by activated airway epithelial cells in response to cigarette smoke (107, 108). Dendritic cells act by endocytosis of inhaled irritants that subsequently are processed into antigen peptides during maturation and further migration to lymph nodes.
Uncommitted T lymphocytes are thereafter primed by the presented antigen. These important cells are activated by IL-12 released from dendritic cells for subsequent commitment into antigen-specific T cell lineages, i.e., T helper 1 (Th1; CD3+CD4+) cells, whereas immature dendritic cells in the airway promote Th2 differentiation (23, 109). Interestingly, in COPD patients, pulmonary Th and cytotoxic T cells (Tc; CD3+CD8+) express more CXCR3 receptors compared to healthy individuals (110, 111). This enhances their migration toward chemoattractants CXCL9, CXCL10, and CXCL11 that are actively released by alveolar macrophages in COPD subjects. Activated CD8+ T cell subset type 1 (Tc1) releases perforins, granzyme B, and TNF-α to induce alveolar cells apoptosis, contributing to the emphysema (112). In parallel, pulmonary Th17 T cells are activated by alveolar macrophage-derived IL-6 and IL-23 to secrete IL-17A and IL-22 causing neutrophilic inflammation (113, 114). Inflammatory cytokines are also released by type 3 innate lymphoid cells (ILC3) (115). The ILCs are involved in the homeostasis of lung immunity and are regulated by epithelially produced IL-33 and TSLP (116, 117), and are further stimulated in response to cell damage.
The accumulation of B lymphocytes in the peripheral airway and within lymphoid follicles is associated with airway autoimmunity in the progression of COPD (118). Airway tissue damage in conjunction with impaired T-regulatory cells (Tregs), both related by cigarette smoke, contributes to the formation of autoantibodies against airway components. Autoantibodies against elastin, epithelial, endothelial, carbonylated, and citrullinated proteins are found in the circulation of COPD patients (119–124). The generation of autoantibodies might activate plasma exudate-derived complement components resulting in a chronic inflammation, and consequently damage of the airways with emphysema progression (124–127).
From a physiological point of view, a modulated inflammatory process is important for a protective and optimal immune response. However, the prolonged airway inflammation in COPD as a results of impaired homeostasis leads to serious side effects since it amplifies the tissue damage and impairs the local immune defenses. The abrogated local immune system may make the airways of COPD patients susceptible for opportunistic or recurrent infections by viruses and bacteria that in turn might exacerbate the disease.
Acute exacerbations of COPD (AECOPD) are episodes of acute symptom worsening that usually are associated with both respiratory (increased airway inflammation) and non-respiratory (system inflammation/co-morbidities) effects (128–130). The typical symptoms of an AECOPD include increased production of purulent sputum, dyspnea, cough, wheezing, and symptoms of a cold that may last from 7 days up to 12 weeks (15, 130, 131). It commonly occurs in patients with advanced COPD and results in additional therapy based on the level of exacerbations. Exacerbations are classified in three levels according to GOLD. There is the mild disease that can be treated with short acting bronchodilators (SAB); moderate disease with SAB combined with antibiotics and/ or oral corticosteroids; and finally severe exacerbations with acute respiratory failure which requires emergency room visit and eventually hospitalization (15, 130).
AECOPD is a complex yet multifactorial consequence of COPD. Most of the exacerbations could be triggered by infectious (up to 80%) or non-infectious agents (~10%) (AECOPD with known etiology), whereas up to 30% of cases are of unknown etiology (132, 133). Respiratory tract infections are the major causes for AECOPD with known etiology and are mainly attributed to infections by viruses, bacteria, and atypical bacteria (not detected with conventional Gram-staining) (11, 134, 135). Non-infectious causes of AECOPD include air pollution, environmental factors, meteorological effects, and comorbidities of the patients, all of which are partially contributing to COPD exacerbations (133, 135, 136).
Respiratory viral infections are often the primary cause in the infection-dependent AECOPD, and virus was identified as single or multiple infecting strains from up to 64% of COPD patients with exacerbations recorded between years 2001–2017 (137–145). The most common infecting viruses are, by far, human rhinovirus, influenza virus A, and respiratory syncytial virus, whereas parainfluenza virus, coronavirus, echovirus, human metapneumovirus, and adenovirus are considerably rare.
Bacterial infections contribute to an average of 50% of infective acute exacerbations with a prevalence being reported ranging from 26 to 81% (132, 135, 146–148). The most commonly pathogenic bacterial species isolated from the lower airway of COPD patients during AECOPD are NTHi, Moraxella catarrhalis, Streptococcus pneumoniae, Staphylococcus aureus, Pseudomonas aeruginosa, and Klebsiella pneumoniae (11, 129, 133, 136, 149–152). It has been suggested that infection with new strains of the infecting species, rather than a new species, is highly associated with an increased risk of exacerbation (11, 153, 154). Atypical bacteria that cause exacerbations are Chlamydia spp., Legionella pneumophilia, and Mycoplasma spp.
In contrast to viral infections that are diagnosed in 5–45% of COPD patients with stable disease and increase to 39.3–64% during COPD exacerbations, bacterial colonization in the airways are more common with the same species during both stable disease (25–86%) and exacerbations (58.8–81%) (11, 132, 136, 137, 142, 155–158). Hence the precise or direct role of bacterial infection as the primary cause in triggering AECOPD remains controversial although a significantly increased bacterial load is observed during exacerbation in several patients. This further suggests that bacteria might be more involved as secondary invaders after an initial viral infection.
Viral infections have been reported to cause several physiological changes in the lung that in turn facilitates secondary bacterial invasion. The mechanism of bacterial superinfection has been described for H. influenzae, S. pneumoniae, S. aureus, and many other airway pathogens (159–161). Firstly, viral infections destroy the tight junctions of the airway epithelial barrier while inducing epithelium apoptosis. This results in the onset of airway epithelium lining repair whereby the sloughed off dead cells would become a rich nutrient source for growth of infecting bacteria. The damaged epithelium lining also enables bacterial adherence to the exposed basement membrane and ECM. Secondly, the demolished ciliated clearance as a result of the virus-damaged airway epithelium lining further promotes bacterial colonization and subsequent epithelial transmigration into deeper tissues (162–164). Lastly, viral infections are also detrimental to the airway immune defense by causing degradation of antimicrobial peptides (AMP), and by triggering IFN-γ secretion by immune cells. This results in suppressed macrophage and neutrophil responses to infecting bacteria, and thus enables bacterial evasion of the airway immune defense (165–168). Nevertheless, viral and bacterial coinfection have greater impact in the AECOPD airway inflammatory responses than bacteria or virus infection alone (168, 169). This is in parallel with the co-isolation of both respiratory viruses and bacteria from 6 to 30% of AECOPD patients (129, 133, 136, 170–174).
Infective AECOPD is also attributed to impaired functions of AMP, macrophages, and neutrophils triggered by inhaled irritants such as tobacco smoke. Expression of microbial-induced AMP (human β-defensin 2) is suppressed in airway epithelial cells when exposed to cigarette smoke (175, 176). Both the alveolar and monocyte-derived macrophages in patients with COPD are defective in phagocytosis of bacteria such as H. influenzae and S. pneumoniae (177, 178), and in efferocytosis of apoptotic neutrophils and epithelial cells. In addition, neutrophils from COPD patients are aberrant in chemotactic response with defective accuracy (179). All these factors contribute to the failure to resolve inflammation in COPD leading to facilitated chronic microbial colonization, also during exacerbations.
The low number of cultivable bacteria found in healthy individuals previously led to the conclusion that healthy and normal lungs are virtually sterile. This hypothesis is currently being revised, since the introduction of 16S rDNA based molecular diagnostics has shown that even healthy lungs have a distinct microbial community, different from that seen in the upper respiratory tract (180, 181). This has led to the concept of a core human lung microbiome which can be altered in COPD stable disease and during exacerbations (182). The role of the lung microbiome in the pathogenesis of COPD by influencing host immune response has also been suggested (151, 183–188).
The stability of the lung microbiome has profound impact on maintaining local immune homeostasis (189). According to the “vicious circle” hypothesis, airway inflammation and impaired immune defenses caused by either viral infections or irritant inhalation have ecological influence on the airway microenvironment and growth conditions that would eventually lead to dysbiosis of the lung microbiota (182, 190). The changed lung microbiome would then cause a maladaptive immunological response resulting in further inflammation and damage of the lung immune defenses, and additional alteration of the lung microbiome. The chain of events thus generates a vicious circle that contributes to COPD progression and exacerbation.
Several studies have documented that COPD progression from stable state to an exacerbation could induce microbiota shift in the lower airway (bronchioles), sputum, and throat (151, 190–196). Alteration in the microbiome complexity or richness is associated with the inflammatory process and changes in ECM protein expression in the lung, as observed in COPD (185, 197). Declined diversity in the lung microbiome has been reported to be related to disease severity, inflammation and decreased lung functions in COPD. This includes the increased emphysematous destruction, bronchial tissue remodeling, lymphoid follicle formation, elevated autoantibodies, and IL-17A production, and finally increased neutrophil extracellular traps (NET) formation in the airway of animal models or AECOPD patients (198–201). It has recently been reported that lung microbiome diversity is also associated with genetic factors. Mannose-binding lectin (MBL) deficiency has also been associated with disease severity and exacerbations in patients with cystic fibrosis and bronchiectasis (202). However, COPD patients with a genetic deficiency in MBL are less susceptible to Haemophilus spp. colonization, lowering the risk of exacerbations while their lung microbiota is more diverse than normal COPD patients (203).
In this review we will focus on NTHi, one of the dominant genera that is relatively abundant in the total COPD-dependent lung microbiome, due to its role of infection in COPD immunological responses (136, 149, 192–195, 198, 204–206).
The microbiology of H. influenzae has recently been reviewed in detail by our group and others (4, 5, 207). It is a Gram-negative coccobacillus that commonly colonizes the human nasopharynx, and is typed as capsulated (type a–f) or non-encapsulated strains (NTHi). H. influenzae may cause both invasive and mucosal disease (208). Since the introduction of capsule polysaccharide conjugate vaccines against type b (Hib), NTHi dominate, followed by capsule type f (Hif) (209, 210). Mucosal infections, including acute otitis media, sinusitis, and exacerbations in COPD, are nowadays mainly associated with NTHi. There has also been a significant shift in the epidemiology of severe invasive disease, from Hib infections in small children to NTHi in adults (210, 211). The most common principal infection focus by H. influenzae is now community acquired pneumonia (CAP), whereas the incidence of historically common diagnoses such as meningitis and epiglottitis have significantly decreased (210, 211). Patients with underlying conditions, notably COPD, seem to be at higher risk for invasive infections (209).
There is consensus that H. influenzae is one of the key bacterial pathogens involved in pathogenesis of both stable COPD disease and acute exacerbations (207). However, the relative abundance and significance of NTHi in COPD varies between different studies. Several factors, such as sampling methodology, choice of microbiological analysis and, if the patient has a stable disease or an exacerbation, or has been subject to previous antibiotic therapies, tend to affect the outcome of the studies (212).
Common sampling methods from the lower respiratory tract include both bronchoscopy techniques such as protected specimen brush (PSB) and collection of BAL fluid as well as non-invasive methods like sputum sampling (213). All of these methods, particularly sputum, are to some extent subject to the risk of contamination from the normal microbial flora of the oro- and nasopharynx, which might reduce their specificity (213). However, several studies still show a distinct association between lower respiratory tract samples and clinical parameters in COPD patients, making the information valuable (214).
Cultivable bacteria are seldom found in the lower airways of healthy individuals (215), whereas COPD patients show bacterial growth in 30–50% of cases even during stable disease (Table 1). On top of that, several studies have shown a significant increase in the Proteobacteria phylum, which includes Haemophilus spp., in individuals with both stable disease and AECOPD (Table 2). NTHi is consistently one of the predominating bacterial species isolated in those cultures; other important pathogens include S. pneumoniae, M. catarrhalis, and P. aeruginosa (219). During AECOPD, the bacterial load is increased even further, and NTHi continues to be the predominating species (208). Furthermore, acquisition of a new NTHi strain has, in one study, been linked to the onset of AECOPD (153). Moreover, the growth and dominance of H. influenzae following rhinovirus infection was observed in the sputum microbiome of patients with COPD (190).
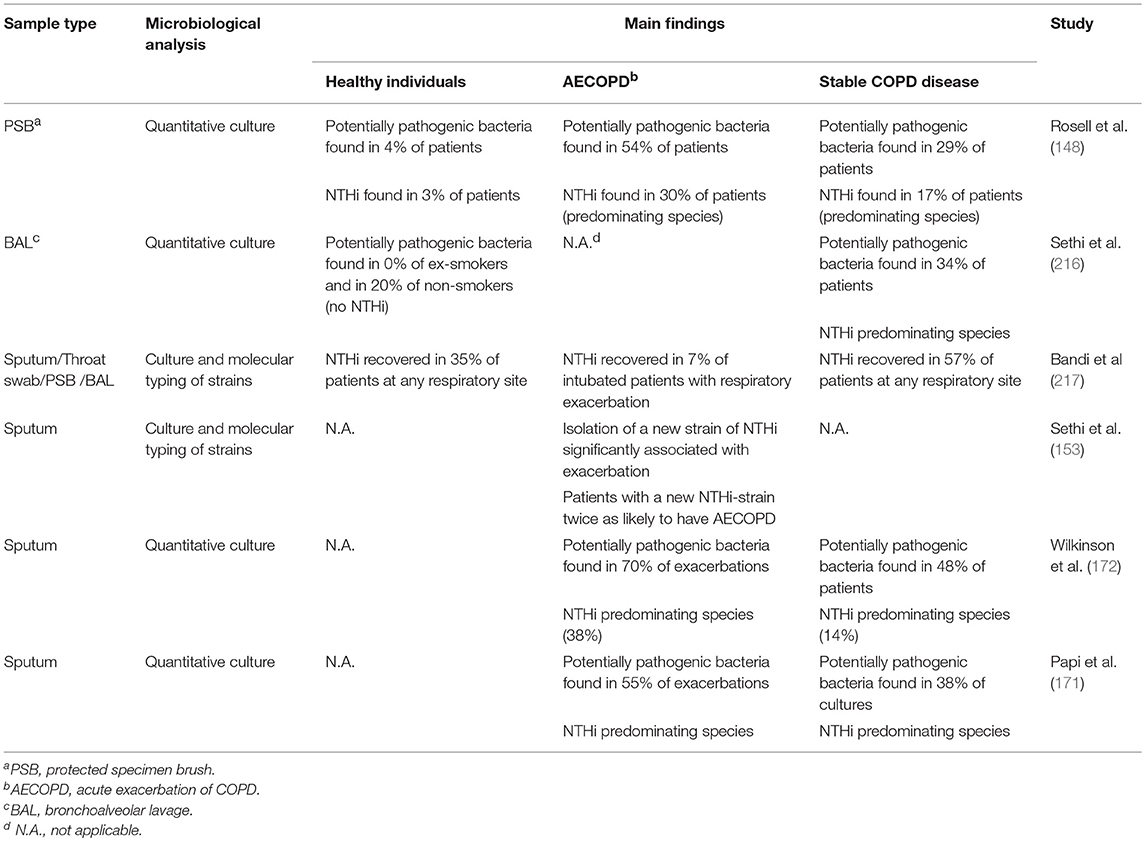
Table 1. Abundance and significance of NTHi and other potentially pathogenic bacteria in healthy individuals and various stages of COPD using culture-based methods.
