 Jessie M. Alperin1
Jessie M. Alperin1 Lourdes Ortiz-Fernández
Lourdes Ortiz-Fernández Amr H. Sawalha
Amr H. Sawalha
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 October 2018
Sec. Autoimmune and Autoinflammatory Disorders
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02496
This article is part of the Research TopicMechanisms by which SLE-Associated Genetic Variants Contribute to SLE PathogenesisView all 11 articles
Monogenic lupus is a form of systemic lupus erythematosus (SLE) that occurs in patients with a single gene defect. This rare variant of lupus generally presents with early onset severe disease, especially affecting the kidneys and central nervous system. To date, a significant number of genes have been implicated in monogenic lupus, providing valuable insights into a very complex disease process. Throughout this review, we will summarize the genes reported to be associated with monogenic lupus or lupus-like diseases, and the pathogenic mechanisms affected by the mutations involved upon inducing autoimmunity.
Systemic lupus erythematosus (SLE or lupus) is a complex multisystem disease whose underlying disease mechanism continues to be a topic of intense research. SLE can affect many organs including the kidneys, skin, joints, lungs, cardiovascular system, central nervous system, and hematopoietic system. As with most complex diseases, the etiology of SLE is incompletely understood, however, cumulative evidence has pointed to the involvement of both genetic and epigenetic mechanisms (1, 2). Multiple genetic variants associated with lupus susceptibility have been identified through genome-wide association studies (GWAS). Support for a genetic component of lupus can be realized from twin studies. Concordance rate of lupus in monozygotic and dizygotic twins has been reported to be 24 and 2%, respectively, demonstrating a role for genetic susceptibility in lupus (3). In parallel, monozygotic twin studies have also provided evidence highlighting the relevance of DNA methylation changes (4). At the same time, non-genetic factors such as viral infections or exposure to ultraviolet (UV) light among others are clearly involved, as suggested by incomplete concordance in monozygotic twins.
Patients with childhood onset SLE usually present with a more severe phenotype and have an increased frequency of glomerulonephritis, cytopenias, neuropsychiatric disease, cutaneous manifestations, anti-dsDNA antibodies, and hemolytic anemia (5). It can be presumed that in early onset disease, genetic factors may play a more important role than environmental and hormonal factors (5). Monogenic lupus is a form of SLE that typically presents early in life, usually at < 5 years of age, with severe disease manifestations. This form of lupus is caused directly by a genetic variant in a specific gene. Monogenic lupus represents a collection of distinct genetic abnormalities causing similar clinical features and resulting in autoantibody production. In particular, consanguinity presents a significant increased risk for monogenic lupus and should prompt consideration in patients with familial SLE. Though monogenic lupus accounts for only a small subset of lupus patients, it provides significant insight into the cause and mechanisms of lupus, and potential treatment strategies.
In the last years, large achievements understanding the genetic component of SLE have been accomplished. More than 80 loci have been reported to be associated with susceptibility in polygenic lupus, and a considerable number of monogenic causes of SLE and lupus-like syndromes are emerging due to the evolution of new sequencing techniques that can identify rare genetic variations across the entire genome. Through this review, we will explore the implications on the disease pathogenesis of the genes that have been found to cause monogenic lupus or lupus-like phenotype (Figure 1, Table 1).
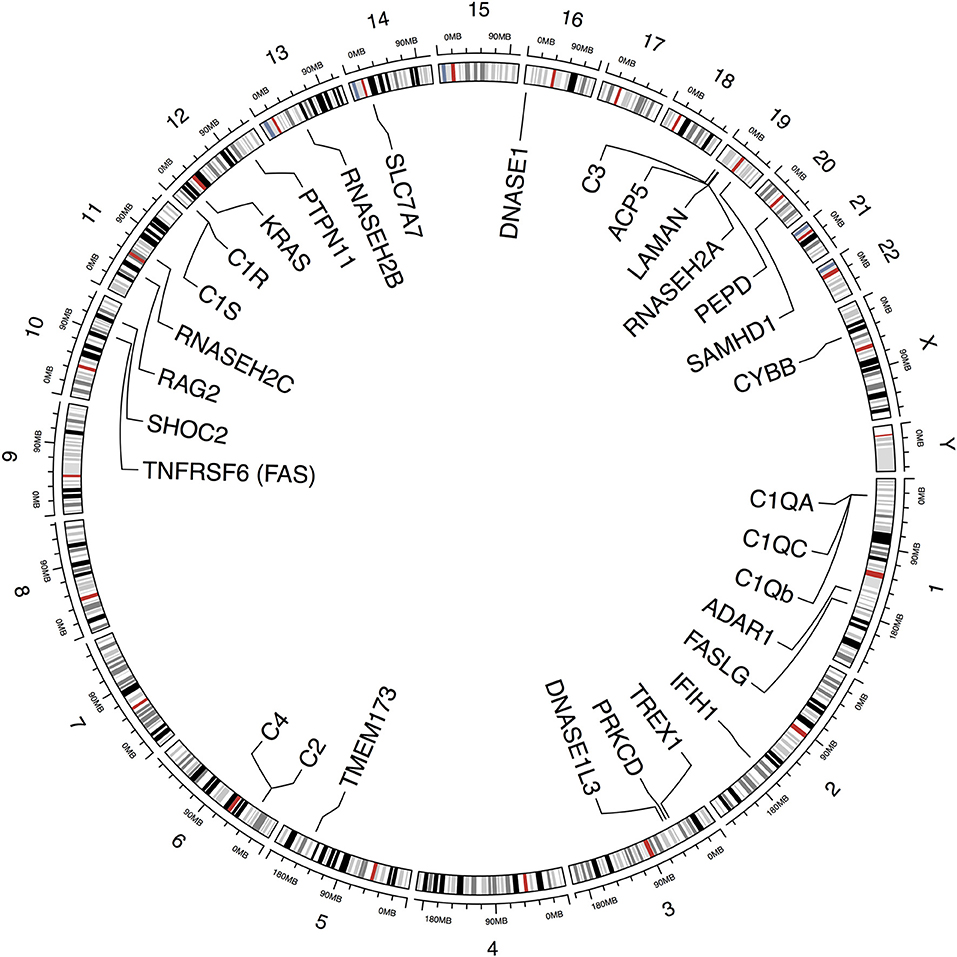
Figure 1. Schematic representation depicting genes and chromosomal locations previously identified to be implicated in monogenic lupus. This figure was produced using ‘circlize' in R (http://cran.r-project.org/web/packages/circlize/).
