 Sonia Guedan1*
Sonia Guedan1* Ramon Alemany2
Ramon Alemany2- 1Department of Hematology and Oncology, Hospital Clinic, Institut d'Investigacions Biomèdiques August Pi i Sunyer, Barcelona, Spain
- 2ProCure Program, IDIBELL-Institut Catala d'Oncologia, L'Hospitalet de Llobregat, Spain
Adoptive transfer of chimeric antigen receptor (CAR)-modified T cells has resulted in unprecedented rates of long-lasting complete responses in patients with leukemia and lymphoma. However, despite the impressive results in patients with hematologic malignancies, CAR-T cells have showed limited effect against solid cancers. New approaches will need to simultaneously overcome the multiple challenges that CAR-T cells encounter in solid tumors, including the immunosuppressive tumor microenvironment and heterogeneity of antigen expression. Oncolytic viruses are lytic and immunogenic anti-cancer agents with the potential to synergize with CAR-T cells for the treatment of solid tumors. In addition, viruses can be further modified to deliver therapeutic transgenes selectively to the tumor microenvironment, which could enhance the effector functions of tumor-specific T cells. This review summarizes the major limitations of CAR-T cells in solid tumors and discusses the potential role for oncolytic viruses as partners for CAR-T cells in the fight against cancer.
Introduction
The recent approval by the US Food and Drug Administration (FDA) of two different CAR-T cell therapies for the treatment of leukemia and lymphoma represents a landmark in the development of cancer immunotherapies. Together with immune checkpoint blockade therapy (1), CAR-T cells are revolutionizing the field of cancer therapy, providing hope for a cure in patients with previously refractory cancers (2–8). However, despite the stunning results of CAR-T cells in patients with hematologic malignancies, this approach has shown little effect in patients with solid tumors. Recent clinical trials demonstrated that CAR-T cells are able to infiltrate the tumor mass and exert antigen-directed activity (9–12). However, with rare exceptions (13, 14), observed responses in patients with solid tumors have been minor and transient.
In order to induce complete responses in patients with solid tumors, CAR-T cells need to overcome several barriers. First, CAR-T cells must traffic from the blood into the tumor, infiltrate the tumor mass, and be able to survive and maintain their effector functions in a tumor microenvironment that is highly immunosuppressed and enriched in stroma. Then, CAR-T cells need to eliminate the totality of the cancer cells, which is extremely difficult due to the heterogeneity of antigen expression in cancer cells and the intrinsic plasticity of tumors that may lead to tumor escape (15, 16). Finally, CAR-T cells are living drugs that can lead to dramatic antitumor responses but can also induce significant toxicities (17–19). New approaches to enhance therapeutic outcome in patients with solid tumors must therefore focus on enhancing potency without increasing toxicity.
Rapid advances in synthetic biology, T cell immunology, and gene editing have fueled the design of next generation CAR-T cells with the potential to overcome some of the hurdles encounter in solid tumors (20). However, it is unlikely that CAR-T cell therapy alone will be sufficient to induce complete responses in the majority of cancers. Combining CAR-T cells with other cancer treatments that have different mechanisms of action and the potential to synergize with T cells may reduce tumor escape and increase the success rates of CAR-T cell therapy. As novel therapies emerge, rational combinations will need to be tested based on an understanding of the mechanisms underlaying tumor resistance to CAR-T cells.
Oncolytic virotherapy is a therapeutic approach to treat cancer that uses native or genetically modified viruses that selectively replicate within cancer cells (21). The field of oncolytic virotherapy has gained renewed attention after the FDA approval of Talimogene laherparepvec (T-VEC), an oncolytic herpes simplex virus type 1 (HSV-1) modified to express GM-CSF (22), and the recent reports of high response rates obtained in patients with advanced melanoma when combining T-VEC with checkpoint blockade (23, 24). Oncolytic viruses (OV) mediate their antitumor effect through a dual mechanism of action, including a direct lytic effect on tumor cells and the induction of anti-cancer adaptive immunity (25, 26). Moreover, OV can be further modified to selectively deliver therapeutic transgenes to the tumor microenvironment to enhance their antitumor potency or boost an antitumor immune response (27). All these characteristics make OV excellent potential partners to synergize with emerging immunotherapies, and several combinatorial approaches are being currently tested in preclinical and clinical trials (26, 28).
This review provides an overview of current barriers that CAR-T cells encounter in solid tumors, summarizes the advances in the field of OV and discusses the preclinical and clinical data that support the clinical testing of OV in combination with CAR-T cells to overcome the solid tumor challenge.
CAR-T Cells in Solid Tumors: Challenges and Limitations
While most of the early trials of CAR-T cells for solid tumors resulted in poor therapeutic outcomes, some case reports of dramatic clinical responses with manageable therapy-related toxic effects provide clear reasons for optimism (13, 14). Recent reports with second generation CAR-T cells suggest that CAR-T cells can traffic, persist, and proliferate in the tumor (9, 10). Moreover, evidence of transient antitumor activity has been observed in patients with difficult-to-treat tumors, such as glioblastoma (13), neuroblastoma (14), pancreatic cancer (12), and sarcoma. Here, we summarize the lessons learned in these clinical trials and discuss the hurdles that CAR-T cells must overcome for effective therapy, focusing on those challenges that OV may help to address.