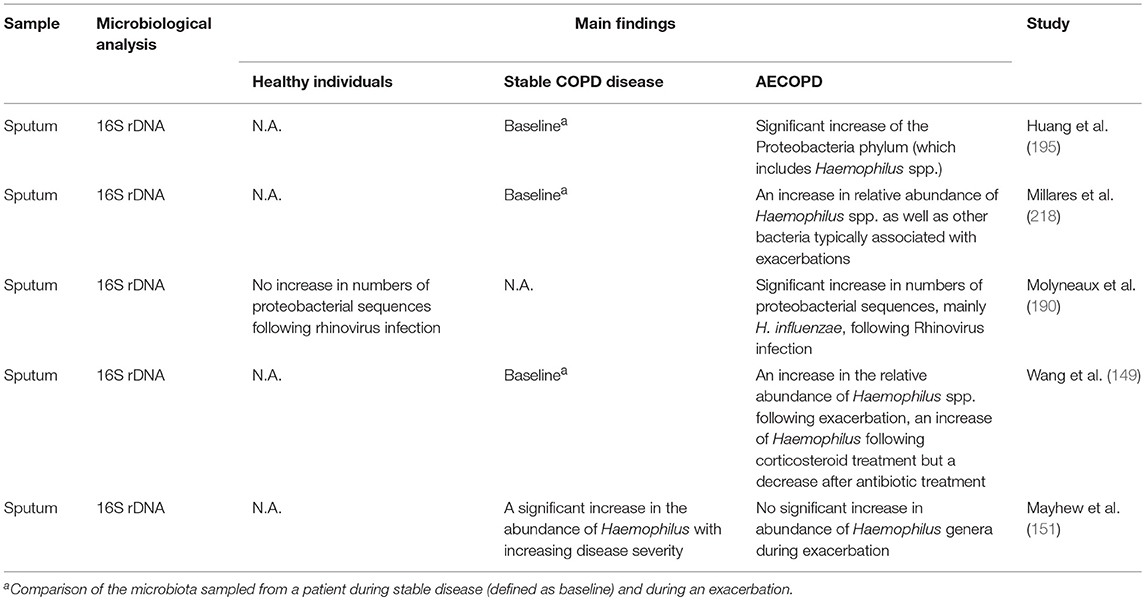
Table 2. Abundance and significance of NTHi and other potentially pathogenic bacteria in healthy individuals and various stages of COPD using molecular methods.
The chronic inflammation that characterizes COPD pathogenesis causes significant changes to the pulmonary tissue. The lower respiratory tract of patients suffering from this disease is marked by epithelial denuding, hypersecretion of mucus, disproportionate phagocyte presence and imbalances in antioxidant/oxidants (220). This altered milieu selects for specific bacterial species that are genetically equipped to competently address these environmental stressors (151, 195, 221). NTHi is the most common pathogen isolated from the sputum of COPD patients, and the primary cause of exacerbations (212), indicating a unique ability to colonize and persist in the chronically inflamed lower respiratory tract.
In recent years, great efforts have been made in understanding how NTHi colonizes the pulmonary tissue. In addition to the regular arsenal of virulence factors associated with NTHi (5), the bacterial pathogen undergoes specific adaptations to increase its fitness in the COPD setting. Specific genetic islands that include ureABCEFGH, lic2b, hgbA, iga, hmw1, and hmw2 have been reported to be enriched in NTHi strains isolated from COPD patients compared to commensal NTHi (222). These genes are involved in raising the pH of the environment, lipooligosaccharide (LOS) synthesis, iron uptake, immune evasion, and attachment to host tissue. The validity of these findings is strengthened by previous work identifying upregulation of many of the same bacterial gene products during growth in COPD sputum (223). Moreover, peroxiredoxin-thioredoxin, an antioxidant enzyme, was found to be one of the most enriched proteins in NTHi during growth in COPD sputum, suggesting that the bacteria upregulate oxidative stress-countermeasures when facing oxidative imbalances in the diseased lung (223). Oxidative stress resistance has previously been shown to be vital for NTHi survival in infection models (224).
In a seminal investigation by Pettigrew et al., whole-genome sequencing (WGS) was conducted to follow the in vivo adaptation of NTHi to the COPD environment over time (225). Several interesting findings were reported in this work. Firstly, the median duration of persistence by the pathogen was found to be 161 days, but it could persist in patients for up to as many as 1,422 days. Secondly, slipped-strand mispairing-mediated phase variation was identified as the primary genetic adaptation to the niche. Poignantly, the genes affected by the regulation mechanism encoded for (among others) the HMW adhesins, LOS biosynthesis, and iron uptake, that is, the same processes identified in the previous studies as important for COPD adaptation (222, 223). Thirdly, and somewhat surprising, it was observed that a very limited number of genes were gained/lost during persistent colonization, meaning that selection for strains that thrive in the inflamed lower airways occurs at the very onset of colonization. Finally, the authors reported that genetic changes occurred in 8 of the 12 investigated vaccine antigens during persistent infections, a fact that might be taken into consideration for potential vaccine development against NTHi.
Another virulence factor that has been reported by Murphy and co-workers to play a pivotal role for NTHi survival in COPD settings is IgA-protease, a hydrolytic enzyme that cleaves secretory IgA (sIgA) antibodies in the mucosal epithelium (226–228). Four genes encode for the same number of different variants of the endopeptidase with various cleavage site specificities: two igaA (igaA1 and igaA2) and two igaB (igaB1 and igaB2). The igaA is present in all NTHi whereas igaB is present in ~40% of the strains (226). The igaB1 gene has been reported to be more prevalent in COPD exacerbation-causing strains, although the in vivo expression levels did not differ from asymptomatic colonization strains that also carried the gene (226). However, IgA-protease B1 and B2 have been found to promote the intracellular survival of NTHi in human epithelial cells, providing a secondary function (in addition to hydrolysis of IgA antibodies) that could facilitate NTHi growth in inflamed environments (227). While a majority of the persistent NTHi strains that dwell in COPD patients continuously express one or more variants of the enzyme, it has recently been found that a phase variation to an OFF-state can occur via slipped-strand mispairing over time (228). This suggests that during certain conditions, there is a fitness benefit in not expressing iga in the airways of COPD patients, albeit the specifics of this process are currently unknown.
Another interesting aspect of NTHi colonization of COPD patients is with regard to biofilm formation (229). NTHi strains that colonize the Eustachian tube causing otitis media are known to build up biofilms in situ (230). However, strains isolated from COPD patients tend to have significantly diminished ability to form biofilm compared to invasive strains or those isolated from otitis media patients (229), suggesting that this mechanism is not important for survival in the COPD niche. As biofilms tend to protect the bacterial community from external assaults, these findings could indicate that the hypermucoid milieu in the COPD airways is severely impaired in its ability to deliver an apt immune response for optimal clearance of residing microorganisms. In light of this impairment, biofilm formation might not be necessary for NTHi to persist in this particular environment.
Infections with NTHi have also been shown to reduce cellular levels of E-cadherin, a protein required for tight junction formation and epithelial cell integrity in human cells (231). Considering that perturbations in the epithelial cell barrier caused by the loss of E-cadherin is a common symptom of COPD, NTHi-mediated exacerbations likely contribute to this step of COPD pathogenesis. The subsequent denuding of the epithelium could facilitate microbial colonization of the basal lamina, a well-established virulence mechanism employed by NTHi and other pathogens (232). It is currently unknown which bacterial virulence factor(s) that induce the reduction of E-cadherin levels in the host.
In summary, investigations from recent years show that the environment of the lower respiratory tract of COPD patients selects for NTHi strains that can upregulate adhesins, modify LOS biosynthesis pathways, increase antioxidant stress responses and cellular invasion strategies, and, finally, trigger tolerance against acidic pH. These important colonization mechanisms thus provide researchers with viable targets for developing novel therapies.
NTHi is a commensal in the nasopharyngeal site but is often associated with strong inflammatory responses in the lower respiratory airways, especially in patients with COPD, bronchiectasis, cystic fibrosis, pneumonia, or idiopathic pulmonary fibrosis (11, 233). Colonization and subsequent infection of NTHi in the lower airways of COPD patients elicits episodes of immune responses orchestrated by both the innate and adaptive immunity. NTHi infection is thus commonly associated with inflammation that is mainly mediated by transcription factor NF-κB-dependent production of proinflammatory mediators. The activation of NF-κB requires induction of cross-signaling networks and cascades via activation of PRRs (pattern recognition receptors) of host innate immune cells (234). Unresolved or prolonged (chronic) inflammation or failure to restore the homeostatic inflammatory status potentially contributes to exacerbations. This is clearly shown in murine COPD simulation models with NTHi-triggered inflammation (235–237). Mice exposed to NTHi lysates display inflamed airways loaded with increased levels of inflammatory mediators and phagocyte infiltrates. Moreover, multiple exposures to bacterial lysates which may represent a chronic NTHi infection caused extremely high infiltration of phagocytes and lymphocytes in the airways of this particular mouse model. In addition, the airway walls of the infected animals were also thickened due to increased collagen deposition (fibrosis) that reflects the typical COPD features. The host immune response and specific interactions during NTHi infection in COPD is summarized in Figure 2.
The epithelium and alveolar macrophages are predominant cell types in the airway compartment. They comprise the first line of defense in the cellular immune response against potential inhaled pathogens and antigens. The sensing of bacteria, and particularly NTHi in the lower airways is initiated via PRRs expressed on innate immune cells and endothelium in addition to epithelial cells (238–240). TLRs are PRRs that sense stimulation by NTHi-derived pathogen-associated molecular patterns (PAMPs), and play a primary role in initiating effector cellular responses and intracellular signaling for NF-κB activation (238). Among the different TLRs, most of the studies on NTHi infection have by far been focused on TLR2 and 4. Lipoproteins including NTHi P6, and LOS are potent immunomodulators for activation of TLR2 and TLR4, respectively, and has been described in several studies on airway epithelial cells and alveolar macrophages (241–244).
Interaction of NTHi lipoprotein P6 with TLR2 on human epithelial cells [type II alveolar A549 and human middle ear epithelial cells (HMEE)] causes NF-κB-dependent activation via two distinct TLR-signaling pathways, that is, the NF-κB translocation-dependent, and -independent pathways (242). The NF-κB nuclear translocation-dependent pathway requires activation of NF-κB-inducing kinase IKK complex. In the second pathway, the MKK3/6-p38 MAPK signaling cascade is recruited for direct nuclear phosphorylation, and thus activation of NF-κB. The branching of both pathways may occur at the TGF-β activated kinase 1 (TAK1) signaling junction. NTHi stimulation via TLR2 and downstream activation of p38 MAPK/NF-κB-dependent pathways result in expression of COX-2 and prostaglandin (E2) (PGE2) that promote inflammatory responses (245).
TLR4 stimulation by NTHi LOS also contributes to the activation of NF-κB via two signaling pathways, the primary activating pathway of MyD88 cascade and the alternative pathway of Toll/IL-1R domain-containing adapter-inducing interferon-β (TRIF). Both pathways activate NF-κB through phosphorylation and degradation of inhibitor IκBα (243, 246). NTHi-TLR4 signaling mediates an effective innate immune response that leads to upregulation of TNF-α, IL-1β, IL-6, macrophage-inflammatory protein (MIP)-1α, MIP-2, and neutrophil infiltration in the airways of mice. The TLR4 response promotes efficient pulmonary clearance of bacteria in TLR4-expressing animals compared to CD14/TLR4 knockout mice (243, 244). A recent study by Jungnickel et al. revealed that, in parallel with the infection-induced pulmonary neutrophilic inflammation, NTHi-dependent stimulation of both TLR2 and TLR4 in a transgenic mouse [(KrasLA1) with oncogenic Kras allele in the lung epithelium] additionally promotes the proliferation of Kras-induced early adenomatous lesion in the lung in an TLR-dependent manner (247). The association or role of NTHi-induced airway inflammation in lung cancer progression, however, is not supported by another recent cohort study showing the lack of differences in NTHi specific-antibodies between cancer- and non-cancer COPD patients (12).
Lastly, Dectin-1 and the epidermal growth factor receptor (EFGR) pathway also have proinflammtory effects upon interaction with NTHi (248, 249). Activation of the Dectin-dependent proinflammatory response requires NTHi-induced phosphorylation of the Dectin-1 hem-immunoreceptor tyrosine-based activation motif (hemITAM) (248). Direct activation of EFGR in alveolar cells and HMEE by NTHi-derived EGF-like factor has been shown to contribute to NF-κB activation. The EFGR-dependent NF-κB activation is mediated via an NF-κB nuclear translocation-independent pathway, which involves both MKK3/6-p38 and PI3K/Akt signaling pathways (249). Surprisingly, the interaction of EFGR and NTHi also results in negative regulation and suppression of the induction of TLR2 via the Src-MKK3/6-p38 α/β MAP kinase-dependent signaling cascade, and this in turn may facilitate NTHi infection (250). The actual components of NTHi that exhibit the EGF-like factor activity have, however, yet to be defined. The EFGR-dependent negative regulation of TLR2 may thus suggest a novel mechanism targeted by NTHi for immune evasion by attenuating the responses of host PRR, despite the contradicted role of EFGR in proinflammatory and innate immune responses of the airway epithelium (251). NTHi infection also upregulates the NRLP3-inflammasome during NTHi-induced inflammation in the airway epithelium and alveolar macrophages, leading to increased secretion of IL-1β and IL-8, and thus neutrophilic influx to the lung (252).
Some of the endogenous inflammatory mediators that are produced in response to NTHi infection, including TNF-α, IL-1α, and TGF-β1, may act synergetically with NTHi on the airway epithelial and immune cells. The synergetic interaction drives a positive feedback loop to amplify the NF-κB transcriptional activity on proinflammatory genes and further augments airway inflammation.
The synergetic activation of NF-κB by NTHi and TNF-α in HMEE and normal human bronchial epithelial (NHBE) cells occurs via NF-κB nuclear translocation-dependent and independent pathways. The latter pathway involves MAPK/extracellular signal regulated kinase kinase kinase 1 (MEKK1)-dependent activation of MAPK kinase 3/6–p38 MAPK pathway (253). However, the synergetic action of NTHi with TGF-β1 is mediated by another mechanism which involves Smad3/4-protein kinase A (PKA)-p300-dependent signaling cascade. The pathway components, PKA and p300, phosphorylates residue Ser276 and acetylates Lys221 of the NF-κB subunit p65, respectively. This results in enhanced DNA-binding activity of NF-κB (254).
The synergetic action of NTHi with both TNF-α and TGF-β1 enhances the production of TNF-α, IL-1β, and IL-8 from airway epithelial cells and interstitial polymorphonuclear infiltrates. Recently, it has been reported that co-infection of human rhinovirus and NTHi on the airway epithelial cells (NHBE cells and the BEAS-2B cell line) also results in synergetic induction of CCL20 and IL-8, albeit the exact mechanism remains to be elucidated (255). Of note, activated macrophages also release increased concentrations of TNF-α and IL-1α (256), further enhancing the inflammatory synergetic effect of surrounding immune cells.
Finally, IL-1α acts synergetically with NTHi to upregulate the expression of AMP β-defensin 2 (DEFB-4) via the p38/MAPK pathway (257). Of note, IL-1α could also act individually to upregulate the expression of DEFB-4 via the Src-dependent MEK1/2-ERK1/2 signaling pathway (258). Taken together, the synergetic action may aid in the expansion of the inflammatory response and in some cases worsen the clinical outcome.
Alveolar macrophages located in the air-parenchyma interface are the primary professional phagocytes in the lung (259, 260). These cells are responsible for infection eradication through its phagolysosomal machinery while releasing a plethora of inflammatory cytokines and chemokines for promoting a local inflammatory response and recruitment of neutrophils. Neutrophils are the first responder cells recruited from circulation to the airway for efficient killing of pathogens through an array of microbicidal strategies (261, 262). During NTHi lung infection, both alveolar macrophages and neutrophils are the main innate immune cells involved in the pulmonary bacterial clearance through phagocytosis. They are also an important source of cytokine secretion required for induction of other immune cells and enhanced bacterial killing. Eradication of NTHi by alveolar macrophages involves adhesion or contact, phagocytosis and phagolysosomal processing of bacteria, in addition to secretion of TNF-α. Phagocytic clearance of NTHi by alveolar macrophages is orchestrated through actin polymerization, plasma membrane lipid rafts, and phosphatidylinositol 3-kinase (PI3K) signaling cascade upon induction of macrophage PRRs by NTHi (256).