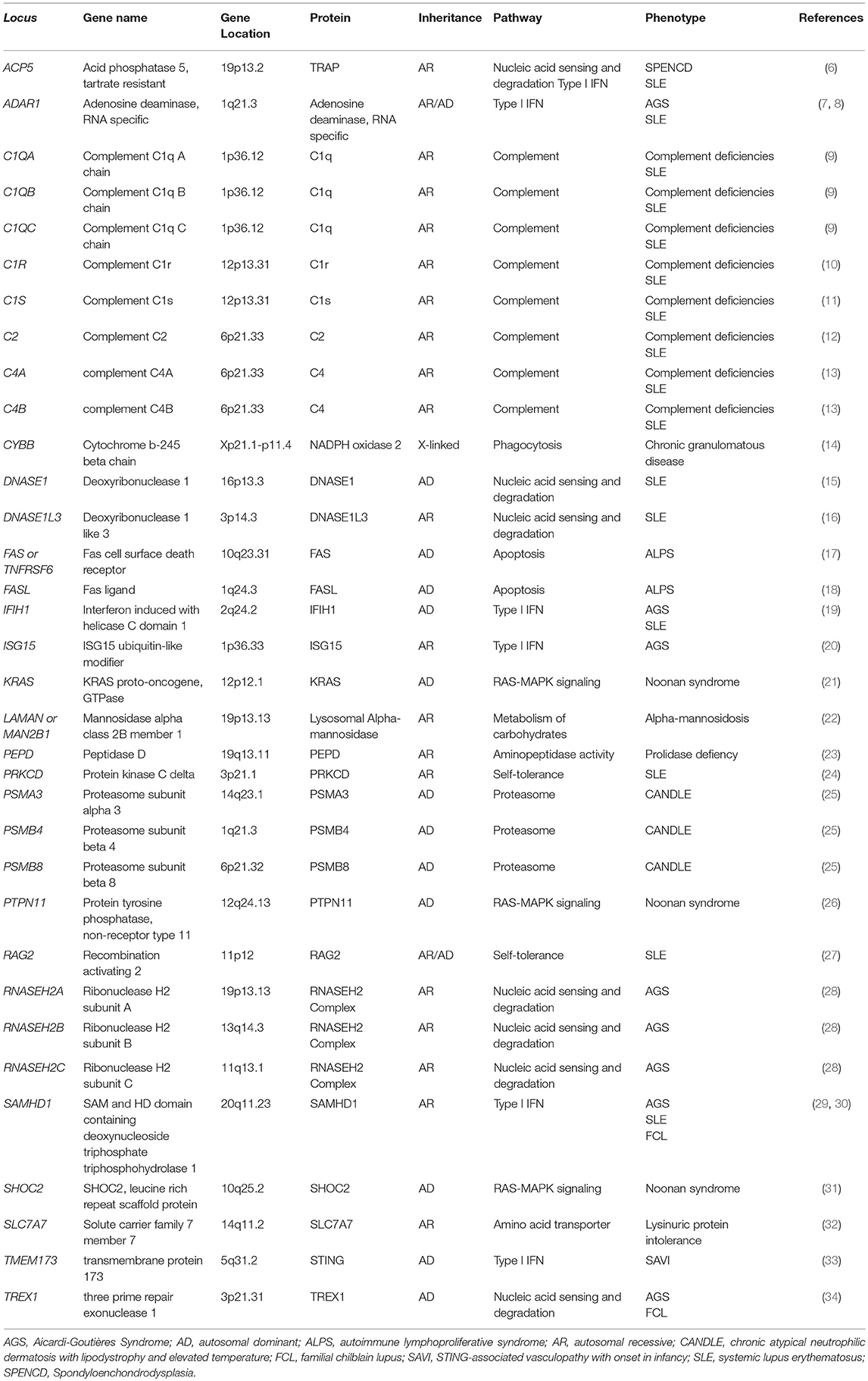
Table 1. List of genes identified to cause monogenic lupus or lupus-like diseases.
Although the clinical manifestations and severity observed are different among patients, the interferonopathies are a wide group of complex genetic disorders with a common pathogenic mechanism characterized by imbalance of interferon (IFN) mediated immune responses. Given that studies have repeatedly identified an increased blood IFN signature in SLE patients (35, 36), it is not surprising that clinical features of some of the diseases classified as interferonopathies overlap with systemic lupus erythematosus. Consequently, some of these diseases, such as Aicardi-Goutières Syndrome (AGS) and Spondyloenchondrodysplasia (SPENCD) can also be considered as forms of monogenic lupus or lupus-like syndromes.
Aicardi-Goutières Syndrome is a genetic syndrome caused by multiple genetic defects. The disease phenotype resembles a congenital viral infection and many patients develop lupus as a feature of the syndrome. AGS develops in young patients, typically before 6 months of age and is characterized by encephalopathy that is usually associated with calcification of the basal ganglia and white matter changes on brain imaging (37). Frequently, there is cutaneous involvement and, in particular, chilblains. Patients with AGS have been shown to have multiple IgG autoantibodies particularly directed against nuclear antigens, basement membrane components, gliadin, and brain endothelial cells and astrocytes. Patient with AGS develop elevated type I IFN levels in both serum and cerebral spinal fluid (7). These observations suggest that genetic defects in RNA or DNA clearance result in increased type I IFN production and interferon stimulated genes (ISGs), and then lead to autoimmunity. AGS is associated with mutations in multiples genes and many of these overlap with various forms of monogenic lupus, suggesting a spectrum of disease likely influenced by the location of the mutation as well as environmental factors (7).