Trafficking, Proliferation, and Persistence
The ability of tumor-specific T cells to traffic to the tumor, proliferate, and persist is considered critical to achieve an effective anti-tumor response (14, 29, 30). While T cells can actively traffic to sites of disease, often tumors present low levels of inflammation and lack of the chemokines required for migration. Also, physical barriers, such as aberrant vasculature, increased stromal stiffness and high interstitial pressure, may impair T-cell infiltration. Once in the tumor, CAR-T cells must efficiently proliferate, and persist until the entirety of the tumor is eliminated. However, T-cell proliferation and persistence are often hampered due to T-cell intrinsic (T-cell fitness) or extrinsic factors (tumor microenvironment). The requirements for proliferation and persistence can be relaxed in some instances if regional delivery and redosing of CAR-T cells is a therapeutic option (31). For example, in a recent clinical trial, multiple intracranial injections of CAR-T targeting IL13Rα2 mediated a transient complete response in a patient with glioblastoma (13). In this patient, two intracranial CAR-T cell delivery routes were tested: intracavitary and intraventricular. While intracavitary therapy was only able to control growth of the local tumor, intraventricular therapy resulted in a dramatic reduction in the size of all intracranial and spinal tumors. These results highlight the importance of trafficking and administration route to achieve the optimal tumor responses. Developing strategies to enhance trafficking and persistence to increase the therapeutic CAR-T cell input in the tumor would represent a vertical advance in the field.
Tumor Immunosuppression
On arrival to the tumor, CAR-T cells encounter an immunosuppressive environment that prevents T-cells from reaching their full therapeutic potential. The main barriers that CAR-T cells need to overcome once in the tumor include: (i) suppression by immunoregulatory cells, including myeloid-derived suppressor cells (MDSC), tumor associated macrophages and neutrophils, and regulatory T cells; (ii) presence of an array of immunosuppressive molecules, such as IL-10, TGF-β, PD-L1, IDO and arginase-1, and (iii) microenvironment factors, such as hypoxia, low pH and nutritional depletion. These conditions, together with chronic antigen exposure, can lead T-cells to distinct stages of functional dysfunction (32–34). Moreover, the stromal microenvironment can actively exclude T cells from the vicinity of cancer cells (35). Finally, a recent clinical report suggests that the tumor microenvironment can become even more immunosuppressive after CAR-T cell activation within the tumor, probably due to an initial production of IFN-γ (10). Finding ways to prevent or reverse T-cell dysfunction by reverting tumor immunosuppression will be key to improving treatment.
Tumor Escape by Loss or Heterogeneity of Antigen Expression
One of the main limitations in the treatment of solid tumors with CAR-T cells is the absence of cancer-restricted antigens that are uniformly expressed in tumor cells and absent in essential organs. Solid tumors exhibit heterogeneity of antigen expression with regards to intensity and distribution. Tumor escape due to heterogeneity or loss of antigen expression is an emerging threat to CAR-T cells, as it can result in overgrowth of target-deficient tumor cells that are invisible to CAR-T cell therapy (36–38). Preclinical studies have demonstrated that tumor cells expressing high levels of the targeted antigen are preferentially eliminated by CAR-T cells, whereas those with the lowest expression may survive (39–41). Decreased expression of the targeted antigen after CAR-T cell therapy has been observed in several clinical trials, including those targeting Her2 (9), EGFRviii (10), IL13Rα2 (11), and mesothelin (12). These results demonstrate the potential of CAR-T cells to eliminate antigen-positive tumor cells, but also highlight the importance of designing new strategies to simultaneously target different antigens. Several groups are designing new CAR constructs able to target more than one antigen simultaneously (39, 42, 43). While reducing the risk of escape, these strategies may also result in increased on-target off-tumor reactivity, as most of the targeted antigens can be expressed in healthy tissue at low levels (17–19). An alternative approach would be to find strategies to activate an endogenous immune response that could partner with CAR-T cells to completely eliminate the tumor. Some reports suggest that CAR-T-cell mediated tumor destruction may lead to the release of other tumor antigens that are cross-presented in a process known as epitope spreading (44, 45). This observation requires further investigation, but it could explain how complete elimination of tumor lesions has been achieved even when the tumors did not uniformly express the target (13).