Interestingly, in response to NTHi infection, human alveolar macrophages, and blood neutrophils produce extensive amount of intracellular and extracellular ROS as a component of the antimicrobial defense. This leads to the formation of macrophage and neutrophil extracellular traps (METs and NETs, respectively), with co-expression of MMP-12 for enhanced bacterial killing (263, 264). Nevertheless, the overexpression of MMPs may adversely result in a protease imbalance and contribute to alveolar emphysematous destruction and bronchiectasis in COPD (265). Moreover, excessive endogenous ROS production could also introduce airway oxidative stress that is detrimental by causing chronic inflammation and tissue damage in the lung, and thus contributing to the COPD exacerbation (266, 267). The NET formation is elicited mainly by NTHi LOS in addition to other Haemophilus PAMPs (264).
Several studies by King et al. have revealed that T cell-mediated adaptive immune responses against NTHi airway infection in patients with idiopathic bronchiectasis and COPD has been predominated by a Th2/Tc2 response (268–270). The activated T cells produce reduced level of the CD40 ligand and IFN-γ, and increased levels of TNF-α, IL-13, and IL-17, as well as altered IgG subclass production by plasma cells. It is to be noted that the Th2/Tc2-mediated immune response is less effective in suppressing NTHi infection. Redirecting the Th2/Tc2-mediated immune response to Th1/Tc1 dominant (which is more protective) by adding the Th1/Tc1 mediators (CD40 ligand and IFN-γ) has helped to restore the T cell-mediated immune killing of NTHi (269). However, a separate study in a COPD mouse model by Lu et al. reported that NTHi infection causes increased production of airway type 1 interferon (1-IFN) (271). It was further reported that DNA of NTHi acts as a PAMP in stimulating the STING/TBK1/IRF3 pathway, and thus the production of 1-IFN. The impact of the bacterial DNA-induced 1-IFN in host immune/inflammatory response, which may potentially induce a Th1/Tc1 response requires further investigations.
COPD patients also have abnormally higher number of Treg cells, myeloid-derived suppressor cells (MDSC), and exhausted effector T cells (PD-1+) than healthy individuals (272, 273). Cigarette smoke-induced anti-inflammatory activity of Tregs in a COPD model is further suppressed by NTHi infection. The pathogen causes downregulation of Foxp3 (biomarker of Tregs), and thus impairs the anti-inflammatory/pro-inflammatory balance of Tregs (273, 274). This may lead to the extensive immunosuppressive activity by Tregs on the proliferation of NTHi P6-specific effector T cells, causing a diminished response of effector T cells to sputum IL-6 and IL-8 induction, and increased levels of IL-10 and TGF-β1 (272, 275). Recently, it has been reported that mucosal-associated invariant T cells (MAIT) from COPD patients are more effective in response to NTHi stimulation and thus produce increased levels of IFN-γ, 3-, to 10-fold more than the COPD Th (CD4+) and Tc (CD8+) cells (276). However, the pulmonary MAIT cell immune responses are compromised in the presence of corticosteroids that are commonly used for the treatment of COPD. This may potentially prone the T cell-mediated immunity to a Th2/Tc2 response in COPD patients treated with corticosteroids (277). Interestingly, antigen-specific Th17 cells from NTHi-immunized non-COPD mice model recognize both homologous and heterologous strains of NTHi, and are able to confer protection upon adoptive transfer (278). However, it is unclear whether the Th17 cell which is prone to the inflammatory response could be “trained” to counteract the NTHi infection in COPD patients, particularly during exacerbations.
During the systemic humoral immune response in NTHi-infected COPD patients, greater concentrations of NTHi-specific IgG, IgA, IgM, and IgE serum antibodies are produced compared to non-infected controls (12, 279–281). Some of the NTHi-specific serum immunoglobulins are specific to P2, P5, and P6 (12, 282, 283). However, decreased mucosal antibodies associated with sIgA deficiency, or decreased total IgG in the small airways have been reported in COPD patients, and might be associated with disease severity (283, 284). Importantly, NTHi-specific mucosal sIgA has been found to be lower in the airways of NTHi-infected COPD patients than the non-colonized patients (285, 286).
The epithelial polymeric immunoglobulin receptor (pIgR) is essential for the generation of mucosal sIgA. It is, however, downregulated in COPD patients with a positive correlation to disease severity and increased level of TGF-β (287). The combinatorial effects of downregulated plgR and elevated TGF-β1 contribute to an impaired mucosal IgA immunity in COPD patients. A mouse model lacking the pIgR (−/−) is therefore devoid of sIgA and are susceptible to airway stimulation by an NTHi lysate resulting in increased inflammation and airway neutrophilia. Interestingly, introduction of exogenously added sIgA mitigated the airway inflammation (288). NTHi-infected COPD patients with greater airway inflammation have also decreased NTHi-specific mucosal IgG1 in the BAL fluid compared to the non-colonized patients (283). Interestingly, the phenomenon with decreased NTHi-specific antibodies seems to be restricted to the airways, since the specific serum antibodies are not affected. Therefore, the reduced mucosal IgG is unlikely to be associated with hypogammaglobulinemia (IgG deficiency), despite the latter was reported as a contributing factor in NTHi infection (289). Decreased airway IgA might be attributed to the expression of IgA proteases by NTHi. The bacterial IgA protease degrades the local airway IgA during airway colonization to avoid immune exclusion by sIgA (226, 228). Reduced mucosal antibodies might promote host immune evasion and resistance to complement-mediated killing of NTHi, thus enable persistent colonization of NTHi in the airways of COPD patients, in addition to a plethora of various other virulence mechanisms (4, 5, 207).
In a cohort study of stable COPD patients, augmented airway inflammation and plasma fibrinogen, but not systemic inflammation, were found to be constantly correlated with the increased bacterial load (233). Higher numbers of NTHi has a greater impact than S. pneumoniae and M. catarrhalis in triggering inflammatory responses as measured by the augmented levels of inflammatory cytokines in sputum including IL-8, MPO, and 1L-1β. The increased inflammatory response in affected patients is potentially attributed to the persistent colonization of NTHi in the lower airway (207, 233). The compromised innate immune response in COPD, particularly the decreased microbicidal activity, has been regarded as one of the culprits for persistent airway colonization by NTHi, and is highly associated with COPD exacerbations (Figure 2).
Whilst the role of macrophage extracellular traps (MET) for killing of NTHi remains unknown, it has been reported that blood neutrophils and NET from COPD patients are defective in the killing of planktonic or biofilm/NET-entrapped NTHi, respectively (263, 264, 290). A series of studies by Berenson et al. revealed that alveolar macrophages derived from COPD patients are basically dysfunctional in eradication of NTHi (177, 291–293). Intriguingly, TLR2 and TLR4 expressed on alveolar macrophages from COPD patients are intrinsically unresponsive to the potent immunomodulatory lipoprotein P6 and LOS, respectively. This causes decreased LOS/P6-induced expression of TLRs, reduced NF-κB nuclear activation and consequently diminished IL-8, TNF-α, and IL-1β responses by alveolar macrophages from COPD patients. The compromised TLR expression and signaling potentially contribute to the defective complement-dependent and independent phagocytosis of NTHi. The defective phagocytosis is greater for NTHi than for M. catarrhalis, and correlates with disease severity. Interestingly, the phagocytosis disability was not detected in monocyte-derived macrophages in COPD. In contrast, however, Taylor et al. reported that monocyte-derived macrophages from COPD patients are also defective in phagocytosis of NTHi and S. pneumoniae. The author also suggested that the defective monocyte-derived macrophages are not attributed to the alteration in cell surface TLR2 or TLR4 expression, macrophage receptor with collagenous structure (MARCO), CD163, CD36 or the mannose receptor (178). The unresponsive TLR2 and TLR4 in COPD alveolar macrophages to NTHi lipoprotein and LOS might be explained by the recently reported phenomenon of TLR tolerance (294). Repetitive stimulation of COPD alveolar macrophages with the same TLR ligands, Pam3CSK4 and LPS desensitizes the TLR2 and TLR4, respectively, and generates TLR tolerance. Moreover, the repetitive TLR stimulation further reduced the production of TNF-α, CCL5, and IL-10 without affecting the constantly augmented level of IL-6 and IL-8 in alveolar macrophages. This may provide alternative explanations for diminished immune responses against the recurrent/repetitive infection by NTHi.
The intrinsically reduced expression of TLRs in COPD patients may also contribute to the impaired pulmonary immune response thus facilitating NTHi persistent colonization. Expression of TLR2 or TLR4 are found to be lower on sputum neutrophils, alveolar macrophages, nasal epithelium, and T cells in COPD patients despite high concentrations of IL-8 and MMP-9 (295–298).
The lack of the more protective Th1/Tc1 immune response in COPD patients against NTHi infection might be attributed to upregulated antagonists (A20, IRAK-M, and MyD88s) of the MyD88/IRAK/MAPK signaling pathway in COPD T cells (295). It should be noted that the MyD88/IRAK/MAPK pathway is required for expression of TLR4 in Th1, whereas production of IFN-γ in Th1/Tc1 is TLR4-dependent via the TLR4/TRIF/IKKe/TBK1 signaling pathway. The antagonists prevent the NTHi LOS-induced TLR4 expression in Th1 and Tc1 and thus a reduced secretion of IFN-γ. In addition, unusual high numbers of Tregs in COPD patients have also contributed to effector T cell dysfunction or a Th2/Tc2 predominant immune response (272). However, Freeman et al. reported that Tc (CD8+) cells from COPD patients have increased expression of TLR1, TLR2, TLR4, TLR6, and TLR2/1 as well as Tc1 cytokines (IFN-γ and TNF-α) compared to healthy individuals that may imply the auto-aggressive response of lung Tc cells in COPD lung inflammation (299). However, the COPD Tc cells can only be stimulated by ligands for TLR2/1 (Pam3CSK4) yet tolerant to other agonists, indicating the dysfunctional TLRs or TLR tolerance on T cells despite their high level of receptor expression.
Inversely, peripheral blood neutrophils isolated from COPD patients have increased expression of TLR2, TLR4, and NLRP3 (298, 300). Nevertheless, the increased TLRs expression might not improve the microbicidal ability of COPD peripheral neutrophils probably due to the inaccurate responses to cytokines (179). In addition, certain types of SNPs (SNPs) in TLR2 and TLR4 have also been associated with decreased lung function, enhanced inflammatory responses and increased immune cell infiltration in COPD (301). Interestingly, the diminished IL-8 responsiveness of COPD alveolar macrophage to NTHi infection has a strong association with the carriage of TLR9 (T1237C) polymorphism instead of TLR2 (Arg753Gln), TLR4 (Thr399Ile; Asp299Gly), and TLR9 (T1486C) (302). The carriage of TLR9 (T1237C) is also positively correlated with diminished lung function. Of note, the activation of TLR9-signaling cascade in pro-inflammatory cytokine response requires stimulation from microbial DNA (303).
The microbicidal malfunction in both innate and adaptive immune cells is also potentially linked to the deleterious effect of tobacco smoke, the major risk factor for COPD. It has been reported that, exposure of tobacco or cigarette smoke can impair phagocytosis/engulfment of NTHi by alveolar macrophages isolated from COPD patients (256, 304). Moreover, the chemical exposure also suppressed the TLR-induced TNF-α, IL-6, and IL-10 production in COPD alveolar macrophages that have been pre-stimulated with TLR2, 4, or 5 ligands (Pam3CSK4, LPS, or phase I flagellin, respectively), or whole NTHi bacteria (305). This may potentially delay the macrophage-dependent bacterial clearance. The suppressive effect of cigarette smoke in macrophage-dependent phagocytosis is due to the suppression of the PI3K signaling cascade which is required for optimal phagocytic activity and movement (256). Meanwhile, the cigarette smoke also inhibits the activation of the p38-ERK signaling pathway and p65/NF-κB, thus dampens the NTHi LOS-induced cytokine production of COPD alveolar macrophages (305). The diminished alveolar macrophage responsiveness could also be related to anticholinergic agents used by COPD patients that results in lower concentrations of NTHi-induced TNF-α (306). Nevertheless, the impaired phagocytosis of NTHi by COPD alveolar macrophages could be improved in the presence of nuclear erythroid related factor 2 and microRNA MiR-328 (307, 308). Interestingly, in addition to the constant exacerbated inflammatory effect observed in different murine model studies, Gaschler et al. observed a rapid pulmonary clearance of NTHi in mice upon exposure to cigarette smoke, and this was positively correlated with an increased neutrophilia in the animal BAL fluid (236, 309–311). However, in other COPD animal studies, cigarette smoke also impaired the IL-22 production that has a potential anti-bacterial activity while delaying the airway clearance of NTHi (311–313). Interestingly, IL-22 might play a protective role in COPD exacerbation as supplementation of IL-22 manages to restore the homeostasis of airway immune response and improve NTHi clearance (313).
The increased airway neutrophilia might be due to the enhanced production of pulmonary IL-17 triggered by cigarette smoke (152, 236, 311, 314). This may imply the important microbicidal role of neutrophils (neutrophilia) in compensating the COPD- or cigarette smoke-associated dysfunctional alveolar macrophages (96, 315). However, such compensation may not be adequate to provide optimal immune defense to eradicate persistent NTHi lower airway colonization, since the cigarette smoke also has profound suppressive effect on the host adaptive immunity, thus constantly risking the COPD patients to episodes of exacerbation and relapsed infection. In adaptive immunity, cigarette smoke impairs the antigen-specific B and T cells responses to NTHi infection. It suppresses the secretion of IFN-γ and IL-4 by NTHi-specific T cells. Antibody production by B cells has also been attenuated, with lower levels of specific anti-P6 antibodies and compromised IgG1, IgG2a, and IgA class switching (311, 312).
A recent and some previous cohort studies revealed that the level of airway antimicrobial cathelicidin (hCAP18/LL-37) in COPD patients increase gradually from the stable disease to exacerbation states (176, 316). Moreover, higher levels of cathelicidin are positively associated with NTHi airway colonization, sputum neutrophilia, and higher concentrations of IL-8, particularly in the NTHi-infected COPD patients. Of note, cathelicidin and other AMPs play important roles in the innate immune defense against different pathogens and persist immunomodulatory properties (317–319). Ironically, it is plausible that the increased level of cathelicidin could diminish or alter the balance in lung microbiota, and the immune/inflammatory response. This might contribute to the “vicious circle,” thus considerably increasing the risk for NTHi infection during COPD exacerbations (214, 320). Moreover, the microbicidal property of cathelicidin could be compromised by the inflammatory conditions in the airway, such as low pH, or the effect of cigarettes that causes peptide citrulination and modification (321, 322). Finally, expression of AMPs (human beta defensin 2 and S100A7) by COPD airway epithelium in response to NTHi infection, is also disturbed by cigarette smoke. The insulted airway cells have also a reduced expression of TLR4 and IL-8, and impaired NTHi-induced NF-κB activation (175, 296). Thus, a large body of evidence exists on the deleterious effects of tobacco smoke.
Antibiotic treatment of AECOPD has been shown to significantly reduce the risk of treatment failure, especially for in-patients with severe exacerbations and patients requiring intensive care (323). The efficacity of antibiotic treatment for out-patients with exacerbations is less clear (323, 324).