Mutations of TREX1 gene have been identified in AGS. Approximately 25% of AGS patients have a TREX1 mutation. Patients with AGS due to a TREX1 mutation have a prototypical disease phenotype. A review of autoimmune features in AGS patients showed that approximately 60% of patients with a TREX1 variant had at least one autoimmune feature: thrombocytopenia, leukopenia, antinuclear antibodies (ANA), skin lesions, oral ulceration, arthritis, anti-dsDNA antibodies, or antibodies to extractable nuclear antigens (ENA) (34). Persisting severe physical and intellectual disability is frequent. In the large majority, patients will have no purposeful gross motor, hand or communication function. About a third of patients with a TREX1-related AGS present in the neonatal period with thrombocytopenia, hepatosplenomegaly, and a transaminitis in addition to neurological disease (7). For reasons that are not understood, these extra-neurological features often resolve within the first year of life. A broad spectrum of mutations across TREX1 have been associated with different immune-mediated diseases. Most patients with AGS show biallelic mutations within TREX1 with autosomal recessive inheritance, which usually causes a complete loss of protein function. However, some heterozygous mutations have also been identified in individuals diagnosed with AGS (34). Early-onset familial chilblain lupus (FCL) is a rare form of cutaneous lupus which results in cold-induced severe discoloration of hands, feet, and ears, where the lesions frequently ulcerate. Most of these patients have heterozygous mutations with autosomal dominant inheritance (34). In addition, heterozygous mutations in TREX1 with autosomal dominant inheritance has been linked to retinal vasculopathy with cerebral leukodystrophy (RVCL) (38). Interestingly, a deleterious homozygous variant of this gene has been recently identified in a patient with cerebral SLE (39), and, it is worth noting that several single nucleotides polymorphisms (SNPs) in TREX1 have been found associated with common forms of SLE in different populations (40–42). This gene, located on chromosome 3, encodes a protein with exonuclease activity which is an IFN-inducible protein responsible for degradation of genomic DNA in response to DNA damage. Therefore, it plays an important role in the immune response to single-stranded (ss)-DNA and dsDNA (43), and maintains immune tolerance to cytosolic self-DNA (44). When TREX1 is dysfunctional, the cytosolic DNA does not get degraded which constitutes a damage-associated molecular pattern (DAMP). This activates the cGAS-STING-mediated type I IFN response and systemic inflammation (44). TREX1 deficiency is thought to trigger autoimmunity through the accumulation of self DNA in the cytosol. These are sensed by cyclic GMP-AMP (cGAMP) synthase (cGAS). cGAMP is a ligand for stimulator of IFN genes protein (STING), which leads to the production of type I IFN (43). On the other hand, TREX1 is a DNase component of the SET complex which is involved, among other mechanisms, in apoptosis (45). Altogether, these data provide different pathomechanisms for the involvement of TREX1 dysfunction in SLE.
Patients with IFIH1 (interferon induced with helicase C domain 1) gain of function mutations can develop early onset SLE and AGS-like disease, including musculoskeletal involvement and Jaccoud's arthropathy (46). IFIH1 gene, which encodes MDA5 (melanoma differentiation-associated protein 5), is a cytoplasmic RNA receptor that binds cytoplasmic double-stranded RNA. IFIH1 belongs to the RIG-I–like family which is part of the pathway responsible for activating type I interferon signaling (19). The gain of function mutation in IFIH1 gene leads to activated dendritic cells and macrophages, which are the primary producers of IFN-alpha in response to nucleic acid (47). This leads to the activation of T cells and production of autoantibodies (48). It is interesting to note that polymorphisms in IFIH1 have been reported in patients with inflammatory myopathies and anti-MDA5 antibodies are seen in some patients with amyopathic dermatomyositis (49). Remarkably, a recent study revealed that these patients showed a high activity of type I IFN system. Although the mechanisms are still unclear, the study detected high levels of transcripts of IFN-associated sensors and several IFN-inducible genes were up-regulated in these patients (50).
SAMHD1 (SAM domain and HD domain-containing protein 1) is a dGTP-dependent triphosphohydrolase responsible for the regulation of intracellular levels of deoxynucleoside triphosphates (dNTPs), the building blocks of DNA synthesis (51, 52). In unaffected individuals, SAMHD1 promotes cell stability and prevents reverse transcription of retroviruses. Deficiency of SAMHD1 results in unbalanced pools of dNTPs. This leads to loss of DNA replication and repair, DNA damage, and apoptosis leading to a sustained IFN production (53). SAMHD1 is upregulated in response to viral infections. It plays a role in the antiviral immune response through initiation of the interferon pathway (51). SAMHD1 disease-causing variants can present with AGS, SLE, and chilblain lupus (29, 30). SAMHD1 has been shown to be reduced or absent in the cells of patients with AGS (54). Cells from patients with SAMHD1 mutations exhibited increased dNTP pools and DNA damage resulting in a failure of the cell cycle and cellular senescence. Increased DNA damage leads to upregulation in IFN-stimulated genes by activation of the innate immune system (53).
RNaseH2 (Ribonuclease H2) is a nucleic acid repair surveillance enzyme expressed in all cells and functions to remove ribonucleotides from DNA hybrid complexes. Three different genes encode the three protein components of the RNaseH2 complex, RNASEH2A, RNASEH2B and RNASEH2C. Mutations in all three genes have been associated with AGS and SLE. A recent study established mice models in which mutations in RNASEH2B trigger an increase of the expression levels of ISGs. The results of this work also proposed that this inflammatory response is dependent upon the cGAS/STING pathway (28). The findings of another study described that these mutations result in accumulation of ribonucleotides in genomic DNA placed during replication, which causes chronic DNA damage triggering the p53 pathway and type I IFN production (55). It has also been reported that fibroblasts from patients with SLE and AGS secondary to RNaseH2 mutations, as well as RNaseH2 deficient mice have significant accumulation of ribonucleotides and increased DNA in the cytoplasm. Furthermore, patient fibroblasts revealed an upregulation in IFN stimulated genes, which was enhanced, among other factors, by UV light exposure. UV light is a major trigger of SLE symptoms and it is thought that UV can raise apoptotic debris containing nucleic acids. Therefore, a deficiency in RNAseH2 in individuals exposed to UV light could be a possible link between genetic and environmental factors in the pathogenesis of SLE (55). Approximately one-third of AGS patients with variants in the RNaseH2 complex have positive ANA. Sequencing of the genes encoding the RNaseH2 complex in 600 SLE subjects identified 18 rare variants. Clinically, these patients mainly showed cutaneous involvement, photosensitivity, arthritis, lymphopenia, and autoantibody formation; internal organ involvement was less frequent (41).