Oncolytic Viruses: Lessons Learned in Clinical Trials
To date, there are three viruses commercially available for the treatment of cancer: T-VEC approved in the USA, H101 approved in China and Rigvir approved in Latvia, Georgia and Armenia. Several other viruses are in clinical trials and may eventually join this short list of marketed viruses (46). Some of the lessons learned from clinical trials that will drive the design of future therapies include: (a) OV can induce a therapeutic benefit in cancer patients, including complete responses, in the absence of severe adverse effects (47–50). Interestingly, some of these complete responses are reached after the virus have been eliminated, suggesting that the complete elimination of the tumor may depend on the activation of an immune-mediated anti-tumor response (48). On line with this observation, a recent clinical trial reported that the overall survival among patients who received a chimeric poliovirus reached a plateau of 21% 1 year after treatment that was sustained for months (51). This plateau in long-term survival is similar to the one observed in Kaplan-Meier curves from cancer patients treated with other cancer immunotherapies and highlights the role of the immune system on the emergence of long-term survivors (52); (b) The antiviral immunity constitutes an obstacle against OV as it sequesters or neutralizes viral particles before they reach their target. A major question is how to deliver the virus to the tumor efficiently; (c) Virus replication has been detected in tumor biopsies a few days after treatment. However, the ability of OV to survive and spread through the tumor is limited by antiviral T cells (47, 48, 53); (d) Tumors treated with OV typically show increased immune cell infiltration, including activated macrophages and cytotoxic T-cells, and pro-inflammatory cytokines (47, 48, 53). Tumor-specific T cells have been detected after treatment with OV (53, 54). While the capacity of OV to expand neoantigen-specific T cells deserves further investigation, the potential of OV for combination with immunotherapies such as immune checkpoint inhibitors has been well-recognized (28, 55–58). Several clinical trials are currently testing the combination of OV with immune checkpoint therapy and initial reports showed promising results (23, 24).
Oncolytic Viruses: The Ideal Allies for CAR-T Cells?
OV have the potential to synergize with CAR-T cells by helping them simultaneously overcome some of the multiple barriers found in solid tumors. First, viruses provide a danger signal that can revert tumor immunosuppression, which could facilitate CAR-T cell trafficking, proliferation, and persistence in the tumor microenvironment. Second, the direct lytic effect of OV on cancer cells results in tumor lysis and release of tumor-associated antigens (TAA), which can induce an anti-tumor adaptive response that could potentially mitigate tumor escape by antigen loss. Third, OV can be armed with therapeutic transgenes that could further enhance the effector functions of T cells. Here, we provide an overview of the biological properties of OV that may be considered when choosing a viral platform for combination with CAR-T cells, and we summarize the recent preclinical strategies that have been explored combining CAR-T cells and OV.
Oncolytic Viruses as Immunotherapy Agents
The immune system is well-equipped to mount an innate inflammatory response to viruses that eventually will induce the infiltration of effector T-cells. In particular, OV have pathogen-associated molecular patterns (PAMPS) detected by pattern-recognition receptors (PRRs) on tumor and epithelial cells as well as macrophages and dendritic cells (59). These PRRs induce danger associated molecular patterns (DAMPs) characteristic of an immunogenic cell death (60, 61). PRRs also signal through NF-kB to induce the expression of cytokines such as TNF-α and IL6, and through IFN Regulatory Factor (IRF) to induce type I interferons and activate caspase 1 that matures IL-1β (62). This pro-immune cytokine environment can facilitate the maturation and function of DC's, macrophages, and epithelial cells that can lead to the recruitment of neutrophils and natural killer (NK) cells, monocytes, and memory T-cells to the site of infection (63–65). Tumor cells dying due to the lytic activity of OV can release TAA. Activated DC's with their MHC loaded with virus and/or tumor epitopes can traffic to the draining lymph nodes to engage specific T-cells and stimulate their proliferation and circulation into the bloodstream. Chemokines of the infected tumors can induce integrin expression on these T-cells and selectin expression on endothelial cells to extravasate them. Under these conditions, T cells can be recruited efficiently to infected tumors, and as discussed above, increased T cell infiltration is generally detected in tumors of patients treated with OV therapy. Interestingly, viral infection has been shown to induce neoantigen-directed T cell responses (53, 54), which could synergize with CAR-T cells and virus-specific T cells to clear the tumor. A mayor limitation to study the impact of the immune-modulating effects of OV on CAR-T cell therapy is the lack of good animal models. However, it can be hypothesized that following the establishment of a more immunogenic intratumoral milieu, killing of target cells may be more efficient due to cooperation between the effector T-cells.
The ability of OV to induce an anti-tumor immune response is now considered a key mechanism of action to obtain long-term antitumor responses. Therefore, most of the current efforts directed at enhancing the therapeutic potential of OV are focused on improving their capacity to induce a systemic antitumor response.