Recommendations on which empirical treatment to use for AECOPD varies between different countries, but common antimicrobial agents that are frequently used as definitive therapy against NTHi include aminopenicillins (with or without a beta-lactamase inhibitor), tetracyclines, trimethoprim-sulfamethoxazole, and fluoroquinolones. In addition, the Clinical and Laboratory Standards Institute (CLSI) has developed clinical breakpoints for the macrolides azithromycin and clarithromycin (325), whereas the European Committee on Antimicrobial Susceptibility Testing (EUCAST) have not set any clinical breakpoints against this class of antibiotics due to lack of clinical data (326). One study shows that NTHi frequently develops resistance to macrolides during prolonged treatment and that treatment failure may occur, making fluoroquinolones more reliable for eradication in COPD-patients (327). As for aminopenicillins, resistance is also common, with up to 10–20% of NTHi isolates expressing beta-lactamases and an additional 10–20% of the isolates having amino acid substitutions in penicillin-binding protein 3 (PBP3), which reduces their susceptibility to these agents (328, 329). The fraction of isolates expressing beta-lactamases has been stable during the last years, whereas an increase has been seen in isolates displaying altered PBP3 (330, 331). This is worrisome, since some of these amino acid substitutions also confer resistance to third generation cephalosporins (332). Moreover, there seems to be a correlation between isolates expressing altered PBP3 and increased invasiveness. Studies have shown that strains that express a mutated PBP3 with certain key amino acid substitution have a significantly higher rate of invasion of bronchial epithelial cells compared to strains with a wild type PBP3 (333). However, when such mutated PBP3 was cloned into a susceptible wild type strain, invasion efficacy did not increase, suggesting that PBP3 is only indirectly linked to invasion (334).
Besides using antibiotics for acute management of COPD exacerbations, some studies have considered the use of continuous prophylactic antibiotics in the management of patients with COPD (212). There is some evidence that continuous administration of macrolide antibiotics would prevent future exacerbations in a selected population of the most severely ill patients, but a Cochrane review revealed no support for a reduced all-cause mortality or less hospital readmissions (335). However, more recent studies have shown a significant decrease in both the frequency of exacerbations and hospitalizations when long-term azithromycin treatment was chosen (336).
The fact that macrolide antibiotics display not only antimicrobial effects, but also have anti-inflammatory and immunomodulatory properties, has made them interesting to use as prophylactic therapy (212). It has been shown that azithromycin inhibits mucus hypersecretion in the respiratory tract by significantly inhibiting TNF-α induction of the MUC5AC mucin secretion from human nasal epithelial cells (337). More specifically, it has been shown that azithromycin can reduce the NTHi-dependent induction of MUC5AC expression by suppressing the transcription factor activator protein-1 (338). Apart from affecting mucus secretion, it also seems that low-dose azithromycin has the ability to improve phagocytosis of bacteria by airway macrophages (339). One study showed that azithromycin concentrations that were unable to kill NTHi still increased the uptake rate of the bacteria into alveolar macrophages by enhancing their phagocytic function (340). However, the risk of development of antimicrobial resistance limits the use of low-dose azithromycin solely for its immunological properties. This has triggered an interest in finding new macrolide substances that lack antibiotic effect and solely interact with the airway immune system (341).
The considerable clinical problems caused by NTHi with regard to COPD exacerbations and otitis media has prompted the scientific community to investigate whether a vaccine can be developed against the pathogen (5, 58, 342). The search has been intensified due to a steady increase in antibiotic resistance and a trend of more invasive infections caused by NTHi over the last decade (5). Whereas, a highly efficient glycoconjugate vaccine has previously been developed against Hib, an identical strategy cannot be employed against NTHi due to the lack of a polysaccharide capsule. Vaccine developments efforts have thus been concentrated on identifying NTHi surface structures that are immunogenic, have low antigenic variability, and are conserved across this genetically highly heterogeneous species. Several promising vaccine candidates have been identified in the last 25 years, as excellently reviewed elsewhere (58, 342).
Two of these antigens, fused into one protein, Protein E-PilA, are together with Protein D currently being tested by GlaxoSmithKline in a phase IIb proof-of-concept clinical trial (randomized, observer-blind, placebo-controlled, and multicentric) for infection prophylaxis in COPD patients (50–70 years old) (5). Notably, the M. catarrhalis ubiquitous surface protein A2 (UspA2) is also included in the vaccine so that an immune response against both exacerbation-causing pathogens could be elicited by the same preparation. This clinical study (NCT03281876) is the only one currently being conducted on NTHi (and M. catarrhalis) according to clinicaltrials.gov, and as the investigations are on-going, the results are currently unknown.
Due to an increase in the difficulty to treat NTHi infections, an efficient and protective NTHi vaccine likely considerably raises the quality of life of COPD patients. Since NTHi-mediated exacerbations contribute to the progression of the disease and a steady deterioration of the pulmonary capacity of those patients, prevention against NTHi infections potentially slows down the debilitating effect of the disease. It is therefore critical to continue this line of research until such a vaccine has been obtained. It could also be worth targeting non-conventional structures with a vaccine, such as the secreted enzymes urease and IgA1-protease that have proven important for NTHi infections in COPD patients in several studies (222).
COPD is a multifaceted airway disease. Several factors influence the clinical outcome of COPD. Importantly, the crosstalk between intrinsic factors (the stability and integrity of the airway immune response and structure in addition to hereditary factors), and the extrinsic factors (lung microbiome, viral and bacterial infections, meteorological factors, and noxious inhalation) determines the fate of lower airway opportunistic infection by H. influenzae. Intriguingly, NTHi has been one of the most isolated pathogens at both stable and exacerbation states of COPD. Such persistent airway colonization of NTHi costs virulence fitness to counteract with the bactericidal effect of the host immune response. Adversely, the impaired defense mechanisms in COPD are not only unable to protect the lung structure from inhaled physical assaults, but they also fail to suppress NTHi infection. The disoriented immune response in COPD instead allows the pathogen to cause more harm and inflammation in the airways. The currently used bronchodilator and inhaled corticosteroid therapies have limited efficacy in preventing disease progression in COPD. Moreover, the inhaled corticosteroid therapies might have side effects that may weaken the immune response. Hence, more investigations are needed to garner a more adequate knowledge regarding the variabilities in immune networking of COPD. This knowledge will be an important platform for a more efficient drug design. In addition, a vaccine targeting NTHi is another important approach in controlling the infective exacerbations in COPD as the antibiotic treatment is also getting dampened by the emergence of NTHi antibiotic resistance.
Y-CS coordinated and drafted the major part of the manuscript, and prepared the figures; FJ participated in the literature study of virulence and vaccine research of NTHi; JT prepared the review section for NTHi epidemiology in COPD and antibiotic studies; Y-CS and KR edited and critically revised the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the following funding agencies for their financial support during the preparation of the manuscript. They are the Alfred Österlund, the Anna and Edwin Berger, the Swedish Medical Research Council (grant number K2015-57X-03163-43-4, www.vr.se), the Physiographical Society (Forssman's Foundation and, Endowments for the Natural Sciences, Medicine and Technology), Skåne County Council's research and development foundation, and Heart Lung Foundation (KR: grant number 20150697, www.hjart-lungfonden.se).
1. Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. (2011) 184:957–63. doi: 10.1164/rccm.201104-0655OC
2. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, et al. Bacterial topography of the healthy human lower respiratory tract. MBio (2017) 8:e02287-16. doi: 10.1128/mBio.02287-16
3. Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. (2017) 15:259–70. doi: 10.1038/nrmicro.2017.14
4. Duell BL, Su YC, Riesbeck K. Host-pathogen interactions of nontypeable Haemophilus influenzae: from commensal to pathogen. FEBS Lett. (2016) 590:3840–53. doi: 10.1002/1873-3468.12351
5. Jalalvand F, Riesbeck K. Update on non-typeable Haemophilus influenzae-mediated disease and vaccine development. Expert Rev Vaccines (2018) 17:503–12. doi: 10.1080/14760584.2018.1484286
6. Slack MPE. The evidence for non-typeable Haemophilus influenzae as a causative agent of childhood pneumonia. Pneumonia (Nathan) (2017) 9:9. doi: 10.1186/s41479-017-0033-2
7. Pizzutto SJ, Hare KM, Upham JW. Bronchiectasis in children: current concepts in immunology and microbiology. Front Pediatr. (2017) 5:123. doi: 10.3389/fped.2017.00123
8. Cardines R, Giufre M, Pompilio A, Fiscarelli E, Ricciotti G, Di Bonaventura G, et al. Haemophilus influenzae in children with cystic fibrosis: antimicrobial susceptibility, molecular epidemiology, distribution of adhesins and biofilm formation. Int J Med Microbiol. (2012) 302:45–52. doi: 10.1016/j.ijmm.2011.08.003
9. Azadeh N, Limper AH, Carmona EM, Ryu JH. The role of infection in interstitial lung diseases: a review. Chest (2017) 152:842–52. doi: 10.1016/j.chest.2017.03.033
10. Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, Dumont P, et al. Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax (2013) 68:1150–6. doi: 10.1136/thoraxjnl-2012-202917
11. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. (2008) 359:2355–65. doi: 10.1056/NEJMra0800353
12. Sriram KB, Cox AJ, Sivakumaran P, Singh M, Watts AM, West NP, et al. Non-typeable Haemophilus influenzae detection in the lower airways of patients with lung cancer and chronic obstructive pulmonary disease. Multidiscip Respir Med. (2018) 13:11. doi: 10.1186/s40248-018-0123-x
13. Ferkol T, Schraufnagel D. The global burden of respiratory disease. Ann Am Thorac Soc. (2014) 11:404–6. doi: 10.1513/AnnalsATS.201311-405PS
14. Diaz-Guzman E, Mannino DM. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin Chest Med. (2014) 35:7–16. doi: 10.1016/j.ccm.2013.10.002
15. GOLD-Report. Global strategy for diagnosis, management, and prevention of chronic obstructive pulmonary disease (2018 Report). in Global Initiative for Chronic Obstructive Lung Disease. Agusti AG, Vogelmeier C, editor (2018). Available online at: http://goldcopd.org
16. Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. (2008) 31:1334–56. doi: 10.1183/09031936.00018908
17. Barbu C, Iordache M, Man MG. Inflammation in COPD: pathogenesis, local and systemic effects. Rom J Morphol Embryol. (2011) 52:21–7.
18. Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers (2015) 1:15076. doi: 10.1038/nrdp.2015.76
19. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet (2017) 389:1931–40. doi: 10.1016/S0140-6736(17)31222-9
20. Fabbri LM, Luppi F, Beghe B, Rabe KF. Complex chronic comorbidities of COPD. Eur Respir J. (2008) 31:204–12. doi: 10.1183/09031936.00114307
21. Huertas A, Palange P. COPD: a multifactorial systemic disease. Ther Adv Respir Dis. (2011) 5:217–24. doi: 10.1177/1753465811400490
22. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet (2007) 370:765–73. doi: 10.1016/S0140-6736(07)61380-4
23. Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Environ Res Public Health (2018) 15:E1033. doi: 10.3390/ijerph15051033
24. Imaoka H, Hoshino T. Bronchial asthma: progress in diagnosis and treatments. Topics: II. Pathogenesis and pathophysiology; 1. The Dutch hypothesis and British hypothesis. Nihon Naika Gakkai Zasshi (2013) 102:1359–64. doi: 10.2169/naika.102.1359
25. Primhak RA, Tanner MS. Alpha-1 antitrypsin deficiency. Arch Dis Child. (2001) 85:2–5. doi: 10.1136/adc.85.1.2
26. Rodriguez-Revenga L, Iranzo P, Badenas C, Puig S, Carrio A, Mila M. A novel elastin gene mutation resulting in an autosomal dominant form of cutis laxa. Arch Dermatol. (2004) 140:1135–9. doi: 10.1001/archderm.140.9.1135
27. Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, Hersh CP, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. (2014) 2:214–25. doi: 10.1016/S2213-2600(14)70002-5
28. Lee SR, Kim WT, Kim TN, Nam JK, Kim WJ, Leem SH. Association between the length of the MUC8-minisatellite 5 region and susceptibility to chronic obstructive pulmonary disease (COPD). Genes Genomics (2018) 40:123–7. doi: 10.1007/s13258-017-0630-8
29. Smith BM, Traboulsi H, Austin JHM, Manichaikul A, Hoffman EA, Bleecker ER, et al. Human airway branch variation and chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. (2018) 115:E974–81. doi: 10.1073/pnas.1715564115
30. Jackson VE, Ntalla I, Sayers I, Morris R, Whincup P, Casas JP, et al. Exome-wide analysis of rare coding variation identifies novel associations with COPD and airflow limitation in MOCS3, IFIT3 and SERPINA12. Thorax (2016) 71:501–9. doi: 10.1136/thoraxjnl-2015-207876
31. D'Anna C, Cigna D, Di Sano C, Di Vincenzo S, Dino P, Ferraro M, et al. Exposure to cigarette smoke extract and lipopolysaccharide modifies cytoskeleton organization in bronchial epithelial cells. Exp Lung Res. (2017) 43:347–58. doi: 10.1080/01902148.2017.1377784
32. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. (2018) 58:157–69. doi: 10.1165/rcmb.2017-0200TR
33. Lapperre TS, Sont JK, van Schadewijk A, Gosman MM, Postma DS, Bajema IM, et al. Smoking cessation and bronchial epithelial remodelling in COPD: a cross-sectional study. Respir Res. (2007) 8:85. doi: 10.1186/1465-9921-8-85
34. Perotin JM, Adam D, Vella-Boucaud J, Delepine G, Sandu S, Jonvel AC, et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. (2014) 15:151. doi: 10.1186/s12931-014-0151-9
35. Papi A, Casoni G, Caramori G, Guzzinati I, Boschetto P, Ravenna F, et al. COPD increases the risk of squamous histological subtype in smokers who develop non-small cell lung carcinoma. Thorax (2004) 59:679–81. doi: 10.1136/thx.2003.018291
36. Cosio M, Ghezzo H, Hogg JC, Corbin R, Loveland M, Dosman J, et al. The relations between structural changes in small airways and pulmonary-function tests. N Engl J Med. (1978) 298:1277–81. doi: 10.1056/NEJM197806082982303
37. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. (2017) 50:1601805. doi: 10.1183/13993003.01805-2016
38. Vogel ER, VanOosten SK, Holman MA, Hohbein DD, Thompson MA, Vassallo R, et al. Cigarette smoke enhances proliferation and extracellular matrix deposition by human fetal airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L978–86. doi: 10.1152/ajplung.00111.2014
39. Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. (2006) 126:725–35. doi: 10.1309/JC477FAEL1YKV54W7
40. van Straaten JF, Coers W, Noordhoek JA, Huitema S, Flipsen JT, Kauffman HF, et al. Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod Pathol. (1999) 12:697–705.