ADAR1 (adenosine deaminase, RNA specific) gene, located on chromosome 1, encodes a widely expressed enzyme which is involved in the editing of long double strand RNA. Mutations in this gene have been reported to cause AGS and SLE (7, 8). A proinflammatory signal upon recognition of viral or cellular dsRNA unedited due to loss of function mutations in ADAR1 has been described (56). Interestingly, mutations in this gene also cause Dyscrhromatosus Symmetrica Hereditaria (DSH1), a rare skin condition which has been associated with SLE (57).
ISG15 is transcriptionally regulated by IFN-alpha and beta. Patients with ISG15 deficiency have increased risk of viral infection. Mutations in ISG15 are associated with central nervous system (CNS) disease including intracranial calcifications and seizures (20). Patients show immunological and clinical signs of enhanced IFN-alpha/beta immunity. Interestingly, mutations in ISG15 have been found in AGS and, in addition, a higher expression of this gene in SLE patients has been reported in several studies (58–60) ISG15 negatively regulates IFN-alpha and beta production (58), and is known as an activator of natural killer (NK) cells and a driver of IFNγ secretion (61). Therefore, ISG15 has emerged as a potentially critical bridge between type I and type II IFN immune responses.
Spondyloenchondrodysplasia (SPENCD) is a rare immuno-osseous disorder which causes skeletal dysplasia as well as variable neurologic manifestations (spacticity, developmental delay, intracranial calcification). In addition, it has been reported that SPENCD patients also may show overlapping features of lupus such as nephritis, thrombocytopenia, and dsDNA antibodies amongst others. Indeed, some of these patients fulfilled the American College of Rheumatology classification criteria for lupus (6, 62). The ACP5 gene encodes tartrate-resistant acid phosphate (TRAP) protein which is mostly expressed in monocytic cells including osteoclasts, macrophages, and dendritic cells (63). Several different biallelic null mutations in ACP5 distributed throughout the protein have been identified in individuals diagnosed with SPENCD-associated lupus. All mutations identified to date, cause a complete loss of enzymatic function. TRAP regulates the phosphorylation levels of osteopontin (OPN) which is a cytokine required for the production of type I IFN by plasmacytoid dendritic cells in response to TLR9 stimulation (64). It has been described that decreased expression of TRAP triggers increased phosphorylation of OPN leading to overproduction of type I interferon (6, 62). In addition, after TLR9 stimulation, a reduced expression of TRAP provokes higher levels of IFN-alpha, interleukin-6, ISGs, and tumor necrosis factor (TNF) (65). It is worth to note that in a recent study ACP5 was sequenced in nearly 1,000 SLE patients and more than 500 healthy controls. The results of this study showed a significant increase in heterozygous ACP5 missense variants in SLE patients compared to healthy individuals (65).
The classical complement pathway begins with C1, which consists of C1q, two C1r molecules, and two C1s molecules. C1 binds to the Fc portion of IgG or IgM antibody which complexes to antigens. The binding results in the activation of C1q which activates C1r, and then activates C1s. C1s cleaves C4 (to C4a and C4b) and C2 (to C2a and C2b). C4b and C2b combine and cleave C3 which is added to the complex resulting in C4bC2bC3b (also known as C5 convertase). This complex will cleave C5 resulting in the assembly of the membrane attack complex (C5bC6C7C8C9). The alternative complement pathway begins with the hydrolysis of C3 to C3(H2O). C3(H2O) is always present to a small degree but is maintained in homeostasis. The alternate pathway is initiated when C3(H2O) binds to factor Bb (factor B having been cleaved by factor D to form factor Ba and Bb) which forms C3b(H2O)Bb. The C3b(H2O)Bb is an alternative C3 convertase (66). Any defect in these complement components might prevent or hinder the removal or clearance of apoptotic cells or immune complexes, thus allowing these potential autoantigens to activate the immune system and lead to a loss of tolerance. Therefore, deficiencies of C1q, C1r, C1s, C2, or C4 have been strongly associated with both immunodeficiency as well as autoimmunity, including lupus-like phenotype (67).
Approximately 90% of people with C1q deficiency develop lupus like-phenotype (9, 68) including clinical characteristics such as photosensitive skin rash, nephritis, oral ulceration, and arthritis. Most of these patients have early onset disease with an age range from 6 months to 42 years, and a median age of onset of 6 years (69). Interestingly, patients with C1q deficiency have normal complement C3 and C4 levels with low total hemolytic complement levels which can be a helpful tool in diagnosis (70). Of interest, there are case reports of treatment with plasma transfusion, which restores C1q levels, leading to resolution of symptoms (71). Another small case series reported successful cure of patients with a C1q deficiency with bone marrow transplantation (72). C1q is encoded by three genes (C1QA, C1QB, and C1QC) which are closely linked on chromosome 1p34-1p36 (69). Multiple causal mutations, in the three genes, resulting in the deficiency of C1q have been identified in individuals with SLE-like phenotype. C1q is directly responsible for identification and opsonization of apoptotic cells which stimulates phagocytosis and activates the classical complement pathway. Apoptosis generates cellular debris, which if not properly cleared, can promote autoimmunity. Thus, deficiency of C1q results in autoantigen presentation with subsequent loss of tolerance (44). In addition, C1q suppresses IFN alpha production by interacting with leucocyte associated Ig-like receptor (LAIR)1 on plasmacytoid dendritic cells, and indirectly through uptake of C1q containing immune complexes by monocytes rather than plasmacytoid dendritic cells which are the primary producers of IFN alpha (68). Furthermore, C1q can inhibit TLR7 and TLR9 induced IFN-alpha production. Consequently, it has been described that patients with C1q deficiency develop increased levels of IFN alpha (9, 68). Taken into consideration the IFN-signature found in these patients, complement deficiencies could be also considered as secondary interferonopathies.
Deficiencies in C1r and C1s are rare and these patients usually die at a young age due to recurrent and severe infections. However, more than a half of these patients develop a lupus-like disease, with skin involvement and ANA positivity being the most noticeable features (73). Both genes encoding C1s and C1r are located on the short arm of chromosome 12 and several deleterious mutations, resulting in no detectable protein in the serum, have been identified in patients with lupus-like phenotype. Remarkably, consistent reduction in the serum protein levels of C1s in patients with C1r deficiency and low levels of C1r in patients with C1s deficiency have been observed (10, 11, 74).