The Oncolytic Virus Armamentarium
Multiple types of viruses are used in cancer virotherapy, each one of them with its unique properties (Table 1) (66). Here we discuss some of the different factors that should be considered when selecting an OV for combination with CAR-T cells. In general terms, viruses that replicate in the cytoplasm (RNA viruses) kill tumor cells faster than nuclear ones (DNA viruses) as they do not need to reach the nucleus of the infected cells. But for the same reason, they offer less opportunities for tumor-selective control. Tumor-selective replication of most oncolytic RNA viruses, such as reovirus, picornaviruses (Coxsackeivirus, Rigavirus), rhabdovirus (Vesiscular Stomatitis Virus [VSV], Maraba Virus), and paramixovirus (Measles Virus, Newcastle disease virus [NDV]), depends on defects of the interferon pathway in tumor cells. Because IFN induction is a central pathway in the innate response to viruses, which potentiates the adaptive T cell responses, the inflammatory response elicited with these viruses is expected to be lower. DNA viruses, such as adenoviruses, have slower replication cycles but are amenable to being controlled in the nucleus of the infected cells using tumor-selective promoters. The presence of an envelope also determines the oncolytic properties of a virus. Enveloped viruses (i.e., Measles virus, NDV, VSV, Herpes simplex virus, and Vaccinia virus) bud from cells and are less “lytic” than naked viruses. The envelope also contributes to the main clearance mechanisms in blood, with complement having a major role for enveloped viruses and antibodies for non-enveloped ones. Size is also an important parameter for the properties of OVs. The smaller the virus, the easier it will be for the virus to penetrate and diffuse throughout the tumor. But a larger virus with a larger genome allows the insertion of non-viral transgenes. Arming OV with therapeutic transgenes offer the opportunity to complement the OV in multiple ways. Among RNA viruses, VSV, Measles virus, and NDV can accept transgenes in contrast to picornaviruses and reoviruses, and for DNA viruses, Adenovirus, Herpes Simplex Virus and Vaccinia virus can be armed with transgenes in contrast to parvovirus. The list of genes that have been included in OV that could be potentially useful for combination with CAR-T cells is long and it has been reviewed recently (67). It includes, among others: (a) inducers of immunogenic cell death (68), (b) transgenes directed to modulate the immune system, such as cytokines (22, 69–71), chemokines (72, 73), co-stimulatory proteins (74–77), bispecific T-cell engagers (BiTEs) (78, 79), and immune checkpoint blockers (80–83), and (c) stroma-degrading proteins that could facilitate the spread of OV and T-cells within the tumors (84, 85). Comparing viruses and transgenes is a very challenging task given the limitations of preclinical immune competent mouse models, where many human viruses present defects in replication and tumors do not edit the immune system in a slow and progressive way as occurs in humans.
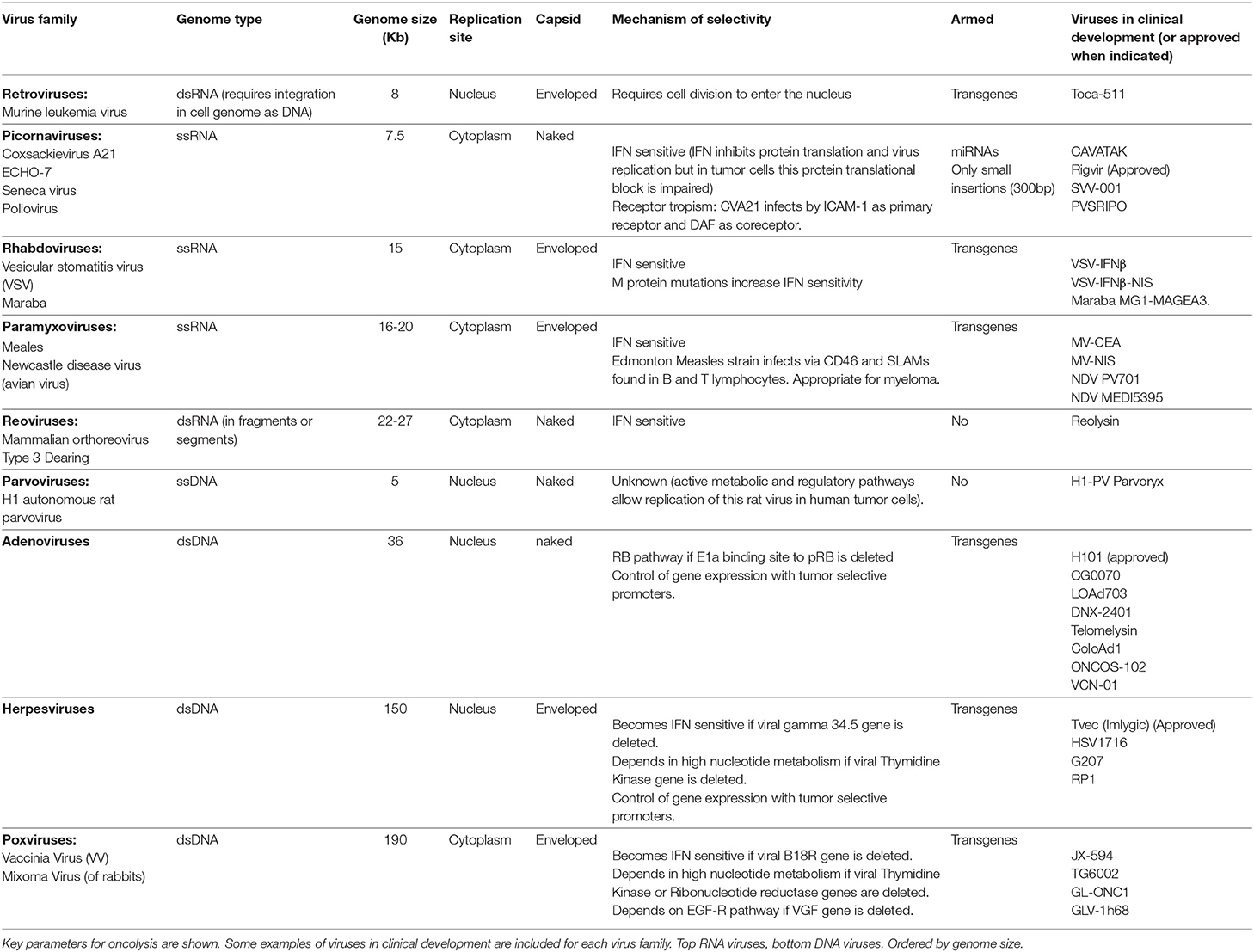
Table 1. Main groups of oncolytic viruses classified by taxonomy families.