41. Ahrman E, Hallgren O, Malmstrom L, Hedstrom U, Malmstrom A, Bjermer L, et al. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J Proteomics (2018) 2018:S1874-3919(18)30084-8. doi: 10.1016/j.jprot.2018.02.027
42. Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur Respir J. (2012) 39:197–209. doi: 10.1183/09031936.00121611
43. Sandhaus RA, Turino G. Neutrophil elastase-mediated lung disease. COPD (2013) 10(Suppl 1):60–3. doi: 10.3109/15412555.2013.764403
44. Moore PJ, Reidel B, Ghosh A, Sesma J, Kesimer M, Tarran R. Cigarette smoke modifies and inactivates SPLUNC1, leading to airway dehydration. FASEB J. (2018) 32:1–16. doi: 10.1096/fj.201800345R
45. Larstad M, Almstrand AC, Larsson P, Bake B, Larsson S, Ljungstrom E, et al. Surfactant protein A in exhaled endogenous particles is decreased in chronic Obstructive pulmonary disease (COPD) patients: a pilot study. PLoS ONE (2015) 10:e0144463. doi: 10.1371/journal.pone.0144463
46. Takamiya R, Uchida K, Shibata T, Maeno T, Kato M, Yamaguchi Y, et al. Disruption of the structural and functional features of surfactant protein A by acrolein in cigarette smoke. Sci Rep. (2017) 7:8304. doi: 10.1038/s41598-017-08588-5
47. Kopa PN, Pawliczak R. Effect of smoking on gene expression profile - overall mechanism, impact on respiratory system function, and reference to electronic cigarettes. Toxicol Mech Methods (2018) 28:397–409. doi: 10.1080/15376516.2018.1461289
48. Kan M, Shumyatcher M, Himes BE. Using omics approaches to understand pulmonary diseases. Respir Res. (2017) 18:149. doi: 10.1186/s12931-017-0631-9
49. Yang M, Kohler M, Heyder T, Forsslund H, Garberg HK, Karimi R, et al. Long-term smoking alters abundance of over half of the proteome in bronchoalveolar lavage cell in smokers with normal spirometry, with effects on molecular pathways associated with COPD. Respir Res. (2018) 19:40. doi: 10.1186/s12931-017-0695-6
50. Damera G, Pham TH, Zhang J, Ward CK, Newbold P, Ranade K, et al. A sputum proteomic signature that associates with increased IL-1β levels and bacterial exacerbations of COPD. Lung (2016) 194:363–9. doi: 10.1007/s00408-016-9877-0
51. Fujii K, Nakamura H, Nishimura T. Recent mass spectrometry-based proteomics for biomarker discovery in lung cancer, COPD, and asthma. Expert Rev Proteomics (2017) 14:373–86. doi: 10.1080/14789450.2017.1304215
52. Philibert RA, Sears RA, Powers LS, Nash E, Bair T, Gerke AK, et al. Coordinated DNA methylation and gene expression changes in smoker alveolar macrophages: specific effects on VEGF receptor 1 expression. J Leukoc Biol. (2012) 92:621–31. doi: 10.1189/jlb.1211632
53. Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. (2012) 21:3073–82. doi: 10.1093/hmg/dds135
54. Zhao J, Harper R, Barchowsky A, Di YP. Identification of multiple MAPK-mediated transcription factors regulated by tobacco smoke in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L480–90. doi: 10.1152/ajplung.00345.2006
55. Geiman TM, Robertson KD. Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem. (2002) 87:117–25. doi: 10.1002/jcb.10286
56. Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. (2005) 1056:218–33. doi: 10.1196/annals.1352.026
57. Sundar IK, Chung S, Hwang JW, Lapek JD Jr, Bulger M, Friedman AE, et al. Mitogen- and stress-activated kinase 1 (MSK1) regulates cigarette smoke-induced histone modifications on NF-kappaB-dependent genes. PLoS ONE (2012) 7:e31378. doi: 10.1371/journal.pone.0031378
58. Khan MN, Ren D, Kaur R, Basha S, Zagursky R, Pichichero ME. Developing a vaccine to prevent otitis media caused by nontypeable Haemophilus influenzae. Expert Rev Vaccines (2016) 15:863–78. doi: 10.1586/14760584.2016.1156539
59. Marumo S, Hoshino Y, Kiyokawa H, Tanabe N, Sato A, Ogawa E, et al. p38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm Med. (2014) 14:79. doi: 10.1186/1471-2466-14-79
60. Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today (1999) 5:439–47.
61. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. (2005) 30:43–52. doi: 10.1016/j.tibs.2004.11.009
62. Yang SR, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, et al. IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol. (2008) 38:689–98. doi: 10.1165/rcmb.2007-0379OC
63. Yao H, Hwang JW, Moscat J, Diaz-Meco MT, Leitges M, Kishore N, et al. Protein kinase C zeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications. J Biol Chem. (2010) 285:5405–16. doi: 10.1074/jbc.M109.041418
64. Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis (2002) 23:1511–8. doi: 10.1093/carcin/23.9.1511
65. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/cr.2010.178
66. Schmidt P, Youhnovski N, Daiber A, Balan A, Arsic M, Bachschmid M, et al. Specific nitration at tyrosine 430 revealed by high resolution mass spectrometry as basis for redox regulation of bovine prostacyclin synthase. J Biol Chem. (2003) 278:12813–9. doi: 10.1074/jbc.M208080200
67. Lee MK, Hong Y, Kim SY, London SJ, Kim WJ. DNA methylation and smoking in Korean adults: epigenome-wide association study. Clin Epigenetics (2016) 8:103. doi: 10.1186/s13148-016-0266-6
68. Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci USA. (2008) 105:16356–61. doi: 10.1073/pnas.0808699105
69. Shen JX, Yakel JL. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol Sin. (2009) 30:673–80. doi: 10.1038/aps.2009.64
71. Daijo H, Hoshino Y, Kai S, Suzuki K, Nishi K, Matsuo Y, et al. Cigarette smoke reversibly activates hypoxia-inducible factor 1 in a reactive oxygen species-dependent manner. Sci Rep. (2016) 6:34424. doi: 10.1038/srep34424
72. Mercado N, Thimmulappa R, Thomas CM, Fenwick PS, Chana KK, Donnelly LE, et al. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun. (2011) 406:292–8. doi: 10.1016/j.bbrc.2011.02.035
73. Shin JM, Park JH, Kim HJ, Park IH, Lee HM. Cigarette smoke extract increases vascular endothelial growth factor production via TLR4/ROS/MAPKs/NF-kappaB pathway in nasal fibroblast. Am J Rhinol Allergy (2017) 31:78–84. doi: 10.2500/ajra.2017.31.4415
74. Reynolds PR, Kasteler SD, Schmitt RE, Hoidal JR. Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am J Respir Cell Mol Biol. (2011) 45:411–8. doi: 10.1165/rcmb.2010-0231OC
75. Alam S, Li Z, Janciauskiene S, Mahadeva R. Oxidation of Z alpha1-antitrypsin by cigarette smoke induces polymerization: a novel mechanism of early-onset emphysema. Am J Respir Cell Mol Biol. (2011) 45:261–9. doi: 10.1165/rcmb.2010-0328OC
76. Pandey KC, De S, Mishra PK. Role of proteases in chronic obstructive pulmonary disease. Front Pharmacol. (2017) 8:512. doi: 10.3389/fphar.2017.00512
77. Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L46–57. doi: 10.1152/ajplung.00241.2005
78. Moretto N, Bertolini S, Iadicicco C, Marchini G, Kaur M, Volpi G, et al. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L929–38. doi: 10.1152/ajplung.00046.2012
79. Hasnis E, Bar-Shai M, Burbea Z, Reznick AZ. Mechanisms underlying cigarette smoke-induced NF-kappaB activation in human lymphocytes: the role of reactive nitrogen species. J Physiol Pharmacol. (2007) 58 (Suppl 5 Pt 1):275–87.
80. Givi ME, Redegeld FA, Folkerts G, Mortaz E. Dendritic cells in pathogenesis of COPD. Curr Pharm Des. (2012) 18:2329–35. doi: 10.2174/138161212800166068
81. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2008) 8:183–92. doi: 10.1038/nri2254
82. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet (2011) 378:1015–26. doi: 10.1016/S0140-6736(11)60988-4
83. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2015) 12(Suppl. 2):S169–75. doi: 10.1513/AnnalsATS.201503-126AW
84. Maes T, Bracke KR, Vermaelen KY, Demedts IK, Joos GF, Pauwels RA, et al. Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. Int Arch Allergy Immunol. (2006) 141:354–68. doi: 10.1159/000095462
85. Mukherjee S, Karmakar S, Babu SP. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis. (2016) 20:193–204. doi: 10.1016/j.bjid.2015.10.011
86. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
87. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. (2012) 91:142–9. doi: 10.1177/0022034511421200
88. Mortaz E, Henricks PA, Kraneveld AD, Givi ME, Garssen J, Folkerts G. Cigarette smoke induces the release of CXCL-8 from human bronchial epithelial cells via TLRs and induction of the inflammasome. Biochim Biophys Acta (2011) 1812:1104–10. doi: 10.1016/j.bbadis.2011.06.002
89. Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. (2013) 123:3967–82. doi: 10.1172/JCI65570
90. Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. (2004) 56:515–48. doi: 10.1124/pr.56.4.2
91. Russell RE, Thorley A, Culpitt SV, Dodd S, Donnelly LE, Demattos C, et al. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L867–73. doi: 10.1152/ajplung.00020.2002
92. Culpitt SV, Rogers DF, Shah P, De Matos C, Russell RE, Donnelly LE, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2003) 167:24–31. doi: 10.1164/rccm.200204-298OC
93. Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax (2002) 57:590–5. doi: 10.1136/thorax.57.7.590
94. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
95. Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. (1998) 158:1277–85. doi: 10.1164/ajrccm.158.4.9802078
96. Liu J, Pang Z, Wang G, Guan X, Fang K, Wang Z, et al. Advanced role of neutrophils in common respiratory diseases. J Immunol Res. (2017) 2017:6710278. doi: 10.1155/2017/6710278
97. Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med. (1994) 149(3 Pt 1):803–10. doi: 10.1164/ajrccm.149.3.7509705
98. Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2007) 119:1065–71. doi: 10.1016/j.jaci.2006.12.640
99. Vaidyanathan A, Damodar KS. Increased metabolic activity of neutrophils in patients with chronic obstructive pulmonary disease. Lung India (2015) 32:589–92. doi: 10.4103/0970-2113.168134
100. Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med. (1997) 155:449–53. doi: 10.1164/ajrccm.155.2.9032177
101. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. (2010) 363:2233–47. doi: 10.1056/NEJMra0910061
102. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
103. Colarusso C, Terlizzi M, Molino A, Pinto A, Sorrentino R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget (2017) 8:81813–24. doi: 10.18632/oncotarget.17850
104. Wang H, Lv C, Wang S, Ying H, Weng Y, Yu W. NLRP3 inflammasome involves in the acute exacerbation of patients with chronic obstructive pulmonary disease. Inflammation (2018) 41:1321–33. doi: 10.1007/s10753-018-0780-0
105. Kim K, Kim HJ, Binas B, Kang JH, Chung IY. Inflammatory mediators ATP and S100A12 activate the NLRP3 inflammasome to induce MUC5AC production in airway epithelial cells. Biochem Biophys Res Commun. (2018). doi: 10.1016/j.bbrc.2018.06.057
106. Uh ST, Koo SM, Kim Y, Kim K, Park S, Jang AS, et al. The activation of NLRP3-inflammsome by stimulation of diesel exhaust particles in lung tissues from emphysema model and RAW 264.7 cell line. Korean J Intern Med. (2017) 32:865–74. doi: 10.3904/kjim.2016.033
107. Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, et al. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2007) 175:998–1005. doi: 10.1164/rccm.200608-1113OC
108. D'Hulst A I, Maes T, Bracke KR, Demedts IK, Tournoy KG, Joos GF, et al. Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir Res. (2005) 6:147. doi: 10.1186/1465-9921-6-147
109. Upham JW, Xi Y. Dendritic cells in human lung disease: recent advances. Chest (2017) 151:668–73. doi: 10.1016/j.chest.2016.09.030
110. Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2002) 165:1404–9. doi: 10.1164/rccm.2107139
111. Costa C, Rufino R, Traves SL, Lapa ESJR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest (2008) 133:26–33. doi: 10.1378/chest.07-0393
112. Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. (2001) 17:946–53. doi: 10.1183/09031936.01.17509460
113. Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. (2009) 157:316–24. doi: 10.1111/j.1365-2249.2009.03965.x
114. Pridgeon C, Bugeon L, Donnelly L, Straschil U, Tudhope SJ, Fenwick P, et al. Regulation of IL-17 in chronic inflammation in the human lung. Clin Sci (Lond.) (2011) 120:515–24. doi: 10.1042/CS20100417
115. De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS ONE (2016) 11:e0145961. doi: 10.1371/journal.pone.0145961
116. Xia J, Zhao J, Shang J, Li M, Zeng Z, Zhao J, et al. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L619–27. doi: 10.1152/ajplung.00305.2014
117. Ying S, O'Connor B, Ratoff J, Meng Q, Fang C, Cousins D, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol. (2008) 181:2790–8. doi: 10.1016/j.jaci.2004.12.051
118. van der Strate BW, Postma DS, Brandsma CA, Melgert BN, Luinge MA, Geerlings M, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. (2006) 173:751–8. doi: 10.1164/rccm.200504-594OC
119. Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. (2007) 13:567–9. doi: 10.1038/nm1583
120. Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, Chida K. Antiendothelial cell antibodies in patients with COPD. Chest (2010) 138:1303–8. doi: 10.1378/chest.10-0863
121. Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2008) 177:156–63. doi: 10.1164/rccm.200701-014OC
122. Koethe SM, Kuhnmuench JR, Becker CG. Neutrophil priming by cigarette smoke condensate and a tobacco anti-idiotypic antibody. Am J Pathol. (2000) 157:1735–43. doi: 10.1016/S0002-9440(10)64810-9
123. Nahm DH, Lee YE, Yim EJ, Park HS, Yim H, Kang Y, et al. Identification of cytokeratin 18 as a bronchial epithelial autoantigen associated with nonallergic asthma. Am J Respir Crit Care Med. (2002) 165:1536–9. doi: 10.1164/rccm.200201-009OC
124. Caramori G, Ruggeri P, Di Stefano A, Mumby S, Girbino G, Adcock IM, et al. Autoimmunity and COPD: clinical implications. Chest (2018) 153:1424–31. doi: 10.1016/j.chest.2017.10.033
125. Cosio MG. Autoimmunity, T-cells and STAT-4 in the pathogenesis of chronic obstructive pulmonary disease. Eur Respir J. (2004) 24:3–5. doi: 10.1183/09031936.04.00043104
126. Agusti A, MacNee W, Donaldson K, Cosio M. Hypothesis: does COPD have an autoimmune component? Thorax (2003) 58:832–4. doi: 10.1136/thorax.58.10.832
127. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:796–802. doi: 10.1164/rccm.201010-1605OC
128. Hurst JR, Wedzicha JA. What is (and what is not) a COPD exacerbation: thoughts from the new GOLD guidelines. Thorax (2007) 62:198–9. doi: 10.1136/thx.2007.077883
129. Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet (2007) 370:786–96. doi: 10.1016/S0140-6736(07)61382-8
130. Pavord ID, Jones PW, Burgel PR, Rabe KF. Exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. (2016) 11:21–30. doi: 10.2147/COPD.S85978
131. Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2000) 161:1608–13. doi: 10.1164/ajrccm.161.5.9908022
132. Sapey E, Stockley RA. COPD exacerbations. 2: aetiology. Thorax (2006) 61:250–8. doi: 10.1136/thx.2005.041822
133. Ko FW, Chan KP, Hui DS, Goddard JR, Shaw JG, Reid DW, et al. Acute exacerbation of COPD. Respirology (2016) 21:1152–65. doi: 10.1111/resp.12780
134. Eldika N, Sethi S. Role of nontypeable Haemophilus influenzae in exacerbations and progression of chronic obstructive pulmonary disease. Curr Opin Pulm Med. (2006) 12:118–24. doi: 10.1097/01.mcp.0000208451.50231.8f
135. Celli BR, Barnes PJ. Exacerbations of chronic obstructive pulmonary disease. Eur Respir J. (2007) 29:1224–38. doi: 10.1183/09031936.00109906
136. Wilkinson TMA, Aris E, Bourne S, Clarke SC, Peeters M, Pascal TG, et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax (2017) 72:919–27. doi: 10.1136/thoraxjnl-2016-209023
137. Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, Message S, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2001) 164:1618–23. doi: 10.1164/ajrccm.164.9.2105011
138. Kurai D, Saraya T, Ishii H, Takizawa H. Virus-induced exacerbations in asthma and COPD. Front Microbiol. (2013) 4:293. doi: 10.3389/fmicb.2013.00293
139. Mohan A, Chandra S, Agarwal D, Guleria R, Broor S, Gaur B, et al. Prevalence of viral infection detected by PCR and RT-PCR in patients with acute exacerbation of COPD: a systematic review. Respirology (2010) 15:536–42. doi: 10.1111/j.1440-1843.2010.01722.x
140. Biancardi E, Fennell M, Rawlinson W, Thomas PS. Viruses are frequently present as the infecting agent in acute exacerbations of chronic obstructive pulmonary disease in patients presenting to hospital. Intern Med J. (2016) 46:1160–5. doi: 10.1111/imj.13213
141. Hewitt R, Farne H, Ritchie A, Luke E, Johnston SL, Mallia P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther Adv Respir Dis. (2016) 10:158–74. doi: 10.1177/1753465815618113
142. Koul PA, Mir H, Akram S, Potdar V, Chadha MS. Respiratory viruses in acute exacerbations of chronic obstructive pulmonary disease. Lung India (2017) 34:29–33. doi: 10.4103/0970-2113.197099
143. Jafarinejad H, Moghoofei M, Mostafaei S, Salimian J, Azimzadeh Jamalkandi S, Ahmadi A. Worldwide prevalence of viral infection in AECOPD patients: a meta-analysis. Microb Pathog. (2017) 113:190–6. doi: 10.1016/j.micpath.2017.10.021
144. Britto CJ, Brady V, Lee S, Dela Cruz CS. Respiratory viral infections in chronic lung diseases. Clin Chest Med. (2017) 38:87–96. doi: 10.1016/j.ccm.2016.11.014
145. Yin T, Zhu Z, Mei Z, Feng J, Zhang W, He Y, et al. Analysis of viral infection and biomarkers in patients with acute exacerbation of chronic obstructive pulmonary disease. Clin Respir J. (2018) 12:1228–39. doi: 10.1111/crj.12656
146. Chang CL, Sullivan GD, Karalus NC, Mills GD, McLachlan JD, Hancox RJ. Predicting early mortality in acute exacerbation of chronic obstructive pulmonary disease using the CURB65 score. Respirology (2011) 16:146–51. doi: 10.1111/j.1440-1843.2010.01866.x
147. Lou P, Zhu Y, Chen P, Zhang P, Yu J, Zhang N, et al. Vulnerability, beliefs, treatments and economic burden of chronic obstructive pulmonary disease in rural areas in China: a cross-sectional study. BMC Public Health (2012) 12:287. doi: 10.1186/1471-2458-12-287
148. Rosell A, Monso E, Soler N, Torres F, Angrill J, Riise G, et al. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med. (2005) 165:891–7. doi: 10.1001/archinte.165.8.891
149. Wang Z, Bafadhel M, Haldar K, Spivak A, Mayhew D, Miller BE, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J. (2016) 47:1082–92. doi: 10.1183/13993003.01406-2015
150. Polverino E, Rosales-Mayor E, Benegas M, Menendez R, Alcaraz-Serrano V, Ansotegui E, et al. Pneumonic and non-pneumonic exacerbations in bronchiectasis: clinical and microbiological differences. J Infect. (2018) 77:99–106. doi: 10.1016/j.jinf.2018.04.006
151. Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax (2018) 73:422–30. doi: 10.1136/thoraxjnl-2017-210408
152. Simpson JL, Baines KJ, Horvat JC, Essilfie AT, Brown AC, Tooze M, et al. COPD is characterized by increased detection of Haemophilus influenzae, Streptococcus pneumoniae and a deficiency of Bacillus species. Respirology (2016) 21:697–704. doi: 10.1111/resp.12734
153. Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. (2002) 347:465–71. doi: 10.1056/NEJMoa012561
154. Rangelov K, Sethi S. Role of infections. Clin Chest Med. (2014) 35:87–100. doi: 10.1016/j.ccm.2013.09.012
155. Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. (2011) 184:662–71. doi: 10.1164/rccm.201104-0597OC
156. George SN, Garcha DS, Mackay AJ, Patel AR, Singh R, Sapsford RJ, et al. Human rhinovirus infection during naturally occurring COPD exacerbations. Eur Respir J. (2014) 44:87–96. doi: 10.1183/09031936.00223113
157. Utokaparch S, Sze MA, Gosselink JV, McDonough JE, Elliott WM, Hogg JC, et al. Respiratory viral detection and small airway inflammation in lung tissue of patients with stable, mild COPD. COPD (2014) 11:197–203. doi: 10.3109/15412555.2013.836166
158. D'Anna SE, Balbi B, Cappello F, Carone M, Di Stefano A. Bacterial-viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. (2016) 11:445–53. doi: 10.2147/COPD.S93398
159. Unger BL, Faris AN, Ganesan S, Comstock AT, Hershenson MB, Sajjan US. Rhinovirus attenuates non-typeable Haemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1. PLoS Pathog. (2012) 8:e1002969. doi: 10.1371/journal.ppat.1002969
160. Gulraiz F, Bellinghausen C, Bruggeman CA, Stassen FR. Haemophilus influenzae increases the susceptibility and inflammatory response of airway epithelial cells to viral infections. FASEB J. (2015) 29:849–58. doi: 10.1096/fj.14-254359
161. Bellinghausen C, Gulraiz F, Heinzmann AC, Dentener MA, Savelkoul PH, Wouters EF, et al. Exposure to common respiratory bacteria alters the airway epithelial response to subsequent viral infection. Respir Res. (2016) 17:68. doi: 10.1186/s12931-016-0382-z
162. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. (2008) 178:1271–81. doi: 10.1164/rccm.200801-136OC
163. Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope (2009) 119:1406–11. doi: 10.1002/lary.20498
164. Jia L, Xie J, Zhao J, Cao D, Liang Y, Hou X, et al. Mechanisms of severe mortality-associated bacterial co-infections following influenza virus infection. Front Cell Infect Microbiol. (2017) 7:338. doi: 10.3389/fcimb.2017.00338
165. Oliver BG, Lim S, Wark P, Laza-Stanca V, King N, Black JL, et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax (2008) 63:519–25. doi: 10.1136/thx.2007.081752
166. Arnason JW, Murphy JC, Kooi C, Wiehler S, Traves SL, Shelfoon C, et al. Human β-defensin-2 production upon viral and bacterial co-infection is attenuated in COPD. PLoS ONE (2017) 12:e0175963. doi: 10.1371/journal.pone.0175963
167. McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. (2014) 12:252–62. doi: 10.1038/nrmicro3231
168. Bellinghausen C, Rohde GGU, Savelkoul PHM, Wouters EFM, Stassen FRM. Viral-bacterial interactions in the respiratory tract. J Gen Virol. (2016) 97:3089–102. doi: 10.1099/jgv.0.000627
169. Welte T, Miravitlles M. Viral, bacterial or both? Regardless, we need to treat infection in COPD. Eur Respir J. (2014) 44:11–3. doi: 10.1183/09031936.00041914
170. Wark PA, Tooze M, Powell H, Parsons K. Viral and bacterial infection in acute asthma and chronic obstructive pulmonary disease increases the risk of readmission. Respirology (2013) 18:996–1002. doi: 10.1111/resp.12099
171. Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. (2006) 173:1114–21. doi: 10.1164/rccm.200506-859OC
172. Wilkinson TM, Hurst JR, Perera WR, Wilks M, Donaldson GC, Wedzicha JA. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest (2006) 129:317–24. doi: 10.1378/chest.129.2.317
173. Perotin JM, Dury S, Renois F, Deslee G, Wolak A, Duval V, et al. Detection of multiple viral and bacterial infections in acute exacerbation of chronic obstructive pulmonary disease: a pilot prospective study. J Med Virol. (2013) 85:866–73. doi: 10.1002/jmv.23495
174. Kawamatawong T, Apiwattanaporn A, Siricharoonwong W. Serum inflammatory biomarkers and clinical outcomes of COPD exacerbation caused by different pathogens. Int J Chron Obstruct Pulmon Dis. (2017) 12:1625–30. doi: 10.2147/COPD.S132132
175. Amatngalim GD, Schrumpf JA, Henic A, Dronkers E, Verhoosel RM, Ordonez SR, et al. Antibacterial defense of human airway epithelial cells from chronic obstructive pulmonary disease patients induced by acute exposure to nontypeable Haemophilus influenzae: modulation by cigarette smoke. J Innate Immun. (2017) 9:359–74. doi: 10.1159/000455193
176. Parameswaran GI, Sethi S, Murphy TF. Effects of bacterial infection on airway antimicrobial peptides and proteins in COPD. Chest (2011) 140:611–7. doi: 10.1378/chest.10-2760
177. Berenson CS, Kruzel RL, Eberhardt E, Sethi S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. (2013) 208:2036–45. doi: 10.1093/infdis/jit400
178. Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. (2010) 35:1039–47. doi: 10.1183/09031936.00036709
179. Sapey E, Stockley JA, Greenwood H, Ahmad A, Bayley D, Lord JM, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 183:1176–86. doi: 10.1164/rccm.201008-1285OC
180. Sze MA, Hogg JC, Sin DD. Bacterial microbiome of lungs in COPD. Int J Chron Obstruct Pulmon Dis. (2014) 9:229–38. doi: 10.2147/COPD.S38932
181. Cookson W, Cox MJ, Moffatt MF. New opportunities for managing acute and chronic lung infections. Nat Rev Microbiol. (2018) 16:111–20. doi: 10.1038/nrmicro.2017.122
182. Mammen MJ, Sethi S. COPD and the microbiome. Respirology (2016) 21:590–9. doi: 10.1111/resp.12732
183. Wang L, Hao K, Yang T, Wang C. Role of the lung microbiome in the pathogenesis of chronic obstructive pulmonary disease. Chin Med J. (Engl.) (2017) 130:2107–11. doi: 10.4103/0366-6999.211452
184. Pienkowska K, Wiehlmann L, Tummler B. Airway microbial metagenomics. Microbes Infect. (2017) 2017:S1286-4579(17)30224-1. doi: 10.1016/j.micinf.2017.12.002
185. Hussell T, Lui S, Jagger C, Morgan D, Brand O. The consequence of matrix dysfunction on lung immunity and the microbiome in COPD. Eur Respir Rev. (2018) 27:180032. doi: 10.1183/16000617.0032-2018
186. O'Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. (2016) 196:4839–47. doi: 10.4049/jimmunol.1600279
187. Monso E. Microbiome in chronic obstructive pulmonary disease. Ann Transl Med. (2017) 5:251. doi: 10.21037/atm.2017.04.20
188. Ghebre MA, Pang PH, Diver S, Desai D, Bafadhel M, Haldar K, et al. Biological exacerbation clusters demonstrate asthma and chronic obstructive pulmonary disease overlap with distinct mediator and microbiome profiles. J Allergy Clin Immunol. (2018) 141:2027.e12–36.e12. doi: 10.1016/j.jaci.2018.04.013
189. Saeedi P, Salimian J, Ahmadi A, Imani Fooladi AA. The transient but not resident (TBNR) microbiome: a Yin Yang model for lung immune system. Inhal Toxicol. (2015) 27:451–61. doi: 10.3109/08958378.2015.1070220
190. Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SA, Homola D, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 188:1224–31. doi: 10.1164/rccm.201302-0341OC
191. Engel M, Endesfelder D, Schloter-Hai B, Kublik S, Granitsiotis MS, Boschetto P, et al. Influence of lung CT changes in chronic obstructive pulmonary disease (COPD) on the human lung microbiome. PLoS ONE (2017) 12:e0180859. doi: 10.1371/journal.pone.0180859
192. Tangedal S, Aanerud M, Gronseth R, Drengenes C, Wiker HG, Bakke PS, et al. Comparing microbiota profiles in induced and spontaneous sputum samples in COPD patients. Respir Res. (2017) 18:164. doi: 10.1186/s12931-017-0645-3
193. Diao W, Shen N, Du Y, Qian K, He B. Characterization of throat microbial flora in smokers with or without COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:1933–46. doi: 10.2147/COPD.S140243
194. Garcia-Nunez M, Millares L, Pomares X, Ferrari R, Perez-Brocal V, Gallego M, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. (2014) 52:4217–23. doi: 10.1128/JCM.01967-14
195. Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. (2014) 52:2813–23. doi: 10.1128/JCM.00035-14
196. Jubinville E, Veillette M, Milot J, Maltais F, Comeau AM, Levesque RC, et al. Exacerbation induces a microbiota shift in sputa of COPD patients. PLoS ONE (2018) 13:e0194355. doi: 10.1371/journal.pone.0194355
197. Shukla SD, Budden KF, Neal R, Hansbro PM. Microbiome effects on immunity, health and disease in the lung. Clin Transl Immunol. (2017) 6:e133. doi: 10.1038/cti.2017.6
198. Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2018) 141:117–27. doi: 10.1016/j.jaci.2017.04.022
199. Yadava K, Pattaroni C, Sichelstiel AK, Trompette A, Gollwitzer ES, Salami O, et al. Microbiota promotes chronic pulmonary inflammation by enhancing IL-17A and autoantibodies. Am J Respir Crit Care Med. (2016) 193:975–87. doi: 10.1164/rccm.201504-0779OC
200. Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2012) 185:1073–80. doi: 10.1164/rccm.201111-2075OC
201. Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:438–45. doi: 10.1164/rccm.201502-0223OC
202. Chalmers JD, McHugh BJ, Doherty C, Smith MP, Govan JR, Kilpatrick DC, et al. Mannose-binding lectin deficiency and disease severity in non-cystic fibrosis bronchiectasis: a prospective study. Lancet Respir Med. (2013) 1:224–32. doi: 10.1016/S2213-2600(13)70001-8
203. Dicker AJ, Crichton ML, Cassidy AJ, Brady G, Hapca A, Tavendale R, et al. Genetic mannose binding lectin deficiency is associated with airway microbiota diversity and reduced exacerbation frequency in COPD. Thorax (2018) 73:510–8. doi: 10.1136/thoraxjnl-2016-209931
204. Chambers DC, Gellatly SL, Hugenholtz P, Hansbro PM. JTD special edition 'Hot topics in COPD'-the microbiome in COPD. J Thorac Dis. (2014) 6:1525–31. doi: 10.3978/j.issn.2072-1439.2014.11.08
205. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE (2011) 6:e16384. doi: 10.1371/journal.pone.0016384
206. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS ONE (2010) 5:e8578. doi: 10.1371/journal.pone.0008578
207. Ahearn CP, Gallo MC, Murphy TF. Insights on persistent airway infection by non-typeable Haemophilus influenzae in chronic obstructive pulmonary disease. Pathog Dis. (2017) 75:ftx042. doi: 10.1093/femspd/ftx042
208. Van Eldere J, Slack MP, Ladhani S, Cripps AW. Non-typeable Haemophilus influenzae, an under-recognised pathogen. Lancet Infect Dis. (2014) 14:1281–92. doi: 10.1016/S1473-3099(14)70734-0
209. Puig C, Grau I, Marti S, Tubau F, Calatayud L, Pallares R, et al. Clinical and molecular epidemiology of Haemophilus influenzae causing invasive disease in adult patients. PLoS ONE (2014) 9:e112711. doi: 10.1371/journal.pone.0112711
210. Resman F, Ristovski M, Ahl J, Forsgren A, Gilsdorf JR, Jasir A, et al. Invasive disease caused by Haemophilus influenzae in Sweden 1997-2009; evidence of increasing incidence and clinical burden of non-type b strains. Clin Microbiol Infect. (2011) 17:1638–45. doi: 10.1111/j.1469-0691.2010.03417.x
211. Langereis JD, de Jonge MI. Invasive disease caused by Nontypeable Haemophilus influenzae. Emerg Infect Dis. (2015) 21:1711–8. doi: 10.3201/eid2110.150004
212. Sriram KB, Cox AJ, Clancy RL, Slack MPE, Cripps AW. Nontypeable Haemophilus influenzae and chronic obstructive pulmonary disease: a review for clinicians. Crit Rev Microbiol. (2018) 44:125–42. doi: 10.1080/1040841X.2017.1329274
213. Loens K, Van Heirstraeten L, Malhotra-Kumar S, Goossens H, Ieven M. Optimal sampling sites and methods for detection of pathogens possibly causing community-acquired lower respiratory tract infections. J Clin Microbiol. (2009) 47:21–31. doi: 10.1128/JCM.02037-08
214. Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet (2014) 384:691–702. doi: 10.1016/S0140-6736(14)61136-3
215. Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. (2012) 7:555–69. doi: 10.2147/COPD.S28286
216. Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2006) 173:991–8. doi: 10.1164/rccm.200509-1525OC
217. Bandi V, Apicella MA, Mason E, Murphy TF, Siddiqi A, Atmar RL, et al. Nontypeable Haemophilus influenzae in the lower respiratory tract of patients with chronic bronchitis. Am J Respir Crit Care Med. (2001) 164:2114–9. doi: 10.1164/ajrccm.164.11.2104093