C2 is the most common complement deficiency, occurring in about 1 in 20,000 individuals of European descent, however lupus develops only in about 10% of patients with C2 deficiency (73). This is likely due to the fact that C2 can be bypassed by the alternative complement pathway and is therefore not required for activation of the complement system (12). Patients with SLE secondary to C2 deficiency are similar in presentation and severity to the general SLE population, with a mean age of onset of 39 years. These patients will typically develop multiple infections at an early age, but are otherwise phenotypically similar to other patients with SLE (75).
C4 is a key component of the classical complement pathway. Homozygous deficiency of C4 results in a dysfunctional immune response which can cause lupus with >75% of penetrance (76). This protein is encoded by two genes, C4A and C4B, closely located within the human major histocompatibility complex (MHC) on chromosome 6. There is a complex pattern of variation in this region and duplications of C4 genes are common. The copy number variation (CNV) of these genes range from two to eight copies (13). Interestingly, the relationship of C4 gene copy number with non-Mendelian SLE has been repeatedly analyzed in different populations and the results of these studies consistently reported that the fewer the number of gene copies the higher the risk of lupus. Conversely, an increased number of C4 gene numbers is protective (77, 78). A deficiency in C4 in mice alters B cell tolerance by increasing the number of self-reactive B cells. These mice develop lupus-like features like glomerulonephritis and high levels of autoantibodies (79).
Autoimmune lymphoproliferative syndrome (ALPS) is a rare autoimmune disease mainly caused by mutations in FAS-mediated apoptotic pathway genes (17, 18). ALPS patients present with clinical features similar to SLE and mutations in these genes have been associated with both diseases (17, 18). The fas cell surface death receptor (FAS) is a protein in the TNF receptor superfamily. It plays a key role in programmed cell death; the binding of this receptor with its ligand results in signaling complex that includes Fas-associated death domain protein (FADD), caspase 8, and caspase 10 (17). Apoptosis is of particular interest in lupus as abnormalities in this process provide a source of autoantigens which are thought to drive the autoimmune response in this disease. Apoptotic cell death results in increased DNA fragments, which if not properly processed can accumulate and result in autoimmunity. FAS gene polymorphisms have been shown to be associated with SLE (80) and variants in the FASL gene have been related to increase apoptosis (81). In this context, it is noteworthy that mice with deficiencies in Fas and FasL develop clinical features similar to SLE and ALPS, thus represent useful murine models to study the pathophysiology of both diseases. Specifically, MRL/lpr mice have been widely used to investigate lupus and these studies significantly contributed to our current knowledge of the pathogenesis of SLE (82, 83).
PRKCD (protein kinase c delta) is a signaling kinase with multiple downstream target proteins which plays a role in regulating B cell development, proliferation, and apoptosis (84). The absence of PRKCD results in chronic B cell receptor signaling, decreased apoptosis, and increased response to stimulation. Conversely, overexpression of PRKCD results in inhibition of cell growth (85). A mutation in the PRKCD gene has been identified in a family with monogenic SLE, and is associated with loss of B cell tolerance and an increased number of immature B cells even in family members heterozygous for the mutation (24). In addition, PRKCD has a negative role in T cell proliferation and a deficiency in PRKCD results in increased T cell activation contributing to T cell autoimmunity (84). Patients with SLE secondary to PRKCD mutation demonstrate typical features of lupus including autoantibody production as well as increased incidence of glomerulonephritis.
Both DNASE1 (deoxyribonuclease 1) and DNASE1L3 (deoxyribonuclease 1 like 3) genes encode proteins involved in the nucleic acid degradation pathway. DNASE1L3 enzyme plays an important role in the clearance of DNA debris from apoptotic cells and exogenous DNA. A loss of function variant in this gene has been identified as the cause of a monogenic form of lupus. Positive ANA, anti-dsDNA, and hypocomplementemia among other features were present in all lupus patients harboring this variant (16). DNASE1L3 variations have also been reported in patients with hypocomplementemic urticarial vasculitis syndrome (86). A heterozygous null allele in DNASE1, which encodes for an endonuclease with certain degree of homology to DNASE1L3, has also been identified in individuals diagnosed with a monogenic form of SLE (15). Further studies have also reported the association of polymorphisms in DNASE1 with non-Mendelian SLE (87). All these findings are consistent with the demonstration that mice deficient in DNASE1 develop a lupus-like phenotype (88). Interestingly, mutations in DNASE2, which encodes another member of the DNAse family, have been identified in three children presenting with severe autoimmune features. Although these patients did not fulfill criteria for a classification of SLE, all of them showed high levels of anti-DNA antibodies among others lupus-like symptoms (89).
Others monogenic disorders presenting rare cases of lupus-like phenotype have been reported. Although in most of them the mechanism causing autoimmunity is unclear, several causal genes have been described (Table 1), including CYBB gene causing chronic granulomatous disease, PTPN11 and SHOC2 genes associated with Noonan Syndrome, among others. Further studies focused on understanding the role of these genes in autoimmunity will help to better understand the pathogenesis of SLE.
Understanding the implications of the genes identified to cause monogenic lupus have enhanced our knowledge of pathways and molecular mechanisms involved in the pathogenesis of SLE. As it has been explored through this review, monogenic lupus results from mutations in genes related to the immune response, either in the innate or in the adaptive immune system. Furthermore, protein-protein interaction analysis suggests that these genes encode proteins with related functions, creating molecular networks (Figure 2). Specifically, these genes are primarily involved in pathways including the complement system, nucleic acid repair, nucleic acid degradation and sensing, apoptosis, and type I interferon regulation. Although we do not yet know the full extent of monogenic lupus, the study of this type of lupus has provided new areas of investigation applicable to non-Mendelian SLE. Many genes have been identified as causes of monogenic lupus and at the same time have been associated with common forms of SLE, such as C4 number variation and polymorphisms in TREX1, among others. Besides, a recent study proposes that a set of rare variants across PRKCD play a role in a wider context of SLE susceptibility (90). Altogether, these findings reinforce the idea of analyzing the genetics of complex diseases by taking into consideration their Mendelian forms, and highlight the potential contribution of rare variants to the heritability of SLE.