Combining CAR-T Cells and Oncolytic Viruses for the Treatment of Solid Tumors
At a preclinical level, several groups have started to test different transgene-armed OV in combination with CAR-T cells (Figure 1). Most of these works assessed the antitumor effects of these therapies in NOD scid gamma (NSG) mice, a mouse strain that is completely deficient in adaptive immunity and severely deficient in innate immunity (86). NSG mice allow the engraftment and persistence of adoptively transferred CAR-T cells, and human tumor xenografts allow the replication of the virus and the delivery of the transgene. Therefore, these studies gave important insights in the antitumor effects of combining CAR-T cells with oncolysis and transgene delivery. An important limitation is that the capacity of OV to induce anti-tumor immunity cannot be assessed using these tumor xenografts.
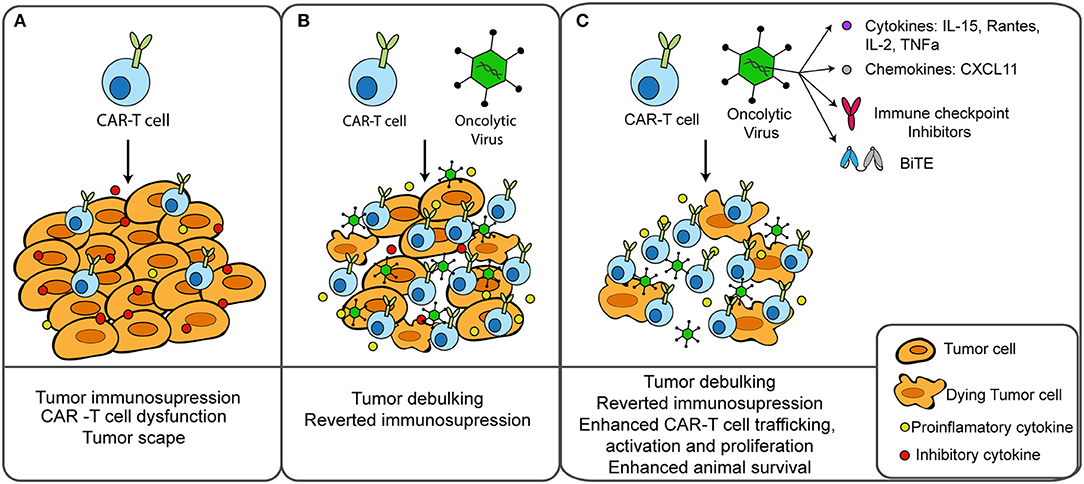
Figure 1. Combination of CAR-T cells and oncolytic virus for the treatment of solid tumors. (A) CAR-T cells find several obstacles in solid tumors, including an immunosuppressive environment that can lead to T cell dysfunction and treatment failure. (B) Cancer treatment with oncolytic viruses prior to CAR-T cell therapy results in tumor debulking, immunogenic cell death and reverted tumor immunosuppression. (C) Oncolytic viruses can be genetically modified to deliver therapeutic transgenes into the tumor microenvironment to enhance T-cell effector functions. Preclinical studies combining CAR-T cells with oncolytic viruses armed with cytokines, chemokines, BiTEs, or immune checkpoint inhibitors resulted in enhanced therapeutic outcomes.
Oncolytic adenoviruses modified to express IL-15 and RANTES (87) or IL-2 and TNF-α (88) have been shown to increase the accumulation and survival of CAR-T cells in the tumor microenvironment. Similarly, with the goal of enhancing the intra-tumoral trafficking of CAR-T cells, a vaccinia virus expressing CXCL11, a CXCR3 ligand, was used to attract effector cells following transfer (89). Another report demonstrated that expression by an oncolytic adenovirus of a BiTE targeting a second tumor antigen could address heterogeneity of antigen expression (40). Combination of a preparation of CAR-T cells with the OV-BiTE induced activation of T cells in the absence of the CAR-targeted antigen or lack of CAR expression (i.e., non-transduced T cell population). In a slightly different approach, combination of an oncolytic adenovirus with a helper-dependent adenovirus expressing a PD-L1 blocking mini-antibody was used to revert T cell dysfunction by preventing PD1:PDL1 interaction (90). Co-expression of IL12p70 and PD-L1 further augmented the therapeutic efficacy of the combination (91). As expected, all these combinations of CAR-T cells and armed-OV resulted in enhanced tumor control and prolonged survival when compared to each agent as monotherapy. An interesting finding by Watanabe et al. is that CAR-T cells as monotherapy failed to control the growth of the primary tumor, while OV could suppress the progression of the primary tumor but mice died from metastatic disease. Combination of CAR-T cells with an OV armed with IL-2 and TNF-a was able to control both the primary tumor and tumor metastasis (88).