218. Millares L, Ferrari R, Gallego M, Garcia-Nunez M, Perez-Brocal V, Espasa M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis. (2014) 33:1101–11. doi: 10.1007/s10096-013-2044-0
219. Finney LJ, Ritchie A, Pollard E, Johnston SL, Mallia P. Lower airway colonization and inflammatory response in COPD: a focus on Haemophilus influenzae. Int J Chron Obstruct Pulmon Dis. (2014) 9:1119–32. doi: 10.2147/COPD.S54477
220. Doring G, Parameswaran IG, Murphy TF. Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol Rev. (2011) 35:124–46. doi: 10.1111/j.1574-6976.2010.00237.x
221. Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens (2015) 4:66–89. doi: 10.3390/pathogens4010066
222. Zhang L, Xie J, Patel M, Bakhtyar A, Ehrlich GD, Ahmed A, et al. Nontypeable Haemophilus influenzae genetic islands associated with chronic pulmonary infection. PLoS ONE (2012) 7:e44730. doi: 10.1371/journal.pone.0044730
223. Qu J, Lesse AJ, Brauer AL, Cao J, Gill SR, Murphy TF. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. (2010) 10:162. doi: 10.1186/1471-2180-10-162
224. Wong SM, Bernui M, Shen H, Akerley BJ. Genome-wide fitness profiling reveals adaptations required by Haemophilus in coinfection with influenza A virus in the murine lung. Proc Natl Acad Sci USA. (2013) 110:15413–8. doi: 10.1073/pnas.1311217110
225. Pettigrew MM, Ahearn CP, Gent JF, Kong Y, Gallo MC, Munro JB, et al. Haemophilus influenzae genome evolution during persistence in the human airways in chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. (2018) 115:E3256–E65. doi: 10.1073/pnas.1719654115
226. Murphy TF, Kirkham C, Jones MM, Sethi S, Kong Y, Pettigrew MM. Expression of IgA proteases by Haemophilus influenzae in the respiratory tract of adults with chronic obstructive pulmonary disease. J Infect Dis. (2015) 212:1798–805. doi: 10.1093/infdis/jiv299
227. Murphy TF, Kirkham C, Gallo MC, Yang Y, Wilding GE, Pettigrew MM. Immunoglobulin A protease variants facilitate intracellular survival in epithelial cells by nontypeable Haemophilus influenzae that persist in the human respiratory tract in chronic obstructive pulmonary disease. J Infect Dis. (2017) 216:1295–302. doi: 10.1093/infdis/jix471
228. Gallo MC, Kirkham C, Eng S, Bebawee RS, Kong Y, Pettigrew MM, et al. Changes in IgA protease expression are conferred by changes in genomes during persistent infection by nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease. Infect Immun. (2018) 86(8). doi: 10.1128/IAI.00313-18
229. Puig C, Domenech A, Garmendia J, Langereis JD, Mayer P, Calatayud L, et al. Increased biofilm formation by nontypeable Haemophilus influenzae isolates from patients with invasive disease or otitis media versus strains recovered from cases of respiratory infections. Appl Environ Microbiol. (2014) 80:7088–95. doi: 10.1128/AEM.02544-14
230. Swords WE. Nontypeable Haemophilus influenzae biofilms: role in chronic airway infections. Front Cell Infect Microbiol. (2012) 2:97. doi: 10.3389/fcimb.2012.00097
231. Kaufhold I, Osbahr S, Shima K, Marwitz S, Rohmann K, Dromann D, et al. Nontypeable Haemophilus influenzae (NTHi) directly interfere with the regulation of E-cadherin in lung epithelial cells. Microbes Infect. (2017) 19:560–6. doi: 10.1016/j.micinf.2017.07.002
232. Singh B, Fleury C, Jalalvand F, Riesbeck K. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev. (2012) 36:1122–80. doi: 10.1111/j.1574-6976.2012.00340.x
233. Singh R, Mackay AJ, Patel AR, Garcha DS, Kowlessar BS, Brill SE, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res. (2014) 15:114. doi: 10.1186/s12931-014-0114-1
234. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:e17023. doi: 10.1038/sigtrans.2017.23
235. Moghaddam SJ, Clement CG, De la Garza MM, Zou X, Travis EL, Young HW, et al. Haemophilus influenzae lysate induces aspects of the chronic obstructive pulmonary disease phenotype. Am J Respir Cell Mol Biol. (2008) 38:629–38. doi: 10.1165/rcmb.2007-0366OC
236. Ganesan S, Comstock AT, Kinker B, Mancuso P, Beck JM, Sajjan US. Combined exposure to cigarette smoke and nontypeable Haemophilus influenzae drives development of a COPD phenotype in mice. Respir Res. (2014) 15:11. doi: 10.1186/1465-9921-15-11
237. Tan WS, Peh HY, Liao W, Pang CH, Chan TK, Lau SH, et al. Cigarette smoke-induced lung disease predisposes to more severe infection with nontypeable Haemophilus influenzae: protective effects of andrographolide. J Nat Prod. (2016) 79:1308–15. doi: 10.1021/acs.jnatprod.5b01006
238. Oliveira-Nascimento L, Massari P, Wetzler LM. The Role of TLR2 in infection and immunity. Front Immunol. (2012) 3:79. doi: 10.3389/fimmu.2012.00079
239. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
240. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. (2014) 5:316. doi: 10.3389/fimmu.2014.00316
241. Berenson CS, Murphy TF, Wrona CT, Sethi S. Outer membrane protein P6 of nontypeable Haemophilus influenzae is a potent and selective inducer of human macrophage proinflammatory cytokines. Infect Immun. (2005) 73:2728–35. doi: 10.1128/IAI.73.5.2728-2735.2005
242. Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, et al. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci USA. (2001) 98:8774–9. doi: 10.1073/pnas.151236098
243. Wang X, Moser C, Louboutin JP, Lysenko ES, Weiner DJ, Weiser JN, et al. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J Immunol. (2002) 168:810–5. doi: 10.4049/jimmunol.168.2.810
244. Wieland CW, Florquin S, Maris NA, Hoebe K, Beutler B, Takeda K, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Haemophilus influenzae from the mouse lung. J Immunol. (2005) 175:6042–9. doi: 10.4049/jimmunol.175.9.6042
245. Xu F, Xu Z, Zhang R, Wu Z, Lim JH, Koga T, et al. Nontypeable Haemophilus influenzae induces COX-2 and PGE2 expression in lung epithelial cells via activation of p38 MAPK and NF-kappa B. Respir Res. (2008) 9:16. doi: 10.1186/1465-9921-9-16
246. Piras V, Selvarajoo K. Beyond MyD88 and TRIF Pathways in Toll-Like Receptor Signaling. Front Immunol. (2014) 5:70. doi: 10.3389/fimmu.2014.00070
247. Jungnickel C, Schnabel PA, Bohle R, Wiewrodt R, Herr C, Bals R, et al. Nontypeable Haemophilus influenzae-promoted proliferation of Kras-induced early adenomatous lesions is completely dependent on Toll-like receptor signaling. Am J Pathol. (2017) 187:973–9. doi: 10.1016/j.ajpath.2017.01.003
248. Heyl KA, Klassert TE, Heinrich A, Muller MM, Klaile E, Dienemann H, et al. Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable Haemophilus influenza. MBio (2014) 5:e01492-14. doi: 10.1128/mBio.01492-14
249. Xu X, Steere RR, Fedorchuk CA, Pang J, Lee JY, Lim JH, et al. Activation of epidermal growth factor receptor is required for NTHi-induced NF-kappaB-dependent inflammation. PLoS ONE (2011) 6:e28216. doi: 10.1371/journal.pone.0028216
250. Mikami F, Gu H, Jono H, Andalibi A, Kai H, Li JD. Epidermal growth factor receptor acts as a negative regulator for bacterium nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via an Src-dependent p38 mitogen-activated protein kinase signaling pathway. J Biol Chem. (2005) 280:36185–94. doi: 10.1074/jbc.M503941200
251. Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L1068–75. doi: 10.1152/ajplung.00025.2008
252. Rotta Detto Loria J, Rohmann K, Droemann D, Kujath P, Rupp J, Goldmann T, et al. Nontypeable Haemophilus influenzae infection upregulates the NLRP3 inflammasome and leads to caspase-1-dependent secretion of interleukin-1β - a possible pathway of exacerbations in COPD. PLoS ONE (2013) 8:e66818. doi: 10.1371/journal.pone.0066818
253. Watanabe T, Jono H, Han J, Lim DJ, Li JD. Synergistic activation of NF-kappaB by nontypeable Haemophilus influenzae and tumor necrosis factor alpha. Proc Natl Acad Sci USA. (2004) 101:3563–8. doi: 10.1073/pnas.0400557101
254. Ishinaga H, Jono H, Lim JH, Kweon SM, Xu H, Ha UH, et al. TGF-beta induces p65 acetylation to enhance bacteria-induced NF-kappaB activation. EMBO J. (2007) 26:1150–62. doi: 10.1038/sj.emboj.7601546
255. Maciejewski BA, Jamieson KC, Arnason JW, Kooi C, Wiehler S, Traves SL, et al. Rhinovirus-bacteria coexposure synergistically induces CCL20 production from human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L731–40. doi: 10.1152/ajplung.00362.2016
256. Marti-Lliteras P, Regueiro V, Morey P, Hood DW, Saus C, Sauleda J, et al. Nontypeable Haemophilus influenzae clearance by alveolar macrophages is impaired by exposure to cigarette smoke. Infect Immun. (2009) 77:4232–42. doi: 10.1128/IAI.00305-09
257. Moon SK, Lee HY, Pan H, Takeshita T, Park R, Cha K, et al. Synergistic effect of interleukin 1 alpha on nontypeable Haemophilus influenzae-induced up-regulation of human beta-defensin 2 in middle ear epithelial cells. BMC Infect Dis. (2006) 6:12. doi: 10.1186/1471-2334-6-12
258. Moon SK, Lee HY, Li JD, Nagura M, Kang SH, Chun YM, et al. Activation of a Src-dependent Raf-MEK1/2-ERK signaling pathway is required for IL-1alpha-induced upregulation of beta-defensin 2 in human middle ear epithelial cells. Biochim Biophys Acta (2002) 1590:41–51. doi: 10.1016/S0167-4889(02)00196-9
259. Sibille Y, Reynolds HY. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis. (1990) 141:471–501. doi: 10.1164/ajrccm/141.2.471
260. Yamasaki K, Eeden SFV. Lung macrophage phenotypes and functional responses: Role in the pathogenesis of COPD. Int J Mol Sci. (2018) 19(2). doi: 10.3390/ijms19020582
261. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. (2015) 11:e1004651. doi: 10.1371/journal.ppat.1004651
262. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
263. King PT, Sharma R, O'Sullivan K, Selemidis S, Lim S, Radhakrishna N, et al. Nontypeable Haemophilus influenzae induces sustained lung oxidative stress and protease expression. PLoS ONE (2015) 10:e0120371. doi: 10.1371/journal.pone.0120371
264. Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE. Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect Immun. (2011) 79:431–8. doi: 10.1128/IAI.00660-10
265. Houghton AM. Matrix metalloproteinases in destructive lung disease. Matrix Biol. (2015) 44–46:167–74. doi: 10.1016/j.matbio.2015.02.002
266. McGuinness AJ, Sapey E. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. (2017) 6:E21. doi: 10.3390/jcm6020021
267. Rogers LK, Cismowski MJ. Oxidative stress in the lung - the essential paradox. Curr Opin Toxicol. (2018) 7:37–43. doi: 10.1016/j.cotox.2017.09.001
268. King PT, Hutchinson PE, Johnson PD, Holmes PW, Freezer NJ, Holdsworth SR. Adaptive immunity to nontypeable Haemophilus influenzae. Am J Respir Crit Care Med. (2003) 167:587–92. doi: 10.1164/rccm.200207-728OC
269. King P, Ngui J, Oppedisano F, Robins-Browne R, Holmes P, Holdsworth S. Effect of interferon gamma and CD40 ligation on intracellular monocyte survival of nontypeable Haemophilus influenzae. APMIS (2008) 116:1043–9. doi: 10.1111/j.1600-0463.2008.01078.x
270. King PT, Lim S, Pick A, Ngui J, Prodanovic Z, Downey W, et al. Lung T-cell responses to nontypeable Haemophilus influenzae in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2013) 131:1314.e14–21.e14. doi: 10.1016/j.jaci.2012.09.030
271. Lu C, Zhang X, Ma C, Xu W, Gan L, Cui J, et al. Nontypeable Haemophilus influenzae DNA stimulates type I interferon expression via STING signaling pathway. Biochim Biophys Acta (2018) 1865:665–73. doi: 10.1016/j.bbamcr.2018.01.011
272. Kalathil SG, Lugade AA, Pradhan V, Miller A, Parameswaran GI, Sethi S, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2014) 190:40–50. doi: 10.1164/rccm.201312-2293OC
273. Guan X, Lu Y, Wang G, Gibson P, Chen F, Fang K, et al. The role of regulatory T cell in nontypeable Haemophilus influenzae-induced acute exacerbation of chronic obstructive pulmonary disease. Mediators Inflamm. (2018) 2018:8387150. doi: 10.1155/2018/8387150
274. McCann JR, Mason SN, Auten RL, St Geme JW III, Seed PC. Early-life intranasal colonization with nontypeable Haemophilus influenzae exacerbates juvenile airway disease in mice. Infect Immun. (2016) 84:2022–30. doi: 10.1128/IAI.01539-15
275. Abe Y, Murphy TF, Sethi S, Faden HS, Dmochowski J, Harabuchi Y, et al. Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2002) 165:967–71. doi: 10.1164/ajrccm.165.7.2109009
276. Hinks TS, Wallington JC, Williams AP, Djukanovic R, Staples KJ, Wilkinson TM. Steroid-induced deficiency of mucosal-associated invariant T cells in the chronic obstructive pulmonary disease lung. Implications for nontypeable Haemophilus influenzae Infection. Am J Respir Crit Care Med. (2016) 194:1208–18. doi: 10.1164/rccm.201601-0002OC
277. Riesbeck K. Inhaled corticosteroids in chronic obstructive pulmonary disease. A two-edged sword. Am J Respir Crit Care Med. (2016) 194:1177–8. doi: 10.1164/rccm.201605-0942ED
278. Li W, Zhang X, Yang Y, Yin Q, Wang Y, Li Y, et al. Recognition of conserved antigens by Th17 cells provides broad protection against pulmonary Haemophilus influenzae infection. Proc Natl Acad Sci USA. (2018) 115:E7149–57. doi: 10.1073/pnas.1802261115
279. Miller RD, Gleich GJ, Offord KP, Dunnette SL. Immunoglobulin concentrations in serum and nasal secretions in chronic obstructive pulmonary disease. A matched-pair study. Am Rev Respir Dis. (1979) 119:229–38. doi: 10.1164/arrd.1979.119.2.229
280. Groeneveld K, Eijk PP, van Alphen L, Jansen HM, Zanen HC. Haemophilus influenzae infections in patients with chronic obstructive pulmonary disease despite specific antibodies in serum and sputum. Am Rev Respir Dis. (1990) 141(5 Pt 1):1316–21. doi: 10.1164/ajrccm/141.5_Pt_1.1316
281. Otczyk DC, Cripps AW. Mucosal immunization: a realistic alternative. Hum Vaccin. (2010) 6:978–1006. doi: 10.4161/hv.6.12.13142
282. Yi K, Murphy TF. Mapping of a strain-specific bactericidal epitope to the surface-exposed loop 5 on the P2 porin protein of non-typeable Haemophilus influenzae. Microb Pathog. (1994) 17:277–82. doi: 10.1006/mpat.1994.1073
283. Staples KJ, Taylor S, Thomas S, Leung S, Cox K, Pascal TG, et al. Relationships between mucosal antibodies, non-typeable Haemophilus influenzae (NTHi) infection and airway inflammation in COPD. PLoS ONE (2016) 11:e0167250. doi: 10.1371/journal.pone.0167250
284. Polosukhin VV, Cates JM, Lawson WE, Zaynagetdinov R, Milstone AP, Massion PP, et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:317–27. doi: 10.1164/rccm.201010-1629OC
285. Millares L, Marin A, Garcia-Aymerich J, Sauleda J, Belda J, Monso E, et al. Specific IgA and metalloproteinase activity in bronchial secretions from stable chronic obstructive pulmonary disease patients colonized by Haemophilus influenzae. Respir Res. (2012) 13:113. doi: 10.1186/1465-9921-13-113
286. Meshi B, Vitalis TZ, Ionescu D, Elliott WM, Liu C, Wang XD, et al. Emphysematous lung destruction by cigarette smoke. The effects of latent adenoviral infection on the lung inflammatory response. Am J Respir Cell Mol Biol. (2002) 26:52–7. doi: 10.1165/ajrcmb.26.1.4253
287. Gohy ST, Detry BR, Lecocq M, Bouzin C, Weynand BA, Amatngalim GD, et al. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease. Persistence in the cultured epithelium and role of transforming growth factor-β. Am J Respir Crit Care Med. (2014) 190:509–21. doi: 10.1164/rccm.201311-1971OC
288. Richmond BW, Brucker RM, Han W, Du RH, Zhang Y, Cheng DS, et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat Commun. (2016) 7:11240. doi: 10.1038/ncomms11240
289. Van der Hilst JC, Smits BW, van der Meer JW. Hypogammaglobulinaemia: cumulative experience in 49 patients in a tertiary care institution. Neth J Med. (2002) 60:140–7.