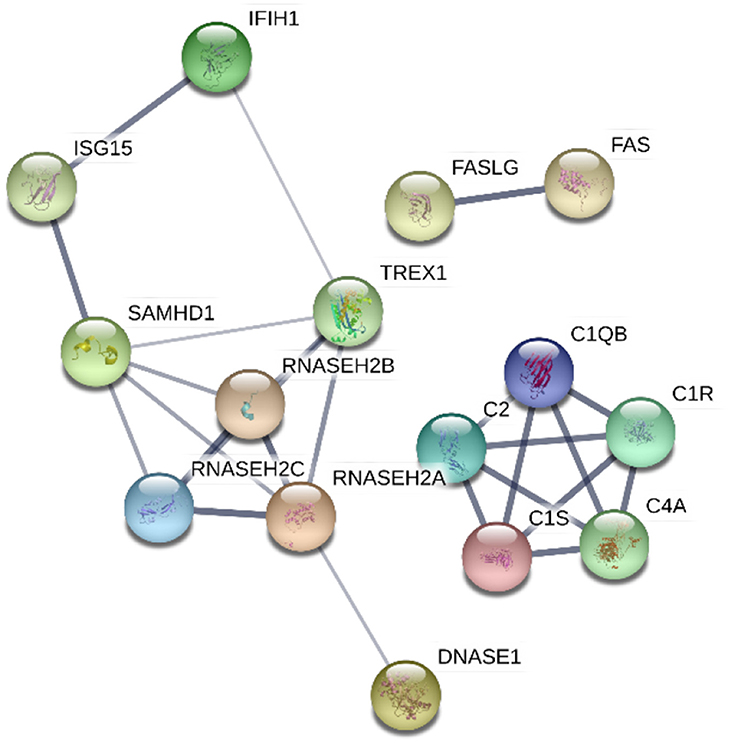
Figure 2. Protein-protein interaction network of proteins encoded by genes found to cause monogenic lupus. The confidence of data supporting these interactions are represented by the thickness of the lines. The analysis was performed using STRING V10.0 (STRING https://string-db.org/).
The molecular complexity of autoimmune diseases and the clinical overlap among them makes accurate diagnosis and specific targeted therapy more challenging. In this context, a better knowledge of the genetic bases may generate insights into biomarker development and new drug targets.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health grants number R01AI097134 and U19AI110502, the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health grant number R01AR070148, and the Lupus Research Alliance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Patrick Coit for graphical support.
1. Wu H, Zhao M, Tan L, Lu Q. The key culprit in the pathogenesis of systemic lupus erythematosus: Aberrant DNA methylation. Autoimmun Rev. (2016) 15:684–9. doi: 10.1016/j.autrev.2016.03.002
2. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol. (2015) 11:329–41. doi: 10.1038/nrneph.2015.33
3. Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. (1992) 35:311–8. doi: 10.1002/art.1780350310
4. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. (2010) 20:170–9. doi: 10.1101/gr.100289.109
5. Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis. (2011) 70:151–6. doi: 10.1136/ard.2010.141697
6. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. (2011) 43:127–31. doi: 10.1038/ng.748
7. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A (2015) 167A:296–312. doi: 10.1002/ajmg.a.36887
8. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. (2012) 44:1243–48. doi: 10.1038/ng.2414
9. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Ronnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. (2009) 60:3081–90. doi: 10.1002/art.24852
10. Demirkaya E, Zhou Q, Smith CK, Ombrello MJ, Deuitch N, Tsai WL, et al. Brief report: deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: the role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol. (2017) 69:1832–39. doi: 10.1002/art.40158
11. Bienaime F, Quartier P, Dragon-Durey MA, Fremeaux-Bacchi V, Bader-Meunier B, Patey N, et al. Lupus nephritis associated with complete C1s deficiency efficiently treated with rituximab: a case report. Arthritis Care Res. (2010) 62:1346–50. doi: 10.1002/acr.20163
12. Miller EC, Atkinson JP. Overcoming C2 deficiency. Clin Immunol. (2012) 144:269–71. doi: 10.1016/j.clim.2012.07.005
13. Blanchong CA, Chung EK, Rupert KL, Yang Y, Yang Z, Zhou B, Moulds JM, Yu CY. Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. Int Immunopharmacol. (2001) 1:365–92. doi: 10.1016/S1567-5769(01)00019-4
14. Battersby AC, Cale AM, Goldblatt D, Gennery AR. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease. J Clin Immunol. (2013) 33:1276–84. doi: 10.1007/s10875-013-9939-5
15. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. (2001) 28:313–4. doi: 10.1038/91070
16. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
17. Agrebi N, Ben-Mustapha I, Matoussi N, Dhouib N, Ben-Ali M, Mekki N, et al. Rare splicing defects of FAS underly severe recessive autoimmune lymphoproliferative syndrome. Clin Immunol. (2017) 183:17–23. doi: 10.1016/j.clim.2017.06.009
18. Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. (1996) 98:1107–13. doi: 10.1172/JCI118892
19. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. (2014) 46:503–9. doi: 10.1038/ng.2933
20. Hermann M, Bogunovic D. ISG15: In Sickness and in Health. Trends Immunol. (2017) 38:79–93. doi: 10.1016/j.it.2016.11.001
21. Leventopoulos G, Denayer E, Makrythanasis P, Papapolychroniou C, Fryssira H. Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS mutation. Clin Exp Rheumatol. (2010) 28:556–7.