Finally, in a totally different and very preliminary approach, CAR-T cells have been used to deliver OV to the tumor (92). Circulating cells such as lymphocytes, monocytes, erythrocytes, or even platelets can bind viruses and have shown tumor-targeting properties (93–96). Loading OV onto tumor-specific T cells (by adhesion to the T-cell surface) can protect the virus from neutralizing antibodies while retaining its antitumor efficacy after release in the tumor microenvironment (96). OV-tumor delivery by CAR-T cells could enhance virus delivery to the tumor and subsequent oncolysis could attract more CAR-T cells, establishing a positive feedback loop.
Remaining Questions and Future Directions
With such a variety of oncolytic viruses it is hard to know which one will be best suited for combination with CAR-T cells. In practical terms, it is difficult to envisage a virus commercially developed solely for the combination with CAR-T cells. Therefore, marketed viruses or viruses under clinical investigation are expected to be the first ones to be used in the clinic in combination with CAR-T cells.
While the general value of the virus to attract T-cells to the tumor is widely accepted (53, 97), practical questions on best delivery routes and dosing schedules are more difficult to predict. Intratumoral administration of the OV provides larger amounts of virus in the injected tumors, but it is technically challenging for visceral tumors or metastases, and non-injected tumor lesions will be less likely to get any virus to change the immunosuppressive microenvironment. Systemic intravenous administration is easier to perform and potentially useful to reach all metastases, but efficient neutralization of the virus in the bloodstream, especially with high titers of neutralizing antibodies raised after the first virus administration, will impose a barrier for repeated delivery. The immune response to the virus may also be very different if the virus is injected intratumorally or systemically. Usually vaccination immunization is performed subcutaneously or intramuscularly as the immune system does not respond aggressively to systemic pathogens, partly due to a lower inflammatory response of liver Kupffer cells compared to tissue-resident dendritic cells and the tolerogenic nature of the liver (98). Therefore, the immune response elicited by an OV replicating in a tumor may be tamed or modulated when the virus has been detected systemically. Timing of the virus and CAR-T cells can also impact the outcome. In principle, the virus should go first to change the immune suppressive tumor microenviroment, induce a direct lytic effect on tumor cells, and create a more appropriate environment that attracts the CAR-T cells. Patient preconditioning should also be considered prior to therapy. Although the immunostimulatory environment generated by the virus may bypass the need to lymphodeplete the patient to promote CAR-T cell expansion, lymphodepletion could still be a good approach to foster virus replication and persistence in the tumor while providing an advantage to the co-administered CAR-T cells (4, 99, 100).
Oncolytic viruses offer a strong inflammatory self-amplification oncolytic mechanism of action that can also result in the release of TAA. However, the ability of OV to induce an anti-tumor immune response is not well-understood. Given the large number of viral non-self-peptides after treatment with OV, it is likely that immune responses to the viral epitopes will dominate the response in a mixture with tumor neoantigens (101–103). New strategies to increase the immunogenicity of tumor epitopes and reduce the immunodominance of viral antigens are needed to promote epitope spreading (104).
Finally, T cells could also be manipulated to become a better partner for oncolytic viruses. Virus-specific T cells have been used as a platform for CAR expression (105). Virus-specific CAR-T cells retain the ability to recognize both virus-infected and tumor targets through their native and chimeric receptors, respectively. Thus, these T-cells could be ideal for a combined treatment with OV, as the presence of the virus could boost the amplification of CAR-T cells in the tumor. A drawback of this approach is that a faster clearance of the OV will occur.
Author Contributions
SG and RA conceptualized, wrote, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anna Wing for extensive revision of this manuscript.