290. Storisteanu DM, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, et al. Evasion of neutrophil extracellular traps by respiratory pathogens. Am J Respir Cell Mol Biol. (2017) 56:423–31. doi: 10.1165/rcmb.2016-0193PS
291. Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. (2006) 194:1375–84. doi: 10.1086/508428
292. Berenson CS, Kruzel RL, Eberhardt E, Dolnick R, Minderman H, Wallace PK, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax (2014) 69:811–8. doi: 10.1136/thoraxjnl-2013-203669
293. Berenson CS, Wrona CT, Grove LJ, Maloney J, Garlipp MA, Wallace PK, et al. Impaired alveolar macrophage response to Haemophilus antigens in chronic obstructive lung disease. Am J Respir Crit Care Med. (2006) 174:31–40. doi: 10.1164/rccm.200509-1461OC
294. Lea SR, Reynolds SL, Kaur M, Simpson KD, Hall SR, Hessel EM, et al. The effects of repeated Toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages. Int J Chron Obstruct Pulmon Dis. (2018) 13:771–80. doi: 10.2147/COPD.S97071
295. Knobloch J, Schild K, Jungck D, Urban K, Muller K, Schweda EK, et al. The T-helper cell type 1 immune response to gram-negative bacterial infections is impaired in COPD. Am J Respir Crit Care Med. (2011) 183:204–14. doi: 10.1164/rccm.201002-0199OC
296. MacRedmond RE, Greene CM, Dorscheid DR, McElvaney NG, O'Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res. (2007) 8:84. doi: 10.1186/1465-9921-8-84
297. von Scheele I, Larsson K, Dahlen B, Billing B, Skedinger M, Lantz AS, et al. Toll-like receptor expression in smokers with and without COPD. Respir Med. (2011) 105:1222–30. doi: 10.1016/j.rmed.2011.02.012
298. Tripathi PM, Kant S, Yadav RS, Kushwaha RAS, Prakash V, Rizvi SHM, et al. Expression of Toll-like receptor 2 and 4 in peripheral blood neutrophil cells from patients with chronic obstructive pulmonary disease. Oman Med J. (2017) 32:477–85. doi: 10.5001/omj.2017.92
299. Freeman CM, Martinez FJ, Han MK, Washko GR Jr, McCubbrey AL, Chensue SW, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. (2013) 14:13. doi: 10.1186/1465-9921-14-13
300. Pouwels SD, van Geffen WH, Jonker MR, Kerstjens HA, Nawijn MC, Heijink IH. Increased neutrophil expression of pattern recognition receptors during COPD exacerbations. Respirology (2017) 22:401–4. doi: 10.1111/resp.12912
301. Budulac SE, Boezen HM, Hiemstra PS, Lapperre TS, Vonk JM, Timens W, et al. Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS ONE (2012) 7:e43124. doi: 10.1371/journal.pone.0043124
302. Berenson CS, Kruzel RL, Wrona CT, Mammen MJ, Sethi S. Impaired innate COPD alveolar macrophage responses and Toll-like receptor-9 polymorphisms. PLoS ONE (2015) 10:e0134209. doi: 10.1371/journal.pone.0134209
303. Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. (2008) 60:795–804. doi: 10.1016/j.addr.2007.12.004
304. Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stampfli MR. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. (2004) 170:1164–71. doi: 10.1164/rccm.200311-1521OC
305. Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott-Banner K, Singh D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol. (2014) 176:461–72. doi: 10.1111/cei.12289
306. Berenson CS, Kruzel RL, Sethi S. The impact of exogenous factors on respiratory pathogen-induced innate alveolar macrophage responses in COPD. Immunol Invest. (2016) 45:130–47. doi: 10.3109/08820139.2015.1113427
307. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. (2011) 3:78ra32. doi: 10.1126/scitranslmed.3002042
308. Tay HL, Kaiko GE, Plank M, Li J, Maltby S, Essilfie AT, et al. Antagonism of miR-328 increases the antimicrobial function of macrophages and neutrophils and rapid clearance of non-typeable Haemophilus influenzae (NTHi) from infected lung. PLoS Pathog. (2015) 11:e1004549. doi: 10.1371/journal.ppat.1004549
309. Gaschler GJ, Skrtic M, Zavitz CC, Lindahl M, Onnervik PO, Murphy TF, et al. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med. (2009) 179:666–75. doi: 10.1164/rccm.200808-1306OC
310. Gaschler GJ, Zavitz CC, Bauer CM, Stampfli MR. Mechanisms of clearance of nontypeable Haemophilus influenzae from cigarette smoke-exposed mouse lungs. Eur Respir J. (2010) 36:1131–42. doi: 10.1183/09031936.00113909
311. Lugade AA, Bogner PN, Thatcher TH, Sime PJ, Phipps RP, Thanavala Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J Immunol. (2014) 192:5226–35. doi: 10.4049/jimmunol.1302584
312. Bhat TA, Kalathil SG, Bogner PN, Miller A, Lehmann PV, Thatcher TH, et al. Secondhand smoke induces inflammation and impairs immunity to respiratory infections. J Immunol. (2018) 200:2927–40. doi: 10.4049/jimmunol.1701417
313. Sharan R, Perez-Cruz M, Kervoaze G, Gosset P, Weynants V, Godfroid F, et al. Interleukin-22 protects against non-typeable Haemophilus influenzae infection: alteration during chronic obstructive pulmonary disease. Mucosal Immunol. (2017) 10:139–49. doi: 10.1038/mi.2016.40
314. Roos AB, Sethi S, Nikota J, Wrona CT, Dorrington MG, Sanden C, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 192:428–37. doi: 10.1164/rccm.201409-1689OC
315. Singh D. Chronic obstructive pulmonary disease, neutrophils and bacterial infection: a complex web involving IL-17 and IL-22 unravels. EBioMed. (2015) 2:1580–1. doi: 10.1016/j.ebiom.2015.10.021
316. Persson LJ, Aanerud M, Hardie JA, Miodini Nilsen R, Bakke PS, Eagan TM, et al. Antimicrobial peptide levels are linked to airway inflammation, bacterial colonisation and exacerbations in chronic obstructive pulmonary disease. Eur Respir J. (2017) 49:1601328. doi: 10.1183/13993003.01328-2016
317. Hiemstra PS, Amatngalim GD, van der Does AM, Taube C. Antimicrobial peptides and innate lung defenses: role in infectious and noninfectious lung diseases and therapeutic applications. Chest (2016) 149:545–51. doi: 10.1378/chest.15-1353
318. Sallenave JM. Secretory leukocyte protease inhibitor and elafin/trappin-2: versatile mucosal antimicrobials and regulators of immunity. Am J Respir Cell Mol Biol. (2010) 42:635–43. doi: 10.1165/rcmb.2010-0095RT
319. Seiler F, Lepper PM, Bals R, Beisswenger C. Regulation and function of antimicrobial peptides in immunity and diseases of the lung. Protein Pept Lett. (2014) 21:341–51. doi: 10.2174/09298665113206660100
320. Marsland BJ, Gollwitzer ES. Host-microorganism interactions in lung diseases. Nat Rev Immunol. (2014) 14:827–35. doi: 10.1038/nri3769
321. Abou Alaiwa MH, Reznikov LR, Gansemer ND, Sheets KA, Horswill AR, Stoltz DA, et al. pH modulates the activity and synergism of the airway surface liquid antimicrobials beta-defensin-3 and LL-37. Proc Natl Acad Sci USA. (2014) 111:18703–8. doi: 10.1073/pnas.1422091112
322. Kilsgard O, Andersson P, Malmsten M, Nordin SL, Linge HM, Eliasson M, et al. Peptidylarginine deiminases present in the airways during tobacco smoking and inflammation can citrullinate the host defense peptide LL-37, resulting in altered activities. Am J Respir Cell Mol Biol. (2012) 46:240–8. doi: 10.1165/rcmb.2010-0500OC
323. Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (2012) 12:CD010257. doi: 10.1002/14651858.CD010257
324. van Velzen P, Ter Riet G, Bresser P, Baars JJ, van den Berg BTJ, van den Berg JWK, et al. Doxycycline for outpatient-treated acute exacerbations of COPD: a randomised double-blind placebo-controlled trial. Lancet Respir Med. (2017) 5:492–9. doi: 10.1016/S2213-2600(17)30165-0
325. Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing, 28th ed. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute (2018). p. 296.
326. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 8.1 (2018). Available online at: http://www.eucast.org/
327. Pettigrew MM, Tsuji BT, Gent JF, Kong Y, Holden PN, Sethi S, et al. Effect of fluoroquinolones and macrolides on eradication and resistance of Haemophilus influenzae in chronic obstructive pulmonary disease. Antimicrob Agents Chemother. (2016) 60:4151–8. doi: 10.1128/AAC.00301-16
328. Skaare D, Anthonisen IL, Caugant DA, Jenkins A, Steinbakk M, Strand L, et al. Multilocus sequence typing and ftsl sequencing: a powerful tool for surveillance of penicillin-binding protein 3-mediated beta-lactam resistance in nontypeable Haemophilus influenzae. BMC Microbiol. (2014) 14:131. doi: 10.1186/1471-2180-14-131
329. Tristram S, Jacobs MR, Appelbaum PC. Antimicrobial resistance in Haemophilus influenzae. Clin Microbiol Rev. (2007) 20:368–89. doi: 10.1128/CMR.00040-06
330. Resman F, Ristovski M, Forsgren A, Kaijser B, Kronvall G, Medstrand P, et al. Increase of β-lactam-resistant invasive Haemophilus influenzae in Sweden, 1997 to 2010. Antimicrob Agents Chemother. (2012) 56:4408–15. doi: 10.1128/AAC.00415-12
331. Skaare D, Anthonisen IL, Kahlmeter G, Matuschek E, Natas OB, Steinbakk M, et al. Emergence of clonally related multidrug resistant Haemophilus influenzae with penicillin-binding protein 3-mediated resistance to extended-spectrum cephalosporins, Norway, 2006 to 2013. Euro Surveill. (2014) 19:20986. doi: 10.2807/1560-7917.es2014.19.49.20986
332. Garcia-Cobos S, Campos J, Lazaro E, Roman F, Cercenado E, Garcia-Rey C, et al. Ampicillin-resistant non-beta-lactamase-producing Haemophilus influenzae in Spain: recent emergence of clonal isolates with increased resistance to cefotaxime and cefixime. Antimicrob Agents Chemother. (2007) 51:2564–73. doi: 10.1128/AAC.00354-07
333. Okabe T, Yamazaki Y, Shiotani M, Suzuki T, Shiohara M, Kasuga E, et al. An amino acid substitution in PBP-3 in Haemophilus influenzae associate with the invasion to bronchial epithelial cells. Microbiol Res. (2010) 165:11–20. doi: 10.1016/j.micres.2008.03.003
334. Atkins NA, Kunde DA, Zosky G, Tristram SG. Genotypically defined beta-lactamase-negative ampicillin-resistant isolates of non-typable Haemophilus influenzae are associated with increased invasion of bronchial epithelial cells in vitro. J Med Microbiol. (2014) 63(Pt 10):1400–3. doi: 10.1099/jmm.0.077966-0
335. Herath SC, Poole P. Prophylactic antibiotic therapy for chronic obstructive pulmonary disease (COPD). Cochrane Database Syst Rev. (2013) CD009764. doi: 10.1002/14651858.CD009764.pub2
336. Pomares X, Monton C, Bullich M, Cuevas O, Oliva JC, Gallego M, et al. Clinical and safety outcomes of long-term azithromycin therapy in severe COPD beyond the first year of treatment. Chest (2018) 153:1125–33. doi: 10.1016/j.chest.2018.01.044
337. Shimizu T, Shimizu S. Azithromycin inhibits mucus hypersecretion from airway epithelial cells. Mediators Inflamm. (2012) 2012:265714. doi: 10.1155/2012/265714
338. Araki N, Yanagihara K, Morinaga Y, Yamada K, Nakamura S, Yamada Y, et al. Azithromycin inhibits nontypeable Haemophilus influenzae-induced MUC5AC expression and secretion via inhibition of activator protein-1 in human airway epithelial cells. Eur J Pharmacol. (2010) 644:209–14. doi: 10.1016/j.ejphar.2010.06.056
339. Hodge S, Reynolds PN. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology (2012) 17:802–7. doi: 10.1111/j.1440-1843.2012.02135.x
340. Euba B, Moleres J, Viadas C, Barberan M, Caballero L, Grillo MJ, et al. Relationship between azithromycin susceptibility and administration efficacy for nontypeable Haemophilus influenzae respiratory infection. Antimicrob Agents Chemother. (2015) 59:2700–12. doi: 10.1128/AAC.04447-14
341. Hodge S, Tran HB, Hamon R, Roscioli E, Hodge G, Jersmann H, et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L678–L87. doi: 10.1152/ajplung.00518.2016
Keywords: airway, COPD, exacerbation, immune response, infection, inflammation, non-typeable Haemophilus influenza
Citation: Su Y-C, Jalalvand F, Thegerström J and Riesbeck K (2018) The Interplay Between Immune Response and Bacterial Infection in COPD: Focus Upon Non-typeable Haemophilus influenzae. Front. Immunol. 9:2530. doi: 10.3389/fimmu.2018.02530
Received: 02 August 2018; Accepted: 15 October 2018;
Published: 05 November 2018.
Edited by:
Junkal Garmendia, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Sara Martí, Hospital Universitario de Bellvitge, SpainCopyright © 2018 Su, Jalalvand, Thegerström and Riesbeck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristian Riesbeck, a3Jpc3RpYW4ucmllc2JlY2tAbWVkLmx1LnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.