22. Urushihara M, Kagami S, Yasutomo K, Ito M, Kondo S, Kitamura A., Sisters with alpha-mannosidosis and systemic lupus erythematosus. Eur J Pediatr. (2004) 163:192–5. doi: 10.1007/s00431-004-1404-2
23. Klar A, Navon-Elkan P, Rubinow A, Branski D, Hurvitz H, Christensen E, et al. Prolidase deficiency: it looks like systemic lupus erythematosus but it is not. Eur J Pediatr. (2010) 169:727–32. doi: 10.1007/s00431-009-1102-1
24. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, S et al. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum. (2013) 65:2161–71. doi: 10.1002/art.38008
25. Al-Mayouf SM, AlSaleem A, AlMutairi N, AlSonbul A, Alzaid T, Alazami AM, et al. Monogenic interferonopathies: Phenotypic and genotypic findings of CANDLE syndrome and its overlap with C1q deficient SLE. Int J Rheum Dis. (2018) 21:208–13. doi: 10.1111/1756-185X.13228
26. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, et al. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med Genet A (2012) 158A:1077–82. doi: 10.1002/ajmg.a.35290
27. Walter JE, Lo MS, Kis-Toth K, Tirosh I, Frugoni F, Lee YN, et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J Allergy Clin Immunol. (2015) 135:272–3. doi: 10.1016/j.jaci.2014.07.063
28. Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. (2016) 35:831–44. doi: 10.15252/embj.201593339
29. Abdel-Salam GM, El-Kamah GY, Rice GI, El-Darouti M, Gornall H, Szynkiewicz M, et al. Chilblains as a diagnostic sign of aicardi-goutieres syndrome. Neuropediatrics (2010) 41:18–23. doi: 10.1055/s-0030-1255059
30. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A (2011) 155A:235–7. doi: 10.1002/ajmg.a.33778
31. Bader-Meunier B, Cave H, Jeremiah N, Magerus A, Lanzarotti N, Rieux-Laucat F, et al. Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Case report and systematic review of the literature. Semin Arthritis Rheum. (2013) 43:217–9. doi: 10.1016/j.semarthrit.2013.04.009
32. Esteve E, Krug P, Hummel A, Arnoux JB, Boyer O, Brassier A, et al. Renal involvement in lysinuric protein intolerance: contribution of pathology to assessment of heterogeneity of renal lesions. Hum Pathol. (2017) 62:160–9. doi: 10.1016/j.humpath.2016.12.021
33. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
34. Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. (2015) 35:235–43. doi: 10.1007/s10875-015-0147-3
35. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
36. Reynier F, Petit F, Paye M, Turrel-Davin F, Imbert PE, Hot A, et al. Importance of correlation between gene expression levels: application to the type I interferon signature in rheumatoid arthritis. PLoS ONE (2011) 6:e24828. doi: 10.1371/journal.pone.0024828
37. Crow YJ. Aicardi-Goutieres Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors GeneReviews((R)). Seattle, WA (1993).
38. DiFrancesco JC, Novara F, Zuffardi O, Forlino A, Gioia R, Cossu F, et al. TREX1 C-terminal frameshift mutations in the systemic variant of retinal vasculopathy with cerebral leukodystrophy. Neurol Sci. (2015) 36:323–30. doi: 10.1007/s10072-014-1944-9
39. Ellyard JI, Jerjen R, Martin JL, Lee AY, Field MA, Jiang SH, et al. Identification of a pathogenic variant in TREX1 in early-onset cerebral systemic lupus erythematosus by Whole-exome sequencing. Arthritis Rheumatol. (2014) 66:3382–6. doi: 10.1002/art.38824
40. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
41. Barizzone N, Monti S, Mellone S, Godi M, Marchini M, Scorza R, et al. Rare variants in the TREX1 gene and susceptibility to autoimmune diseases. Biomed Res Int. (2013) 2013:471703. doi: 10.1155/2013/471703
42. Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. (2011) 12:270–9. doi: 10.1038/gene.2010.73
43. Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin North Am. (2017) 43:415–34. doi: 10.1016/j.rdc.2017.04.005
44. Costa-Reis P, Sullivan KE. Monogenic lupus: it's all new! Curr Opin Immunol. (2017) 49:87–95. doi: 10.1016/j.coi.2017.10.008
45. Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, et al. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell (2006) 23:133–42. doi: 10.1016/j.molcel.2006.06.005
46. de Carvalho LM, Ngoumou G, Park JW, Ehmke N, Deigendesch N, Kitabayashi N, et al. Musculoskeletal disease in MDA5-related type I interferonopathy: a mendelian mimic of Jaccoud's arthropathy. Arthritis Rheumatol. (2017) 69:2081–91. doi: 10.1002/art.40179
47. Lo MS, Tsokos GC. Recent developments in systemic lupus erythematosus pathogenesis and applications for therapy. Curr Opin Rheumatol. (2018) 30:222–8. doi: 10.1097/BOR.0000000000000474
48. Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity (2014) 40:199–212. doi: 10.1016/j.immuni.2013.12.014
49. Gono T, Kawaguchi Y, Sugiura T, Furuya T, Kawamoto M, Hanaoka M, et al. Interferon-induced helicase (IFIH1) polymorphism with systemic lupus erythematosus and dermatomyositis/polymyositis. Mod Rheumatol. (2010) 20:466–70. doi: 10.3109/s10165-010-0311-9
50. Zhang SH, Zhao Y, Xie QB, Jiang Y, Wu YK, Yan B. Aberrant activation of type I interferon system may contribute to the pathogenesis of anti-MDA5 dermatomyositis. Br J Dermatol. (2018). doi: 10.1111/bjd.16917. [Epub ahead of print].
51. Ballana E, Este JA. SAMHD1: at the crossroads of cell proliferation, immune responses, and virus restriction. Trends Microbiol. (2015) 23:680–92. doi: 10.1016/j.tim.2015.08.002
52. Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, et al. SAMHD1-dependent retroviral control and escape in mice. EMBO J. (2013) 32:2454–62. doi: 10.1038/emboj.2013.163
53. Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M, et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis. (2015) 74:e17. doi: 10.1136/annrheumdis-2013-204845
54. Tungler V, Staroske W, Kind B, Dobrick M, Kretschmer S, Schmidt F, et al. Single-stranded nucleic acids promote SAMHD1 complex formation. J Mol Med. (2013) 91:759–70. doi: 10.1007/s00109-013-0995-3
55. Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest. (2015) 125:413–24. doi: 10.1172/JCI78001
56. Gallo A, Vukic D, Michalik D, O'Connell MA, Keegan LP. ADAR RNA editing in human disease; more to it than meets the I. Hum Genet. (2017) 136:1265–78. doi: 10.1007/s00439-017-1837-0
57. Al-Saif F, Alhumidi A, Alhallaf RA. Dyschromatosis symmetrica hereditaria with cutaneous lupus erythematosus and hyperthyroidism. Int Med Case Rep J. (2017) 10:149–52. doi: 10.2147/IMCRJ.S132489
58. Yuan Y, Ma H, Ye Z, Jing W, Jiang Z. Interferon-stimulated gene 15 expression in systemic lupus erythematosus : Diagnostic value and association with lymphocytopenia. Z Rheumatol. (2017) 77:256–62. doi: 10.1007/s00393-017-0274-8.