References
1. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell Lymphoblastic Leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
3. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
4. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
5. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute Lymphoblastic Leukemia. N Eng J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
6. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
7. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
8. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Eng J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
9. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
10. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
11. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
12. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
13. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
14. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
15. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature (2013) 501:338–45. doi: 10.1038/nature12625
16. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell (2015) 27:15–26. doi: 10.1016/j.ccell.2014.12.001
17. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
18. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–e22. doi: 10.1200/JCO.2006.05.9964
19. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
20. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
21. Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell (2018) 33:599–605. doi: 10.1016/j.ccell.2018.03.011
22. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
23. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 Immunotherapy. Cell (2017) 170:1109–19 e10. doi: 10.1016/j.cell.2017.08.027
24. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. (2018) 36:1658–67. doi: 10.1200/JCO.2017.73.7379
25. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front Immunol. (2018) 9:866. doi: 10.3389/fimmu.2018.00866
26. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. (2018) 18(Suppl. 4):1. doi: 10.1038/s41577-018-0014-6
27. Bauzon M, Hermiston T. Armed therapeutic viruses - a disruptive therapy on the horizon of cancer immunotherapy. Front Immunol. (2014) 5:74. doi: 10.3389/fimmu.2014.00074
28. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer (2018) 18:419–32. doi: 10.1038/s41568-018-0009-4
29. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
30. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–571. doi: 10.1038/s41591-018-0010-1
31. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
32. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
33. Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. (2012) 33:364–72. doi: 10.1016/j.it.2012.02.006
34. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
35. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348:74–80. doi: 10.1126/science.aaa6204
36. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
37. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
38. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
39. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. (2014) 22:623–33. doi: 10.1038/mt.2013.262
40. Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. (2018) 6:605–16. doi: 10.1158/2326-6066.CIR-17-0314
41. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ, et al. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol. (2016) 9:56. doi: 10.1186/s13045-016-0285-y
42. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
43. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2016) 126:3036–52. doi: 10.1172/JCI83416
44. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
45. Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. (2014) 20:972–84. doi: 10.1158/1078-0432.CCR-13-0709
46. Russell L, Peng KW. The emerging role of oncolytic virus therapy against cancer. Chin Clin Oncol. (2018) 7:16. doi: 10.21037/cco.2018.04.04
47. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther. (2017) 25:2620–34. doi: 10.1016/j.ymthe.2017.08.016
48. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus, replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
49. Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. (2014) 89:926–33. doi: 10.1016/j.mayocp.2014.04.003
50. Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. (2016) 8:341ra75. doi: 10.1126/scitranslmed.aad9784
51. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Eng J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
52. McDermott D, Lebbé C, Hodi FS, Maio M, Weber JS, Wolchok JD, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev. (2014) 40:1056–64. doi: 10.1016/j.ctrv.2014.06.012
53. Garcia-Carbonero R, Salazar R, Duran I, Osman-Garcia I, Paz-Ares L, Bozada JM, et al. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J ImmunoTherapy Cancer (2017) 5:71. doi: 10.1186/s40425-017-0277-7
54. Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. (2015) 23:1630–40. doi: 10.1038/mt.2015.115
55. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. (2014) 6:226ra32. doi: 10.1126/scitranslmed.3008095
56. Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. (2015) 21:5543–51. doi: 10.1158/1078-0432.CCR-14-2009
57. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:eaam7577. doi: 10.1126/scitranslmed.aam7577
58. Bourgeois-Daigneault MC, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. (2018) 10:eaao1641. doi: 10.1126/scitranslmed.aao1641
59. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73, Table of Contents. doi: 10.1128/CMR.00046-08
60. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology (2014) 3:e955691. doi: 10.4161/21624011.2014.955691
61. Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. (2014) 4:74. doi: 10.3389/fonc.2014.00074
62. Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science (2006) 312:879–82. doi: 10.1126/science.1125676
63. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. (2016) 22:1048–54. doi: 10.1158/1078-0432.CCR-15-2667
64. Benencia F, Courreges MC, Conejo-Garcia JR, Mohamed-Hadley A, Zhang L, Buckanovich RJ, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther. (2005) 12:789–802. doi: 10.1016/j.ymthe.2005.03.026
65. Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, et al. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer (2011) 10:20. doi: 10.1186/1476-4598-10-20
66. Russell SJ, Peng KW. Oncolytic virotherapy: a contest between apples and oranges. Mol Ther. (2017) 25:1107–16. doi: 10.1016/j.ymthe.2017.03.026
67. Ajina A, Maher J. Prospects for combined use of oncolytic viruses and CAR T-cells. J ImmunoTherapy Cancer (2017) 5:90. doi: 10.1186/s40425-017-0294-6
68. Guedan S, Grases D, Rojas JJ, Gros A, Vilardell F, Vile R, et al. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. (2012) 19:1048–57. doi: 10.1038/gt.2011.184
69. Carew JF, Kooby DA, Halterman MW, Kim SH, Federoff HJ, Fong Y. A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Mol Ther. (2001) 4:250–6. doi: 10.1006/mthe.2001.0448
70. Stephenson KB, Barra NG, Davies E, Ashkar AA, Lichty BD. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. (2012) 19:238–46. doi: 10.1038/cgt.2011.81
71. Wang P, Li X, Wang J, Gao D, Li Y, Li H, et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat Commun. (2017) 8:1395. doi: 10.1038/s41467-017-01385-8
72. Lapteva N, Aldrich M, Weksberg D, Rollins L, Goltsova T, Chen SY, et al. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J Immunother. (2009) 32:145–56. doi: 10.1097/CJI.0b013e318193d31e
73. Li J, O'Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. (2011) 19:650–7. doi: 10.1038/mt.2010.312
74. Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. (2017) 8:14340. doi: 10.1038/ncomms14340
75. Kim HS, Kim-Schulze S, Kim DW, Kaufman HL. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. (2009) 69:8516–25. doi: 10.1158/0008-5472.CAN-09-2522
76. Andarini S, Kikuchi T, Nukiwa M, Pradono P, Suzuki T, Ohkouchi S, et al. Adenovirus vector-mediatedin vivogene transfer of OX40 ligand to tumor cells enhances antitumor immunity of tumor-bearing hosts. Cancer Res. (2004) 64:3281–7. doi: 10.1158/0008-5472.CAN-03-3911
77. Ullenhag G, Loskog AS. AdCD40L–crossing the valley of death? Int Rev Immunol. (2012) 31:289–98. doi: 10.3109/08830185.2012.692844
78. Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, et al. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. (2017) 77:2052–63. doi: 10.1158/0008-5472.CAN-16-1708
79. Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. (2014) 22:102–11. doi: 10.1038/mt.2013.240
80. Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. (2012) 19:988–98. doi: 10.1038/gt.2011.176
81. Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. (2014) 22:1949–59. doi: 10.1038/mt.2014.160
82. Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. (2017) 77:2952–63. doi: 10.1158/0008-5472.CAN-16-1638
83. Kleinpeter P, Fend L, Thioudellet C, Geist M, Sfrontato N, Koerper V, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death−1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology (2016) 5:e1220467. doi: 10.1080/2162402X.2016.1220467
84. Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. (2010) 18:1275–83. doi: 10.1038/mt.2010.79
85. Ganesh S, Gonzalez Edick M, Idamakanti N, Abramova M, Vanroey M, Robinson M, et al. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. (2007) 67:4399–407. doi: 10.1158/0008-5472.CAN-06-4260
86. Morgan RA. Human tumor xenografts, the good, the bad, and the ugly. Mol Ther. (2012) 20:882–4. doi: 10.1038/mt.2012.73
87. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
88. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:99573. doi: 10.1172/jci.insight.99573
89. Moon EK, Wang LS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology (2018) 7:e1395997. doi: 10.1080/2162402X.2017.1395997
90. Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, et al. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. (2017) 77:2040–51. doi: 10.1158/0008-5472.CAN-16-1577
91. Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Therapy (2017) 25:2440–51. doi: 10.1016/j.ymthe.2017.09.010
92. VanSeggelen H, Tantalo DG, Afsahi A, Hammill JA, Bramson JL. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol Ther Oncolytics (2015) 2:15014. doi: 10.1038/mto.2015.14
93. Agrahari V, Agrahari V, Mitra AK. Next generation drug delivery, circulatory cells-mediated nanotherapeutic approaches. Exp Opin Drug Deliv. (2016) 14:285–9. doi: 10.1080/17425247.2017.1254614
94. Yotnda P. Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood (2004) 104:2272–80. doi: 10.1182/blood-2003-11-3803
95. Jennings VA, Ilett EJ, Scott KJ, West EJ, Vile R, Pandha H, et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int J Cancer (2014) 134:1091–101. doi: 10.1002/ijc.28450
96. Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. (2005) 11:1073–81. doi: 10.1038/nm1297
97. Heinzerling L, Kunzi V, Oberholzer PA, Kundig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood (2005) 106:2287–94. doi: 10.1182/blood-2004-11-4558
98. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. (2016) 13:267–76. doi: 10.1038/cmi.2016.3
99. Peng KW, Myers R, Greenslade A, Mader E, Greiner S, Federspiel MJ, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. (2013) 20:255–61. doi: 10.1038/gt.2012.31
100. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12. doi: 10.1084/jem.20050732
101. Akram A, Inman RD. Immunodominance: A pivotal principle in host response to viral infections. Clin Immunol. (2012) 143:99–115. doi: 10.1016/j.clim.2012.01.015
102. Frahm N, DeCamp AC, Friedrich DP, Carter DK, Defawe OD, Kublin JG, et al. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest. (2012) 122:359–67. doi: 10.1172/JCI60202
103. Galivo F, Diaz RM, Thanarajasingam U, Jevremovic D, Wongthida P, Thompson J, et al. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum Gene Ther. (2010) 21:439–50. doi: 10.1089/hum.2009.143
104. Rodriguez-Garcia A, Svensson E, Gil-Hoyos R, Fajardo CA, Rojas LA, Arias-Badia M, et al. Insertion of exogenous epitopes in the E3-19K of oncolytic adenoviruses to enhance TAP-independent presentation and immunogenicity. Gene Ther. (2015) 22:596–601. doi: 10.1038/gt.2015.41
Keywords: chimeric antigen receptors (CAR), oncolytic viruses, solid tumors, immunotherapy, immunosuppressive tumor microenvironment, adoptive cell transfer (ACT)
Citation: Guedan S and Alemany R (2018) CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 9:2460. doi: 10.3389/fimmu.2018.02460
Received: 01 August 2018; Accepted: 04 October 2018;
Published: 23 October 2018.
Edited by:
John Maher, King's College London, United KingdomReviewed by:
Fabian Benencia, Ohio University, United StatesAmorette Barber, Longwood University, United States
Bryan Choi, Harvard Medical School, United States
Copyright © 2018 Guedan and Alemany. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia Guedan, c2d1ZWRhbkBjbGluaWMuY2F0