59. Feng X, Huang J, Liu Y, Xiao L, Wang D, Hua B, et al. Identification of interferon-inducible genes as diagnostic biomarker for systemic lupus erythematosus. Clin Rheumatol. (2015) 34:71–9. doi: 10.1007/s10067-014-2799-4
60. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature (2015) 517:89–93. doi: 10.1038/nature13801
61. D'Cunha J, Knight E Jr, Haas AL, Truitt RL, Borden EC. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc Natl Acad Sci USA. (1996) 93:211–5. doi: 10.1073/pnas.93.1.211
62. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. (2011) 43:132–7. doi: 10.1038/ng.749
63. Oddie GW, Schenk G, Angel NZ, Walsh N, Guddat LW, de Jersey J, et al. Structure, function, and regulation of tartrate-resistant acid phosphatase. Bone (2000) 27:575–84. doi: 10.1016/S8756-3282(00)00368-9
64. Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, et al. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol. (2006) 7:498–506. doi: 10.1038/ni1327
65. An J, Briggs TA, Dumax-Vorzet A, Alarcon-Riquelme ME, Belot A, Beresford M, et al. Tartrate-resistant acid phosphatase deficiency in the predisposition to systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:131–42. doi: 10.1002/art.39810
66. Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH. Rheumatology E-Book. Elsevier Health Sciences (2014).
67. Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. (2010) 34:J276–86. doi: 10.1016/j.jaut.2009.11.014
68. Hagberg N, Rönnblom L. Systemic lupus erythematosus–a disease with a dysregulated type i interferon system. Scandinav J Immunol. (2015) 82:199–207. doi: 10.1111/sji.12330
69. Lipsker D, Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus (2010) 19:1096–106. doi: 10.1177/0961203310373370
70. Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol. (2016) 7:36. doi: 10.3389/fimmu.2016.00036
71. Mehta P, Norsworthy PJ, Hall AE, Kelly SJ, Walport MJ, Botto M, et al. SLE with C1q deficiency treated with fresh frozen plasma: a 10-year experience. Rheumatology (2010) 49:823–4. doi: 10.1093/rheumatology/kep387
72. Arkwright PD, Riley P, Hughes SM, Alachkar H, Wynn RF. Successful cure of C1q deficiency in human subjects treated with hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2014) 133:265–7. doi: 10.1016/j.jaci.2013.07.035
73. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. (2000) 76:227–324. doi: 10.1016/S0065-2776(01)76021-X
74. Wu YL, Brookshire BP, Verani RR, Arnett FC, Yu CY. Clinical presentations and molecular basis of complement C1r deficiency in a male African-American patient with systemic lupus erythematosus. Lupus (2011) 20:1126–34. doi: 10.1177/0961203311404914
75. Jönsson G, Sjöholm AG, Truedsson L, Bengtsson AA, Braconier JH, Sturfelt G. Rheumatological manifestations, organ damage and autoimmunity in hereditary C2 deficiency. Rheumatology (2007) 46:1133–9. doi: 10.1093/rheumatology/kem023
76. Macedo ACL, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. (2016) 7:55. doi: 10.3389/fimmu.2016.00055
77. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. (2007) 80:1037–54. doi: 10.1086/518257
78. Chen JY, Wu YL, Mok MY, Wu YJ, Lintner KE, Wang CM, et al. Effects of complement C4 gene copy number variations, size dichotomy, and C4A deficiency on genetic risk and clinical presentation of systemic lupus erythematosus in east asian populations. Arthritis Rheumatol. (2016) 68:1442–53. doi: 10.1002/art.39589
79. Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, et al. A critical role for complement in maintenance of self-tolerance. Immunity (1998) 9:721–31. doi: 10.1016/S1074-7613(00)80669-X
80. Xiang N, Li XM, Wang GS, Tao JH, Li XP. Association of Fas gene polymorphisms with systemic lupus erythematosus: a meta-analysis. Mol Biol Rep. (2013) 40:407–15. doi: 10.1007/s11033-012-2075-0
81. Glesse N, Vianna P, Paim LMG, Matte MCC, Aguiar AKK, Palhano PL, et al. Evaluation of polymorphic variants in apoptotic genes and their role in susceptibility and clinical progression to systemic lupus erythematosus. Lupus (2017) 26:746–55. doi: 10.1177/0961203316678671
82. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. (2010) 184:1840–8. doi: 10.4049/jimmunol.0902592
83. Tilstra JS, Avery L, Menk AV, Gordon RA, Smita S, Kane LP, et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J Clin Invest. (2018) 120859. doi: 10.1172/JCI120859. [Epub ahead of print].
84. Salzer E, Santos-Valente E, Keller B, Warnatz K, Boztug K. Protein Kinase C delta: a Gatekeeper of Immune Homeostasis. J Clin Immunol. (2016) 36:631–40. doi: 10.1007/s10875-016-0323-0
85. Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature (2002) 416:865. doi: 10.1038/416865a
86. Ozcakar ZB, Foster J II, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. (2013) 65:2183–9. doi: 10.1002/art.38010
87. Bodano A, Gonzalez A, Ferreiros-Vidal I, Balada E, Ordi J, Carreira P, et al. Association of a non-synonymous single-nucleotide polymorphism of DNASEI with SLE susceptibility. Rheumatology (2006) 45:819–23. doi: 10.1093/rheumatology/kel019
88. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. (2000) 25:177–81. doi: 10.1038/76032
89. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. (2017) 8:2176. doi: 10.1038/s41467-017-01932-3
Keywords: lupus, monogenic, familial, genetic, mendelian
Citation: Alperin JM, Ortiz-Fernández L and Sawalha AH (2018) Monogenic Lupus: A Developing Paradigm of Disease. Front. Immunol. 9:2496. doi: 10.3389/fimmu.2018.02496
Received: 28 July 2018; Accepted: 09 October 2018;
Published: 30 October 2018.
Edited by:
José Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, MexicoReviewed by:
Christian Michael Hedrich, University of Liverpool, United KingdomCopyright © 2018 Alperin, Ortiz-Fernández and Sawalha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amr H. Sawalha, YXNhd2FsaGFAdW1pY2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.